
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDinuclear manganese alkoxide complexes as catalysts for C–N bond cleavage of simple tertiary N,N-dialkylamides to give esters†
Haruki
Nagae
 ,
Takahiro
Hirai
,
Daiki
Kato
,
Shusei
Soma
,
Shin-ya
Akebi
and
Kazushi
Mashima
*
,
Takahiro
Hirai
,
Daiki
Kato
,
Shusei
Soma
,
Shin-ya
Akebi
and
Kazushi
Mashima
*
Department of Chemistry, Graduate School of Engineering Science, Osaka University, Toyonaka, Osaka 560-8531, Japan. E-mail: mashima@chem.es.osaka-u.ac.jp
First published on 29th January 2019
Abstract
Amide bonds are stable due to the resonance between the nitrogen lone pair and the carbonyl moiety, and therefore the chemical transformation of amides, especially tertiary amides, involving C–N bond fission is considered one of the most difficult organic reactions, unavoidably requiring harsh reaction conditions and strong acids or bases. We report the catalytic C–N bond cleavage of simple tertiary N,N-dialkylamides to give corresponding esters using a catalyst system (2 mol% based on Mn atoms) of a tetranuclear manganese alkoxide, [Mn(acac)(OEt)(EtOH)]4 (1c), combined with four equivalents of 4,7-bis(dimethylamino)-1,10-phenanthroline (L1: Me2N-Phen). Regarding the reaction mechanism, we isolated a dinuclear manganese complex, [Mn(acac)(OEt)(Phen)]2 (6c), which was revealed as the catalytically active species for the esterification of tertiary amides.
Introduction
Amides are ubiquitous and abundant in a huge number of natural and synthetic organic compounds, such as proteins, poly(amides), and pharmaceuticals, due to the extraordinarily stable nature of the amide bond.1–5 The stability of the amide bond is attributed to the resonance between the nitrogen lone pair and the carbonyl moiety, and many researchers have attempted to diminish the resonance structure of the C–N bond by introducing the coordination of transition metals to the nitrogen atom, as exemplified by the complexation of N,N-(2-pyridylmethyl)amides with Cu(II) sources,6,7 and constructing a twisted structure around the nitrogen atom,8,9 whose amide bonds were readily cleaved under mild conditions. Nonetheless, cleavage of the amide bond requires harsh reaction conditions such as using stoichiometric amounts of strong acids or bases at a high temperature. TiCl4- and FeCl3-catalyzed alcoholysis of inactivated primary amides was independently reported by Fisher et al. and Sun et al. but more than one equivalent of aqueous HCl was necessary.10 Shimizu et al. reported catalytic esterification of primary amides without using any additives by utilizing CeO2 as a heterogeneous catalyst under a high reaction temperature.11 Thus, catalytic transformation of amides to the corresponding esters or acids in an atom- and step-economical manner is in high demand. Recently, we reported a catalytic C–N bond cleavage of primary and secondary amides using a catalyst mixture of Sc(OTf)3 and boronic esters,12 and Atkinson et al. also reported Sc(OTf)3-catalyzed esterification of secondary amides.13 In addition, catalytic esterification of 8-aminoquinoline amides was achieved with Ni(tmhd)2 (TMHD = 2,2,6,6-tetramethyl-3,5-heptanedionate) in methanol.14In contrast, the catalytic transformation of tertiary amides remains a challenging task, and only a few catalyst systems developed to date are able to break the stable C–N bond of tertiary amides. Recently, Garg and Houk reported that nickel(0) complexes served as catalysts to activate the C–N bond of tertiary amides bearing functional groups such as Ph or Boc via oxidative addition as a key step to afford the corresponding esters (Fig. 1A),15–17 and Danoun and Gosmini reported that low-valent cobalt species supported by 2,2′-bipyridine using metallic manganese as a reductant catalytically activated the C–N bond of activated amides bearing a Boc group to give the corresponding esters (Fig. 1B),18 though the oxidative addition of the C–N bond of activated amides bearing functional groups such as Ph, Boc, and Ts on the nitrogen atom into low-valent transition metals was reported to be involved in several catalytic reactions.17,19–26 On the other hand, we reported a catalytic C–N bond cleavage of N-alkyl-N-β-hydroxyethylamides via N,O-acyl rearrangement by manganese complexes supported by an N^N-bidentate ligand27 and zinc complexes (Fig. 1C),28 though these catalysts required substrates with functional groups, such as a β-hydroxyethyl group on the nitrogen atom. Very recently, Shimizu and Siddiki reported the first example of the esterification of N,N-dialkylamides using CeO2 as a reusable heterogeneous catalyst, though the catalyst system requires a high reaction temperature and large amount of catalyst. In this reaction, Lewis acid and base sites of the CeO2 surface cooperatively activate the carbonyl moiety and alcohol (Fig. 1D).29 To achieve the esterification of simple N,N-dialkyl tertiary amides under milder reaction conditions, we focused our attention on cooperative activation of multinuclear complexes whose multi-metallic structure is often found in metalloenzymes. Herein, we report a new and efficient catalyst system of [Mn(acac)(OEt)(EtOH)]4 (1c), as Mn(II) precursors to evaluate the general tendency that metal-alkoxide species accelerate the nucleophilic attack of the alkoxide group on the carbonyl group (2 mol% based on Mn atom), and 4,7-bis(dimethylamino)-1,10-phenanthroline (L1: Me2N-Phen) (2 mol%) for the esterification of simple tertiary N,N-dialkylamides to give the corresponding esters with broad substrate scopes including halogenated amides (Fig. 1E). Furthermore, we determined a reaction mechanism by isolating an alkoxide-bridged manganese dinuclear complex [Mn(acac)(OEt)(Phen)]2 (6c) as the key catalyst, and measuring kinetic studies in n-butanol which showed first-order rate dependence on the concentrations of N,N-dimethyl-2-naphthamide (2a) and 6c, indicating that the dinuclear alkoxide complex acted as a catalytically active species.
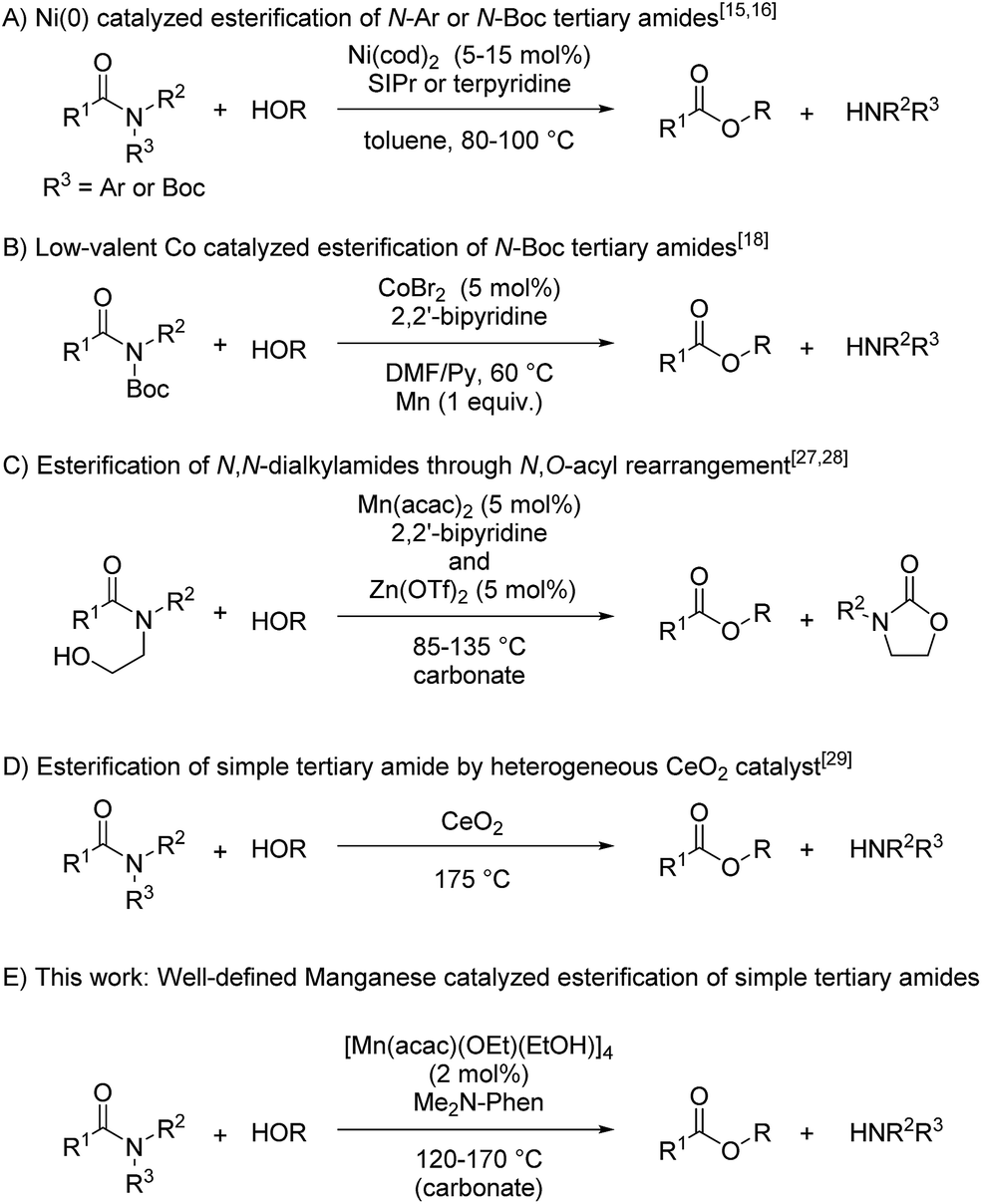 | ||
| Fig. 1 Catalytic esterification via the C–N bond cleavage of tertiary amides. | ||
Results and discussion
We began by searching for the suitable catalyst among the reported alkoxy-bridged tetranuclear complexes, [M(tmhd)(OMe)(MeOH)x]4 (1; M = Mn,30a Fe,30b Co,31 and Cu;32 1.0 mol%; x = 1 or 0; TMHD = 2,2,6,6-tetramethyl-3,5-heptanedionate), as catalyst precursors with 4,7-bis(dimethylamino)-1,10-phenanthroline (L1: Me2N-Phen, 1 mol%) for the catalytic esterification of N,N-dimethyl-2-naphthamide (2a) under the conditions of using n-butanol (0.25 mL) at reflux temperature for 18 h, and the results are shown in Table 1.33 The use of the manganese tetranuclear complex, [Mn(tmhd)(OMe)(MeOH)] (1a), afforded 3a in 80% yield (entry 1). Iron and cobalt tetranuclear complexes exhibited lower catalytic activities (16% and 42% yields, respectively, entries 2 and 3) than the manganese complex. The copper tetranuclear complex, [Cu(tmhd)(OMe)]4, which does not have methanol as a ligand, also showed low catalytic activity (49% yield, entry 4). Accordingly, we selected the manganese complex as the best catalyst precursor among the tetranuclear complexes, [M(tmhd)(OMe)(MeOH)x]4, we tested.| a Determined by GC analysis with dodecane as an internal standard. |
|---|
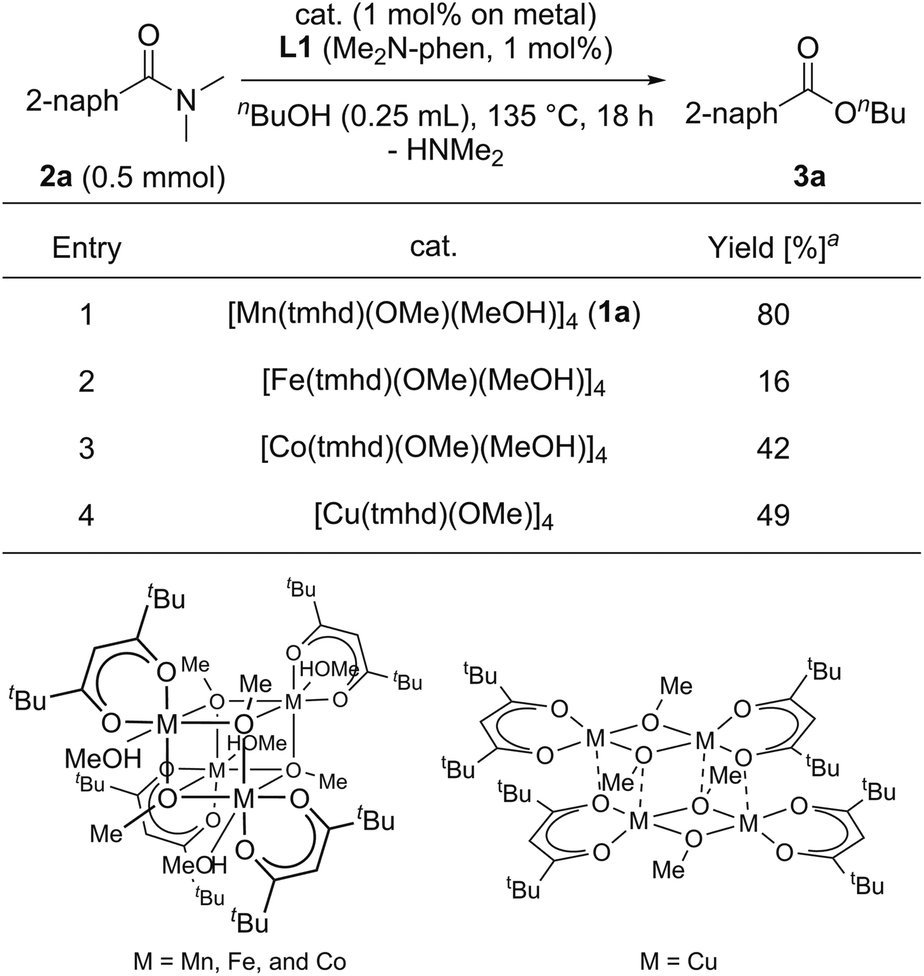
|
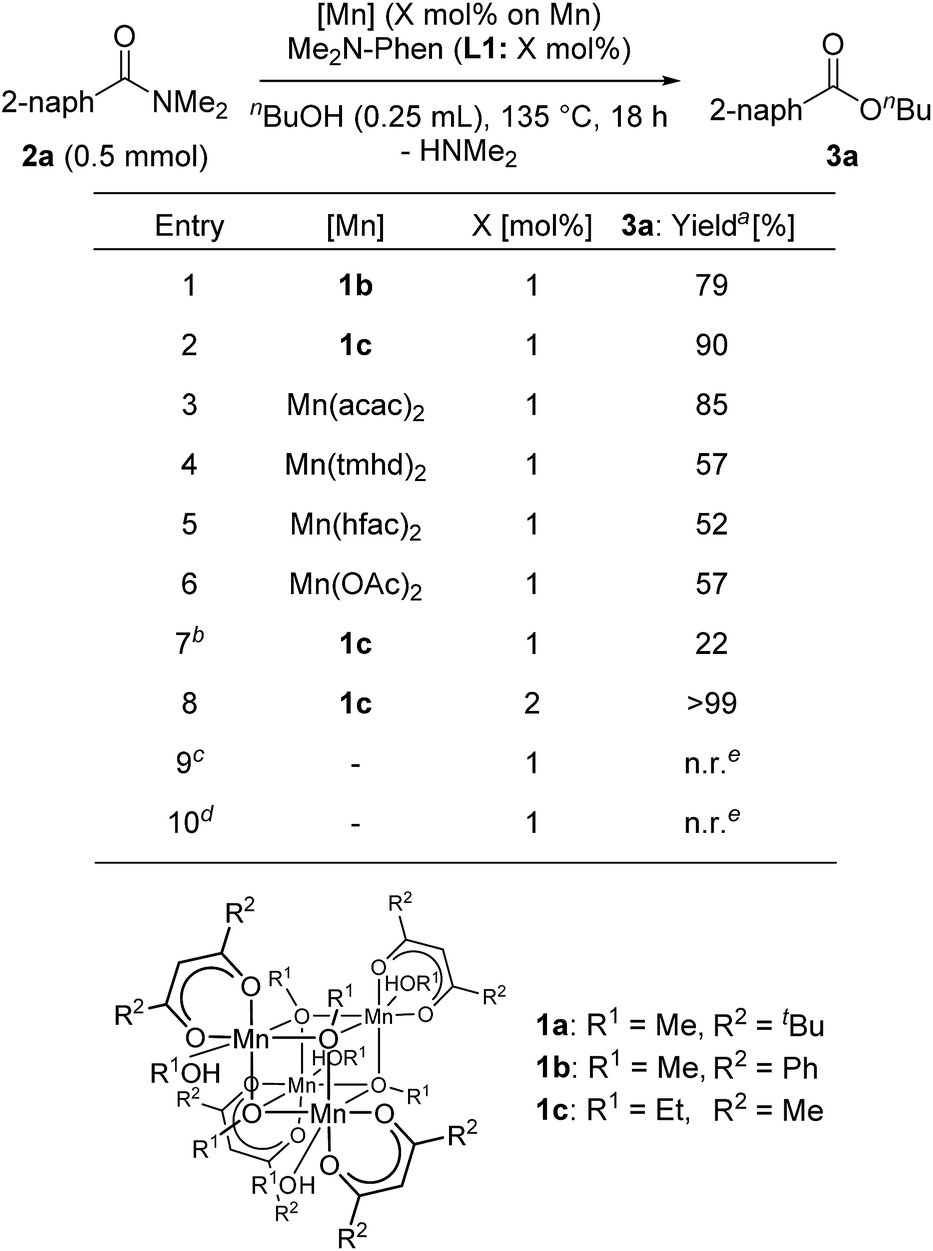
Next, we searched for the best manganese precursor for the catalytic esterification of 2a with n-butanol under the conditions of Mn precursors (1 mol% based on Mn atoms) and L1 (1 mol%) at reflux temperature for 18 h, and the results are shown in Table 2. The manganese tetranuclear alkoxide complex [Mn(dbm)(OMe)(MeOH)]4 (1b: DBM = dibenzoylmethanate)30a exhibited almost the same catalytic activity as 1a to give the corresponding ester 3a in 79% yield (entry 1), and a newly synthesized manganese complex, [Mn(acac)(OEt)(EtOH)]4 (1c), showed the highest catalytic activity among the alkoxy-bridged tetranuclear complexes to yield 3a in 90% yield (entry 2). We also surveyed other simple Mn precursors such as Mn(acac)2, Mn(tmhd)2, Mn(hfac)2 (HFAC = hexafluoroacetylacetonate), and Mn(OAc)2 but these catalyst precursors showed moderate activity to afford 3a in 85%, 57%, 52%, and 57% yields, respectively (entries 3–6). In the absence of the L1 ligand, 1c exhibited low catalytic activity (entry 7, 22% yield). The yield of 3a was increased to >99% when increasing the catalyst loading of 1c to 2 mol% (entry 8). The use of 1 mol% of strong bases, such as KOMe and NaOMe, did not result in catalytic activity (entries 9 and 10). Thus, we selected catalyst 1c as the best precursor, and optimized the conditions using 2 mol% of 1c and L1 in n-butanol at reflux temperature for 18 h. Under the optimized reaction conditions, we detected signals of cationic manganese dinuclear complexes, [Mn(acac)(OnBu)(Me2N-Phen)]2+ (m/z = 986.4) and [Mn(acac)(OnBu)(Me2N-Phen)]2 + H+ (m/z = 987.4), by ESI-MS spectroscopy. In addition, we isolated a dinuclear complex, [Mn(acac)(OEt)(Phen)]2, and characterized it by single crystal X-ray analysis (vide infra).
We then searched for a suitable chelating nitrogen ligand for the catalytic esterification of N,N-dimethyl-2-naphthamide (2a) under the conditions of using [Mn(acac)(OEt)(EtOH)]4 (1c: 1.0 mol%) and a nitrogen ligand (1.0 mol%) in refluxing n-butanol (5.5 equiv.) for 18 h, and the results are shown in Table 3. We first used 1,10-phenanthroline derivatives L2–L7. Electron-donating groups at 4,7-positions on 1,10-phenanthroline exhibited remarkable substituent effects for increasing catalytic activities: L2 with methoxy groups, L3 with methyl groups, and L5 with phenyl groups produced 3a in moderate yields (67%, 54%, and 68%, respectively; entries 1, 2, and 4). In contrast, 1,10-phenanthroline (L4) and L6 with electron-withdrawing chloro groups exhibited decreased catalytic activity, affording 36% and 27% yields of 3a, respectively (entries 3 and 5). The sterically congested chelating nitrogen ligand 2,9-dimethyl-1,10-phenanthroline (L7) gave 3a in 49% yield (entry 6). 4,4′-Substituted 2,2′-bipyridine derivatives, such as L8 with methoxy groups, L9 with tert-butyl groups, L10 with methyl groups, 2,2′-bipyridine (L11), and L12 with methyl ester groups, exhibited low catalytic activities (entries 7–11). In addition, 5,5′-dimethyl-2,2′-bipyridine (L13), 5,5′-bis(trifluoromethyl)-2,2′-bipyridine (L14), and 6,6′-dimethyl-2,2′-bipyridine (L15) produced 3a in low yields (28%, 19%, and 14%, respectively, entries 12–14). Monodentate pyridine derivatives L16–L20 afforded 3a in 16%, 18%, 19%, 29%, and 20% yields, respectively (entries 15–19), which was almost the same as the catalytic activity of 1c without any ligand (Table 2, entry 7). Consequently, we selected L1 as the best ligand for [Mn(acac)(OEt)(EtOH)]4 (1c) with 2 mol% catalyst loading.
| a Determined by GC analysis with dodecane as an internal standard. |
|---|
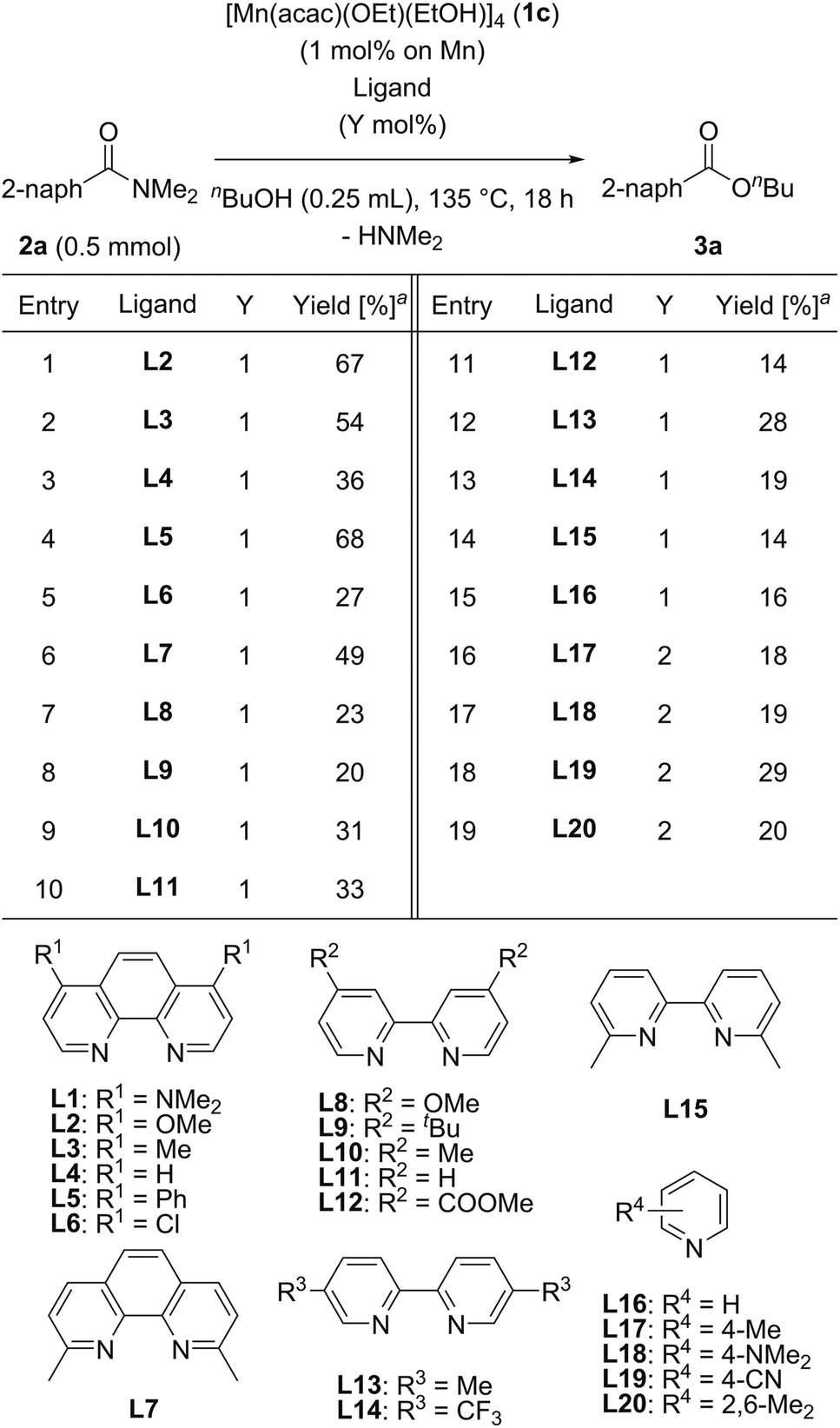
|
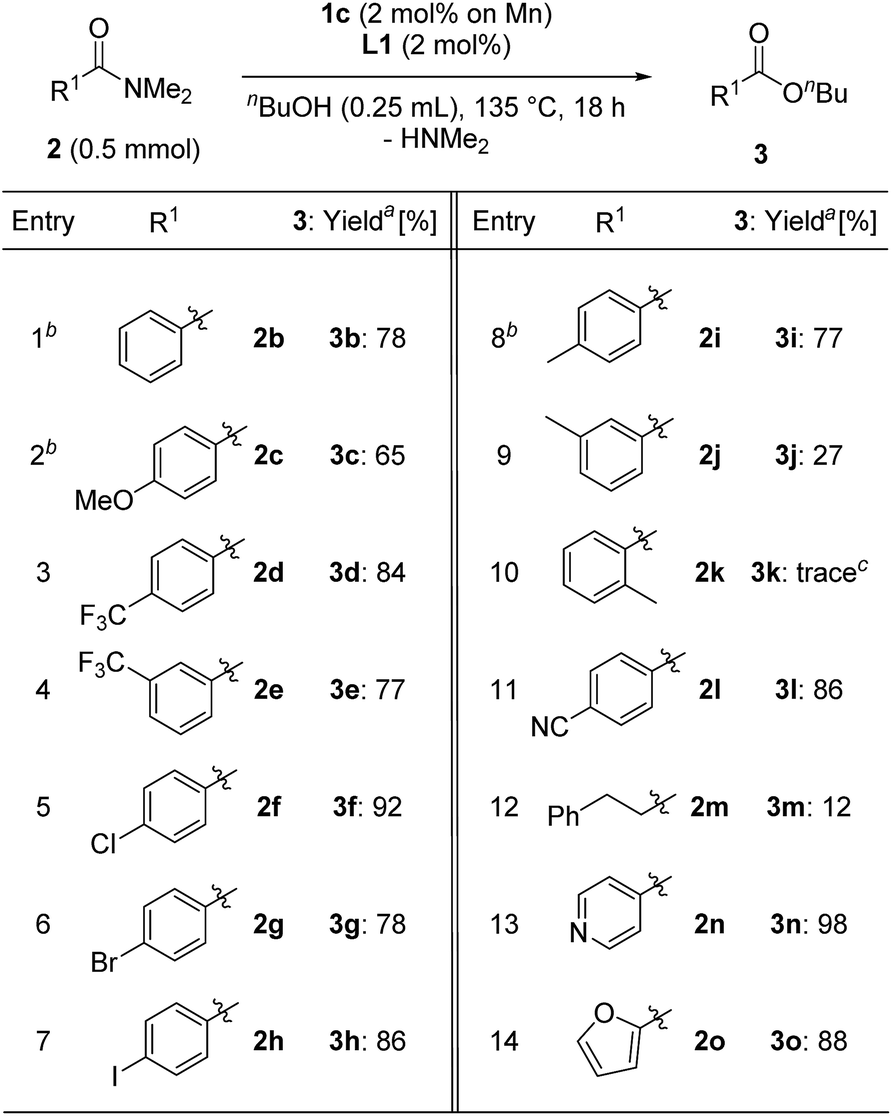
We next evaluated the substrate scope for the esterification of N,N-dimethylamides (Table 4). N,N-dimethylbenzamide (2b) afforded n-butyl benzoate (3b) in a good yield (78%, entry 1). The electronic effects of the para-position on N,N-dimethylbenzamide were examined: 2c having a methoxy group slightly decreased the yield to 65% (entry 2), while substrates 2d and 2e with a trifluoromethyl group at the para- and meta-positions afforded 3d and 3e in the same high yield as 3b (3d: 84%, 3e: 77% yields, entries 3 and 4). Benzamides 2f–h having chloro, bromo, and iodo atoms at the para-position gave the corresponding products 3f–h in high yield without any loss of the C–X bond (entries 5–7). Notably, in the case of methyl substituted compounds, the steric congestion around the carbonyl moiety of the amides was crucial: 4-methyl- and 3-methyl derivatives 2i and 2j produced 3i in 77% yield and 3j in 27% yield, while N,N-dimethyl-2-methylbenzamide (2k) led to a trace amount of 3k (entries 8–10). 4-Cyano-N,N-dimethylbenzamide (2l) gave 3l in 86% yield (entry 11). On the other hand, the use of an aliphatic substituted amide, N,N-dimethyl-3-phenylpropanamide, afforded 12% of the corresponding ester 3m (entry 12). N,N-Dimethylisonicotinamide (2n) and N,N-dimethyl-2-furamide (2o) were converted to the corresponding esters 3n and 3o in 98% and 88% yields, respectively (entries 13 and 14).
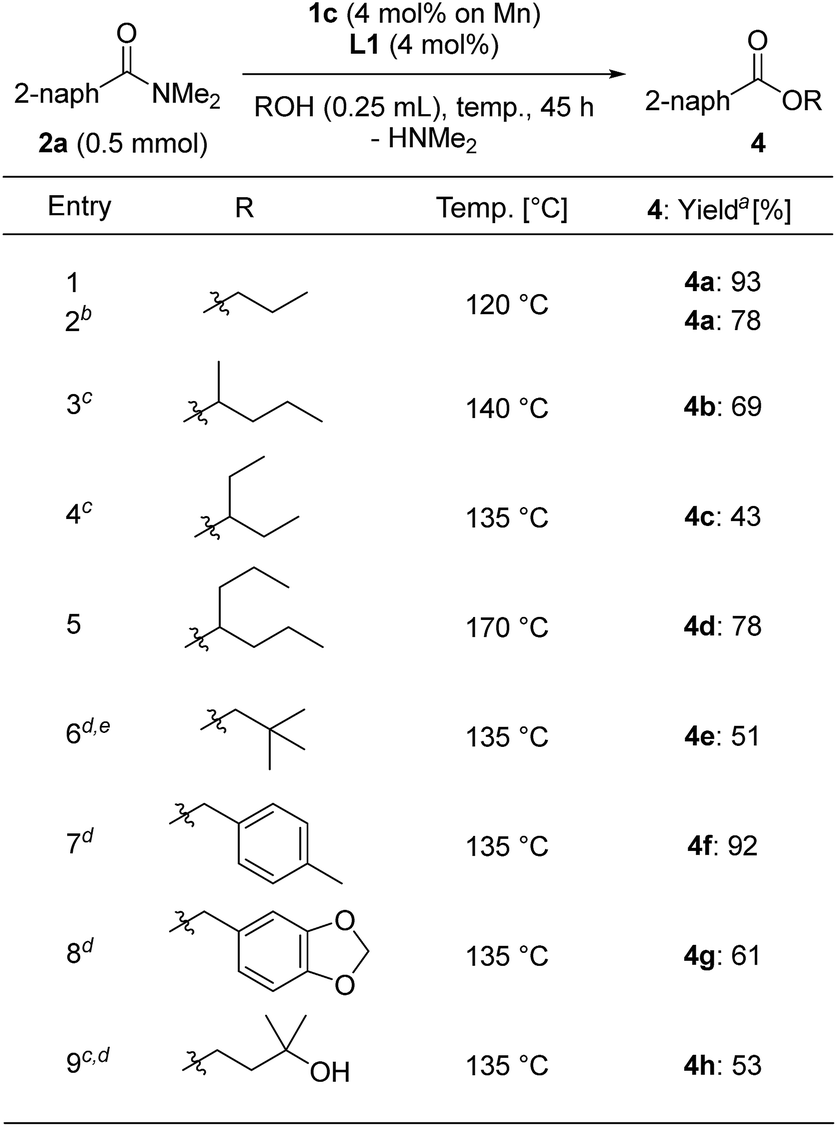
We varied the alcohols used for the esterification of 2a (Table 5). We used alcohols with a low boiling point and sterically bulky secondary alcohols, and modified the reaction conditions by increasing the loading of 1c to 4 mol% based on Mn metal with L1 (4 mol%) and using a longer reaction time depending on the alcohols we assessed. The reaction with n-propanol at reflux temperature (120 °C) for 45 h gave n-propyl benzoate (4a) in 93% yield, although the reaction of n-propanol with 2 mol% of 1c and L1 for 45 h afforded 4a in lower yields (78%) (entries 1 and 2). Secondary alcohols such as 2-pentanol and 3-pentanol, respectively, afforded 4b and 4c in 69% and 43% yields at reflux temperature (140 °C and 135 °C, respectively) for 72 h (entries 3 and 4), while 4-heptanol produced the corresponding ester 4d in 78% yield at reflux temperature (170 °C) for 72 h (entry 5). Solid alcohols such as 2,2-dimethylpropanol, (4-methylphenyl)methanol, and piperonyl alcohol could be used for the esterification of 2a in toluene at 135 °C, giving the corresponding esters 4e (51% yield for 18 h reaction time), 4f (92% yield), and 4g (61% yield) (entries 6–8). When 3-methyl-1,3-butanediol was used for the esterification, 3-hydroxy-3-methylbutyl-2-naphthoate (4h) was selectively obtained in a moderate yield (53% yield, entry 8).
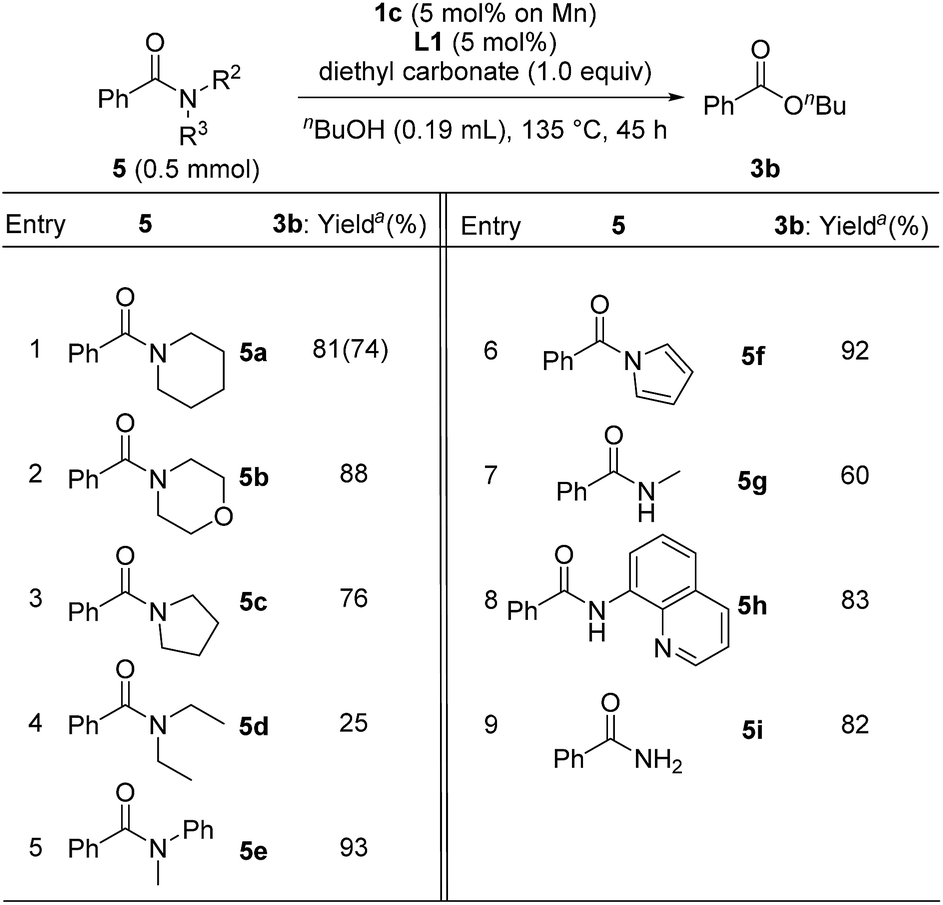
In contrast to the catalytic esterification of N,N-dimethylamides, in which volatile dimethylamine was readily removed, the addition of 1 equiv. of diethyl carbonate was required to trap liberated amines to accomplish smooth esterification of various substituents on the nitrogen atom of benzamide under conditions of using 1c (5 mol% based on Mn atom) with L1 (5 mol%) in refluxing n-butanol for 45 h (Table 6). Cyclic derivatives of benzamide with piperidine 5a, morpholine 5b, and pyrrolidine 5c were converted into n-butyl benzoate 3b in 81%, 88%, and 76% yields, respectively (entries 1–3), though acyclic N,N-diethylbenzamide (5d) significantly decreased the yield of 3b, (25%, entry 4), probably due to the steric hindrance around the carbonyl group of 5d. Esterification of N-methyl-N-phenylbenzamide (5e) and 1-benzoyl pyrrole (5f) gave 3b in 93% and 92% yields, respectively (entries 5 and 6). Moreover, the manganese catalyst system effectively mediated the esterification of secondary and primary amides, such as N-methylbenzamide (5g: 60% yield), N-(quinolin-8-yl)benzamide (5h: 83% yield), and benzamide (5i: 82% yield) (entries 7–9).
| a Determined by GC analysis with dodecane as an internal standard. Isolated yield is given in parentheses. |
|---|
|
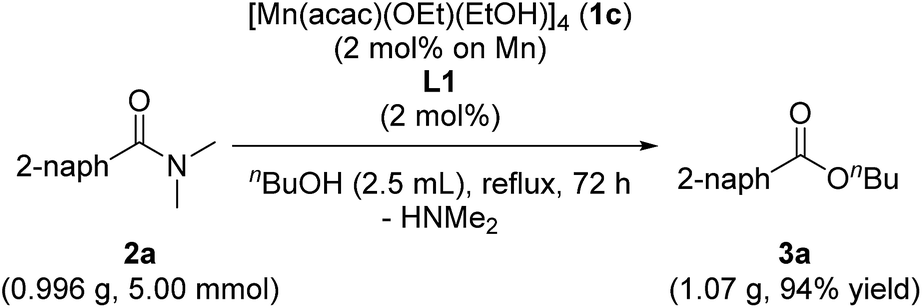
When we conducted a large scale reaction by using 0.996 g of 2a under the conditions of using 1c (2.0 mol%) and L1 (2.0 mol%) in refluxing n-butanol (2.5 mL, 5.5 equiv.) for 72 h, we obtained 1.07 g of corresponding 3a in 94% yield (eqn (1)).
 | (1) |
When radical scavengers, such as TEMPO and BHT, were added to the reaction mixture, the yields of 3a were slightly decreased to 83% and 73%, respectively, and by-products containing TEMPO or BHT moieties were not detected (eqn (2)). Thus, we proposed that the reaction mechanism is not a radical pathway.
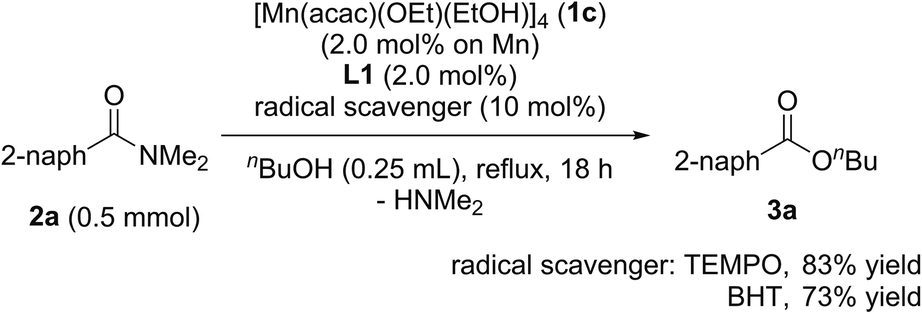 | (2) |
Regarding the reaction mechanism, we isolated an alkoxide-bridged Mn(II) dinuclear complex, [Mn(acac)(OEt)(Phen)]2 (6c: 47% yield), by treating complex 1c with 1,10-phenanthroline (L4) (eqn (3)), as the corresponding manganese complex containing Me2N-Phen (L1) provided no single crystals suitable for X-ray analysis. Complex 6c was characterized by elemental analysis and X-ray analysis due to its paramagnetic nature. In the solid state, complex 6c has a dimeric structure, in which two manganese centers are doubly bridged by two μ-ethoxy groups and each manganese atom adopts a distorted octahedral structure (Fig. 2).
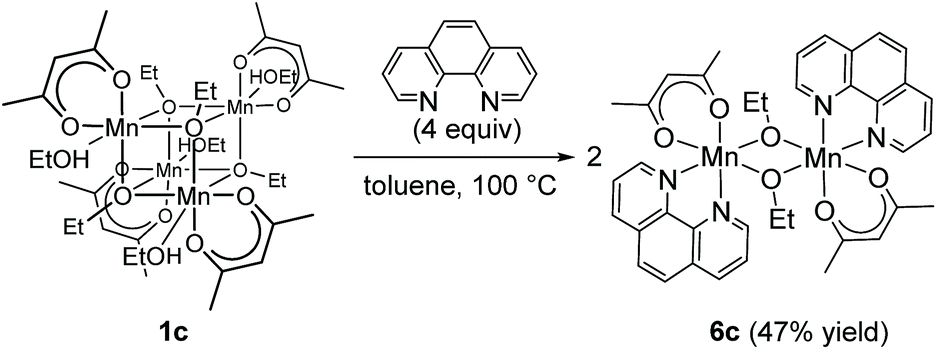 | (3) |
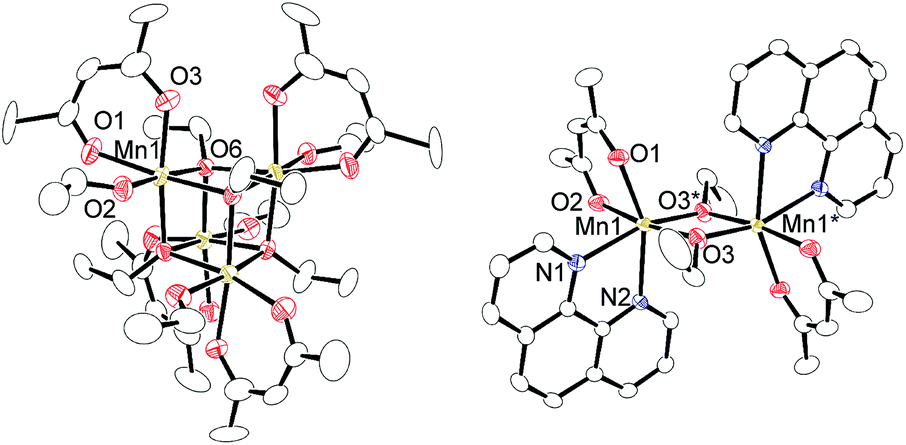 | ||
| Fig. 2 Molecular structures of complex 1c and 6c with 50% thermal ellipsoids. All hydrogen atoms and solvent molecule are omitted for clarity. | ||
We determined a power value on the concentration of substrate 2a using time normalization analysis.34a A mixture of 2a (0.600, 0.800, and 1.00 M) and [Mn(acac)(OEt)(Phen)]2 (6c, 60.7 mg, 80 μmol, 40 mM) in n-butanol (2.00 mL) with dodecane as an internal standard was refluxed at 145 °C and the time course of the yield of 3a was determined by GC analysis. As shown in Fig. 3, the concentration of 3a was plotted against a normalized time scale, ∑[2a]aΔt, and we adjusted the power value, a, until all the corrected conversion curves overlay. As a result, we determined that the value of a was 1 (Fig. 3c). We also determined a power value on the concentration of the catalyst [Mn(acac)(OEt)(Phen)]2 (6c) by using a normalized time scale analysis.34b A mixture of 2a (398.5 mg, 2.00 mmol, 2.00 M) and 6c (30 mM, 50 mM, and 70 mM) in n-butanol (1.00 mL) with dodecane as an internal standard was refluxed at 145 °C. As shown in Fig. 4, the concentration of 3a was plotted against a normalized time scale, t[6c]n, and we determined that the value of n was 1 (Fig. 4c). The first-order rate dependency of 6c suggested that the dinuclear manganese complex 6c acted as a catalytically active species, whose dinuclear function was comparable to that found for an alkoxide-bridged cobalt dinuclear complex active for catalytic transesterification.35
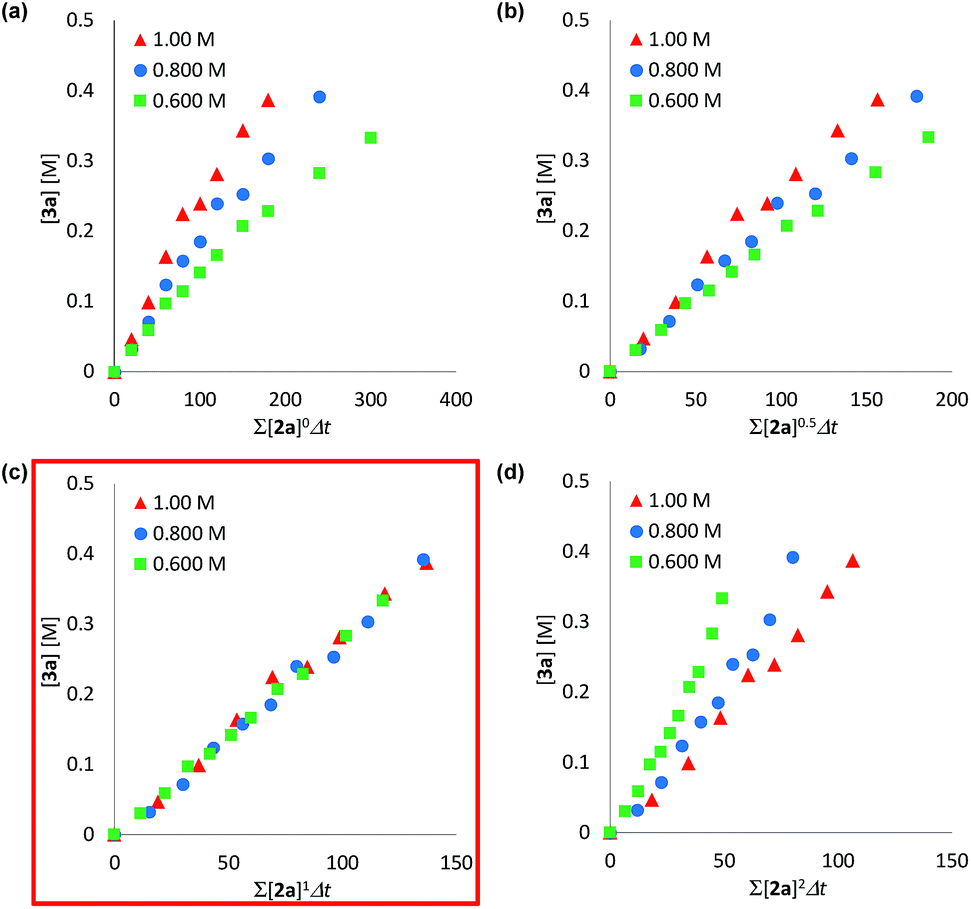 | ||
| Fig. 3 Time normalization analysis to determine the order in amide 2a; (a) a = 0, (b) a = 0.5, (c) a = 1, (d) a = 2. | ||
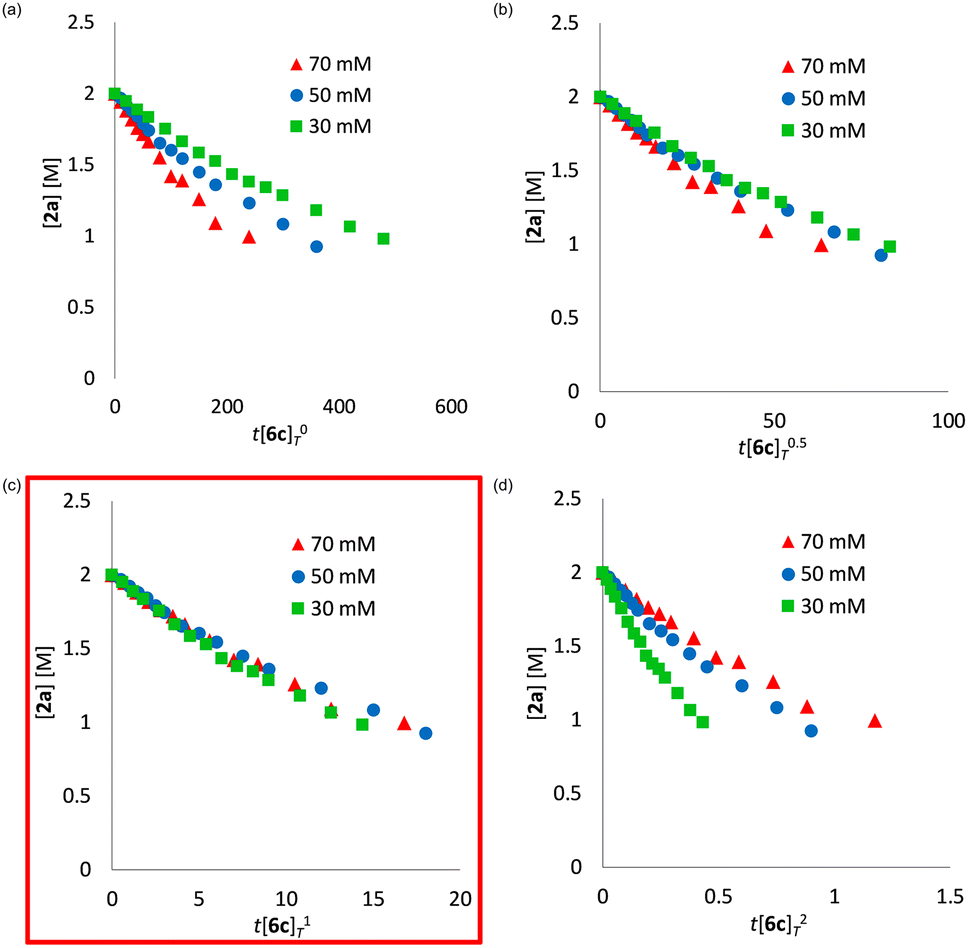 | ||
| Fig. 4 Time normalization analysis to determine the order in dinuclear manganese complex 6c; (a) a = 0, (b) a = 0.5, (c) a = 1, (d) a = 2. | ||
On the basis of these results, we propose a reaction mechanism of the catalytic esterification of simple tertiary N,N-dimethylamides described in Scheme 1. The first step is the coordination of amide 2 onto a manganese atom of complex 6c, affording dinuclear complex A. The alkoxy group bound to the manganese center in A attacks the carbonyl group of the amide moiety to cleave a C–N bond via intermediate B, in which an N^N-bidentate ligand with electron-donating groups at the para-positions accelerated the nucleophilic attack. According to the kinetics, the nucleophilic attack of the alkoxide on the carbonyl group is the rate-determining step. The intermediate B is transformed to C, forming a manganese complex D, where the resulting ester coordinates to the manganese atom bearing a dimethylamido moiety. Finally, protonolysis of the manganese-amido moiety using alcohol produces dimethylamine and the dinuclear Mn(II)-alkoxide complex together with the release of ester 3.
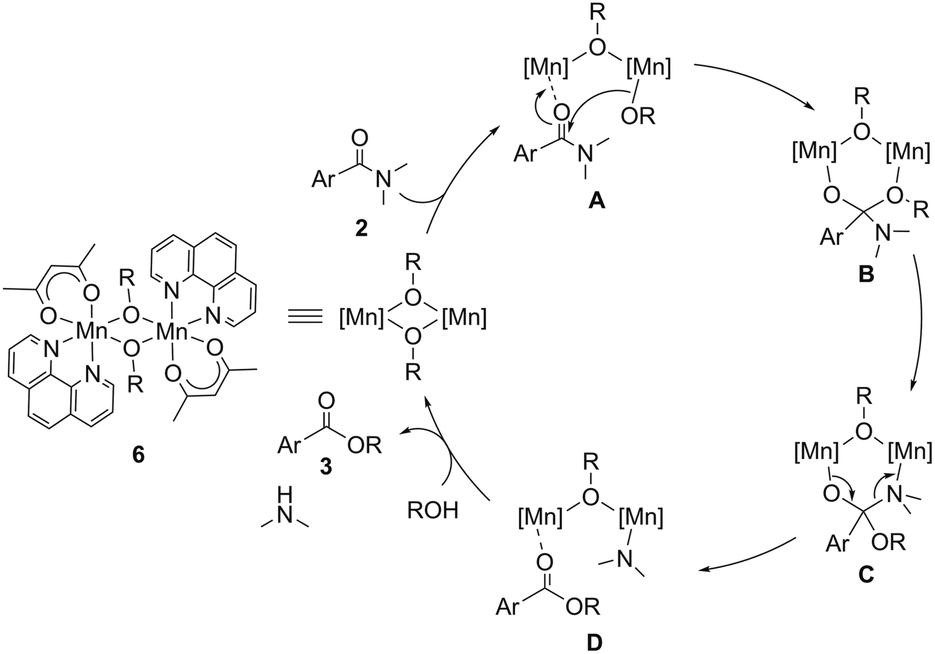 | ||
| Scheme 1 A plausible reaction mechanism for the catalytic esterification of simple tertiary N,N-dimethylamides. | ||
Conclusions
In summary, we provide the first example of catalytic esterification of simple tertiary N,N-dialkylamides under neutral conditions using the catalyst system [Mn(acac)(OEt)(EtOH)]4 (1c, 2 mol% based on Mn metal) and 4,7-bis(dimethylamino)-1,10-phenanthroline (L1: Me2N-Phen, 2 mol%). Regarding the reaction mechanism, we isolated the alkoxide-bridged manganese dinuclear complex [Mn(acac)(OEt)(Phen)]2 (6c) as the key catalyst, and conducted kinetic studies in which the velocity obeys kobs[2a]1[6c]1. It is notable that such a catalytic performance of the dinuclear manganese complex 6c is related to the mechanism of metalloenzymes that regulate the transformation of carboxylic derivatives. Further mechanistic studies and catalytic application of a Mn-based catalyst system are ongoing in our laboratory.Experimental section
All manipulations involving air- and moisture-sensitive organometallic compounds were carried out under argon using the standard Schlenk technique or an argon-filled glovebox. n-Propanol, 2-propanol, n-butanol, 2-pentanol, 3-pentanol, 4-heptanol, and diethyl carbonate were distilled under an argon atmosphere from CaH2. 3-Methyl-1,3-butanediol was distilled under an argon atmosphere by using Kugelrohr. 2,4-Pentadione was distilled under an argon atmosphere from P4O10. THF, diethyl ether, hexane, and toluene were dried and deoxygenated using a Grubbs column (Glass Contour Solvent Dispensing System, Nikko Hansen & Co., Ltd.).36 Ethanol and methanol were distilled under an argon atmosphere from the corresponding magnesium alkoxide. DMSO-d6 was dried over MS4A in a J-Young Schlenk under an argon atmosphere. Wako super dehydrated DMF was degassed and kept in a J-Young Schlenk with MS4A under an argon atmosphere. Lithium ethoxide was prepared by treating metal lithium with ethanol in hexane suspension. All other reagents were purchased from commercial resources and used without further purification. NMR spectra were recorded on a Bruker AV 400M spectrometer operating at 400 MHz (1H NMR), at 100 MHz (13C{1H} NMR) and at 376 MHz (19F{1H} NMR) in 5 mm NMR tubes. All 1H NMR chemical shifts were reported in ppm relative to the TMS at 0.00 ppm or residual solvent protons in DMSO-d6 at δ 2.50. All 13C{1H} NMR chemical shifts were reported in ppm relative to carbon resonance in chloroform-d1 at δ 77.16 and DMSO-d6 at δ 39.52. All 19F{1H} NMR chemical shifts were reported in ppm relative to the external reference α,α,α-trifluorotoluene at δ −63.9. Melting points were recorded using a BUCHI Melting Point M-565 and YANACO MP-J3. IR spectra were recorded on a JASCO FT/IR 4000 spectrometer. Mass spectra (MS) and high resolution mass spectra (HRMS) were recorded using a JEOL JMS-700. ESI-mass spectrometric data were obtained using a BRUKER microTOF-II spectrometer. GC analyses were recorded on a Shimadzu GC-2014 gas chromatograph with J&W Scientific DB-5 and Shimadzu SH-Rtx-50 columns. Flash column chromatography was performed using silica gel 60 (0.040–0.063 mm, 230–400 mesh ASTM).General procedure for catalytic esterification of N,N-dimethyl-2-naphthamide (1.0 mol%)
The mixture of catalyst precursor (5.0 μmol based on Mn atoms), ligand (5.0 μmol), and N,N-dimethyl-2-naphthamide (99.6 mg, 5.00 × 10−1 mmol) in n-butanol (2.50 × 10−1 mL) was refluxed at 135 °C for 18 hours under an argon atmosphere in a slim Schlenk (ϕ 10 mm). After cooling to room temperature, yields were determined using the following procedures.(1) GC yield: metal salts were removed by filtration through silica gel with ethyl acetate. The yield was determined by GC analysis with dodecane as an internal standard.
(2) NMR yield: metal complexes were removed by filtration through silica gel with CDCl3 twice. The yield was determined by 1H NMR analysis with phenanthrene as an internal standard.
General procedure for preparation of amides
Amides were synthesized by standard condensation reaction using the corresponding acyl chlorides and amines. Products were purified by silica gel flash column chromatography or distillation under reduced pressure using Kugelrohr, and characterized by 1H and 13C{1H} NMR spectroscopies.Preparation of ligands, substrate amides, and manganese complexes
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOAc = 20:1), and distillation using Kugelrohr (>0.05 mmHg, 125–135 °C) to give a colorless liquid (1.23 g, 7.18 mmol, 72% yield).
:EtOAc = 20:1), and distillation using Kugelrohr (>0.05 mmHg, 125–135 °C) to give a colorless liquid (1.23 g, 7.18 mmol, 72% yield).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI Grant Numbers JP26248028 and JP16H06934, Grant-in-Aid for Scientific Research(A) and Grant-in-Aid for Young Scientists (Start-up). We thank Ms Shoko Akiyama for her experimental contribution in the early stage of the project.Note and references
- A. Greenberg, C. M. Berneman and J. F. Liebman, The Amide Linkage: Structural Significance in Chemistry and Biotechnology and Materials Science, John Wiley & Sons, Ltd., 2003 Search PubMed.
- H. Lundberg, F. Tinnis, N. Selander and H. Adolfsson, Chem. Soc. Rev., 2014, 43, 2714–2742 RSC.
- J. M. García, F. C. García, F. Serna and J. L. de la Peña, Prog. Polym. Sci., 2010, 35, 623–686 CrossRef.
- R. Das, G. S. Kumar and M. Kapur, Eur. J. Org. Chem., 2017, 5439–5459 CrossRef CAS.
- S. A. Glover and A. A. Rosser, J. Org. Chem., 2012, 77, 5492–5502 CrossRef CAS.
- R. P. Houghton and R. R. Puttner, J. Chem. Soc. D, 1970, 1270–1271 RSC.
- M. C. Bröhmer, S. Mundinger, S. Bräse and W. Bannwarth, Angew. Chem., Int. Ed., 2011, 50, 6175–6177 CrossRef PubMed.
- A. J. Kirby, I. V. Komarov, P. D. Wothers and N. Feeder, Angew. Chem., Int. Ed., 1998, 37, 785–786 CrossRef CAS.
- M. Szoztak and J. Aubé, Chem. Rev., 2013, 113, 5701–5765 CrossRef PubMed.
- (a) L. E. Fisher, J. M. Caroon, S. R. Stabler, S. Lundberg, S. Zaidi, C. M. Sorensen, M. L. Sparacino and J. M. Muchowski, Can. J. Chem., 1994, 72, 142–145 CrossRef CAS; (b) X. Chen, S. Hu, R. Chen, J. Wang, M. Wu, H. Guo and S. Sun, RSC Adv., 2018, 8, 4571–4576 RSC.
- S. M. A. H. Siddiki, A. S. Touchy, M. Tamura and K. Shimizu, RSC Adv., 2014, 4, 35803–35807 RSC.
- Y. Kita, Y. Nishii, A. Onoue and K. Mashima, Adv. Synth. Catal., 2013, 355, 3391–3395 CrossRef CAS.
- B. N. Atkinson and J. M. J. Williams, Tetrahedron Lett., 2014, 55, 6935–6938 CrossRef CAS.
- T. Deguchi, H.-L. Xin, H. Morimoto and T. Ohshima, ACS Catal., 2017, 7, 3157–3161 CrossRef CAS.
- L. Hie, N. F. Fine Nathel, T. K. Shah, E. L. Baker, X. Hong, Y.-F. Yang, P. Liu, K. N. Houk and N. K. Garg, Nature, 2015, 524, 79–83 CrossRef CAS PubMed.
- L. Hie, E. L. Baker, S. M. Anthony, J.-N. Desrosiers, C. Senanayake and N. K. Garg, Angew. Chem., Int. Ed., 2016, 55, 15129–15132 CrossRef CAS PubMed.
- J. E. Dander and N. K. Garg, ACS Catal., 2017, 7, 1413–1423 CrossRef CAS PubMed.
- Y. Bourne-Branchu, C. Gosmini and G. Danoun, Chem.–Eur. J., 2017, 23, 10043–10047 CrossRef CAS PubMed.
- X. Li and G. Zou, Chem. Commun., 2015, 51, 5089–5092 RSC.
- G. Meng and M. Szostak, Org. Lett., 2015, 17, 4364–4367 CrossRef CAS PubMed.
- C. Liu and M. Szostak, Chem.–Eur. J., 2017, 23, 7157–7173 CrossRef CAS PubMed.
- J. Hu, M. Wang, X. Pu and Z. Shi, Nat. Commun., 2017, 8, 14993 CrossRef PubMed.
- S.-C. Lee, L. Guo, H. Yue, H.-H. Liao and M. Rueping, Synlett, 2017, 28, 2594–2598 CrossRef CAS.
- A. Dey, S. Sasmal, K. Seth, G. K. Lahiri and D. Maiti, ACS Catal., 2017, 7, 433–437 CrossRef CAS.
- J. A. Walker Jr, K. L. Vickerman, J. N. Humke and L. M. Stanley, J. Am. Chem. Soc., 2017, 139, 10228–10231 CrossRef PubMed.
- C. W. Cheung, J.-A. Ma and X. Hu, J. Am. Chem. Soc., 2018, 140, 6789–6792 CrossRef CAS PubMed.
- Y. Nishii, S. Akiyama, Y. Kita and K. Mashima, Synlett, 2015, 26, 1831–1834 CrossRef CAS.
- Y. Nishii, T. Hirai, S. Fernandez, P. Knochel and K. Mashima, Eur. J. Org. Chem., 2017, 5010–5014 CrossRef CAS.
- T. Toyano, M. N. Rashed, Y. Morita, T. Kamichi, S. M. A. H. Siddiki, M. A. Ali, A. S. Touchy, K. Kon, Z. Maeno, K. Yoshizawa and K. Shimizu, ChemCatChem, 2019, 11, 449–456 CrossRef.
- (a) L. E. Pence, A. Caneschi and S. J. Lippard, Inorg. Chem., 1996, 35, 3069–3072 CrossRef CAS; (b) K. L. Taft, A. Caneschi, L. E. Pence, C. D. Delfs, G. C. Papaefthymiou and S. J. Lippard, J. Am. Chem. Soc., 1993, 115, 11753–11766 CrossRef CAS.
- J. F. Berry, F. A. Cotton, C. Y. Liu, T. Lu, C. A. Murillo, B. S. Tsukerblat, D. Villagrán and X. Wang, J. Am. Chem. Soc., 2005, 127, 4895–4902 CrossRef CAS PubMed.
- W. H. Watson and W. W. Holley, Croat. Chem. Acta, 1984, 57, 467–476 CAS.
- N,N-Dimethylamide could not be used as a substrate in Ni(0) catalysed esterification (see ref. 15). In addition, the esterification of 2a by the catalyst system Mn(acac)2 (10 mol%) with 2,2′-bipyridine (10 mol%), which is the same as our previous catalyst system (see ref. 27), in nBuOH at reflux temperature for 18 h afforded the corresponding ester 3a in 68% yield.
- (a) J. Burés, Angew. Chem., Int. Ed., 2016, 55, 2028–2031 CrossRef PubMed; (b) J. Burés, Angew. Chem., Int. Ed., 2016, 55, 16084–16087 CrossRef PubMed.
- Y. Hayashi, S. Santoro, Y. Azuma, F. Himo, T. Ohshima and K. Mashima, J. Am. Chem. Soc., 2013, 135, 6192–6199 CrossRef CAS PubMed.
- A. B. Pangborn, M. A. Giardello, R. H. Grubbs, R. K. Rosen and F. J. Timmers, Organometallics, 1996, 15, 1518–1520 CrossRef CAS.
- C. K. Lee, J. H. Jun and J. S. Yu, J. Heterocycl. Chem., 2000, 37, 15–24 CrossRef CAS.
- P. Wehman, V. E. Kaasjager, F. Hartl, P. C. J. Kamer, P. W. N. M. van Leeuwen, J. Fraanje and K. Goubitz, Organometallics, 1995, 14, 3751–3761 CrossRef CAS.
- A. Altomare, G. Cascarano, C. Giacovazzo and A. Guagliardi, J. Appl. Crystallogr., 1993, 26, 343–350 CrossRef.
- CrystalClear: Data Collection and Processing Software, Rigaku Corporation, 1998–2015, Tokyo 196-8666, Japan Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1865079, 1865080. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc05819a |
| This journal is © The Royal Society of Chemistry 2019 |