
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Quinone methide dimers lacking labile hydrogen atoms are surprisingly excellent radical-trapping antioxidants†
Mark A. R.
Raycroft
 a,
Jean-Philippe R.
Chauvin
a,
Matthew S.
Galliher
b,
Kevin J.
Romero
b,
Corey R. J.
Stephenson
*b and
Derek A.
Pratt
*a
a,
Jean-Philippe R.
Chauvin
a,
Matthew S.
Galliher
b,
Kevin J.
Romero
b,
Corey R. J.
Stephenson
*b and
Derek A.
Pratt
*a
aDepartment of Chemistry and Biomolecular Sciences, University of Ottawa, Ottawa, ON K1N 6N5, Canada. E-mail: dpratt@uottawa.ca
bDepartment of Chemistry, University of Michigan, Ann Arbor, MI 48109, USA. E-mail: crjsteph@umich.edu
First published on 6th May 2020
Abstract
Hydrogen atom transfer (HAT) is the mechanism by which the vast majority of radical-trapping antioxidants (RTAs), such as hindered phenols, inhibit autoxidation. As such, at least one weak O–H bond is the key structural feature which underlies the reactivity of phenolic RTAs. We recently observed that quinone methide dimers (QMDs) synthesized from hindered phenols are significantly more reactive RTAs than the phenols themselves despite lacking O–H bonds. Herein we describe our efforts to elucidate the mechanism by which they inhibit autoxidation. Four possible reaction paths were considered: (1) HAT from the C–H bonds on the carbon atoms which link the quinone methide moieties; (2) tautomerization or hydration of the quinone methide(s) in situ followed by HAT from the resultant phenolic O–H; (3) direct addition of peroxyl radicals to the quinone methide(s), and (4) homolysis of the weak central C–C bond in the QMD followed by combination of the resultant persistent phenoxyl radicals with peroxyl radicals. The insensitivity of the reactivity of the QMDs to substituent effects, solvent effects and a lack of kinetic isotope effects rule out the HAT reactions (mechanisms 1 and 2). Simple (monomeric) quinone methides, to which peroxyl radicals add, were found to be ca. 100-fold less reactive than the QMDs, ruling out mechanism 3. These facts, combined with the poor RTA activity we observe for a QMD with a stronger central C–C bond, support mechanism 4. The lack of solvent effects on the RTA activity of QMDs suggests that they may find application as additives to materials which contain H-bonding accepting moieties that can dramatically suppress the reactivity of conventional RTAs, such as phenols. This reactivity does not extend to biological membranes owing to the increased microviscosity of the phospholipid bilayer, which suppresses QMD dissociation in favour of recombination. Interestingly, the simple QMs were found to be very good RTAs in phospholipid bilayers – besting even the most potent form of vitamin E.
Introduction
The efficient inhibition of autoxidation continues to be a widely-pursued objective, given the indispensability of hydrocarbon-based products (either petroleum-derived or plant-derived) in day-to-day life and their propensity to undergo autoxidation. Nevertheless, the inhibitors which are commonly added to these products have remained essentially the same for decades.1,2 The most ubiquitous radical-trapping antioxidants (RTAs) are hindered phenols, such as butylated hydroxytoluene (BHT). BHT and related phenols react with the peroxyl radicals (ROO˙) that propagate the autoxidation chain reaction. The mechanism is well understood to involve hydrogen atom transfer (HAT) from the phenolic O–H to a chain-carrying peroxyl radical followed by rapid combination of the resultant phenoxyl radical with a second peroxyl radical (Fig. 1A).2 A vast literature has established that substituents can have a substantial impact on the rate of the initial H-atom transfer, which is characterized by the inhibition rate constant (kinh).2,3 Electron-donating groups accelerate the rate as they stabilize the electron-deficient phenoxyl radical4 and the transition state leading to its formation, whereas electron-withdrawing groups have the opposite effect. | ||
| Fig. 1 (A) Conventional reactivity of RTAs; (B) oxidation of stilbenoid phenols to form quinone methide dimers, followed by their base-mediated isomerization and Lewis acid-catalyzed Friedel–Crafts cyclization to form quadrangularin A analogues; (C) mechanisms by which QMDs may trap peroxyls. For clarity, half of the dimer is represented as R. | ||
As part of ongoing efforts to better understand the antioxidant activity of resveratrol and its oligomers,5 we found that the dimeric quinone methides which we employed as the key synthetic intermediates en route to resveratrol dimers are significantly better RTAs than their phenolic precursors or products (Fig. 1B).6 This was entirely unexpected given that the quinone methide dimers (hereafter QMDs) lack phenolic H-atoms. In our preliminary report, we suggested four possible reaction paths that would account for this observation (Fig. 1C): (1) HAT from the C–H bonds on the carbon atoms which link the quinone methide moieties; (2) tautomerization or hydration of the quinone methide(s) followed by HAT from the resultant phenolic O–H; (3) direct addition of peroxyl radicals to the quinone methide to form persistent phenoxyl radicals, and (4) homolysis of the weak central C–C bond in the QMDs followed by combination of the resultant persistent phenoxyl radicals with peroxyl radicals. Herein, we offer the details of our efforts to elucidate the mechanism.
Results
Synthetic procedures
A set of hindered resveratrol analogues (1a–j) were synthesized wherein the resorcinol ring was replaced with aryl rings bearing substituents of differing electronics (Chart 1). These phenols were dimerized via anodic oxidation in the presence of 2,6-lutidine to produce quinone methide dimers (2a–j).6 After base-mediated isomerization of one quinone methide, Lewis acid activation and Friedel–Crafts cyclization afforded a series of substituted quadrangularin (quad) A analogues (3a–e).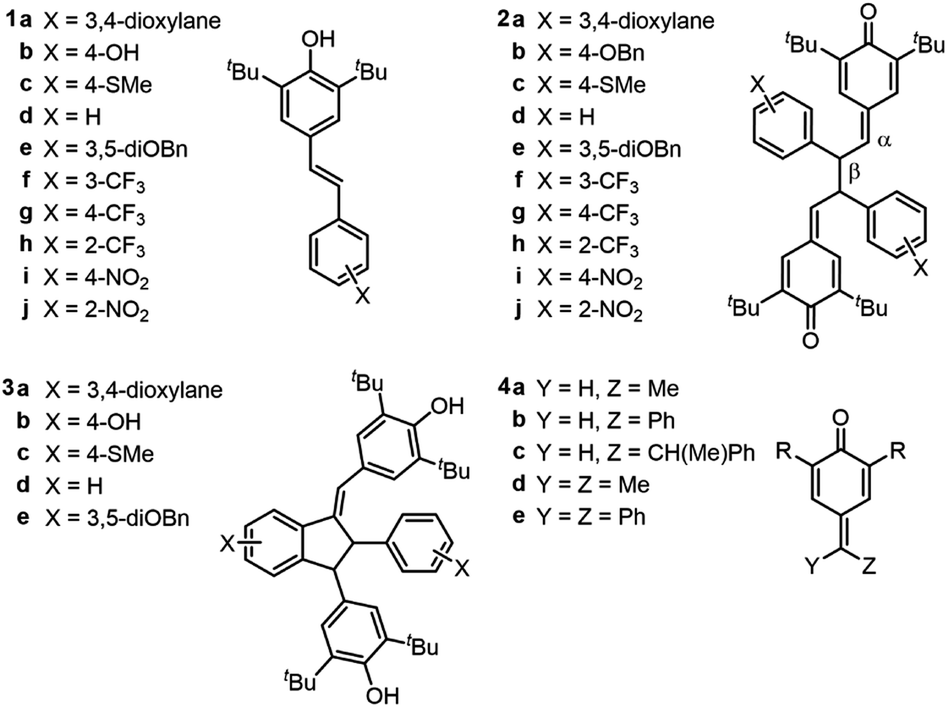 | ||
| Chart 1 Relevant stilbenoid phenols (1), quinone methide dimers (QMDs, 2), quadrangularin A analogues (3), and quinone methides (4). | ||
Inhibited autoxidations
The reactivities of the QMDs as RTAs were determined by the classical inhibited autoxidation approach7 utilizing a co-autoxidation of 1-hexadecene and PBD-BODIPY (Fig. 2A).8 Inclusion of the latter enables monitoring of reaction progress via conventional spectrophotometry by loss of its absorbance at λmax = 588 nm upon addition of peroxyl radicals to its 1-phenylbutadiene moiety. Rate constants for the reaction of peroxyl radicals with the 10 QMDs (2), their 10 precursor stilbenoid phenols (1), and 5 quadrangularin A analogues derived therefrom (3) (kinh) and corresponding reaction stoichiometries (n) were determined from the initial rate and inhibited period (tinh) of the inhibited autoxidations, respectively, according to the expressions in Fig. 2B. Representative results are shown in Fig. 2C–E for series of equivalently-substituted 1, 2 and 3, respectively. | ||
| Fig. 2 (A) PBD-BODIPY serves as the signal carrier in 1-hexadecene autoxidations; (B) determination of inhibition rate constants (kinh) and stoichiometries (n) for reactions of inhibitors with chain-carrying peroxyl radicals from initial rates and inhibition periods (tinh). Co-autoxidations of 1-hexadecene (2.9 M) and PBD-BODIPY (10 μM) initiated by AIBN (6 mM) in chlorobenzene at 37 °C (dashed black traces in (C–E)) and inhibited by 5 μM 1 (C), 1 μM 2 (D), and 5 μM 3 (E) (colour traces); (F) linear free energy relationships for 1, 2, and 3 at 37 °C. | ||
All 10 of the QMDs studied were determined to be excellent RTAs, with kinh ∼ 4 × 106 M−1 s−1 at 37 °C. The striking difference in reactivity between the QMDs and the equivalently-substituted precursor stilbenoid phenols and product quadrangularin A analogues is evident simply upon consideration of the raw inhibited autoxidation reaction progress data (compare the prominent inhibited periods in Fig. 2D to the retarded autoxidations in Fig. 2C and E, respectively), which amount to a difference in kinh of >10-fold. This is even more impressive given that the stilbenoid phenols from which the QMDs are derived are already ∼10-fold more reactive than the archetype hindered phenolic RTAs, such as BHT (kinh = 2 × 104 M−1 s−1).9 Indeed, the reactivity of the QMDs under these conditions is on par with that of α-tocopherol, the most potent form of vitamin E and among the most reactive RTAs .9
The observed inhibition periods correspond to the trapping of ∼2 radicals by the QMDs, which is similar to the precursor phenols, but roughly half that observed for the quadrangularin A analogues. The stoichiometries of the phenols are consistent with precedent (cf.Fig. 1A),9 and since the quadrangularin A analogues contain two phenolic moieties, it follows that they trap twice as many peroxyl radicals. We also carried out inhibited autoxidations at 70 °C and found that the superiority of QMDs compared to their precursor and product phenols is maintained (see ESI for the raw data†). However, pushing the temperature to 100 °C reveals a noticeable drop in reactivity. The stoichiometry exhibited by the QMDs was also found to attenuate at progressively higher temperatures; see the kinetic data summarized in Table 1.
| QMDs | 37 °C | 70 °C | 100 °C | ||||
|---|---|---|---|---|---|---|---|
| k inh (10 5 M −1 s −1 ) | n | k inh (10 5 M −1 s −1 ) | n | k inh (10 5 M −1 s −1 ) | n | ||
| 3,4-Dioxyl | 2a | 48 ± 9 | 1.8 ± 0.1 | 59 ± 10 | 1.6 ± 0.1 | 9.9 ± 0.6 | 0.44 ± 0.01 |
| 4-OBn | 2b | 48 ± 8 | 1.7 ± 0.1 | 44 ± 2 | 1.6 ± 0.1 | ND | ND |
| 4-SMe | 2c | 46 ± 6 | 1.8 ± 0.1 | 59 ± 4 | 1.4 ± 0.1 | 10 ± 1 | 0.49 ± 0.01 |
| H | 2d | 45 ± 3 | 1.8 ± 0.1 | 86 ± 6 | 1.7 ± 0.1 | 17 ± 2 | 0.52 ± 0.01 |
| 3,5-(OBn)2 | 2e | 40 ± 8 | 2.0 ± 0.1 | 120 ± 10 | 1.9 ± 0.1 | 22 ± 2 | 0.61 ± 0.05 |
| 3-CF3 | 2f | 41 ± 4 | 1.8 ± 0.1 | 110 ± 10 | 1.6 ± 0.1 | ND | ND |
| 4-CF3 | 2g | 43 ± 9 | 1.8 ± 0.1 | 98 ± 10 | 1.6 ± 0.1 | 14 ± 2 | 0.50 ± 0.02 |
| 2-CF3 | 2h | 45 ± 3 | 1.9 ± 0.1 | 62 ± 8 | 1.4 ± 0.1 | ND | ND |
| 4-NO2 | 2i | 45 ± 6 | 1.3 ± 0.1 | 40 ± 10 | 1.2 ± 0.1 | ND | ND |
| 2-NO2 | 2j | 42 ± 9 | 1.5 ± 0.1 | 47 ± 10 | 1.0 ± 0.1 | ND | ND |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||||
| Stilbenoid phenols | k inh (10 5 M −1 s −1 ) | n | k inh (10 5 M −1 s −1 ) | n | k inh (10 5 M −1 s −1 ) | n | |
| 3,4-Dioxyl | 1a | 2.1 ± 0.2 | 2.2 ± 0.1 | 6.2 ± 0.3 | 1.6 ± 0.1 | 2.9 ± 0.1 | 1.1 ± 0.1 |
| 4-OH | 1b | 3.1 ± 0.2 | 2.1 ± 0.1 | 11 ± 1 | 1.9 ± 0.1 | ND | ND |
| 4-SMe | 1c | 1.7 ± 0.1 | 2.2 ± 0.1 | 5.9 ± 0.7 | 1.5 ± 0.1 | 2.8 ± 0.3 | 1.1 ± 0.1 |
| H | 1d | 1.6 ± 0.1 | 2.0 ± 0.3 | 4.6 ± 0.4 | 1.5 ± 0.1 | 2.6 ± 0.2 | 1.0 ± 0.1 |
| 3,5-(OBn)2 | 1e | 1.4 ± 0.1 | 2.3 ± 0.1 | 19 ± 5 | 1.7 ± 0.1 | 2.5 ± 0.1 | 1.3 ± 0.1 |
| 3-CF3 | 1f | 1.1 ± 0.2 | 2.3 ± 0.3 | 3.7 ± 0.1 | 1.5 ± 0.1 | ND | ND |
| 4-CF3 | 1g | 1.3 ± 0.1 | 2.3 ± 0.1 | 3.5 ± 0.4 | 1.6 ± 0.2 | 2.3 ± 0.2 | 1.2 ± 0.1 |
| 2-CF3 | 1h | 1.0 ± 0.1 | 2.3 ± 0.3 | 3.6 ± 0.4 | 1.3 ± 0.1 | ND | ND |
| 4-NO2 | 1i | 1.0 ± 0.1 | 2.3 ± 0.1 | 2.8 ± 0.3 | 1.6 ± 0.1 | ND | ND |
| 2-NO2 | 1j | 1.0 ± 0.1 | 2.3 ± 0.1 | 2.7 ± 0.2 | 1.4 ± 0.1 | ND | ND |
|
|||||||
| Quad A analogues | k inh (10 5 M −1 s −1 ) | n | k inh (10 5 M −1 s −1 ) | n | k inh (10 5 M −1 s −1 ) | n | |
| 3,4-Dioxyl | 3a | 1.3 ± 0.1 | 4.1 ± 0.1 | 2.8 ± 0.3 | 4.2 ± 0.4 | 1.9 ± 0.2 | 3.6 ± 0.1 |
| 4-OH | 3b | 2.6 ± 0.4 | 5.5 ± 0.5 | 16 ± 1 | 6.3 ± 0.4 | ND | ND |
| 4-SMe | 3c | 0.8 ± 0.1 | 3.8 ± 0.1 | 1.2 ± 0.1 | 3.5 ± 0.3 | 1.6 ± 0.1 | 2.0 ± 0.1 |
| H | 3d | 1.0 ± 0.1 | 4.1 ± 0.3 | 2.2 ± 0.2 | 4.0 ± 0.3 | 1.7 ± 0.1 | 2.9 ± 0.1 |
| 3,5-(OBn)2 | 3e | 1.1 ± 0.1 | 3.8 ± 0.3 | 2.7 ± 0.4 | 3.5 ± 0.4 | 1.9 ± 0.1 | 2.8 ± 0.1 |
Mechanistic studies
Without a labile H-atom, the mechanism by which QMDs act as RTAs was not immediately obvious. The lack of substituent effects on the reactivity of the QMDs (ρ+ ∼ 0 at 37 °C, Fig. 2F) is in direct contrast to the sensitivity observed for the precursor phenols (ρ+ = −0.18 at 37 °C), which is consistent with known structure–reactivity relationships for phenolic RTAs, in general (vide supra). The reactivities of the quadrangularin A analogues, which are also phenols, are lower than the precursor stilbenoid phenols and essentially independent of their substitution. This difference can be rationalized based upon the reduced conjugation between the substituted phenyl rings and the reactive hindered phenolic moieties in the quadrangularin A analogues relative to the stilbenoid phenols, which are fully conjugated (see ESI for the calculated minimum energy structures†).10 Upon increasing the temperature, the trends among the two sets of phenols remain consistent (see ESI for the plots†), but those for the QMDs change – demonstrating a slight positive correlation at 70 °C which increases slightly along with the diminution of reactivity at 100 °C (Fig. 3A).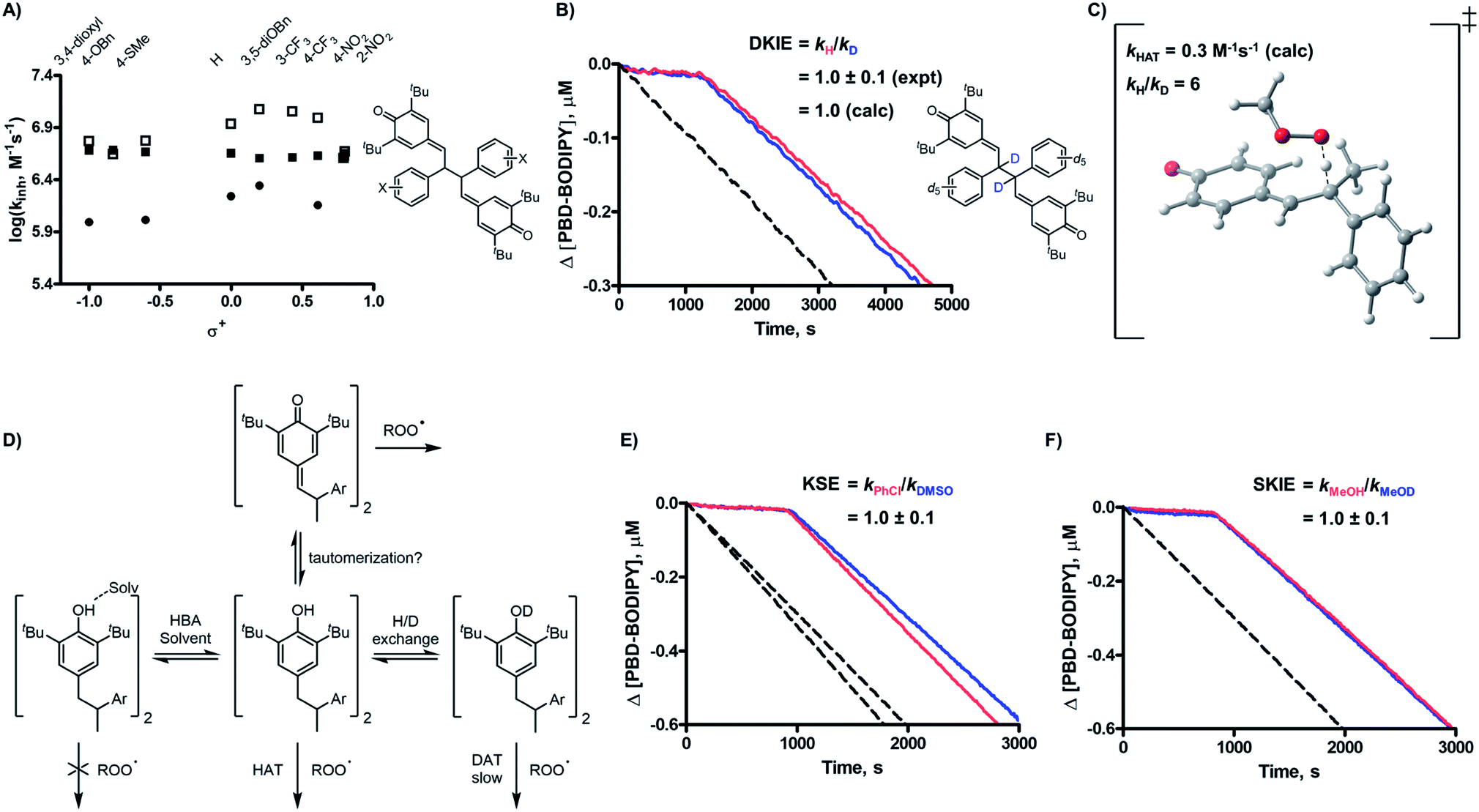 | ||
| Fig. 3 (A) Linear free energy relationships (log(kinh) vs. σ+) at 37 (■), 70 (□), and 100 (●) °C; (B) absence of deuterium kinetic isotope effect (DKIE) on the reactivity of 2d in hexadecene/PBD-BODIPY co-autoxidations in PhCl at 37 °C, [2d] and [2d-d12] = 1 μM; (C) computed TS structure for hydrogen atom transfer and corresponding computed DKIE; (D) scheme depicting the effect of H-bond-accepting solvents (Solv) and deuterated solvent on activity of phenolic RTAs; (E) absence of kinetic solvent effect (KSE) in PhCl vs. 2:1 PhCl–DMSO (dioxane/PhCl, 37 °C, [2d] = 1 μM); (F) absence of solvent kinetic isotope effect (SKIE) in 1% MeOH vs. 1% MeOD (dioxane/PhCl, 37 °C, [2d] = 1 μM). | ||
The first mechanistic possibility we considered involves H-atom transfer from the benzylic positions adjacent to the methide carbons of the QMD. Although HAT from a benzylic carbon to a peroxyl radical is generally a sluggish reaction (k ∼ 1 M−1 s−1),11 in this case HAT restores the aromaticity of one of the aryl rings, which suggests it may be significantly enhanced. To probe this mechanism, an analogue of 2d was synthesized wherein both benzylic H-atoms were replaced with D-atoms (structure shown in Fig. 3B; for synthetic details see Experimental section), and inhibited autoxidations were carried out as above.12 The results of this experiment (shown in Fig. 3B) revealed no kinetic isotope effect. Corresponding DFT computations on a model reaction (HAT from an analogous monomeric quinone methide to a methylperoxyl radical, shown in Fig. 3C) yield kHAT = 0.3 M−1 s−1 from ΔG‡ = 20.9 kcal mol−1 and kH/kD = 6.13 Clearly, this mechanism fails to account for the reactivity of the QMDs.
The second mechanistic possibility we considered involves in situ tautomerization of the QMD to a stilbenoid phenol that can react with peroxyl radicals (Fig. 1C). The simple fact that the kinh values for the QMDs exceed that of the precursor stilbenoid phenols by more than an order of magnitude strongly suggests that a phenol is not involved in the mechanism;6 however, we sought corroborating evidence from kinetic solvent effect (KSE14) and solvent kinetic isotope effect (SKIE) experiments (see Fig. 3D). Thus, we carried out co-autoxidations of dioxane and PBD-BODIPY in chlorobenzene (PhCl) and a 2:1 PhCl:DMSO mixture. Previous studies15 conducted under identical conditions have shown a predictable attenuation of kinh for HAT from X–H groups to peroxyl radicals due to a combination of the H-bond accepting capacities of the substrate (1,4-dioxane; βH2 = 0.41) and co-solvent (DMSO; βH2 = 0.78).15 The results are shown in Fig. 3E, which reveal no suppression in the reactivity of the QMD in the presence of H-bond accepting (HBA) solvents. Furthermore, QMD-inhibited co-autoxidations of dioxane and PBD-BODIPY in PhCl to which 1% v/v MeOH or 1% v/v MeOD were added were indistinguishable (Fig. 3F), suggesting no exchangeable protons are involved in the RTA activity.16
The third mechanistic possibility, addition of a peroxyl radical to one of the methide carbons of the QMD (Fig. 1C), has some precedent. Volodkin found quinone methides to be modest antioxidants (kinh ∼ 103 M−1 s−1),17 but since the rate constants they determined were much lower than those we found for the QMDs, and they investigated a limited number of structurally similar compounds, it is possible that quinone methide reactivity is enhanced when part of a QMD.18 When we calculated the TS for addition of a peroxyl radical to a model QMD (non-tert-butylated 2d, see Fig. 4A), we found a barrier of ΔG‡ = 16.0 kcal mol−1, which corresponds to kadd = 9 × 102 M−1 s−1 upon application of transition state theory – close to the reported values for simple QMs, but about 4500-fold lower than the kinh values derived from the inhibited autoxidations.19 Aside from this discrepancy between theory and experiment, the addition mechanism is fully consistent with the lack of substituent effects on the reactivity at ambient temperature and the absence of kinetic solvent effects and either O–H/O–D or C–H/C–D kinetic isotope effects.
