
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupramolecular and biomacromolecular enhancement of metal-free magnetic resonance imaging contrast agents†
Hamilton
Lee
 a,
Arezoo
Shahrivarkevishahi
a,
Jenica L.
Lumata
a,
Michael A.
Luzuriaga
a,
Laurel M.
Hagge
a,
Candace E.
Benjamin
a,
Olivia R.
Brohlin
a,
Christopher R.
Parish
b,
Hamid R.
Firouzi
a,
Steven O.
Nielsen
a,
Lloyd L.
Lumata
b and
Jeremiah J.
Gassensmith
*ac
a,
Arezoo
Shahrivarkevishahi
a,
Jenica L.
Lumata
a,
Michael A.
Luzuriaga
a,
Laurel M.
Hagge
a,
Candace E.
Benjamin
a,
Olivia R.
Brohlin
a,
Christopher R.
Parish
b,
Hamid R.
Firouzi
a,
Steven O.
Nielsen
a,
Lloyd L.
Lumata
b and
Jeremiah J.
Gassensmith
*ac
aDepartment of Chemistry and Biochemistry, The University of Texas at Dallas, 800 West Campbell Rd., Richardson, TX 75080, USA. E-mail: gassensmith@utdallas.edu; Web: www.twitter.com/gassensmith
bDepartment of Physics, The University of Texas at Dallas, 800 West Campbell Rd., Richardson, TX 75080, USA
cDepartment of Bioengineering, The University of Texas at Dallas, 800 West Campbell Rd., Richardson, TX 75080, USA
First published on 5th February 2020
Abstract
Many contrast agents for magnetic resonance imaging are based on gadolinium, however side effects limit their use in some patients. Organic radical contrast agents (ORCAs) are potential alternatives, but are reduced rapidly in physiological conditions and have low relaxivities as single molecule contrast agents. Herein, we use a supramolecular strategy where cucurbit[8]uril binds with nanomolar affinities to ORCAs and protects them against biological reductants to create a stable radical in vivo. We further overcame the weak contrast by conjugating this complex on the surface of a self-assembled biomacromolecule derived from the tobacco mosaic virus.
Introduction
Of the many spatially resolved biomedical imaging techniques available, magnetic resonance imaging (MRI) is of particular importance in modern medicine due to its non-invasive nature, potential for high spatial resolution, tissue penetration, and lack of ionizing radiation.1 MRI relies on detecting the energy released over time by water protons returning to magnetic equilibrium after a radio frequency pulse has been applied. This relaxation rate—or relaxivity—is highly dependent on the chemical environment of the water. Since water intrinsically possesses low sensitivity to magnetic fields, contrast agents are used to increase the contrast between different features in the final image2–4 with the majority of modern MRI contrast agents based on Gd3+ complexes.5–7 Although Gd has performed8–10 remarkably in clinical settings, concerns about its toxicity,11–15 especially for patients with impaired renal functionality, have catalysed efforts to design alternatives to Gd and other metal-based MRI contrast agents. Several types of metal-free contrast agents have been investigated16–23 with organic radical contrast agents (ORCAs) based on paramagnetic aminoxyl moieties showing significant promise. Aminoxyl ORCAs are distinguished by their compatibility with existing MRI techniques, enabling facile implementation in current clinical settings, and have low cytotoxicity and high biodegradability, reducing the potential for side effects.24,25 However, two major issues prevent aminoxyl-based ORCAs from replacing traditional contrast agents based on Gd: (i) their single unpaired electron provides weaker contrast compared to the seven unpaired electrons of Gd and (ii) they are reduced rapidly to MRI-silent hydroxylamines in physiological conditions by compounds including ascorbate, saccharides, and cysteine-rich proteins.24Contrast issues in ORCAs have improved greatly in recent years by attaching them to polymeric or biomacromolecular nanoparticle systems26–44 that both create high local concentrations of aminoxyl moieties and decrease the diffusional and rotational motion of the attached ORCAs. Because rotational correlation time is inversely proportional to relaxivity, attaching contrast agents onto large macromolecules will—in general—improve their performance. On the other hand, sensitivity to reduction in vivo has been more difficult to address as the reduction–oxidation (REDOX) potentials of aminoxyl radicals are such that they are quickly reduced in the high physiological concentrations of ascorbate. Strategies to overcome this have primarily focused on mitigating reduction by installing sterically hindered moieties around the aminoxyl radical and incorporating aminoxyl-containing molecules into macromolecular systems that shield the radical.45 Problematically, creating more steric bulk may also preclude water from interacting with the free electron, which is detrimental to good contrast; consequently, synthetic strategies to prevent reductants from reacting with the aminoxyl radical must conceptually titrate good shielding of the relatively large reductants while not greatly inhibiting access of water.
In this work, we utilize a supramolecular strategy to overcome ORCAs' poor relaxivity and high sensitivity toward reduction by fabricating a viral nanorod-based ORCA inclusion complex—specifically a semirotaxane—wherein the macrocycle cucurbit[8]uril (CB[8]) binds with nanomolar affinities to TEMPO moieties that have been conjugated onto the exterior surface of an anisotropic virus-like particle (VLP). We also show that this architecture is effective at shielding the radicals from reduction by ascorbate while still allowing the exchange of water and providing high contrast in vivo. These results indicate that shielding of aminoxyl radicals with macrocycles is a promising strategy in the pursuit of a persistent ORCA viable for clinical applications.
Results and discussion
To achieve maximum contrast, many types of nanomaterials have been investigated for their potential as platforms for aminoxyl-based MRI contrast agents. Relative to silica, synthetic polymer, or metallic nanoparticles, VLPs offer the advantages of monodispersity, high functionalizability, and high biodegradability.46–52 VLPs are self-assembled macromolecular structures composed of tens to thousands of individual protein subunits. The particular VLP utilized in this study is the tobacco mosaic virus (TMV), a 300 × 18 nm rod-shaped plant virus with a central 4 nm pore along its axis of symmetry. The ease of synthetic modification and its resilience under a wide range of temperatures, solvents, and pH values have allowed TMV to function as a versatile platform in VLP technology for applications involving biomedicine and stimulus-responsive materials. Each virus is composed of 2130 identical self-assembled 17.5 kDa coat proteins53–55 (Scheme 1) of which, tyrosine 139 (Y139), located on the outer surface of the rod is solvent exposed and available for functionalization.56–59 To this exposed residue, we planned to conjugate a derivative of the aminoxyl radical TEMPO, compound 6, featuring an ammonium for enhanced binding of CB[8].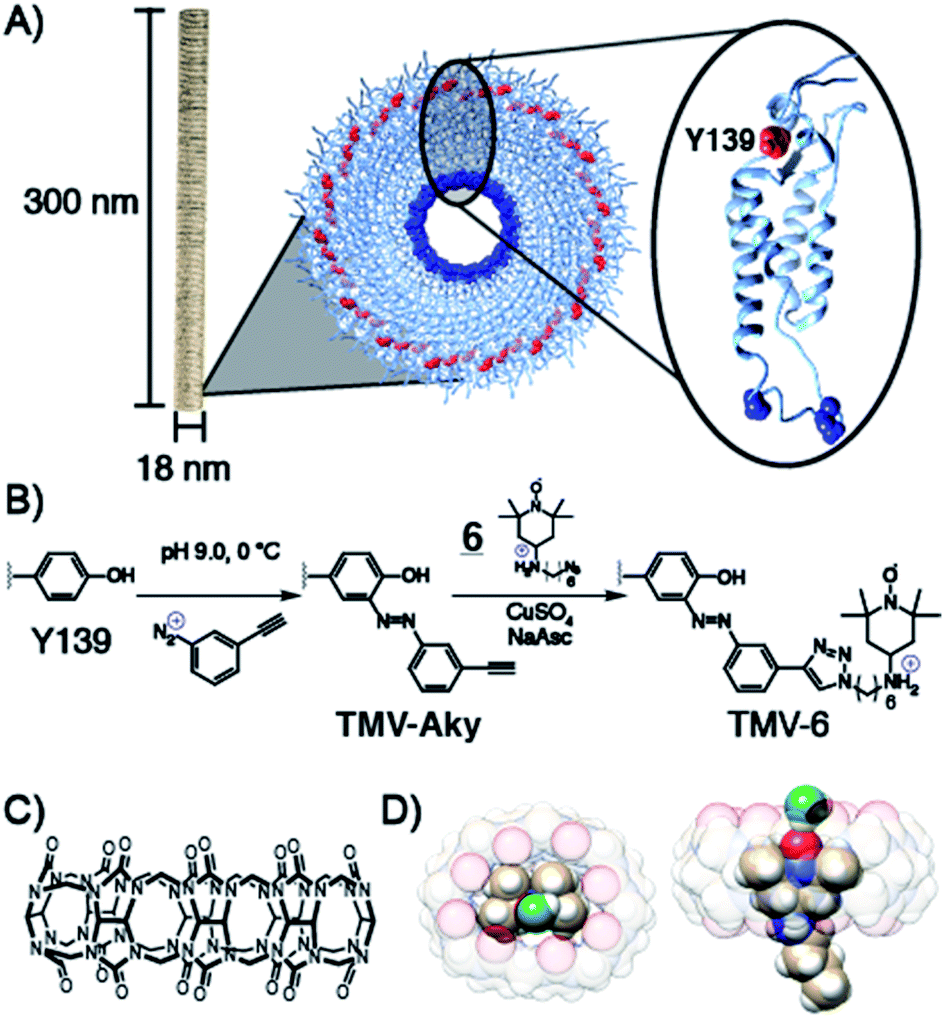 | ||
| Scheme 1 (A) Structure of TMV highlighting the solvent exposed amino acid residues of a single coat protein. (B) Installation of alkyne functionality on Y139 via diazonium coupling followed by the conjugation of 6via CuAAC. (C) Structural formula of CB[8], (D) CB[8]⊃6 inclusion complex showing the TEMPO oxygen is accessible by water (green) but still embedded within the macrocycle and protected from reduction. | ||
Various derivatives of TEMPO have been shown60–65 to bind CB[8] and we predicted that the amine in 6 would offer enhanced binding through an extra ion–dipole interaction with the oxygens in the crown of the macrocycle. Molecular dynamics equilibrium and free energy simulations of the CB[8]⊃6 inclusion complex establish the stability and precise location of TEMPO within the CB[8] cavity and quantify the accessibility to solvent water molecules of the TEMPO oxygen radical. Through equilibrium, adaptive biasing force, and umbrella sampling simulations we computed the free energy to reversibly remove TEMPO from the CB[8] cavity (Fig. S13†). From these data the equilibrium position of the TEMPO ring is 0.85 Å above the plane of the CB[8] ring and centred within it. From equilibrium simulations we observe that water hydrogen atoms are found preferentially a distance of 2 Å from the TEMPO oxygen radical (Fig. S14†). On average, one water hydrogen atom is found within 2.6 Å of the TEMPO oxygen radical. A representative snapshot of the CB[8] and TEMPO molecules along with the water molecule containing this hydrogen atom is shown in Scheme 1D and Fig. S15.† Since this water molecule is surrounded by other solvent water molecules, the water exchange needed to generate the MRI contrast is readily apparent. Prior to the conjugation of 6 to TMV, the binding of 6 to CB[8] was probed via isothermal titration calorimetry (ITC) (Fig. S9†). The Kd value for the CB[8]⊃6 complex was determined to be 5.8 × 10−7 ([4.1 × 10−7 to 7.9 × 10−7] M at 68.3% confidence interval as determined using error-surface projection see ESI†)—this nanomolar affinity between 6 and CB[8] is on par with many antibody-substrate binding affinities. This suggests CB[8]⊃6 should remain associated under concentrations and time scales relevant for MRI contrast agents in murine models.
Compound 6 was attached to TMV by first conjugating an alkyne handle to Y139 (Scheme 1) via a diazonium coupling reaction to produce TMV-Aky. Following this, a copper-catalyzed azide–alkyne cycloaddition (CuAAC) between TMV-Aky and 6 produced TMV-6. As seen in Fig. 1B, ESI-MS confirmed quantitative conversions of the TMV coat proteins while TEM (Fig. 1A) and SEC (Fig. 1C) show that the size and morphology of the TMV rods were unaltered following bioconjugation. Finally, the EPR spectrum of the TMV-6 conjugate (Fig. 1D) shows a characteristic triplet centred at a g-value of 2.007 corresponding to 14N (I = 1; A ∼ 45 MHz). The spectrum of 6 contains sharp peaks and isotropic g/A values that are characteristic of the rotational averaging found in small molecules. The spectrum of TMV-6 contains broad peaks, anisotropic g/A values, and a lower S/N ratio relative to the spectrum of 6. These differences between the spectra are attributed to the reduction of rotational and translational mobility upon attachment of 6 to the TMV rod. The high density of aminoxyl radicals on the surface of TMV can also allow for dipole spin exchange, which also results in peak broadening.66,67
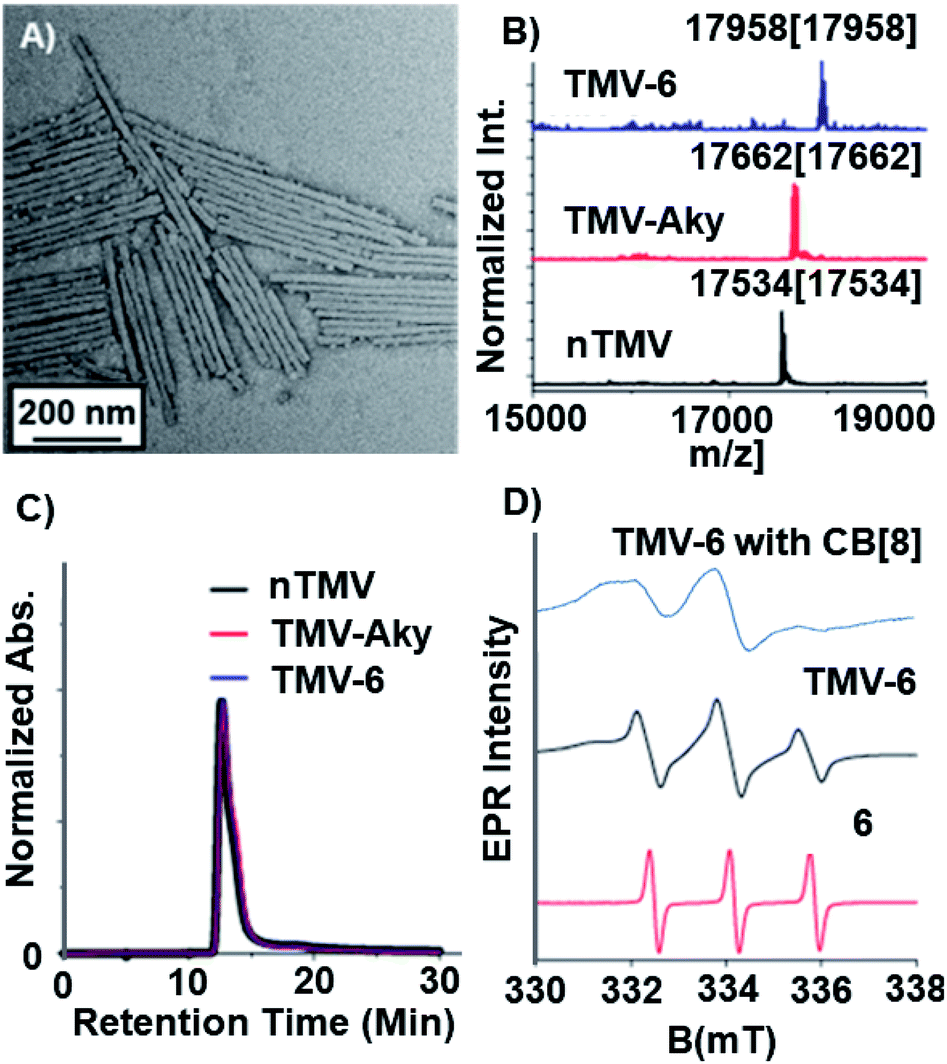 | ||
Fig. 1 Characterization of TMV after bioconjugation reactions. (A) TEM image of TMV-6. (B) Bioconjugation of the TEMPO radical to TMV was confirmed via ESI-MS. The peak at 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 534 m/z corresponds to native TMV. The peak at 17662 m/z corresponds to TMV-Aky. The peak at 17958 m/z corresponds to TMV-6. The spectrum corresponding to TMV-6 confirms complete conversion of TMV-Aky to TMV-6. (C) The integrity of the TMV rods after modification was confirmed by SEC. The single peak in the chromatograms (@ 260 nm) of the modified TMV samples demonstrates that the bioconjugation reactions did not compromise the morphology of the TMV rods. (D) X-Band EPR spectra of 6, TMV-6, and TMV-6 with CB[8]. The samples were prepared in capillary tubes to minimize interactions between high dielectric aqueous solvent and the electric field of the incident microwave radiation. 534 m/z corresponds to native TMV. The peak at 17662 m/z corresponds to TMV-Aky. The peak at 17958 m/z corresponds to TMV-6. The spectrum corresponding to TMV-6 confirms complete conversion of TMV-Aky to TMV-6. (C) The integrity of the TMV rods after modification was confirmed by SEC. The single peak in the chromatograms (@ 260 nm) of the modified TMV samples demonstrates that the bioconjugation reactions did not compromise the morphology of the TMV rods. (D) X-Band EPR spectra of 6, TMV-6, and TMV-6 with CB[8]. The samples were prepared in capillary tubes to minimize interactions between high dielectric aqueous solvent and the electric field of the incident microwave radiation. | ||
Since the properties of small molecules can change upon conjugation to proteins, we investigated the binding of CB[8] to TMV-6 following the confirmation of the TMV-6 conjugate. Since the solutions of TMV-6 at concentrations required for ITC characterization were subject to viscosity and adhesion issues, a fluorescence titration was used instead to determine a dissociation constant. In this case, neither TMV-6 nor CB[8] contain fluorophores, so a competitive binding strategy incorporating a fluorophore was implemented. Acridine-3,6-diamine, or proflavin (PF), is an acridine derivative that is fluorescent as a free molecule in aqueous solutions, but its fluorescence becomes quenched68,69 upon binding inside the cavity of CB[8]. A fluorescence titration of increasing concentrations of TMV-6 (0–20 μM relative to TEMPO) titrated into solutions with a constant concentration of the CB[8]⊃PF (0.2 μM) complex was performed. Upon the addition of increasing concentrations of TMV-6, the observed fluorescence of PF increased (Fig. S12†). Upon fitting these results to the observed fluorescence intensities, the Kd value for the CB[8]⊃TMV-6 complex was determined to be 3.8 ± 0.5 × 10−7 M. The reasons for the modest increase in binding affinity of the CB[8]⊃TMV-6 complex relative to the CB[8]⊃6 complex is not yet fully known, but the binding is still high enough to be relevant for the purposes of the ORCA design. Taken as a whole, the fluorescence titration experiments suggest that CB[8] molecules can bind to the TEMPO moieties conjugated onto the exterior surface of TMV to form a semirotaxane.
After establishing that CB[8] binds strongly to TMV-6, the relaxation behaviour of the TEMPO moieties was evaluated to determine the suitability of the semirotaxane as an ORCA. Relaxation behaviour is dependent on several factors, with magnetic field strength and solvent exchange being major examples. While maintaining a constant magnetic field strength of 1 T, longitudinal (T1) and transverse (T2) relaxation values were obtained for varying concentrations of TMV-6 in the presence and absence of CB[8] (Fig. 2). Relaxivity values (r1 and r2) were derived from a linear fit of the inverse relaxation data. When compared to the small molecule TEMPO control, the TMV-6 conjugate exhibits a five-fold increase in r1 going from a modest 0.6 mM−1 s−1 to 2.9 mM−1 s−1—rivalling that of clinically available Gd-DOTA (Table S2†) at the field strengths tested. For the supramolecular systems, the small molecule complex CB[8]⊃6 has very poor r1 values, which are similar to that of bulk water70 (0.2 ± 0.1 mM−1 s−1); however, conjugating CB[8]⊃6 to TMV yields an order of magnitude enhancement in relaxivity (to 1.9 ± 0.1 mM−1 s−1). These enhancements from conjugation to TMV are readily explained by the limited molecular motion provided by the attachment of TEMPO moieties to TMV. The modest difference in r1 values between TMV-6 and CB[8]⊃TMV-6 likely arises from restriction of the exchange of bulk water to the TEMPO radical by the CB[8]. Likewise, r2 values were enhanced by more than an order of magnitude from conjugation of CB[8]⊃6 to TMV (Table S2†). Curiously, the CB[8] had a much more pronounced attenuating effect on r2, relative to r1. Nevertheless, the transverse and longitudinal relaxivities obtained by our experiments are comparable to clinically available Gd-DOTA, demonstrating that our semirotaxane provides the contrast required to function in vivo.
