
Conventional Type II photoinitiators as activators for photoinduced metal-free atom transfer radical polymerization
Andrit
Allushi
,
Ceren
Kutahya
,
Cansu
Aydogan
,
Johannes
Kreutzer
*,
Gorkem
Yilmaz
* and
Yusuf
Yagci
 *
*
Department of Chemistry, Istanbul Technical University, 34469 Maslak, Istanbul, Turkey. E-mail: yusuf@itu.edu.tr; a.gorkemyilmaz@gmail.com; johannes.kreutzer@yahoo.com
First published on 6th March 2017
Abstract
A novel methodology for photoinduced metal-free Atom Transfer Radical Polymerization (ATRP) by using conventional Type II photoinitiators such as benzophenone, thioxanthone, isopropyl thioxanthone and camphorquinone as sensitizers is presented. These sensitizers efficiently activate the ATRP of vinyl monomers when used in conjunction with co-initiators and alkyl halides under light irradiation. DFT calculations were performed to reveal mechanistic aspects and showed that the initiation of the controlled ATRP reaction pathway from the triplet state is energetically favoured for all sensitizers. In the case of benzophenone, however, the hydrogen abstraction pathway, which leads to free radical polymerization, is a possible side reaction. The strategy applied was proved to yield polymers with narrow molecular distribution. The chain-end fidelity of the polymer obtained was approved by chain extension and block copolymerization experiments, whereas the irradiation dependency of polymerization was confirmed by light on/off experiments.
Introduction
Photoinitiators are the most important elements of photopolymerization processes as they are responsible for the transformation of light energy to chemical energy.1 Among the various modes of photopolymerization,2–6 free radical polymerization is in a more advanced state due to the wavelength flexibility and broad availability of monomer formulations. Free radical photoinitiators are divided into two classes depending on their decomposition mechanism. Type I or α-cleavage photoinitiators undergo decomposition directly after irradiation to yield radicals that can initiate polymerization. Type II photoinitiators generate radicals in the presence of co-initiators in a multi-step reaction mechanism. If an amine is used as a co-initiator, generally an electron is transferred to the triplet state of the Type II initiator, followed by a proton release to generate radicals.7 Typical examples of commercially available Type II initiators include benzophenone (BP), thioxanthone (TX) and their derivatives, and camphorquinone (CQ), which have a broad wavelength absorption in the UV-vis range of the electromagnetic spectrum. Among them, TXs are the most widely used photoinitiators due to their absorption characteristics and high quantum efficiency.7 These photoinitiators are often employed for free radical8–16 and cationic polymerization17 and as catalysts for photoinduced click reactions18,19 and also as anticancer agents.20Scheme 1 shows the initiation of the radical polymerization process with TX as an example. Notably, ketyl radicals formed on the photoinitiator skeleton do not initiate polymerization due to resonance stability and steric effects, whereas radicals from the co-initiator are responsible for the initiation.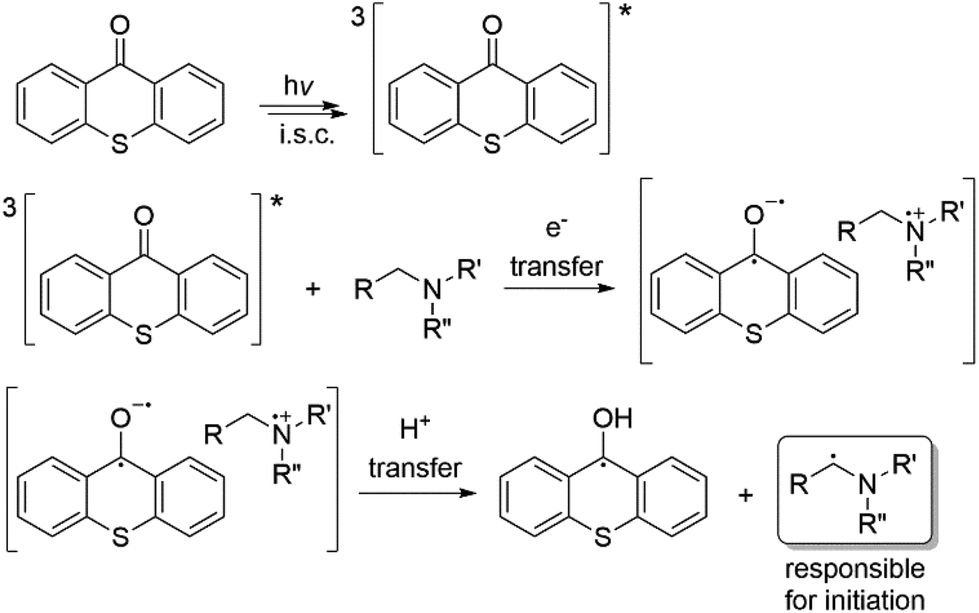 | ||
| Scheme 1 Photoinitiated radical formation in a thioxanthone/amine system. | ||