 | ||
| Fig. 4 (A) Comparison of the transition state structures for addition to QM 4a and QMD 2d (tBu groups omitted); (B) experimental evidence for the enhanced reactivity of QMDs over QMs (hexadecene/PhCl, 37 °C, [2d] = [4a] = 5 μM); (C) kinetic solvent effect (KSE) data in PhCl vs. 2:1 PhCl–DMSO for 4a and 4a-H2O (dioxane/PhCl, 37 °C, [4a] = [4a-H2O] = 50 μM); (D) kinetic solvent effect (KSE) data in PhCl vs. 2:1 PhCl–DMSO for 5a and 5b (dioxane/PhCl, 37 °C, [5a] = [5b] = 50 μM). | ||
To provide insight on the origin of the possibility of an enhancement of quinone methide reactivity when part of a dimer, we calculated the TS for the addition of a peroxyl radical to a simple QM for comparison to the calculated data for the model QMD (Fig. 4A). To our surprise, the reaction was predicted to be much faster (ΔG‡ = 13.0 kcal mol−1, which was used to derive kadd = 1 × 105 M−1 s−1). The discrepancy between the calculated rate constant for the reaction of 4a and the reported experimental values (e.g. 1.4 × 103 and 5.8 × 102 M−1 s−1 at 60 °C in styrene and cumene, respectively)17 prompted us to synthesize it to corroborate Volodkin's results using our own approach and under the same conditions that the reactivity of the QMDs had been determined. Inhibited co-autoxidations of PBD-BODIPY in 1-hexadecene and of STY-BODIPY in cumene (both in chlorobenzene) yielded kinh = 8.2 × 104 and 7.2 × 103 M−1 s−1, respectively, at 37 °C (see Fig. 4B for a direct comparison of the reactivity of 4a and 2d, tabulated in Table 2).20 We believe that these values are slightly higher than those obtained by Volodkin due to differences in the experimental conditions employed.21 Nevertheless, these results confirm that simple QMs are much less reactive than the QMDs.22
| Y | Z | k calcadd (104 M−1 s−1) | k litinh (104 M−1 s−1) | n | k hexadeceneinh (104 M−1 s−1) | n | k cumeneinh (104 M−1 s−1) | n | |
|---|---|---|---|---|---|---|---|---|---|
| a Data from ref. 17. b Stoichiometry set to n = 2.0 to determine kinh, [4a-H2O] = 5 μM. c The data primarily reflect HAT. | |||||||||
| 4e | Me | Me | 80 | 1.6 | 0.9 | ND | ND | ||
| 4a | H | Me | 10 | 0.058 | 1.1 | 8.2 ± 0.2 | 2.0 ± 0.1 | 0.72 ± 0.03 | 2.2 ± 0.1 |
| 4a-H2O | NA | ND | 8.3 ± 0.6 | 2.0b | 0.84 ± 0.09 | 2.1 ± 0.1 | |||
| 4b | H | Ph | 0.01 | 0.018 | 0.5 | Retards | 0.34 ± 0.03 | 1.9 ± 0.1 | |
| 4f | Ph | Ph | 0.0002 | Does not inhibit | ND | ND | |||
| 5a | ND | 8.1 ± 0.2 | 2.2 ± 0.1 | 0.75 ± 0.06 | 2.4 ± 0.1 | ||||
| 5b | 0.01 | 6.4 ± 0.1 | 2.4 ± 0.1 | 1.10 ± 0.03 | 2.4 ± 0.1 | ||||
Given that the observed reactivity of 4a was highly coincident with the corresponding hindered phenols, we considered that the simple QMs undergo hydration in situ to produce hindered phenols that are the active RTAs, and that the QMDs are simply more resistant to hydration – making them appear more reactive. Thus, we prepared the hydrated form of 4a and evaluated its reactivity in a 1-hexadecene/PBD-BODIPY co-autoxidation to find nearly indistinguishable reactivity to that of 4a. To confirm that 4a and hydrated 4a react via different mechanisms, we carried out additional experiments wherein DMSO was added as a co-solvent. While the reactivity of 4a was unchanged, a significant suppression in the reactivity of the hydrated 4a was observed, consistent with the sequestration of the phenolic H-atom as part of an H-bonded complex with DMSO (Fig. 4C). We were also able to monitor the QM chromophore (λmax ∼ 300 nm) by UV-vis spectrophotometry and found that it disappeared steadily during the inhibited period of the autoxidation (Fig. 6D).
The confirmation of the lackluster – but authentic – RTA activity of the quinone methide 4a suggests that there is something special about the QMD structure which confers greater reactivity compared to monomeric quinone methides. To provide further insight on this point, we carried out autoxidations inhibited by the tautomerized QMD precursor to quadrangularin A analogue 5a and an isomer thereof (5b) – both of which contain one hindered phenol and one quinone methide moiety. Both compounds exhibited kinetics consistent with the phenolic moiety (refer to Table 2 and Fig. 4D), in agreement with the larger kinh of the phenols compared to the quinone methides.
The fourth mechanistic possibility (Fig. 1C) involves trapping of peroxyl radicals by combination with the small amount of persistent phenoxyl radical that is in equilibrium with the QMD (Fig. 5A).23,24 We had previously shown that QMD 2e does not possess the weakest C–C bond (with a BDE of 17.0 vs. 6.1 kcal mol−1),25 it is sufficiently weak that a relevant concentration of phenoxyl radical exists at ambient temperatures. Based upon the equilibrium constant we previously reported for 2e (Keq = 5.5 × 10−10 M at 37 °C),24 the phenoxyl radical concentration at the beginning of an autoxidation inhibited by 1 μM of the QMD is ∼23 nM. If establishment of this equilibrium is fast relative to propagation of the autoxidation, then kinh[QMD] = kcomb[phenoxyl], such that kcomb ∼ 43(4.0 × 106 M−1 s−1) ∼ 1.7 × 108 M−1 s−1. This value is in excellent agreement with findings of Jonsson et al., who determined rate constants ranging from 1 × 108 M−1 s−1 to 5 × 108 M−1 s−1 for the reactions of various stabilized phenoxyl radicals with peroxyl radicals derived from iPrOH in water (the effects of non-viscous solvents are generally negligible on radical combination reactions).26
 | ||
| Fig. 5 (A) Dissociation equilibrium for substituted QMDs (2) and key resonance structures of the resultant radical monomers; (B) UV-visible absorbance spectra of the phenoxyl radical derived from 2d as a function of temperature; (C) representative Van't Hoff plots used to determine ΔHC–C; (D) relationship between log(kinh) and ΔHC–C at 37 (■), 70 (□), and 100 (●) °C for meta- and para-substituted QMDs; (E) EPR spectra of the radicals present in solutions of 2d (1 mM, blue) and 6 (10 mM, red) in benzene at 20 °C; (F) comparison of diaryl (6R=Ph, blue) vs. dialkyl (6R=Me, red) QMDs (5 μM) as inhibitiors of the AIBN-initiated autoxidation of cumene in PhCl at 37 °C. | ||
Since we have no information on how substituents impact the QMD-phenoxyl radical equilibrium (Fig. 5A), we investigated it by UV-vis spectrophotometry at different temperatures (representative plot shown in Fig. 5B), enabling determination of the C–C BDEs (ΔHC–C, tabulated in the ESI†) from the resultant Van't Hoff plots (e.g.Fig. 5C).27 The BDEs of a broader series of QMDs were also computed using DFT and dispersion-corrected DFT (see ESI†), and we find that the trend in the sensitivity of ΔHC–C to the substituent is similar to that of the experimental values, with derivatives substituted with electron-donating groups possessing weaker bonds. These data imply that electron-donating substituents should enhance RTA activity, but this is not observed. In fact, the relationship between logkinh and ΔHC–C (Fig. 5D) shows the opposite trend – at elevated temperatures, at least.
Nevertheless, to probe whether this pre-equilibrium is relevant, we synthesized a QMD bearing alkyl substituents in lieu of aryl substituents (6, structure shown in Fig. 5F) in the anticipation that it would have a stronger central C–C bond. Indeed, we could find no evidence for phenoxyl radicals in the UV/vis spectra of the QMD up to 100 °C even though the predicted spectrum of the radical suggests λmax = 325 nm (see ESI†). Furthermore, we were unable to identify crossover products when equivalent amounts of 2d and 6 were heated together (see ESI for spectral data†). Indeed, only poorly resolved (low intensity) spectra were obtained when concentrated samples of 6 (10 mM) were placed in the cavity of an EPR spectrometer, in contrast to the strong signals observed from samples of 2d (1 mM, see Fig. 5E), which can be readily identified as being fully consistent with the radicals derived therefrom (see ESI†). Nonetheless, integration of the spectra observed from samples of 6 yields Keq = 4.2 × 10−13 M at 20 °C. This can be directly compared with a value of Keq = 1.7 × 10−10 M at 20 °C that we obtained with 2d, and assuming ΔS is similar for C–C bond homolysis in 2d and 6, suggests that the difference in their C–C BDEs is at least 3.5 kcal mol−1. Most importantly, this compound is a very poor RTA (kinh ∼ 2 × 103 M−1 s−1)28 compared to the other QMDs (see Fig. 5F) and is even several-fold worse than the simple QM 4a.
Discussion
We recently prepared QMD 2e as the key intermediate in the total synthesis of pallidol and quadrangularin A – natural products resulting from the oxidative dimerization of resveratrol.6 We were very surprised to find that 2e and related QMDs are potent RTAs (kinh ∼ 4 × 106 M−1 s−1 at 37 °C); ca. 10-fold more reactive than the phenols from which they are derived. In fact, the QMDs were similarly reactive to α-tocopherol – the most biologically active form of vitamin E and the standard to which all other RTAs are compared – despite lacking labile H-atoms, the key structural feature of canonical RTAs.The preparation of 2e and its subsequent stereoselective conversion to pallidol and quadrangularin A was enabled by its reversible fragmentation to two persistent phenoxyl radicals.5,24 The foregoing mechanistic investigations suggest that this equilibrium is also responsible for the impressive RTA activity of 2e and related QMDs. Although the persistent phenoxyl radicals are present in small quantities at equilibrium (ca. 1% of the QMD under the conditions which were investigated here), their combination with peroxyl radicals is quite rapid (kinh ∼ 2 × 108 M−1 s−1 at 37 °C). Thus, one molecule of QMD can be expected to trap two peroxyl radicals – consistent with the inhibition times observed in the QMD-inhibited autoxidations, from which n ∼ 2 were derived.
It is noteworthy that the resultant peroxidic adducts still contain QM moieties which can, in principle, react with additional peroxyl radicals. Indeed, simple QMs such as 4a react with peroxyl radicals with rate constants similar to those of the hindered phenols from which they are derived (e.g.4a-H2O), kinh ∼ 8 × 104 M−1 s−1 at 37 °C. Since the initial addition of a peroxyl radical forms a phenoxyl that can subsequently combine with a peroxyl radical (as do the phenoxyl radicals in equilibrium with the QMDs), the simple QMs were also found to trap 2 peroxyl radicals (see Fig. 6B). Thus, in principle, QMDs should trap a total of 6 peroxyl radicals, as is shown in the overall mechanistic scheme in Fig. 6A.
 | ||
| Fig. 6 (A) Proposed mechanism for peroxyl radical-trapping by aryl-containing QMDs; (B) mechanism by which QMs trap peroxyls and the dependence of stoichiometry on [QM]; (C) spectral changes associated with inhibitors 2d (blue and red) and (D) 4a (green) in the co-autoxidations of 1-hexadecene (2.9 M) and PBD-BODIPY (black traces); (E) co-autoxidations of cumene (3.6 M) and STY-BODIPY (10 μM) initiated by AIBN (6 mM) in chlorobenzene at 37 °C inhibited by 2d (1–50 μM) with an inset showing the second phase of inhibition translated to the uninhibited trace (broken line). | ||
The inhibited autoxidation data for the isomerized QMD structures 5a and 5b (Fig. 4D) provide some insight to why the QMDs only appear to trap 2 peroxyl radicals in our experiments. Each of these compounds, which feature one phenolic moiety and one QM moiety react as if only the phenol were present with n ∼ 2 and kinh = 8.1 × 104 M−1 s−1 and 6.4 × 104 M−1 s−1, respectively, which are subject to the same substantial KSEs. The lack of retardation of oxidation due to the QM moiety implies that it is significantly less reactive to peroxyl radicals than a simple monomeric QM. Indeed, when we monitored the QMD-inhibited autoxidations by looking at the whole spectrum rather than simply λmax of PBD-BODIPY at 588 nm, we noticed consumption of the characteristic absorbance of the QM moiety at ∼300 nm only after the initial inhibited period, which coincided with consumption of the characteristic absorbance of the QMD moiety at ∼330 nm (Fig. 6C). This slower phase is consistent with the loss at ∼300 nm in autoxidations of the simple QM 4a (Fig. 6D). In an attempt to quantify the slower reactivity of the more hindered QM moieties in the peroxyl adducts derived from the QMD (or 5a or 5b), we carried out inhibited autoxidations of cumene in PhCl with much higher concentrations of QMD (up to 50 μM). Upon doing so, we could indeed see a retardation of autoxidation beyond the initial inhibited period (Fig. 6E), which yielded kinh ∼ 4 × 103 M−1 s−1 – on par with a kinh for a simple QM measured in cumene/PhCl at 37 °C. Ultimately, the most compelling result in support of the dissociation/combination mechanism is the observation that QMD 6, which has a much stronger central C–C bond than 2, is >1000-fold less reactive as an RTA than 2. The reactivity of 6 is even lower than the simple QM 4a, implying some steric effects on the addition of peroxyl radicals. This is consistent with computational predictions22 as well as the slower reactivity of the QM moieties in the tautomerized QMD precursor to quad A (5a) and isomer thereof (5b) relative to 4a.
The one piece of mechanistic data that is not, at first glance, fully consistent with the dissociation/combination mechanism is the erosion in RTA activity with increasing temperature. Increasing the temperature shifts the QMD/radical equilibrium toward phenoxyl radical formation, which should improve peroxyl radical-trapping, but instead only a slight increase is observed on going from 37 °C to 70 °C and then a noticeable drop on going from 70 °C to 100 °C. We believe this simply reflects the reversible nature of the phenoxyl-peroxyl radical combination. It is well known that the activities of hindered phenolic RTAs diminish at elevated temperatures and that n values erode from 2 to 1 due to the reversible nature of the phenoxyl-peroxyl radical combination step that follows the initial H-atom transfer from phenol to peroxyl radical.29 The same phenoxyl-peroxyl combination step features here. The fact that the erosion in reactivity is not as severe for the more electron-poor QMDs (cf.Fig. 5D) presumably reflects a stronger C–O bond in the phenoxyl-peroxyl adduct.
The step-wise homolytic substitution mechanism of the QMDs is reminiscent of that by which the dimeric form of the Ciba (now BASF) antioxidant Irganox HP-136 reacts. The HP-136 dimer was determined to have largely solvent-independent RTA kinetics,30 with kinh = 4.3 × 105 M−1 s−1 and n = 0.8 from styrene autoxidations in chlorobenzene at 30 °C.31 The QMDs are therefore roughly 10-fold more reactive than the HP-136 dimer, presumably due to their weaker central C–C bond: 14–17 kcal mol−1vs. 23 kcal mol−1 for (HP-136)2. Thus, the QMDs add to a short list of molecules which lack labile H-atoms but are nevertheless reactive RTAs. Two of the few other examples of which we are aware are tetrasulfides32 and trisulfide-1-oxides.33 These compounds undergo concerted (bimolecular) homolytic substitution by peroxyl radicals to produce stable and persistent perthiyl radicals that do not propagate the autoxidation, but combine to give tetrasulfides. Although these substitution reactions are also insensitive to solvent effects, they are much slower at ambient temperatures (kinh ∼ 103 and 104 M−1 s−1 for the tetrasulfides and trisulfide-1-oxides, respectively, compared to kinh ∼ 106 M−1 s−1 for the QMDs at 37 °C), limiting their applications to elevated temperatures.
