
Chlorosulfonic acid supported diethylamine ionic liquid catalyzed green synthesis of novel 2-mercaptonaphthalen-1-yl)methyl)-3-hydroxy-5,5-dimethylcyclohex-2-enones under neat conditions†
Mudumala Veeranarayana Reddya,
Gangireddy Chandra Sekhar Reddyb,
Reddi Mohan Naidu Kallac and
Yeon Tae Jeong*a
aDepartment of Image Science and Engineering, Pukyong National University, Busan 608-737, Korea. E-mail: ytjeong@pknu.ac.kr; Fax: +82-51-629-6408; Tel: +82-51-629-6411
bDepartment of Chemistry, Sri Venkateswara Degree & PG College, Balaji Nagar, Kadapa – 516003, AP, India
cDepartment of Chemistry, Yogananda Inistitute of Technology and Science, 517520 Tirupati, India
First published on 1st April 2015
Abstract
Chlorosulfonic acid supported diethylamine ionic liquid (DEACSA IL) was prepared and characterized by FT-IR, elemental analysis, NMR spectroscopy and thermogravimetric analysis. The DEACSA IL functions as an efficient heterogeneous, environmentally benign, and very active catalyst and can be recycled for several runs without any significant loss of activity with short reaction times, good to excellent yields of the desired products and high catalytic activity for the synthesis of a variety of novel 2-mercaptonaphthalen-1-yl)methyl)-3-hydroxy-5,5-dimethylcyclohex-2-enones under neat conditions. This is the first example of the condensation of naphthalene-2-thiol, aldehydes and 5,5-dimethylcyclohexane-1,3-dione to provide a novel series of thiol derivatives, which were characterized by 2D-NMR data.
Sulfur plays a major role in biology, and is found in numerous peptides, proteins and low molecular weight compounds.1 The sulfur compounds found in plants, bacteria, fungi and animals have unique chemical and biochemical properties linked to redox processes, metal binding and catalytic reactions.2 The antibiotic and anticancer activities of thiols, thiosulfinates, thiosulfonates, isothiocyanates, sulfoxides, sulfones and polysulfides make them interesting from the pharmacological perspective.3 In particular aryl thiols are important building blocks in the synthesis of sulfur-containing natural products and pharmaceutical compounds. Further thiols and hydroxydimethylcyclohex-2-enone are a very important group of compounds since they play a very unique role in biological systems, medicine, food, pharmaceutical and aroma industries. These compounds are also useful as key synthons for the synthesis of bioactive thiopyran derivatives.4
The ionic liquid (IL) conditions have attracted much interest for organic chemists from the green chemistry point of view.5 The possibility of performing multi-component reactions (MCR) under IL conditions could enhance their efficiency from an economic as well as ecological point of view. In addition one of the most important and highly applicable category is the use of supported ionic liquid catalyst for effecting numerous organic transformations.6 In this connection functionalized sulfonic acid ionic liquids received great attention due to their unique properties like environmental compatibility, reusability, greater selectivity, and ease of product isolation. They serve as both solvent and catalyst for several organic reactions.7
Diethylamine (DEA) base is works as an efficient catalyst in various organic transformations. However, its drawbacks, non-reusability and difficulties in its separation from reaction mixture are limiting its usage in synthetic organic chemistry. But chlorosulfonic acid supported diethylamine liquids (DEACSA IL) are identified as one of the best catalyst and solvent. The use of the DEACSA IL complex catalyst has several advantages over a conventional base catalyst, such as its ease of handling, stability, inexpensive, greater selectivity, dual solvent-catalyst, easy recyclability, eco-friendly, nontoxic enhanced reaction rates, cleaner products, manipulative simplicity, and improved product selectivity.
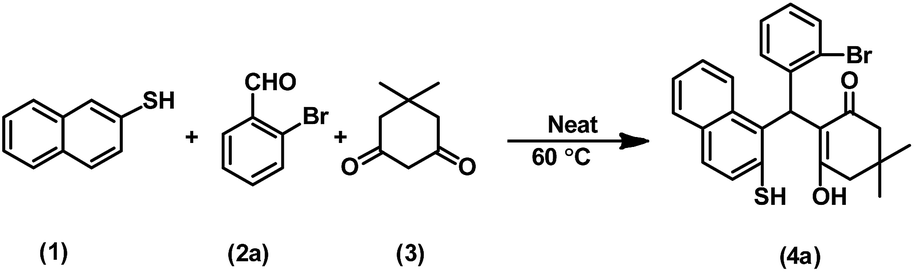
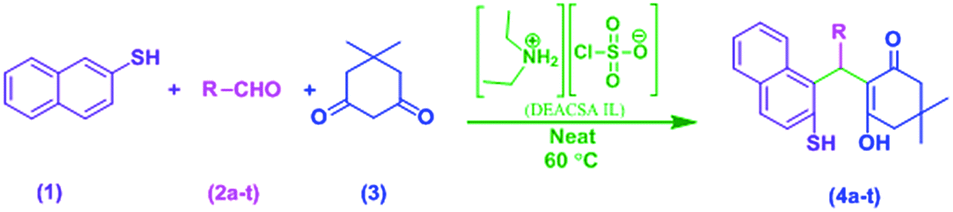
In continuation of our efforts to develop green chemical synthetic approaches,5e,f herein, we report a novel and efficient one-pot method for the synthesis of 2-mercaptonaphthalen-1-yl)methyl)-3-hydroxy-5,5-dimethylcyclohex-2-enones (4a–t) via three-component condensation reaction of naphthalene-2-thiol (1), various aldehydes (2a–t) and 5,5-dimethylcyclohexane-1,3-dione (3) by reusable DEACSA IL catalyst at 60 °C (Scheme 1).
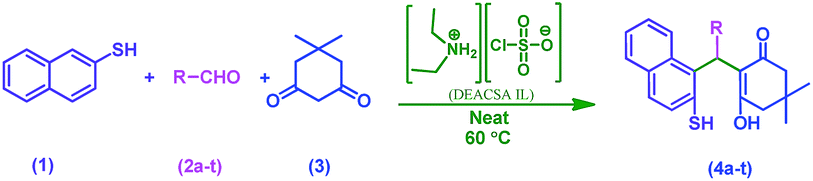 | ||
| Scheme 1 Synthesis of 2-((substituted) (2-mercaptonaphthalen-1-yl) methyl)-3-hydroxy-5,5-dimethylcyclohex-2-enones (4a–t). | ||
Results and discussion
Characterization of the new DEACSA IL
The IR spectrum (Fig. 1) showed O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) SO asymmetric and symmetric stretching peaks at 1185 and 1056 cm−1, respectively, and an S–O stretching peak at 763 cm−1, which precisely confirmed the SO3 group linkage. The amino group N–H vibrations observed at 3315 and 3302 cm−1 are shown two hydrogens on nitrogen.
SO asymmetric and symmetric stretching peaks at 1185 and 1056 cm−1, respectively, and an S–O stretching peak at 763 cm−1, which precisely confirmed the SO3 group linkage. The amino group N–H vibrations observed at 3315 and 3302 cm−1 are shown two hydrogens on nitrogen.
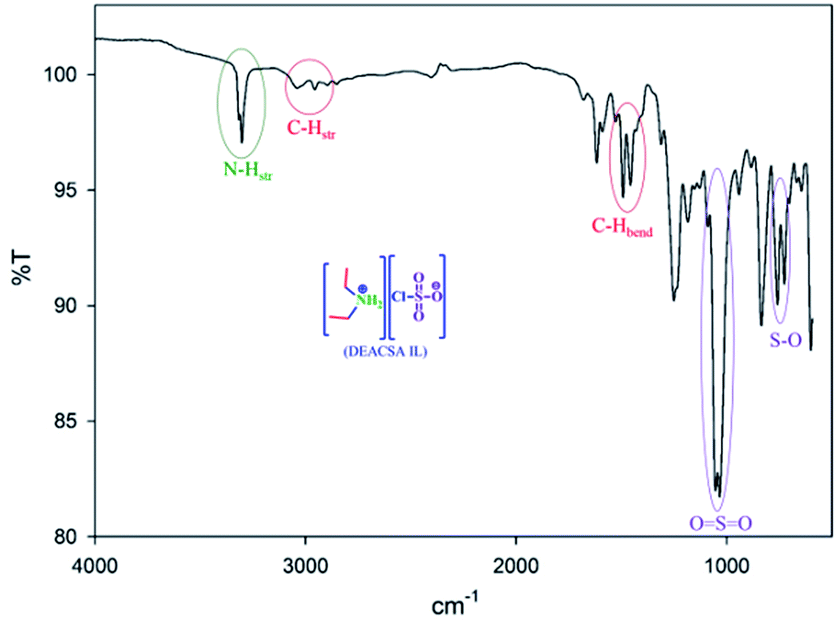 | ||
| Fig. 1 FT-IR spectra of DEACSA IL. | ||
The 1H NMR spectrum (Fig. 2(a)) of the DEACSA IL shows two distinguished triplet peaks at δ 1.12 and 1.16 assigned to methyl protons. At the same time the two methylene group protons also showed two distinguished quartets at δ 2.78 and 3.17. It indicates that the two ethyl groups are asymmetrically arranged around the nitrogen. The two amino protons are observed δ 8.23 and while run the spectrum in D2O it disappeared because due H → D chemical exchange (Fig. 2(b)). The 13C NMR spectra (Fig. 2(c) and (d)) also shown distinguishable peaks for both ethyl groups on nitrogen at δ 10.2 and 11.3 for methyl group and δ 42.1 and 44.7 for methylene group carbons.
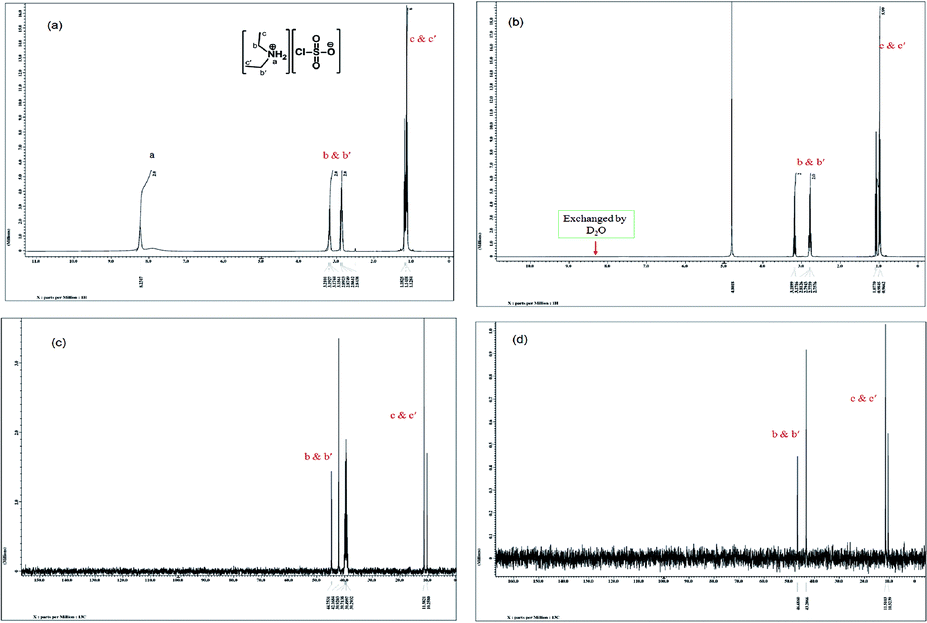 | ||
| Fig. 2 1H and 13CNMR spectra of DEACSA IL measured in D2O [(a) and (c)] and DMSO-d6 [(b) and (d)]. | ||
The thermal stability of the DEACSA IL was measured by TGA (Fig. 3) and the complete degradation of it occurred between 287 and 470 °C. It shown that the degradation start from 260 °C and uniform dissociation rate observed between these ranges of temperature. It indicating that the stability of catalyst is up to 260 °C.
 | ||
| Fig. 3 TGA patterns of DEACSA IL. | ||
Application of DBA IL for the synthesis of 2-mercaptonaphthalen-1-yl)methyl)-3-hydroxy-5,5-dimethylcyclohex-2-enones
The three-component condensation reaction of naphthalene-2-thiol (1, 1 mmol), 2-bromobenzaldehyde (2a, 1 mmol) and 5,5-dimethylcyclohexane-1,3-dione (3, 1 mmol) in the presence of DEACSA IL (10 mol%) at 60 °C afforded interestingly the product 2-((2-bromophenyl) (2-mercaptonaphthalen-1-yl)methyl)-3-hydroxy-5,5-dimethylcyclohex-2-enone (4a) in 90% yield rather than the expected cyclisation product, 12-(2-bromophenyl)-9,9-dimethyl-9,10-dihydro-8H-benzo[a]thioxanthen-11(12H)-one (5a) (Scheme 2). | ||
| Scheme 2 Condensation of naphthalene-2-thiol (1), 2-bromobenzaldehyde (2a) and 5,5-dimethylcyclohexane-1,3-dione (3). | ||
The structure of 4a was confirmed by IR and NMR analysis. The NMR performed on it standard one-dimensional 1H and 13C-NMR measurements and the two-dimensional NMR experiments, COSY, NOESY, HMQC and HMBC. Data were in agreement with the proposed structure for 4a and this data suggested that the structure 4a exists in to two conformers as 4a′&4a′′ (Scheme 3).
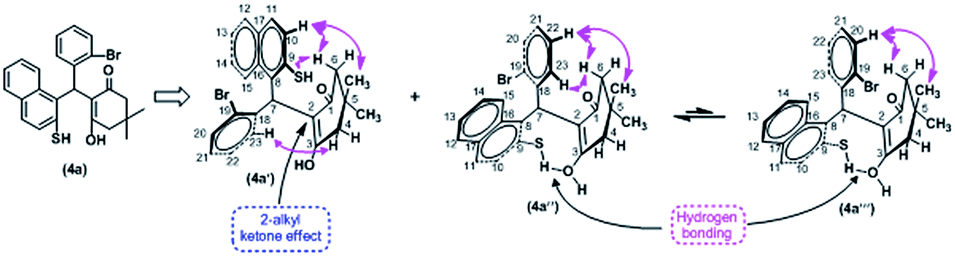 | ||
| Scheme 3 Conformation analysis of compound 4a. | ||
The IR spectrum (Fig. 4) shows the λmax at 3431 and 2615 cm−1 indicating that the free –OH and –SH group respectively. On the other hand an absorption band at 1706 cm−1 indicating that the aliphatic conjugated carbonyl group. It also evidenced that the λmax at 3050 and 1575 cm−1 showing the CC group in conjugation. Therefore, these are evidenced that the non-cyclic nature of 4a.
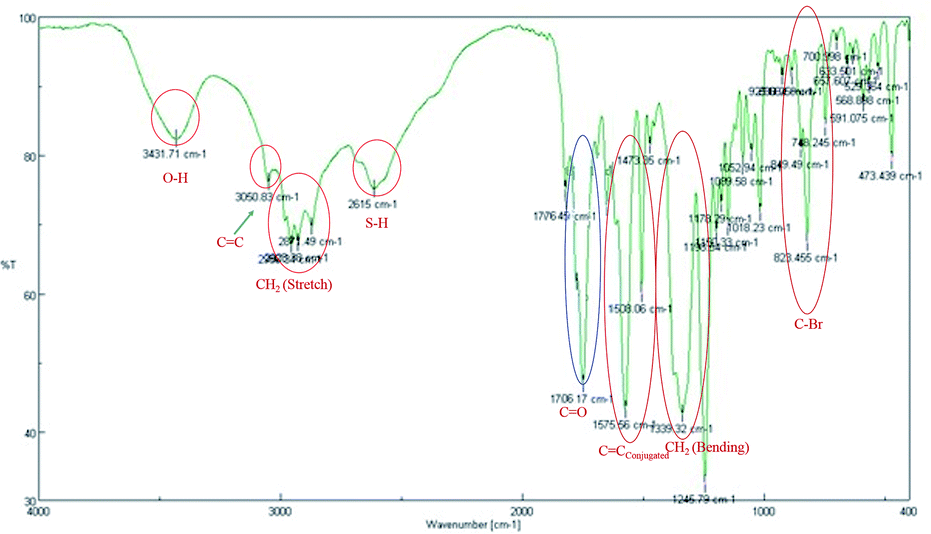 | ||
| Fig. 4 FT-IR spectrum of compound 4a. | ||
The NOESY spectrum (Fig. 5) shows that the conformer 4a′ is major one due to 2-alkyl ketone effect because to this effect, possibility of the hydrogen bonding at acidic proton of either OH or SH is less probable. In the NOESY spectrum, through space interactions of the two methyl group protons and protons at C-4 & C-6 in alicyclic ring show as cross peaks with both aromatic ring protons at ortho and/or meta position which are closer proximity to each them in space.
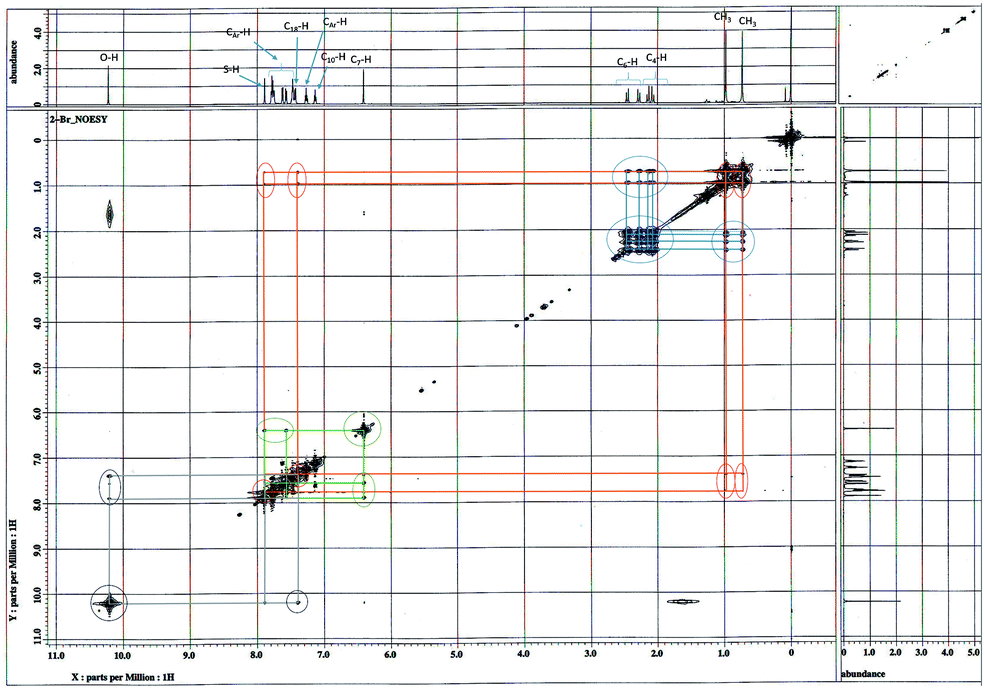 | ||
| Fig. 5 2D-1H–1H NOESY spectrum of compound 4a in DMSO-d6. | ||
The protonated and quaternary carbons were assigned under good experimental conditions by means of HMQC and HMBC experiments. In HMQC spectrum (Fig. 6) showing the short range interaction of H and C at respective positions. In HMBC spectrum (Fig. 7) two types of connections of residual and long range interactions are clearly visible. According to our experience, such connections are observable, for example, in structural fragment, –HC7– with C2 (residual) and C1, C3, C9, C18 and C23 (long-range) interactions.
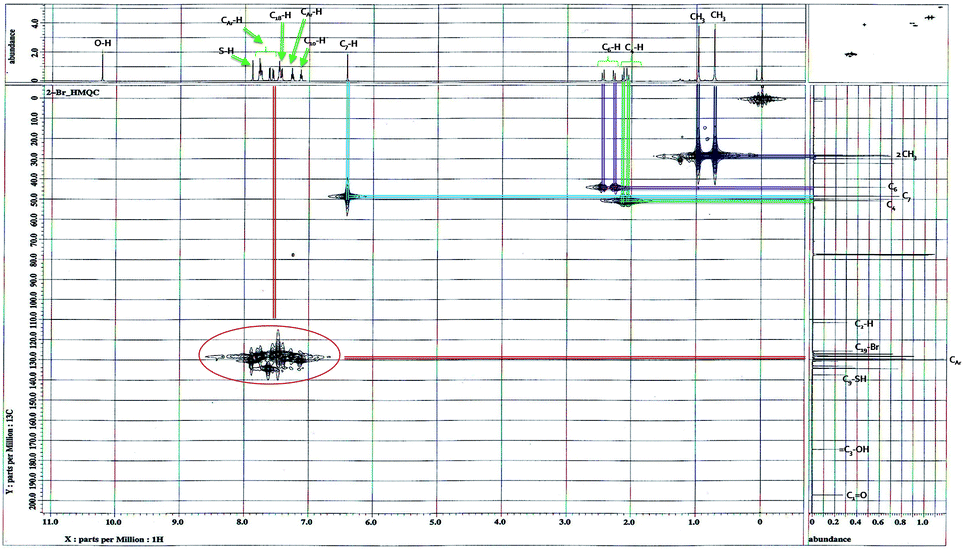 | ||
| Fig. 6 2D-1H–13C HMQC spectrum of compound 4a in DMSO-d6. | ||
 | ||
| Fig. 7 2D-1H–13C HMBC spectrum of compound 4a in DMSO-d6. | ||
From above analysis the inability of formation of 5a may be due to probably 2-alkyl ketone effect and extending conjugation of the double bond with carbonyl group in alicyclic ring.
Encouraged by the result we chosen this reaction as a model reaction and standardized the reaction conditions further for the synthesis of titled compounds (4a–t). At first the model reaction was carried under neat condition at 60 °C in the absence of catalyst. Desired product, 4a, is not obtained even after 10 h stirring. Only a trace amount of this product was formed even after 15 h of stirring (Table 1, entry 1). The same reaction when performed in the presence of 10 mol% of the Et2NH 4a is formed in 32% yield within 8 h (Table 1, entry 2).
|
|||
|---|---|---|---|
| Entry | Catalyst (mol%) | Time (min) | Yieldb (%) |
| a Reaction of naphthalene-2-thiol (1, 1 mmol), 2-bromobenzaldehyde (2a, 1 mmol) and 5,5-dimethylcyclohexane-1,3-dione (3, 1 mmol) under neat condition at 60 °C.b Isolated yield.c Catalyst was reused four times. | |||
| 1 | Neat | 900 | Trace |
| 2 | Et2NH (10) | 680 | 32 |
| 3 | Et3N (10) | 675 | 33 |
| 4 | Dibutylamine (10) | 530 | 35 |
| 5 | TMG (10) | 550 | 35 |
| 6 | Piperazine (10) | 700 | 25 |
| 7 | InCl3 (10) | 750 | 20 |
| 8 | PTSA (10) | 550 | 42 |
| 9 | H2SO4 (10) | 500 | 45 |
| 10 | Al2O3 (10) | 600 | 22 |
| 11 | FeCl3 (10) | 450 | 30 |
| 12 | PSA (10) | 200 | 68 |
| 13 | MSA (10) | 220 | 60 |
| 14 | TSA (10) | 225 | 63 |
| 15 | DBA IL (10) | 150 | 75 |
| 16 | [TMGTf] (10) | 180 | 72 |
| 17 | [TMGT] (10) | 150 | 81 |
| 18 | [TMGT] (10) | 120 | 80 |
| 19 | TMG IL (10) | 170 | 81 |
| 20c | DEACSA IL (10) | 120 | 90, 88, 87, 85 |
| 21 | DEACSA IL (2) | 190 | 60 |
| 22 | DEACSA IL (5) | 140 | 75 |
| 23 | DEACSA IL (20) | 120 | 91 |
To optimize the product yield, the same reaction was conducted using various basic catalysts such as Et3N, dibutylamine, TMG and piperazine and obtained very low product yields (Table 1, entries 3–6). Although Lewis acid catalysts such as InCl3, PTSA, H2SO4, Al2O3, and FeCl3 promoted the reaction to certain extent but the yields of the compounds are not satisfactory (Table 1, entries 7–11). Sulfonic acid-supported catalysts such as PSA, MSA, and TSA (Table 1, entries 12–14) promoted the reaction effectively and produced the desired product (4a) in moderate yields. But sulfonic acid-supported secondary amine based ILs such as [DBA IL], [TMGTf], [TMGT], [TMG IL] and [DEACSA IL] exhibited moderate to good catalytic properties (Table 1, entry 15–20). Among all the screened catalysts DEACSA IL was found to be superior with respect to reaction time and product yield (Table 1, entry 20). These results clearly demonstrated that DEACSA IL is a more efficient catalyst for the synthesis of title products.
The quantity of the catalyst required for obtaining maximum product yield was studied by using 2 mol%, 5 mol%,10 mol% and 20 mol% of DEACSA IL and the obtained yields were 60, 75, 90 and 90%, respectively (Table 1, entries 20–23). This showed that 10 mol% of DEACSA IL was sufficient and further increases in the quantity of catalyst, did not show significant improvement in the reaction rate and product yields. It shows that the rate of the reaction and product yield is depending on the nature and quantity of the catalyst.
The influence of various organic solvents at different reaction temperatures on the model reaction with 10 mol% of DEACSA IL and without catalyst was investigated. In DMF, 1,4 dioxane, methanol, and chloro benzene rate of the reaction was slow and lower product yields were obtained (Table 2, entries 1–5). The same reaction in ethanol improved both the reaction rate as well as product yield (Table 2, entry 6). But highest product yield was observed in solvent-free conditions at 60 °C (Table 2, entry 7) and this could be attributed to a uniform distribution of the eutectic mixture of reactants in it.
|
||||
|---|---|---|---|---|
| Entry | Solvent | Temp (°C) | Yieldb (%) | |
| Catalyst-free | DEACSA IL (10 mol%) | |||
| a Reaction conditions: naphthalene-2-thiol (1, 1 mmol), 2-bromobenzaldehyde (2a, 1 mmol) and 5,5-dimethylcyclohexane-1,3-dione (3, 1 mmol) at 120 min.b Isolated yield. | ||||
| 1 | DMF | 120 | 25.3 | 48.3 |
| 2 | 1,4-Dioxane | 140 | 35.6 | 45.3 |
| 3 | Methanol | 100 | 39.2 | 55.4 |
| 4 | Methanol | 60 | 30.4 | 45.1 |
| 5 | Chloro benzene | 120 | 20.2 | 52.2 |
| 6 | Ethanol | 70 | 45.5 | 65.3 |
| 7 | Solvent-free | 60 | 25.3 | 92.0 |
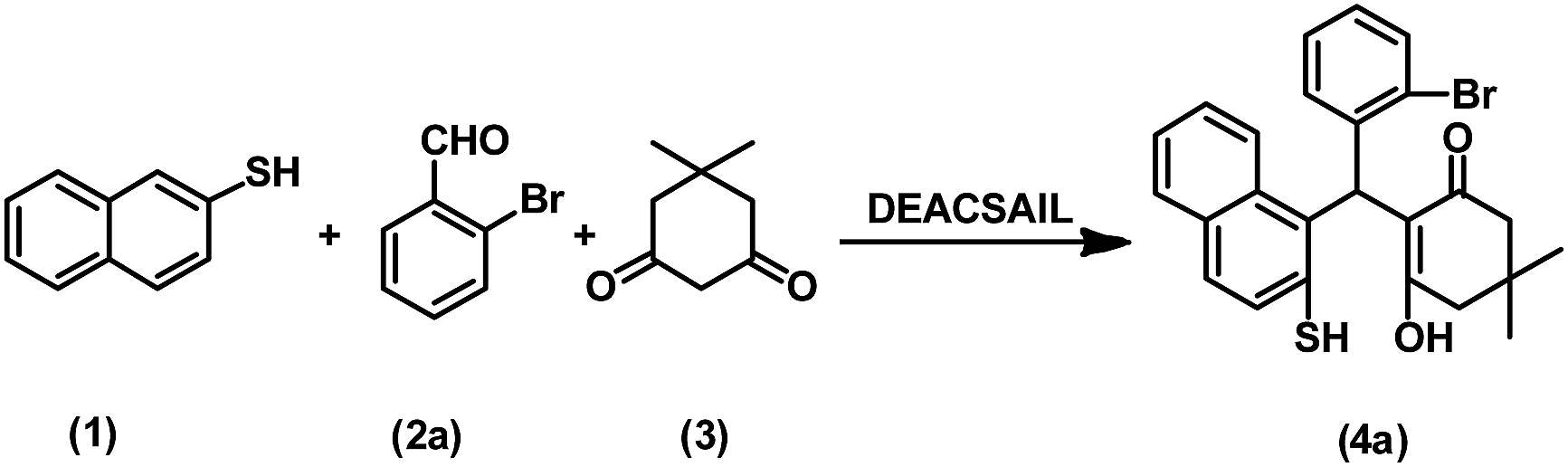
In order to investigate the catalytic activity and the possibility of the IL recyclability and reusability, the DEACSA IL was recovered by the addition of ethyl acetate to the reaction mixture; the insoluble DEACSA IL was separated by filtration, washed twice with ethyl acetate (30 mL), and dried in vacuum at 50 °C and reused for subsequent experiments under similar reaction conditions (Table 1, entry 20). The results showed that the catalyst could be effectively reused for at least four consecutive cycles without much appreciable loss in its catalytic activity. The recyclability data demonstrate the high stability of the catalyst under the reaction conditions (Fig. 8).
 | ||
| Fig. 8 Reusability of the DEACSA IL catalyst. | ||
The effect of temperature on the reaction in various solvents and without solvent was also tested (Table 2, entries 1–6). Irrespective of the nature of the solvent and even in high boiling solvent the yield was poor. But under solvent-free conditions higher product yield (80%) was observed at 60 °C (Table 2, entry 7). Salvation of the substrates with solvents which reduced their effective interaction might be the primary cause for the drop in the product yield.
Having optimized the reaction conditions for the synthesis of 2-mercaptonaphthalen-1-yl)methyl)-3-hydroxy-5,5-dimethylcyclohex-2-enones (4a–t) the procedure is extended to various aldehydes (2b–t) to explore its generality (Table 3). A wide range of aldehydes bearing electron donating and electron-withdrawing groups were reacted as coupling partners with naphthalene-2-thiol (1) with dimedone (3) and they were smoothly transformed to the corresponding 2-mercaptonaphthalen-1-yl)methyl)-3-hydroxy-5,5-dimethylcyclohex-2-enones (4b–t) in good to moderate yields. All the substituted aldehydes were easily converted into the desired products indicating that steric factors did not significantly affect the reactivity. Even though, the electron donor groups on aldehydes resulted higher product yields and the electron withdrawing groups on aldehydes shown little bit lesser product yields. Overall all type of aldehydes are afforded good to excellent product yields. In this procedure pure products were isolated by simple filtration of the reaction mixture without chromatography or a cumbersome work-up procedure. After the reaction, the catalyst was separated from the product and reused without significant decrease in its catalytic activity. The structures of the novel compounds were determined by 1H NMR, 13C NMR and HRMS.
|
|||||
|---|---|---|---|---|---|
| S. No. | Product | Aldehydes (R) | Time (h) | Yieldb (%) | mp (°C) |
| a Reaction conditions: naphthalene-2-thiol (1, 1 mmol), aldehydes (2a–t, 1 mmol) and 5,5-dimethylcyclohexane-1,3-dione (3, 1 mmol) in the presence of DEACSA IL under solvent-free condition.b Isolated yield. | |||||
| 1 | 4a | 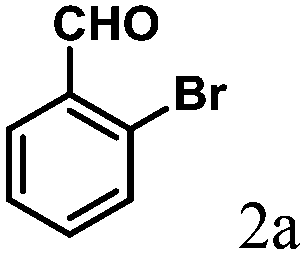 |
2.0 | 90 | 180–182 |
| 2 | 4b | 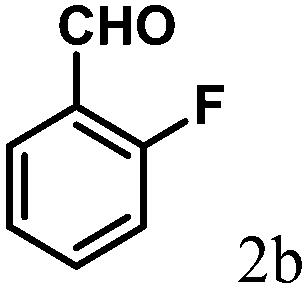 |
2.1 | 89 | 157–159 |
| 3 | 4c |  |
2.0 | 90 | 116–118 |
| 4 | 4d |  |
1.8 | 91 | 128–130 |
| 5 | 4e |  |
2.2 | 88 | 145–147 |
| 6 | 4f |  |
2.4 | 90 | 151–153 |
| 7 | 4g | 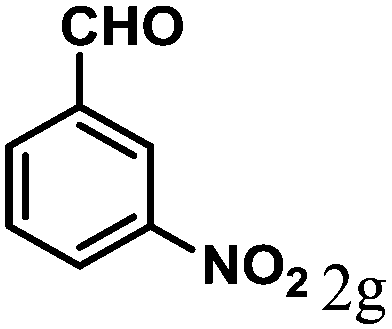 |
2.1 | 89 | 167–169 |
| 8 | 4h | 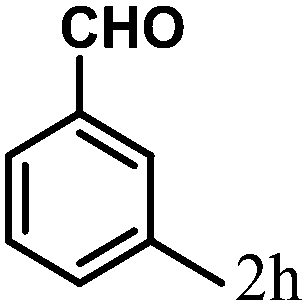 |
2.1 | 94 | 138–140 |
| 9 | 4i | 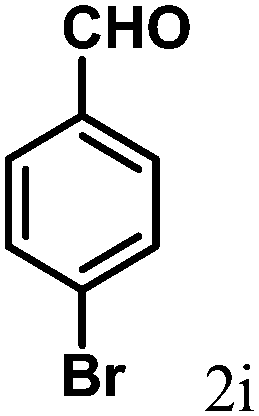 |
2.0 | 92 | 189–181 |
| 10 | 4j | 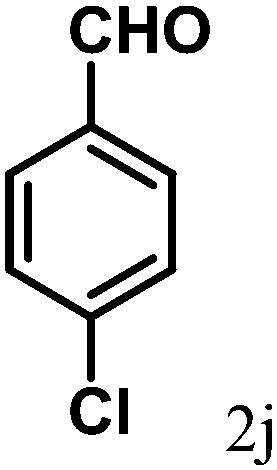 |
1.9 | 91 | 168–170 |
| 11 | 4k | 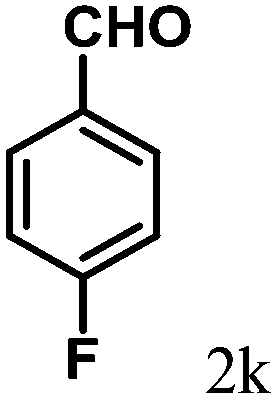 |
2.0 | 92 | 139–141 |
| 12 | 4l | 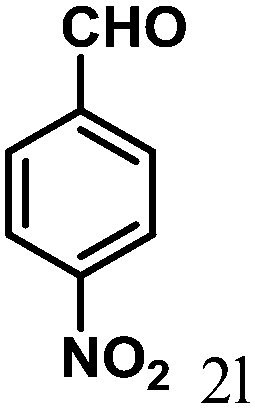 |
2.2 | 90 | 163–165 |
| 13 | 4m | 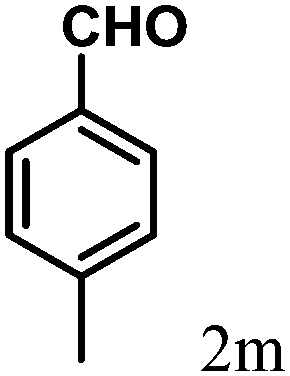 |
1.9 | 93 | 185–187 |
| 14 | 4n | 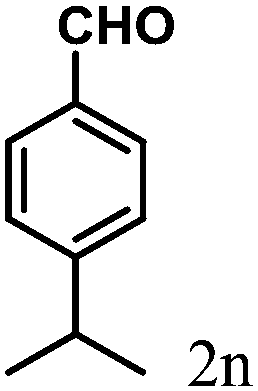 |
2.1 | 91 | 138–140 |
| 15 | 4o |  |
2.5 | 92 | 146–148 |
| 16 | 4p |  |
1.9 | 91 | 142–144 |
| 17 | 4q |  |
2.4 | 91 | 159–161 |
| 18 | 4r |  |
1.8 | 94 | 183–185 |
| 19 | 4s |  |
1.8 | 90 | 168–170 |
| 20 | 4t | 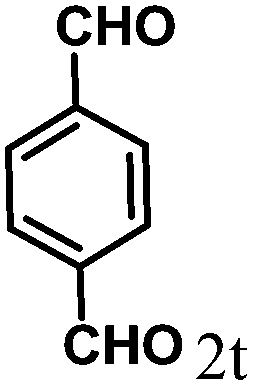 |
2.0 | 90 | 213–215 |
The probable sequence of events of mechanistic justification portraying is shown in Scheme 4. The multi-component reaction should be proceeding via Knoevenagel condensation between enolic form of dimedone (3) and aldehyde (2a–t) in the presence of DEACSA IL to form the intermediate, 5a–t.In subsequent addition of naphthalene-2-thiol (1) to the intermediate (5a–t) forms the addition product (6a–t). Which is undergo enolising as 4′and followed by rotation of α-sigma bond due to 2-alkyl ketone effect leads to 4′′. With this effect the possibility of cyclisation is less because of the existence of keto group is not closer to the thiol function and afforded the final product 4a–t.
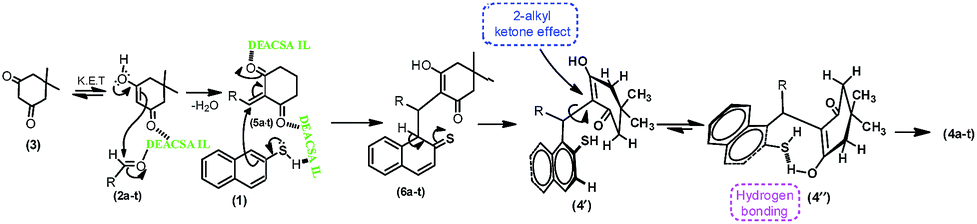 | ||
| Scheme 4 Mechanistic presentation of synthesis of 2-mercaptonaphthalen-1-yl)methyl)-3-hydroxy-5,5-dimethylcyclohex-2-enones (4a–t). | ||
Conclusion
We have reported a green and efficient method for the synthesis of a series of 2-mercaptonaphthalen-1-yl)methyl)-3-hydroxy-5,5-dimethylcyclohex-2-enones (4a–t) in excellent yields via one-pot multi-component reaction by using a catalytic amount of DEACSA IL under solvent-free condition. The environmental acceptability, economic viability, less reaction time, high product yields, and easy purification of products, cleaner reaction profile, higher atom efficiency and reusability of DEACSA IL qualify this method as the best one for the synthesis of these compounds.Experimental
Synthesis of DEACSA IL
The solution of chlorosulfonic acid (2 mmol) in CH2Cl2 (10 mL) was added to a solution of DEA (2 mmol) in CH2Cl2 (15 mL) in a 50 mL reaction flask over a period of 5 min at 0 °C (Scheme 5). After completion of the addition, the reaction mixture was stirred for 40 min at room temperature and then concentrated it by rotary evaporation. The obtained residue was washed with CH2Cl2 (30 mL) and dried at 70 °C by rotary evaporation to afford DEACSA IL as a colorless semi solid without using any further specific purification procedures.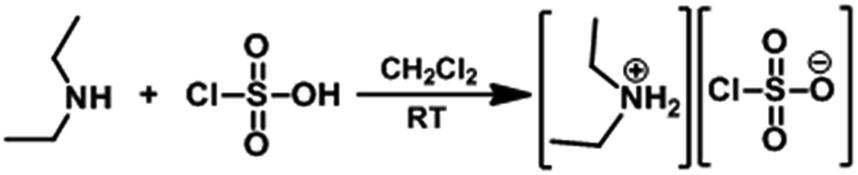 | ||
| Scheme 5 Preparation of DEACSA IL. | ||
DEACSA IL![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :IR (KBr): ν (cm−1) 3317, 3302, 3036, 2955, 1616, 1492, 1455, 1251, 1054,1033, 830, 758, 727, 600; 1H NMR (400 MHz, DMSO-d6): ν (ppm) 8.23 (s, 2H, –NH2), 3.20–3.16 (q, 2H), 2.88–2.84 (q, 2H), 1.16 (t, J = 7.3 Hz, 3H), 1.12 (t, J = 6.9 Hz, 3H); 13C NMR (100 MHz, DMSO-d6): δ (ppm) 44.7, 42.1, 11.3, 10.2. 1H NMR (400 MHz, 400 MHz, D2O): δ (ppm) 3.18–3.14 (q, 2H), 2.79–2.75 (q, 2H), 1.07 (t, J = 7.3 Hz, 3H), 0.96 (t, J = 6.9 Hz, 3H); 13C NMR (100 MHz, 400 MHz, D2O): δ (ppm) 46.6, 43.2, 11.5, 10.5. Anal. cacld for C4H12ClNO3S:C, 25.332; H, 6.377; N, 7.385; S, 16.928. Found: C, 25.001; H, 6.707; N, 8.01: S, 16.22. Decomposition point (by thermogravimetric analysis (TGA)) 230.5 °C.
:IR (KBr): ν (cm−1) 3317, 3302, 3036, 2955, 1616, 1492, 1455, 1251, 1054,1033, 830, 758, 727, 600; 1H NMR (400 MHz, DMSO-d6): ν (ppm) 8.23 (s, 2H, –NH2), 3.20–3.16 (q, 2H), 2.88–2.84 (q, 2H), 1.16 (t, J = 7.3 Hz, 3H), 1.12 (t, J = 6.9 Hz, 3H); 13C NMR (100 MHz, DMSO-d6): δ (ppm) 44.7, 42.1, 11.3, 10.2. 1H NMR (400 MHz, 400 MHz, D2O): δ (ppm) 3.18–3.14 (q, 2H), 2.79–2.75 (q, 2H), 1.07 (t, J = 7.3 Hz, 3H), 0.96 (t, J = 6.9 Hz, 3H); 13C NMR (100 MHz, 400 MHz, D2O): δ (ppm) 46.6, 43.2, 11.5, 10.5. Anal. cacld for C4H12ClNO3S:C, 25.332; H, 6.377; N, 7.385; S, 16.928. Found: C, 25.001; H, 6.707; N, 8.01: S, 16.22. Decomposition point (by thermogravimetric analysis (TGA)) 230.5 °C.
References
- (a) A. H. Ingall, in Comprehensive Heterocyclic Chemistry II, ed. A. S. Boulton and A. McKkillop, Pergamon Press, Oxford, 1996, vol. 5, p. 501 Search PubMed; (b) S. W. Schneller, Adv. Heterocycl. Chem., 1975, 18, 59–97 CrossRef CAS; (c) A. R. Katritzky and A. J. Bonlton, Adv. Heterocycl. Chem., 1975, 18, 76 CrossRef; (d) C. Jacob, G. I. Giles, N. M. Giles and H. Sies, Angew. Chem., Int. Ed., 2003, 42, 4742–4758 CrossRef CAS PubMed; (e) N. M. Giles, A. B. Watts, G. I. Giles, F. H. Fry, J. A. Littlechild and C. Jacob, Chem. Biol., 2003, 10, 677–693 CrossRef CAS.
- (a) W. Quaglia, M. Pigini, A. Piergentili, M. Giannella, F. Gentili, G. Marucci, A. Carrieri, A. Carotti, E. Poggesi, A. Leonardi and C. Melchiorre, J. Med. Chem., 2002, 45, 1633–1643 CrossRef CAS PubMed; (b) E. N. Chauvel, C. Llorens-Cartes, P. Coric, S. Wilk, B. P. Roques and M. C. Fournie-Zaluski, J. Med. Chem., 1994, 37, 2950–2957 CrossRef CAS; (c) G. I. Giles and C. Jacob, Biol. Chem., 2002, 383, 375–388 CrossRef CAS PubMed; (d) P. Vicini, A. Geronikaki, K. Anastasia, M. Incerti and F. Zani, Bioorg. Med. Chem., 2006, 14, 3859–3864 CrossRef CAS PubMed; (e) S. Bae, H. G. Hahn, K. D. Nam and H. Mah, J. Comb. Chem., 2005, 7, 7–9 CrossRef CAS PubMed.
- (a) M. T. Heafield, S. Fearn, G. B. Stevenson, R. H. Waring, A. C. Williams and S. G. Sturman, Neurosci. Lett., 1990, 110, 216–220 CrossRef CAS; (b) H. Refsum, P. M. Ueland, O. Nygard and S. E. Vollset, Annu. Rev. Med., 1998, 49, 31–62 CrossRef CAS PubMed; (c) D. W. Jacobsen, Clin. Chem., 1998, 44, 1833–1843 CAS; (d) S. Seshadri, A. Beiser, J. Selhub, P. Jacques, I. Rosenberg, R. D'Agostino and P. Wilson, N. Engl. J. Med., 2002, 346, 476–483 CrossRef CAS PubMed.
- (a) P. Vink, NL 6411477, 1965, Chem. Abstr., 1965, 63, 13265; (b) H. I. El-Subbagh, A. A. El-Emam, M. B. El-Ashmawy and I. A. Shehata, Arch. Pharmacal Res., 1990, 13, 24–27 CrossRef CAS; (c) M. J. Brown, P. S. Carter, A. E. Fenwick, A. P. Fosberry, D. W. Hamprecht, M. J. Hibbs, R. L. Jarvest, L. Mensah, P. H. Milner, P. J. O'Hanlon, A. J. Pope, C. M. Richardson, A. West and D. R. Witty, Bioorg. Med. Chem. Lett., 2002, 12, 3171–3174 CrossRef CAS; (d) D. J. Rogier Jr, J. S. Carter and J. J. Talley, WO 2001049675, 2001; (e) K. D. Berlin, D. M. Benbrook and E. C. Nelson, US pat. 6586460, 2003; (f) Y. Sugita, H. Hosoya, K. Terasawa, I. Yokoe, S. Fujisawa and H. Sakagami, Anticancer Res., 2001, 21, 2629–2632 CAS; (g) J. J. Hollick, B. T. Golding, I. R. Hardcastle, N. Martin, C. Richardson, L. J. Rigoreau, G. C. Smith and R. J. Griffin, Bioorg. Med. Chem. Lett., 2003, 13, 3083–3086 CrossRef CAS.
- (a) K. Erfurt, I. Wandzik, K. Walczak, K. Matuszek and A. Chrobok, Green Chem., 2014, 16, 3508–3514 RSC; (b) V. Fuente, N. Fleury-Brégeot, S. Castillón and C. Claver, Green Chem., 2012, 14, 2715–2718 RSC; (c) D. Arup Kumar, P. Gogoia and R. Borah, RSC Adv., 2014, 4, 41287–41291 RSC; (d) Q. Yan, H. Zang, C. Wu, J. Feng, M. Li, M. Zhang, L. Wang and B. Cheng, J. Mol. Liq., 2015, 204, 156–161 CrossRef CAS PubMed; (e) M. Veeranarayana Reddy, S. D. Dindulkar and Y. T. Jeong, Tetrahedron Lett., 2011, 52, 4764–4767 CrossRef PubMed; (f) A. S. Amarasekara and M. A. Hasan, Tetrahedron Lett., 2014, 55, 3319–3321 CrossRef CAS PubMed; (g) S. Koguchi and K. Nakamura, Synlett, 2013, 24, 2305–2309 CrossRef CAS PubMed; (h) Y. Zhang, A. Zhu, Q. Li, L. Li, Y. Zhao and J. Wang, RSC Adv., 2014, 4, 22946–22950 RSC.
- (a) H. Li, P. S. Bhadury, B. Song and S. Yang, RSC Adv., 2012, 2, 12525–12551 RSC; (b) M. Bagherzadeh and S. G. Esfahani, Tetrahedron Lett., 2013, 54, 3765–3768 CrossRef CAS PubMed; (c) K. P. Boroujeni and P. Ghasemi, Catal. Commun., 2013, 37, 50–54 CrossRef PubMed; (d) P. H. Li, L. Bao-Le, H. Hai-Chuan, X. N. Zhao and Z. H. Zhang, Catal. Commun., 2014, 46, 118–122 CrossRef CAS PubMed; (e) J. Isaad, RSC Adv., 2014, 4, 49333–49341 RSC; (f) A. Khalafi-Nezhad and M. Somayeh, RSC Adv., 2014, 4, 13782–13787 RSC; (g) H. Zhou, J. Song, X. Kang, J. Hu, Y. Yang, H. Fan, Q. Menga and B. Han, RSC Adv., 2015, 5, 15267–15273 RSC; (h) K. Ali Reza, Z. Dalirnasab and M. Karimi, Synthesis, 2014, 46, 917–922 CrossRef PubMed.
- (a) K. Rajkumar and R. Srivastava, Catal. Commun., 2011, 12, 1420–1424 CrossRef PubMed; (b) G. Wang, Z. Zhangb and L. Song, Green Chem., 2014, 16, 1436–1443 RSC; (c) A. M. Hengne, S. B. Kamble and C. V. Rode, Green Chem., 2013, 15, 2540–2547 RSC; (d) G. A. Kraus and T. Guney, Green Chem., 2012, 14, 1593–1596 RSC; (e) Q. Peng, K. Mahmood, Y. Wu, L. Wang, Y. Liang, J. Shen and Z. Liu, Green Chem., 2014, 16, 2234–2241 RSC; (f) M. Veeranarayana Reddy, K. Reddi Mohan Naidu, L. S. Dong and Y. T. Jeong, Catal. Commun., 2015, 61, 102–106 CrossRef PubMed; (g) L. Yang, L. Wen Xu and C. Gu Xia, Synthesis, 2009, 12, 1969–1974 Search PubMed; (h) M. A. Naik, D. Sachdev and A. Dubey, Catal. Commun., 2010, 11, 1148–1153 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Analytical and spectral data and NMR spectra were provided as supplementary data for all compounds. See DOI: 10.1039/c5ra04277a |
| This journal is © The Royal Society of Chemistry 2015 |