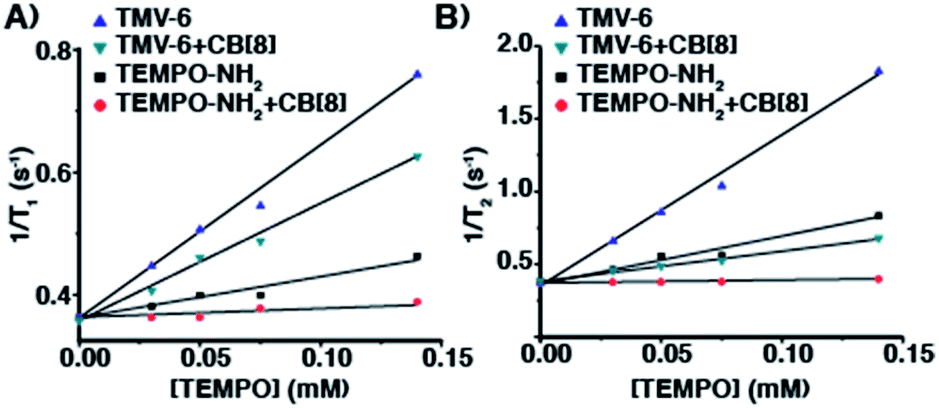 | ||
| Fig. 2 Plots of (A) 1/T1 (s−1) and (B) 1/T2 (s−1) versus [TEMPO] (mM) for TMV-6 and compound 6 in the absence and presence of CB[8] at 43 MHz in KP buffer (0.1 M, pH 7.4) @ 310 K. | ||
Having characterized the NMR relaxation properties of the semirotaxane and establishing its ability to function as an ORCA, we sought to investigate the shielding performance of the semirotaxane architecture. The rapid reduction of aminoxyl radicals to hydroxylamines in the presence of physiologically relevant reducing agents is well known. Ascorbate is one example of these reducing agents and is commonly utilized for reduction experiments owing to its ubiquity in the human body and its extensively studied redox properties. Upon the addition of a sodium ascorbate (10 equivalents per TEMPO moiety) solution in KP buffer (0.1 M, pH 7.4) to solutions of TMV-6 in the absence and presence of CB[8] (10 equivalents per TEMPO moiety), EPR spectra of the TMV conjugates were collected over 2 h (Fig. 3). EPR intensities were fitted under pseudo-first-order conditions. The pseudo-first-order rate constant for the reduction of TEMPO, k′, was determined to be 1.6 ± 0.1 × 10−3 s−1 (t1/2 = 7.2 min) for TMV-6 in the absence of CB[8] and 2.0 ± 0.1 × 10−5 s−1 (t1/2 = 577.6 min) for TMV-6 in the presence of CB[8]. To the best of our knowledge, this two-order of magnitude rate reduction in the semirotaxane is 20-fold lower than any measured initial rates of reduction reported for aminoxyl-based ORCAs thus far (Table S1†). The substantial decrease in the reduction rate of TEMPO suggests strongly that the CB[8] in the semirotaxane architecture effectively shields the aminoxyl radical from ascorbate. We were able to fully demonstrate the high contrast enhancement and stability of ORCA in vivo as shown in Fig. 3C. We injected equal concentrations of CB[8]⊃TMV-6 (green circle) and TMV-6 (orange circle) into the thigh of a Balb/C mouse. Both probes initially showed bright contrast in the muscle; however, TMV-6 began to lose contrast quickly, with most signal gone after 2 h, in agreement with our EPR results.
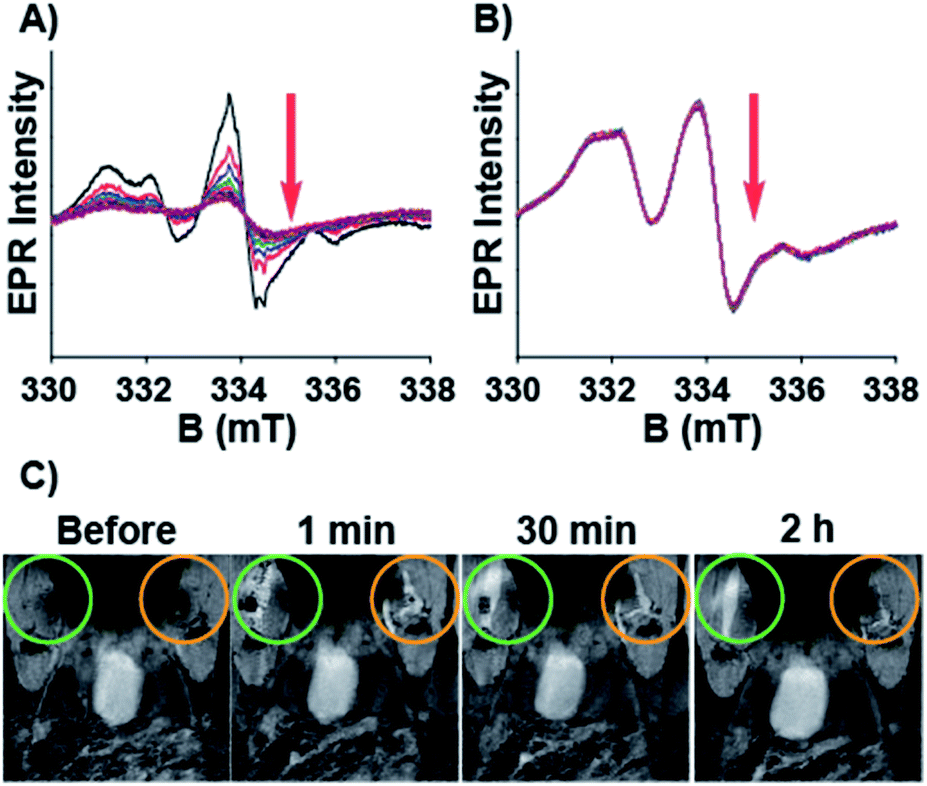 | ||
| Fig. 3 EPR spectra for the reduction of TMV-6 (2.6 mg mL−1) with sodium ascorbate (10 equivalents per TEMPO moiety) in the (A) absence and (B) presence of CB[8] (10 equivalents per TEMPO moiety). Data were collected at 10 min intervals over 2 h. (C) T1 weighted images of CB[8]⊃TMV-6 (green circle) vs. TMV-6 (orange circle) injected into the thigh muscle of a mouse. | ||
Conclusion
In conclusion, we have demonstrated the enhancement of the survivability of aminoxyl radicals in the presence of ascorbate via shielding with CB[8]. The attachment of TEMPO derivatives onto the exterior surface of TMV results in a conjugate that provides enhanced T1 and T2 relaxation properties relative to small molecule aminoxyl radicals. The addition of CB[8] to the TMV conjugate forms a semirotaxane ORCA where the TEMPO moieties are encaged by CB[8]. The CB[8] can sterically shield the aminoxyl radical from reduction by ascorbate while still allowing for the exchange of water. Although the CB[8] also reduces the contrast strength of the ORCA compared to its unshielded form, the contrast strength is still higher than that of small molecule aminoxyl radicals and close to that of Gd-DOTA (Table S2†). Most importantly, the ORCA can survive in the presence of ascorbate for periods of time that exceed—to the best of our knowledge (Table S1†)—currently known aminoxyl-based ORCAs by 20-fold.24,40–45 Our results indicate that with the combined approach of (i) utilizing macrocycles for steric shielding to inhibit reduction in vivo along with (ii) enhancing T1 relaxivity by conjugating these supramolecular agents to biomacromolecular scaffolds, ORCAs are moving closer to clinically viable contrast agents.Ethical statement
All animal procedures were reviewed by the UT Southwestern IACUC committee and accepted under protocol #2016-101780.Author contributions
JJG conceived the project and wrote the manuscript with HL. HL, AS, JLL, HRF and LMH synthesized all compounds. HL conducted all EPR reduction experiments and fluorescence titration experiments with TMV-6. JLL and CRP conducted relaxometry measurements. MAL conducted MRI imaging studies. CEB performed preliminary ITC measurements. ORB imaged TMV-6 by TEM. SON did computational modeling. LLL supervised CRP.Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
J. J. G. would like to thank the National Science Foundation [CAREER DMR-1654405] and the Welch Foundation [AT-1989-20190330]. C. E. B. thanks the National Science Foundation Graduate Research Fellows Program (1746053). L. L. L. would like to thank the Welch Foundation [AT-1877-20180324]. S. O. N. acknowledges that this project was partially funded by the University of Texas at Dallas Office of Research through the COBRA program. J. J. G. would also like to thank Darby Ball who conducted MS studies under Sheena D'Arcy. ITC was conducted at UT Southwestern's Macromolecular Biophysics Core facility.Notes and references
- W. Y. Huang and J. J. Davis, Dalton Trans., 2011, 40, 6087 RSC.
- P. Caravan, Chem. Soc. Rev., 2006, 35, 512 RSC.
- S. A. Corr, S. J. Byrne, R. Tekoriute, C. J. Meledandri, D. F. Brougham, M. Lynch, C. Kerskens, L. O'Dwyer and Y. K. Gun'ko, J. Am. Chem. Soc., 2008, 130, 4214–4215 CrossRef CAS PubMed.
- G. L. Davies, S. A. Corr, C. J. Meledandri, L. Briode, D. F. Brougham and Y. K. Gun'ko, ChemPhysChem, 2011, 12, 772–776 CrossRef CAS PubMed.
- P. Caravan, J. J. Ellison, T. J. McMurry and R. B. Lauffer, Chem. Rev., 1999, 99, 2293–2352 CrossRef CAS PubMed.
- E. J. Werner, A. Datta, C. J. Jocher and K. N. Raymond, Angew. Chem., Int. Ed., 2008, 47, 8568–8580 CrossRef CAS PubMed.
- P. Hermann, J. Kotek, V. Kubicek and I. Lukes, Dalton Trans., 2008, 23, 3027 RSC.
- Z. Zhou and Z.-R. Lu, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2012, 5, 1–18 Search PubMed.
- Y. Li, M. Beija, S. Laurent, L. v. Elst, R. N. Muller, H. T. T. Duong, A. B. Lowe, T. P. Davis and C. Boyer, Macromolecules, 2012, 45, 4196–4204 CrossRef CAS.
- G. L. Davies, I. Kramberger and J. J. Davis, Chem. Commun., 2013, 49, 9704 RSC.
- I. A. Mendichovszky, S. D. Marks, C. M. Simcock and O. E. Olsen, Pediatr. Radiol., 2007, 38, 489–496 CrossRef PubMed.
- F. G. Shellock and E. Kanal, J. Magn. Reson. Imaging, 1999, 10, 477–484 CrossRef CAS PubMed.
- S. Swaminathan, T. D. Horn, D. Pellowski, S. Abul-Ezz, J. A. Bornhorst, S. Viswamitra and S. V. Shah, N. Engl. J. Med., 2007, 357, 720–722 CrossRef CAS PubMed.
- T.-H. Shin, Y. Choi, S. Kim and J. Cheon, Chem. Soc. Rev., 2015, 44, 4501–4516 RSC.
- P. Verwilst, S. Park, B. Yoon and J. S. Kim, Chem. Soc. Rev., 2015, 44, 1791–1806 RSC.
- E. Terreno, D. D. Castelli, A. Viale and S. Aime, Chem. Rev., 2010, 110, 3019–3042 CrossRef CAS PubMed.
- K. Glunde, D. Artemov, M.-F. Penet, M. A. Jacobs and Z. M. Bhujwalla, Chem. Rev., 2010, 110, 3043–3059 CrossRef CAS PubMed.
- B. R. Smith and S. S. Gambhir, Chem. Rev., 2017, 117, 901–986 CrossRef CAS PubMed.
- P. Harvey, I. Kuprov and D. Parker, Eur. J. Inorg. Chem., 2012, 2012, 2015–2022 CrossRef CAS.
- N. R. B. Boase, I. Blakey and K. J. Thurecht, Polym. Chem., 2012, 3, 1384 RSC.
- I. Tirotta, V. Dichiarante, C. Pigliacelli, G. Cavallo, G. Terraneo, F. B. Bombelli, P. Metrangolo and G. Resnati, Chem. Rev., 2015, 115, 1106–1129 CrossRef CAS PubMed.
- S. Aime, D. D. Castelli, S. G. Crich, E. Gianolio and E. Terreno, Acc. Chem. Res., 2009, 42, 822–831 CrossRef CAS PubMed.
- G. Liu, X. Song, K. W. Y. Chan and M. T. McMahon, NMR Biomed., 2013, 26, 810–828 CrossRef CAS PubMed.
- J. T. Paletta, M. Pink, B. Foley, S. Rajca and A. Rajca, Org. Lett., 2012, 14, 5322–5325 CrossRef CAS PubMed.
- Y. Wang, J. T. Paletta, K. Berg, E. Reinhart, S. Rajca and A. Rajca, Org. Lett., 2014, 16, 5298–5300 CrossRef CAS PubMed.
- E. A. Anderson, S. Isaacman, D. S. Peabody, E. Y. Wang, J. W. Canary and K. Kirshenbaum, Nano Lett., 2006, 6, 1160–1164 CrossRef CAS PubMed.
- A. Datta, J. M. Hooker, M. Botta, M. B. Francis, S. Aime and K. N. Raymond, J. Am. Chem. Soc., 2008, 130, 2546–2552 CrossRef CAS PubMed.
- M. A. Bruckman, L. N. Randolph, N. M. Gulati, P. L. Stewart and N. F. Steinmetz, J. Mater. Chem. B, 2015, 3, 7503–7510 RSC.
- N. Bye, O. E. Hutt, T. M. Hinton, D. P. Acharya, L. J. Waddington, B. A. Moffat, D. K. Wright, H. X. Wang, X. Mulet and B. W. Muir, Langmuir, 2014, 30, 8898–8906 CrossRef CAS PubMed.
- H. Hu, Y. Zhang, S. Shukla, Y. Gu, X. Yu and N. F. Steinmetz, ACS Nano, 2017, 11, 9249–9258 CrossRef CAS PubMed.
- M. A. Bruckman, S. Hern, K. Jiang, C. A. Flask, X. Yu and N. F. Steinmetz, J. Mater. Chem. B, 2013, 1, 1482 RSC.
- M. A. Bruckman, K. Jiang, E. J. Simpson, L. N. Randolph, L. G. Luyt, X. Yu and N. F. Steinmetz, Nano Lett., 2014, 14, 1551–1558 CrossRef CAS PubMed.
- J. Prasuhn, E. Duane, R. M. Yeh, A. Obenaus, M. Manchester and M. G. Finn, Chem. Commun., 2007, 1269–1271, 10.1039/b615084e.
- L. Liepold, S. Anderson, D. Willits, L. Oltrogge, J. A. Frank, T. Douglas and M. Young, Magn. Reson. Med., 2007, 58, 871–879 CrossRef CAS PubMed.
- P. D. Garimella, A. Datta, D. W. Romanini, K. N. Raymond and M. B. Francis, J. Am. Chem. Soc., 2011, 133, 14704–14709 CrossRef CAS PubMed.
- J. M. Hooker, A. Datta, M. Botta, K. N. Raymond and M. B. Francis, Nano Lett., 2007, 7, 2207–2210 CrossRef CAS PubMed.
- K. N. Raymond and V. C. Pierre, Bioconjugate Chem., 2005, 16, 3–8 CrossRef CAS PubMed.
- M. Allen, J. W. M. Bulte, L. Liepold, G. Basu, H. A. Zywicke, J. A. Frank, M. Young and T. Douglas, Magn. Reson. Med., 2005, 54, 807–812 CrossRef CAS PubMed.
- S. Qazi, L. O. Liepold, M. J. Abedin, B. Johnson, P. Prevelige, J. A. Frank and T. Douglas, Mol. Pharm., 2013, 10, 11–17 CrossRef CAS PubMed.
- M. Dharmarwardana, A. F. Martins, Z. Chen, P. M. Palacios, C. M. Nowak, R. P. Welch, S. Li, M. A. Luzuriaga, L. Bleris, B. S. Pierce, A. D. Sherry and J. J. Gassensmith, Mol. Pharm., 2018, 15, 2973–2983 CrossRef CAS PubMed.
- A. O. Burts, Y. Li, A. V. Zhukhovitskiy, P. R. Patel, R. H. Grubbs, M. F. Ottaviani, N. J. Turro and J. A. Johnson, Macromolecules, 2012, 45, 8310–8318 CrossRef CAS.
- M. A. Sowers, J. R. McCombs, Y. Wang, J. T. Paletta, S. W. Morton, E. C. Dreaden, M. D. Boska, M. F. Ottaviani, P. T. Hammond, A. Rajca and J. A. Johnson, Nat. Commun., 2014, 5, 5460 CrossRef PubMed.
- H. V. T. Nguyen, Q. Chen, J. T. Paletta, P. Harvey, Y. Jiang, H. Zhang, M. D. Boska, M. F. Ottaviani, A. Jasanoff, A. Rajca and J. A. Johnson, ACS Cent. Sci., 2017, 3, 800–811 CrossRef CAS PubMed.
- H. V. T. Nguyen, A. Detappe, N. M. Gallagher, H. Zhang, P. Harvey, C. Yan, C. Mathieu, M. R. Golder, Y. Jiang, M. F. Ottaviani, A. Jasanoff, A. Rajca, I. Ghobrial, P. P. Ghoroghchian and J. A. Johnson, ACS Nano, 2018, 12, 11343–11354 CrossRef CAS PubMed.
- A. Rajca, Y. Wang, M. Boska, J. T. Paletta, A. Olankitwanit, M. A. Swanson, D. G. Mitchell, S. S. Eaton, G. R. Eaton and S. Rajca, J. Am. Chem. Soc., 2012, 134, 15724–15727 CrossRef CAS PubMed.
- J. K. Pokorski, K. Breitenkamp, L. O. Liepold, S. Qazi and M. G. Finn, J. Am. Chem. Soc., 2011, 133, 9242–9245 CrossRef CAS PubMed.
- A. M. Wen and N. F. Steinmetz, Chem. Soc. Rev., 2016, 45, 4074–4126 RSC.
- Z. Chen, N. Li, S. Li, M. Dharmarwardana, A. Schlimme and J. J. Gassensmith, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2016, 8, 512–534 CrossRef PubMed.
- I. Yildiz, S. Shukla and N. F. Steinmetz, Curr. Opin. Biotechnol., 2011, 22, 901–908 CrossRef CAS PubMed.
- K. J. Koudelka, A. S. Pitek, M. Manchester and N. F. Steinmetz, Annu. Rev. Virol., 2015, 2, 379–401 CrossRef CAS PubMed.
- K. L. Lee, S. Shukla, M. Wu, N. R. Ayat, C. E. El Sanadi, A. M. Wen, J. F. Edelbrock, J. K. Pokorski, U. Commandeur, G. R. Dubyak and N. F. Steinmetz, Acta Biomater., 2015, 19, 166–179 CrossRef CAS PubMed.
- M. Longmire, P. L. Choyke and H. Kobayashi, Nanomedicine, 2008, 3, 703–717 CrossRef CAS PubMed.
- K. B. G. Scholthof, S. Adkins, H. Czosnek, P. Palukaitis, E. Jacquot, T. Hohn, B. Hohn, K. Saunders, T. Candresse, P. Ahlquist, C. Hemenway and G. D. Foster, Mol. Plant Pathol., 2011, 12, 938–954 CrossRef CAS PubMed.
- B. D. Harrison and T. M. A. Wilson, Philos. Trans. R. Soc. London, Ser. B, 1999, 354, 521–529 CrossRef CAS PubMed.
- A. Klug, Philos. Trans. R. Soc. London, Ser. B, 1999, 354, 531–535 CrossRef CAS PubMed.
- P. Sitasuwan, L. A. Lee, K. Li, H. G. Nguyen and Q. Wang, Front. Chem., 2014, 2, 31 CrossRef PubMed.
- T. L. Schlick, Z. Ding, E. W. Kovacs and M. B. Francis, J. Am. Chem. Soc., 2005, 127, 3718–3723 CrossRef CAS PubMed.
- S. Li, M. Dharmarwardana, R. P. Welch, Y. Ren, C. M. Thompson, R. A. Smaldone and J. J. Gassensmith, Angew. Chem., Int. Ed., 2016, 55, 10691–10696 CrossRef CAS PubMed.
- S. Li, M. Dharmarwardana, R. P. Welch, C. E. Benjamin, A. M. Shamir, S. O. Nielsen and J. J. Gassensmith, ACS Appl. Mater. Interfaces, 2018, 10, 18161–18169 CrossRef CAS PubMed.
- D. Bardelang, K. Banaszak, H. Karoui, A. Rockenbauer, M. Waite, K. Udachin, J. A. Ripmeester, C. I. Ratcliffe, O. Ouari and P. Tordo, J. Am. Chem. Soc., 2009, 131, 5402–5404 CrossRef CAS PubMed.
- N. Jayaraj, M. Porel, M. F. Ottaviani, M. V. S. N. Maddipatla, A. Modelli, J. P. Da Silva, B. R. Bhogala, B. Captain, S. Jockusch, N. J. Turro and V. Ramamurthy, Langmuir, 2009, 25, 13820–13832 CrossRef CAS PubMed.
- E. Mileo, E. Mezzina, F. Grepioni, G. F. Pedulli and M. Lucarini, Chem.–Eur. J., 2009, 15, 7859–7862 CrossRef CAS PubMed.
- E. V. Peresypkina, V. P. Fedin, V. Maurel, A. Grand, P. Rey and K. E. Vostrikova, Chem.–Eur. J., 2010, 16, 12481–12487 CrossRef CAS PubMed.
- Z. Rinkevicius, B. Frecuş, N. A. Murugan, O. Vahtras, J. Kongsted and H. Ågren, J. Chem. Theory Comput., 2012, 8, 257–263 CrossRef CAS PubMed.
- S. Yi, B. Captain, M. F. Ottaviani and A. E. Kaifer, Langmuir, 2011, 27, 5624–5632 CrossRef CAS PubMed.
- M. Peric, B. L. Bales and M. Peric, J. Phys. Chem. A, 2012, 116, 2855–2866 CrossRef CAS PubMed.
- K. M. Salikhov, Appl. Magn. Reson., 2010, 38, 237–256 CrossRef CAS.
- S. Kemp, N. J. Wheate, F. H. Stootman and J. R. Aldrich-Wright, Supramol. Chem., 2007, 19, 475–484 CrossRef CAS.
- R. N. Dsouza, U. Pischel and W. M. Nau, Chem. Rev., 2011, 111, 7941–7980 CrossRef CAS PubMed.
- W. Li, K. Grgac, A. Huang, N. Yadav, Q. Qin and P. C. M. van Zijl, Magn. Reson. Med., 2016, 76, 270–281 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc05510j |
| This journal is © The Royal Society of Chemistry 2020 |