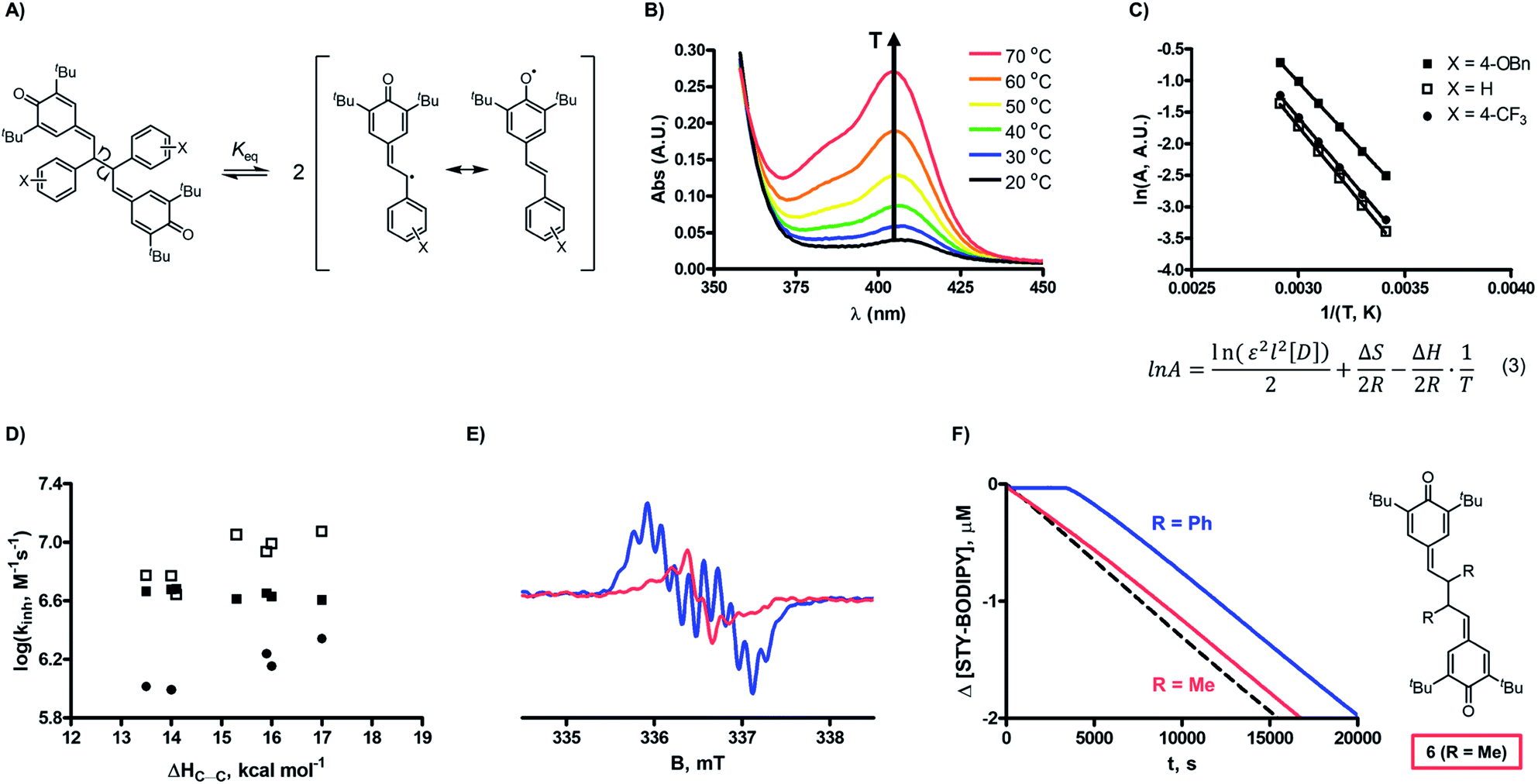
The lack of solvent effects and high reactivity of the QMDs at ambient temperatures prompted us to carry out one additional set of experiments (see Fig. 7). It has recently come to light that the poor performance of phenolic antioxidants as inhibitors of lipid peroxidation in biological membranes is the result of strong H-bonding interactions between phenols and the phosphate diester moiety of the membrane phospholipids (Fig. 8A).34 Indeed, the reactivity of α-tocopherol (α-TOH), Nature's premier lipophilic RTA, toward peroxyl radicals is suppressed almost 1000-fold on going from chlorobenzene to a phospholipid bilayer (kinh ∼ 106vs. 103 M−1 s−1). As such, we wondered if QMDs would be effective RTAs in this context, and evaluated their ability to suppress (phospho)lipid peroxidation using our recently described FENIX assay.34a Disappointingly, we found that they were quite poor inhibitors. Alas, the large microviscosity of the phospholipid bilayer35 presumably suppresses cage escape and combination of the QMD-derived phenoxyl radicals with lipid peroxyl radicals (Fig. 8B). This result further supports a stepwise substitution by peroxyl radicals on the QMDs in contrast to the concerted substitution favoured for the tetrasulfides and trisulfide-1-oxides, which do not depend on the viscosity of the medium. Alongside the foregoing experiment, we evaluated the RTA activity of a simple QM (4a) as a control. In fact, the QM turned out to be a much better RTA under these conditions, even besting α-TOH (see Fig. 7).36 The apparent kinh value measured for 4a in the phospholipid bilayer is scarcely different from that obtained in chlorobenzene – reinforcing that addition of peroxyl radicals is operative and medium independent (Fig. 8C). As far as we are aware, this is the first instance of the inhibition of (phospho)lipid peroxidation by a non-canonical (HAT)-like RTA, and is compelling indirect evidence underlining the deleterious role of H-bonding on the reactivity of canonical RTAs.
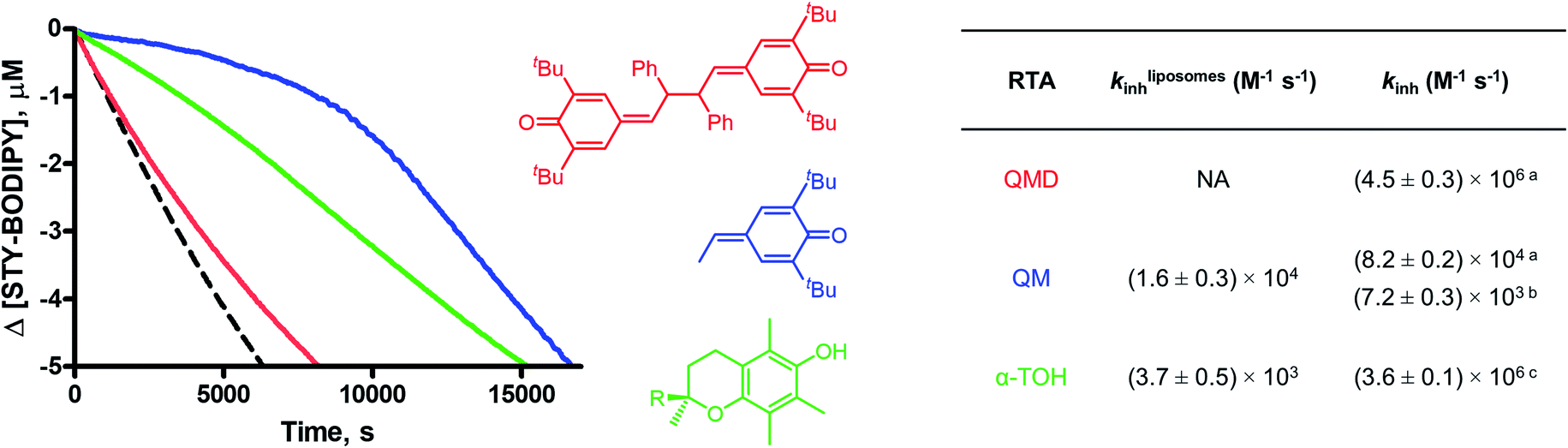 | ||
| Fig. 7 Co-autoxidations of unilammelar (100 nm) liposomes of egg phosphatidylcholine (1 mM) and STY-BODIPY (10 μM) initiated by DTUN (0.2 mM) in PBS buffer (pH = 7.4) at 37 °C (dashed black trace) and inhibited by 10 μM 2d (red), 4a (blue), and α-tocopherol (R = C16H33, green). The table compares kinh values obtained from co-autoxidations for 2d (QMD), 4a (QM), and α-tocopherol (α-TOH, R = C16H33) in this system with those determined in hexadecene/PhCl (a), cumene/PhCl (b), and/or styrene/PhCl (c) at 37 °C. | ||
 | ||
| Fig. 8 (A) Hydrogen-bonding of the labile H-atom of phenols such as α-TOH to the phosphate diester of phospholipids suppresses their reactivity to HAT; (B) the high microviscosity of the lipid environment promotes in-cage recombination of QMD-derived radicals, thereby suppressing the peroxyl radical substitution pathway. Addition of peroxyls to the QM moieties of the QMD is slowed by steric congestion imposed by the QMD scaffold; (C) simple QMs are more sterically accessible and highly effective RTAs. | ||
Conclusions
This work establishes the mechanistic basis which underpins the surprisingly potent RTA activity of quinone methide dimers (QMDs) of the general formula 2. These dimers, which are formed by the oxidative coupling of hindered stilbenoid phenols, have previously served as synthetic precursors to resveratrol oligomer natural products. Despite lacking any conventional RTA motif (e.g. a phenolic O–H bond), these QMDs are not only reactive in that capacity, but best both the phenolic compounds from which they are derived and to which they are converted. Our work corroborates previous data which shows that monomeric quinone methides are modest RTAs at best, and shows that the diaryl QMD scaffold is privileged in that it opens up a new mode of reactivity via its reversible fragmentation. The resultant persistent phenoxyl radicals can combine with peroxyl radicals leaving the second quinone methide moiety to react with additional peroxyl radicals. This mode of reactivity is not influenced by solvent, in contrast to traditional phenolic RTAs, whose H-atom transfer reactions are slowed by H-bonding interactions. This fact suggests that QMDs may be useful RTAs for applications in non-viscous H-bonding media. Experiments in phospholipid bilayers reveal that the high microviscosity of the lipid phase suppresses the reactivity of QMDs, but that the lacklustre reactivity of simple QMs, which undergo addition, is fully consistent with what is observed in solution – making it the only class of RTA reported to date whose solution phase reactivity directly translates to lipid bilayers.Experimental section
General
All chemicals and solvents obtained from commercial suppliers were used as received unless indicated otherwise. 1-Hexadecene and cumene were purified and stored as previously described.8,32 The synthesis, purification, and characterization of substituted stilbenes (1), QMDs (2), and quad A analogues (3) as well as hybrid phenol–QMs (5 and 6) used in this study are described in previous reports.5,6 The synthesis, purification, and characterization of 4a,374a-H2O,384b39 and S440 were carried out according to literature reports.
:1 dr). 1H NMR (500 MHz, chloroform-d) δ 7.22 (d, J = 2.4 Hz, 1H, major diastereomer), 7.19 (d, J = 2.4 Hz, 1H, minor diastereomer), 6.84 (d, J = 2.4 Hz, 1H, minor diastereomer), 6.80 (d, J = 2.4 Hz, 1H, diastereomer), 6.10 (d, J = 10.1 Hz, 1H, major + minor diastereomers overlapped), 3.02 (ddd, J = 9.7, 8.0, 5.2 Hz, 1H, major diastereomer), 2.98–2.91 (m, 1H, minor diastereomer), 1.31 (s, 9H), 1.27 (s, 9H), 1.19 (d, J = 6.2 Hz, 3H). 13C NMR (176 MHz, chloroform-d) δ 152.22, 152.21, 145.37, 145.10, 137.79, 137.16, 136.19, 135.45, 134.56, 132.58, 127.44, 126.93, 126.40, 124.26, 124.01, 122.45, 116.78, 115.01, 113.50, 53.72, 50.45, 34.50, 34.45, 30.48, 30.45. HRMS (ESI) m/z calculated for C34H51O2 ([M + H]+) 491.3884, found 491.3879.
:2.5:2.5 to 90:5:5 hexanes/EtOAc/DCM to obtain the final product as a yellow solid (19 mg, 76%). 1H-NMR (600 MHz; CDCl3): δ 7.15 (d, J = 2.4 Hz, 2H), 7.12 (d, J = 2.4 Hz, 2H), β-H's of quinone methides: 6.81 (minor diastereomer, d, J = 2.4 Hz, 2H), 6.69 (major diastereomer, d, J = 2.4 Hz, 2H), δ-H's of quinone methides: 6.49 (minor diastereomer, s, 2H), 6.38 (major diasteromer, s, 2H), tBu signals: 1.25 (s, 18H), 1.24 (s, 18H), 1.23 (s, 18H), 1.21 (s, 18H). 13C NMR (151 MHz; CDCl3): δ 186.6, 186.5, 149.1, 148.9, 147.6, 147.3, 145.4, 144.9, 140.9, 140.3, 134.7, 132.7, 132.0, 126.1, 125.9, 35.5, 35.5, 35.0, 34.9, 29.6, 29.6, 29.5. HRMS (ESI, [M + Na]+): m/z calcd for C44H42O2D12Na 649.4775, found 649.4774.
General procedures for inhibited Co-autoxidations
The inhibited co-autoxidations were carried out following our reported methods.8,34a Autoxidations of 1-hexadecene (2.9 M) and PBD-BODIPY (10 μM) in chlorobenzene were initiated by AIBN (6 mM) at 37 °C, tBuOOtBu (87 mM) at 70 °C, or dicumyl peroxide (1 mM) at 100 °C. A 3.5 mL quartz cuvette was charged with PhCl (0.44 mL) and 1-hexadecene (2.00 mL). The cuvette was preheated to the desired temperature in a thermostated sample holder of a UV-vis spectrophotometer and allowed to equilibrate for approximately 15 min. To the cuvette was added PBD-BODIPY (12.5 μL of a 2.00 mM stock solution in 1,2,4-trichlorobenzene) and initiator (50 μL of a 300 mM stock solution of AIBN in chlorobenzene, 40 μL of neat tBuOOtBu, or 50 μL of a 50 mM stock solution of dicumyl peroxide in chlorobenzene). The solution was thoroughly mixed prior to monitoring the uninhibited co-autoxidation via the disappearance of PBD-BODIPY at 588 nm (37 °C), 587 nm (70 °C), or 586 nm (100 °C) for 5–10 min to ensure the reaction was proceeding at a constant rate. Finally, the antioxidant under investigation was added (5.0–10.0 μL of a 0.25 or 2.5 mM solution in chlorobenzene), the solution was mixed thoroughly, and the absorbance readings were resumed. The resulting Abs vs. time data were processed as previously reported.8 The rate of initiation (Ri = 1.30 × 10−9 M s−1 (37 °C), 1.26 × 10−9 M s−1 (70 °C), 6.58 × 10−9 M s−1 (100 °C)) and second order rate constant for propagation of the dye (kPBD-BODIPY = 3792 M−1 s−1 (37 °C), 7633 M−1 s−1 (70 °C), 8283 M−1 s−1 (100 °C)) necessary to compute stoichiometric data (n) and inhibition rate constants (kinh) were determined using 2,2,5,7,8-pentamethylchromanol (PMC) as a standard, which has an established stoichiometry of 2.42 Similar experiments were conducted at 37 °C employing cumene (3.6 M) and STY-BODIPY (10 μM) initiated by AIBN (6 mM) in chlorobenzene. Reaction progress was monitored at 571 nm. The rate of initiation, also determined using PMC, was measured to be Ri = 2.28 × 10−9 M s−1 and the second order rate constant for propagation had been determined previously (kSTY-BODIPY = 141 M−1 s−1) (37 °C). Inhibited autoxidation experiments involving liposomes were conducted at 37 °C employing egg-phosphatidylcholine liposomes (1 mM), STY-BODIPY (10 μM), initiated by DTUN34a (0.2 mM) in PBS buffer (10 mM). Reaction progress was monitored at 565 nm. The rate of initiation, determined using PMC, was measured to be Ri = 2.29 × 10−9 M s−1 and the second order rate constant for propagation had been determined previously (kSTY-BODIPY = 894 M−1 s−1) (37 °C).000 M−1 cm−1 in PhCl, 118200 M−1 cm−1 in 2:1 PhCl/DMSO). The rate of initiation in PhCl (Ri = 2.40 × 10−9 M s−1) and in 2:1 PhCl/DMSO were previously standardized by PMC. The second order rate constants for propagation are kPBD-BODIPY = 5310 M−1 s−1 (PhCl) and 5900 M−1 s−1 (2:1 PhCl/DMSO).15
EPR spectra
Electron paramagnetic resonance (EPR) spectra were recorded using a Bruker EMXplus (X-band) spectrometer equipped with an ER 4119HS cavity at 20 °C. The samples were 0.1–10 mM in benzene and degassed (3 cycles of freeze–pump–thaw) and placed under an atmosphere of N2 prior to acquisition. The radical concentration was determined using the quantitative EPR package of the Bruker Xenon software. Spectral simulations were performed using EasySpin.43Calculations
Calculations were conducted using the B3LYP method44 (CBSB7 basis set) and CBS-QB3 (complete basis set) method45 as implemented in the Gaussian 16 suite of programs.46 Rate constants were calculated using transition state theory at 37 °C. Dispersion corrections were applied using Grimme's D3 approach.47 Hyperfine coupling constants were predicted by spin density distributions at the B3LYP/TZVP level of theory.48Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the Natural Sciences and Engineering Research Council of Canada and the Canada Foundation for Innovation and through generous access to the computational resources of the Centre for Advanced Computing (http://cac.queensu.ca). J. P. C. acknowledges the support of the Ontario Graduate Scholarships Program. We also acknowledge the financial support for this research from the NIH NIGMS (R01-GM121656), the Camille Dreyfus Teacher–Scholar Award Program, and the University of Michigan. This material is based upon work supported by the National Science Foundation Graduate Research Fellowship under Grant No. DGE 1256260 (for K. J. R.). Finally, we would like to acknowledge Luke A. Farmer for his technical assistance in acquiring EPR spectra.Notes and references
- K. U. Ingold, Chem. Rev., 1961, 61, 563 CrossRef CAS.
- K. U. Ingold and D. A. Pratt, Chem. Rev., 2014, 114, 9022 CrossRef CAS PubMed.
- G. W. Burton and K. U. Ingold, Acc. Chem. Res., 1986, 19, 194 CrossRef CAS.
- D. A. Pratt, G. A. DiLabio, P. Mulder and K. U. Ingold, Acc. Chem. Res., 2004, 37, 334 CrossRef CAS.
- B. S. Matsuura, M. H. Keylor, B. Li, Y. Lin, S. Allison, D. A. Pratt and C. R. J. Stephenson, Angew. Chem., Int. Ed., 2015, 54, 3754 CrossRef CAS.
- K. J. Romero, M. S. Galliher, M. A. R. Raycroft, J.-P. R Chauvin, I. Bosque, D. A. Pratt and C. R. J. Stephenson, Angew. Chem., Int. Ed., 2018, 57, 17125 CrossRef CAS.
- J. A. Howard and K. U. Ingold, Can. J. Chem., 1962, 40, 1851 CrossRef CAS.
- E. A. Haidasz, A. T. M. Van Kessel and D. A. Pratt, J. Org. Chem., 2016, 81, 737 CrossRef CAS PubMed.
- G. W. Burton, T. Doba, E. Gabe, L. Hughes, F. L. Lee, L. Prasad and K. U. Ingold, J. Am. Chem. Soc., 1985, 107, 7053 CrossRef CAS.
- DFT optimization of 3d reveals that the hindered phenol and resorcinol rings are not coplanar, with a dihedral angle of 32°. See the ESI† for a comparison to 1d..
- J. A. Howard and K. U. Ingold, Can. J. Chem., 1967, 45, 973 Search PubMed.
- The presence of perdeuterated phenyl rings on 2d-d12 is not expected to have an effect on kinh at 37 °C..
- Employing CBS-QB3, ΔG‡ = 19.1 kcal mol−1, which corresponds to k = 6 M−1 s−1 and kH/kD = 6..
- G. Litwinienko and K. U. Ingold, Acc. Chem. Res., 2007, 40, 222 CrossRef CAS.
- L. A. Farmer, E. A. Haidasz, M. Griesser and D. A. Pratt, J. Org. Chem., 2017, 82, 10523 CrossRef CAS PubMed.
- We have also used 1H NMR to explore the effect of H2O and MeOH on 2e in benzene-d6. Incubated at 37 °C, we observed neither tautomerization of, nor addition to, 2e on the timescale of the inhibited co-autoxidations..
- N. V. Zolotova, F. A. Galieva, M. B. Tokareva, E. T. Denisov and A. A. Volodkin, Kinet. Katal., 1979, 20, 48 CAS.
- One QMD was studied as part of ref. 17 (structure 6 where R = C(O)OCH3); however, its reactivity (4 × 104 M−1 s−1 in cumene at 60 °C) was reported to be on par with hindered phenols. Interestingly, the observed reactivity is intermediate to that which we have determined for diaryl QMDs (106 M−1 s−1) and a dialkyl QMD (103 M−1 s−1, vide infra) and may reflect an intermediate central C–C BDE. Consistent with our results, the authors report n = 1.8 for this dicarboxymethyl QMD..
- We also found a higher energy TS structure involving the QMD that is analogous to the low energy TS involving the QM shown in Fig. 4A, but it is higher in energy by 1.7 kcal mol−1. We also employed CBS-QB3 in the case of a truncated QMD (4c, R = H) and found ΔG‡ = 10.8 kcal mol−1, which corresponds to kadd = 4 × 106 M−1 s−1 – in startlingly good agreement with rate constants derived from the inhibited co-autoxidation experiments..
- The ∼11-fold reduction in kinh in going from 1-hexadecene to cumene is consistent with computed results indicating kadd of t-butylperoxyl radicals to an analogue of 4a is ∼37-fold slower than addition of i-propyl peroxyl radicals..
- The kinetic data acquired by Volodkin et al. was collected at much higher concentrations of QM (>100 μM, which is at least 100-fold larger than the concentrations used here). We have found that higher [QM] can give rise to smaller measured kinh and n values (e.g., increasing [4a] by 10-fold led to a ∼2-fold reduction in kinh and n (hexadecane/PhCl, 37 °C); see ESI† for the raw data and Fig. S55† for the dependence of n on [QM])..
- To understand the basis of the discrepancy in the computed and experimental values we computed transitions states for higher-substituted peroxyl radicals to find a progressive increase in the barrier and corresponding decrease in the predicted rate constant from 1 × 105 M−1 s−1 to 1 × 103 M−1 s−1 on going from methylperoxyl to t-butylperoxyl radicals. Moreover, incorporation of the effects of bulk solvation increased the barrier and decreased the predicted rate constant further, exhibiting a range of 1 × 104 M−1 s−1 to 1 × 102 M−1 s−1, in good agreement with the experimental results. See the ESI† for further details..
- H. D. Becker, J. Org. Chem., 1969, 34, 1211 CrossRef CAS.
- M. H. Keylor, B. S. Matsuura, M. Griesser, J.-P. R. Chauvin, R. A. Harding, M. S. Kirillova, X. Zhu, O. J. Fischer, D. A. Pratt and C. R. J. Stephenson, Science, 2016, 354, 1260 CrossRef CAS PubMed.
- J. M. Wittman, R. Hayoun, W. Kaminsky, M. K. Coggins and J. M. Mayer, J. Am. Chem. Soc., 2013, 135, 12956 CrossRef CAS PubMed.
- J. Jonsson, J. Lind, T. Reitberger, T. E. Eriksen and G. Merenyi, J. Phys. Chem., 1993, 97, 8229 CrossRef.
- M. Frenette, C. Aliaga, E. Font-Sanchis and J. C. Scaiano, Org. Lett., 2004, 6, 2579 CrossRef CAS PubMed.
- Triplicate measurements of kinh and n are reported in Table S3 of the ESI.† The stoichiometry of this QMD is ∼2 which may be attributable to a 5-exo-trig cyclization of the phenoxyl formed subsequent to the first peroxyl addition followed by combination with a second peroxyl. A similar cyclization has been reported previously: A. P. Krysin, A. M. Genaev, L. M. Pokrovskii and M. M. Shakirov, Russ. J. Org. Chem., 2014, 50, 367 CrossRef CAS.
- (a) W. G. Lloyd and R. G. Zimmerman, Ind. Eng. Chem. Prod. Res. Dev., 1965, 4, 180 CrossRef CAS; (b) E. C. Horswill, J. A. Howard and K. U. Ingold, Can. J. Chem., 1966, 44, 985 CrossRef CAS; (c) J. Igarashi, R. K. Jensen, J. Lusztyk, S. Korcek and K. U. Ingold, J. Am. Chem. Soc., 1992, 114, 7727 CrossRef CAS.
- V. Filippenko, M. Frenette and J. C. Scaiano, Org. Lett., 2009, 11, 3634 CrossRef CAS PubMed.
- M. Frenette, P. D. MacLean, R. C. Barclay and J. C. Scaiano, J. Am. Chem. Soc., 2006, 128, 16432 CrossRef CAS PubMed.
- J.-P. R. Chauvin, M. Griesser and D. A. Pratt, Chem. Sci., 2019, 10, 4999 RSC.
- J.-P. R. Chauvin, E. A. Haidasz, M. Griesser and D. A. Pratt, Chem. Sci., 2016, 7, 6347 RSC.
- (a) R. Shah, L. A. Farmer, O. Zilka, A. T. M. Van Kessel and D. A. Pratt, Cell Chem. Biol., 2019, 26, 1594 CrossRef CAS PubMed; (b) M. Conrad and D. A. Pratt, Nat. Chem. Biol., 2019, 15, 1137 CrossRef CAS PubMed.
- U. Cogan, M. Shinitzky, G. Weber and T. Nishida, Biochemistry, 1973, 12, 521 CrossRef CAS PubMed.
- Data for α-TOH in liposomes/PBS buffer comes from J.-F. Poon, O. Zilka, D. A. Pratt and that for α-TOH in styrene/PhCl comes from O. Zilka, R. Shah, B. Li, J. P. Friedmann Angeli, M. Griesser, M. Conrad and D. A. Pratt, ACS Cent. Sci., 2017, 3, 232 CrossRef CAS PubMed.
- (a) C. D. Cook and B. E. Norcross, J. Am. Chem. Soc., 1956, 78, 3797 CrossRef CAS; (b) C. E. Cook and B. E. Norcross, J. Am. Chem. Soc., 1958, 81, 1176 CrossRef; (c) L. K. Dyall and S. Winstein, J. Am. Chem. Soc., 1972, 94, 2196 CrossRef CAS; (d) J. A. Richards and D. H. Evans, J. Electroanal. Chem., 1977, 81, 171 CrossRef CAS.
- (a) A. P. Krysin and A. N. Fedorenko, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1989, 38, 1533 ( Izv. Akad. Nauk SSSR, Ser. Khim. , 1989 , 1671 ) CrossRef; (b) T. Matsuura and Y. Kitaura, Tetrahedron, 1969, 25, 4501 CrossRef CAS; (c) A. A. Volodkin, et al. , Bull. Acad. Sci. USSR, Div. Chem. Sci., 1969, 1467 ( Izv. Akad. Nauk SSSR, Ser. Khim. , 1969 , 1583 ) CrossRef.
- (a) D. Richter, N. Hampel, T. Singer, A. R. Ofial and H. Mayr, Eur. J. Org. Chem., 2009, 3203 CrossRef CAS; (b) V. Reddy and R. V. Anand, Org. Lett., 2015, 17, 3390 CrossRef CAS.
- A. Nishinaga, H. Iwasaki, T. Shimizu, Y. Toyoda and T. Matsuura, J. Org. Chem., 1986, 51, 2257 CrossRef CAS.
- Y. Jaiswal, Y. Kumar and A. Kumar, J. Org. Chem., 2018, 83, 1223 CrossRef CAS PubMed.
- G. W. Burton, T. Doba, E. J. Gabe, L. Hughes, F. L. Lee, L. Prasad and K. U. Ingold, J. Am. Chem. Soc., 1985, 107, 7053 CrossRef CAS.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- J. A. Montgomery Jr, M. J. Frisch, J. W. Ochterski and G. A. Petersson, J. Chem. Phys., 1999, 110, 2822 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 CrossRef CAS PubMed.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Additional kinetic and thermodynamic data and analyses, synthesis and characterization data, computed optimized geometries, energies, and Cartesian coordinates. See DOI: 10.1039/d0sc02020f |
| This journal is © The Royal Society of Chemistry 2020 |