
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRelative orientation of the carbonyl groups determines the nature of orbital interactions in carbonyl–carbonyl short contacts†
Biswajit
Sahariah
 and
Bani Kanta
Sarma
*
and
Bani Kanta
Sarma
*
Department of Chemistry, School of Natural Sciences, Shiv Nadar University, Dadri, Uttar Pradesh-201314, India. E-mail: banikanta.sarma@snu.edu.in
First published on 29th October 2018
Abstract
Carbonyl–carbonyl (CO⋯CO) interactions are emerging noncovalent interactions found in many small molecules, polyesters, peptides and proteins. However, little is known about the effect of the relative orientation of the two carbonyl groups on the nature of these interactions. Herein, we first show that simple homodimers of acetone and formaldehyde can serve as models to understand the effect of relative orientations of the two carbonyl groups on the nature of CO⋯CO interactions. Further, from a comprehensive statistical analysis of molecules having inter- or intramolecular CO⋯CO interactions, we show that the molecules can be broadly categorized into six different structural motifs (I–VI). The analysis of pyramidality of the acceptor carbon atoms in these motifs and natural bond orbital (NBO) analysis suggest that the relative orientation of the two interacting carbonyl groups determines whether the orbital interaction between the two carbonyl groups would be n → π* or π → π* or a combination of both.
Introduction
Noncovalent interactions mediated by carbonyl groups are important in both chemistry and biology. The carbonyl group is polar and has a strong dipole moment. Therefore, it can participate in electrostatic interactions through its positively charged carbon and negatively charged oxygen centers as well as can involve in dipolar interactions. Further, the lone pairs of oxygen and the antibonding π orbital of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond of the carbonyl group can be involved in orbital interactions with appropriate acceptor and donor atom orbitals, respectively. These properties make the carbonyl group unique and it can participate in many distinct noncovalent interactions such as hydrogen bonding1 (CO⋯H), carbonyl–chalcogen interactions2 (CO⋯X; X = S, Se, and Te) and nucleophile–carbonyl interactions3 (Nu⋯CO) in chemical and biological systems. While hydrogen bonding and carbonyl–chalcogen interactions are well studied, relatively less is known about Nu⋯CO interactions. In their seminal work,4 Burgi et al. showed that various nucleophilic atoms noncovalently interact with the carbonyl carbon atom in a specific trajectory [∠Nu–CO ∼ 109°; Burgi–Dunitz (BD) trajectory], which laid the foundation for our current understanding of the mechanisms of nucleophilic addition and substitution reactions at the carbonyl centers. When the nucleophile is another carbonyl group, the interaction is termed as a carbonyl–carbonyl (CO⋯CO) interaction. This interaction is important as it can control the geometries and functions of molecules, especially when they have multiple carbonyl groups such as polyesters, peptoids, peptides and proteins.
O bond of the carbonyl group can be involved in orbital interactions with appropriate acceptor and donor atom orbitals, respectively. These properties make the carbonyl group unique and it can participate in many distinct noncovalent interactions such as hydrogen bonding1 (CO⋯H), carbonyl–chalcogen interactions2 (CO⋯X; X = S, Se, and Te) and nucleophile–carbonyl interactions3 (Nu⋯CO) in chemical and biological systems. While hydrogen bonding and carbonyl–chalcogen interactions are well studied, relatively less is known about Nu⋯CO interactions. In their seminal work,4 Burgi et al. showed that various nucleophilic atoms noncovalently interact with the carbonyl carbon atom in a specific trajectory [∠Nu–CO ∼ 109°; Burgi–Dunitz (BD) trajectory], which laid the foundation for our current understanding of the mechanisms of nucleophilic addition and substitution reactions at the carbonyl centers. When the nucleophile is another carbonyl group, the interaction is termed as a carbonyl–carbonyl (CO⋯CO) interaction. This interaction is important as it can control the geometries and functions of molecules, especially when they have multiple carbonyl groups such as polyesters, peptoids, peptides and proteins.
CO⋯CO short contacts were first observed by Bolton in a glucose analogue named alloxan.5 He reported intermolecular O⋯C distances much shorter than the sum of van der Waals radii (3.22 Å)6 between the oxygen and carbon atoms of the carbonyl groups of two neighboring alloxan molecules in the solid state. Interestingly, in alloxan, the CO⋯CO interaction determined the crystal packing even in the presence of strong hydrogen bond donor (NH) and acceptor (CO) moieties. Several subsequent structural studies established that two carbonyl groups can interact with each other in three possible ways leading to structural motifs I–III shown in Fig. 1A–C.7 Maccallum et al. demonstrated the importance of the “intramolecular” CO⋯CO interaction between proximal amide groups as an important factor in stabilizing α-helices, β-sheets and the right-hand twist often observed in β-strands of proteins.8 Later on, Raines and co-workers showed that perturbations of CO⋯CO interactions could affect the stability of proteins.9 Since then, there have been extensive studies on intramolecular CO⋯CO interactions in small molecules, peptides, polyesters and proteins.10 It is now well established that intramolecular CO⋯CO interactions are predominant in α-helices.10j,k Recently, the “reciprocal” variant of the CO⋯CO interaction has been discovered in small molecules and proteins.11
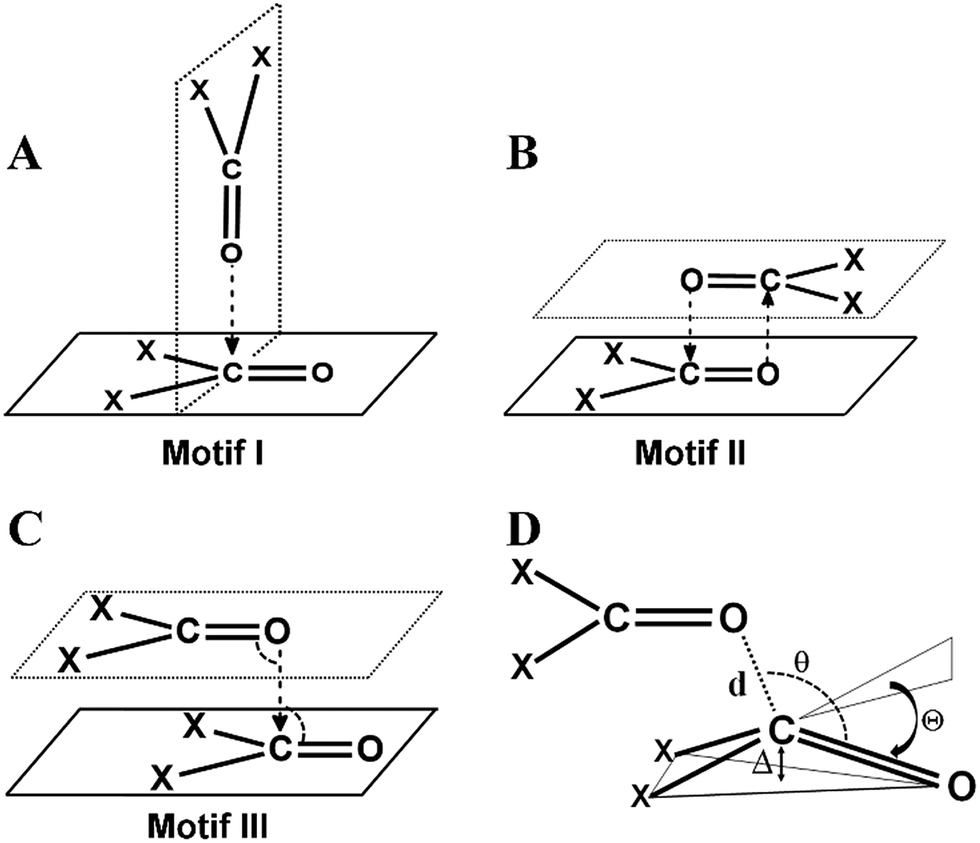 | ||
| Fig. 1 Different structural motifs previously observed by Allen et al. in intermolecular CO⋯CO interactions in ketone dimers (A) perpendicular motif I, (B) antiparallel motif II, (C) sheared parallel motif III and (D) various structural parameters that are indicative of n → π* interactions. | ||
There are two schools of thought on the nature of CO⋯CO interactions. Bolton, Allen and others considered intermolecular CO⋯CO interactions as dipolar interactions.12 Maccallum et al. also considered intramolecular CO⋯CO interactions in proteins and peptides to be dipolar in nature. However, Raines and co-workers showed that intramolecular CO⋯CO interactions are stabilized by n → π* interactions where electron delocalization occurs between the p-character lone pair of a donor carbonyl oxygen atom and the π*CO orbital of the CO bond of a nearby acceptor carbonyl group.13 The n → π* nature of the CO⋯CO interaction in an intramolecular system was subsequently supported by many other researchers working in this area.10 The reciprocal variants of intramolecular CO⋯CO interactions were reported to have contributions from both n → π* and π → π* orbital interactions.11
The questions that then arise are “Are inter- and intramolecular CO⋯CO interactions different in nature? Do the relative orientations of the two interacting carbonyl groups in inter- and intramolecular systems affect the nature of CO⋯CO interactions?” To address these questions, it would be necessary to first find out all the possible structural motifs in molecules having CO⋯CO interactions in both inter- and intramolecular systems. The recent discovery of a reciprocal variant of the CO⋯CO interaction indicates that we may not yet completely know all the possible structural motifs of CO⋯CO interactions for inter- and intramolecular systems. However, to the best of our knowledge, there are no systematic studies on CO⋯CO interactions in both inter- and intramolecular systems. In this paper, we have studied both inter- and intramolecular CO⋯CO interactions using statistical and theoretical methods. Herein, we have first studied the homodimers of acetone and formaldehyde to understand how different orientations of the two interacting carbonyl groups affect the nature of CO⋯CO interactions. Further, we have identified the various structural motifs of inter- and intramolecular CO⋯CO interactions and showed that the nature of orbital interactions for the stabilization of CO⋯CO interactions varies with the relative geometrical orientations of the two interacting carbonyl groups in these motifs. Further, from gas-phase computational and solid-state structural studies we show that the nature of orbital interactions in these motifs can be effectively modelled by using appropriate conformations of homodimers of acetone and formaldehyde.
Results and discussion
1 CO⋯CO interactions in acetone and formaldehyde homodimers
This work is inspired by our observation that the two simple homodimeric carbonyl systems, the acetone dimer and the formaldehyde dimer, have very different arrangements of the carbonyl groups in their most stable conformations in the gas phase. Previous studies have shown that the antiparallel C2h symmetric structure of the acetone dimer that resembles motif II is the global minimum.14a For the formaldehyde dimer, the global minimum is a Cs symmetric structure where the molecular plane of one molecule of formaldehyde is orthogonal to the molecular plane of the second formaldehyde molecule.14b–d In view of their well-established global minimum structures, we avoided carrying out detailed conformational analysis of these dimeric systems and looked only into the C2h symmetric structure of the acetone dimer and the Cs symmetric structure of the formaldehyde dimer for further studies. We optimized these two dimer structures using the MP2/6-311+G(2d,p) level of theory (Fig. 2A–E). Natural bond orbital (NBO)15 analyses were carried out to investigate the orbital interactions in these molecules. We observed that the major orbital interaction for the stabilization of the acetone dimer comes from the reciprocal π → π* orbital interactions (2.06 kcal mol−1) of the filled π orbital of one acetone molecule with the vacant π* orbital of the other. There is a relatively small contribution from n → π* interactions (0.14 kcal mol−1) to the stabilization of the acetone dimer. The BSSE-corrected16 binding energy of the acetone dimer (5.65 kcal mol−1) is much higher than the sum of orbital interaction energies, which suggests that other interactions such as dipole–dipole or electrostatic interactions may also be important for the stabilization of the system. In the formaldehyde dimer on the other hand, the major orbital contribution to the CO⋯CO interaction comes from n → π* electron delocalization of the lone pair of oxygen of one formaldehyde to the π* orbital of the other formaldehyde CO bond (3.9 kcal mol−1) and a relatively small contribution from π → π* orbital interactions (0.26 kcal mol−1). In addition, there is a C–H⋯O hydrogen bond that holds the two formaldehyde molecules together (0.27 kcal mol−1). The low value of the BSSE-corrected binding energy (3.52 kcal mol−1) of the formaldehyde dimer suggests that other interaction energies such as dipole–dipole and electrostatic interactions should contribute little to the stabilization of the system. Atoms in molecules (AIM)17 analysis of the two dimer complexes clearly shows that the bond path in the acetone dimer is similar to that of a typical π → π* interaction and that of the formaldehyde dimer is similar to that of the n → π* interaction (Fig. 2C and D). In the acetone dimer, the n → π* interaction involves the sp-character lone pair on the carbonyl oxygen atom. On the other hand, in the formaldehyde dimer, the participating lone pair of the carbonyl oxygen atom is of p-character as seen from the NBO calculations.
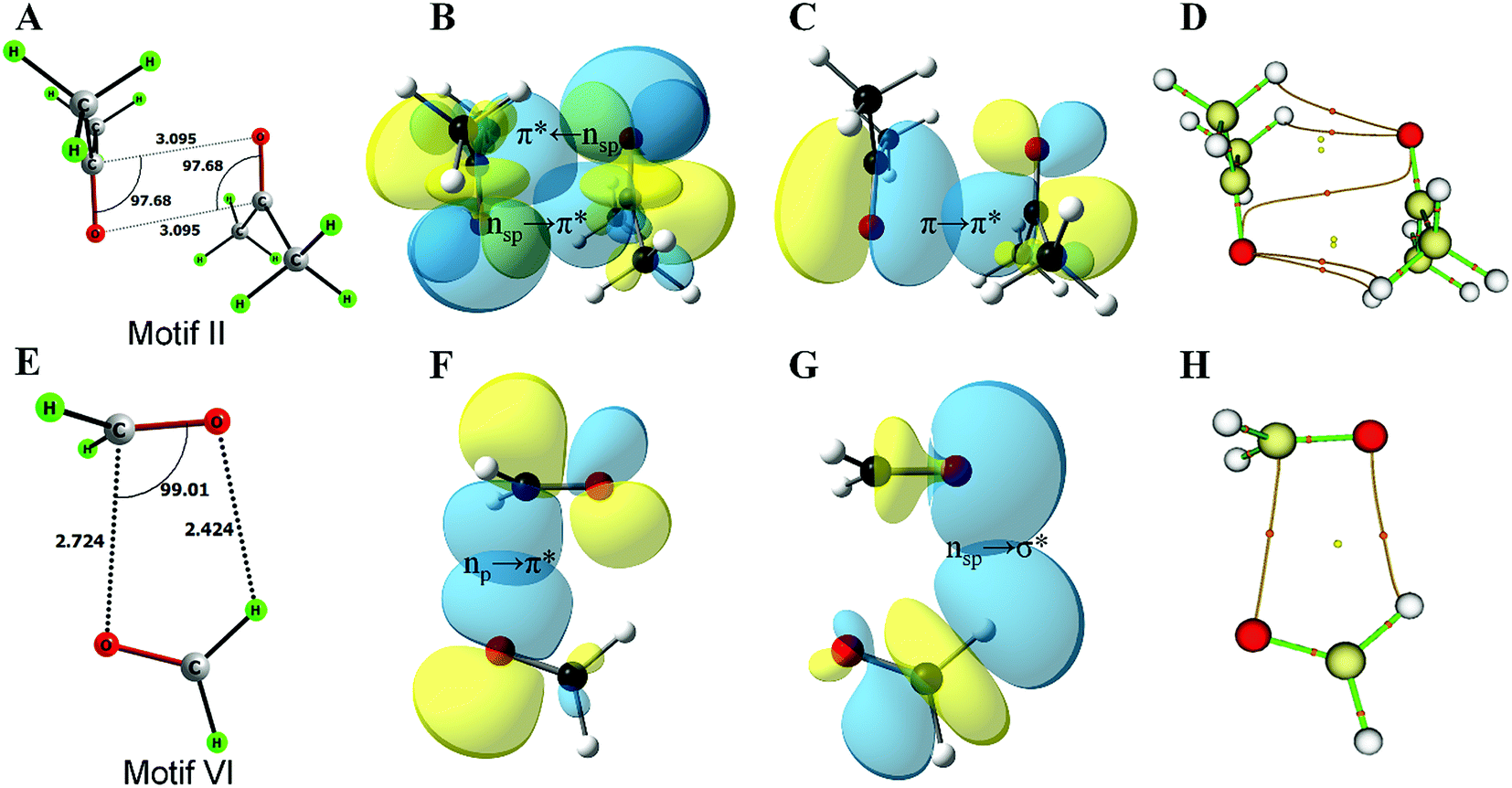 | ||
| Fig. 2 Optimized structures, NBO overlap diagrams and AIM structures of the homodimers of acetone and formaldehyde. (A) Optimized structure of the acetone dimer; (B) n → π* interaction in the acetone dimer; (C) π → π* interaction in the acetone dimer; (D) AIM structure of the acetone dimer showing the bond paths and various critical points; (E) optimized structure of the formaldehyde dimer; (F) n → π* interaction in the formaldehyde dimer; (G) n → σ* interaction in the formaldehyde dimer; (H) AIM structure of the formaldehyde dimer showing the bond paths and various critical points. | ||
These observations suggest that the nature of the orbital interaction in the CO⋯CO interaction is quite different in the homodimers of acetone and formaldehyde and it is clearly dependent on the orientation of the monomers with respect to each other. To understand how the nature of orbital interactions changes with change in relative orientation of the two carbonyl groups, we carried out relaxed potential energy surface (PES) scans on the acetone dimer to assess geometries resembling motifs I, II and III. Further, we looked into the orbital nature of the CO⋯CO interaction in every geometry in the interconversion pathways for motifs I, II and III. The motif II geometry of acetone (X = CH3) can be converted to motif III by changing the CO⋯CO dihedral angle (T) whereas the motif III geometry can be converted to motif I by changing the bond angle θ3 or θ4 (Fig. 3; X = CH3). First, we scanned the CO⋯CO dihedral angle (T) starting from the antiparallel geometry [motif II; T = 0°] to the parallel geometry [motif III; T = 180°] at an increment of 20° at the MP2/6-311+G(2d,p) level of theory. The scans were done for three different intermolecular O⋯C distances (d) of 2.90 Å, 3.00 Å, and 3.09 Å and orbital interactions for each intermediate geometry were analyzed by NBO. NBO analyses indicated that during the interconversion of II to III, π → π* orbital interactions gradually decreased with a concomitant increase in the n → π* orbital interactions as shown in Fig. 3B. This study indicated that the orientation of the two acetone monomers dictates whether the carbonyl groups would be involved in π → π* or n → π* orbital interactions. We then carried out a relaxed potential energy scan by changing the CO⋯C bond angle (θ3 or θ4) from 90° (motif I) to 170° (motif III) for three O⋯C distances (d) of 2.90 Å, 3.00 Å, and 3.10 Å and carried out NBO analysis on each geometry along the path. As expected from the geometry, NBO analyses indicated that the π → π* interaction present in the parallel motif III diminished completely in the T-shaped motif I and the n → π* orbital interaction became the sole contributor to the stabilization of the CO⋯CO interaction (Fig. 3C). Taken together, the PES scan and NBO analyses suggest that the π → π* interaction gradually decreases and the n → π* interaction increases from the antiparallel (II) to the parallel (III) motif and it becomes solely the n → π* interaction in T-shaped motif I.
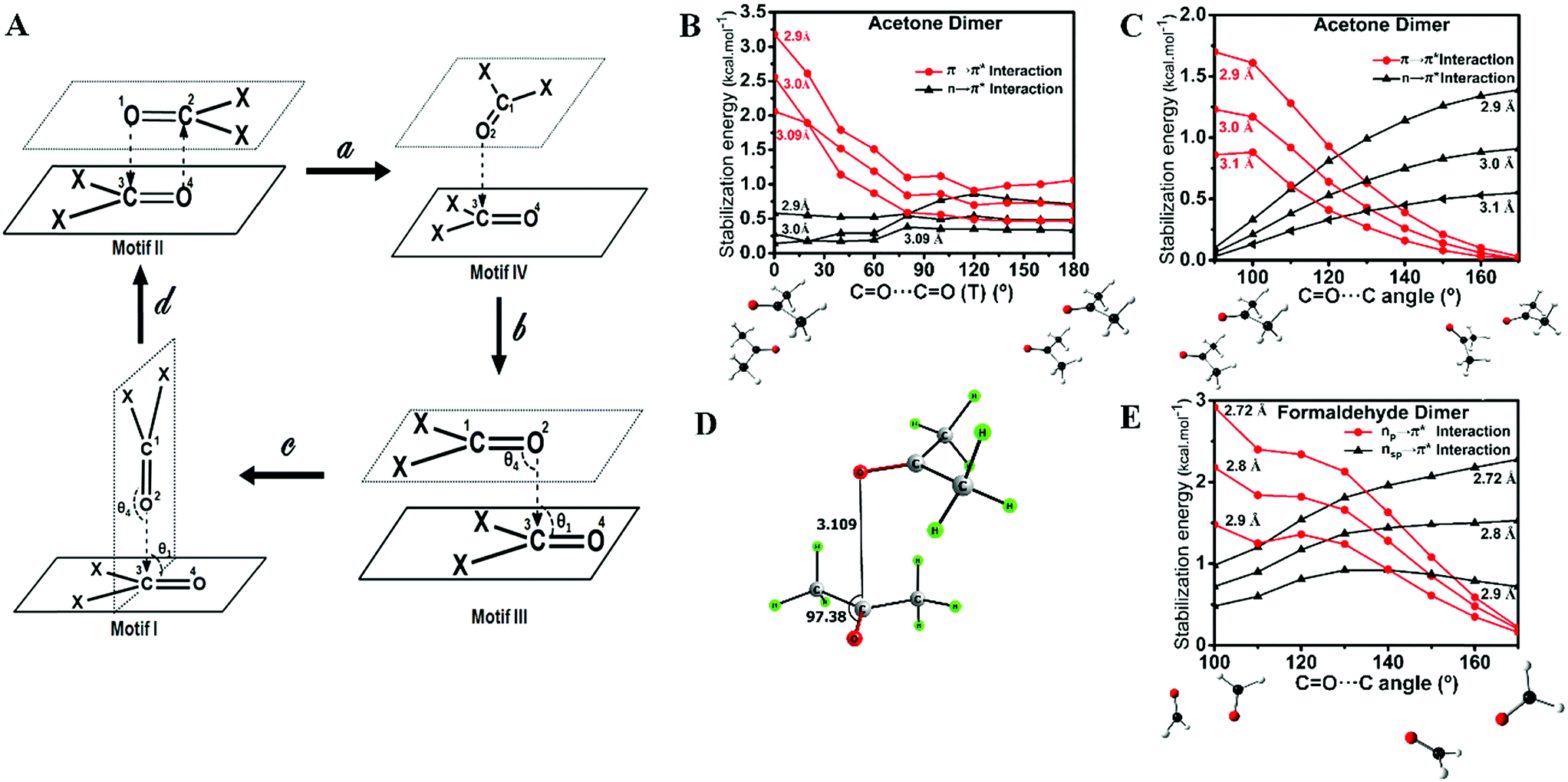 | ||
| Fig. 3 Relaxed potential energy surface (PES) scans and NBO analyses of the homodimers of acetone and formaldehyde and evaluation of orbital interactions. (A) Mechanism of interconversion between motifs I–III. We observed a unique “L-shaped” structure at an intermediate CO⋯CO dihedral angle of T = 90°. (B)O⋯CO (T) show that the π → π* interaction gradually decreases and the n → π* interaction gradually increases from the antiparallel (II) to the parallel motif (III). (C) NBO analyses of the various geometries of the acetone dimer obtained during relaxed scan of CO⋯C angle show that the π → π* interaction gradually decreases and the n → π* interaction gradually increases from the perpendicular T-shaped geometry (I) to the parallel geometry (III). (D) Novel L-shaped geometry of the acetone dimer observed during the relaxed scan of T (motif IV). (E) NBO analyses of various geometries obtained during the relaxed scan of the CO⋯C angle of the formaldehyde dimer show that the character of the oxygen lone pair participating in the n → π* interaction gradually changes from p to sp-character. | ||
During the PES scan of the acetone dimer from the antiparallel (II) to the parallel (III) geometry [T = 0° to T = 180°], we observed a unique “L-shaped” structure at an intermediate CO⋯CO dihedral angle of T = 90° (Fig. 3A, motif IV). To get better insight into this geometry, we carried out geometry optimization of the acetone dimer by freezing the dihedral angle (T) at 90°. The L-shaped local minimum for T = 90° was obtained with one O⋯C (d) contact of 3.109 Å and θ = 97.4° (Fig. 3D). NBO analysis of the structure showed the presence of both n → π* [0.30 kcal mol−1] and π → π* [0.61 kcal mol−1] interactions. This could be a unique dimeric structure for ketones, however, Allen et al. in their CSD analysis of ketone dimers did not observe such a motif.
The formaldehyde dimer structure is unique due to the presence of both n → π* interactions and C–H⋯O hydrogen bonding. As their study was restricted to ketone dimers that lacked hydrogen bond donors, Allen et al. did not observe such a geometry.7d We envisage that aldehydes, carboxylic acids and amides that have hydrogen bond donors may form structures similar to that of the formaldehyde dimer. The relaxed potential energy surface scan to convert the formaldehyde dimer to a T-shaped motif (II) brought out an interesting observation. NBO analyses indicated that the nature of the oxygen lone pair involved in n → π* orbital interactions gradually changed from p- to sp-character (Fig. 3E) suggesting that n → π* orbital interactions can involve either the sp- or p-character lone pair of the donor oxygen atom depending on the orientation of the two monomers that participate in interactions.
2 Cambridge structural database analysis
Inspired by the above results, we carried out a Cambridge Structural Database18 (CSD) search and extracted all the structures having inter- and intramolecular CO⋯CO interactions for all kinds of carbonyl groups. We searched for organic molecules having inter- and intramolecular CO⋯CO short contacts by using a restriction of 3.20 Å to the nonbonded C⋯O atomic distances [sum of the van der Waals radii of C and O is 3.22 Å].6 We looked for two different modes of CO⋯CO short contacts (a) one-sided interaction where one carbonyl donates and the other carbonyl accepts [only one O⋯C distance (either d1 or d2) is ≤3.20 Å] and (b) reciprocal CO⋯CO interaction where both the carbonyl groups work as donors and acceptors (both d1 and d2 are ≤3.20 Å) (Fig. 4A). For one-sided interaction, the ‘group–group (G–G)’ search facility of the CSD was utilized. Atoms C1 and O2 were defined as G1, and C3 and O4 were defined as G2 (Fig. 4A). The search for the nonbonded distance between the centroids of G1 and G2 was repeated three times using d(G1⋯G2) ≤ 3.20 Å, 4.00 Å and 5.50 Å. The data were combined and duplicate values were omitted. Only those molecules having the nonbonded distance O2⋯C3 (d1) or O4⋯C1 (d2) ≤ 3.20 Å were considered for this study. For reciprocal interactions, both the nonbonded distance d1 and d2 were kept ≤3.20 Å and the search was carried out for both intermolecular and intramolecular cases. For the intramolecular system, the software considers the minimum bond path between C and O atoms.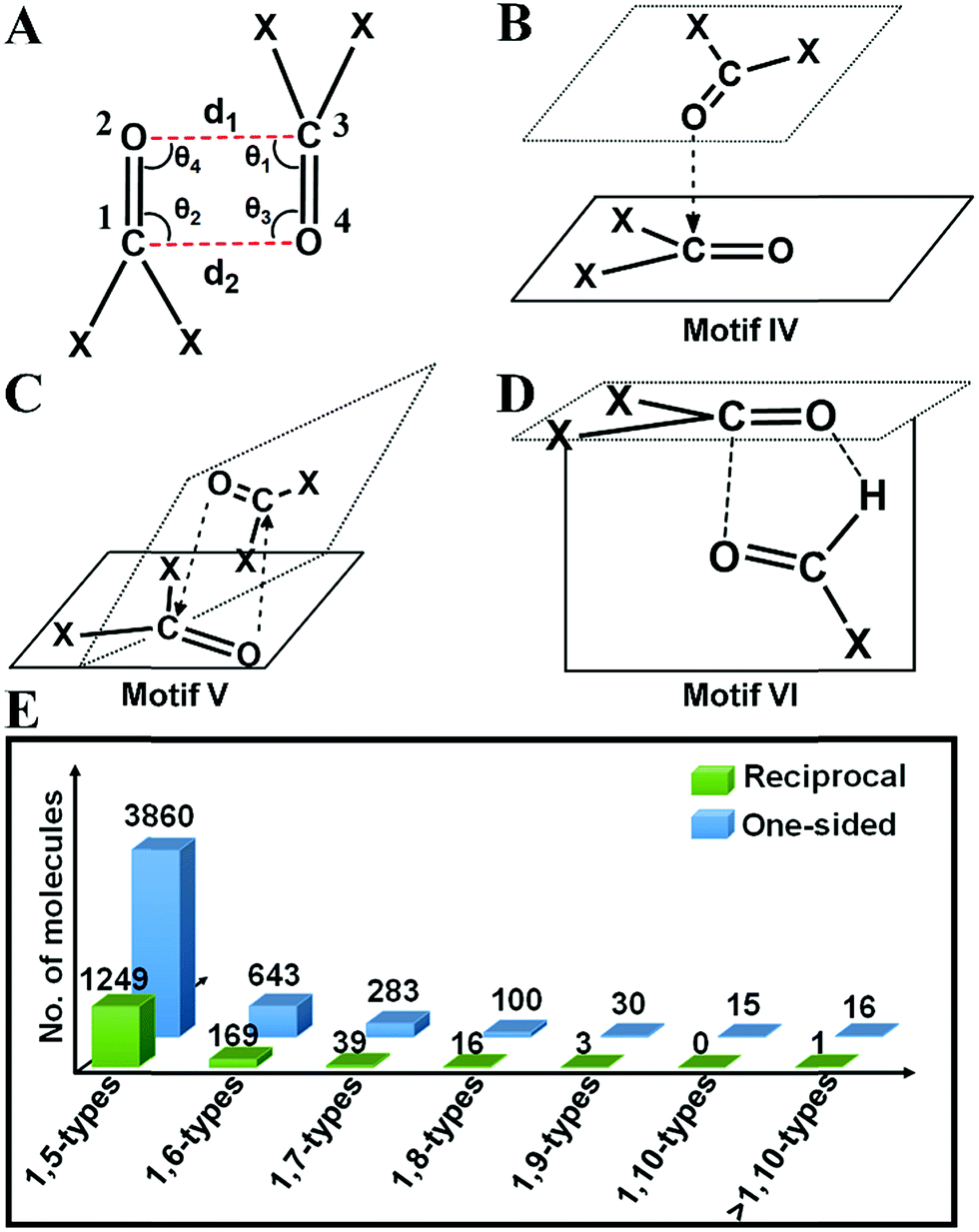 | ||
| Fig. 4 Chemical structures of some motifs newly discovered in this study. (A) Fragment having CO⋯CO short contacts used for the CSD search. Torsion angle, T is defined as C1O2⋯C3O4. Chemical structures of newly discovered (B) motif IV (C) motif V and (D) motif VI. (E) Plot showing how the number of molecules decreases for intramolecular one-sided and reciprocal interactions as the bond separation between the two carbonyl groups increases. In 1,x-type interaction x represents the size of the ring to be formed due the interaction. For example, 1,5-type interaction means there are 3 bonds separating the carbonyl groups and a 5-membered ring will be formed due to the noncovalent interaction. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 858 molecules out of which 4237 contained intermolecular and 6621 contained intramolecular nonbonded C⋯O contacts of ≤3.20 Å. For the intermolecular case, we observed that out of 4237 molecules, 2897 (68%) contained one-sided interactions where only one of the C⋯O distances is within 3.20 Å while 1340 (32%) molecules contained reciprocal interactions where both C⋯O distances were within 3.20 Å (Table 1). In the intramolecular case, 4749 (76%) molecules contained one-sided interactions while 1472 (24%) molecules were found to have reciprocal interactions. The shortest O⋯C distance in the intermolecular one-sided interactions is ∼2.72 Å, whereas the shortest O⋯C distance in the intramolecular one-sided interactions is ∼2.44 Å. The average O⋯C distance (2.91 Å) is shorter for intramolecular interactions compared to intermolecular interactions (3.06 Å), which suggests that intramolecular interactions are generally stronger than intermolecular interactions. Also, the average O⋯C distance in reciprocal interactions is longer than that in one-sided interactions (Table 1). Therefore, the individual interaction of one carbonyl group with the other should be relatively weak for reciprocal interactions compared to one-sided interactions. However, the overall effect of the back and forth interactions between the two carbonyl groups should contribute a significant amount to the stabilization of the molecules. The statistical data are summarized in Fig. S2–S6.† The average value of the O⋯CO angle in the intermolecular case is 92.6° whereas for the intramolecular case it is 94.7°, which are smaller than that in the Burgi–Dunitz trajectory.
858 molecules out of which 4237 contained intermolecular and 6621 contained intramolecular nonbonded C⋯O contacts of ≤3.20 Å. For the intermolecular case, we observed that out of 4237 molecules, 2897 (68%) contained one-sided interactions where only one of the C⋯O distances is within 3.20 Å while 1340 (32%) molecules contained reciprocal interactions where both C⋯O distances were within 3.20 Å (Table 1). In the intramolecular case, 4749 (76%) molecules contained one-sided interactions while 1472 (24%) molecules were found to have reciprocal interactions. The shortest O⋯C distance in the intermolecular one-sided interactions is ∼2.72 Å, whereas the shortest O⋯C distance in the intramolecular one-sided interactions is ∼2.44 Å. The average O⋯C distance (2.91 Å) is shorter for intramolecular interactions compared to intermolecular interactions (3.06 Å), which suggests that intramolecular interactions are generally stronger than intermolecular interactions. Also, the average O⋯C distance in reciprocal interactions is longer than that in one-sided interactions (Table 1). Therefore, the individual interaction of one carbonyl group with the other should be relatively weak for reciprocal interactions compared to one-sided interactions. However, the overall effect of the back and forth interactions between the two carbonyl groups should contribute a significant amount to the stabilization of the molecules. The statistical data are summarized in Fig. S2–S6.† The average value of the O⋯CO angle in the intermolecular case is 92.6° whereas for the intramolecular case it is 94.7°, which are smaller than that in the Burgi–Dunitz trajectory.
| Total | One-sided | d min (Å) | d avg (Å) | θ avg (deg) | Reciprocal | d min (Å) | d avg (Å) | θ avg (deg) | |
|---|---|---|---|---|---|---|---|---|---|
|
a
d
min – minimum O⋯C distance; davg – average O⋯C distance; θavg – average O⋯CO angle.
|
|||||||||
| Intermolecular | 4237 | 2897 | 2.72 | 3.06 | 95.5 | 1340 | 2.81 | 3.10 | 89.8 |
| Intramolecular | 6221 | 4749 | 2.44 | 2.91 | 103.8 | 1472 | 2.31 | 3.00 | 85.7 |
The motif structures (motifs I–V) were categorized based on the CO⋯CO dihedral angle T, and the two CO⋯C angles θ3 and θ4. For motif VI, the carbonyl planes were also taken into account. The different parameters for an ideal geometry of each motif type are shown in Table S2† and the criteria for their categorization into various motifs are discussed in the ESI.† In the following section, molecules from the motifs I–VI will be divided into two major classes (i) molecules having one-sided CO⋯CO interactions and molecules with (ii) reciprocal CO⋯CO interactions.
2(a) (i) One-sided interaction (motifs I, III and IV). Molecules having one-sided interactions could be categorized into three different structural motifs I, III and IV, irrespective of whether the interactions are inter- or intramolecular. In intermolecular cases, most of the 2897 molecules with one-sided CO⋯CO interactions have geometries represented by either perpendicular motif I [∠C
O⋯C ∼ 150–180°] or parallel motif III [CO⋯CO (T) = 180° and ∠CO⋯C = 90° or 50°] observed previously by Allen and co-workers for ketone dimers. These motifs are often found in the crystal structures where the carbonyl group(s) are attached to bulky groups to avoid steric clashes. We also observed many molecules with a novel L-shaped geometry with CO⋯CO torsion angles (T) ∼ 90° as discussed above for the acetone dimer. These molecules cannot be categorized into motif I or III. We identified ∼400 such molecules, represented by the structural motif IV. In motif IV, one carbonyl group sits over the other carbonyl group in an “L-shaped” manner facilitating the nonbonded O⋯C short contact. These dimeric structures have angles (θ1–θ4) similar to that of motif III but unlike motif III where the CO⋯CO torsion angle (T) is ∼180°, they have torsion angles of ∼90°.
For intramolecular cases, 1059 molecules with one-sided interactions were found to have a torsion angle |T| ∼ 180°, which resembles that of the sheared parallel motif III. In contrast to the intermolecular case, only a few molecules (14) with a T-shaped geometry (∠CO⋯C = 150–180°) were observed having one-sided intramolecular interactions. This is expected given the steric crowding and bond constraints that arise in such geometries. The majority of the molecules (1762) can be categorized into the “L-shaped” motif IV with the CO⋯CO torsion angle |T| close to 90°. Interestingly, most of the intramolecular interactions are 1,5-type in nature where the two carbonyl groups were separated by three covalent bonds. It should be noted that all the backbone carbonyl–carbonyl interactions in proteins are also 1,5-type in nature. As the number of bonds separating the two carbonyl groups increases, the number of molecules steeply decreases (Fig. 4E).
2(a) (ii) Reciprocal interaction (motifs II and V). In the intermolecular system, the majority of the molecules have reciprocal interactions (motif II; |T| ∼ 0° and ∠C
O⋯C ∼ 90°) with sheared antiparallel orientation, which is similar to the global minimum geometry of the acetone dimer. In intramolecular cases, the reciprocal interactions were observed with a CO⋯CO dihedral angle (T) of ∼70° and two ∠CO⋯C angles ∼ 75° (motif V). Rahim et al. recently reported that donor oxygen atoms approach the acceptor carbonyl carbon atoms significantly away from the BD trajectory (∠O⋯CO is ∼85°) in these cases.11 Motif V has a close resemblance to motif IV, the major difference being, in motif V, both O⋯C distances are shorter than 3.20 Å whereas only one of the O⋯C distances is shorter than 3.20 Å in motif IV. Structurally motif V is more twisted than motif IV due to the restriction on both the O⋯C distances in motif V (<3.20 Å).
Note that there is a striking difference in the geometrical arrangements of the two carbonyl groups in intramolecular and intermolecular reciprocal interactions. In the intermolecular case, reciprocal interactions were observed in the antiparallel motif II (T ∼ 0°) whereas intramolecular reciprocal interactions are observed in “twisted” geometries with T = ∼70° that we have represented as motif V in this paper. Similar to one-sided interactions, reciprocal interactions can also be categorized as separate 1,x-types in the intramolecular case (Fig. 4E). Interestingly, as the bond separation between the two carbonyl groups increases, the number of molecules decreases and the “twisted” bond type motif V tends to slowly convert to an antiparallel motif II (ESI Fig. S10†).
2(a) (iii) Unprecedented structural motif VI that resembles the formaldehyde dimer. As discussed above, the formaldehyde dimer does not fall in any of the aforementioned categories. We were curious to see if such molecules are also present in the CSD. We could envisage such a motif in aldehydes, carboxylic acids or amides wherein a carbonyl group can engage in one O⋯C short contact and one O⋯H short contact as shown in Fig. 4D and ESI Fig. S7.† We have represented this type of structure as motif VI in this paper. Though the carbonyl groups are antiparallel to each other the H-bonding imposes restriction on the O atom to interact with the C attached to the hydrogen producing a separate geometry, where ∠C
O⋯C is neither 90° nor 180°. For this kind of structure, the carbonyl planes are perpendicular to each other. A total of 17 aldehydes [motif VI(a)], 7 carboxylic acids [motif VI(b)] and 24 amides [motif VI(c)] were found in the CSD search that have a geometry similar to that of motif VI. In the search, the software was allowed to normalize the hydrogens and one of the two carbonyl groups was considered as aldehyde or carboxylic acid or amide in the CSD search fragment. We observed that motif VI is unique to intermolecular systems and we did not find any molecule with the intramolecular CO⋯CO interaction having a geometry similar to that of motif VI. This could be due to the probable steric restrictions associated with such an intramolecular system.
We observed that motifs III and IV are common to both inter- and intramolecular systems. However, there are very few molecules having intramolecular CO⋯CO interactions that can be categorized into motif I or II whereas there is no molecule of motif VI type with intramolecular CO⋯CO interactions. On the other hand, motif V is only present in the intramolecular system and there are very few molecules with the intermolecular CO⋯CO interaction that could be categorized into motif V.
O (θ) angle in inter- and intramolecular cases is shown in Fig. 5A and B. As can be seen from Fig. 5A, θ has a maximum of ∼90° in the intermolecular cases, while the maximum of θ lies close to ∼100° for intramolecular interactions (Fig. 5B) suggesting that the approach of donor oxygen to acceptor carbon is closer to the Burgi–Dunitz trajectory for intramolecular cases. A closer inspection of the θ values in individual motifs (I–VI) shows that the majority of the molecules in each motif have a θ value less than 100 except for motif III (Fig. 5E). However, for all types of motifs, as the O⋯C distance gets shorter (interaction gets stronger), the value of θ tends to approach closer to the BD trajectory (∼109°) (Fig. 6A–F). This signifies that it is important that the donor atom approaches the acceptor atom in a trajectory closer to the BD trajectory for stronger CO⋯CO interactions. Interestingly, for the motifs II and V having reciprocal CO⋯CO interactions, the angle of approach (θ) of the donor oxygen atom to the acceptor carbon atom mostly falls in the 80–90° range, which is much smaller than that of the BD trajectory.
 | ||
| Fig. 5 Distribution of the O⋯CO angle (θ) of all the molecules obtained from the CSD search. (A) all molecules having intermolecular CO⋯CO interactions; (B) for all molecules having intramolecular CO⋯CO interactions; (C) motif I; (D) motif II; (E) motif III; (F) motif IV; (G) motif V; (H) motif VI. | ||
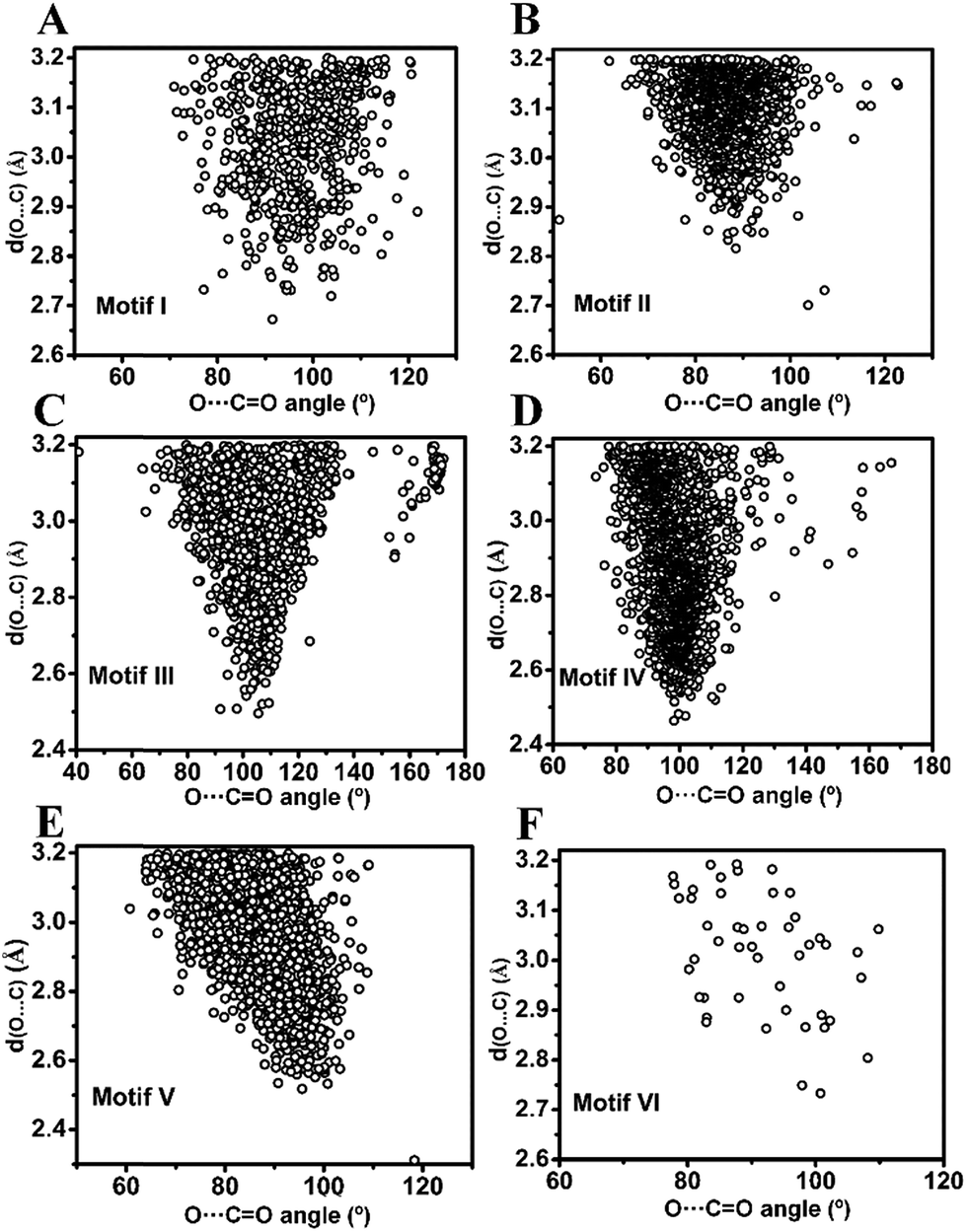 | ||
| Fig. 6 O⋯CO angle (θ) versus O⋯C distance (dO⋯C) scatter plot of molecules obtained from the CSD search. (A)–(F) motifs I–VI. | ||
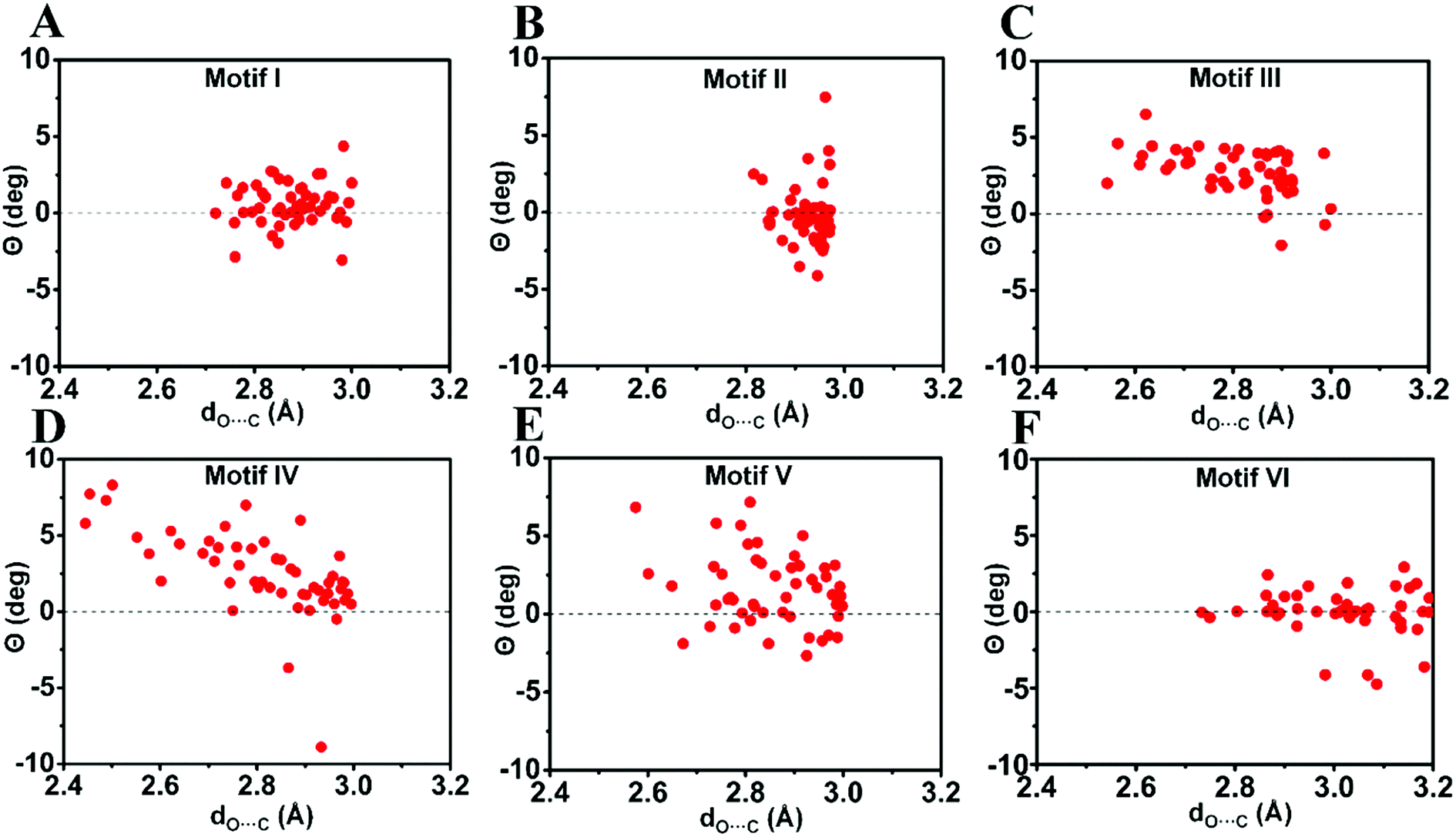 | ||
| Fig. 7 Pyramidality (Θ) of the acceptor carbonyl carbon in motifs I–VI. (A)–(F) Motifs (I)–(VI). Only molecules with dO⋯C ≤ 3.0 Å [dO⋯C ≤ 3.2 Å for motif VI] were considered. See ESI† for details. | ||
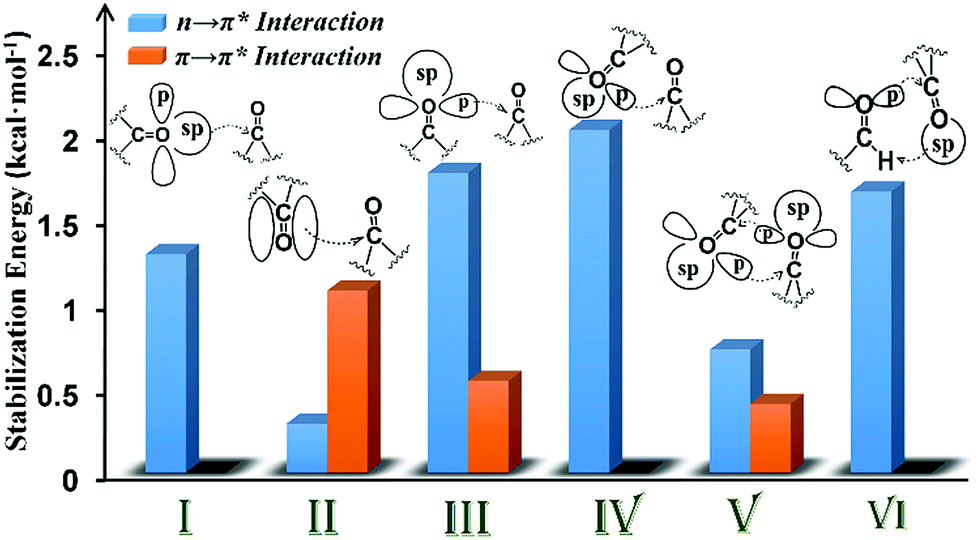 | ||
| Fig. 8 Plot showing the various orbital interactions in various motifs (I–VI) having CO⋯CO interactions and the NBO second-order perturbation energy of representative molecules of each motif. | ||
In motif V, significant reciprocal n → π* interactions were observed. These interactions can be viewed as donation from the first carbonyl group to the second and back donation from the second carbonyl group to the first. Both p- and sp-character oxygen lone pairs participate in this interaction. We also observed some π → π* interactions in motif V. This interaction is unique to intramolecular systems and, therefore, cannot be mimicked by using a dimeric model system.
The CO⋯CO orbital interactions in molecules from motif VI can be explained by using the formaldehyde dimer model wherein the p-character oxygen lone pair donates electrons to the π* orbital of the acceptor carbonyl to form an n → π* interaction. The acceptor carbonyl oxygen is also involved in a hydrogen bonding interaction with the donor XH group (X = C, N, and O).
In conclusion, we have shown that the molecules with inter- and intramolecular CO⋯CO interactions can be categorized into six different structural motifs (I–VI). Both n → π* and π → π* interactions may contribute to the stabilization of CO⋯CO interactions. The majority of the intermolecular CO⋯CO interactions resemble antiparallel motif II that are driven by π → π* orbital interactions whereas intramolecular CO⋯CO interactions are dominated by motifs III and IV with strong n → π* interactions and motif V with both n → π* and π → π* interactions. We observed that the relative geometrical arrangement of the two interacting carbonyl groups determines the nature of the orbital interaction for the stabilization of the CO⋯CO interaction. The nature of the donor oxygen lone pair (sp- or p-character) in the n → π* orbital interaction also depends on the relative orientation of the two carbonyl groups. Interestingly, CO⋯CO interactions in motifs I–VI in both inter- and intramolecular systems can be modelled by using simple homodimeric models of acetone and formaldehyde. As intramolecular CO⋯CO interactions in large systems are often modelled by using small dimeric systems having intermolecular CO⋯CO interactions, our study should provide guidance in choosing the correct model system based on the similarity of the nature of CO⋯CO interactions in the system under investigation and the system used to model it. Although theoretically all six structural motifs I–VI can exist in proteins, the presence of only two of these structural motifs III and V is known to date. As long range CO⋯CO interactions are probable in proteins, it would be interesting to see if any of the other motifs reported herein is also observed in proteins in the future.
Author contributions
All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank Dr Mausumi Goswami for helpful discussions. This project was funded by Shiv Nadar University (SNU) and Early Career Research Grant (ECR/2015/000337) from the Science and Engineering Research Board (SERB), Department of Science and Technology (DST), Government of India. We acknowledge the MAGUS supercomputing facility at SNU.References
- (a) T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS; (b) W. Kabsch and C. Sander, Biopolymers, 1983, 22, 2577–2637 CrossRef CAS PubMed; (c) L. Pauling and R. B. Corey, Proc. Natl. Acad. Sci. U. S. A., 1951, 37, 251–256 CrossRef CAS PubMed; (d) L. Pauling, R. B. Corey and H. R. Branson, Proc. Natl. Acad. Sci. U. S. A., 1951, 37, 205–211 CrossRef CAS PubMed.
- (a) D. J. Pascoe, K. B. Ling and S. L. Cockroft, J. Am. Chem. Soc., 2017, 139, 15160–15167 CrossRef CAS PubMed; (b) B. R. Beno, K. S. Yeung, M. D. Bartberger, L. D. Pennington and N. A. Meanwell, J. Med. Chem., 2015, 58, 4383–4438 CrossRef CAS PubMed; (c) B. K. Sarma, J. Mol. Struct., 2013, 1048, 410–419 CrossRef CAS; (d) B. K. Sarma, D. Manna, M. Minoura and G. Mugesh, J. Am. Chem. Soc., 2010, 132, 5364–5374 CrossRef CAS PubMed; (e) B. K. Sarma and G. Mugesh, Chem.–Eur. J., 2008, 14, 10603–10614 CrossRef CAS PubMed; (f) B. K. Sarma and G. Mugesh, J. Am. Chem. Soc., 2007, 129, 8872–8881 CrossRef CAS PubMed; (g) B. K. Sarma and G. Mugesh, J. Am. Chem. Soc., 2005, 127, 11477–11485 CrossRef CAS PubMed; (h) M. Iwaoka, H. Komatsu, T. Katsuda and S. Tomoda, J. Am. Chem. Soc., 2004, 126, 5309–5317 CrossRef CAS PubMed; (i) M. Iwaoka, S. Takemoto and S. Tomoda, J. Am. Chem. Soc., 2002, 124, 10613–10620 CrossRef CAS PubMed.
- (a) R. W. Newberry and R. T. Raines, Acc. Chem. Res., 2017, 50, 1838–1846 CrossRef CAS PubMed; (b) S. K. Singh and A. Das, Phys. Chem. Chem. Phys., 2015, 17, 9596–9612 RSC; (c) S. Blanco and J. C. Lopez, J. Phys. Chem. Lett., 2018, 9, 4632–4637 CrossRef CAS PubMed.
- (a) H. B. Bürgi, J. D. Dunitz and E. J. Shefter, J. Am. Chem. Soc., 1973, 95, 5065–5067 CrossRef; (b) H. B. Bürgi, J. D. Dunitz and E. Shefter, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 1517–1527 CrossRef; (c) H. B. Bürgi, J. D. Dunitz and J. M. Lehn, Tetrahedron, 1974, 30, 1563–1572 CrossRef.
- (a) W. Bolton, Acta Crystallogr., 1963, 16, 166–173 CrossRef CAS; (b) W. Bolton, Acta Crystallogr., 1964, 17, 147–152 CrossRef CAS; (c) W. Bolton, Acta Crystallogr., 1965, 18, 5–10 CrossRef CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- (a) J. Bernstein, M. D. Cohen and L. Leiserowitz, in The Chemistry of Quinonoid Compounds, ed. S. Patai, Wiley, London, 1974 Search PubMed; (b) R. Taylor, A. Mullaley and G. W. Mullier, Pestic. Sci., 1990, 29, 197–213 CrossRef CAS; (c) A. Gavezzotti, J. Phys. Chem., 1990, 94, 4319–4325 CrossRef CAS; (d) F. H. Allen, C. A. Baalham, J. P. M. Lommerse and P. R. Raithby, Acta Crystallogr., Sect. B: Struct. Sci., 1998, 54, 320–329 CrossRef.
- (a) P. H. Maccallum, R. Poet and E. J. Milner-White, J. Mol. Biol., 1995, 248, 361–373 CAS; (b) P. H. Maccallum, R. Poet and E. J. Milner-White, J. Mol. Biol., 1995, 248, 374–384 CAS.
- L. E. Bretcher, C. L. Jenkins, K. M. Taylor, M. L. DeRider and R. T. Raines, J. Am. Chem. Soc., 2001, 123, 777–778 CrossRef.
- (a) R. W. Newberry, G. J. Bartlett, B. VanVeller, D. N. Woolfson and R. T. Raines, Protein Sci., 2014, 23, 284–288 CrossRef CAS PubMed; (b) R. W. Newberry and R. T. Raines, ACS Chem. Biol., 2014, 9, 880–883 CrossRef CAS PubMed; (c) G. J. Bartlett, R. W. Newberry, B. VanVeller, R. T. Raines and D. N. Woolfson, J. Am. Chem. Soc., 2013, 135, 18682–18688 CrossRef CAS PubMed; (d) R. W. Newberry and R. T. Raines, Chem. Commun., 2013, 49, 7699–7701 RSC; (e) R. W. Newberry, B. VanVeller, I. A. Guzei and R. T. Raines, J. Am. Chem. Soc., 2013, 135, 7843–7846 CrossRef CAS PubMed; (f) J. S. Laursen, J. Engel-Andreasen, P. Fristrup, P. Harris and C. A. Olsen, J. Am. Chem. Soc., 2013, 135, 2835–2844 CrossRef CAS PubMed; (g) A. Choudhary, K. J. Kamer and R. T. Raines, J. Org. Chem., 2011, 76, 7933–7937 CrossRef CAS PubMed; (h) A. Choudhary and R. T. Raines, Protein Sci., 2011, 20, 1077–1081 CrossRef CAS PubMed; (i) J. R. Stringer, J. A. Crapster, I. A. Guzei and H. E. Blackwell, J. Am. Chem. Soc., 2011, 133, 15559–15567 CrossRef CAS PubMed; (j) G. J. Bartlett, A. Choudhary, R. T. Raines and D. N. Woolfson, Nat. Chem. Biol., 2010, 6, 615–620 CrossRef CAS PubMed; (k) C. Fufezan, Proteins: Struct., Funct., Bioinf., 2010, 78, 2831–2838 CrossRef CAS PubMed; (l) B. C. Gorske, J. R. Stringer, B. L. Bastian, S. A. Fowler and H. E. Blackwell, J. Am. Chem. Soc., 2009, 131, 16555–16567 CrossRef CAS PubMed; (m) B. C. Gorske, B. L. Bastian, G. D. Geske and H. E. Blackwell, J. Am. Chem. Soc., 2007, 129, 8928–8929 CrossRef CAS PubMed.
- (a) A. Rahim, P. Saha, K. K. Jha, N. Sukumar and B. K. Sarma, Nat. Commun., 2017, 8, 78 CrossRef PubMed; (b) A. Rahim, B. Sahariah and B. K. Sarma, Org. Lett., 2018, 20, 5743–5746 CrossRef CAS PubMed.
- (a) F. R. Fischer, P. A. Wood, F. H. Allen and F. Diederich, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17290–17294 CrossRef CAS PubMed; (b) R. Paulini, K. Müller and F. Diederich, Angew. Chem., Int. Ed., 2005, 44, 1788–1805 CrossRef CAS PubMed.
- (a) K. J. Kamer, A. Choudhary and R. T. Raines, J. Org. Chem., 2013, 78, 2099–2103 CrossRef CAS PubMed; (b) A. Choudhary, D. Gandla, G. R. Krow and R. T. Raines, J. Am. Chem. Soc., 2009, 131, 7244–7246 CrossRef CAS PubMed.
- (a) J. M. Hermida-Ramón and M. A. Ríos, J. Phys. Chem. A, 1998, 102, 2594–2602 CrossRef; (b) E. V. Dornshuld, C. M. Holy and G. S. Tschumper, J. Phys. Chem. A, 2014, 118, 3376–3385 CrossRef PubMed; (c) G. A. Dolgonos, Chem. Phys. Lett., 2013, 585, 37–41 CrossRef CAS; (d) J. M. Hermida-Ramón and M. A. Ríos, J. Phys. Chem. A, 1998, 102, 10818–10827 CrossRef.
- E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO Version 3.1 Search PubMed.
- M. Head-Gordon, J. A. Pople and M. Frisch, Chem. Phys. Lett., 1988, 153, 503–506 CrossRef CAS.
- (a) R. F. W. Bader, Atoms in Molecules A Quantum Theory, Oxford University Press, New York, 1990 Search PubMed; (b) P. Popelier, Atoms in Molecules. An Introduction, Pearson Education, Harlow, Great Britain, 2000 Search PubMed; (c) R. J. Gillespie and P. L. A. Popelier, Chemical Bonding and Molecular Geometry from Lewis to Electron Densities, Oxford University Press, New York, 2001 Search PubMed.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 389–397 CrossRef.
- L. Esposito, L. Vitagliano, A. Zagari and L. Mazzarella, Protein Sci., 2000, 9, 2038–2042 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic and computational data (PDF) and X-ray crystallographic data (excel). See DOI: 10.1039/c8sc04221g |
| This journal is © The Royal Society of Chemistry 2019 |