
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Applicability of electro-osmotic flow for the analysis of the surface zeta potential†
Olivija Plohl *a,
Lidija Fras Zemljiča,
Sanja Potrčab and
Thomas Luxbacherc
*a,
Lidija Fras Zemljiča,
Sanja Potrčab and
Thomas Luxbacherc
aUniversity of Maribor, Faculty of Mechanical Engineering, Laboratory for Characterization and Processing of Polymers, Smetanova 17, 2000 Maribor, Slovenia. E-mail: olivija.plohl@um.si
bUniversity of Maribor, Faculty of Chemistry and Chemical Engineering, Smetanova 17, 2000 Maribor, Slovenia
cAnton-Paar GmbH, Anton-Paar-Str. 20, A-8054 Graz, Austria
First published on 13th February 2020
Abstract
The analysis of the surface zeta potential (SZP) opens up new possibilities in the characterization of various materials used for scientific or industrial applications. It provides at the same time insight into the material surface chemistry and elucidates the interactions with charged species in the aqueous test solution. For this purpose, an accurate, reliable and repeatable analysis of the SZP is the key factor. This work focuses on a detailed and systematic comparison of two electrokinetic techniques, i.e. the mapping of the electro-osmotic flow (EOF) and the measurement of the streaming potential (SP), for the surface zeta potential (SZP) determination of several materials with varying properties. Both techniques have advantages as well as drawbacks. The applicability of latex polymer material and inorganic tracer particles at varying ionic strength, the interaction between oppositely charged tracer particles and solid surfaces, the assessment of the pH dependence of the SZP and the isoelectric point (IEP), and the effects of sample porosity and conductance have been investigated. Although in some cases the EOF method gives a SZP similar to the streaming potential measurement, especially when the tracer particle exhibits the same charge as the solid surface, it was revealed that reliable results were only obtained with the streaming potential and streaming current method. Several obstacles such as elevated conductivity at higher ionic strength, the applied voltage for the EM measurement, and the nature of tracer particles lower the accuracy and reliability of the SZP determined by the EOF method. It was shown that the EOF method is not applicable to oppositely charged surface and tracer particles and also limited to low salinity conditions especially when using polymeric tracer particles. Although the EOF method does not require the formation of a capillary flow channel, it disables a non-destructive SZP of fragile or valuable samples, such as QCM-D sensors, in comparison to the SP approach.
1. Introduction
When materials are brought into contact with an aqueous solution, they acquire a surface electric charge by different processes such as ionization, ion adsorption or ion dissolution. The need for charge compensation leads to the formation of an interfacial charge distribution in the aqueous phase that is described by the model of the electric double layer. Electrokinetic phenomena are induced by the movement of one of the phases (solid or liquid) relative to the second phase. The electrokinetic behaviour depends on the electric potential at the shear plane between the charged surface and the electrolyte solution. This potential at the shear plane is called the electrokinetic or ζ potential (ZP).1 Different electrokinetic effects exist depending on the way how the movement is induced. Electrophoresis, electro-osmosis, the streaming potential (SP) and electroacoustic represent the four electrokinetic measurement techniques from which zeta potential (ZP) is derived.1,2In general, ZP represents the charge at the solid–water interface that affects materials functionality and at the same time is the crucial parameter for the determination of the material's isoelectric point. Surface zeta potential (SZP) analysis is a vital method for qualifying important features of new materials in technical (e.g., effects of fouling and cleaning of membranes used for water treatment,3,4 textile industry5–7) and biomedical applications (e.g. biofilm formation, haemocompatible implants8–11). Furthermore, it enables to gain insights into modification processes that result from surface treatment or surface interactions with biological or natural environments under near-ambient conditions.1,2 Thus, SZP is a key parameter for understanding surface properties and for developing new specialized materials, e.g., biomaterials that get in contact with blood, or virus retention filters in biopharmaceutical segments that specifically require accurate, reliable and reproducible macroscopic surface SZP analyses.
Materials macroscopic SZP is commonly determined using the established streaming potential technique. The measurement of the SP (and alternatively of the streaming current) is the direct approach to the SZP, where liquid flow through a capillary generates an electric potential. A pressure gradient is applied between both ends of a capillary flow channel, which generates liquid flow and the streaming potential signal. This electrokinetic effect is used to assess the surface charge of macroscopic solids with a flat surface, but also with more complex surfaces such as porous material, fibers, and granular media.12 Recently, an alternative indirect measurement technique for SZP of flat surfaces was introduced,13 and is comprising phase analysis light scattering (PALS) using mostly polymeric tracer particles through electro-osmotic flow (EOF) mapping in a simple dip cell arrangement. Several studies were reported on using EOF for SZP determination in a dip cell,13–21 in a coated microchannel that measures the mobility of known ZP of tracer particles close to the surface,22 in a quartz cell having mostly non-ionic hydroxypropyl-coated polystyrene tracer particles23–35 or with microelectrophoresis instrumentation modified to accommodate quartz capillaries using sulphated polystyrene latex particles.36 It relies on the principle that the electrophoretic mobility of tracer particles dispersed in a liquid is affected by the surface when these particles approach the solid sample. Each technique shows advantages and drawbacks. The EOF method requires an apparently smaller sample size compared to the SP method but disables a non-destructive zeta potential analysis of fragile or valuable samples, e.g., QCM-D sensors.37 The solid sample is held in such way that if tracer particles sediment they do not deposit on the sample surface.13–15 The SP method is very competitive due to its versatility to determine the surface zeta potential for various geometries of solid samples such as flat surfaces, fibers, or granular media. Despite of the known reputation of the SP method, there the following drawbacks are proposed in the literature when using this method. These were correlated to the needs for a careful sealing of the sample in the measuring cell to accept the applied pressure gradient, to the limited sensitivity at higher salinity of the aqueous test solution, and that the SP technique typically requires an instrument that is solely dedicated to the SZP analysis.13,14 However, for a state-of-the-art SP analyser, these drawbacks have been overcome. On the other hand several authors have reported SZP results for various material types (13–21,23–35) obtained by the EOF method (in different configurations) using mostly polymeric tracer particles. The problems observed in these studies were associated with a high measurement uncertainty that consequently lowers the quality of the data15,16,24,27,38–40 especially for more complex sample surfaces (nanofibers, membranes), difficulties in determining the surface IEP when oppositely charged tracer particles were exposed,15,18,19,26,29 or the observation of polymeric tracer particles sedimentation and degradation (colour change of tracer particle dispersion), which is even more pronounced under elevated ionic strength,15,19,24,28 that limits the applicability of the EOF method at high salinity conditions. Moreover, results from the EOF method showed also bad reproducibility and questionable reliability.17,28–31,33 Results were of varying quality, for instance with 14% difference in the SZP when using two different tracer particles under otherwise same measurement conditions in the dip cell19 and frequently compared with results obtained by the SP method, just to mention a few.13–21 Additionally, research showed that EOF is not applicable to oppositely charged surface and tracer particles.22 In another paper the need for the selection of non-ionic tracer particles was also pointed out in order to avoid electrostatic adsorption.20 As an example it was not possible to observe a positive zeta potential below the IEP of glass (negatively charged at neutral pH) using negatively charged tracer particles.26,34 Taking all these limitations into account, the necessity for a reliable, repeatable and reproducible SZP with minor measurement uncertainty is of particular concern, especially for the characterization of surface properties of more delicate materials such as membranes or biomaterials.
In spite of the already available investigations that compared the SP method with the EOF technique in the dip cell arrangement with few types of materials, such as different polymeric membranes,15 glass13,14 and PVDF foil,13 a comparison between both techniques for a wider range of materials and the study of the individual influences on the measurements is still missing. To our knowledge, no such study on the detailed comparison of the EOF and the SP methods for a range of inert, conductive and highly porous materials with diverse properties has been undertaken yet.
The comparison of both electrokinetic techniques, EOF and SP, was conducted on various materials (i.e. polyamide thin-film composite membrane, pristine and chitosan-coated polypropylene foil, cellulose acetate filter, silicon oxide wafer and Ni-based alloy) that cover a wide range of possible applications (microfilters for biopharmaceutical applications, membranes for desalination, food packaging, wet processing of semiconductor wafers, etc) and challenge both measurement techniques. In this way, the role of the chemical nature, the size and the charge of tracer particles as a function of ionic strength, pH, solid material's IEP, porosity and conductivity was examined. The SZP obtained with the EOF method was compared to the SZP determined from the SP measurement. For all measuring conditions we focused on the reliability, reproducibility and accuracy of the obtained SZP from both measuring techniques.
2. Materials and methods
FeSO4·7H2O was purchased from Riedel-De Haen, Fe2(SO4)3·7H2O, HCl (≥37%), tetraethylorthosilicate (TEOS, ≥98%) were all purchased from Sigma-Aldrich. NH4OH (25% aqueous solution), NaOH (>98%) and acetone (≥99.5%) were purchased from Honeywell. Absolute EtOH (anhydrous) was obtained from CarloErba and citric acid (≥99.5%, water free), KCl, HCl, and KOH were purchased from Roth. Albumin V from bovine serum (BSA) was purchased from Merck KgaA (USA). All chemicals were used as received, without any further purification. Ultrapure water (with a resistivity of 18.2 MΩ cm obtained from Milli-Q, Millipore Corporation, Massachusetts, USA) was used throughout the experiments. A latex dispersion standard with a conductivity of 0.4 mS cm−1 and pH 9 was provided by Anton Paar GmbH. A silica nanoparticle dispersion was prepared from TM-40 colloidal silica, 40 wt% suspension in H2O (Ludox, Sigma-Aldrich), with a conductivity of 4.85 mS cm−1 and pH 8.5–9.5.2.1 Solid samples
One of the initial goals to use the SZP analysis was the characterization of the surface and interfacial charge of flat sheet polymer membranes.3 We therefore included a polyamide thin-film composite membrane for reverse osmosis (SW30-HR, Dow Chemical, USA). For equilibration the RO membrane was soaked in Milli-Q water prior to the SZP analysis. A polypropylene film with 50 μm thickness was obtained from Goodfellow (Huntingdon, UK). A single-side polished silicon wafer (150 mm, thickness 675 μm) with a 1000 Å thick silicon oxide coating was cut into pieces of 20 mm × 10 mm (Siegert Wafer, Aachen, Germany). Cellulose acetate microfiltration membranes with 0.2 μm and 0.45 μm pore size were obtained from Sartorius (Göttingen, Germany). A foil of Hastelloy C-276 (NiMoCr alloy, thickness 1 mm) was purchased from GoodFellow, UK.2.2 Preparation of chitosan-based PP foil
To achieve positive charge of the surface, the PP foil was functionalized with coating based on polysaccharide chitosan. The surface of PP was first cleaned ultrasonically in a bath of 70% ethanolic solution for 5 min, afterwards dried in an oven at 40 °C and finally activated for 20 min using ultraviolet-ozone surface treatment; during the activation process, the surface becomes more hydrophilic and consequently better adhesion of dispersions is obtained. The PP foil was functionalized in two layers (layer-by-layer composition): as a (1) layer 2 wt% chitosan macromolecular solution was applied and (2) layer was formed with dispersion of chitosan nanoparticles with embedded cinnamon extract. All the details of a solution preparation are presented in our recently published paper.41 After application of each layer the foil was dried at room temperature. For modification of the surface, the method of printing using a magnet was used (roll printing).2.3 Preparation of tracer particle dispersion
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :n(Fe3+) = 2.4:1 ratio was prepared by dissolving iron sulphate in ultrapure water. Afterwards, diluted aqueous ammonia solution was slowly added to iron salts solution at pH 3 to precipitate iron hydroxides. For the formation of MNPs, 125 mL of ammonia solution (25%) was added to the above mixture and additionally agitated with a magnetic stirrer for 30 min. As-prepared MNPs were cleaned several times with diluted ammonia solution and ultrapure water. Then, the stable colloidal dispersion of MNPs was prepared using adsorption of citric acid. Here, 0.6 g of as-prepared bare MNPs were redispersed in 30 mL of ultrapure water and 2.5 mL (0.5 g mL−1) citric acid aqueous solution was added. The pH was raised to 5.2 with diluted ammonia solution and put under reflux for 1.5 h at 80 °C. After refluxing, the pH of cooled dispersion was set to ∼10 with ammonia solution (25%). Stable MNPs were then coated with thin silica shell (MNPs@SiO2). NH4OH was added to stable MNPs dispersion (15 mg mL−1, pH = 10.6). The mixture was agitated for 15 min and added rapidly to the solution of EtOH and TEOS (10 mg mL−1). This was followed with pH setting to 10.6, using 25% NH4OH. The coating process was left to proceed for 2 h under continuous stirring. The obtained core–shell MNPs@SiO2 were cleaned to remove the excess reagents using absolute EtOH and ultrapure water. The details about the preparation of the MNPs@SiO2 and about their characteristics can be found in the following ref. 42 and 43. For the EOF analysis, 0.1 wt% dispersions of MNPs@SiO2 in 2 mM KCl were prepared with the pH adjusted to pH 3, 5, 7 and 9. Prior to be used as a tracer particle dispersion and to remove possible agglomerates, the dispersion was additionally filtered using a 1 μm filter.
:n(Fe3+) = 2.4:1 ratio was prepared by dissolving iron sulphate in ultrapure water. Afterwards, diluted aqueous ammonia solution was slowly added to iron salts solution at pH 3 to precipitate iron hydroxides. For the formation of MNPs, 125 mL of ammonia solution (25%) was added to the above mixture and additionally agitated with a magnetic stirrer for 30 min. As-prepared MNPs were cleaned several times with diluted ammonia solution and ultrapure water. Then, the stable colloidal dispersion of MNPs was prepared using adsorption of citric acid. Here, 0.6 g of as-prepared bare MNPs were redispersed in 30 mL of ultrapure water and 2.5 mL (0.5 g mL−1) citric acid aqueous solution was added. The pH was raised to 5.2 with diluted ammonia solution and put under reflux for 1.5 h at 80 °C. After refluxing, the pH of cooled dispersion was set to ∼10 with ammonia solution (25%). Stable MNPs were then coated with thin silica shell (MNPs@SiO2). NH4OH was added to stable MNPs dispersion (15 mg mL−1, pH = 10.6). The mixture was agitated for 15 min and added rapidly to the solution of EtOH and TEOS (10 mg mL−1). This was followed with pH setting to 10.6, using 25% NH4OH. The coating process was left to proceed for 2 h under continuous stirring. The obtained core–shell MNPs@SiO2 were cleaned to remove the excess reagents using absolute EtOH and ultrapure water. The details about the preparation of the MNPs@SiO2 and about their characteristics can be found in the following ref. 42 and 43. For the EOF analysis, 0.1 wt% dispersions of MNPs@SiO2 in 2 mM KCl were prepared with the pH adjusted to pH 3, 5, 7 and 9. Prior to be used as a tracer particle dispersion and to remove possible agglomerates, the dispersion was additionally filtered using a 1 μm filter.2.4 Electro-osmotic flow mapping
The EOF experiments were performed with a Zetasizer Nano ZS equipped with a He–Ne laser (λ = 633 nm) and the SZP accessories (Malvern, UK). The signal for dynamic light scattering (DLS) was detected at 173° and for the electrophoretic mobility measurement at 13°. The SZP analysis were conducted by the method described by the Corbett et al.13 Briefly, for the SZP analysis the sample was cut into rectangular pieces not larger than 7 mm × 4 mm (L × W) and attached via a double-sided adhesive tape to the sample holder (7 mm × 4 mm, poly(ether ether ketone), PEEK) perpendicular between the electrodes of the dip cell. The completed SZP cell was then placed into the electrophoretic light scattering (ELS) instrument. It should be noted that special care was taken not to damage the sample surface during the attachment to the sample holder. A coarse alignment of the zero position of the sample stage was performed using the height alignment tool. A disposable cuvette was filled with 1.2 mL of the prepared aqueous tracer particle dispersion and then the SZP cell was inserted. The latter should be done by tilting the cuvette in order to avoid any bubbles being caught between sample and electrodes, and to ensure that the sample plate was entirely submerged. The final alignment of the sample stage was performed using the count rate meter of the Zetasizer software (version 7.12). It should be noted that special attention has to be paid for the fine alignment since hitting the sample surface with the laser beam gives a false indication about the zero-position location that significantly lowers the quality of the data. The attenuator was set to 11 (i.e., the full intensity of the laser beam was used) and forward scatter was selected for monitoring the ELS signal. The number of ELS runs for the EM measurement was selected automatically and the measurements of the EM for each surface displacement was repeated 3 times. The SZP was evaluated from the measurement of the EM at 4 displacements with a step size of 125 μm. For the EM measurement the applied voltage, the attenuation of the laser beam, and the number of consecutive runs were adjusted automatically with 3 repetitions at 1500 μm displacement. The apparent EM of the tracer particles (which may be converted to an apparent zeta potential) was measured at surface displacements of 125, 250, 375 and 500 μm using the slow field reversal mode while the tracer particle mobility itself was measured at 1500 μm using the fast field reversal mode. The measurement relies on the assumption that the EOF at the solid surface decays with increasing distance from the surface. Close to the surface the velocity of the tracer particles will be dominated by the electroosmotic flow, while at distances far away from the surface it will be dominated by its EM.The apparent – zeta potential ζapparent is calculated at each displacement from the mobility measurement by
| ζapparent = μapparentη/(εrelε0) | (1) |
The SZP is then calculated by eqn (2) using the zeta potential of the tracer particle (determined at a distance far from the surface) and the intercept on the y-axis, which is obtained by a linear extrapolation of the experimental data for the particle mobility at various distances from the solid surface.
| Surface zeta potential = −intercept + tracer zeta potential | (2) |
2.5 SP and streaming current method
The measurement of the SP Ustr (and alternatively of the streaming current Istr) is the direct approach to the SZP. Liquid flow through a capillary generates an electric potential. This electrokinetic effect is used to assess the SZP for macroscopic solids with a flat surface. A pressure gradient Δp is applied between both ends of a rectangular flow channel, which generates liquid flow and the SP signal. The SP is recorded within a range of pressure differences and the slope of the linear dependence (the SP coupling coefficient dUstr/dΔp) is used to calculate the SZP according to eqn (3):| ς = dUstr/dΔp × η/(εrel × ε0)κB | (3) |
The SP measurements were performed with SurPASS 3 (Anton Paar GmbH, Austria) using the adjustable gap cell for mounting samples with a flat surface. A pair of each sample with a size of 20 mm × 10 mm was mounted on the sample holder using double-sided adhesive tape. The distance between sample surfaces was adjusted to 110 ± 10 μm. The electrolyte solutions were 2 and 10 mM KCl and the pH was automatically adjusted with 0.05 M KOH and 0.05 M HCl. Prior to the measurement the solid sample was equilibrated at neutral pH with several rinsing steps and then pH was set to the alkaline range. A pressure gradient of 200–600 mbar was applied to generate the SP or the streaming current, which was measured using a pair of AgCl electrodes. For each individual pH, 3 measurements were performed and the average SZP is reported. The electrolyte pH and conductivity were monitored using pH and conductivity probe, and all the experiments were done at room temperature. Between individual sample analyses, the electrolyte system was thoroughly rinsed with ultra-pure water to ensure that any prior solution was removed.
3. Results and discussion
3.1 Verification of the EOF method
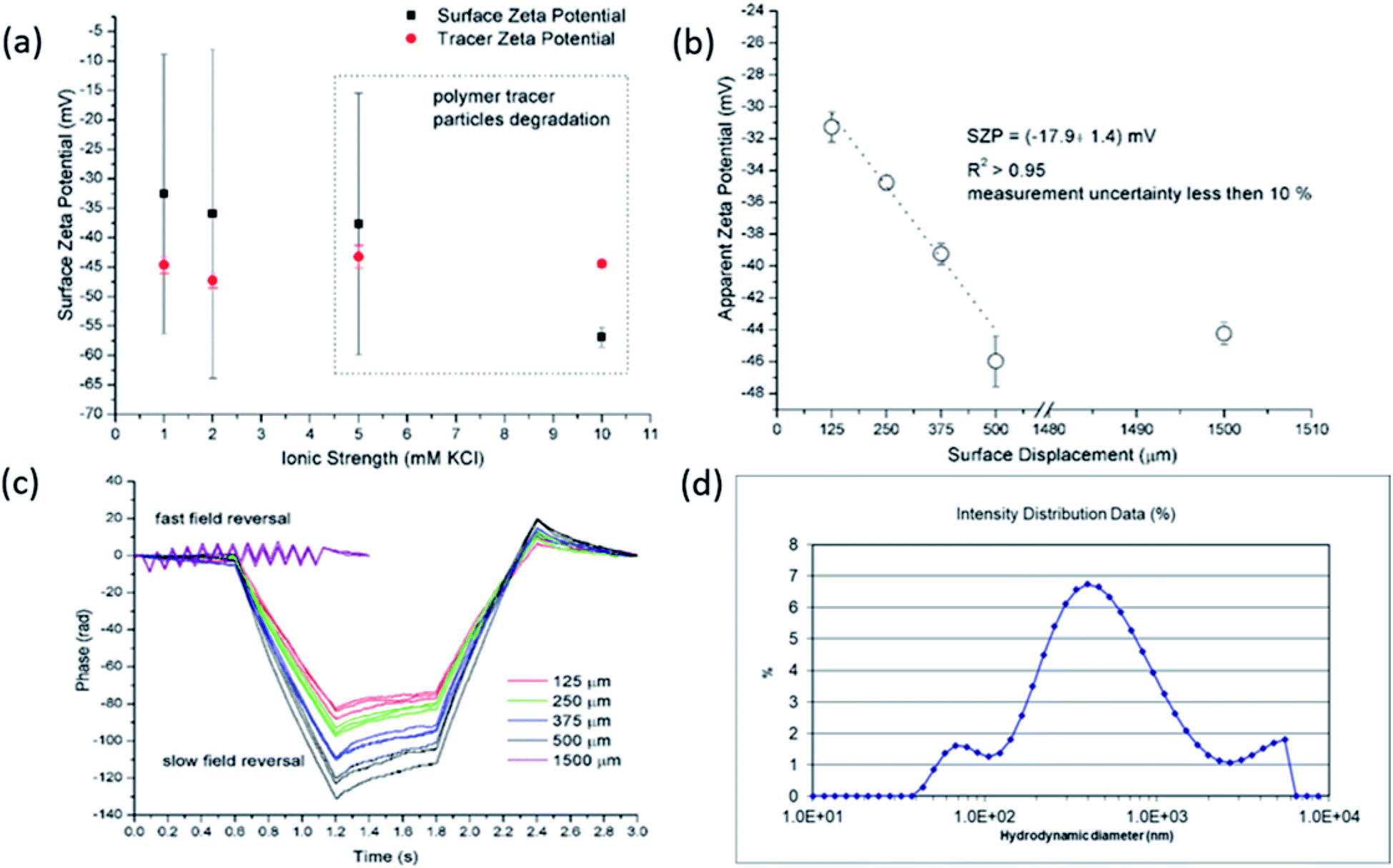 | ||
| Fig. 1 SZP results from EOF measurements for RO membrane using latex tracer particles as a function of the ionic strength (a). Exemplarity apparent zeta potential at different displacement in 2 mM KCl (b) and corresponding phase plots (c). Size distribution of the latex tracer particles after EOF measurement at 5 mM ionic strength (d). | ||
Preliminary experiments clearly revealed that the standard polymeric latex tracer particles are not suitable to be used as tracer particles for the determination of the SZP using the EOF mode under certain conditions – especially in media close to ambient conditions, where the materials' surfaces are exposed to even more complex aqueous environments. However, this is of paramount importance for measurements that allow for the phenomenological assessment of the solid materials' properties that are mainly used for industrial purposes. Here, the accurate and reliable SZP analysis is important for the prediction of, e.g., membrane performance. The lack of proper measurement can be in the first place attributed to polymeric tracer particle degradation (the degradation of the electrodes that are integrated in the dip cell was excluded) that significantly influences the absolute value and interpretation of the SZP. Further research was therefore focused on finding more inert, stable and appropriate inorganic tracer particles with a suitable size and a negative charge. It should be also pointed out that further experiments were limited to 2 mM ionic strength in order to achieve reliable data with EOF mapping.
3.2 Comparison of EOF and SP: influence of different “novel” effects
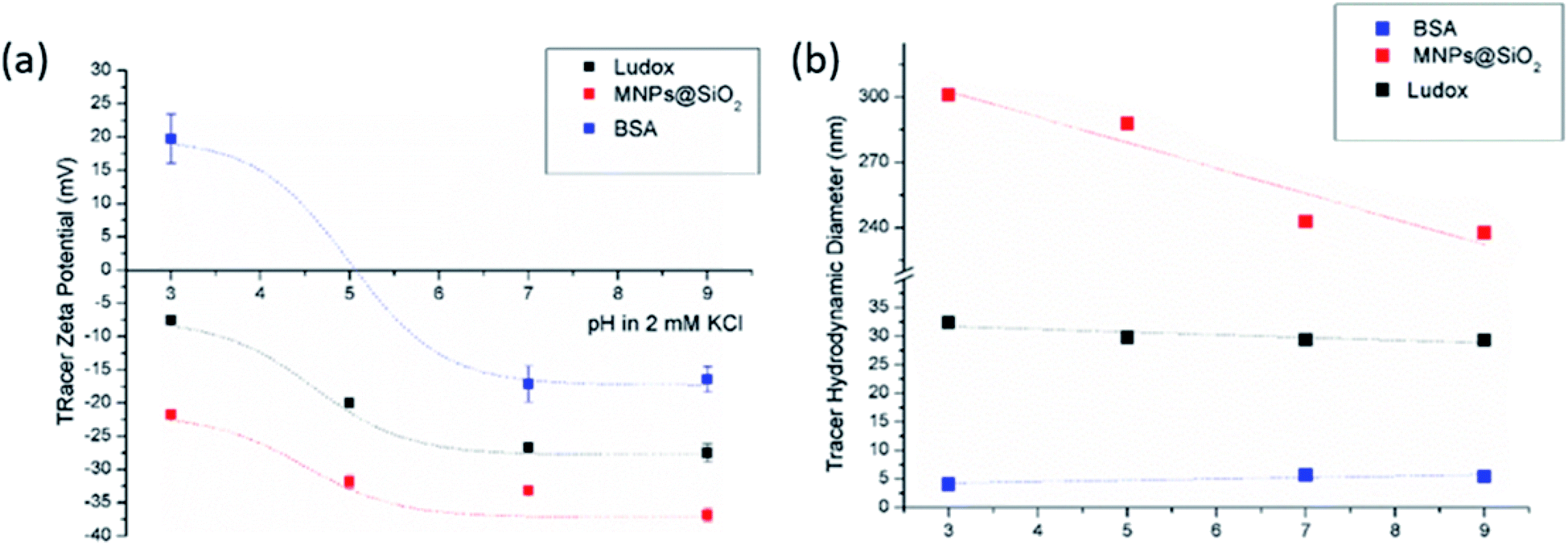 | ||
| Fig. 2 Tracer ZP (a) and hydrodynamic diameter (b) as a pH function at initial 2 mM KCl electrolyte solution. | ||
The size effect of two different inorganic tracer particles (MNPs@SiO2 and Ludox) with similar surface chemistry together and of a protein (BSA) was studied on a polypropylene (PP) foil, which exhibits a simple, non-complex flat and inert polymer surface (Fig. 3a), and was suggested as a reference material for the non-destructive SZP analysis by the SP method.45–47 The SP data for the PP foil show minimum measurement uncertainty (less than 10%), good repeatability (each point is the mean value of three repetitions) and the IEP at pH 3.8, which is typical for polymers.48 Oppositely, larger deviations were observed for the indirect EOF method for all three types of tracer particles. A significantly larger measurement uncertainty was determined using the MNPs@SiO2 particles, which exhibit the largest particle size. Otherwise all three results follow a similar trend as seen in the SP measurement. For instance, at pH 7 the measurement uncertainty was 29% with magnetic particles, 24% with Ludox and 6% with BSA (Fig. 3a). In comparison the SZP determined from SP measurements shows a repeatability of 1.1%. Although smaller in size, BSA showed the smallest deviation in comparison to the SP measurements, however, problems were observed with the EOF method that were associated with the protein deposition onto the palladium electrodes of the dip cell. Ludox and magnetic tracer particles were therefore selected to study solid materials with a more complex surface behavior, such as the RO membrane (Fig. 3b). Similarly as in the case of the PP foil, SP data show reliable results with minor measurement uncertainty exhibiting an IEP and the magnitude of the SZP that are common for such kind of polyamide thin-film composite membranes.49 On the contrary, results provided from EOF mapping showed a significant deviation from the SP results, but with a smaller measurement uncertainty as in the case of the PP foil. For instance, at pH 7 a standard deviation of 16% was determined with Ludox tracer particles, and 11% with MNPs@SiO2. Both types of tracer particles showed negative phase plots at all surface displacements at pH > IEP similarly as represented in Fig. 1c for the latex tracer particles. These results indicate that also small tracer particles can be used opposite to the suggestion by Mateos et al.14 who concluded that smaller tracer particles with a small absolute value of the ZP are not applicable for the EOF method due to the high effect of the EOF onto tracer mobility. Besides this, one should also take into account the effect of ionic strength that contributes to the EM of tracer particles. Interestingly, regardless of the sample surface tested, with negatively charged tracer particles neither the IEP nor the oppositive sign of the SZP for positively charged surfaces was achieved (Fig. 3) and can be related to possible Ludox particles attachment to positively charged surface. Moreover, if we compare the ZP trend of the tracer particles (Fig. 2a) with the SZP determined by the EOF method, a similar behaviour can be seen. In the case of the RO membrane larger deviations in the SZP were observed for magnetic nanoparticles when compared to SP data, thus in continuation, Ludox was chosen as an optimal tracer particle, which provided more accurate data.
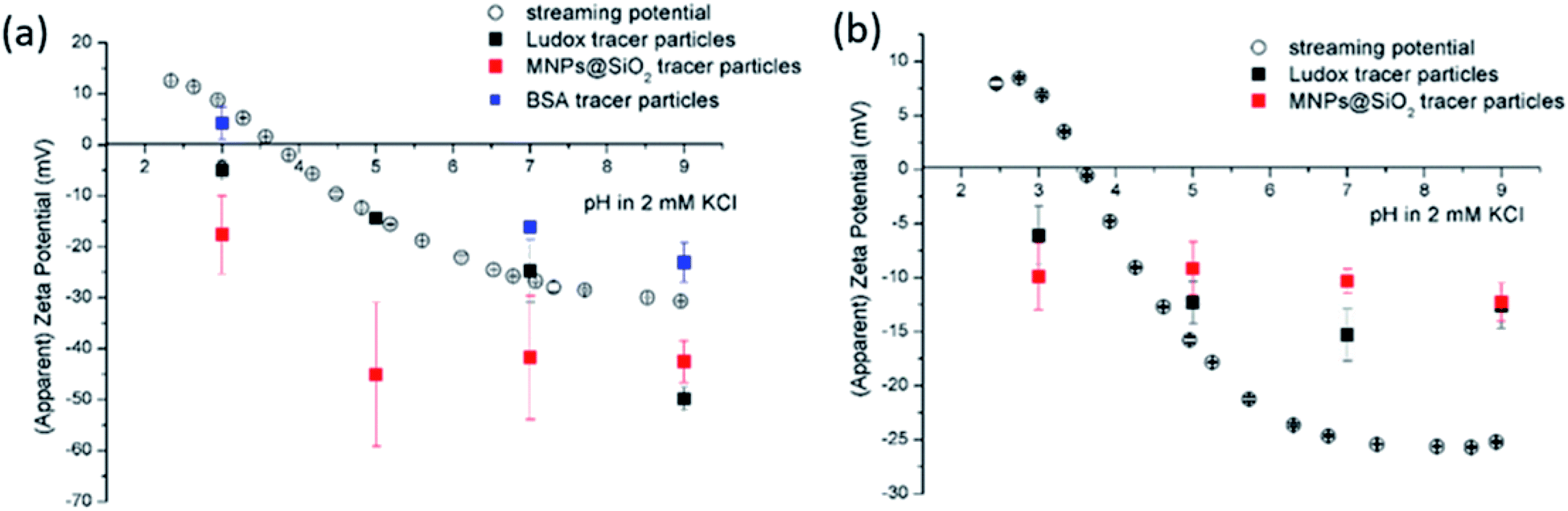 | ||
| Fig. 3 Surface zeta potential for (a) a polypropylene foil and (b) a reverse osmosis membrane determined from streaming potential measurement (empty circles) and EOF mapping (filled squares) using different tracer particles (black: Ludox, red: MNP@SiO2, blue: BSA). The symbols represent the average zeta potential of three measurements and the error bars the corresponding standard deviation. For the colour code the reader may refer to the electronic version. | ||
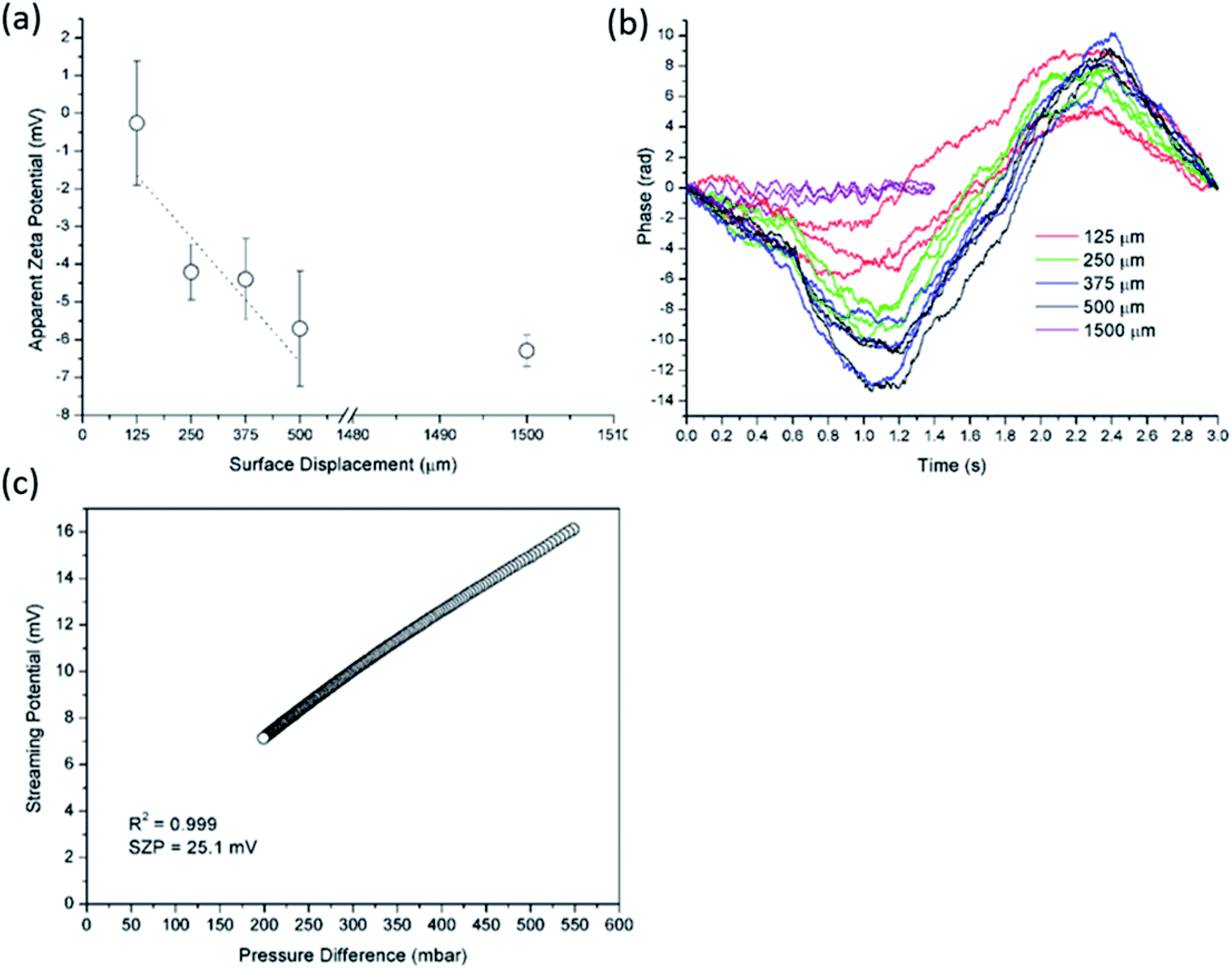 | ||
| Fig. 4 Apparent zeta potential versus surface displacement for EOF method using Ludox tracer particles at pH 3 and initial 2 mM ionic strength for PP foil (a). In (b) corresponding phase plots are illustrated. The pressure ramp at pH 3 for PP foil obtained from SP measurement is shown in (c). | ||
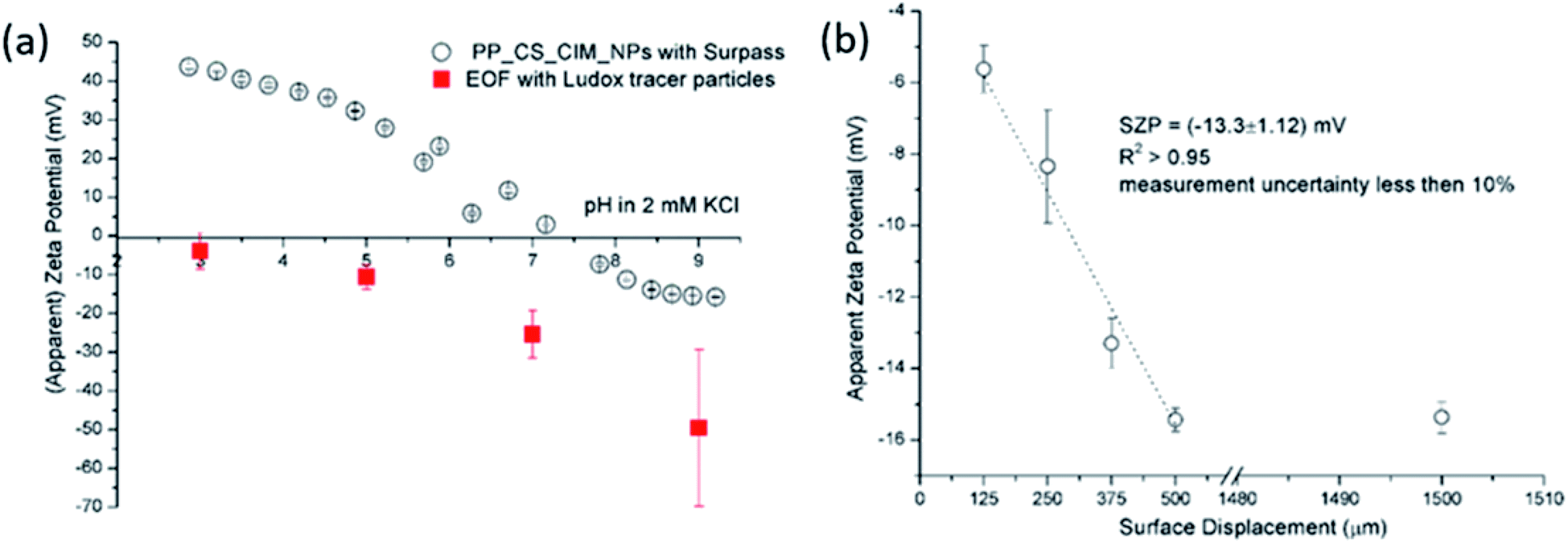 | ||
| Fig. 5 SZP of chitosan-based functional coating on PP foil determined with the SP and EOF methods using Ludox tracer particles (a). Exemplarily shown apparent zeta potential versus surface displacement for EOF measurement at pH 5 in 2 mM ionic strength using Ludox tracer particles (b). | ||
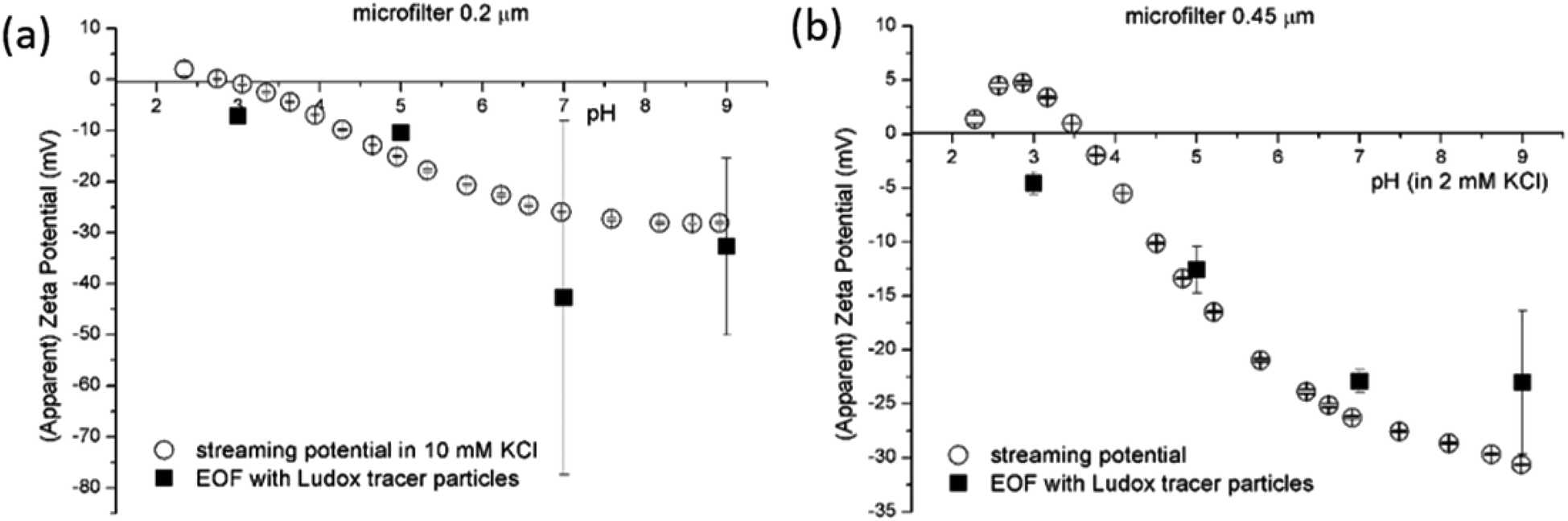 | ||
| Fig. 6 SP and EOF methods for SZP determination for microfilters with pore sizes 0.2 μm (a) and 0.45 μm (b). | ||
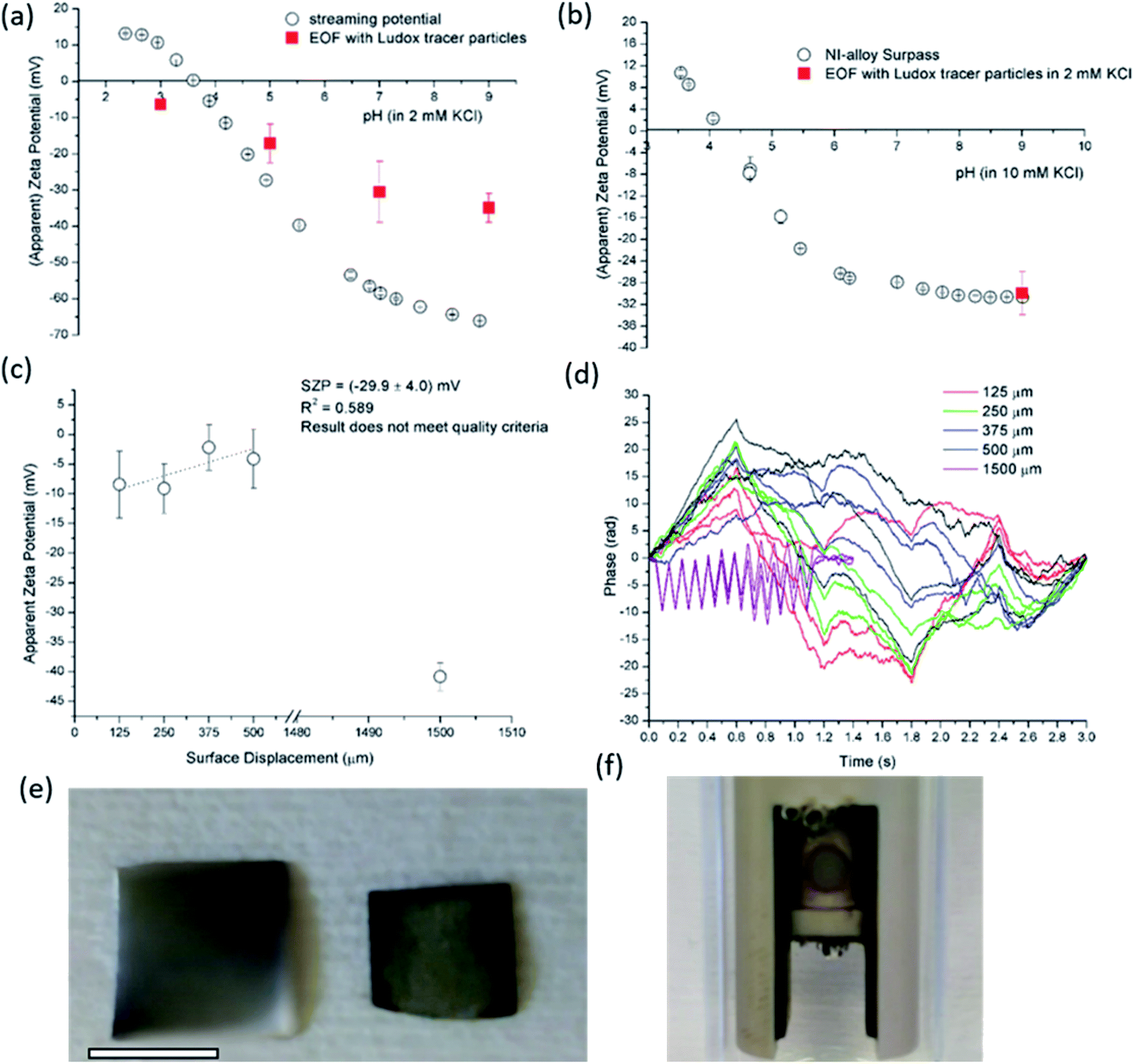 | ||
| Fig. 7 SP and EOF data using Ludox tracer particles for semiconductive silicon wafer (a). EOF measurement and SP data in all pH range for Ni-based conductive alloy (b). Apparent zeta potential versus surface displacements for Ni-based alloy at pH 9 and 2 mM ionic strength (c) with corresponding phase plots (d). The damage caused on the Ni-based alloy after EOF measurement in shown in ((e); right Ni-based alloy), where bar is 4 mm. Observation of possible electrochemical reaction with bubbles formation is presented in (f). | ||
3.3 Repeatability, reproducibility and reliability of EOF and SP
| EOF method | SP method | ||||
|---|---|---|---|---|---|
| SZP (mV) | Relative uncertainty (%) | R2 | SZP (mV) | Relative uncertainty (%) | R2 |
| −12.1 ± 4.5 | 37 | 0.716 | −29.5 ± 0.4 | 1.3 | 0.999 |
| −14.9 ± 2.5 | 17 | 0.881 | −29.1 ± 0.3 | 1.2 | 0.999 |
| −10.8 ± 3.6 | 33 | 0.806 | −28.8 ± 0.4 | 1.2 | 0.999 |
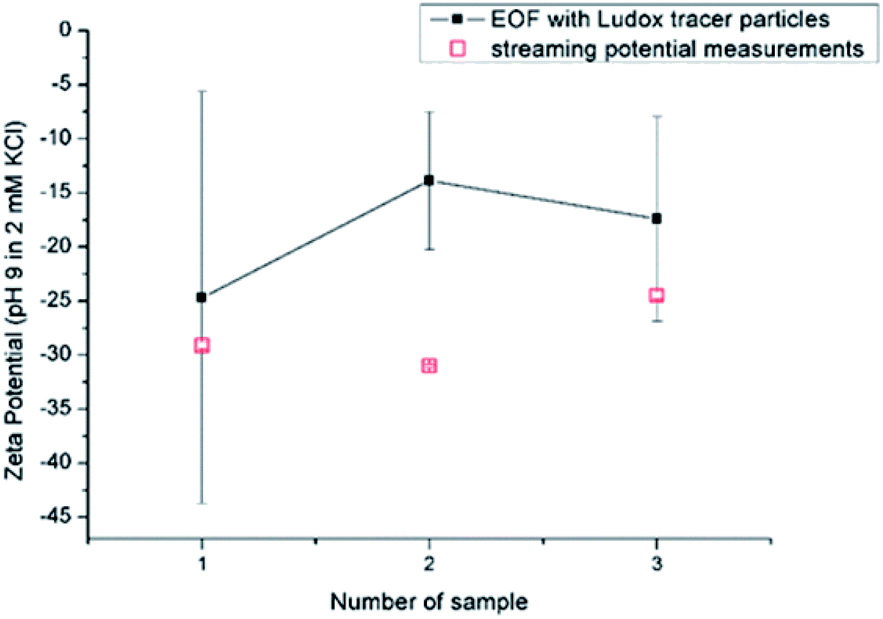 | ||
| Fig. 8 Reproducibility for RO membrane using EOF mapping with Ludox tracer particles and SP at pH 9 in 2 mM KCl. | ||
4. Conclusions
In this report we compared in detail two different electrokinetic phenomena for the SZP determination, i.e. the electro-osmotic flow using tracer particles in a dip cell arrangement and the SP method. We took into account materials with different surface and bulk properties (roughness, porosity, electric conductivity) that are expected to influence the SZP analysis. For the validation of the EOF mapping method, we investigated the effects of the type of tracer particles, of the ionic strength, and of the applied voltage. The results revealed the unsuitability of the standard latex dispersion to be used as tracer particles at an electrolyte concentration exceeding 5 mM KCl, and at an applied voltage between palladium electrodes of 5 V. Two different silica-based tracer particles of different sizes were thus compared, and more reliable data for the EOF method were obtained with Ludox tracer particles. In general the indirect SZP analysis by the EOF method showed a large difference and significantly higher measurement uncertainty when compared to the SP method. Interestingly, for both negatively charged tracer particles neither the IEP nor a positive sign of the SZP were achieved for all solid samples studied. This was not the case for the SP method. We explain the failure to obtain a positive SZP with negatively charged tracer particles by a possible electrostatic attraction of particles to the solid surface. The effect of sample porosity was tested on using microfiltration membranes with different pore size, which lead to large measurement uncertainties for the EOF method, while minor error bars were obtained for the SP method. While the streaming current measurement allowed for a reasonable and reliable SZP for conductive samples, several obstacles were observed during EOF mapping that resulted even in the surface damage of a stainless metal sample. Measurement repeatability, reproducibility and reliability for selected samples are satisfactory when using the SP method, confidence in for the SZP obtained by EOF mapping is rather low. The EOF method disables the use of the environmentally relevant ionic strength due to tracer particle degradation.The benefit of the EOF method is a smaller investment in the SZP accessory provided that a specific ELS instrument is already available but the disadvantages are as obvious. We observe a significant consumption of tracer particles, which likely degrade during the EOF measurement and require an exchange for every single measurement. Moreover, longer measurement times are required correlated with the high labour cost and longer measurement times. The manual adjustment of the surface displacements using the dip cell and the preparation of individual dispersions of tracer particles at each pH require a longer measurement time and the permanent attention of the operator. On the other hand, only a fraction of the measuring time and minimal user attention are required for the SP method.
In conclusion the reliability of SZP results obtained with the EOF method is recognized only by a comparison with the corresponding SP measurement.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support of Ministry of Higher Education, Science and Sport of the Republic of Slovenia under contract C3330-17-529004 and from the Slovenian Research Agency (research core funding no. P2-0118) is gratefully acknowledged.References
- R. J. Hunter, Zeta potential in colloid science: Principles and applications, Academic Press, London, 1981 Search PubMed
.
- D. J. Shaw, Introduction to colloid & surface chemistry, Butterworth-Heinemann, Oxford, 4th edn, 1992 Search PubMed
- M. Elimelech, W. H. Chen and J. J. Waypa, Desalination, 1994, 95, 269–286 CrossRef CAS
- A. Imbrogno, A. Tiraferri, S. Abbenante, S. Weyand, R. Schwaiger, T. Luxbacher and A. I. Schäfer, J. Membr. Sci., 2018, 549, 474–485 CrossRef CAS
- K. Stana-Kleinschek and V. Ribitsch, Colloids Surf., A, 1998, 140, 127–138 CrossRef CAS
- A. M. Grancaric, A. Tarbuk and T. Pusic, Color. Technol., 2005, 121, 221–227 CAS
- I. Petrinić, H. Bukšek, T. Luxbacher, T. Pušić and S. Bischof, J. Appl. Polym. Sci., 2018, 135, 1–8 CrossRef
- M. Espanol, G. Mestres, T. Luxbacher, J. B. Dory and M. P. Ginebra, ACS Appl. Mater. Interfaces, 2016, 8, 908–917 CrossRef CAS PubMed
- M. Lorenzetti, G. Bernardini, T. Luxbacher, A. Santucci, S. Kobe and S. Novak, Biomed. Mater., 2015, 10, 45012 CrossRef CAS PubMed
- B. J. Pedimonte, T. Moest, T. Luxbacher, C. Von Wilmowsky, T. Fey, K. A. Schlegel and P. Greil, Acta Biomater., 2014, 10, 968–974 CrossRef CAS PubMed
- M. S. Niepel, D. Peschel, X. Sisquella, J. A. Planell and T. Groth, Biomaterials, 2009, 30, 4939–4947 CrossRef CAS PubMed
- V. Ribitsch, C. Jorde, J. Schurz and H. J. Jacobasch, Prog. Colloid Polym. Sci., 1988, 77, 49–54 CAS
- J. C. W. Corbett, F. Mcneil-watson, R. O. Jack and M. Howarth, Colloids Surf., A, 2012, 396, 169–176 CrossRef CAS
- H. Mateos, A. Valentini, E. Robles, A. Brooker, N. Cioffi and G. Palazzo, Colloids Surf., A, 2019, 576, 82–90 CrossRef CAS
- T. E. Thomas, S. Al Aani, D. L. Oatley-Radcliffe, P. M. Williams and N. Hilal, J. Membr. Sci., 2017, 523, 524–532 CrossRef CAS
- M. A. E. Cruz, R. M. de Souza, L. G. Dias and A. P. Ramos, Thin Solid Films, 2017, 638, 433–440 CrossRef CAS
- S. Al, C. J. Wright and N. Hilal, Desalination, 2018, 432, 115–127 CrossRef
- Y. Shim, H.-J. Lee, S. Lee, S.-H. Moon and J. Cho, Environ. Sci. Technol., 2002, 36, 3864–3871 CrossRef CAS PubMed
- J. M. Vasconcelos, F. Zen, S. N. Stamatin, J. A. Behan and P. E. Colavita, Surf. Interface Anal., 2017, 49, 781–787 CrossRef CAS
- N. J. W. Penfold, A. J. Parnell, M. Molina, P. Verstraete, J. Smets and S. P. Armes, Langmuir, 2017, 33, 14425–14436 CrossRef CAS PubMed
- C. Zhao, G. Hu, D. Hou, L. Yu, Y. Zhao, J. Wang, A. Cao and Y. Zhai, Sep. Purif. Technol., 2018, 202, 385–396 CrossRef CAS
- K. Hiratsuka, T. Suzuki, E. Dzieminska and M. Ichiyanagi, J. Fluid Sci. Technol., 2018, 13, 1–14 Search PubMed
- Y. Kakihana, L. Cheng, L. Fang, S. Wang, S. Jeon and D. Saeki, Colloids Surf., A, 2017, 533, 133–139 CrossRef CAS
- H. Rho, K. Chon and J. Cho, Desalination, 2018, 427, 19–26 CrossRef CAS
- S. R. Suwarno, S. Hanada, T. H. Chong, S. Goto, M. Henmi and A. G. Fane, Desalination, 2016, 387, 1–13 CrossRef CAS
- M. Kukizaki, Sep. Purif. Technol., 2009, 69, 87–96 CrossRef CAS
- T. Ishigami, K. Amano, A. Fujii, Y. Ohmukai, E. Kamio, T. Maruyama and H. Matsuyama, Sep. Purif. Technol., 2012, 99, 1–7 CrossRef CAS
- T. Hongo-Hirasaki, M. Komuro and S. Ide, Biotechnol. Prog., 2010, 26(4), 1080–1087 CAS
- A. Hozumi, H. Inagai, Y. Yokogawa and T. Kameyama, Thin Solid Films, 2003, 437, 89–94 CrossRef CAS
- M. J. Han, G. N. B. Baroña and B. Jung, Desalination, 2011, 270, 76–83 CrossRef CAS
- N. Park, S. Lee, S. Yoon Ro, Y. Hoon Kim and J. Cho, Desalination, 2007, 202, 231–238 CrossRef CAS
- J. Jang and W. Go, Fibers Polym., 2008, 9, 375–379 CrossRef CAS
- H. K. Shon, S. H. Kim, S. Vigneswaran, R. Ben Aim, S. Lee and J. Cho, Desalination, 2009, 238, 10–21 CrossRef CAS
- P. Xu, E. Drewes, T.-U. Kim, C. Bellona and G. Amy, J. Membr. Sci., 2006, 279, 165–175 CrossRef CAS
- S. Lee and J. Cho, Desalination, 2004, 160, 223–232 CrossRef CAS
- N. L. Burns, J. M. Van Alstine and J. M. Harris, Langmuir, 1995, 11, 2768–2776 CrossRef CAS
- B. Jachimska, S. Świątek, J. I. Loch, K. Lewiński and T. Luxbacher, Bioelectrochemistry, 2018, 121, 95–104 CrossRef CAS
- M. Vandenbossche, J. Dorst, M. Amberg, U. Schütz, P. Rupper, M. Heuberger and D. Hegemann, Polym. Degrad. Stab., 2018, 156, 259–268 CrossRef CAS
- F. Croisier, P. Sibret, C. C. Dupont-Gillain, M. J. Genet, C. Detrembleur and C. Jerome, J. Mater. Chem. B, 2015, 3, 3508–3517 RSC
- F. Croisier, G. Atanasova, Y. Poumay and C. Jérôme, Adv. Healthcare Mater., 2014, 3, 2032–2039 CrossRef CAS PubMed
- T. K. Glaser, O. Plohl, A. Vesel, U. Ajdnik, N. P. Ulrih, M. K. Hrnčič, U. Bren and L. F. Zemljič, Materials, 2019, 12, 2118 CrossRef PubMed
- O. Plohl, U. Ajdnik, S. Gyergyek, I. Ban, A. Vesel, T. K. Glaser and L. F. Zemljič, J. Environ. Chem. Eng., 2019, 7(1), 102913 CrossRef
- O. Plohl, M. Finšgar, S. Gyergyek, U. Ajdnik, I. Ban and L. Fras Zemljič, Nanomaterials, 2019, 9, 209 CrossRef CAS PubMed
- E. E. Uzgiris, Prog. Surf. Sci., 1981, 10, 53–164 CrossRef CAS
- M. Bauman, A. Košak, A. Lobnik, I. Petrinić and T. Luxbacher, Colloids Surf., A, 2013, 422, 110–117 CrossRef CAS
- K. M. Ashraf, D. Giri, K. J. Wynne, D. A. Higgins and M. M. Collinson, Langmuir, 2016, 32, 3836–3847 CrossRef CAS PubMed
- C. Wang, O. Zolotarskaya, K. M. Ashraf, X. Wen, D. E. Ohman and K. J. Wynne, ACS Appl. Mater. Interfaces, 2019, 11, 20699–20714 CrossRef CAS PubMed
- R. Zimmermann, U. Freudenberg, R. Schweiß, D. Küttner and C. Werner, Curr. Opin. Colloid Interface Sci., 2010, 15, 196–202 CrossRef CAS
- I. Owusu-Agyeman, A. Jeihanipour, T. Luxbacher and A. I. Schäfer, J. Membr. Sci., 2017, 528, 82–94 CrossRef CAS
- T. Tkavc, I. Petrinič, T. Luxbacher, A. Vesel, T. Ristić and L. F. Zemljič, Int. J. Adhes. Adhes., 2014, 48, 168–176 CrossRef CAS
- T. Ristić, Z. Persin, M. Kralj Kuncic, I. Kosalec and L. F. Zemljic, Text. Res. J., 2019, 89, 748–761 CrossRef
- L. Fras-Zemljič, I. Kosalec, M. Munda, S. Strnad, M. Kolar, M. Bračič and O. Šauperl, Cellulose, 2015, 22, 1933–1942 CrossRef
- U. Ajdnik, L. F. Zemljič, M. Bračič, U. Maver, O. Plohl and J. Rebol, Materials, 2019, 12, 1–20 CrossRef PubMed
- L. F. Zemljič, O. Plohl, A. Vesel, T. Luxbacher and S. Potrč, Int. J. Mol. Sci., 2020, 21, 495 CrossRef PubMed
- A. Yaroshchuk and T. Luxbacher, Langmuir, 2010, 26, 10882–10889 CrossRef CAS PubMed
- A. E. Childress and S. S. Deshmukh, Desalination, 1998, 118, 167–174 CrossRef
- S. Schwarz, K. J. Eichhorn, E. Wischerhoff and A. Laschewsky, Colloids Surf., A, 1999, 159, 491–501 CrossRef CAS
- B. Martin Cabanas, J. Lützenkirchen, S. Leclercq, P. Barboux and G. Lefèvre, J. Nucl. Mater., 2012, 430, 150–155 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Zeta potential and hydrodynamic diameter of latex tracer particles at different ionic strength; image of latex tracer particle degradation; size distribution of tracer latex particles at differently applied voltages together with determined surface zeta potential and effect of sample onto latex tracer particle stability. See DOI: 10.1039/c9ra10414c |
| This journal is © The Royal Society of Chemistry 2020 |