Besides such applications, these photoinitiators were also applied in atom transfer radical polymerization (ATRP), starting from high oxidation state copper halides.21 In conventional ATRP, Cu(I) halides together with a suitable ligand system and an alkyl halide are used as redox active species.22 The air sensitivity of these metal catalysts and limitations concerning the bio-applications of the obtained polymers led to the development of approaches to generate Cu(I) in situ and where ppm levels of Cu catalysts are sufficient to initiate the polymerization. Reported strategies include chemical,23,24 electrochemical,25 and photochemical reduction of Cu(II) species26–28 in the polymerization system. Recent work concentrated on the in situ generation of Cu(I) catalyst by direct29 and indirect photoinduced electron transfer processes utilizing photoinitiators,30,31 sensitizers,32 nanoparticles33 and fullerene derivatives.34 More recently, certain photosensitizers (PS) such as phenothiazine derivatives,35,36 perylene,37 pyrene38 and diaryldihydrophenazines39 were found to realize photoinduced ATRP even in the absence of copper catalysts through an oxidative quenching pathway. In the proposed mechanism, an alkyl halide is reduced by photoinduced electron transfer from the excited state to generate radicals capable of initiating polymerization and the control over polymerization is achieved by the reversibility of activation/deactivation steps as depicted in Scheme 2.
 | ||
| Scheme 2 Mechanism of photoinduced metal-free ATRP using PSs. | ||
Similar photo-redox reactions were also proposed for reducible dyes.40,41 It seemed, therefore, appropriate to investigate whether Type II photoinitiators would act as activators for metal-free photo ATRP. Theoretical studies were performed to confirm the validity of the proposed mechanism.
As part of our continuous interest in developing photochemical systems for both conventional and controlled/living polymerization processes, we now report the application of commercially available Type II photoinitiators having different absorption characteristics and excited state properties for photoinduced metal-free ATRP. For this purpose, TX, BP, isopropyl thioxanthone (ITX) and CQ were used as PSs together with N,N,N′,N″,N″-pentamethyldiethylenetriamine (PMDETA) and alkyl halides as the electron donor source and initiator, respectively, for the polymerization of various monomers.
Experimental part
Materials
Thioxanthone (TX, 98%, Sigma Aldrich) was crystallized from ethanol prior to use. 2-Isopropylthioxanthone (ITX, Ward Blenkinsop and Co. Ltd), benzophenone (BP, 98%, Sigma Aldrich), and camphorquinone (CQ, 97%, Sigma Aldrich) were used as received. N,N-Dimethylformamide (DMF, EMPARTA, HPLC grade) was stored over activated molecular sieves (4 Å). N,N,N′,N″,N″-Pentamethyldiethylenetriamine (PMDETA; Aldrich, 99%) was distilled before use. Ethyl α-bromoisobutyrate (EBI, 98%, Sigma Aldrich), ethyl 2-bromopropionate (EBP, 99%, Sigma Aldrich), and (1-bromoethyl)benzene (BEB, 97%, Sigma Aldrich) were used as received. Methyl methacrylate (MMA, 99%, Sigma Aldrich) and butyl acrylate (BA, 99.9%, Sigma Aldrich) were passed through basic alumina and stored under a nitrogen atmosphere in cold before use.Instrumentation
1H NMR spectra of the intermediates and final polymers were recorded at room temperature at 500 MHz on an Agilent VNMRS 500 spectrometer. UV spectra were recorded on a Shimadzu UV-1601 spectrometer. The resolution was 4 cm−1 and 24 scans were performed with a 0.2 cm s−1 scan speed. Gel permeation chromatography (GPC) measurements were performed from a Viscotek GPCmax autosampler system consisting of a pump, a Viscotek UV detector, and a Viscotek differential refractive index (RI) detector. Three ViscoGEL GPC columns (G2000H HR, G3000H HR, and G4000H HR, 7.8 mm internal diameter, 300 mm length) were used in series. THF was used as an eluent at a flow rate of 1.0 mL min−1 at 30 °C. Both detectors were calibrated with polystyrene standards having narrow-molecular-weight distribution. Data were analyzed using Viscotek OmniSEC Omni-01 software.Calculations
All calculations were performed with the Gaussian09 program package.42 Geometries were optimized with the B3LYP functional43–45 in combination with the 6-31G(d) basis set. Dimer structures were optimized with the long range corrected CAM-B3LYP functional46 in combination with the 6-31G(d) basis set. All geometries were confirmed as minimum structures displaying all frequencies real. Single point calculations were performed with the M06-2X functional and the 6-311++G(3df,2p) basis set. The combination of the M06-2X functional47 and 6-311++G(3df,2p) basis set showed good performance for calculations of reaction enthalpies and reaction rates for electron transfer processes before.48 The optimized structures, as well as single point energies were calculated with the Solvation Model based on Density (SMD) in DMF. Gibbs free energies and enthalpies include thermal corrections at 298 K.General procedure for visible light induced metal-free ATRP of MMA
In a typical experiment, MMA (1 mL, 200 eq.), alkyl halide (1 eq.), DMF (1 mL), PMDETA (5 eq.) and photosensitizer (TX, BP, ITX or CQ, 1 eq.) were put into a Schlenk tube and the reaction mixture was degassed by freeze–pump–thaw cycles and left in a vacuum (∼10−3 bar). The stirring solution was irradiated with a light source emitting light nominally at 350 nm with a light intensity of 3.0 mW cm−2 for 4 h (in the case of CQ, the reaction mixture was irradiated at 400–500 nm with a light intensity of 45 mW cm−2). At the end of the irradiation, the resulted polymers were precipitated in methanol and then dried under reduced pressure. Conversions were determined gravimetrically.Results and discussion
TXs, BP and CQ as typical Type II photoinitiators appeared to be suitable candidates for UV-vis induced electron transfer processes because of their broad absorption spectra, covering the UV and visible spectral region and high molar absorptivities (Fig. 1). Initially, we examined the use of TX and BP as PSs for the polymerization of methyl methacrylate (MMA) in the presence of PMDETA and various alkyl halide sources, namely ethyl α-bromoisobutyrate (EBI), ethyl 2-bromopropionate (EBP) and (1-bromoethyl)benzene (BEB) as initiators at ca. 350 nm. As can be seen in Table 1 all yield polymers with low dispersities (<1.5), although at different conversions. | ||
| Fig. 1 UV-vis spectra of thioxanthone, benzophenone, isopropyl thioxanthone and camphorquinone in DMF. | ||
| Sensitizer | Initiator | Conv.b (%) |
M
n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c (g mol−1) c (g mol−1) |
M
w/Mnc |
|---|---|---|---|---|
| a V MMA = 1 mL, VDMF = 1 mL, [MMA]/[RX]/[PMDETA]/[S]: 200/1/5/1, λ = 350 nm, time = 4 h. b Determined gravimetrically. c Determined by gel permeation chromatography using polystyrene standards. | ||||
| TX | EBI | 14.3 | 8900 | 1.38 |
| BP | EBI | 21.2 | 4750 | 1.42 |
| TX | BEB | 7.6 | 14500 |
1.43 |
| BP | BEB | 8.1 | 11700 |
1.34 |
| TX | EBP | 13.4 | 15000 |
1.45 |
| BP | EBP | 1.7 | 12300 |
1.50 |
Apparently, with both sensitizers, longer polymerization times lead to higher conversions. With TX, an increase in polymerization time yields polymers with lower PDI values, whereas in the case of BP, a similar trend was not observed. This can be explained by the participation of two different polymerization mechanisms as discussed below (vide infra) (Table 2).
| Sensitizer | Time (h) | Conv.b (%) |
M
nc (g mol−1) |
M
w/Mnc |
|---|---|---|---|---|
| a V MMA = 1 mL, VDMF = 1 mL, [MMA]/[RX]/[PMDETA]/[S]: 200/1/5/1, λ = 350 nm, time = 4 h. b Determined gravimetrically. c Determined by gel permeation chromatography using polystyrene standards. | ||||
| TX | 4 | 14.3 | 8900 | 1.38 |
| TX | 8 | 24.5 | 15100 |
1.33 |
| TX | 12 | 32.5 | 17000 |
1.28 |
| BP | 4 | 21.2 | 4750 | 1.42 |
| BP | 8 | 47.0 | 6500 | 1.55 |
| BP | 12 | 57.3 | 9200 | 1.53 |
We then extended our experiments to other long wavelength absorbing PSs, namely ITX and CQ. Notably, higher conversions in the case of ITX indicate that this PS activates the polymerization more efficiently than the others do. In accordance with the absorption characteristics, the polymerization can be applied under visible light irradiation when CQ is used as a sensitizer (Table 3).
| Sensitizer | Irradiation wavelength (nm) | Conv.b (%) |
M
nc (g mol−1) |
M
w/Mnc |
|---|---|---|---|---|
| a V MMA = 1 mL, VDMF = 1 mL, [MMA]/[RX]/[PMDETA]/[S]: 200/1/5/1, time = 4 h. b Determined gravimetrically. c Determined by gel permeation chromatography using polystyrene standards. | ||||
| ITX | 350 | 59.0 | 11200 |
1.74 |
| CQ | 400–500 | 17.0 | 8700 | 1.56 |
In addition, we were interested in the effect of the solvent on the polymerization and the results are depicted in Table 4. Expectedly, polymerizations in more polar solvents give higher conversions and polymers with narrower dispersities further confirming involvement of electron transfer reactions and the ionic nature of the process. The ionic intermediates formed in the course of metal-free photo ATRP can be more easily stabilized in polar solvents.
| Solvent | Polarity index | Conv.b (%) |
M
nc (g mol−1) |
M
w/Mnc |
|---|---|---|---|---|
| a V MMA = 1 mL, Vsolvent = 1 mL, [MMA]/[RX]/[PMDETA]/[ITX]: 200/1/5/1, λ = 350 nm, time = 4 h. b Determined gravimetrically. c Determined by gel permeation chromatography using polystyrene standards. | ||||
| ACN | 5.8 | 35.2 | 10700 |
1.97 |
| DMF | 6.4 | 59.0 | 11200 |
1.74 |
| DMSO | 7.2 | 65.8 | 12500 |
1.56 |
In order to examine the chain-end fidelity of the polymers obtained, chain extension and block copolymerization experiments were performed. For this purpose, bromide chain-end functional PMMAs, which were obtained by the aforementioned process, were used as the halide source and identical polymerization conditions were applied as described in Table 1. In the chain extension process, MMA was used as a monomer, whereas for block copolymer synthesis, styrene was applied as a monomer. GPC analyses showed that there are clear shifts to lower retention volumes, which indicates the success of the polymerizations from the chain ends of the precursor PMMA in either case (Fig. 2).
 | ||
| Fig. 2 Comparison of the GPC traces of precursor PMMA with (a) PMMA-b-PBA and (b) chain extended PMMA. | ||
Light on/off experiments were performed under identical experimental conditions as indicated in Table 3 at multiple quantities to test the dark reaction behavior. The polymerization mixture was placed in a Schlenk tube under a nitrogen atmosphere and subjected to repeated cycles of light exposure (λ = 350 nm, 1 h illumination followed by a dark time of 30 min respectively). At the end of each step, equivalent volumes of the solution were precipitated into excess methanol to gravimetrically determine the conversion. The results demonstrate that the polymerization is irradiation dependent, and almost no polymerization occurred when the solutions were kept in the dark (Fig. 3). The minor increase in the conversion during the “dark” periods may be attributed to the slow deactivation rates of the propagating chains.
 | ||
| Fig. 3 Monomer conversion (%) vs. time using ITX to determine the dependency of propagation on irradiation: light on (yellow regions) light off (white regions). | ||
In the light of these studies and the general photoexcited state behavior of these sensitizers, the following mechanism can be proposed for the polymerizations on the example of TX (Scheme 3). The excited state of TX undergoes an electron transfer with electron donor amines. The formed radical anion form of TX reduces the initiator alkyl halide to yield radicals responsible for the initiation. A back electron transfer from the halide anion to the amine radical cation concludes the formation of the dormant macroalkyl halides that return to the polymerization cycle.
 | ||
| Scheme 3 Proposed mechanism of photoinduced metal-free ATRP using the TX/amine initiating system. | ||
Mechanism
DFT calculations were performed to support the proposed mechanism. Reaction of the PSs under light irradiation and in the absence of the amine component PMDETA as well as the reaction without light irradiation did not occur. Thus, a direct electron transfer reaction between the sensitizer and the alkyl bromide, or an electron transfer reaction between the singlet ground state of the sensitizer (S0) with either alkyl bromide or PMDETA can be disregarded. From this, it follows that in the reaction mechanism PMDETA must be first oxidized by an excited state of the PS. Upon excitation, the first excited singlet (S1) or the triplet state (T) can theoretically be reduced to give the PS radical anion (PS°−) and PMDETA radical cation (PMDETA°+). Half reaction potentials of S0, S1 and T for the reduction of the PS were calculated and are summarized in Table 5. The oxidizing ability of the PS increases in the order S1 < S0 < T which clearly indicates that the triplet state is the active species in the redox reaction with PMDETA.| Sensitizer | State | E° vs. SHE [V] |
|---|---|---|
| ITX | S0 | −1.71 |
| ITX | S1 | −1.86 |
| ITX | T | 1.22 |
| BP | S0 | −1.64 |
| BP | S1 | −1.85 |
| BP | T | 1.46 |
| CQ | S0 | −1.51 |
| CQ | S1 | −1.31 |
| CQ | T | 0.76 |
The reduced sensitizer can react either with EIB or with PMDETA to form an alkyl radical or a PMDETA radical which will eventually start the polymerization reaction (Scheme 4, reaction pathways II and III).
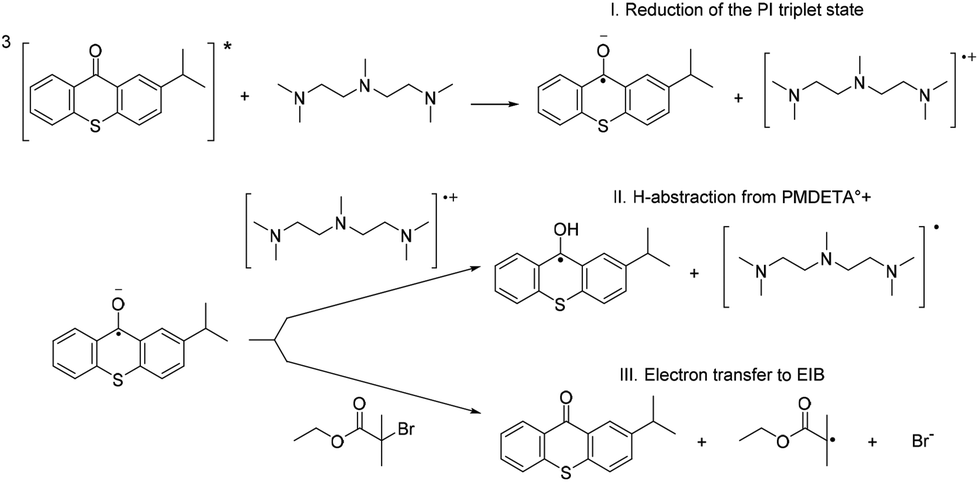 | ||
| Scheme 4 Possible pathways for the reaction mechanisms. | ||
In the first case, PS°− undergoes hydrogen abstraction from PMDETA°+ to give the protonated PS radical (PS-OH°) and a PMDETA radical (PMDETA°). In this pathway the PMDETA° radical will initiate the polymerization and control over the polymerization reaction will be lost (see Scheme 4, pathway II). In the second case (Scheme 4, pathway III) PS°− reduces EIB to give the PS in its S0 ground state, an alkyl radical and a bromine anion. Considering this reaction pathway, a controlled polymerization reaction according to ATRP is expected.
To gain more insight into the kinetics and thermodynamics of the possible pathways, Gibbs free energies and reaction enthalpies were calculated and are compared in Table 6.
| Pathway | ΔH [kcal mol−1] | ΔG [kcal mol−1] | ||||
|---|---|---|---|---|---|---|
| ITX | BP | CQ | ITX | BP | CQ | |
| I | −11.82 | −16.31 | 0.52 | −11.15 | −16.58 | −0.68 |
| II | −5.42 | −13.03 | −6.05 | −6.56 | −12.87 | −5.97 |
| III | −15.79 | −15.77 | −12.21 | −15.02 | −13.6 | −10.42 |
The first step of the reaction, which is the reduction of the T state of the PS to give PS°− and PMDETA°+, is thermodynamically favored for all three photoinitiators, with BP being the most favored followed by ITX and CQ. BP and CQ show negative reaction enthalpies, but not so CQ, which shows a slightly positive reaction enthalpy.
For all PS both possible consecutive reactions, the generation of the PMDETA radical (pathway II in Scheme 4) and the formation of the alkyl radical (pathway III in Scheme 4) are thermodynamically favored. Thus, free radical polymerization (reaction path II) as well as controlled ATRP (reaction path III) can potentially occur in a competing manner. However, the free energy and reaction enthalpy data suggest that reduction of the EIB compound will occur more likely than the proton transfer from PMDETA. In the case of BP the values are close to each other, which might make a potential contribution of the H-abstraction pathway possible with the consequence of loss over control of the polymerization reaction. This is well reflected in the higher PDI values of BP initiated polymerizations, when compared to e.g. ITX initiated polymerizations (see Table 2).
Conclusions
In conclusion, Type II photoinitiators were shown to activate metal-free ATRP under UV-vis light irradiation. The computational results give evidence that oxidation of the amine compound occurs from the triplet state of the PSs. In all cases the reaction kinetics for the controlled polymerization reaction are energetically favored. However, in the case of benzophenone, initiation of ATRP and free-radical polymerization pathway lie close to each other. Therefore, the free radical polymerization pathway cannot be excluded. Experimentally, this results in higher PDI values in comparison to TX. Light on/off experiments demonstrate the complete dependency of polymerization on irradiation, whereas chain extension and block copolymerization experiments approve the chain-end fidelity of the polymers obtained.Acknowledgements
The authors acknowledge the National Center for High Performance Computing (UHEM) for computation time under the project number 4004142016. JK gratefully acknowledges TÜBITAK for financial support in the framework of the project 116C038.References
- Y. Yagci, S. Jockusch and N. J. Turro, Macromolecules, 2010, 43, 6245–6260 CrossRef CAS.
- J. P. Fouassier, X. Allonas and D. Burget, Prog. Org. Coat., 2003, 47, 16–36 CrossRef CAS.
- J. V. Crivello, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 4241–4254 CrossRef CAS.
- J. Lalevee, N. Blanchard, M. A. Tehfe, M. Peter, F. Morlet-Savary, D. Gigmes and J. P. Fouassier, Polym. Chem., 2011, 2, 1986–1991 RSC.
- P. Xiao, J. Zhang, F. Dumur, M. A. Tehfe, F. Morlet-Savary, B. Graff, D. Gigmes, J. P. Fouassier and J. Lalevee, Prog. Polym. Sci., 2015, 41, 32–66 CrossRef CAS.
- J. P. Fouassier and J. Lalevee, RSC Adv., 2012, 2, 2621–2629 RSC.
- J.-P. Fouassier, Photoinitiation, photopolymerization, and photocuring: fundamentals and applications, Munich; NewYork; Cincinnati, 1995 Search PubMed.
- M. Aydin, N. Arsu and Y. Yagci, Macromol. Rapid Commun., 2003, 24, 718–723 CrossRef CAS.
- D. K. Balta, N. Arsu, Y. Yagci, S. Jockusch and N. J. Turro, Macromolecules, 2007, 40, 4138–4141 CrossRef CAS.
- L. Cokbaglan, N. Arsu, Y. Yagci, S. Jockusch and N. J. Turro, Macromolecules, 2003, 36, 2649–2653 CrossRef CAS.
- G. Yilmaz, B. Aydogan, G. Temel, N. Arsu, N. Moszner and Y. Yagci, Macromolecules, 2010, 43, 4520–4526 CrossRef CAS.
- D. Tunc and Y. Yagci, Polym. Chem., 2011, 2, 2557–2563 RSC.
- G. Yilmaz, G. Acik and Y. Yagci, Macromolecules, 2012, 45, 2219–2224 CrossRef CAS.
- G. Yilmaz, A. Tuzun and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5120–5125 CrossRef CAS.
- S. Dadashi-Silab, C. Aydogan and Y. Yagci, Polym. Chem., 2015, 6, 6595–6615 RSC.
- S. Kork, G. Yilmaz and Y. Yagci, Macromol. Rapid Commun., 2015, 36, 923–928 CrossRef CAS PubMed.
- G. Yilmaz, S. Beyazit and Y. Yagci, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1591–1596 CrossRef CAS.
- M. Uygun, M. A. Tasdelen and Y. Yagci, Macromol. Chem. Phys., 2010, 211, 103–110 CrossRef CAS.
- S. Dadashi-Silab and Y. Yagci, Tetrahedron Lett., 2015, 56, 6440–6443 CrossRef CAS.
- G. Yilmaz, E. Guler, F. B. Barlas, S. Timur and Y. Yagci, Macromol. Rapid Commun., 2016, 37, 1046–1051 CrossRef CAS PubMed.
- O. S. Taskin, G. Yilmaz, M. A. Tasdelen and Y. Yagci, Polym. Int., 2014, 63, 902–907 CrossRef CAS.
- K. Matyjaszewski and J. H. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed.
- Y. Gnanou and G. Hizal, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 351–359 CrossRef CAS.
- K. Min, H. F. Gao and K. Matyjaszewski, Macromolecules, 2007, 40, 1789–1791 CrossRef CAS.
- T. S. Hansen, J. U. Lind, A. E. Daugaard, S. Hvilsted, T. L. Andresen and N. B. Larsen, Langmuir, 2010, 26, 16171–16177 CrossRef CAS PubMed.
- Z. B. Guan and B. Smart, Macromolecules, 2000, 33, 6904–6906 CrossRef CAS.
- M. A. Tasdelen and Y. Yagci, Angew. Chem., Int. Ed., 2013, 52, 5930–5938 CrossRef CAS PubMed.
- M. A. Tasdelen, G. Yilmaz, B. Iskin and Y. Yagci, Macromolecules, 2012, 45, 56–61 CrossRef CAS.
- M. A. Tasdelen, M. Uygun and Y. Yagci, Macromol. Rapid Commun., 2011, 32, 58–62 CrossRef CAS PubMed.
- M. Ciftci, M. A. Tasdelen and Y. Yagci, Polym. Chem., 2014, 5, 600–606 RSC.
- M. A. Tasdelen, M. Uygun and Y. Yagci, Macromol. Chem. Phys., 2011, 212, 2036–2042 CrossRef CAS.
- M. A. Tasdelen, M. Ciftci and Y. Yagci, Macromol. Chem. Phys., 2012, 213, 1391–1396 CrossRef CAS.
- S. Dadashi-Silab, M. A. Tasdelen, A. M. Asiri, S. B. Khan and Y. Yagci, Macromol. Rapid Commun., 2014, 35, 454–459 CrossRef CAS PubMed.
- O. S. Taskin, G. Yilmaz and Y. Yagci, ACS Macro Lett., 2016, 5, 103–107 CrossRef CAS.
- N. J. Treat, H. Sprafke, J. W. Kramer, P. G. Clark, B. E. Barton, J. R. de Alaniz, B. P. Fors and C. J. Hawker, J. Am. Chem. Soc., 2014, 136, 16096–16101 CrossRef CAS PubMed.
- X. C. Pan, M. Lamson, J. J. Yan and K. Matyjaszewski, ACS Macro Lett., 2015, 4, 192–196 CrossRef CAS.
- G. M. Miyake and J. C. Theriot, Macromolecules, 2014, 47, 8255–8261 CrossRef CAS.
- A. Allushi, S. Jockusch, G. Yilmaz and Y. Yagci, Macromolecules, 2016, 49, 7785–7792 CrossRef CAS.
- J. C. Theriot, C. H. Lim, H. Yang, M. D. Ryan, C. B. Musgrave and G. M. Miyake, Science, 2016, 352, 1082–1086 CrossRef CAS PubMed.
- C. Kutahya, F. S. Aykac, G. Yilmaz and Y. Yagci, Polym. Chem., 2016, 7, 6094–6098 RSC.
- X. D. Liu, L. F. Zhang, Z. P. Cheng and X. L. Zhu, Polym. Chem., 2016, 7, 689–700 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian Inc., Wallingford, CT, 2016.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS.
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef CAS.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- V. G. Zakrzewski, J. V. Ortiz, J. A. Nichols, D. Heryadi, D. L. Yeager and J. T. Golab, Int. J. Quantum Chem., 1996, 60, 29–36 CrossRef.
- X. C. Pan, C. Fang, M. Fantin, N. Malhotra, W. Y. So, L. A. Peteanu, A. A. Isse, A. Gennaro, P. Liu and K. Matyjaszewskit, J. Am. Chem. Soc., 2016, 138, 2411–2425 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |