
Is a medium-range order pre-peak possible for ionic liquids without an aliphatic chain?†
Marco Campetella‡a,
Serena De Santis‡a,
Ruggero Caminiti‡*a,
Paolo Ballirano‡b,
Claudia Sadun‡a,
Luana Tanzi‡c and
Lorenzo Gontrani‡a
aDepartment of Chemistry, University of Rome “La Sapienza”, P.le A. Moro 5, Roma, Italy. E-mail: ruggero.caminiti@uniroma1.it; Fax: +39 06490631
bDepartment of Earth Sciences, University of Rome “La Sapienza”, P.le A. Moro 5, Roma, Italy
cDepartment of Physical and Chemical Sciences, University of L’Aquila, Via Vetoio (Coppito 1), 67100 Coppito, AQ, Italy
First published on 1st June 2015
Abstract
The combination of amino acids anions with a choline cation gives origin to a new and potentially important class of organic ionic liquids that might represent a viable and bio-compatible alternative with respect to the traditional ones. We present here a combined experimental and theoretical study of a choline–proline ionic liquid, using both large and small angle X-ray diffraction (WAXS–SAXS), and classical and ab initio molecular dynamics calculations, in which we are able to point out for the first time the existence of a low Q peak in the X-ray patterns in the absence of linear or branched alkyl chains. From the calculations, we can obtain theoretical scattering patterns that reproduce very nicely the experimental spectra in all Q ranges, and from detailed analysis of the radial distribution functions (RDFs) and hydrogen bond patterns, we can state that very strong ion pairs are established in the liquid and the observed pre-peak can be ascribed to the interactions between atoms belonging to different ion pairs.
Ionic liquids (ILs) have been one of the most investigated research fields in material science in recent decades, owing to their outstanding chemical and technological properties.1–7 An interesting subset of ionic liquids prepared recently8 is composed of cations or anions derived from biomaterials obtainable from renewable sources, such as amino acids, ammonium, guanidinium and choline. More specifically, the combination of choline with amino-acid anions has been recently exploited to provide ionic liquids with very low toxicity to humans1,5,9 and to the environment, though endowed of very desirable solvent properties, such as the ability to selectively dissolve lignin and cellulose from biomasses.10,11 Owing to the relative novelty of these materials, the literature on their structural properties is still rather scarce and is limited to general theoretical studies of choline liquids12 and to a recently developed force field, for which, though the systems examined contained aminoacids as the anions and a well-studied class of imidazolium compounds as the cations,13 no experimental structural studies exist, to our knowledge. Among all the liquids of this class, we chose to report the results obtained for an interesting system synthesised in our lab (see ESI†), choline–proline (see Fig. 1), since this combination exhibits an unexpected feature, namely the presence of a pre-peak or First Strong Diffraction Peak (FSDP) in the X-ray scattering patterns collected at small angles (Small Angle X-ray Scattering, SAXS), i.e. a peak falling at shorter Q values than the principal peak of the liquid, analogous to what is generally observed for ionic liquids with medium-long alkyl chains.14,15 The origin of this feature has been largely debated in several studies16,17 reaching the generally agreed hypothesis according to which the pre-peak is related to the existence of a medium-range order (MRO), mostly assignable to the 2nd and higher coordination shells (10–20 Å) in the sample,18,19 that in ionic liquids is reflected in the alternation of domains of different polarity.20,21 Such a mesoscopic separation tends to disappear when a polar group is introduced into the alkyl chain,17,22 or when the fragment can undergo some kind of intramolecular interaction that induces a partial folding of the chain.23 The latter phenomenon could be invoked for proline as well, since the four carbon atoms are arranged in a cycle, thus decreasing the available contact surface for hydrophobic interactions. The absence of well separated domains with cycloalkyl substitution was correlated to some thermodynamic data by our group24 and was described in a previous structure–activity relationship study by Tao et al.25 Nevertheless, a clear pre-peak is visible in the small-angle X-ray scattering curve (Fig. 2), whose origin is therefore presumably different from the segregation of apolar domains within the charged matrix.
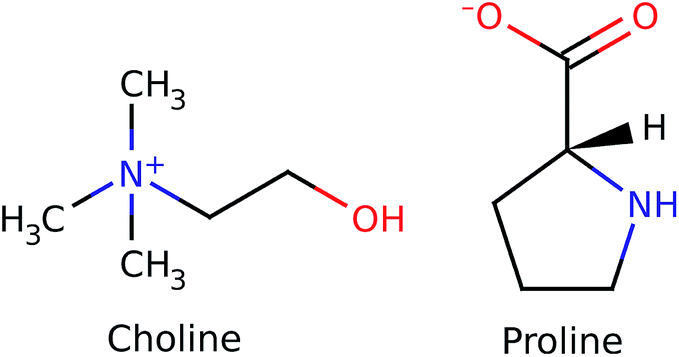 | ||
| Fig. 1 Representation of the choline cation and proline anion. | ||
 | ||
| Fig. 2 Small angle X-ray scattering curve. Experimental: black dash; model: red line. | ||
The first scattering peak of Fig. 2 falls at about 0.4 Å−1 and is therefore contributed by medium-range structural correlations in the bulk liquid.18 The spatial dimension of a suitable interpretion model must therefore be twice as much as this value, owing to the minimum image convention of the Periodic Boundary Conditions (PBC) algorithm.26 Given the large dimension of the system (more than 10 K atoms) we decided to perform classical molecular dynamics simulations using the GPU-accelerated version of Amber software27 (see ESI† for details), with the two-body force field Gaff28 and properly tuned point charges.29 The agreement found using this model is very satisfactory for the low Q range of Fig. 2, in particular regarding the position of the peaks. To further assess the model reliability, we confronted the theoretical structure function30–32 and the relative radial distribution function with the experimental analogues in the medium-long Q range (Wide Angle X-ray Scattering, WAXS, i.e. short distances), and with an alternative higher accuracy theoretical model obtained from first-principle calculations (ab initio molecular dynamics).33 The scattering function reported in this Q-range (structure function QIQM(Q)) is not suitable for highlighting the low-Q peaks contributed by long distance correlations, but is generally preferred when radial patterns descriptive of the system are sought (see ESI, eqn (2)†). Very good agreements between the experimental and calculated patterns (structure functions and radial distribution functions) were found for all Q and r values, see Fig. 3, for both models, though the QM results are clearly superior in all spatial ranges, up to their allowed distance limit (half box length).
