
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Boosting interlayer charge transfer in polymeric carbon nitride by Mo ions for efficient photocatalytic H2 evolution†
Feng
Liu‡
,
Wencheng
Li‡
,
Xi
Rao
 *,
Shaohui
Zheng
* and
Yongping
Zhang
*
*,
Shaohui
Zheng
* and
Yongping
Zhang
*
School of Materials and Energy, Southwest University, Chongqing 400715, China. E-mail: raoxiemail@swu.edu.cn; shaohuizheng@swu.edu.cn; zhangyyping6@swu.edu.cn
First published on 22nd December 2023
Abstract
Tuning the band structure by doping metal elements is an effective way to boost charge transfer and thus improve the photocatalytic activity of polymeric carbon nitride (CN). Herein, Mo-doped carbon nitride (Mo–CN) was prepared by calcining melamine, cyanuric acid and sodium molybdate at elevated temperature. Under visible light at λ ≥ 420 nm, the optimal hydrogen production rate of 3Mo–CN reaches 16.7 μmol h−1, about eight times that of pristine CN. Experimental results demonstrate that Mo doping reduces the band gap, increases the specific surface area, extends the visible light harvesting range, and enhances the separation and transfer of photogenerated carriers. Theoretical simulation verifies that the intercalated Mo acts as a bridging channel for interlayer charge transfer, increases the intramolecular electron transition distance (Dct) and charge transfer quantity, and enhances the localization of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO). All these factors are interwoven to contribute to the enhanced photocatalytic performance of polymeric carbon nitride.
1. Introduction
With the increase of energy demand and environmental deterioration, the search for renewable and clean energy has aroused widespread concern around the world. The conversion of solar energy into chemical energy is a very important and promising clean energy technology. As one of the ideal “solar energy fuels”, hydrogen energy has aroused considerable attention due to its advantages of high calorie capacity and environmentally friendly green energy. Semiconductor photocatalysts were applied to achieve sustainable energy production under the premise of fully utilizing solar energy to generate hydrogen or value-added chemicals, providing a potential solution to energy and environment issues.1–3 Polymeric carbon nitride (g-C3N4), as a two-dimensional layered n-type semiconductor photocatalyst, has the advantages of low preparation cost, non-toxicity, suitable band gap and excellent physical and chemical stability. However, g-C3N4 has a relatively narrow response range to visible light, and easy recombination of photogenerated electrons and holes, which limits its catalytic performance.4–8 Several strategies have been employed to improve the photocatalytic efficiency, including heterostructure construction with other semiconductors,9–11 nanostructure formation,12–14 metal doping15–17 and non-metallic doping.18,19 Among all of those modification techniques, intercalation of metal ions in the triangle vacancies can not only effectively inhibit the recombination of photogenerated electron/hole pairs, but also increase the surface reaction sites of g-C3N4, since the triangle vacancy is an ideal site to anchor the metal ion by forming coordinated bonds with N atoms with lone electrons.20–22 For example, Na doping reduced the band gap and effectively improved the separation and transfer of photoinduced carriers of g-C3N4.23 Mn introduced into g-C3N4 reduced the band gap of g-C3N4 and improved the absorption of visible light.24 Indium doped in the g-C3N4 catalyst improved the separation efficiency and migration ability of the photogenerated carrier.25Molybdenum (Mo) doped in the g-C3N4 molecular layer enhanced an uneven distribution of charge density, facilitating the separation of photoexcited charge carriers.26–30 Mo single atoms and Mo-based compound catalysts were widely investigated due to its unique electrical, optical and catalytic properties.31–35 For example, Che et al. prepared Mo-doped g-C3N4 nanosheets by in situ thermal condensation polymerization, with hydrogen evolution efficiency nine times higher than that of bulk phase g-C3N4.36 Guo et al. introduced Mo into g-C3N4 by a hydrothermal method, which improved the hydrogen production performance by six times compared to that of pristine g-C3N4.37 Wang et al. successfully realized Mo doping of g-C3N4 by a simple pyrolysis method, and increased the reduction rate of CO2 by seven times under visible light irradiation.38 Although the above works demonstrated that Mo doping played a positive role in improving the performance of g-C3N4, there still existed three key problems to be solved: firstly, relatively little research has been conducted on the optimal reaction conditions of the catalytic reaction system; secondly, the coordination mode of transition metal atoms in carbon nitride molecules remains undecided; thirdly, how metal doping changes the energy band structure and the active site of photocatalysis is unknown. We proposed a pathway to synthesize Mo-doped g-C3N4 by directly calcining melamine, cyanuric acid and sodium molybdate at elevated temperature. Experimental and theoretical studies on the doping behavior of Mo in g-C3N4 can provide a new understanding of the effect of transition metal ions on the photocatalytic activity of g-C3N4, which is of great significance for exploring new efficient catalysts.
Herein, Mo-doped g-C3N4 was prepared by calcining melamine, cyanuric acid and sodium molybdate at elevated temperature. Experimental results showed that Mo-doped g-C3N4 exhibits excellent hydrogen evolution performance under visible light irradiation. Density functional theory revealed that the intercalated Mo acts as a bridging channel for interlayer charge transfer, and a built-in electric field, thus improving the absorption range of visible light and the separation efficiency of photogenerated electron/hole pairs.
2. Experimental details
2.1 Catalyst preparation
1 g melamine, 5 g cyanuric acid and a certain amount (0.02 g, 0.03 g, 0.04 g) of sodium molybdate (Na2MoO4) were fully ground with a mortar and pestle. Then the powders were transferred to a ceramic boat, and calcined in a tubular furnace under a N2 environment at 550 °C for 4 hours with a heating rate of 5 °C min−1. The prepared photocatalysts were marked as 2Mo–CN, 3Mo–CN, and 4Mo–CN respectively, according to the amount of sodium molybdate. Porous g-C3N4 was prepared by calcining 1 g melamine and 5 g cyanuric acid under the same conditions, denoted as PCN. Pristine g-C3N4 was obtained by calcining 5 g melamine under the same conditions, recorded as CN.2.2 Catalyst characterization
X-ray photoelectron spectroscopy (XPS, Thermo Scientific K-alpha) was carried out to characterize the chemical states of the catalysts. Nitrogen adsorption–desorption isotherms were obtained by a Quadrasorb evo 2QDS-MP-30 surface area analyzer. The absorption property was measured by a UV-vis diffuse reflectance spectrometer (Agilent Cary 5000 UV-vis NIR), using BaSO4 as the reference. The photoluminescence (PL) spectra were recorded on a Hitachi F-7000 spectrophotometer with 150 W xenon light as the excitation source. The transient photocurrent response curve (I–t), and electrochemical impedance spectroscopy (EIS) were performed using an AUTOLAB (model PGSTAT302 N) electrochemical workstation. 0.25 M Na2SO4 solution was used as the electrolyte and a 500 W xenon lamp as the light source.2.3 Photocatalytic reaction
Photocatalytic hydrogen evolution was carried out in a reactor cell connected to a sealed gas circulation and evaluation system (Suncat Instrument, Beijing). Typically, 10 mg of the photocatalyst was dispersed in 30 ml aqueous solution containing the sacrificial agent 17 vol% triethanolamine (TEOA) and co-catalyst 3 wt% Pt ion (H2PtCl6·H2O), which was continuously stirred during the reaction process. The reactor was evacuated to a high vacuum with pressure of 10−8 torr, and then filled with argon, preceding the photocatalytic reaction. Then, the solution was irradiated by a 500 W Xenon lamp (zolix, gloria-x500a) with a 420 nm cut off filter. The amount of H2 production was analyzed by an online gas chromatograph (GC-2018, TCD detector, Shimadzu) using Ar as a carrier.3. Results and discussion
SEM was used to observe the morphology and microstructure of the catalyst, as shown in Fig. 1. Pristine g-C3N4 appeared as a thick blocky stacked structure (Fig. 1a), while PCN (Fig. 1b) and 3Mo–CN (Fig. 1c) appeared as a two-dimensional lamellar structure, and the sample surface was dispersed by an uneven microporous structure, which may be caused by the continuous release of gases generated during the calcination process, such as NH3 and CO2,39,40 which will greatly increase the specific surface area. The relative contents of nitrogen, carbon and molybdenum in 3Mo–CN determined by EDS are 61.5 wt%, 37.8 wt% and 0.7 wt%, respectively, as presented in Fig. 1(d). EDS electron image and element mapping in Fig. 1(e) show that the g-C3N4 doped with Mo is composed of rich carbon and nitrogen elements and dispersed Mo elements. Mo element is dispersed in the background of C and N elements, which confirms that the Mo element is successfully introduced into g-C3N4. The TEM images demonstrated that PCN appeared as thick layers, while 3Mo–CN appeared as thin layers with curved profiles, as shown in Fig. 1(f and g). | ||
| Fig. 1 SEM images of CN (a), PCN (b), and 3Mo–CN (c). EDS spectrum (d) and electron image and corresponding EDS element mapping (e) of 3Mo–CN. TEM images of PCN (f) and 3Mo–CN (g). | ||
The XRD patterns in Fig. 2(a) showed two different diffraction peaks at 13.0° and 27.1°. The diffraction peak at 13.0° belongs to the g-C3N4 (100) crystal plane, representing the in-plane structural arrangement of triazine units. The diffraction peak at 27.1° belongs to the g-C3N4 (002) crystal plane, representing the interlayer accumulation of the aromatic system.41,42 The intensity of the diffraction peak corresponding to the (002) crystal plane decreases with Mo doping, indicating that Mo doping destroys the stable stacking between layers and reduces the crystallinity. The FT-IR spectra in Fig. 2(b) demonstrated that the peak at 810 cm−1 corresponds to the bending vibration of the triazine structure, the peak at 1232 cm−1, 1313 cm−1, 1399 cm−1, 1455 cm−1, 1539 cm−1, and 1633 cm−1 corresponds to the tensile vibration of the C–N skeleton structure of conjugated aromatic ring, and the wide peak at 3000–3500 cm−1 corresponds to the stretching vibration of N–H caused by incomplete condensation of amino groups.43 For different Mo doping amounts, the structure of g-C3N4 remains unchanged, which is consistent with the XRD results.
 | ||
| Fig. 2 Nitrogen adsorption–desorption isotherms (a), and pore size distribution (b) of the prepared photocatalysts. | ||
The specific surface area and pore size distribution of all samples were measured by N2 absorption desorption. As shown in Fig. 2(c), the samples all showed Type IV isotherms with H3 hysteresis loops, which confirmed that the materials were mesoporous materials. The specific surface area of pristine CN is 19.1 m2 g−1, and the specific surface area of PCN is 36.7 m2 g−1. The specific surface areas of 4Mo–CN, 3Mo–CN and 2Mo–CN are 54.8 m2 g−1, 129.3 m2 g−1 and 105.9 m2 g−1, respectively. Mo doping resulted in a larger specific surface area of CN, providing more active reaction sites for catalytic reaction. As shown in Fig. 2(d), the mesopore size is mainly distributed between 1–5 nm, and more pore size distribution on the material surface contributes to increasing the specific surface area and accelerating the photocatalytic reaction.40 The pore volumes of 4Mo–CN, 3Mo–CN, 2Mo–CN and PCN are 0.43 cm3 g−1, 0.99 cm3 g−1, 0.52 cm3 g−1 and 0.18 cm3 g−1, respectively, which are higher than 0.11 cm3 g−1 of pristine CN. This is consistent with SEM observation.
X-ray photoelectron spectroscopy (XPS) was carried out to further analyze the composition and chemical states of CN, PCN and 3Mo–CN. The survey spectra in Fig. 3(a) showed C 1s, N 1s, and O 1s signals for the as-prepared samples. A new peak at 232 eV can be allocated to Mo 3d for 3Mo–CN. The high-resolution C 1s spectra in Fig. 3(b) demonstrated that the C 1s spectra can be fitted into three peaks with the binding energy at 284.4 eV, 285.9 eV and 287.8 eV. The peak at 287.8 eV is ascribed to the sp2 hybridized carbon in the aromatic C–N skeleton structure (N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), the peak at 285.9 eV is assigned to the C atom at the edge of the g-C3N4 plane (C–NH2), and the peak at 284.4 eV is assigned to the surface adventitious C atom (C–C, CC).41,42 High-resolution N 1s spectra in Fig. 3(c) can be fitted into three peaks at 398.3 eV, 399.7 eV and 400.8 eV, being ascribed to the sp2 bonded N atom in the aromatic ring (C–NC), the sp3 hybridized N atom (N–[C]3) and the N atom (C–NHx) in the amino group at the end of the aromatic ring plane.43,44 Compared with CN, the ratio of N atom in C–NHx increased to 13.6% from 9.3% of CN, indicating that the polymerization degree of PCN is decreased. The N 1s peak shift to low binding energy may be caused by breaking some C–N bonds during the calcination process of melamine and cyanuric acid. Mo doping induces N 1s peak shifts towards low binding energy, which may be caused by the Coulomb interaction of coordination bonds between Mo atoms and the lone electrons on the N atoms at the edge of the aromatic ring. The Mo 3d spectrum in Fig. 4(d) can be fitted to 228.6 eV and 231.5 eV corresponding to 3d5/2 and 3d3/2 of Mo(VI), while the peaks at 231.6 eV and 234.5 eV correspond to 3d5/2 and 3d3/2 of Mo(IV).45–47 The structure of g-C3N4 contains a large number of vacancies composed of six nitrogen atoms with a lone electron, providing a suitable site for the anchoring of Mo6+. A small amount of Mo exists in the form of Mo4+. The reason for its existence may be that g-C3N4 releases a large amount of NH3 during the calcination process, reducing the small amount of Mo6+ exposed on the surface to Mo4+.31 The results support that the Mo atoms mainly exist in the form of Mo6+ and form a coordination bond with the N atoms at the edge of the aromatic ring when intercalated into the hollow triangular holes of the g-C3N4 aromatic ring plane. The atomic ratio of 3Mo–CN is 44.08%, 55.74%, 0.11%, and 0.07% for C, N, Mo6+, and Mo4+, respectively. The Mo atomic ratio is 0.18%, corresponding to (C + N)/Mo = 300
N), the peak at 285.9 eV is assigned to the C atom at the edge of the g-C3N4 plane (C–NH2), and the peak at 284.4 eV is assigned to the surface adventitious C atom (C–C, CC).41,42 High-resolution N 1s spectra in Fig. 3(c) can be fitted into three peaks at 398.3 eV, 399.7 eV and 400.8 eV, being ascribed to the sp2 bonded N atom in the aromatic ring (C–NC), the sp3 hybridized N atom (N–[C]3) and the N atom (C–NHx) in the amino group at the end of the aromatic ring plane.43,44 Compared with CN, the ratio of N atom in C–NHx increased to 13.6% from 9.3% of CN, indicating that the polymerization degree of PCN is decreased. The N 1s peak shift to low binding energy may be caused by breaking some C–N bonds during the calcination process of melamine and cyanuric acid. Mo doping induces N 1s peak shifts towards low binding energy, which may be caused by the Coulomb interaction of coordination bonds between Mo atoms and the lone electrons on the N atoms at the edge of the aromatic ring. The Mo 3d spectrum in Fig. 4(d) can be fitted to 228.6 eV and 231.5 eV corresponding to 3d5/2 and 3d3/2 of Mo(VI), while the peaks at 231.6 eV and 234.5 eV correspond to 3d5/2 and 3d3/2 of Mo(IV).45–47 The structure of g-C3N4 contains a large number of vacancies composed of six nitrogen atoms with a lone electron, providing a suitable site for the anchoring of Mo6+. A small amount of Mo exists in the form of Mo4+. The reason for its existence may be that g-C3N4 releases a large amount of NH3 during the calcination process, reducing the small amount of Mo6+ exposed on the surface to Mo4+.31 The results support that the Mo atoms mainly exist in the form of Mo6+ and form a coordination bond with the N atoms at the edge of the aromatic ring when intercalated into the hollow triangular holes of the g-C3N4 aromatic ring plane. The atomic ratio of 3Mo–CN is 44.08%, 55.74%, 0.11%, and 0.07% for C, N, Mo6+, and Mo4+, respectively. The Mo atomic ratio is 0.18%, corresponding to (C + N)/Mo = 300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Since the atomic number of (C + N) is 16 in the unit cell of g-C3N4, the saturated doping content should be (C + N)/Mo = 16:1. It was estimated that there is likely only one Mo ion in every 19 unit cells for 3Mo–CN. The unsaturated doping leads to the formation of an interplanar heterojunction between the Mo-doped and undoped domains. The doping ratio deviating from this optimal value may affect the transfer and separation of the photoinduced carriers for 2Mo–CN and 4Mo–CN.
:1. Since the atomic number of (C + N) is 16 in the unit cell of g-C3N4, the saturated doping content should be (C + N)/Mo = 16:1. It was estimated that there is likely only one Mo ion in every 19 unit cells for 3Mo–CN. The unsaturated doping leads to the formation of an interplanar heterojunction between the Mo-doped and undoped domains. The doping ratio deviating from this optimal value may affect the transfer and separation of the photoinduced carriers for 2Mo–CN and 4Mo–CN.
 | ||
| Fig. 3 XPS survey scan (a), and high resolution C 1s (b), N 1s (c) and Mo 3d (d) spectra of CN, PCN and 3Mo–CN. | ||
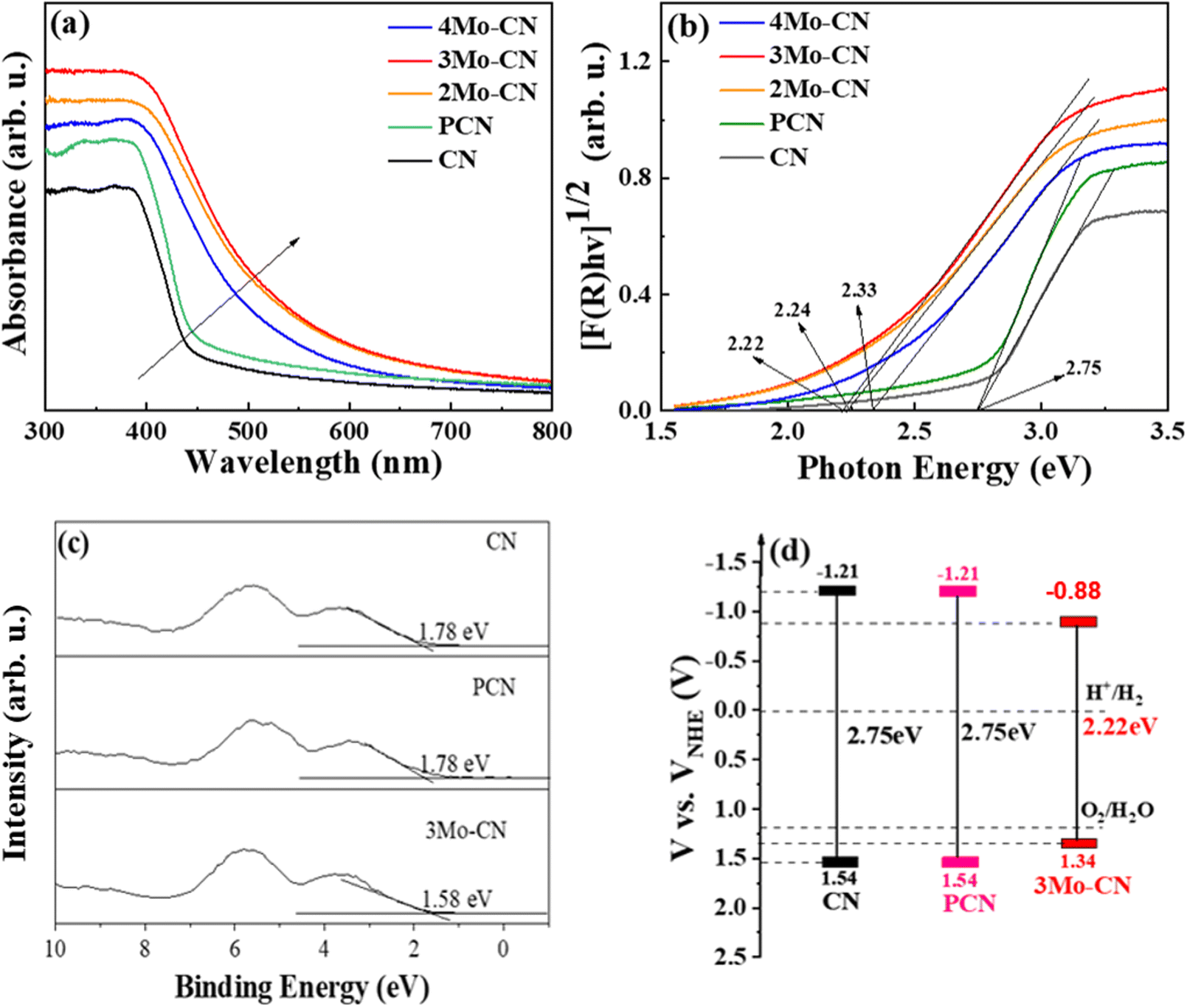 | ||
| Fig. 4 UV-vis spectra (a), and the estimated band gaps (b) of CN, PCN, and Mo-doped CN. XPS valence band spectra (c), and schematic illustration of the band gap structure (d) of CN and 3Mo–CN. | ||
UV visible diffuse reflectance spectra were carried out to investigate the light absorption property of these samples, as presented in Fig. 4(a). Compared with the pristine g-C3N4, Mo doped g-C3N4 exhibits enhanced visible light absorption, and the absorption edges of Mo doped g-C3N4 undergo obvious red shifts. UV-vis spectra demonstrated that Mo doping significantly enhanced the visible light absorption of g-C3N4, and 3Mo–CN shows the strongest visible light absorption among all these samples. The stronger visible light absorption favors better visible light catalytic activity. The band gap of the CN, PCN, 2Mo–CN, 3Mo–CN, and 4Mo–CN is 2.75 eV, 2.75 eV, 2.24 eV, 2.22 eV, and 2.33 eV, respectively, estimated from the intercept of the tangents to the plots of [F(R)hv]1/2 vs photon energy, as indicated in the Kubelka–Munk function curve in Fig. 4(b). The narrowed band gap facilitates the absorption of visible light, and the light absorption enhancement plays an extremely important role in improving the hydrogen production performance.48
VB-XPS spectra in Fig. 4(c) showed that the VB edges (EVB) of pristine CN, PCN and 3Mo–CN are estimated to be 1.78 eV, 1.78 eV and 1.58 eV, respectively, versus the Fermi level. The potential of standard hydrogen electrode (NHE) can be calculated according to the following formula: NHE = φ + EVB − 4.44,49 where φ is the work function of the analyzer (4.20 eV), and thus the NHE potentials of the pristine CN, PCN and 3Mo–CN are 1.54 eV, 1.54 eV and 1.34 eV, respectively. Combined with the band gap values of CN, PCN and 3Mo–CN being 2.75 eV, 2.75 eV and 2.22 eV, respectively, and the VB position, the energy band structure of CN, PCN and 3Mo–CN is schematically illustrated in Fig. 4(d).50 Compared with CN, 3Mo–CN has the CD position still more negative than the H+/H2 potential, and the VB position more positive than O2/H2O potential.
The transient photocurrent response curve in Fig. 5(a) revealed that all catalysts underwent a stable light response under visible light irradiation, with no obvious decrease after 5 cycles. Moreover, under the same bias voltage, the photocurrent intensity of the Mo-doped samples is higher than that of pristine CN, and the photocurrent of the 3Mo–CN samples is about four times that of pristine CN. The results show that doping Mo facilitates photoelectron generation. The photocurrent density bias voltage curve of the sample under visible light is shown in Fig. 5(b). Under continuous bias voltage, all samples can continuously and stably respond, indicating that the samples have good stability, and the photocurrent response of 3Mo–CN is the highest. The electrochemical impedance spectrum of the sample is shown in Fig. 5(c). The EIS arc radius of the 3Mo–CN samples is significantly smaller than that of other samples, indicating that the interface charge transfer resistance of Mo-doped g-C3N4 is smaller, which can effectively promote the transport and separation of photogenerated carriers in the catalytic reaction. The photoluminescence spectra in Fig. 5(d) depicted that the photoluminescence intensity of the samples doped with Mo atoms decreased significantly, indicating that Mo doping significantly reduced the recombination of photogenerated electron hole pairs of g-C3N4 and promoted the photocatalytic reaction. In general, the stronger the fluorescence peak of a semiconductor photocatalyst, the greater the recombination rate of photogenerated electron hole pairs.48 In addition, due to the influence of Mo atom doping, the PL spectrum of the samples had a certain red shift.
 | ||
| Fig. 5 Transient photocurrent response (a), photocurrent density–bias voltage curves (b), EIS Nyquist plots (c), and PL spectra (d) of the prepared samples. | ||
The photocatalytic performance of CN and Mo intercalated CN samples was tested toward the photocatalytic hydrogen evolution in a xenon lamp simulating sunlight (λ > 420 nm). 10 mg of photocatalyst was dispersed in 30 ml of aqueous solution containing 3 wt% Pt ion (H2PtCl6·6H2O) and 17 vol% TEOA. Fig. 6(a) indicates that the hydrogen evolution rate of Mo intercalated g-C3N4 is higher than that of pristine g-C3N4, and the 3Mo–CN has the highest hydrogen evolution rate of 16.7 μmol h−1, about 7.9 times as much as the pristine g-C3N4. The H2 evolution rate is higher than the reported data listed in Table S1 (ESI†). As shown in Fig. 6(b), the hydrogen evolution stability of the 3Mo–CN photocatalyst was tested. The results showed that after five times of photocatalytic hydrogen evolution cycle tests, the photocatalytic activity of the sample did not decrease significantly, indicating that the photocatalytic performance of the sample was stable and it could be reused. The XRD patterns of 3Mo–CN before and after the reaction did not vary obviously, indicating that the 3Mo–CN catalyst is stable during the irradiation process, as represented in Fig. 6(c).
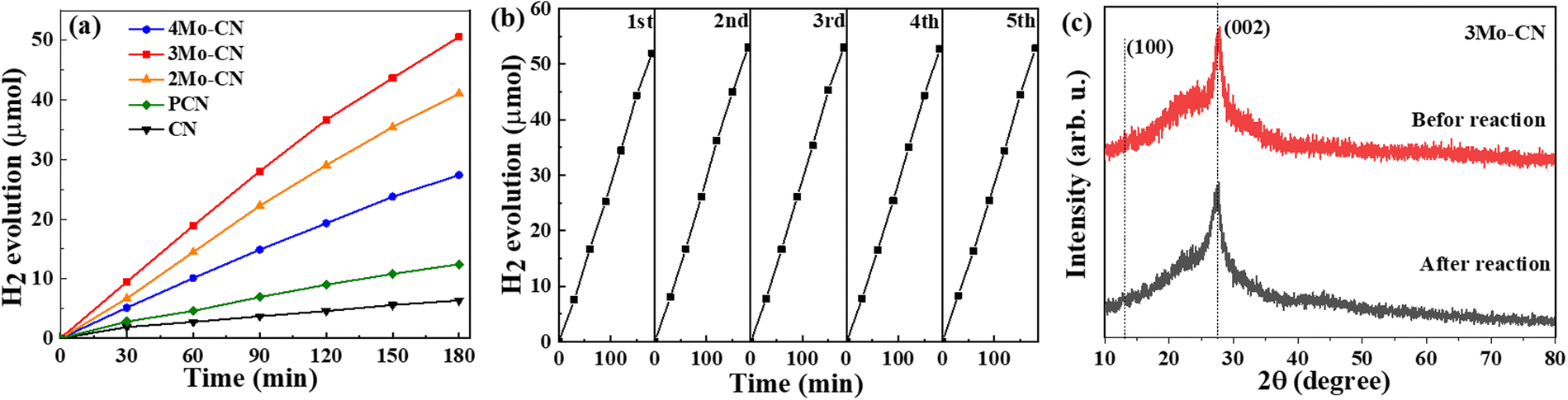 | ||
| Fig. 6 Photocatalytic hydrogen evolution (a) of CN, PCN, and modoped CN. Stability test of 3Mo–CN (b). XRD patterns of 3Mo–CN before and after light irradiation (c). | ||
Based on density functional theory (DFT), Gaussian 09 and Revision E.01 are used to simulate CN and BCN. B3LYP/6-31G(d) is used to calculate the structure optimization, density of states (DOS), molecular orbital (HOMO/LUMO) and electronic transition. To generate the density of states and spin density, we used the Multiwfn Rev 3.7 software package.51 The optimal structure of CN and Mo intercalated CN was illustrated in the bottom part of Fig. 7. The MPP (molecular planarity parameter) and SDP (span of deviation from plane) parameters are used to quantitatively measure the flatness. The smaller the value, the stronger the flatness and the closer the structure is to the plane. The MPP reflects the overall flatness of the area under consideration, while the SDP measures the maximum deviation from the fitting plane in the area under consideration, that is, the maximum span perpendicular to the fitting plane. The simulation results show that the flatness of g-C3N4 becomes worse due to the doping of Mo atoms, which is consistent with the results of BET, and increases the specific surface area of g-C3N4, thus greatly improving the photocatalytic performance.
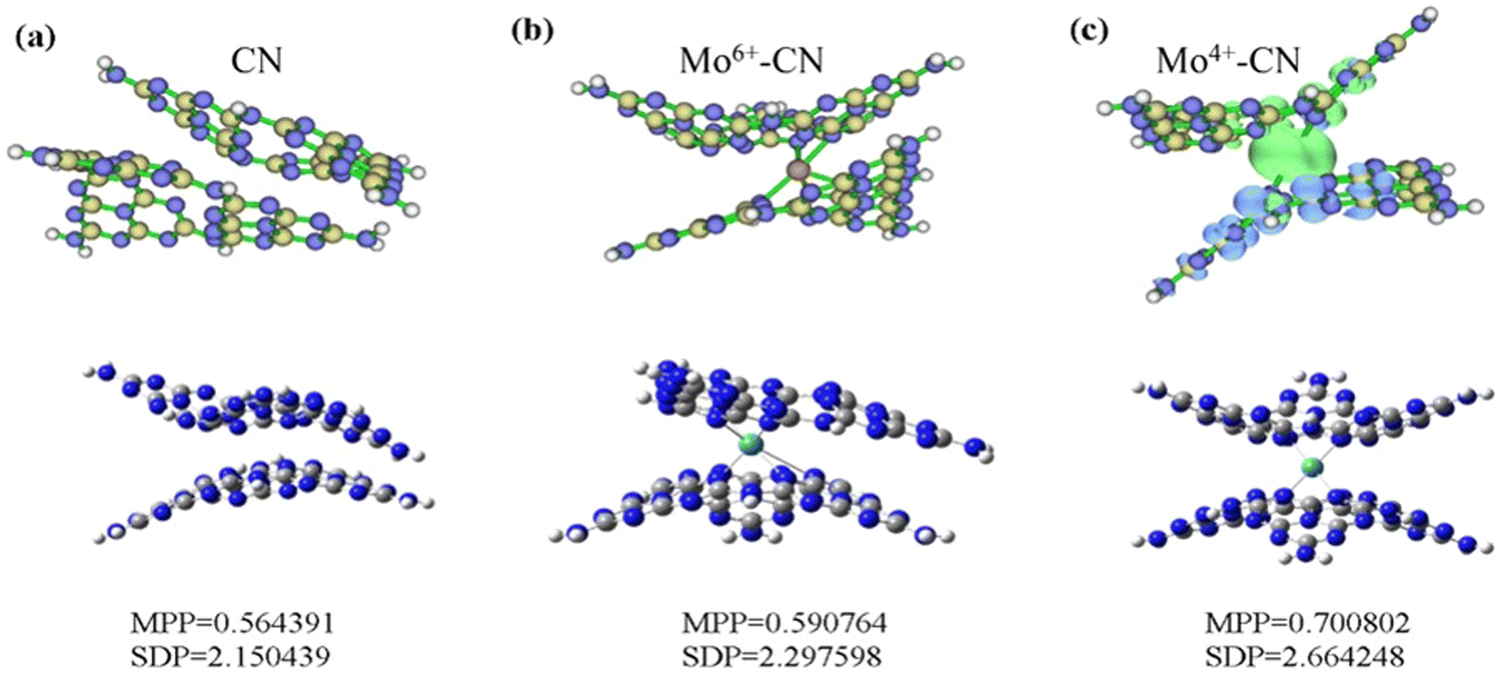 | ||
| Fig. 7 The optimized structures (a) and spin density (b) of pristine CN, and Mo-doped CN. | ||
The spin density of CN and Mo-doped CN was depicted in the upper part of Fig. 7. The spin density of Mo4+-doped g-C3N4 (with two unpaired electrons and a spin multiplicity of 3) is mainly located around Mo (green represents positive value) and adjacent g-C3N4 layers (blue represents negative value). Both pristine phase and Mo6+-doped g-C3N4 are closed shell systems. Since the alpha and beta electrons are the same everywhere and perfectly matched, there is no spin density.
The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) are calculated to theoretically study the photocatalytic active sites of g-C3N4, as shown in Fig. 8(a). For pristine g-C3N4, holes (HOMO) and electrons (LUMO) are generated after being excited by light, and N and C atoms are located at the edge of the aromatic ring, respectively. The HOMO provides an oxidation site for oxygen atoms in water, and the LUMO provides a reduction site for hydrogen ions. It can be speculated that after g-C3N4 is excited by light, the electrons on the 2p orbital of the N atom at the edge of the aromatic ring will transition to the 2p orbital of the C atom at the edge of the ring and the N atom at the center of the ring. At this time, the photogenerated electron Pt ions on the C atom at the edge of the aromatic ring and the N atom at the center of the ring can directly reduce H+ in water to H2. However, the HOMO and LUMO of the pristine phase g-C3N4 show an alternating distribution, leading to the easy recombination of the photogenerated electron hole pairs, and the short lifetime reduces the photocatalytic efficiency. It is worth noting that doping Mo atoms intensifies the separation of the HOMO and LUMO. Mo atoms act as reduction sites, and photoinduced electrons easily transition from the valence band to the conduction band, which improves the mobility and lifetime of g-C3N4 photogenerated carriers, thus enhancing the photocatalytic reaction. The corresponding band gap is now at the bottom of the figure. The band gaps of pristine CN, Mo6+ and Mo4+-doped CN are 3.78 eV, 0.66 eV, and 0.99 eV, respectively. The reduction of the band gap can promote the absorption of absorbable light by the photocatalyst, thus promoting the photocatalytic reaction.
 | ||
| Fig. 8 HOMO, LUMO, and band gap of CN (a), Mo6+–CN (b) and Mo4+–CN (c). The iso-surface value is set at 0.03 e bohr−3. Denotation: C atom – gray, N atom – dark blue. | ||
As shown in Fig. 9, the intercalated Mo ions act as a charge transfer bridge of the g-C3N4 interlayer, while there exists no interlayer charge transfer for CN. The intramolecular electron transition distance (Dct) and charge transfer quantity of Mo6+ and Mo4+ doped g-C3N4 are larger than those of pristine g-C3N4, indicating the easier charge separation of Mo-doped g-C3N4. Meanwhile, doping Mo6+ will cause a larger dipole moment (1.5257D → 4.5980D) of the molecule, thereby increasing the push–pull effect of the intramolecular electrons, which facilitates the transfer and separation of photogenerated carriers and accelerates the photocatalytic reaction.
 | ||
| Fig. 9 First excited state of the electron/hole distribution of CN (a), Mo6+–CN (b), and Mo4+–CN (c). The intramolecular electron transition distance (Dct) and the intramolecular charge transfer amount (qct) are given. | ||
Taking the above results into consideration, we could gain a deeper understanding of the mechanism for the efficient photocatalytic hydrogen evolution of Mo intercalated CN. Firstly, introducing Mo in the g-C3N4 results in strong light harvesting in the visible light range by narrowing the band gap, as confirmed by the UV-vis absorption spectra and DFT calculation. Secondly, the efficient photocatalytic performance can be accredited to the increased specific surface area and pore volume by Mo-doped CN, which provided more active sites for the photocatalytic reaction. Thirdly, Mo doping resulted in a higher photocurrent density, and lower impedance and PL intensity, which indicates the efficient transfer and separation of photoinduced charge carriers. DFT calculation confirmed that Mo doping leads to electron delocalization and molecular bending.
4. Conclusion
In summary, a facile method was employed to prepare the Mo-doped g-C3N4 catalyst by calcining melamine, cyanuric acid and sodium molybdate at elevated temperature. The specific surface area of 3Mo–CN reaches 129.3 m2 g−1, 6.8 times that of pristine CN. Meanwhile, Mo doping reduced the band gap, increased the visible light harvesting, and promoted the effective separation and migration of carriers. These factors synergistically contributed to improving the photocatalytic performance. The optimized photocatalytic hydrogen evolution rate of 3Mo–CN reached 16.7 μmol h−1, 7.9 times that of pristine g-C3N4. DFT simulation supported that Mo intercalated in g-C3N4 facilitated the interlayer charge transfer and narrowed the band gap. This study provided new insight into the understanding of Mo ions on the photocatalytic activity.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (52271250), the Natural Science Foundation of Chongqing, China (cstc2021jcyj-msxmX0964).References
- X. Wang, G. Sun, P. Routh, D. H. Kim, W. Huang and P. Chen, Heteroatom-doped graphene materials: syntheses, properties and applications, Chem. Soc. Rev., 2014, 43, 7067–7098 RSC.
- F. Wang, Q. Li and D. Xu, Recent Progress in Semiconductor-Based Nanocomposite Photocatalysts for Solar-to-Chemical Energy Conversion, Adv. Energy Mater., 2017, 7, 1700529 CrossRef.
- N. Liu, S. Zhou and J. Zhao, Visible-light overall water splitting on g-C3N4 decorated by subnanometer oxide clusters, Mater. Today Phys., 2021, 16, 100312 CrossRef CAS.
- B. Chai, T. Peng, J. Mao, K. Li and L. Zan, Graphitic carbon nitride (g-C3N4)-Pt-TiO2 nanocomposite as an efficient photocatalyst for hydrogen production under visible light irradiation, Phys. Chem. Chem. Phys., 2012, 14, 16745–16752 RSC.
- P. Chen, P. Xing, Z. Chen, H. Lin and Y. He, Rapid and energy-efficient preparation of boron doped g-C3N4 with excellent performance in photocatalytic H2-evolution, Int. J. Hydrogen Energy, 2018, 43, 19984–19989 CrossRef CAS.
- J. Ding, L. Song, X. Li, L. Chen, X. Li, J. Sun, X. Zhang, Y. Wang and X. Tian, Interfacial Engineering of the Platinum/Molybdenum Disulfide/graphitic Carbon Nitride Composite for Enhanced Photocatalytic Hydrogen Production, ACS Appl. Energy Mater., 2022, 5, 8800–8811 CrossRef CAS.
- J. Ran, J. Zhang, J. Yu, M. Jaroniec and S. Z. Qiao, Earth-abundant cocatalysts for semiconductor-based photocatalytic water splitting, Chem. Soc. Rev., 2014, 43, 7787–7812 RSC.
- Z. Yu, F. Li, Q. Yang, H. Shi, Q. Chen and M. Xu, Nature-Mimic Method To Fabricate Polydopamine/Graphitic Carbon Nitride for Enhancing Photocatalytic Degradation Performance, ACS Sustainable Chem. Eng., 2017, 5, 7840–7850 CrossRef CAS.
- F. Liu, T.-T. Xu and Z.-Y. Jiang, Insights into the photocatalytic mechanism of S-scheme g-C3N4/BiOBr heterojunction, Inorganic, Chem. Commun., 2022, 143, 109735 Search PubMed.
- J. Li, H. Yuan and Z. Zhu, Improved photoelectrochemical performance of Z-scheme g-C3N4/Bi2O3/BiPO4 heterostructure and degradation property, Appl. Surf. Sci., 2016, 385, 34–41 CrossRef CAS.
- Y. Yang, C. Lu, J. Ren, X. Li, Y. Ma, W. Huang and X. Zhao, Enhanced photocatalytic hydrogen evolution over TiO2/g-C3N4 2D heterojunction coupled with plasmon Ag nanoparticles, Ceram. Int., 2020, 46, 5725–5732 CrossRef CAS.
- X. Wu, C. Liu, X. Li, X. Zhang, C. Wang and Y. Liu, Effect of morphology on the photocatalytic activity of g-C3N4 photocatalysts under visible-light irradiation, Mater. Sci. Semicond. Process., 2015, 32, 76–81 CrossRef CAS.
- Y. Zhang, H. Li, L. Zhang, R. Gao and W.-L. Dai, Construction of Highly Efficient 3D/2D MnO2/g-C3N4 Nanocomposite in the Epoxidation of Styrene with TBHP, ACS Sustainable Chem. Eng., 2019, 7, 17008–17019 CrossRef CAS.
- P. H. Linh, P. Do Chung, N. Van Khien, L. T. M. Oanh, V. T. Thu, T. N. Bach, L. T. Hang, N. M. Hung and V. D. Lam, A simple approach for controlling the morphology of g-C3N4 nanosheets with enhanced photocatalytic properties, Diamond Relat. Mater., 2021, 111, 108214 CrossRef CAS.
- X. Dong, S. Zhang, H. Wu, Z. Kang and L. Wang, Facile one-pot synthesis of Mg-doped g-C3N4 for photocatalytic reduction of CO2, RSC Adv., 2019, 9, 28894–28901 RSC.
- M. Molaei, S. M. Mousavi-Khoshdel and M. Ghiasi, Exploring the effect of phosphorus doping on the utility of g-C3N4 as an electrode material in Na-ion batteries using DFT method, J. Mol. Model., 2019, 25, 256 CrossRef PubMed.
- Y. Wang, Y. Li, X. Bai, Q. Cai, C. Liu, Y. Zuo, S. Kang and L. Cui, Facile synthesis of Y-doped graphitic carbon nitride with enhanced photocatalytic performance, Catal. Commun., 2016, 84, 179–182 CrossRef CAS.
- K. Ding, L. Wen, M. Huang, Y. Zhang, Y. Lu and Z. Chen, How does the B,F-monodoping and B/F-codoping affect the photocatalytic water-splitting performance of g-C3N4?, Phys. Chem. Chem. Phys., 2016, 18, 19217–19226 RSC.
- H. S. H. Mohamed, L. Wu, C. F. Li, Z. Y. Hu, J. Liu, Z. Deng, L. H. Chen, Y. Li and B. L. Su, In-Situ Growing Mesoporous CuO/O-Doped g-C3N4 Nanospheres for Highly Enhanced Lithium Storage, ACS Appl. Mater. Interfaces, 2019, 11, 32957–32968 CrossRef CAS PubMed.
- D. Long, W. Chen, S. Zheng, X. Rao and Y. Zhang, Barium- and Phosphorus-Codoped g-C3N4 Microtubes with Efficient Photocatalytic H2 Evolution under Visible Light Irradiation, Ind. Eng. Chem. Res., 2020, 59, 4549–4556 CrossRef CAS.
- W. Yan, L. Yan and C. Jing, Impact of doped metals on urea-derived g-C3N4 for photocatalytic degradation of antibiotics: Structure, photoactivity and degradation mechanisms, Appl. Catal., B, 2019, 244, 475–485 CrossRef CAS.
- I. Neelakanta Reddy, A. Sreedhar, V. Manjunath, J. Shim and J.-S. Noh, Water splitting kinetics of Sr-doped g-C3N4 edge-wrinkled nanosheets under visible light, Mater. Sci. Semicond. Process., 2021, 132, 105918 CrossRef CAS.
- X. Liu, X. Wu, D. Long, X. Rao and Y. Zhang, Template-free synthesis of tetragonal graphitic carbon nitride microtubes doped by sodium chloride for enhanced photocatalytic H2 performance under visible light irradiation, J. Photochem. Photobiol., A, 2020, 391, 112337 CrossRef CAS.
- S. Wang, C. Wan, F. Chen, M. Chong and D.-G. Cheng, Regulating the bandgap of graphitic carbon nitride via Mn doping for boosting visible-light-driven water reduction, J. Phys. D: Appl. Phys., 2022, 55, 284002 CrossRef.
- X. Yang, Z. Guo, X. Zhang, Y. Han, Z. Xue, T. Xie and W. Yang, The effect of indium doping on the hydrogen evolution performance of g-C3N4 based photocatalysts, New J. Chem., 2021, 45, 544–550 RSC.
- J. Liao, B. Sa, J. Zhou, R. Ahuja and Z. Sun, Design of High-Efficiency Visible-Light Photocatalysts for Water Splitting: MoS2/AlN(GaN) Heterostructures, J. Phys. Chem. C, 2014, 118, 17594–17599 CrossRef CAS.
- Y. Lu, X. Yao, J. Yin, G. Peng, P. Cui and X. Xu, MoS2 nanoflowers consisting of nanosheets with a controllable interlayer distance as high-performance lithium ion battery anodes, RSC Adv., 2015, 5, 7938–7943 RSC.
- Q. Xiang, J. Yu and M. Jaroniec, Synergetic effect of MoS2 and graphene as cocatalysts for enhanced photocatalytic H2 production activity of TiO2 nanoparticles, J. Am. Chem. Soc., 2012, 134, 6575–6578 CrossRef CAS PubMed.
- M.-Q. Yang, C. Han and Y.-J. Xu, Insight into the Effect of Highly Dispersed MoS2 versus Layer-Structured MoS2 on the Photocorrosion and Photoactivity of CdS in Graphene–CdS–MoS2 Composites, J. Phys. Chem. C, 2015, 119, 27234–27246 CrossRef CAS.
- Y.-X. Zeng, X.-W. Zhong, Z.-Q. Liu, S. Chen and N. Li, Preparation and Enhancement of Thermal Conductivity of Heat Transfer Oil-Based MoS2 Nanofluids, J. Nanomater., 2013, 2013, 1–6 Search PubMed.
- J. Dong, Y. Shi, C. Huang, Q. Wu, T. Zeng and W. Yao, A New and stable Mo-Mo2C modified g-C3N4 photocatalyst for efficient visible light photocatalytic H2 production, Appl. Catal., B, 2019, 243, 27–35 CrossRef CAS.
- X.-W. Guo, S.-M. Chen, H.-J. Wang, Z.-M. Zhang, H. Lin, L. Song and T.-B. Lu, Single-atom molybdenum immobilized on photoactive carbon nitride as efficient photocatalysts for ambient nitrogen fixation in pure water, J. Mater. Chem. A, 2019, 7, 19831–19837 RSC.
- J. Sun, S. Yang, Z. Liang, X. Liu, P. Qiu, H. Cui and J. Tian, Two-dimensional/one-dimensional molybdenum sulfide (MoS2) nanoflake/graphitic carbon nitride (g-C3N4) hollow nanotube photocatalyst for enhanced photocatalytic hydrogen production activity, J. Colloid Interface Sci., 2020, 567, 300–307 CrossRef CAS PubMed.
- Y. Wang, Y. Zhang, S. Zhao, Z. Huang, W. Chen, Y. Zhou, X. Lv and S. Yuan, Bio-template synthesis of Mo-doped polymer carbon nitride for photocatalytic hydrogen evolution, Appl. Catal., B, 2019, 248, 44–53 CrossRef CAS.
- H. Zhao, S. Sun, Y. Wu, P. Jiang, Y. Dong and Z. J. Xu, Ternary graphitic carbon nitride/red phosphorus/molybdenum disulfide heterostructure: An efficient and low cost photocatalyst for visible-light-driven H2 evolution from water, Carbon, 2017, 119, 56–61 CrossRef CAS.
- H. Che, C. K. Ngaw, P. Hu, J. Wang, Y. Li, X. Wang and W. Teng, Fabrication of molybdenum doped carbon nitride nanosheets for efficiently photocatalytic water splitting, J. Alloys Compd., 2020, 849, 156440 CrossRef CAS.
- C. Guo, K. Tian, L. Wang, F. Liang, F. Wang, D. Chen, J. Ning, Y. Zhong and Y. Hu, Approach of Fermi level and electron-trap level in cadmium sulfide nanorods via molybdenum doping with enhanced carrier separation for boosted photocatalytic hydrogen production, J. Colloid Interface Sci., 2021, 583, 661–671 CrossRef CAS PubMed.
- Y. Wang, Y. Xu, Y. Wang, H. Qin, X. Li, Y. Zuo, S. Kang and L. Cui, Synthesis of Mo-doped graphitic carbon nitride catalysts and their photocatalytic activity in the reduction of CO2 with H2O, Catal. Commun., 2016, 74, 75–79 CrossRef CAS.
- X. Li, K. Pan, Y. Qu and G. Wang, One-dimension carbon self-doping g-C3N4 nanotubes: Synthesis and application in dye-sensitized solar cells, Nano Res., 2018, 11, 1322–1330 CrossRef CAS.
- D. Long, L. Wang, H. Cai, X. Rao and Y. Zhang, Sulfur Doped Carbon-Rich g-C3N4 for Enhanced Photocatalytic H2 Evolution: Morphology and Crystallinity Effect, Catal. Lett., 2020, 150, 2487–2496 CrossRef CAS.
- D. Gao, Y. Liu, P. Liu, M. Si and D. Xue, Atomically Thin B doped g-C3N4 Nanosheets: High-Temperature Ferromagnetism and calculated Half-Metallicity, Sci. Rep., 2016, 6, 35768 CrossRef CAS PubMed.
- J. Li, B. Shen, Z. Hong, B. Lin, B. Gao and Y. Chen, A facile approach to synthesize novel oxygen-doped g-C3N4 with superior visible-light photoreactivity, Chem. Commun., 2012, 48, 12017–12019 RSC.
- L. Shi, L. Liang, F. Wang, M. Liu, S. Zhong and J. Sun, Tetraethylorthosilicate induced preparation of mesoporous graphitic carbon nitride with improved visible light photocatalytic activity, Catal. Commun., 2015, 59, 131–135 CrossRef CAS.
- J. Liao, W. Cui, J. Li, J. Sheng, H. Wang, X. A. Dong, P. Chen, G. Jiang, Z. Wang and F. Dong, Nitrogen defect structure and NO+ intermediate promoted photocatalytic NO removal on H2 treated g-C3N4, Chem. Eng. J., 2020, 379, 122282 CrossRef CAS.
- N. Iqbal, A. Afzal, I. Khan, M. S. Khan and A. Qurashi, Molybdenum impregnated g-C3N4 nanotubes as potentially active photocatalyst for renewable energy applications, Sci. Rep., 2021, 11, 16886 CrossRef CAS PubMed.
- B. Singh, M. B. Gawande, A. D. Kute, R. S. Varma, P. Fornasiero, P. McNeice, R. V. Jagadeesh, M. Beller and R. Zboril, Single-Atom (Iron-Based) Catalysts: Synthesis and Applications, Chem. Rev., 2021, 121, 13620–13697 CrossRef CAS PubMed.
- L. Zhao, J. Jia, Z. Yang, J. Yu, A. Wang, Y. Sang, W. Zhou and H. Liu, One-step synthesis of CdS nanoparticles/MoS2 nanosheets heterostructure on porous molybdenum sheet for enhanced photocatalytic H2 evolution, Appl. Catal., B, 2017, 210, 290–296 CrossRef CAS.
- M. Zhou, G. Dong, F. Yu and Y. Huang, The deep oxidation of NO was realized by Sr multi-site doped g-C3N4 via photocatalytic method, Appl. Catal., B, 2019, 256, 117825 CrossRef CAS.
- X. Li, B. Kang, F. Dong, Z. Zhang, X. Luo, L. Han, J. Huang, Z. Feng, Z. Chen, J. Xu, B. Peng and Z. L. Wang, Enhanced photocatalytic degradation and H2/H2O2 production performance of S-pCN/WO2.72 S-scheme heterojunction with appropriate surface oxygen vacancies, Nano Energy, 2021, 81, 105671 CrossRef CAS.
- D. Long, H. Dou, X. Rao, Z. Chen and Y. Zhang, Z-Scheme Ag3PO4/g-C3N4 Nanocomposites for Robust Cocatalyst-Free Photocatalytic H2 Evolution Under Visible Light Irradiation, Catal. Lett., 2019, 149, 1154–1166 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, et al., Gaussian 09, Revision E.01, 2013 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional information including XRD, and FTIR spectra, and calculated total, partial, and overlapped density of states of pristine, and Mo-doped g-C3N4. See DOI: https://doi.org/10.1039/d3ya00485f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |