
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A shape changing tandem Rh(CNC) catalyst: preparation of bicyclo[4.2.0]octa-1,5,7-trienes from terminal aryl alkynes†
Caroline M.
Storey
 a,
Audrius
Kalpokas
b,
Matthew R.
Gyton
a,
Tobias
Krämer
*bc and
Adrian B.
Chaplin
*a
a,
Audrius
Kalpokas
b,
Matthew R.
Gyton
a,
Tobias
Krämer
*bc and
Adrian B.
Chaplin
*a
aDepartment of Chemistry, University of Warwick, Coventry CV4 7AL, UK. E-mail: a.b.chaplin@warwick.ac.uk
bDepartment of Chemistry, Maynooth University, Maynooth, Co. Kildare, Ireland. E-mail: tobias.kraemer@mu.ie
cHamilton Institute, Maynooth University, Maynooth, Co. Kildare, Ireland
First published on 22nd January 2020
Abstract
The preparation of a range of tetraaryl-substituted bicyclo[4.2.0]octa-1,5,7-trienes using a one-pot procedure starting from terminal aryl alkynes and catalysed by a rhodium(I) complex is reported. This synthesis proceeds by a reaction sequence involving head-to-tail homocoupling of the terminal alkyne and zipper annulation of the resulting gem-enyne. The rhodium catalyst employed is notable for the incorporation of a flexible NHC-based pincer ligand, which is suggested to interconvert between mer- and fac-coordination modes to fulfil the orthogonal mechanistic demands of the two transformations. Evidence for this interesting auto-tandem action of the catalyst is provided by reactions of the precatalyst with model substrates, corroborating proposed intermediates in both component cycles, and norbornadiene, which reversibly captures the change in pincer ligand coordination mode, along with a DFT-based computational analysis.
Introduction
In pursuit of more efficient synthetic procedures tandem catalysis has emerged as a powerful approach, enabling complex molecules to be assembled in one pot through a precisely choreographed sequence of catalytic steps, reducing waste and saving time (Fig. 1).1–3 Contrasting cascade or domino reaction manifolds, tandem catalysis involves orchestration of functionally distinct transformations using a single or multiple catalyst precursor(s) (orthogonal tandem catalysis).2 The former variation is operationally the simplest and harnesses catalyst productivity most efficiently, however, sequencing multiple operations with high fidelity using a common catalyst precursor can be a formidable challenge. Whilst such temporal control can be accomplished through deliberate intervention to transform the catalyst in situ after a suitable time period (assisted tandem catalysis), this solution lacks the practical simplicity of autonomous counterparts (auto-tandem catalysis) that do not require reaction monitoring nor additional experimental operations.3 | ||
| Fig. 1 Sequential reaction protocols and bicyclo[4.2.0]octa-1,5,7-triene synthesis. | ||
As part of our work exploring terminal alkyne coupling reactions promoted by rhodium complexes of NHC-based pincer ligands,4 we serendipitously discovered that [Rh(CNC-Me)(C2H4)][BArF4] (1·C2H4, ArF = 3,5-(CF3)2C6H3) is a highly effective auto-tandem precatalyst for the preparation of bicyclo[4.2.0]octa-1,5,7-trienes from terminal aryl alkynes with high selectivity (2 → 3 → 4, Fig. 1). There are only two literature precedents for isobenzenes of this type, with the most pertinent example involving a single-step nickel(0) catalysed annulation of isolated (electron deficient) gem-enynes.5,6 Despite extensive investigation, the selective head-to-tail coupling of terminal alkynes (2 → 3) invoked in the formation of 4 remains a challenging task for transition metal catalysts and is typically accompanied with mechanistically interconnected E-enynes that are products of head-to-head coupling.7 We have, however, previously demonstrated that 1·C2H4 is a remarkably regioselective precatalyst for the dimerisation of terminal aryl alkynes (2a, R = 3,5-tBu2C6H3![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Ar′; 2b, R = Ph) into the corresponding gem-enynes (3a and 3b) under mild conditions in dichloromethane solution.4 Upon switching to more weakly coordinating solvent 1,2-F2C6H4 (DFB),8 catalyst stability and activity are significantly enhanced with no loss of selectivity, but subsequent metal catalysed conversion into 4 became more apparent. Although metal-catalysed reactions of terminal alkynes have been extensively investigated,9 to the best of our knowledge, this one-pot reaction sequence is unprecedented: a paucity that we attribute to the orthogonal mechanistic demands of the component steps.
Ar′; 2b, R = Ph) into the corresponding gem-enynes (3a and 3b) under mild conditions in dichloromethane solution.4 Upon switching to more weakly coordinating solvent 1,2-F2C6H4 (DFB),8 catalyst stability and activity are significantly enhanced with no loss of selectivity, but subsequent metal catalysed conversion into 4 became more apparent. Although metal-catalysed reactions of terminal alkynes have been extensively investigated,9 to the best of our knowledge, this one-pot reaction sequence is unprecedented: a paucity that we attribute to the orthogonal mechanistic demands of the component steps.
After briefly overviewing the scope of this unique one-pot reaction, we herein present mechanistic and computational studies that suggest the unique catalytic behaviour of 1 is enabled by the ability of the flexible CNC ligand to adopt both mer- and fac-coordination modes.10 This is a potentially widely applicable concept for tandem catalysis.
Results and discussion
Scope of tandem reaction
We have previously shown the homocoupling of 2a catalysed by 1·C2H4 (5 mol%) in dichloromethane proceeds at 25 °C with a TOF = 2.5 h−1 and exclusive formation of the corresponding gem-enyne 3a (until conversion reached ca. 90%), which was readily isolated by quenching the reaction at high alkyne conversion with carbon monoxide.4 We now report the use of DFB as a solvent, which results in an order of magnitude faster homocoupling under otherwise equivalent conditions (TOF = 26 h−1). On the considerably shorter time frames associated with these experiments, the ensuing metal-catalysed reaction of 3a into balance tetramer 4a was more readily apparent nearing complete consumption of 2a by 1H NMR spectroscopy, with the appearance of a characteristic 4H singlet resonance at δ 2.87 alongside two new 36H tBu resonances at δ 1.03 and 1.11 (Fig. 2). Under these conditions, 4a was obtained in quantitative spectroscopic yield within 4 h. Subsequent isolation and characterisation in solution and the solid state enabled unambiguous assignment of 4a as a bicyclo[4.2.0]octa-1,5,7-triene and motivated us to explore the scope of this unique sequential reaction.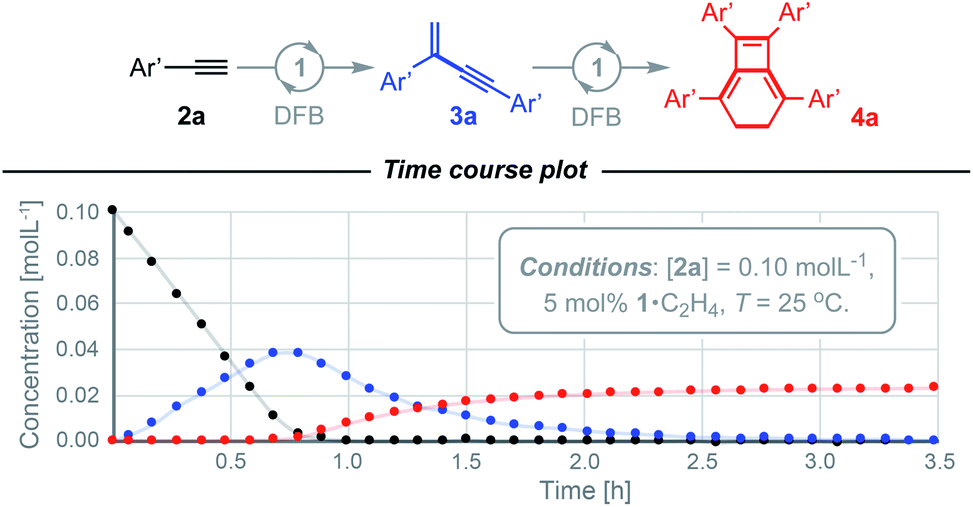 | ||
| Fig. 2 Time course analysis of the formation 4a from 2a catalysed by 1·C2H4. | ||
After ascertaining the intermediacy of gem-enynes, using in situ experiments monitored by 1H NMR spectroscopy (see ESI†), a straightforward one-pot protocol was developed for the preparation of a range of 2,5,7,8-tetraarylbicyclo-[4.2.0]octa-1,5,7-trienes (Fig. 3). Analytically pure samples of 4a–g were obtained, with unoptimised yields ranging from 64–94%, and fully characterised. This range of products demonstrates the compatibility of the tandem process for both electron withdrawing and donating groups in the para or meta positions of the aryl alkyne. Attempts at employing the bulky mesityl substituted alkyne 2h, however, yielded only an 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 mixture of gem- and E-enyne dimerisation products under these conditions. The tandem reaction also appears to be limited to aryl substituted alkynes, with alkyl alkynes 2i (R = nBu) and 2j (R = tBu) failing to afford the respective bicyclo[4.2.0]octa-1,5,7-trienes, even on prolonged heating at 65 °C. Homocoupling of these substrates was nevertheless observed, with the latter notable for proceeding with the slow, but exclusive, formation of E-tBuC
:20 mixture of gem- and E-enyne dimerisation products under these conditions. The tandem reaction also appears to be limited to aryl substituted alkynes, with alkyl alkynes 2i (R = nBu) and 2j (R = tBu) failing to afford the respective bicyclo[4.2.0]octa-1,5,7-trienes, even on prolonged heating at 65 °C. Homocoupling of these substrates was nevertheless observed, with the latter notable for proceeding with the slow, but exclusive, formation of E-tBuC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CCHCHtBu at 25 °C. Monitoring this reaction in situ using 1H NMR spectroscopy suggests catalytic turnover is impeded by a significant degree of product inhibition. Indeed, supporting this assertion the corresponding rhodium E-enyne complex [Rh(CNC-Me)(E-tBuCCCHCHtBu)][BArF4] 5 observed in situ was independently synthesised and fully characterised (see ESI†).
CCHCHtBu at 25 °C. Monitoring this reaction in situ using 1H NMR spectroscopy suggests catalytic turnover is impeded by a significant degree of product inhibition. Indeed, supporting this assertion the corresponding rhodium E-enyne complex [Rh(CNC-Me)(E-tBuCCCHCHtBu)][BArF4] 5 observed in situ was independently synthesised and fully characterised (see ESI†).
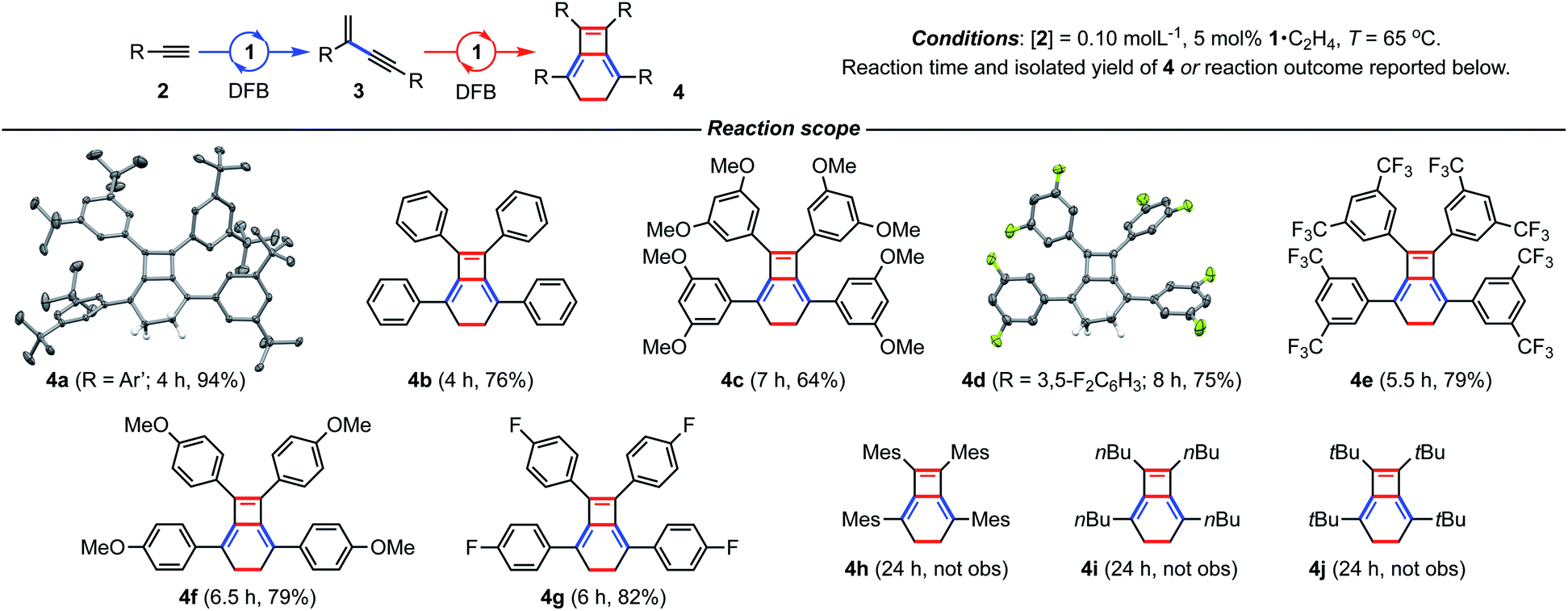 | ||
| Fig. 3 Preparation of bicyclo[4.2.0]octa-1,5,7-trienes from terminal alkynes. Solid-state structures of 4a (not unique, Z′ = 2) and 4d shown with 50% probability thermal ellipsoids. | ||
Mechanistic proposal and supporting organometallic chemistry
To reconcile the formation of 4 the auto-tandem scheme outlined in Fig. 4 is hypothesised, which involves asynchronous homocoupling of 2 into 3 followed by annulation of 3 into 4: the latter enabled by the capacity of CNC-Me to shuttle between mer- and fac-coordination modes.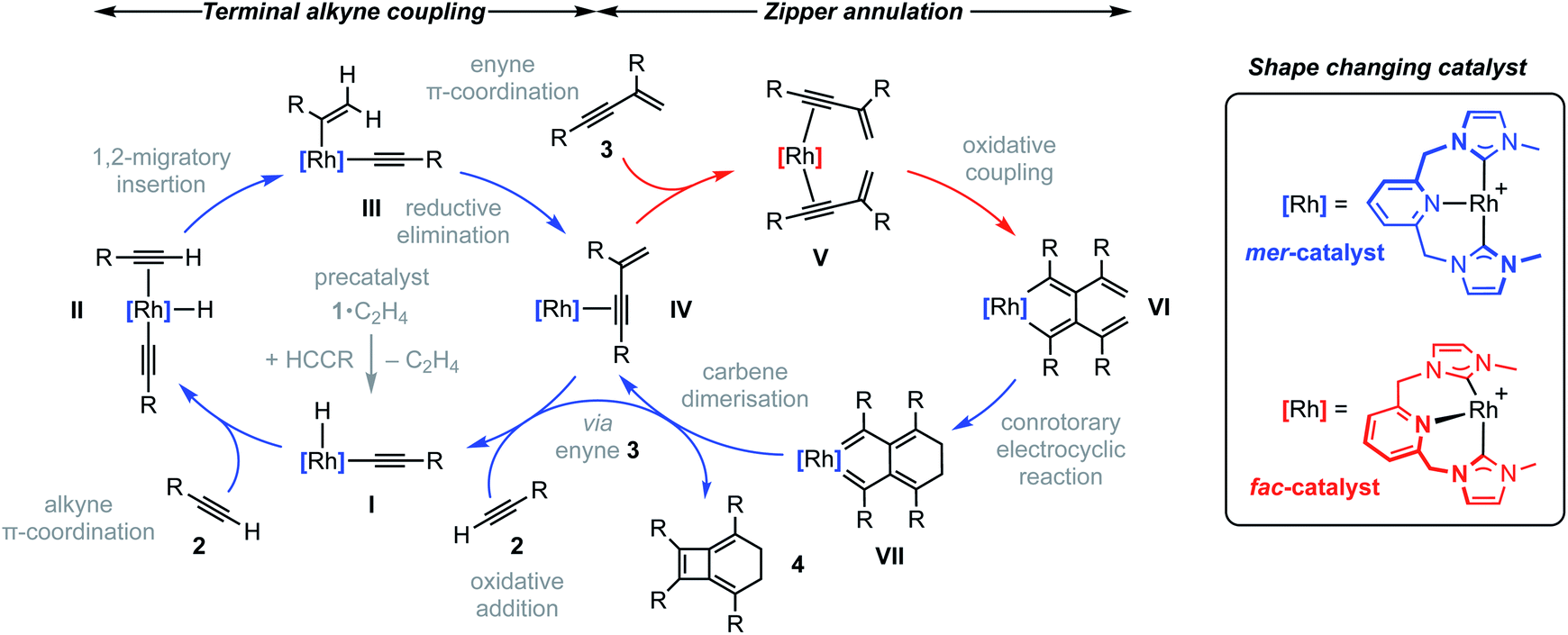 | ||
| Fig. 4 Proposed auto-tandem catalysed preparation of bicyclo[4.2.0]octa-1,5,7-trienes from terminal alkynes. | ||
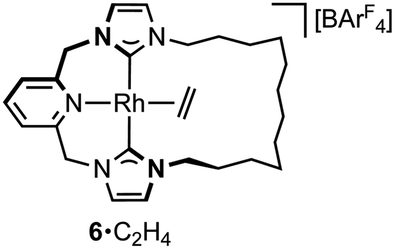
A hydrometallation mechanism, which comprises C–H bond oxidative addition of the terminal alkyne (1/IV → I), selectivity determining 1,2-migratory insertion of the second alkyne into the resulting metal hydride (II → III), and rate determining C–C bond reductive elimination to generate the gem-enyne (III → IV), is proposed for the homocoupling based on literature precedents and our previous interrogation of the 1/2a system.4,7 Pincer complexes have been shown to be effective catalysts for terminal alkyne coupling reactions and these component steps are fully compatible with mer-coordination of CNC-Me throughout the cycle.11 Indeed, we have previously shown that terminal alkyne coupling of 2a by an analogue of 1, bearing instead a macrocyclic NHC-based pincer ligand [Rh(CNC-12)(C2H4)][BArF4] 6·C2H4, results exclusively in entrapment of the enyne product within the annulus of the ligand.4 This outcome is only possible if the NHC-based pincer ligand maintains a mer-coordination mode during the homocoupling.
To help provide further evidence for the hydrometallation mechanism, the reaction between 1·C2H4 and 2-ethynylpyridine was studied in DFB (Fig. 5). Heating at 65 °C overnight resulted in exclusive formation of Rh(III) gem-alkenyl alkynyl 7 (cf.III),4,12 where chelation of the pyridine completes the coordination sphere and encumbers onward reductive elimination. Complex 7 was subsequently isolated from solution in 82% yield and fully characterised, including structural elucidation in the solid state using single crystal X-ray diffraction (1.2 Å resolution). Key spectroscopic features of 7 include C1 symmetry, geminal alkene 1H resonances at δ 5.97 and 6.48, and 13C resonances at δ 98.8 (Rh![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) C, 1JRhC = 56 Hz) and 137.0 (Rh(CH2), 1JRhC = 25 Hz) that display large 103Rh coupling.
C, 1JRhC = 56 Hz) and 137.0 (Rh(CH2), 1JRhC = 25 Hz) that display large 103Rh coupling.
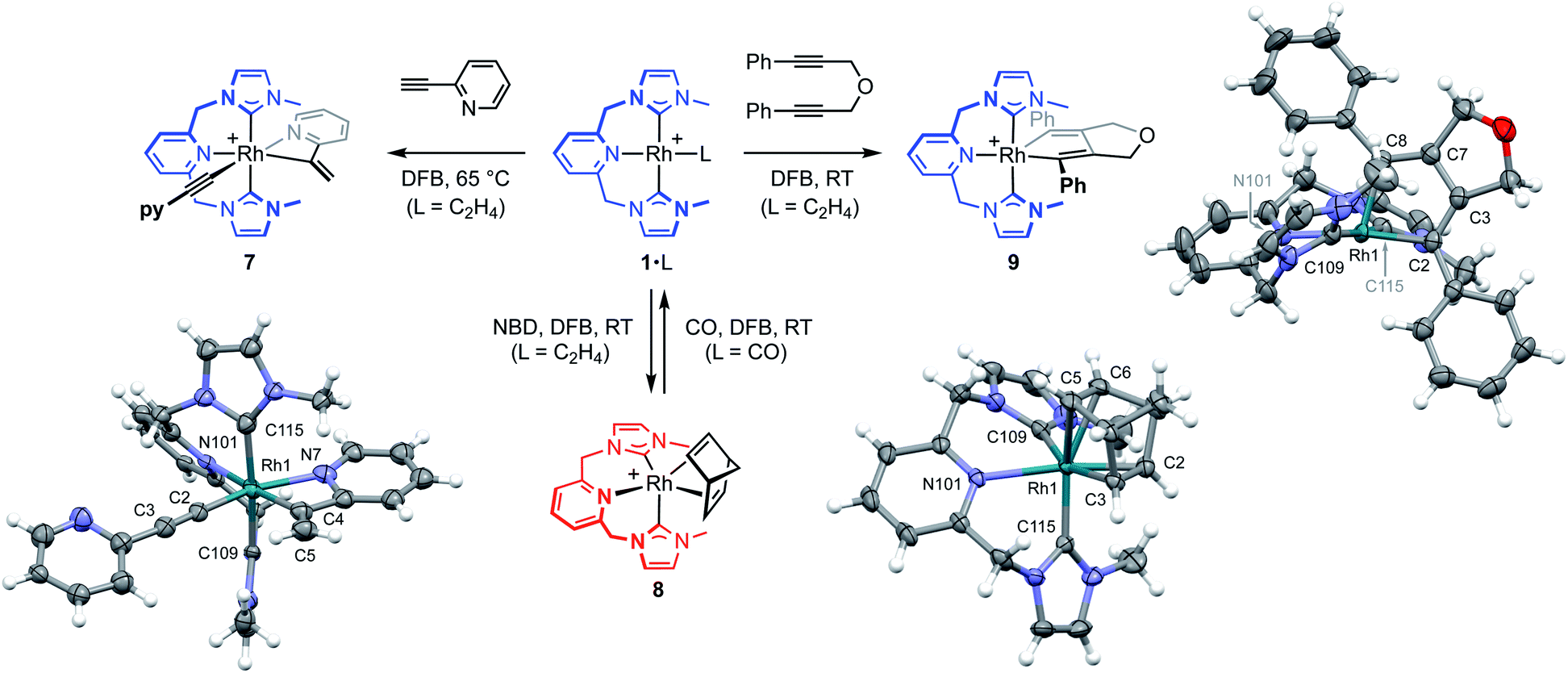 | ||
| Fig. 5 Reactivity of 1·C2H4 relevant to the proposed mechanism ([BArF4]− anions omitted). Solid-state structures of the cations of 7, 8 and 9 (not unique, Z′ = 2) shown with 30%, 50% and 50% probability thermal ellipsoids, respectively. Selected data: 7, Rh1–C2, 1.92(2) Å; C2–C3, 1.21(2) Å; Rh1–C2–C3, 179(2)°; Rh1–C4, 2.02(2) Å; C4–C5, 1.30(3) Å; Rh1–N7, 2.15(2) Å; Rh1–C4–C5, 140(2)°; Rh1–N101, 2.25(2) Å; Rh1–C109, 2.01(2) Å; Rh1–C115, 2.01(2) Å; C109–Rh1–C115, 174.5(8)°; 8, Rh1–Cnt(C2,C3), 1.964(2) Å; Rh1–Cnt(C5,C6), 2.144(2) Å; C2–C3, 1.431(4) Å; C5–C6, 1.369(4) Å; N101–Rh1–Cnt(C2,C3), 135.48(8)°; C109–Rh1–Cnt(C2,C3), 140.01(9)°; C115–Rh1–Cnt(C5,C6), 160.86(10)°; Rh1–N101, 2.327(2) Å; Rh1–C109, 2.118(3) Å; Rh1–C115, 2.026(2) Å; C109–Rh1–C115, 103.60(10)°; 9, Rh1–C2, 2.029(2) Å; Rh1–C8, 2.024(2) Å, N101–Rh1–C2, 171.69(8)°; C2–C3, 1.354(3) Å; C3–C7, 1.433(3) Å; C7–C8, 1.345(3) Å; Rh1–N101, 2.242(2) Å; Rh1–C109, 2.049(2) Å; Rh1–C115, 2.063(2) Å; C109–Rh1–C115, 171.92(8)°. | ||
Although the annulation of 3 has little direct precedent, parallels can be drawn with oxidative coupling reactions of internal alkynes, which afford metallacyclopentadienes and, in some cases, thereafter the corresponding cyclobutadiene complexes.13 In these well-established reactions, cis-parallel coordination of the alkynes is a prerequisite for efficient mixing of the alkyne frontier molecular orbitals,14 and correspondingly the majority of examples are associated with fac-coordinating ancillary ligands (e.g. cyclopentadienyls) that promote these geometries. On this basis we suggest the annulation of 3 commences with distortion of the catalyst geometry in such a way as to place the CNC-Me ligand in a fac-coordination mode and therefore enable binding of two enynes in a cis-parallel arrangement (IV → V). Subsequent oxidative coupling would then afford metallacyclopentadiene VI, with the alkyne carbon atom bearing the most sterically demanding substituents forming the σ-bond with the metal centre in accordance with Wakatsuki's rule.14 A metal promoted conrotatory electrocyclic reaction (VI → VII) and product elimination, involving formal alkylidene dimerisation, is thereafter proposed to afford 4. The latter step is related, by the principle of microscopic reversibility, to the insertion of metals into tetraaminoethylenes for which there is precedent for rhodium(I).15 An alternative product releasing route, where formation of the four-membered ring of the isobenzene precedes that of the six, through formation of the corresponding cyclobutadiene complex of VI and then electrocyclic reaction, was also considered but ultimately discounted (vide infra).
The key requirement of the annulation conjecture is the ability of the CNC ligand to adopt a fac-coordination mode and this was confirmed by reaction of 1·C2H4 with 1 equivalent of norbornadiene (NBD) in DFB at RT, which resulted in quantitative formation of 8 within 3 h in a sealed tube (Fig. 5, 76% isolated yield). Such behaviour is uncharacteristic for pincer ligands, but some examples can be found in the literature.16 The formation of 8 was established by NMR spectroscopy and X-ray diffraction. In solution, 8 is notable is for the adoption of time averaged Cs symmetry of the pincer and fast rotation of the NBD ligand on the NMR time scale (298 K, 500 MHz), carbene resonances at δ 187.4 that show enhanced 1JRhC coupling compared to 1·C2H4 (51 vs. 40 Hz), and an alkene 13C signal at δ 43.6 (1JRhC = 8 Hz). In the solid state the metal adopts a distorted trigonal bipyramidal geometry (C109–Rh1–C115 = 103.60(10)°), with the appreciably shorter axial Rh–NHC (2.026(2) vs. 2.118(3) Å) and equatorial Rh–alkene (1.964(2) vs. 2.144(2) Å) contacts in line with a structurally related complexes.17 Reaction of isolated 8 with carbon monoxide (1 atm) generated the known square planar carbonyl derivative of 1 within 5 h at RT.4 Macrocyclic 6·C2H4 also reacts reversibly with NBD and, in strong support of the underlying hypotheses, catalyses the formation of 4a from 3a, albeit under considerably more forcing reaction conditions than its acyclic congener (TON = 2, after 45 days at 50 °C; details provided in the ESI†).
We next turned to probe the capacity for 1 to promote the oxidative coupling of two alkynes, for which di(3-phenylprop-2-ynyl)ether was identified from the literature.18 Gratifyingly, reaction between 1·C2H4 and the propargyl ether in DFB at RT afforded five-coordinate metallacyclopentadiene 9 within 30 min, as marked visually by its characteristic dark green colour (Fig. 5). Complex 9 was subsequently isolated in 97% yield and fully characterised. In solution 9 displays time averaged C2 symmetry (298 K, 500 MHz), with the metallacyclopentadiene 13C resonances located at δ 150.8 (Rh(Ph)C, 1JRhC = 41 Hz) and 155.5 (RhC(Ph), 2JRhC = 3 Hz). Five-coordinate 9 adopts a distorted square pyramidal structure in the solid state, with the pincer ligand in the expected mer-coordination mode. We have previously reported a 2,2′-biphenyl complex of 6, which shows a similar geometry and the metal-based metrics are in good agreement.19 As a structural analogue of VI, we sought to ascertain if the corresponding cyclobutadiene complex could be obtained. As no reaction was evident on extended thermolysis of 9 in DFB (85 °C, 24 h) we discount this possibility.
Computational evaluation
To substantiate the proposed auto-tandem reaction and help elucidate the mechanistic intricacies, DFT calculations at the B3PW91-D3/SDD/6-31G** level of theory were employed for the most computationally amenable phenyl-substituted system (Fig. 6). Coordination and C(sp)–H bond oxidative addition of 2b proceeds with a low barrier, but endergonic isomerisation (ΔG = +7.0 kcal mol−1) of the resulting alkynyl hydride I is required to bind the second equivalent trans to the alkynyl (II).20 The two possible regioisomers of II are nearly isoenergetic, but it is only for the head-to-tail configuration that 1,2-migratory insertion is productive with respect to dissociation. As the reaction thereafter proceeds thermodynamically downhill, this step is irreversible and selectively determining (ΔΔG‡ = −2.5 kcal mol−1).21 Consistent with the experimental findings, gem-alkenyl alkynyl III is calculated to be the resting state. Subsequent reductive elimination affording IV is turnover limiting (ΔG‡ = 22.0 kcal mol−1) and completes the production of 3b, which is formed in ΔG = −36.6 kcal mol−1 overall.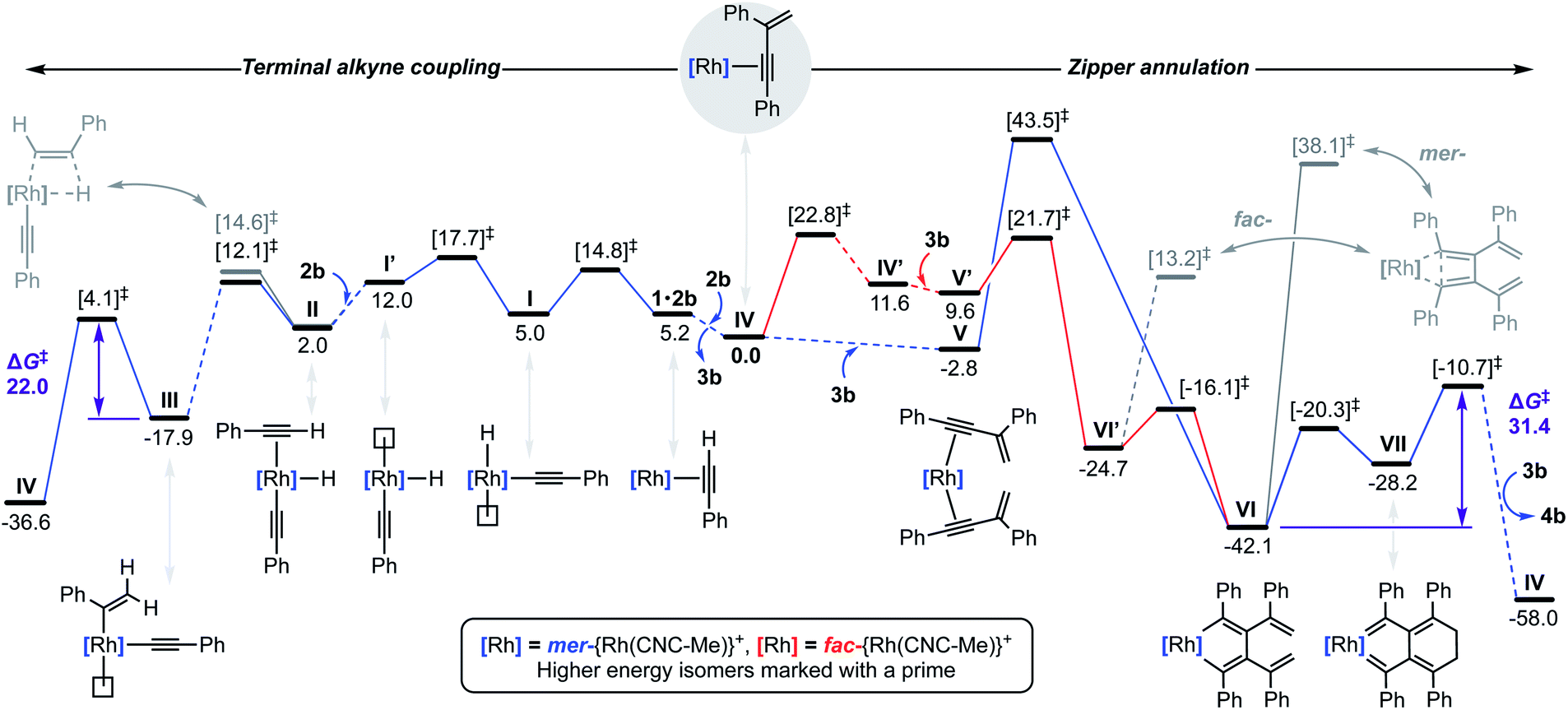 | ||
| Fig. 6 Calculated reaction profile (B3PW91-D3/SDD/6-31G**) for the terminal alkyne coupling of 2b and zipper annulation of 3b. Relative Gibbs free energies (kcal mol−1) are corrected for DFB solvent. Solid traces for elementary steps for which transition states have been calculated. | ||
Two pathways for the annulation have been computationally evaluated: the lowest energy route commences with ring flipping of one of the bridging methylene groups of the pincer backbone,22 which distorts the metal coordination geometry and ultimately causes CNC-Me to adopt a fac-coordination mode (IV′). This process is associated with a small thermodynamic penalty of ΔG = +11.6 kcal mol−1, but appreciable barrier of ΔG‡ = 22.8 kcal mol−1. The latter is consistent with the highly asynchronous nature of the tandem reaction, with the homocoupling proceeding with a lower barrier of ΔG‡ = 17.7 kcal mol−1 with respect to IV. The fac-configuration is stabilised by coordination of an additional equivalent of 3b, forming V′ and from which facile and irreversible oxidative coupling is possible: ΔG‡ = 21.7 kcal mol−1 relative to IV, but only 12.1 kcal mol−1 with respect to V′. Formation of a cyclobutadiene complex from the resulting fac-metallacyclopentadiene VI′ was examined, but the associated barrier is considerably higher than formation of the corresponding and thermodynamically preferred mer-isomer VI (ΔG = −42.1 kcal mol−1). The alternative higher energy annulation pathway involves retention of CNC-Me in a mer-coordination mode and converges at VI, but is associated with a prohibitively high activation barrier of ΔG‡ = 43.5 kcal mol−1 for the oxidative coupling and is consequently discounted. In line with the experimentally established stability of 9, subsequent C–C bond reductive elimination from VI is prohibitively high in energy (ΔG‡ = 55.3/80.2 kcal mol−1), however the postulated conrotatory electrocyclic reaction (VI → VII) and alkylidene dimerisation (VII → IV) appears energetically feasible. The latter is predicted to be turnover limiting in the case of the phenyl-substituted system (ΔG‡ = +31.4 kcal mol−1), producing 4b in ΔG = −58.0 kcal mol−1 overall.23
Conclusions
An atom efficient and operationally simple procedure for the synthesis of unusual bicyclic isobenzenes from terminal alkynes is reported. Using this one-pot protocol seven novel tetraaryl-substituted bicyclo[4.2.0]octa-1,5,7-trienes were successfully prepared, with the aryl substituents bearing a range of electron withdrawing and donating groups in the para or meta positions.This synthesis proceeds by a reaction sequence involving head-to-tail homocoupling of the terminal alkyne and annulation of the resulting gem-enyne. Both are catalysed with remarkably high fidelity by a common rhodium(I) catalyst, which features a flexible NHC-based pincer ligand that is hypothesised to interconvert between mer- and fac-coordination modes to fulfil the orthogonal mechanistic demands of the two transformations. Experimental evidence for this interesting auto-tandem action of the catalyst is provided by reactions of the precatalyst with model substrates, corroborating the formation of gem-alkenyl alkynyl and metallacyclopentadiene intermediates in the homocoupling and annulation steps respectively, and norbornadiene, which reversibly captures the change in the pincer ligand coordination mode. This work is supplemented by a detailed DFT-based computational analysis, which supports a hydrometallation-based homocoupling and a zipper-type annulation reaction that proceeds by oxidative coupling of the two gem-enynes, metal promoted conrotatory electrocyclic reaction and a product releasing formal alkylidene dimerisation.
The capacity of the NHC-based pincer ligand to adopt both mer- and fac-coordination modes appears to be central to the success of this one-pot procedure and this concept may prove to be a fruitful for the design of new tandem catalytic reactions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the European Research Council (ERC, grant agreement 637313; C. M. S, M. R. G, A. B. C.), Maynooth Department of Chemistry (T. K.), and Royal Society (UF100592, UF150675; A. B. C.) for financial support. T. K. acknowledges the DJEI/DES/SFI/HEA Irish Centre for High-End Computing (ICHEC) for the provision of computational facilities and support. Laura Scally, Adrian Deacon and Sean O. Dowd are thanked for generating preliminary DFT results. High-resolution mass-spectrometry data were collected using instruments purchased through support from Advantage West Midlands and the European Regional Development Fund. Crystallographic data were collected using an instrument that received funding from the ERC under the European Union's Horizon 2020 research and innovation programme (grant agreement No 637313).Notes and references
- (a) D. E. Fogg and E. N. dos Santos, Coord. Chem. Rev., 2004, 248, 2365–2379 CrossRef CAS; (b) J.-C. Wasilke, S. J. Obrey, R. T. Baker and G. C. Bazan, Chem. Rev., 2005, 105, 1001–1020 CrossRef CAS PubMed.
- T. L. Lohr and T. J. Marks, Nat. Chem., 2015, 7, 477–482 CrossRef CAS PubMed.
- J. E. Camp, Eur. J. Org. Chem., 2016, 425–433 Search PubMed; N. Shindoh, Y. Takemoto and K. Takasu, Chem.–Eur. J., 2009, 15, 12168–12179 CrossRef CAS PubMed.
- C. M. Storey, M. R. Gyton, R. E. Andrew and A. B. Chaplin, Angew. Chem., Int. Ed., 2018, 57, 12003–12006 CrossRef CAS PubMed.
- (a) S. Saito, T. Tanaka, T. Koizumi, N. Tsuboya, H. Itagaki, T. Kawasaki, S. Endo and Y. Yamamoto, J. Am. Chem. Soc., 2000, 122, 1810–1811 CrossRef CAS; (b) H. Hopf, H. Berger, G. Zimmermann, U. Nüchter, P. G. Jones and I. Dix, Angew. Chem., Int. Ed., 1997, 36, 1187–1190 CrossRef CAS.
- Parallels may be drawn with cycloaddition reactions of bisallenes (and related substrates), see for example: (a) B. Alcaide, P. Almendros and C. Aragoncillo, Chem. Soc. Rev., 2014, 43, 3106–3135 RSC; (b) C. Aubert, L. Fensterbank, P. Garcia, M. Malacria and A. Simonneau, Chem. Rev., 2011, 111, 1954–1993 CrossRef CAS PubMed; (c) S. Kitagaki, M. Kajita, S. Narita and C. Mukai, Org. Lett., 2012, 14, 1366–1369 CrossRef CAS PubMed; (d) X. Jiang, X. Cheng and S. Ma, Angew. Chem., Int. Ed., 2006, 45, 8009–8013 CrossRef CAS PubMed.
- B. M. Trost and J. T. Masters, Chem. Soc. Rev., 2016, 45, 2212–2238 RSC.
- S. D. Pike, M. R. Crimmin and A. B. Chaplin, Chem. Commun., 2017, 53, 3615–3633 RSC.
- (a) L. Rubio-Pérez, R. Azpíroz, A. Di Giuseppe, V. Polo, R. Castarlenas, J. J. Pérez-Torrente and L. A. Oro, Chem.–Eur. J., 2013, 19, 15304–15314 CrossRef PubMed; (b) L. Leroyer, V. Maraval and R. Chauvin, Chem. Rev., 2012, 112, 1310–1343 CrossRef CAS PubMed; (c) G. Domínguez and J. Pérez-Castells, Chem. Soc. Rev., 2011, 40, 3430–3444 RSC; (d) B. R. Galan and T. Rovis, Angew. Chem., Int. Ed., 2009, 48, 2830–2834 CrossRef CAS PubMed; (e) J. Liu, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2009, 109, 5799–5867 CrossRef CAS PubMed; (f) S. Saito and Y. Yamamoto, Chem. Rev., 2000, 100, 2901–2916 CrossRef CAS PubMed; (g) M. Lautens, W. Klute and W. Tam, Chem. Rev., 1996, 96, 49–92 CrossRef CAS PubMed.
- (a) R. E. Andrew, L. González-Sebastián and A. B. Chaplin, Dalton Trans., 2016, 45, 1299–1305 RSC; (b) C. M. Storey, H. P. Cook and A. B. Chaplin, in Complexes of NHC-Based CEC Pincer Ligands, ed. D. Morales-Morales, Elsevier, 2018, vol. 1, pp. 173–189 Search PubMed.
- (a) N. Gorgas, B. Stöger, L. F. Veiros and K. Kirchner, ACS Catal., 2018, 8, 7973–7982 CrossRef CAS; (b) N. Gorgas, L. G. Alves, B. Stöger, A. M. Martins, L. F. Veiros and K. Kirchner, J. Am. Chem. Soc., 2017, 139, 8130–8133 CrossRef CAS PubMed; (c) G. Kleinhans, G. Guisado-Barrios, D. C. Liles, G. Bertrand and D. I. Bezuidenhout, Chem. Commun., 2016, 52, 3504–3507 RSC; (d) O. Rivada-Wheelaghan, S. Chakraborty, L. J. W. Shimon, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2016, 55, 6942–6945 CrossRef CAS PubMed; (e) C. J. Pell and O. V. Ozerov, ACS Catal., 2014, 4, 3470–3480 CrossRef CAS; (f) W. Weng, C. Guo, R. Çelenligil-Çetin, B. M. Foxman and O. V. Ozerov, Chem. Commun., 2006, 197–199 RSC.
- M. Schafer, J. Wolf and H. Werner, Organometallics, 2004, 23, 5713–5728 CrossRef.
- (a) N. V. Shvydkiy and D. S. Perekalin, Coord. Chem. Rev., 2017, 349, 156–168 CrossRef CAS; (b) W. Ma, C. Yu, T. Chen, L. Xu, W.-X. Zhang and Z. Xi, Chem. Soc. Rev., 2017, 46, 1160–1192 RSC; (c) A. Steffen, R. M. Ward, W. D. Jones and T. B. Marder, Coord. Chem. Rev., 2010, 254, 1950–1976 CrossRef CAS; (d) A. Efraty, Chem. Rev., 1977, 77, 691–744 CrossRef CAS.
- Y. Wakatsuki, O. Nomura, K. Kitaura, K. Morokuma and H. Yamazaki, J. Am. Chem. Soc., 1983, 105, 1907–1912 CrossRef CAS.
- P. B. Hitchcock, M. F. Lappert, P. Terreros and K. P. Wainwright, J. Chem. Soc., Chem. Commun., 1980, 1180–1181 RSC.
- (a) E. Peris and R. H. Crabtree, Chem. Soc. Rev., 2018, 47, 1959–1968 RSC; (b) E. Suárez, P. Plou, D. G. Gusev, M. Martín and E. Sola, Inorg. Chem., 2017, 56, 7190–7199 CrossRef PubMed; (c) M. Hernández-Juárez, J. López-Serrano, P. Lara, J. P. Morales-Cerón, M. Vaquero, E. Álvarez, V. Salazar and A. Suárez, Chem.–Eur. J., 2015, 21, 7540–7555 CrossRef PubMed; (d) D. M. Roddick and D. Zargarian, Inorg. Chim. Acta, 2014, 422, 251–264 CrossRef CAS; (e) J. J. Adams, N. Arulsamy and D. M. Roddick, Organometallics, 2011, 30, 697–711 CrossRef CAS.
- (a) M. F. Cain, D. S. Glueck, J. A. Golen and A. L. Rheingold, Organometallics, 2012, 31, 775–778 CrossRef CAS; (b) C. J. Adams, K. M. Anderson, J. P. H. Charmant, N. G. Connelly, B. A. Field, A. J. Hallett and M. Horne, Dalton Trans., 2008, 2680–2692 RSC; (c) T. Nishioka, Y. Onishi, K. Nakajo, J. Guo-Xin, R. Tanaka and I. Kinoshita, Dalton Trans., 2005, 2130–2137 RSC; (d) C. M. Beck, S. E. Rathmill, Y. J. Park, J. Chen, R. H. Crabtree, L. M. Liable-Sands and A. L. Rheingold, Organometallics, 1999, 18, 5311–5317 CrossRef CAS; (e) S.-T. Liu, M.-C. Cheng and S.-M. Peng, J. Organomet. Chem., 1989, 368, C38–C40 CrossRef CAS.
- (a) S. Moulin, H. Dentel, A. Pagnoux-Ozherelyeva, S. Gaillard, A. Poater, L. Cavallo, J.-F. Lohier and J.-L. Renaud, Chem.–Eur. J., 2013, 19, 17881–17890 CrossRef CAS PubMed; (b) Y. Yamamoto, Y. Miyabe and K. Itoh, Eur. J. Inorg. Chem., 2004, 3651–3661 CAS; (c) A. Scheller, W. Winter and E. Müller, Justus Liebigs Ann. Chem., 1976, 1448–1454 CrossRef CAS.
- M. R. Gyton, B. Leforestier and A. B. Chaplin, Organometallics, 2018, 37, 3963–3971 CrossRef CAS PubMed.
- Formation of 3b by coordination of 2b directly to I and a carbometallation mechanism was also calculated, but the barrier for 1,2-migratory insertion is prohibitively high (ΔG‡ = +28.0 kcal mol−1, with respect to IV). Details are provided in the ESI.†.
- As the homocoupling of 2j proceeds with orthogonal regioselectivity, affording E-tBuCCCHCHtBu, we have also considered this mechanism computationally. The 1,2-migratory insertion is also predicted to be the selectivity determining step, favouring the head-to-head product with ΔΔG‡ = −1.4 kcal mol−1. Moreover, this step is reversible with respect to formation of 3b. Details are provided in the ESI.†.
- (a) T. M. Hood, B. Leforestier, M. R. Gyton and A. B. Chaplin, Inorg. Chem., 2019, 58, 7593–7601 CrossRef CAS PubMed; (b) H. Li, J. V. Obligacion, P. J. Chirik and M. B. Hall, ACS Catal., 2018, 8, 10606–10618 CrossRef CAS PubMed.
- This suggestion would infer the annulation rate is independent of the gem-enyne concentration. From inspection of the time course data recorded for 4a this does not appear to be the case for the Ar′ substituted-system, where instead turnover limiting oxidative coupling would be more consistent with the data. The reactivity of macrocyclic 6·C2H4 with 3a notably provides experimental evidence the latter step takes place with a fac- and not mer-coordinated CNC ligand.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details, NMR and ESI spectra (PDF), primary NMR data (MNOVA), and optimised geometries (XYZ). CCDC 1970203–1970209. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc06153c |
| This journal is © The Royal Society of Chemistry 2020 |