
Oriented porous LLZO 3D structures obtained by freeze casting for battery applications†
Hao
Shen
ab,
Eongyu
Yi
b,
Marco
Amores
 bc,
Lei
Cheng
bd,
Nobumichi
Tamura
e,
Dilworth Y.
Parkinson
e,
Guoying
Chen
b,
Kai
Chen
*a and
Marca
Doeff
*b
bc,
Lei
Cheng
bd,
Nobumichi
Tamura
e,
Dilworth Y.
Parkinson
e,
Guoying
Chen
b,
Kai
Chen
*a and
Marca
Doeff
*b
aCenter for Advancing Materials Performance from the Nanoscale (CAMP-Nano), State Key Laboratory for Mechanical Behavior of Materials, Xi'an Jiaotong University, Xi'an, Shaanxi 710049, China. E-mail: kchenlbl@gmail.com
bEnergy Storage and Distributed Resources Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA. E-mail: mmdoeff@lbl.gov
cDepartment of Chemistry, Graduate School of Science, The University of Tokyo, 7-3-1, Hongo, Bunkyo-Ku, Tokyo, 113-0033, Japan
dRobert Bosch LLC, Research and Technology Center, Sunnyvale, CA 94085, USA
eAdvanced Light Source, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA
First published on 20th August 2019
Abstract
All solid-state lithium batteries are, potentially, higher energy density and safer alternatives to conventional lithium-ion batteries (LIBs). These are particularly attractive characteristics for large-scale applications such as electric vehicles and grid energy storage systems. However, the thin film deposition techniques used to make current devices are not readily scalable, and result in low areal capacities, which translate to low practical energy densities. To overcome these deficiencies, it is necessary to design thicker electrodes similar to what are used in LIBs (30–100 μm), in which the active material is composited with an ionic conductor and an electronically conducting additive, to overcome transport limitations. In this paper, we propose a method for making such an electrode, starting with a porous scaffold, i.e. Li7La3Zr2O12 (LLZO), made by freeze casting, which is then infiltrated with the active material LiNi0.6Mn0.2Co0.2O2 (NMC-622) and other components. The freeze casting technique results in the formation of oriented channels with low tortuosity, which run roughly parallel to the direction of the current. The scaffolds were characterized with synchrotron X-ray micro-tomography for structural analysis, as well as synchrotron X-ray fluorescence to map the elemental distribution in the infiltrated composite. A hybrid half-cell was constructed and cycled as proof of principle, and it showed good stability. In addition, a bilayer structure consisting of a porous layer combined with a dense LLZO film was successfully made as a prototype of an all solid-state battery. A mathematical model was established to propose optimized scaffold structures for battery performance.
Introduction
The widespread use of Li-ion batteries (LIBs) in electronic devices and electric vehicles has highlighted the need for both improved safety and energy density. The use of lithium metal as an anode can potentially increase both specific energy and energy density due to the high gravimetric capacity (3869 mA h g−1) and low density (0.534 g cm−3).1–3 Unfortunately, safety issues preclude its use in conventional battery configurations because of mossy lithium deposition or Li-dendrite induced cell shorting when Li metal is cycled in cells with flammable liquid electrolytic solutions.4 To overcome this, solid state electrolytes have been proposed as safer alternatives to liquids. In particular, solid-state garnet type ceramic electrolytes, such as cubic Al-substituted Li7La3Zr2O12 (LLZO), are promising, due to their kinetic stability against Li,5–8 large potential window (0 ∼ 6 V), and ionic conductivities up to 1 mS cm−1.9–15 Although Li deposition or dendrite growth along the LLZO grain boundaries in polycrystalline dense films has recently been recognized as a source of shorting in cells, modifications of the LLZO/Li interface and cell architecture have successfully improved the stability and cycling behavior of symmetrical cells.11,16–19Another challenge, particularly if solid-state batteries are to be used for large-scale applications such as vehicles, has to do with the design of the electrodes and cells.20 Most solid-state batteries are fabricated in thin film configurations to overcome the transport limitations of cathode materials.21–24 Not only does this usually require the use of vacuum deposition techniques, which are costly and difficult to scale, but it also results in low cathode areal capacity. For this reason, practical energy densities are low, in spite of the use of metallic lithium anodes. Several attempts have been made to build composite electrodes for solid-state batteries, in which the active material and ionic conductor are combined to improve ionic transport. In some cases, composites may be fabricated by cold-pressing the active material and electrolyte together.25–27 This is most easily accomplished with soft electrolytes such as sulfides. The low compressibility of garnets, in contrast, makes this an impractical approach. Instead, Fu et al. constructed a porous garnet layer by including a sacrificial polymer, which was burned away to create pores and then infiltrated these with other components to make a composite.28
Sander et al. recently demonstrated the advantages of low tortuosity pores in battery electrodes using a magnetic alignment technique on thick LiCoO2 electrodes.29 There was a noticeable increase of usable capacity of the aligned LiCoO2 electrodes with low tortuosity pores oriented in the direction of transport, compared to those with isotropic porosity, when cycled in half cells with liquid electrolytic solutions. To this end, McOwen et al. 3D printed LLZO scaffolds by using LLZO inks, which allows good control of the electrode structure.30 The effect of tortuosity on the electrochemical behavior of LLZO electrolytes has recently been described, with lower tortuosity potentially providing a means for achieving higher critical current densities and power densities in solid-state batteries.31 Another attractive method for producing low-tortuosity pores in ceramics is that of freeze casting.32 There has already been a report on freeze-casting LLZO to make ceramic/polymer composite electrolytes.33 Here we report our preliminary results using freeze casting to produce LLZO scaffolds with low tortuosity pores, which were subsequently infiltrated with the active material and used as electrodes in hybrid half-cells. A prototype all solid-state cell was also assembled. We used synchrotron radiation micro-computed tomography to visualize the three-dimensional (3D) models of the porous LLZO scaffolds and X-ray fluorescence to map distributions of the components. A simple mathematical model is also presented to provide guidance on the design of porous scaffolds.
Experimental
1. Preparation of porous LLZO scaffolds
Sub-micron-sized commercial Al-doped LLZO powders were obtained from MSE Supplies, Inc. for the freeze casting experiments. A slurry containing 20 wt% LLZO powder and 78 wt% tert-butyl alcohol (TBA) was mixed with 1 wt% polyvinyl butyral (PVB) and 1 wt% deionized water (to adjust the freezing point). Afterwards, another 5 wt% Li2CO3 was added to the slurry to compensate for Li loss during sintering. The mixture was ball-milled with ZrO2 grinding media for 8 h to form a stable LLZO slurry at room temperature. Although water is known to cause slow decomposition of LLZO, the amount of decomposed LLZO is negligible after several hours of ball milling. Moreover, during the sintering process, the decomposed LLZO is recovered through addition of extra Li2CO3 and heating. A house-made freeze casting system was used to prepare samples at various temperatures as illustrated in Fig. 1a. A PVC mold 2 cm in diameter and 1.5 cm in height was attached to a liquid nitrogen cooling bed with a polished copper plate in between to ensure excellent thermal conductivity. The top surface of the slurry is still exposed to ambient temperature resulting in a temperature gradient from the bottom (cold) to the top (warm). To control the freezing rate, the temperature of the slurry was adjusted and monitored using an embedded heating element and thermocouple in the cooling bed. For this work, the cooling plate temperature was set at −20 °C and −50 °C. After the temperature stabilized, the LLZO slurry was poured into the mold and maintained at the desired temperature until fully frozen. Then the solidified TBA with LLZO was unmolded and transferred to a freeze drier at −40 °C and kept under vacuum overnight to sublime the TBA crystals (Fig. 1b). The removal of TBA through this process results in a porous green body structure (inset of Fig. 1b). Some samples were also prepared using water as the solvent as follows: 15–30 wt% LLZO was mixed with 1 wt% Darvan, 1 wt% poly(vinyl alcohol) (PVA) with the remainder being deionized water. The samples were then were ball milled overnight and frozen at a cooling rate about of ∼20 °C min−1. One sample was also prepared by substituting gelatin for PVA.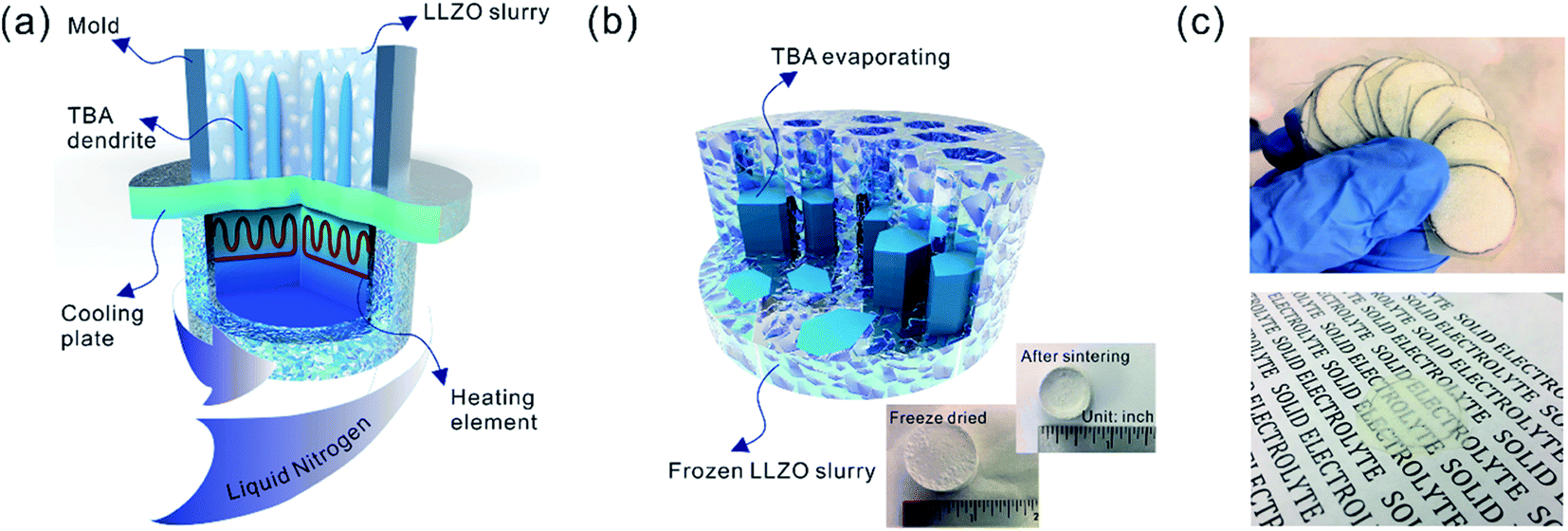 | ||
| Fig. 1 Schematics of (a) the freeze casting apparatus and (b) formation of the porous green body by freeze drying. Insets are the bulk scaffold before and after sintering, with rulers serving as scale bars. (c) Slices cut from the sintered scaffold mounted in epoxy. A 200 μm thick slice is transparent, as displayed in the lower part of (c). | ||
The scaffolds were then sintered at 1050 °C for 2 h in an Ar atmosphere. Ambient air was avoided because a large amount of lithium was lost, resulting in the decomposition of LLZO and crumbling of the structure (ESI, Fig. S1†). The sintered scaffolds decreased in volume substantially (inset of Fig. 1b). The scaffolds were then encapsulated with epoxy (Allied High Tech Products, Inc.) to avoid destroying the scaffold during the following operations. The epoxy had an insignificant volume change after curing. The samples were then sliced into thin sections using a diamond saw. The LLZO section with a thickness of 200 μm showed high optical transparency (Fig. 1c). Freestanding porous LLZO scaffold films were obtained after burning away the epoxy in dry air at 800 °C for 30 min.
2 Preparation of dense/porous LLZO bilayer structures
The oriented scaffold becomes fragile and hard to handle after sintering, especially when sliced thin. Adding the PVDF binder to the NMC slurry helps to maintain the integrity of the composite electrode. Another solution to avoid damage and fracture during assembly is to support the porous layers with a dense layer to form a bilayer structure. In this study, two types of dense LLZO structures with thicknesses of approximately 100 μm and 20 μm were used for the proof of principle study and the prototype half-cells, respectively. The thick dense films were fabricated using a tape casting method. The LLZO slurry for the dense layer preparation was a mixture of 30 wt% LLZO powder, 3 wt% Li2CO3 (32 wt% extra elemental Li in LLZO), 3 wt% PVA and 64 wt% analytically pure isopropyl alcohol. The slurry was introduced into a ZrO2 container and ball milled with ZrO2 milling media using a SPEX 8000M mixer for 30 minutes to guarantee a stable suspension of LLZO. Then the slurry was tape cast onto conventional household polyethylene membranes using a doctor blade, and dried at room temperature. The dried LLZO film was peeled off from the polyethylene membrane, cut into pieces and cold pressed using a stainless-steel die.The ultrathin LLZO dense films (∼20 μm thick) were fabricated by adopting a previously reported approach.34 LLZO, Li2CO3, polyvinyl butyral (binder), and benzyl butyl phthalate (plasticizer) were dissolved/dispersed in an alcohol/acetone mixed solvent system by ball-milling for 48 h using 3 mm ZrO2 beads. The suspension was formulated to result in ∼55 vol% LLZO/Li2CO3 excluding the solvent. The amount of Li2CO3 was selected to equal 50 wt% excess elemental Li in LLZO. The suspension was tape cast onto a Mylar substrate and left to dry for several hours. The dried green tapes were peeled off and punched to desired sizes for subsequent sintering.
The bilayer structure was prepared by co-sintering an LLZO porous scaffold (after epoxy burnout) and a dense film (green) at 1070 °C for 3 h in an Ar atmosphere between two pieces of alumina disks, each of which was 2 mm thick. No extra pressure was applied onto the LLZO scaffold and film besides the weight of the alumina disks. In order to prevent unwanted diffusion and reaction with the alumina, graphite foil pieces or Ni meshes were placed between the LLZO scaffold and the disks.
3 Active material infiltration and electrochemical testing
In this study, the NMC-622 particles (3–12 μm) provided by Umicore, Inc. were introduced into the LLZO scaffolds as the active material. A slurry was prepared by mixing 84 wt% NMC-622, 8 wt% carbon black, and 8 wt% PVDF in N-methyl-2-pyrrolidone (NMP). During the stirring, several more drops of NMP were added to adjust the viscosity of the slurry. The slurry was then drop-cast onto the LLZO scaffolds. The success of the infiltration process of NMC particles could be readily indicated by a color change from natural ivory to black. It usually took several rounds of infiltration to fully fill the pores, as confirmed by scanning electron microscope (SEM) observation of the bottom surface of the infiltrated sample. The addition of polyaniline was also demonstrated in freeze cast structures (see ESI, Fig. S2e†). A partially infiltrated sample was also prepared using smaller (submicron diameter) NMC particles (ESI, Fig. S3†). Smaller NMC particles were obtained by ball-milling the as-received NMC for 48 h in EtOH using 2 mm ZrO2 beads which resulted in fracturing of secondary particles to primary particles. After infiltration, samples were kept in a vacuum oven at 80 °C overnight.The composite structures were then transferred to an argon-filled glove box for assembly into coin cells. For the hybrid cells, Celgard 2400 polypropylene membranes wet with 1 M LiPF6 in ethylene carbonate![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :diethyl carbonate (1:1 vol%) were employed as separators, and Li foil pieces (Alfa-Aesar) were used as the anodes. The coin cells were then galvanostatically cycled using a VMP3 multichannel potentiostat/galvanostat equipped with a frequency response analyzer between 2.5 and 4.7 V at a current density of 0.3 mA cm−2 (approximately C/10) after 12 hours of rest. The impedance was measured from 1 mHz to 100 kHz every 10 cycles. The bilayer structures were also assembled into coin cells with lithium anodes, without polymeric separators or liquid electrolyte. The open circuit voltage was monitored for 12 h.
:diethyl carbonate (1:1 vol%) were employed as separators, and Li foil pieces (Alfa-Aesar) were used as the anodes. The coin cells were then galvanostatically cycled using a VMP3 multichannel potentiostat/galvanostat equipped with a frequency response analyzer between 2.5 and 4.7 V at a current density of 0.3 mA cm−2 (approximately C/10) after 12 hours of rest. The impedance was measured from 1 mHz to 100 kHz every 10 cycles. The bilayer structures were also assembled into coin cells with lithium anodes, without polymeric separators or liquid electrolyte. The open circuit voltage was monitored for 12 h.
4 Structure characterization
The morphologies of the selected samples were examined using a Hitachi TM-1000 tabletop scanning electron microscope (SEM). Filters were applied to binarize the SEM images (ESI, Fig. S4†) to enable measurement of pore size and estimate porosity by calculating the dark area. X-ray powder diffraction (XRD) patterns were acquired on a Bruker D2 PHASER diffractometer with monochromatic Cu Kα radiation to check the phase purity of the samples.Because of the fragility of the LLZO samples after sintering, the freeze cast pellet was first mounted in epoxy prior to sample characterization. Both the LLZO films and longitudinal sections were cut from the same sample. Films were cut perpendicular to the TBA channel growing direction, while the longitudinal sections were cut parallel to the channel direction. The thicknesses of the films and the diameters of the longitudinal sections were controlled to be 200–300 μm. Then synchrotron micro-computed tomography (SR-μCT) experiments were performed at Hard X-ray Micro-Tomography Beamline 8.3.2 of the Advanced Light Source (ALS) on the samples. Images were collected over 180 degrees in 0.072 degree steps, with 24 keV X-rays. Detection was accomplished with a 50 micrometer thick LuAG:Ce scintillator, a 10× Olympus optical lens in an optical system from Optique Peter, and a PCO.edge sCMOS detector, with 500 ms exposure time. Dark field images (with the X-ray shutter closed) were collected to subtract detector dark counts, and bright field images were collected before and after the sample scan to normalize for variations in the incident illumination. The voxel dimension with this setup was approximately 0.64 microns. Tomographic reconstruction was done with TomoPy35 and Xi-CAM.36 Visualization and analysis were carried out with Avizo, from FEI.
The elemental distribution of the NMC particle-infiltrated LLZO scaffolds was mapped using the synchrotron radiation based X-ray micro-fluorescence (SR-μXRF) technique, which was conducted at Beamline 12.3.2 of the ALS. The specimen cross-section along the thickness direction was raster scanned using a micro-focused polychromatic X-ray beam (5–24 keV). At each scanning position the fluorescence signal was collected with a silicon drift detector. In this study, the Zr K-edge and Mn K-edge intensities were recorded, and thus the concentration distribution of these two elements was obtained.
Results and discussion
The fracture surface of a green body made using TBA frozen at −20 °C is shown in Fig. 2a and b. The long channels exhibit clean and sharp edges, with minimal bridging. After sintering, the structure shrank by about 35% in diameter and became fragile. The fracture surface of the sintered scaffold in Fig. 2c and d displays walls with high density and low thickness, composed of grains about 2 μm in diameter. The walls in the scaffold are only a few grains thick, and the pores are prismatic in shape and well faceted due to the directional growth behavior of the TBA. | ||
| Fig. 2 SEM images of the fractured LLZO scaffolds (a and b) before and (c and d) after sintering. | ||
The 3D image of the sintered scaffold was collected using SR-μCT. A sub-volume with the size of 700 μm × 700 μm × 200 μm of the LLZO film is shown in Fig. 3a. A video of the reconstructed model is included in the ESI,† and one slice of the top view and left view from this video are shown in Fig. 3b and c, respectively, indicating that the pore size is around 50 μm and that the pores are uniformly distributed throughout the whole structure. Better contrast is obtained from the 3D model of an LLZO longitudinal section shown in Fig. 3d. By rotating the image, it is found that smooth and empty channels with the same orientation are present and there is no bridging over the length range of several hundred microns (Fig. 3e and f), which by far exceeds the thickness of the scaffolds (∼100 μm). Moreover, the shape and size of the channels remain comparatively unvaried throughout the thickness of the sample. The long-range bridge-free structure provides distinct advantages for the infiltration process compared to the slit-like or layer-stacked structures fabricated using water as the solvent for freeze casting (ESI, Fig. S2 and S5†).
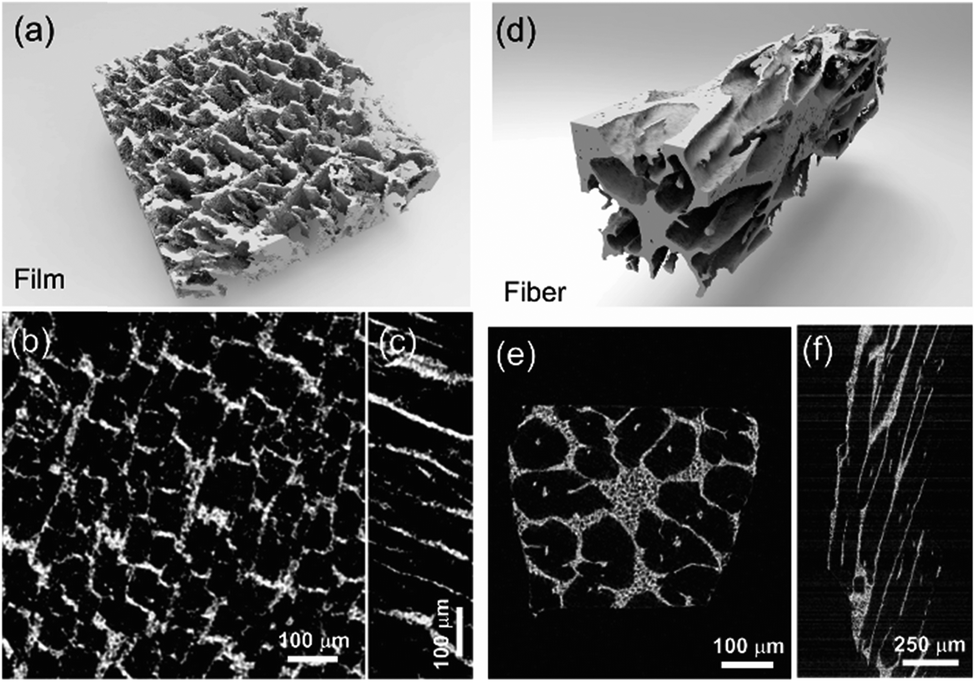 | ||
| Fig. 3 The 3D reconstructed structure from the SR-μCT data of (a) the LLZO film and (d) longitudinal section. The cross-sections for the film are shown in (b) top and (c) left views. (e) and (f) are the top and left views of the LLZO longitudinal section. | ||
The temperature of the cooling bed in the freeze cast apparatus is one of the key processing parameters for optimizing the pore size and porosity of the LLZO scaffold. A lower cooling temperature leads to more nucleation sites and higher solidification velocity, and thus finer TBA dendrites are formed, resulting in more, smaller pores than when the temperature is higher. However, if the temperature is too low, the sample may crack; if it is too high, the slurry will not freeze completely in a reasonable period of time. The SEM images of the top cross-sections of samples which are frozen at −20 and −50 °C are compared in Fig. 4. Both samples were sintered before taking the SEM photos. The average pore size of the sample processed at −20 °C is about 52 μm, twice as large as the one fabricated at −50 °C (∼23 μm). A statistical analysis of the white (LLZO scaffold) and black areas (pores) indicates that the porosity of the −20 °C sample is 73 vol% and the −50 °C sample has a lower porosity of about 60 vol%. The same trend was also reported by Xu et al. for a freeze cast lead zirconate titanate (PZT) system.37 As the cooling bed temperature decreases, a higher volume fraction of pore walls is observed, and pore size decreases.
 | ||
| Fig. 4 SEM top views of the sintered LLZO scaffolds, frozen at −20 °C and −50 °C, respectively, at low and high magnifications. | ||
After the initial sintering but prior to epoxy burnout, the excess Li2CO3 used to compensate for Li loss results in the formation of some tetragonal LLZO, evidenced by the weak peak splitting observed in the XRD pattern (Fig. 5a). After the epoxy removal heat treatment, the tetragonal LLZO signals disappear and the structure fully converts to cubic LLZO, with traces of LiAlO2 and La2Zr2O7 impurities associated with Li loss due to the high temperature exposure. Although increasing the amount of excess of Li2CO3 helps to compensate for the Li loss, it also accelerates the sintering process and generates cracks in the bulk of the LLZO scaffold, which is undesirable.
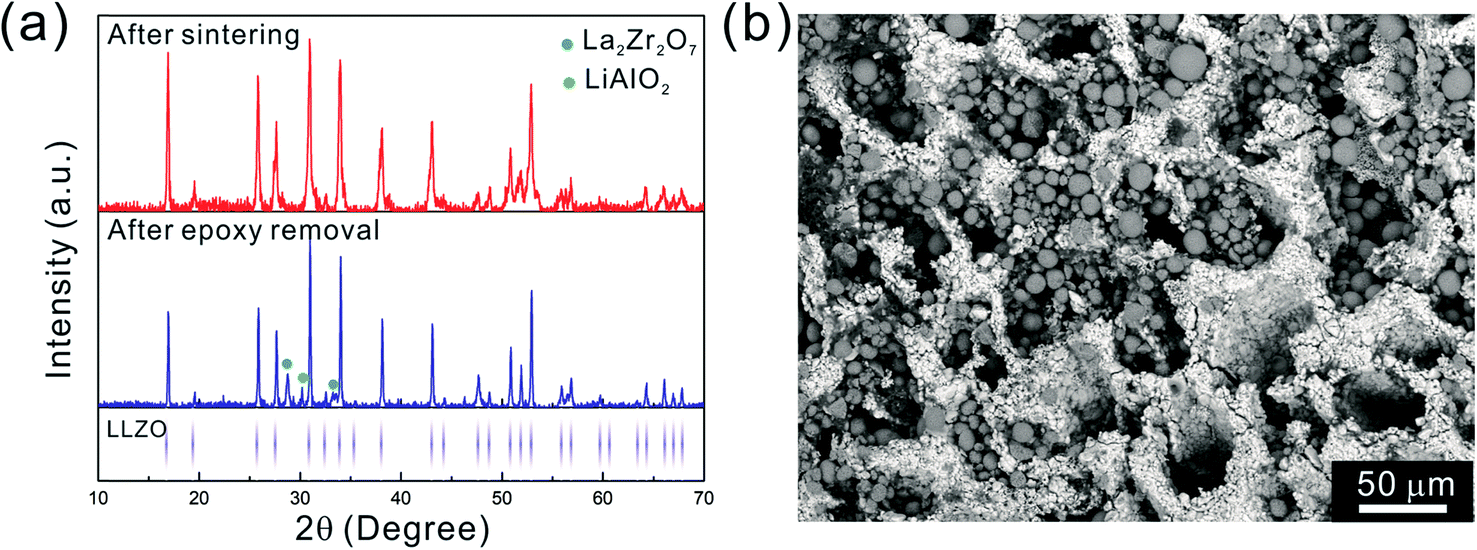 | ||
| Fig. 5 (a) XRD pattern of the scaffold after sintering and after epoxy removal, respectively. The LLZO XRD peaks are indicated in the lower part. (b) SEM image of the scaffold flipped over after drop casting of NMC particles from the other side. | ||
As the NMC particles are comparable in size to the pore size obtained at −50 °C, the infiltration would be challenging. Only a few point contacts would be formed between LLZO and NMC, which is harmful to the cell performance. Thus, the scaffold made at −20 °C is preferable for cell testing. The bottom view of the LLZO scaffold freeze cast at −20 °C infiltrated with NMC particles, PVDF, and carbon suggests that a fraction of the pores are not completely filled (Fig. 5b). A scaffold about 8 mm in diameter and 1 mm in thickness was infiltrated using the same process and contains 7.9 mg of NMC-622, corresponding to an active material loading of 15.7 mg cm−2 or about 3.14 mA h cm−2 (based on the practical specific capacity of 200 mA h g−1), similar to loadings used in commercial lithium-ion batteries. It was then incorporated into a hybrid cell with a Celgard separator, electrolytic solution, and Li anode, and was cycled at a current density of 0.3 mA cm−2 between 4.7 and 2.5 V (approximately C/10). The charge and discharge profiles of every tenth cycle as a function of cycle number are shown in Fig. 6a, together with the coulombic efficiency and capacity for each cycle (Fig. 6b). The initial capacity of NMC-622 is similar to previously reported values, which is around 200 mA h g−1 when charged to over 4.5 V.38,39 The areal capacity is also comparable to that of the reported high areal loading liquid Li/NMC-622 cell of about 3 mA h cm−2 with a current density of 0.5 mA cm−2.40,41 There is some capacity fading observed, but it is similar to what is seen in conventional NMC half-cells cycled under similar conditions.42 The fading can be attributed to the increased interfacial impedance (Fig. 6c) caused by the formation of a resistive cathode/electrolyte film and surface reconstruction to rock salt, similar to what is seen in conventional cells. This has been documented in many reports using surface sensitive characterization techniques.39,42–46 These results can be taken as proof of principle that infiltrated LLZO scaffolds can be used as cathodes in cells, although further optimization is required.
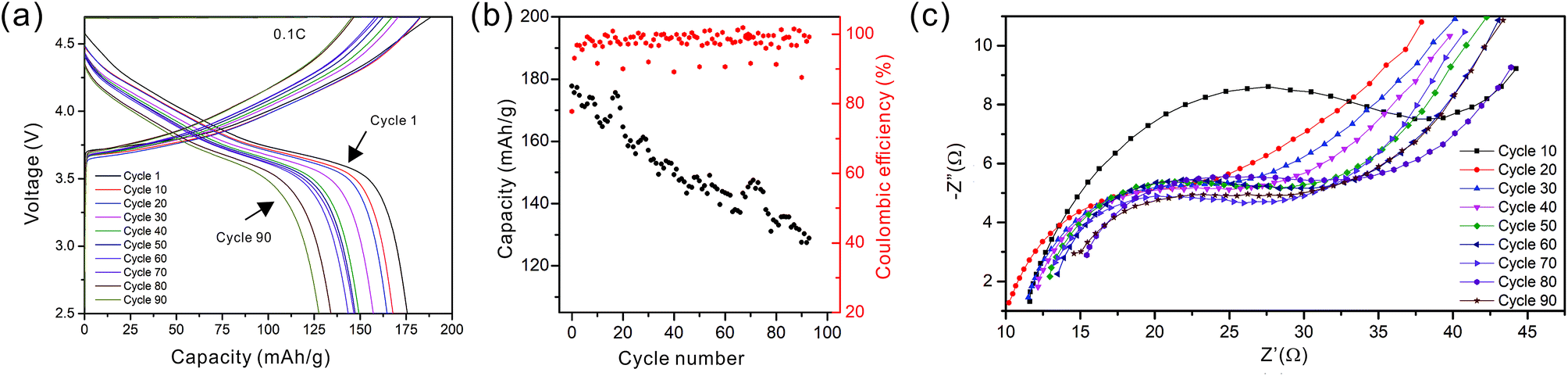 | ||
| Fig. 6 (a) The cycling curves, (b) capacity and coulombic efficiency, and (c) impedance curves of the hybrid cell from the 1st to the 90th cycle. | ||
Although the specific area loading of the active material NMC demonstrated in this example, 15.7 mg cm−2, is fairly high, the pores are not completely filled using the drop casting method. The density of NMC-622 provided by the manufacturer is about 2 g cm−1.3 From the SEM images, the porosity of the LLZO scaffold is estimated to be approximately 70% of the total volume. Therefore, only 11% of the pore volume is filled by NMC-622. Thinner scaffolds, similar to what is used in lithium ion batteries (about 1/10 of this thickness or ∼100 μm), would be easier to infiltrate and would most likely perform better, particularly if a completely solid state configuration is used. Work on making thinner scaffolds and optimizing the infiltration process is currently underway in our laboratories.
The ultimate goal is to use the scaffolds in a totally solid state configuration. To this end, we built bilayer structures, in which a dense LLZO layer is stacked with a porous scaffold and sintered together (Fig. 7a). In other words, the dense LLZO layer is employed in this configuration to play the role that both the separator and liquid electrolytic solution play in a hybrid cell. From the XRD patterns in Fig. 7b it is observed that fewer impurities are formed in the scaffold upon sintering in a bilayer configuration than those shown in Fig. 5a, because the dense layer acts as an additional source of lithium. Fig. 7c and d show the fracture surfaces of the bilayer structures with 100 μm and 20 μm thick dense layers fabricated using the two different approaches explained in the Experimental section, respectively. In both structures, the porous scaffold is about 150 μm thick. A smooth fracture surface will be formed if the scaffold and dense layer are well connected, which is important for ion transfer. Combining the results from the SEM photos and the fracture surfaces, it appears that the connections are partly formed, but there are still some gaps between the scaffold and dense layer, which may be caused by the different shrinkage rates of the two parts during the sintering procedure. The overall differences in shrinkage rates can be caused by the differences in porosities and additive concentrations. In addition, once a local connection is formed, the area around will be pinned (ESI, Fig. S6b†), and the uneven shrinkage may result in gaps between the layers and even pinholes in the dense layer (ESI, Fig. S6c†). It can also be seen that the channels in the scaffold are oriented slightly off the normal direction of the interface.
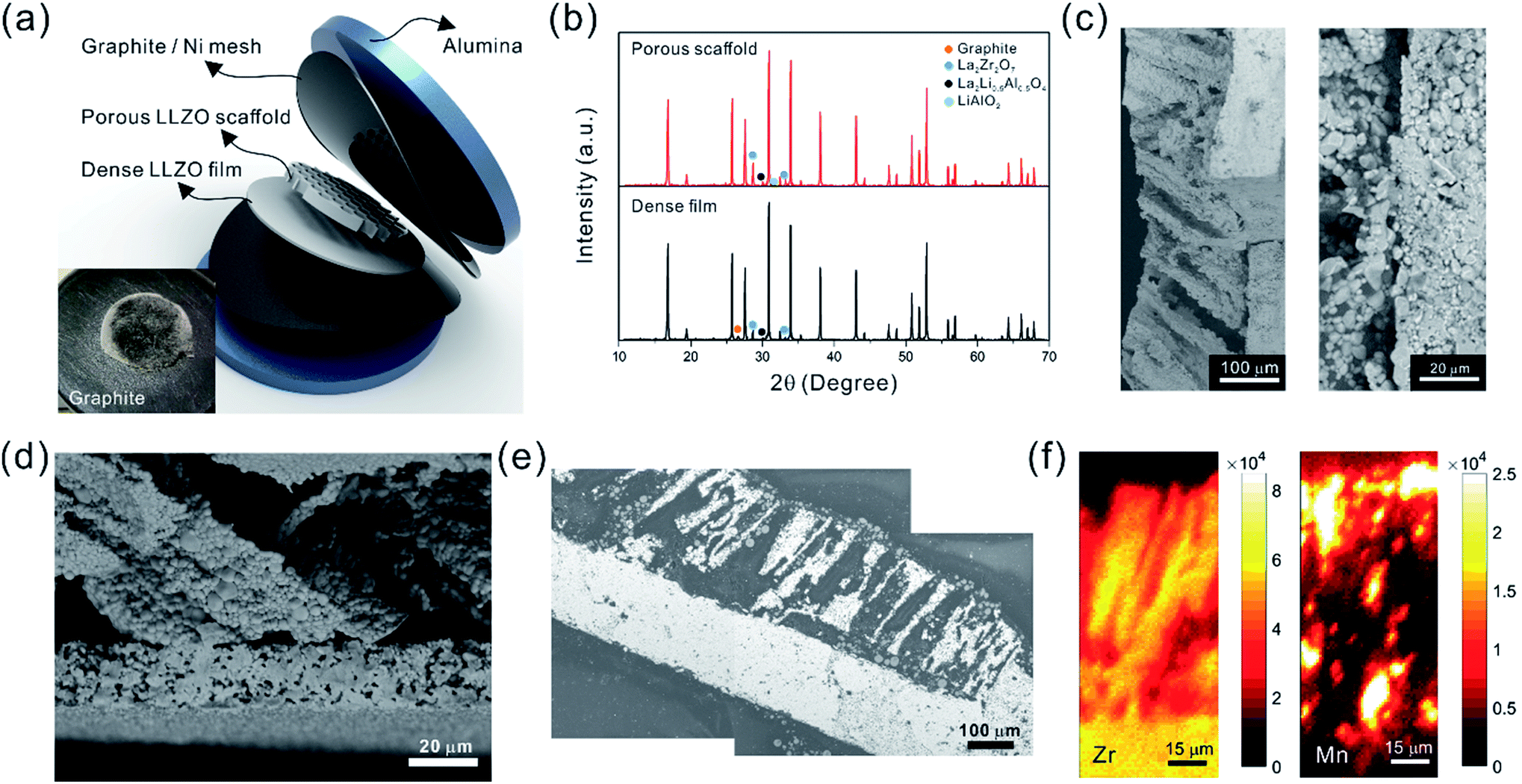 | ||
| Fig. 7 (a) Schematic of the bilayer fabrication. The inset shows a bilayer structure on graphite, with carbon remaining on the top surface after sintering. The porous scaffold and dense film XRD patterns are displayed in (b). Fractured bilayer structure with (c) thick and (d) thin dense films are presented. The SEM backscattered electron image (e) and its synchrotron radiation X-ray fluorescence maps (f) of the infiltrated bilayer structure indicate well-distributed NMC particles in areas where LLZO is absent. | ||
After the active material NMC-622 was infiltrated into the scaffold, the cross-section morphology of the bilayer structure was first observed in SEM in backscattered electron (BSE) mode (Fig. 7e), and then the elemental distribution was mapped using the SR-μXRF method, focusing on the Mn K-edge signals arising from the NMC and the Zr K-edge signals from LLZO (Fig. 7f). The Mn signal is primarily located in the areas where Zr is absent and almost fills these spaces, even though many of the NMC particles cannot be seen in the corresponding SEM images. From the map as well as the SEM images, a thin layer of NMC particles is observed on top of the scaffold after infiltration, indicating some heterogeneity in the distribution. A cell with a lithium foil anode and a bilayer with a 20 μm thick dense layer as the separator was assembled and the open circuit voltage (OCV) monitored over a period of about twelve hours (ESI, Fig. S7†). Initially, the OCV was close to 0 V, indicating shorting of the cell. However, the potential rose to 2 V over 2 hours, and then reached 2.8 V after 12 hours, the expected potential for a Li/NMC cell in the discharged state. This indicates that there was a soft short that could have been caused by pinholes in the dense film, which allowed some carbon particles to cross over and caused initial voltage instability (ESI, Fig. S8†). Although thicker dense layers might mitigate the soft short problem, this would come at the cost of higher cell resistance and reduced energy density due to the weight of the excess LLZO. Therefore, it remains a particular challenge when very thin dense layers are used in solid-state configurations along with extremely fine particles of carbon. The tri-layer LLZO reported by G. T. Hitz et al. offers a successful example for densifying multi-layer LLZO structures through laminating green LLZO layers as porous–dense–porous sandwich structures.47 This method decreases the difference in shrinkage rates between the scaffolds and dense layers and also balances the stress on both sides. However, for the porous structure made by freeze casting in the current study, the bulk scaffold needed to be sliced into thin sections, and the fragile ceramic structure needed to be stabilized. Thus sintering the structure for a short time was required. We are currently exploring making trilayers and using a related technique, which yields thinner structures (freeze tape casting), and which will simplify this process.
A number of technical questions arise for this approach. The oriented low-tortuosity channels in the freeze cast scaffolds should be advantageous because they minimize ion diffusion distances, but it is not clear what the optimized channel sizes, wall thicknesses, and porosities should be. To attempt to answer these questions, we established a simple model for semi-quantitative estimation of these parameters. For the model, the geometric configuration of the porous scaffold shown in Fig. 8a was assumed. The channels are assumed to run through the thickness of the scaffold and have smooth walls, have the same constant diameter, and are uniformly distributed with no bridges to form a regular hexagonal honeycomb. The top view in Fig. 8b shows various parameters such as the pore diameter (dp), wall thickness (dw), porosity, and number density of the pores. Taking the number density of the pores as N (per unit area), the side length a (distance between the centers of adjacent pores) of the regular hexagonal base is calculated to be:
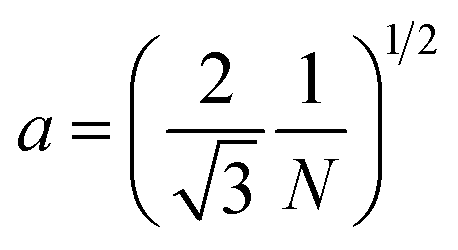 | (1) |
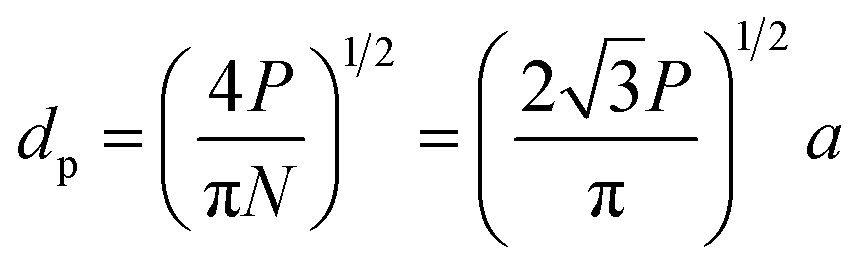 | (2) |
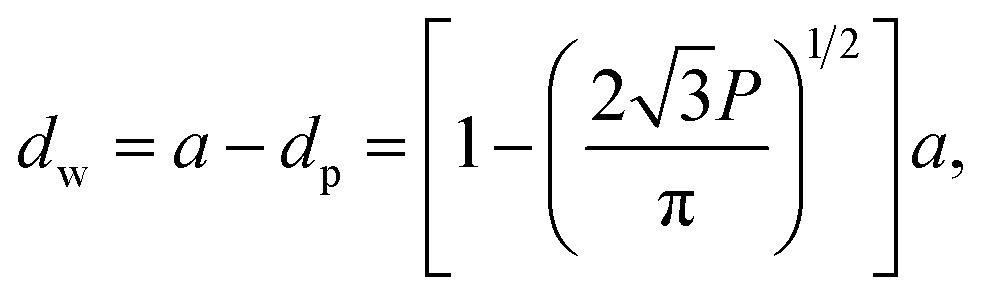 | (3) |
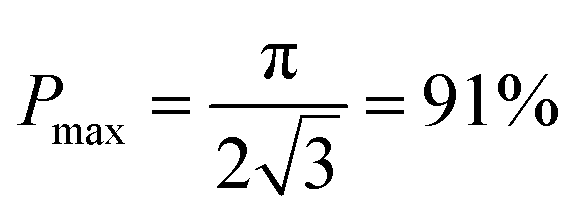 , will be obtained.
, will be obtained.
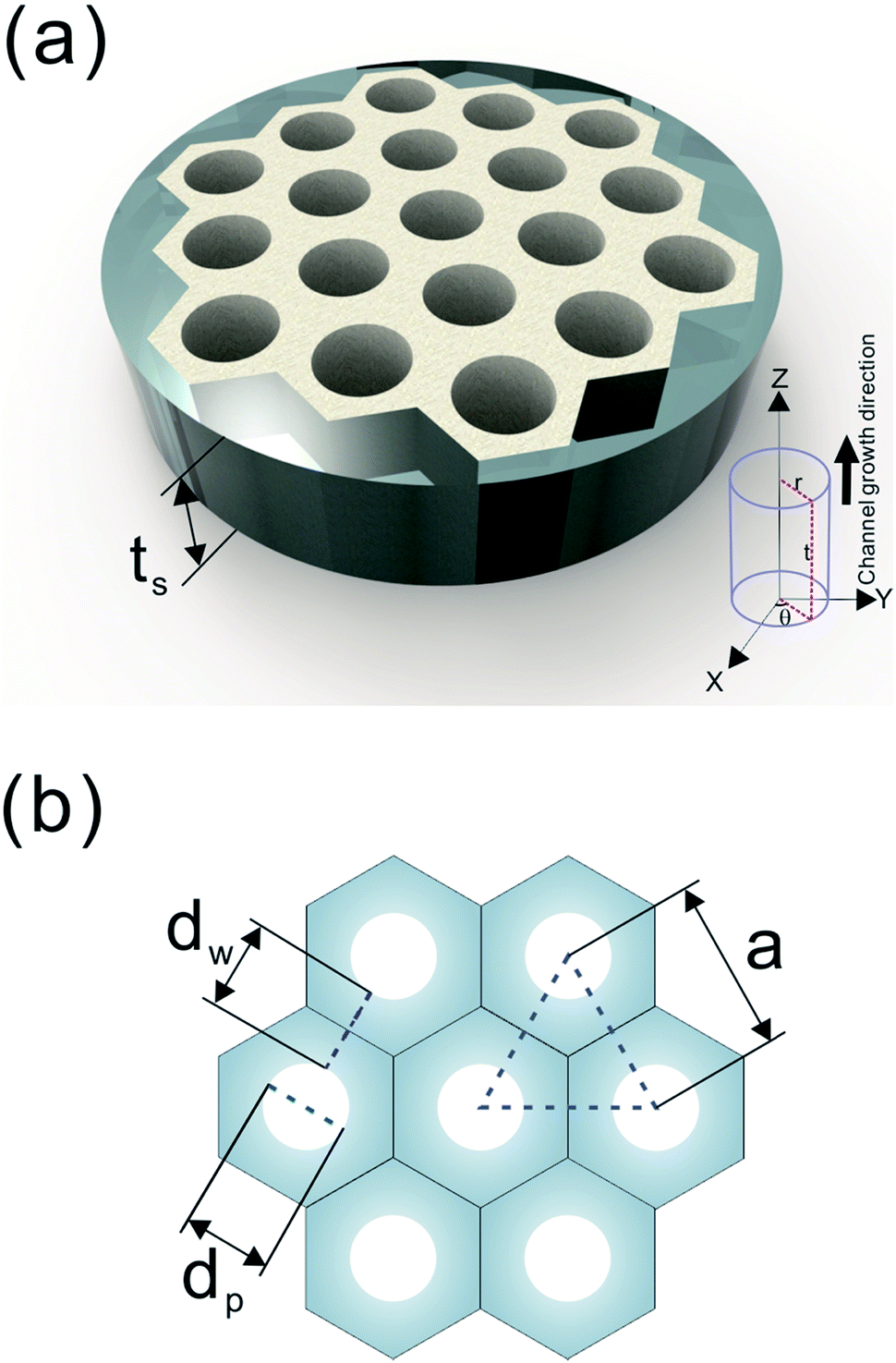 | ||
| Fig. 8 (a) The 3D model and (b) top view of the simplified honeycomb structure of the porous LLZO scaffold. | ||
When using this type of scaffold structure with the active material infiltrated into the channels, which run approximately parallel to the current direction, the lithium diffusion path length is shortened and the effective NMC/LLZO contact area is increased, compared to the case in which dense LLZO films are employed. A semi-quantitative evaluation of the improved performance is estimated. Considering that Li ions diffuse in LLZO and in the active material at different rates, we can define the constant K as the diffusivity ratio: 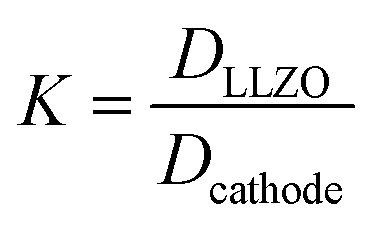 . From Einstein's random walk theory,48 we know that in a certain period of time the Li ion diffusion distances in LLZO and in the active material follow a square root relationship. In other words, in the same period of time, Li ions will migrate the length LNMC in NMC as
. From Einstein's random walk theory,48 we know that in a certain period of time the Li ion diffusion distances in LLZO and in the active material follow a square root relationship. In other words, in the same period of time, Li ions will migrate the length LNMC in NMC as 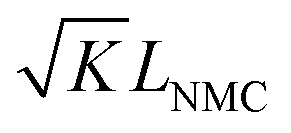 in LLZO. The equivalent Li ion diffusion path in the porous/dense bilayer structure Lbilayer, assuming that the channels are all fully infiltrated by active materials, and that Li ions need to first diffuse from NMC to LLZO and then migrate through LLZO, is expressed as:
in LLZO. The equivalent Li ion diffusion path in the porous/dense bilayer structure Lbilayer, assuming that the channels are all fully infiltrated by active materials, and that Li ions need to first diffuse from NMC to LLZO and then migrate through LLZO, is expressed as:
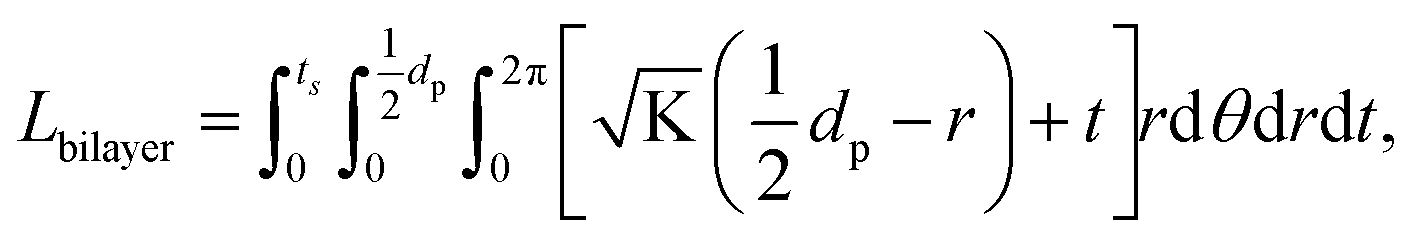 | (4) |
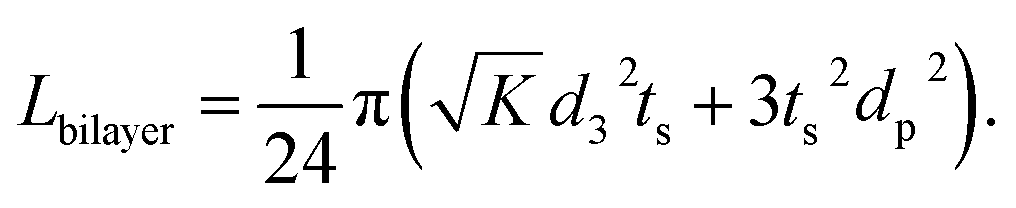 | (5) |
It is called the equivalent diffusion path because the diffusivity difference between Li-in-NMC and Li-in-LLZO is considered, and the diffusion path length in NMC is normalized by calculation to its equivalent length in LLZO.
To consider a cell in which only a dense LLZO film and a dense layer of active material are used, assuming that the thickness of the LLZO film and the amount of the NMC active material are the same as those used in the bilayer structure, the thickness of the active material layer is ta = Pts, and in this situation the total equivalent diffusion path length Ldense is calculated to be:
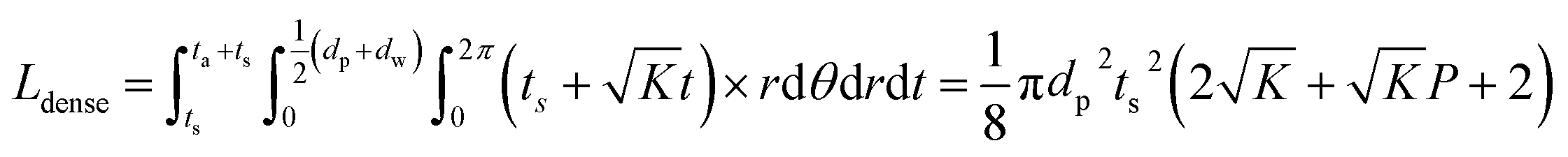 | (6) |
To compare the equivalent diffusion path lengths of porous and dense LLZO films, the Lbilayer to Ldense ratio is computed to be
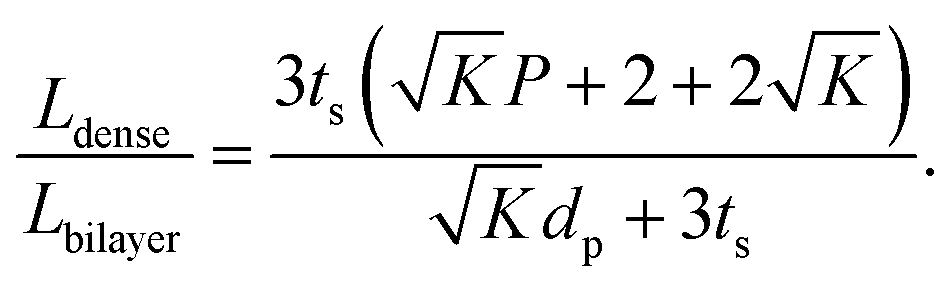 | (7) |
In most cases, the pore diameter dp is much smaller than the thickness of the scaffold ts. Thus, the ratio is approximated to be:
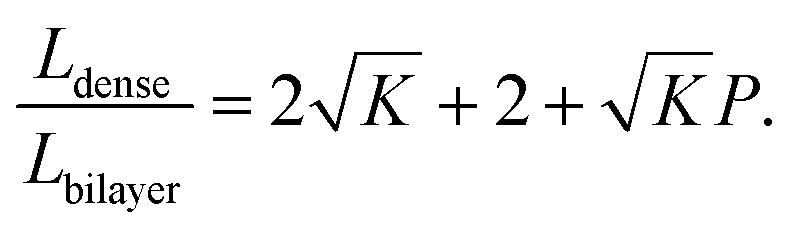 | (8) |
From this point of view, the higher the porosity is in the porous structure, the better the performance that can be obtained, and the faster the diffusion is in LLZO compared to the active materials, the more the enhancement that can be achieved. With NMC particles infiltrated in the LLZO scaffold, K can be estimated to be about 10, according to the reported Li ion diffusivity around 10−10 cm2 s−1 in NMC49 and 10−9 cm2 s−1 in LLZO.50 Since porosity P ranges between 0 and 0.91, the equivalent diffusion path length of the bilayer structure is 88% to 91% shorter than that of the dense LLZO for Li ions. Although there is considerable variation in the literature values of Li ion diffusivities in LLZO50–53 and NMC49,54–57 it is still true that the equivalent diffusion path length of the bilayer structure is much shorter than that of the dense LLZO configuration.
In practice, this will be highly dependent upon how intimately the active material and LLZO are in contact with each other, and will require careful optimization of the fabrication parameters. Thus a dimensionless quantity, Acontact, which is defined as the contact area per unit LLZO film cross-sectional area, is calculated by summing up the contribution from two parts – the base Ab and the side-walls Aw:
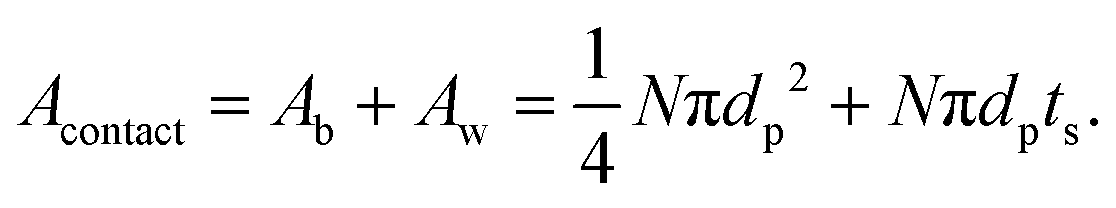 | (9) |
Substituting eqn (2) and (3) in (9), we get:
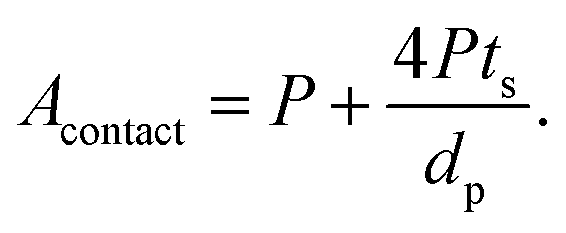 | (10) |
From eqn (10), it is apparent that not only the diameter of the channels should be as small as possible to increase the contact area, but also the porosity should be as high as possible. To decrease the channel diameter, lower cooling temperatures and higher solidification rates in the freeze casting experiment are preferred. However, as revealed in the experimental study, higher freezing rates also decrease the wall thickness. Moreover, the wall thickness thinning rate is usually slower than the channel diameter shrinking rate, resulting in lower porosity, which is not desirable in terms of battery capacity, diffusion path length, and contact area. Therefore, it is not sufficient to tune the microstructure by changing the cooling rate only. To obtain small pore diameter and high porosity simultaneously, the LLZO slurry mass loading has to be decreased as well.
It must be emphasized that the model presented above primarily considers the Li ion diffusion path length. However, in reality, more factors may need to be taken into account. Charge transfer occurs at the cathode/LLZO interface during cell cycling and the kinetics will have a strong impact on the performance of the cell. Besides the contact area, the selection and matchup of the morphology and size of the active material particles with respect to the pores in the LLZO films are equally important. Large active material particles are more difficult to infiltrate into the pores and channels and may result in relatively fewer points of contact between the active material and LLZO than when smaller particles are used. However, if small particles are infiltrated into pores much larger than their diameters, there will also be particle-to-particle charge transfer resistance. Moreover, volume changes during redox processes will affect the amount of contact among all the components in the composite, and thus the electrochemistry.
Conclusion
Here we propose a novel configuration for thick composite electrodes in solid-state batteries utilizing a freeze casting approach to make ionically conductive porous scaffolds, which are then infiltrated with the active material and other components. To illustrate the principle, we prepared scaffolds using LLZO with TBA as the solvent. By changing the cooling bed temperature and thus the solidification rate, the porosities and channel diameters can be varied. Sintering then strengthens the structure and densifies the pore walls. 3D models of the LLZO scaffolds and longitudinal sections were examined using monochromatic SR-μCT, and they show that the low-tortuosity channels are uniformly distributed parallel to the thickness of the scaffold, with pore diameters almost constant from the top to bottom. The open and oriented channels with few bridges make it possible for large commercial cathode particles to be infiltrated into the structure readily. As an example, a composite NMC622/LLZO electrode was successfully cycled in a hybrid cell configuration. Bilayer structures were also fabricated by co-sintering a dense layer and a porous layer together. SR-μXRF shows that the porous layer was successfully infiltrated with the active material. It was possible to read an open circuit potential on a totally solid state half-cell consisting of a lithium anode and the infiltrated bilayer structure. Theoretical calculations were performed to model the porosity of the scaffolds, indicating that the porosity cannot exceed 91%. Higher porosity results in higher energy density and should result in improved rate capability, by offering shorter diffusion path lengths and larger contact areas. However, smaller channel diameters result in increased contact area between the ionic conductor and active material, which also benefits performance. While lowering the temperature of the freeze casting experiment results in smaller pore sizes, it also decreases porosity. With these design considerations in mind, it is suggested that the loading of LLZO in slurries for freeze casting experiments should be lowered to meet the simultaneous goals of increased porosity and smaller pores.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 51671154 and 91860109), the National Key Research and Development Program of China (Grant No. 2016YFB0700404), and the Assistant Secretary for Energy Efficiency and Renewable Energy, Office of Vehicle Technologies of the U.S. Department of Energy, under Contract No. DE-AC02-05CH11231. This research used resources of the Advanced Light Source, which is a DOE Office of Science User Facility under contract no. DE-AC02-05CH11231. H. S. would like to acknowledge the financial support from the program of China Scholarships Council (No. 201606280062) from Oct. 2016 to Sep. 2017 and from the ALS Doctoral Fellowship in Residence from Oct. 2017 to Sep. 2018. K. C. appreciates the support from the International Joint Laboratory for Micro/Nano Manufacturing and Measurement Technologies and the Collaborative Innovation Center of High-End Manufacturing Equipment. This document was prepared as an account of work sponsored by the United States Government. While this document is believed to contain correct information, neither the United States Government nor any agency thereof, nor the Regents of the University of California, nor any of their employees, makes any warranty, express or implied, or assumes any legal responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by its trade name, trademark, manufacturer, or otherwise, does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof, or the Regents of the University of California. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof or the Regents of the University of California.References
- J. Zheng, M. H. Engelhard, D. Mei, S. Jiao, B. J. Polzin, J.-G. Zhang and W. Xu, Nat. Energy, 2017, 2, 17012 CrossRef CAS.
- D. Deng, Energy Science Engineering, 2015, 3, 385–418 CrossRef.
- D. Lin, Y. Liu and Y. Cui, Nat. Nanotechnol., 2017, 12, 194–206 CrossRef CAS PubMed.
- P. Bai, J. Li, F. R. Brushett and M. Z. Bazant, Energy Environ. Sci., 2016, 9, 3221–3229 RSC.
- C. Ma, Y. Cheng, K. Yin, J. Luo, A. Sharafi, J. Sakamoto, J. Li, K. L. More, N. J. Dudney and M. Chi, Nano Lett., 2016, 16, 7030–7036 CrossRef CAS PubMed.
- Y. Zhu, X. He and Y. Mo, J. Mater. Chem. A, 2016, 4, 3253–3266 RSC.
- W. D. Richards, L. J. Miara, Y. Wang, J. C. Kim and G. Ceder, Chem. Mater., 2015, 28, 266–273 CrossRef.
- J. Wolfenstine, J. L. Allen, J. Read and J. Sakamoto, J. Mater. Sci., 2013, 48, 5846–5851 CrossRef CAS.
- T. Thompson, A. Sharafi, M. D. Johannes, A. Huq, J. L. Allen, J. Wolfenstine and J. Sakamoto, Adv. Energy Mater., 2015, 5, 1500096 CrossRef.
- R. Murugan, V. Thangadurai and W. Weppner, Angew. Chem., 2007, 119, 7925–7928 CrossRef.
- A. Sharafi, H. M. Meyer, J. Nanda, J. Wolfenstine and J. Sakamoto, J. Power Sources, 2016, 302, 135–139 CrossRef CAS.
- S. Yu, R. D. Schmidt, R. Garcia-Mendez, E. Herbert, N. J. Dudney, J. B. Wolfenstine, J. Sakamoto and D. J. Siegel, Chem. Mater., 2016, 28, 197–206 CrossRef CAS.
- D. Rettenwander, G. Redhammer, F. Preishuber-Pflugl, L. Cheng, L. Miara, R. Wagner, A. Welzl, E. Suard, M. M. Doeff, M. Wilkening, J. Fleig and G. Amthauer, Chem. Mater., 2016, 28, 2384–2392 CrossRef CAS PubMed.
- L. Buannic, B. Orayech, J.-M. López Del Amo, J. Carrasco, N. A. Katcho, F. Aguesse, W. Manalastas, W. Zhang, J. Kilner and A. Llordés, Chem. Mater., 2017, 29, 1769–1778 CrossRef CAS.
- Z. Zhang, Y. Shao, B. V. Lotsch, Y.-S. Hu, H. Li, J. Janek, C. Nan, L. Nazar, J. Maier, M. Armand and L. Chen, Energy Environ. Sci., 2018, 11, 1945–1976 RSC.
- K. Fu, Y. Gong, G. T. Hitz, D. W. McOwen, Y. Li, S. Xu, Y. Wen, L. Zhang, C. Wang, G. Pastel, J. Dai, B. Liu, H. Xie, Y. Yao, E. D. Wachsman and L. Hu, Energy Environ. Sci., 2017, 10, 1568–1575 RSC.
- X. Han, Y. Gong, K. Fu, X. He, G. T. Hitz, J. Dai, A. Pearse, B. Liu, H. Wang, G. Rubloff, Y. Mo, V. Thangadurai, E. D. Wachsman and L. Hu, Nat. Mater., 2016, 16, 572 CrossRef PubMed.
- L. Porz, T. Swamy, B. W. Sheldon, D. Rettenwander, T. Frömling, H. L. Thaman, S. Berendts, R. Uecker, W. C. Carter and Y.-M. Chiang, Adv. Energy Mater., 2017, 7, 1701003 CrossRef.
- F. Shen, M. B. Dixit, X. Xiao and K. B. Hatzell, ACS Energy Lett., 2018, 3, 1056–1061 CrossRef CAS.
- H. Shen, E. Yi, L. Cheng, M. Amores, G. Chen, S. Sofie and M. Doeff, Sustainable Energy Fuels, 2019, 3, 1647–1659 RSC.
- M. Kotobuki, H. Munakata, K. Kanamura, Y. Sato and T. Yoshida, J. Electrochem. Soc., 2010, 157, A1076–A1079 CrossRef CAS.
- J. van den Broek, S. Afyon and J. L. M. Rupp, Adv. Energy Mater., 2016, 6, 1600736 CrossRef.
- E. Dumont-Botto, C. Bourbon, S. Patoux, P. Rozier and M. Dolle, J. Power Sources, 2011, 196, 2274–2278 CrossRef CAS.
- G. Delaizir, V. Viallet, A. Aboulaich, R. Bouchet, L. Tortet, V. Seznec, M. Morcrette, J.-M. Tarascon, P. Rozier and M. Dollé, Adv. Funct. Mater., 2012, 22, 2140–2147 CrossRef CAS.
- X. Yao, N. Huang, F. Han, Q. Zhang, H. Wan, J. P. Mwizerwa, C. Wang and X. Xu, Adv. Energy Mater., 2017, 7, 1602923 CrossRef.
- X. Yao, D. Liu, C. Wang, P. Long, G. Peng, Y. S. Hu, H. Li, L. Chen and X. Xu, Nano Lett., 2016, 16, 7148–7154 CrossRef CAS PubMed.
- Q. Zhang, H. Wan, G. Liu, Z. Ding, J. P. Mwizerwa and X. Yao, Nano Energy, 2019, 57, 771–782 CrossRef CAS.
- K. Fu, Y. Gong, B. Liu, Y. Zhu, S. Xu, Y. Yao, W. Luo, C. Wang, S. D. Lacey, J. Dai, Y. Chen, Y. Mo, E. Wachsman and L. Hu, Sci. Adv., 2017, 3, e1601659 CrossRef PubMed.
- J. S. Sander, R. M. Erb, L. Li, A. Gurijala and Y. M. Chiang, Nat. Energy, 2016, 1, 16099 CrossRef CAS.
- D. W. McOwen, S. Xu, Y. Gong, Y. Wen, G. L. Godbey, J. E. Gritton, T. R. Hamann, J. Dai, G. T. Hitz, L. Hu and E. D. Wachsman, Adv. Mater., 2018, 30, e1707132 CrossRef PubMed.
- M. B. Dixit, M. Regala, F. Shen, X. Xiao and K. B. Hatzell, ACS Appl. Mater. Interfaces, 2019, 11, 2022–2030 CrossRef CAS PubMed.
- S. Deville, Adv. Eng. Mater., 2008, 10, 155–169 CrossRef CAS.
- L. Buannic, M. Naviroj, S. M. Miller, J. Zagorski, K. T. Faber and A. Llordes, J. Am. Ceram. Soc., 2019, 102, 1021–1029 CrossRef CAS.
- E. Yi, W. Wang, J. Kieffer and R. M. Laine, J. Mater. Chem. A, 2016, 4, 12947–12954 RSC.
- D. Gursoy, F. De Carlo, X. Xiao and C. Jacobsen, J. Synchrotron Radiat., 2014, 21, 1188–1193 CrossRef PubMed.
- R. J. Pandolfi, D. B. Allan, E. Arenholz, L. Barroso-Luque, S. I. Campbell, T. A. Caswell, A. Blair, F. De Carlo, S. Fackler, A. P. Fournier, G. Freychet, M. Fukuto, D. Gursoy, Z. Jiang, H. Krishnan, D. Kumar, R. J. Kline, R. Li, C. Liman, S. Marchesini, A. Mehta, A. T. N'Diaye, D. Y. Parkinson, H. Parks, L. A. Pellouchoud, T. Perciano, F. Ren, S. Sahoo, J. Strzalka, D. Sunday, C. J. Tassone, D. Ushizima, S. Venkatakrishnan, K. G. Yager, P. Zwart, J. A. Sethian and A. Hexemer, J. Synchrotron Radiat., 2018, 25, 1261–1270 CrossRef PubMed.
- T. Xu and C.-A. Wang, Mater. Des., 2016, 91, 242–247 CrossRef CAS.
- Z. Wu, S. Ji, Z. Hu, J. Zheng, S. Xiao, Y. Lin, K. Xu, K. Amine and F. Pan, ACS Appl. Mater. Interfaces, 2016, 8, 15361–15368 CrossRef CAS PubMed.
- Y. Ruan, X. Song, Y. Fu, C. Song and V. Battaglia, J. Power Sources, 2018, 400, 539–548 CrossRef CAS.
- E. Markevich, G. Salitra, F. Chesneau, M. Schmidt and D. Aurbach, ACS Energy Lett., 2017, 2, 1321–1326 CrossRef CAS.
- G. Salitra, E. Markevich, M. Afri, Y. Talyosef, P. Hartmann, J. Kulisch, Y. K. Sun and D. Aurbach, ACS Appl. Mater. Interfaces, 2018, 10, 19773–19782 CrossRef CAS PubMed.
- F. Lin, I. M. Markus, D. Nordlund, T. C. Weng, M. D. Asta, H. L. Xin and M. M. Doeff, Nat. Commun., 2014, 5, 3529 CrossRef PubMed.
- F. Lin, D. Nordlund, I. M. Markus, T.-C. Weng, H. L. Xin and M. M. Doeff, Energy Environ. Sci., 2014, 7, 3077 RSC.
- J. Zhu and G. Chen, J. Mater. Chem. A, 2019, 7, 5463 RSC.
- C. Tian, D. Nordlund, H. L. Xin, Y. Xu, Y. Liu, D. Sokaras, F. Lin and M. M. Doeff, J. Electrochem. Soc., 2018, 165, A696–A704 CrossRef CAS.
- S.-K. Jung, H. Gwon, J. Hong, K.-Y. Park, D.-H. Seo, H. Kim, J. Hyun, W. Yang and K. Kang, Adv. Energy Mater., 2013, 4, 1300787 CrossRef.
- L. Zou, W. Zhao, Z. Liu, H. Jia, J. Zheng, G. Wang, Y. Yang, J.-G. Zhang and C. Wang, ACS Energy Lett., 2018, 3, 2433–2440 CrossRef CAS.
- G. T. Hitz, D. W. McOwen, L. Zhang, Z. Ma, Z. Fu, Y. Wen, Y. Gong, J. Dai, T. R. Hamann, L. Hu and E. D. Wachsman, Mater. Today, 2019, 22, 50–57 CrossRef CAS.
- A. Einstein, Ann. Phys., 1905, 17, 549–560 CrossRef CAS.
- P.-C. Tsai, B. Wen, M. Wolfman, M.-J. Choe, M. S. Pan, L. Su, K. Thornton, J. Cabana and Y.-M. Chiang, Energy Environ. Sci., 2018, 11, 860–871 RSC.
- M. Månsson, H. Nozaki, J. M. Wikberg, K. Prša, Y. Sassa, M. Dahbi, K. Kamazawa, K. Sedlak, I. Watanabe and J. Sugiyama, J. Phys. Conf. Ser., 2014, 551, 012037 CrossRef.
- M. Amores, T. E. Ashton, P. J. Baker, E. J. Cussen and S. A. Corr, J. Mater. Chem. A, 2016, 4, 1729–1736 RSC.
- K. Hayamizu, S. Seki and T. Haishi, J. Chem. Phys., 2017, 146, 024701 CrossRef.
- C. Chen, Z. Lu and F. Ciucci, Sci. Rep., 2017, 7, 40769 CrossRef CAS PubMed.
- Y. Wei, J. Zheng, S. Cui, X. Song, Y. Su, W. Deng, Z. Wu, X. Wang, W. Wang, M. Rao, Y. Lin, C. Wang, K. Amine and F. Pan, J. Am. Chem. Soc., 2015, 137, 8364–8367 CrossRef CAS.
- S. Cui, Y. Wei, T. Liu, W. Deng, Z. Hu, Y. Su, H. Li, M. Li, H. Guo, Y. Duan, W. Wang, M. Rao, J. Zheng, X. Wang and F. Pan, Adv. Energy Mater., 2016, 6, 1501309 CrossRef.
- S.-L. Wu, W. Zhang, X. Song, A. K. Shukla, G. Liu, V. Battaglia and V. Srinivasan, J. Electrochem. Soc., 2012, 159, A438–A444 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ta06520b |
| This journal is © The Royal Society of Chemistry 2019 |