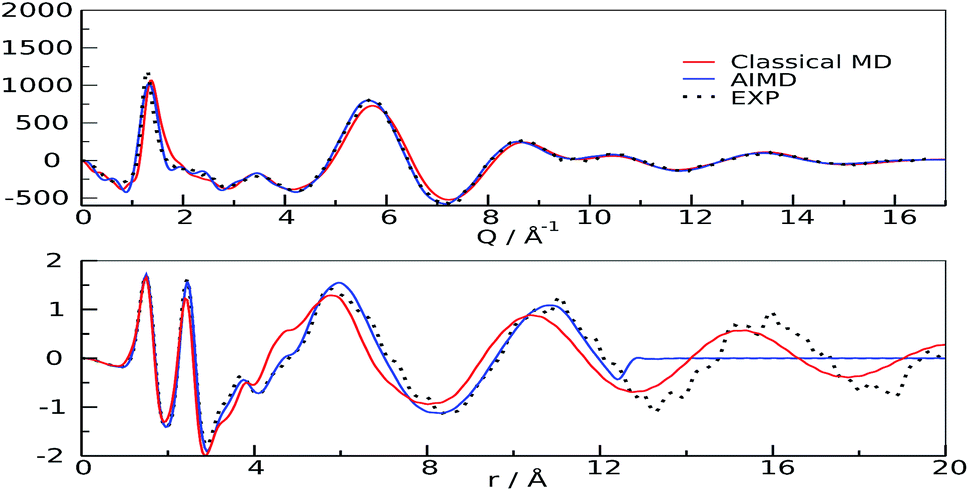 | ||
| Fig. 3 Structure functions (upper panel) and radial distribution functions (lower panel). Experimental: black dots; classical MD: red; AIMD: blue | ||
The quality of the agreement with the experimental data made us confident that our calculations were representative of the system, and could be used for further analysis of the microscopic structure of the liquid. One of the most important rheological features of choline–aminoacid liquids is the very high viscosity they possess, that can be ascribed to the existence of very strong interactions between the ions. Among all the possible forms of interactions, hydrogen bonds play a major role, as already pointed out in the study by Benedetto et al.12 In fact, the relative radial distribution function (between Cho and Pro oxygen atoms), reported in Fig. 4-panel A, shows a sharp peak whose maximum value (2.65 Å and 2.62 Å, for classical and QM models, respectively), falls at significantly shorter distance than the equilibrium value of 2.75–2.8 Å of liquid water.34,35 This observation suggests that cations and anions give origin to strong ion pairs. To better characterize the cation–anion interaction we have calculated the relative Combined Distribution Function (CDF),36 which correlates the distance between the cation and anion oxygen atoms and the distribution of the angle formed by two vectors, the former corresponding to the O–H bond and the latter connecting the two hetero atoms; the relative function is shown in Fig. 4-panel B. The region of maximum occurrence in the contour plot corresponds to the distance range from 2.5 to 2.9 Å and to the angular interval of 160–180 degrees; these distance and angle values were used as threshold criteria to classify the O–H⋯O contacts as hydrogen bonds.37–39
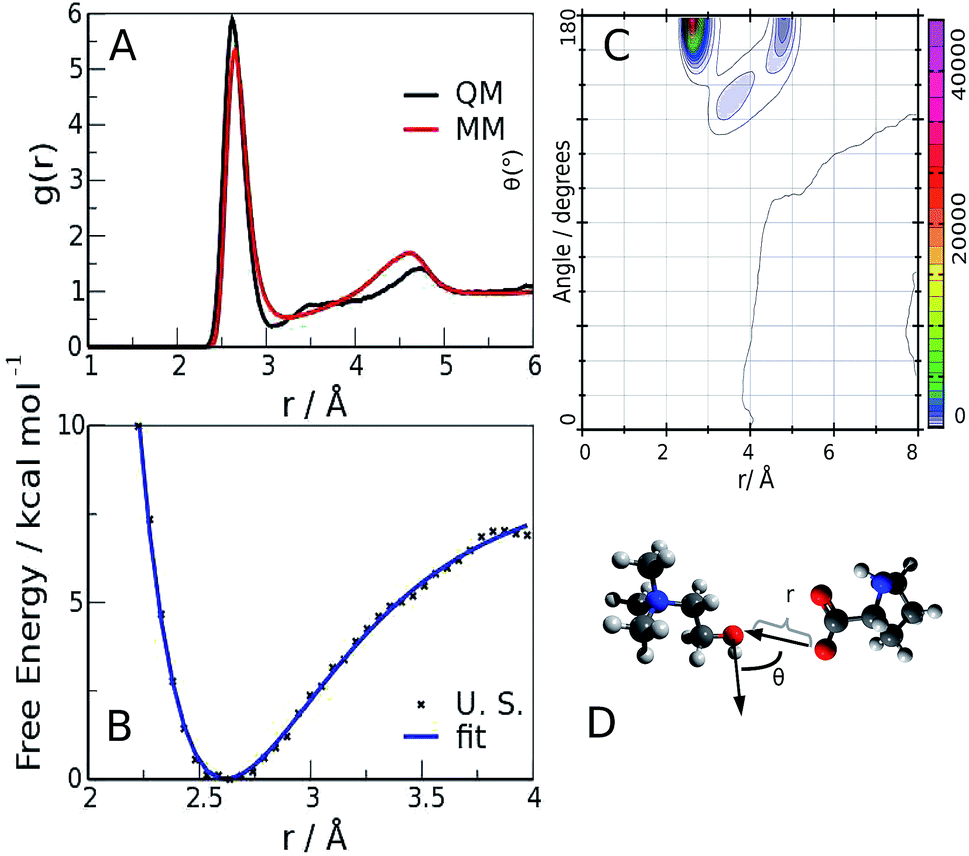 | ||
| Fig. 4 O(Cho)⋯O(Pro) radial distribution function (panel A), combined radial–angular distribution function (panel B), free energy profile of the H-bond interaction (panel C) and sketch of the H-bond (panel D). The blue line is a fit of the free energy data with a Morse-type function. | ||
The occurrence and time duration of these hydrogen bonds were derived from a detailed analysis of the AIMD trajectory (see ESI, computational details†), whose shorter time step and more correct description of the interaction potential was considered more appropriate for investigating the H-bond spatial features and appraising its time evolution. Over a total of 50 ion pairs, we found that 38 cations and 30 anions form persistent bonds during the observed time (40 ps); such cations and anions remain bonded for 35.9 and 38.7 ps, respectively. The lower number of anions involved is compliant with the presence of two H-bond sites in the carboxylate. To quantify such an interaction, we performed an Umbrella Sampling (U. S.) calculation40 (ESI, computational details†) on the QM trajectory using the O(Cho)⋯O(Pro) distance as the reaction coordinate; the resulting potential of the mean force,41 shown in Fig. 4-panel C, has a minimum of about 7 kcal mol−1 located at 2.62 Å. Such a value is similar to the values reported for the carboxylic acid dimers in ref. 42–44 and is stronger than for liquid water (about 5 kcal mol−1), in compliance with the shorter O⋯O distance observed in the radial distribution function.
From these results, it can be stated that this ion pair interaction is largely responsible for the strong structural ordering at low distances; the “structure” of the liquid stems from the packing of the fundamental units, the ion pairs. The overall disposition of the ions, through the interference of their scattered beams, gives origin to the observed spectra Fig. 3 and 4. The total scattering pattern can be divided into the individual contributions coming from each pair of scatterers (see ESI, eqn (3)†), which can be, in turn, clustered into group terms, like anion and cation. This analysis is shown in Fig. 5, where the whole pattern (black) is decomposed into anion–anion, cation–cation and cation–anion contributions. It can be seen that homologous correlations (cation–cation and anion–anion) lead to the positive peaks at around 0.5 Å−1 and are counterbalanced by the intense negative trough due to the mixed cation–anion terms. The resultant curve shows the presence of a pre-peak located around 0.4 Å−1 that is also observed in the experimental data. The structural correlations underlying the two peaks can be related to an average “effective distance” of about 2π/Q ≈ 12.5 Å, that can be easily appreciated in the g(r)′s between the nitrogen and oxygen atoms of the cations and anions, respectively, and in their corresponding Fourier-transform H(Q), the partial contributions to the scattering function (see ESI, eqn (3)†) that show a consensus peak at 0.5 Å−1. All the curves are reported in Fig. 6, together with the typical geometrical arrangement of the ions taken from a trajectory frame, in which the cation–cation contacts are highlighted.
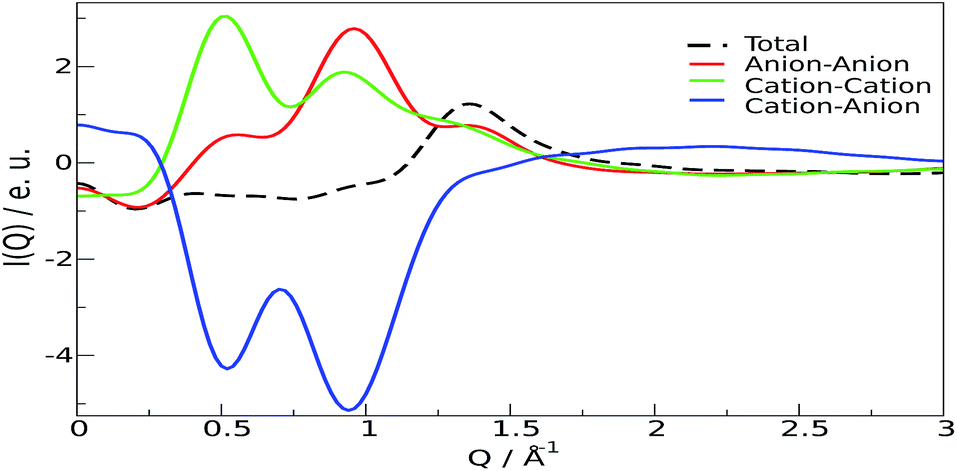 | ||
| Fig. 5 Individual contributions to the low Q diffraction pattern. Black: total; red: cation–cation; green: anion–anion; blue: cation–anion. | ||
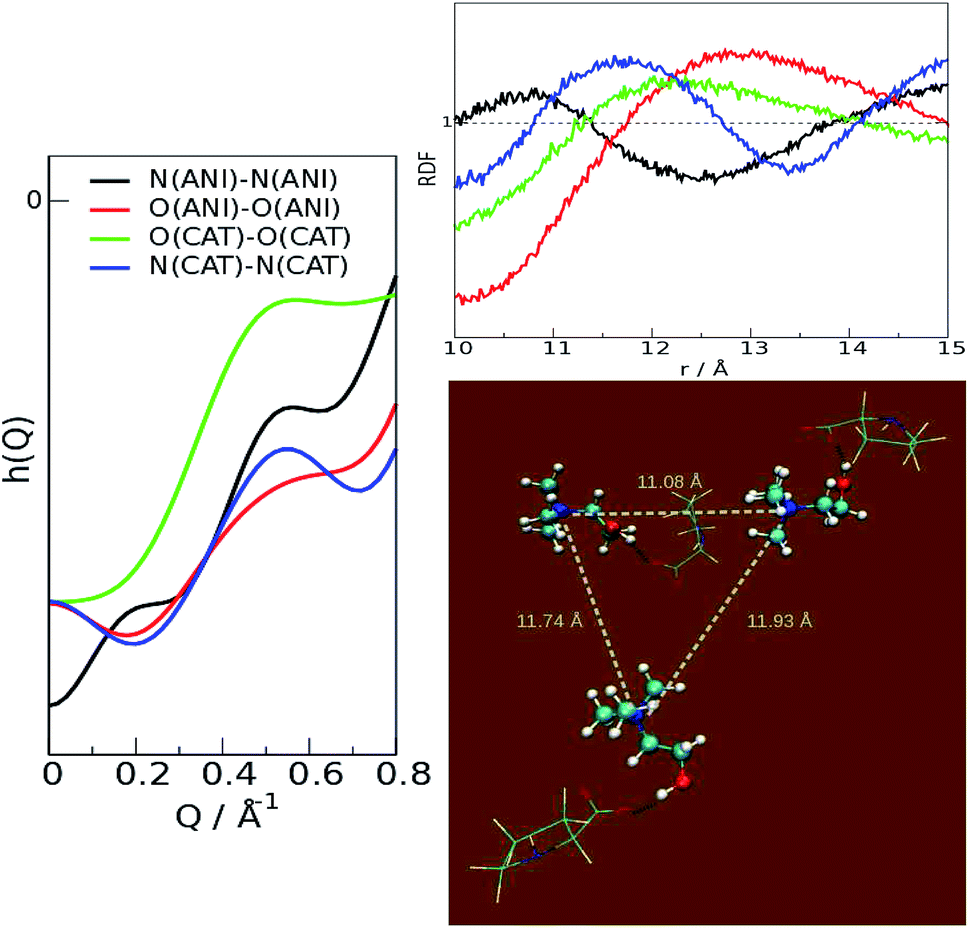 | ||
| Fig. 6 Partial contributions of the heteroatoms. Left: scattering function H(Q); right, top: radial distribution functions; right, bottom: typical second-shell arrangement of the ion pairs. | ||
Summarizing, the results shown indicate that in the choline–proline ionic liquid the most important structural feature is the existence of strong ion pairs that mutually solvate one another. The diffraction pre-peak observed can be ascribed to the scattering of second-neighbour groups. In future work, we plan to investigate if this motif can be recognized in other systems lacking hydrophobic moieties, e.g. aromatic rings.
Acknowledgements
We are deeply grateful to Enrico Bodo, for his support and for the very helpful discussions. We also thank the Sapienza University of Rome for financial support through grant C26H14P9R2, Prof. Ruggero Caminiti (Sapienza University of Rome Chemistry Department) for providing free usage of the Narten computing cluster facility and the Sapienza Nanotechnology Center (CNIS) for providing access to the D8 Diffractometer. Computational support from PRACE (grant no. 2013091962) is also acknowledged.References
- P. Nockemann, B. Thijs, K. Driesen, C. R. Janssen, K. V. Hecke, L. V. Meervelt, S. Kossmann, B. Kirchner and K. Binnemans, J. Phys. Chem. B, 2007, 111, 5254–5263 CrossRef CAS PubMed.
- Y. Fukaya, Y. Iizuka, K. Sekikawa and H. Ohno, Green Chem., 2007, 9, 1155–1157 RSC.
- Y. Yu, X. Lu, Q. Zhou, K. Dong, H. Yao and S. Zhang, Chem.–Eur. J., 2008, 14, 11174–11182 CrossRef CAS PubMed.
- J.-C. Plaquevent, J. Levillain, F. Guillen, C. Malhiac and A.-C. Gaumont, Chem. Rev., 2008, 108, 5035–5060 CrossRef CAS PubMed.
- M. Petkovic, J. L. Ferguson, H. Q. N. Gunaratne, R. Ferreira, M. C. Leitao, K. R. Seddon, L. P. N. Rebelo and C. S. Pereira, Green Chem., 2010, 12, 643–649 RSC.
- A. J. L. Costa, M. R. C. Soromenho, K. Shimizu, I. M. Marrucho, J. M. S. S. Esperança, J. N. C. Lopes and L. P. N. Rebelo, ChemPhysChem, 2012, 13, 1902–1909 CrossRef CAS PubMed.
- Y. Gao, S. W. Arritt, B. Twamley and J. M. Shreeve, Inorg. Chem., 2005, 44, 1704–1712 CrossRef CAS PubMed.
- C. Rüss and B. Konig, Green Chem., 2012, 14, 2969–2982 RSC.
- K. D. Weaver, H. J. Kim, J. Sun, D. R. MacFarlane and G. D. Elliott, Green Chem., 2010, 12, 507–513 RSC.
- Q.-P. Liu, X.-D. Hou, N. Li and M.-H. Zong, Green Chem., 2012, 14, 304–307 RSC.
- X.-D. Hou, T. J. Smith, N. Li and M.-H. Zong, Biotechnol. Bioeng., 2012, 109, 2484–2493 CrossRef CAS PubMed.
- A. Benedetto, E. Bodo, L. Gontrani, P. Ballone and R. Caminiti, J. Phys. Chem. B, 2014, 118, 2471–2486 CrossRef CAS PubMed.
- V. V. Chaban and E. E. Fileti, J. Phys. Chem. B, 2015, 119, 3824–3828 CrossRef CAS PubMed.
- O. Russina, A. Triolo, L. Gontrani and R. Caminiti, J. Phys. Chem. Lett., 2012, 3, 27–33 CrossRef CAS.
- M. Campetella, L. Gontrani, F. Leonelli, L. Bencivenni and R. Caminiti, ChemPhysChem, 2015, 16, 197–203 CrossRef CAS PubMed.
- C. Hardacre, J. D. Holbrey, C. L. Mullan, T. G. A. Youngs and D. T. Bowron, J. Chem. Phys., 2010, 133, 074510 CrossRef PubMed.
- O. Russina, A. Triolo, L. Gontrani, R. Caminiti, D. Xiao, L. G. Hines Jr, R. A. Bartsch, E. L. Quitevis, N. Plechkova and K. R. Seddon, J. Phys.: Condens. Matter, 2009, 21, 424121 CrossRef.
- P. H. Gaskell and D. J. Wallis, Phys. Rev. Lett., 1996, 76, 66–69 CrossRef CAS.
- G. Lucovsky and J. C. Phillips, Phys. Status Solidi B, 2009, 246, 1806–1812 CrossRef CAS PubMed.
- H. K. Kashyap, C. S. Santos, R. P. Daly, J. J. Hettige, N. S. Murthy, H. Shirota, E. W. Castner and C. J. Margulis, J. Phys. Chem. B, 2013, 117, 1130–1135 CrossRef CAS PubMed.
- A. Triolo, O. Russina, H.-J. Bleif and E. Di Cola, J. Phys. Chem. B, 2007, 111, 4641–4644 CrossRef CAS PubMed.
- A. Triolo, O. Russina, R. Caminiti, H. Shirota, H. Y. Lee, C. S. Santos, N. S. Murthy and E. W. Castner Jr, Chem. Commun., 2012, 48, 4959–4961 RSC.
- M. Campetella, L. Gontrani, E. Bodo, F. Ceccacci, F. C. Marincola and R. Caminiti, J. Chem. Phys., 2013, 138, 184506 CrossRef PubMed.
- S. De Santis, G. Masci, F. Casciotta, R. Caminiti, E. Scarpellini, M. Campetella and L. Gontrani, Phys. Chem. Chem. Phys., 2015 Search PubMed , in revision.
- D.-J. Tao, Z. Cheng, F.-F. Chen, Z.-M. Li, N. Hu and X.-S. Chen, J. Chem. Eng. Data, 2013, 58, 1542–1548 CrossRef CAS.
- M. P. Allen and D. J. Tildesley, Computer Simulation of Liquids, Oxford University Press, 1987 Search PubMed.
- A. W. Goetz, M. J. Williamson, X. Dong, P. Duncan, S. Le Grand and R. C. Walker, J. Chem. Theory Comput., 2012, 8, 1542–1555 CrossRef PubMed.
- J. Wang, W. Wang, P. A. Kollman and D. A. Case, J. Mol. Graphics Modell., 2006, 25, 247–260 CrossRef CAS PubMed.
- Y. Zhang and E. J. Maginn, J. Phys. Chem. B, 2012, 116, 10036–10048 CrossRef CAS PubMed.
- L. Gontrani, O. Russina, F. C. Marincola and R. Caminiti, J. Chem. Phys., 2009, 131, 244503 CrossRef PubMed.
- V. R. Albertini, L. Bencivenni, R. Caminiti, F. Cilloco and C. Sadun, J. Macromol. Sci., Part B: Phys., 1996, 35, 199–213 CrossRef PubMed.
- The structure of Ionic Liquids, ed. R. Caminiti and L. Gontrani, Springer, 2014, vol. 193 Search PubMed.
- J. Hutter, M. Iannuzzi, F. Schiffmann and J. VandeVondele, WIREs Comput. Mol. Sci., 2014, 4, 15–25 CrossRef CAS PubMed.
- T. Head-Gordon and G. Hura, Chem. Rev., 2002, 102, 2651–2670 CrossRef CAS PubMed.
- A. K. Soper, ISRN Phys. Chem., 2013, 2013, 67 Search PubMed.
- M. Brehm and B. Kirchner, J. Chem. Inf. Model., 2011, 51, 2007–2023 CrossRef CAS PubMed.
- K. Dong and S. Zhang, Chem.–Eur. J., 2012, 18, 2748–2761 CrossRef CAS PubMed.
- E. Bodo, A. Sferrazza, R. Caminiti, S. Mangialardo and P. Postorino, J. Chem. Phys., 2013, 139, 144309 CrossRef CAS PubMed.
- L. Gontrani, E. Bodo, A. Triolo, F. Leonelli, P. D’angelo, V. Migliorati and R. Caminiti, J. Phys. Chem. B, 2012, 116, 13024–13032 CrossRef CAS PubMed.
- G. Torrie and J. Valleau, J. Comput. Phys., 1977, 23, 187–199 CrossRef.
- S. Kumar, J. M. Rosenberg, D. Bouzida, R. H. Swendsen and P. A. Kollman, J. Comput. Chem., 1995, 16, 1339–1350 CrossRef CAS PubMed.
- T. Neuheuser, B. A. Hess, C. Reutel and E. Weber, J. Phys. Chem., 1994, 98, 6459–6467 CrossRef CAS.
- Y. Gu, T. Kar and S. Scheiner, J. Am. Chem. Soc., 1999, 121, 9411–9422 CrossRef CAS.
- M. W. Feyereisen, D. Feller and D. A. Dixon, J. Phys. Chem., 1996, 100, 2993–2997 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and computational details. See DOI: 10.1039/c5ra07567j |
| ‡ All the authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |