
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent advancements in graphitic carbon nitride based direct Z- and S-scheme heterostructures for photocatalytic H2O2 production
Subrat Kumar
Sahoo
 ,
Lopamudra
Acharya
,
Lijarani
Biswal
,
Priyanka
Priyadarshini
and
Kulamani
Parida
*
,
Lopamudra
Acharya
,
Lijarani
Biswal
,
Priyanka
Priyadarshini
and
Kulamani
Parida
*
Centre for Nanoscience and Nanotechnology, Siksha ‘O’ Anusandhan (Deemed to be University), Bhubaneswar, Odisha 751030, India. E-mail: kulamaniparida@soa.ac.in; Fax: +91-674-235064; Tel: +91-674-2351777
First published on 20th June 2024
Abstract
Escalating global energy demands and the pressing need for sustainable and environmentally friendly energy sources have intensified research in the field of renewable energy, particularly solar energy. Hydrogen peroxide (H2O2), as a green and sustainable oxidant, is important for environmental remediation, chemical synthesis, and as a next-generation energy fuel. Solar energy harnessed in photocatalysis enables light-driven H2O2 production, offering an eco-friendly synthesis method. High-performance photocatalysts are essential for achieving viable solar H2O2 synthesis. Photocatalysis, particularly using g-C3N4, a visible-light-responsive metal-free semiconductor, presents a promising avenue for future large-scale H2O2 production. This is due to its unique properties, such as its oxygen-reduction-friendly conduction band, tuneable molecular structure, stability, cost-effectiveness, Earth abundance, facile synthesis, non-toxicity, numerous active sites, surface imperfections and high selectivity for H2O2 generation, making it a vital material in the renewable energy sector. However, challenges like rapid exciton recombination, limited light absorption capacity, suboptimal electrical conductivity, low specific surface area, and slow water oxidation kinetics need to be addressed to enhance its catalytic efficiency. Hence, the development of direct Z- or more relevant S-scheme heterostructures of g-C3N4 could promote the charge carrier separation efficiency, optimize the redox potential and improve the photocatalytic activity significantly. This review focuses on g-C3N4 as a photocatalyst, emphasizing its properties and the potential of direct Z- and S-scheme heterojunctions in photocatalytic H2O2 production. It introduces the background and surge in research on these heterojunctions, covers design principles, charge transfer mechanisms, advanced characterization methods, driving force enhancement strategies, Fermi level adjustment tactics, and principles of H2O2 production, including formation pathways, kinetics, detection, and performance evaluation. It offers insights into g-C3N4-based heterostructures’ potential for application in H2O2 production and concludes with future prospects and challenges, focusing on strategies to enhance yields and suggesting new research directions.
 Subrat Kumar Sahoo | Subrat Kumar Sahoo received his M.Sc. degree in Chemistry from Siksha ‘O’ Anusandhan (Deemed to be University), Bhubaneswar in 2019. Since November 2022, he has been working as a research scholar under the supervision of Prof. Kulamani Parida at the Centre for Nanoscience and Nanotechnology (CNSNT), Institute of Technical Education and Research (ITER), Siksha ‘O’ Anusandhan (Deemed to be University). His current research focuses on the development of carbon nitride based heterostructures for solar H2O2 synthesis. |
 Lopamudra Acharya | Dr Lopamudra Acharya completed her M.Sc. degree in Chemistry at Centurion University of Technology and Management, Jatni, Odisha in 2018. Then, she received her Ph.D. in chemistry in 2023 from Siksha ‘O’ Anusandhan (Deemed to be University), Bhubaneswar under the supervision of Prof. Kulamani Parida. Currently, she is working as a Post-Doctoral Fellow in the Centre for Nano Science and Nano Technology (CNSNT), Institute of Technical Education and Research (ITER), Siksha ‘O’ Anusandhan (Deemed to be University). Her current research interests focus on modified g-C3N4-based heterostructures for energy and environmental applications. |
 Lijarani Biswal | Dr Lijarani Biswal completed her M.Sc. degree in Chemistry at Siksha ‘O’ Anusandhan (Deemed to be University), Bhubaneswar in 2019. Then, she received her Ph.D. in chemistry in 2023 from Siksha ‘O’ Anusandhan (Deemed to be University), Bhubaneswar under the supervision of Prof. Kulamani Parida. Currently, she is working as a research associate in the Centre for Nano Science and Nano Technology (CNSNT), Institute of Technical Education and Research (ITER), Siksha ‘O’ Anusandhan (Deemed to be University). Her current research interests focus on MXene-based nanomaterials for energy and environmental applications. |
 Priyanka Priyadarshini | Priyanka Priyadarshini received her M.Sc. degree in Chemistry from Siksha ‘O’ Anusandhan (Deemed to be University), Bhubaneswar in 2022. In November 2022, she started working as a research scholar under the supervision of Prof. Kulamani Parida at the Centre for Nano Science and Nano Technology (CNSNT), Institute of Technical Education and Research (ITER), Siksha ‘O’ Anusandhan (Deemed to be University). Her current research interests are focused on the development of MOF-based functional nanomaterials towards energy generation and storage applications. |
 Kulamani Parida | Prof. Kulamani Parida is a globally distinguished scientist in the areas of materials science, catalysis and nanotechnology. He superannuated as a Chief Scientist and a Professor of Academy of Scientific and Innovative Research (AcSIR) from CSIR-IMMT, Bhubaneswar in 2014. His adoration for Chemistry and passion towards research would not let Prof. Parida be idle after retirement. With the same devotion and dedication, Prof. Parida (Director) started the ‘Centre for Nano Science and Nano Technology’, a research unit of Siksha ‘O’ Anusandhan (Deemed to be University), Bhubaneswar. His research interests include the design and development of advanced materials encompassing fine chemical, energy generation/storage and environmental applications. He has published more than 509 research articles and review articles in renowned international journals. He has 30 |
1. Introduction
The search for renewable energy sources has become a critical focus for society, as on-going reliance on non-renewable fossil fuels has led to significant environmental and energy challenges.1 Among the various renewable options such as wind, tidal, ocean currents, and hydroelectric energy, solar energy stands out for its almost limitless potential and environmental benefits.2,3 Sunlight provides an impressive 1.19 × 1017 J to the Earth per second, offering a clean and abundant source for energy production.4 Research into solar fuels, particularly since the pioneering work of Fujishima and Honda on water splitting, has gained widespread attention.5 These fuels provide a way to convert sunlight into other forms of energy or valuable chemicals, a concept inspired by nature's photosynthesis where plants and organisms turn CO2 into carbohydrates.6 This biological process has served as a model for green solar-to-chemical conversions, including ammonia synthesis, hydrogen (H2) evolution, and hydrocarbon production. However, despite their promise, the current state of solar fuels is not yet cost-competitive with fossil fuels.7 One widely considered approach to cost-effective solar fuel production is the generation of H2 through the water splitting reaction.8,9 Unfortunately, water-splitting systems face multiple challenges, including high production costs, complex synthesis conditions, and the low energy density of H2 generated.10 Therefore, H2O2 as a liquid is easier to store and transport than H2 gas, making it a promising fuel for rockets and new energy vehicles like jet cars.11–13 Additionally, H2O2 as a safe and efficient green oxidant has received immense research interest in sectors such as pulp and textile manufacturing for decolourization processes,14 as well as in chemical production,15 remediation of wastewater,16 metal mining,17 detergents,18 and fuel cell technologies,19 as illustrated in Fig. 2. The anticipated worth of H2O2 is projected to rise from $3.86 billion in the year 2023 to $4.09 billion in 2024; with an estimated yearly expansion rate of 6.16%, the market is expected to attain a valuation of $5.87 billion by 2030.20 Its high active oxygen content and safe byproducts, i.e. water and oxygen, make it ideal for environmental clean-up. Unlike traditional disinfectants, H2O2 does not produce harmful byproducts, a feature increasingly valued during times of high demand like the COVID-19 crisis.21 In environmental remediation, it sterilizes bacteria and breaks down organic pollutants through advanced oxidation processes (AOPs), generating hydroxyl radicals via Fenton-like reactions or UV exposure.22 Overall, its efficiency, safety, and versatility make H2O2 a go-to for both cleaning and potential energy storage. Presently, the anthraquinone (AQ) oxidation method dominates industrial H2O2 production and is responsible for the vast majority of the total output.23,24 However, this process has inherent limitations, such as high energy requirements, resource-intensive reactions, and complex operation, purification and transportation steps. Additionally, it generates a considerable amount of hazardous byproducts. Given these constraints, alternative methods like photocatalysis and electrocatalysis are attracting increasing research attention.25–30 Although electrocatalysis yields more H2O2, its high energy input restricts its broad application. On the other hand, semiconductor based photocatalysis operates under ambient conditions and offers a more sustainable approach by converting water (H2O) and oxygen (O2) into H2O2, using cost-effective and abundant raw materials, solely powered by renewable solar energy.31–33 Moreover, it offers the advantage of on-demand, on-site production, thereby mitigating the risks associated with the handling and distribution of concentrated H2O2. This makes photocatalysis an increasingly attractive route for decentralized and environmentally friendly H2O2 production. With its promise for a greener and more cost-effective H2O2 synthesis, the field of photocatalytic H2O2 production is experiencing rapid growth. The concept of generating H2O2 through photocatalysis was originally introduced by Baur and Neuweiler in 1927.34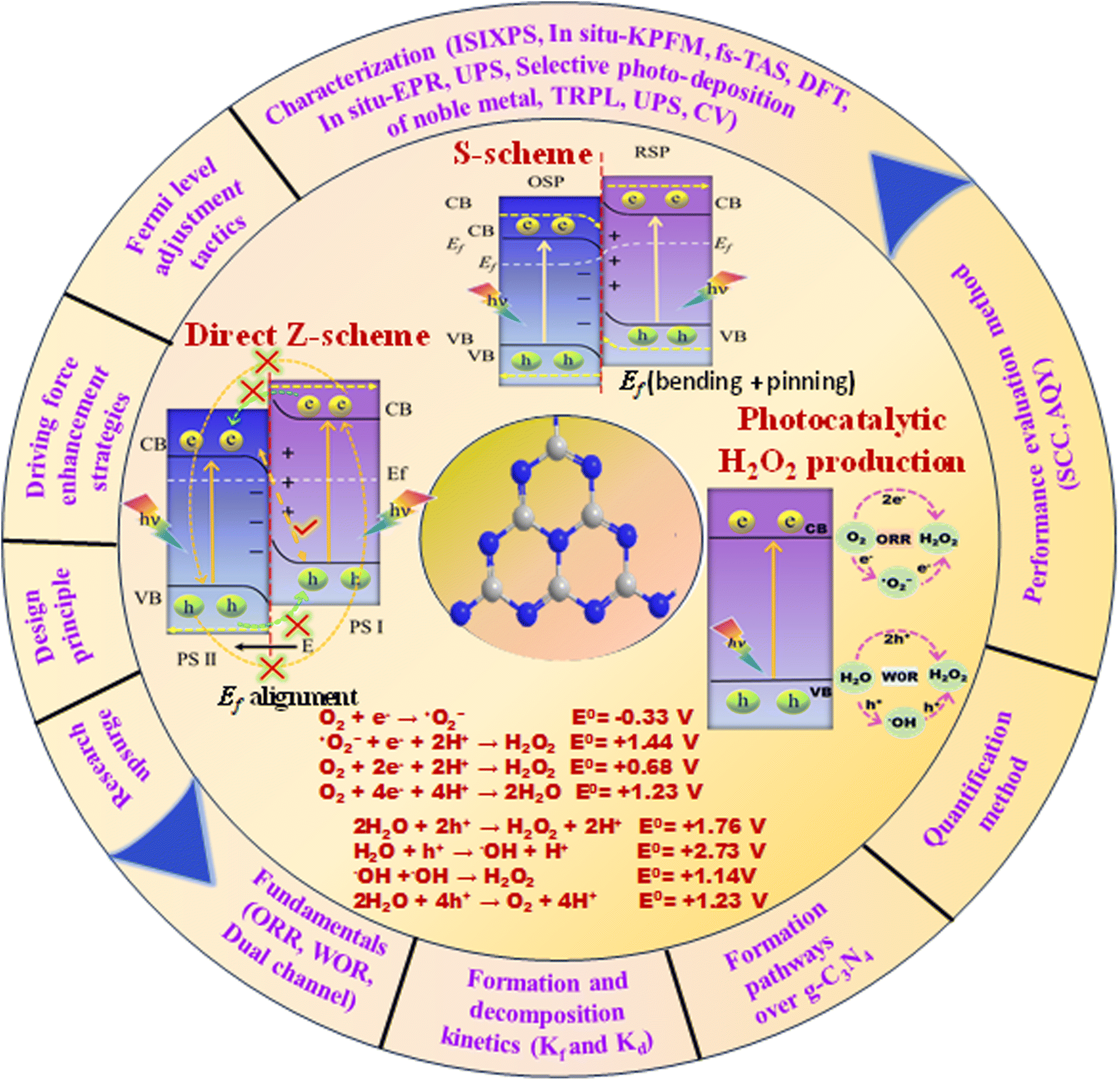 | ||
| Fig. 1 Comprehensive overview of the key themes and topics covered in the review. | ||
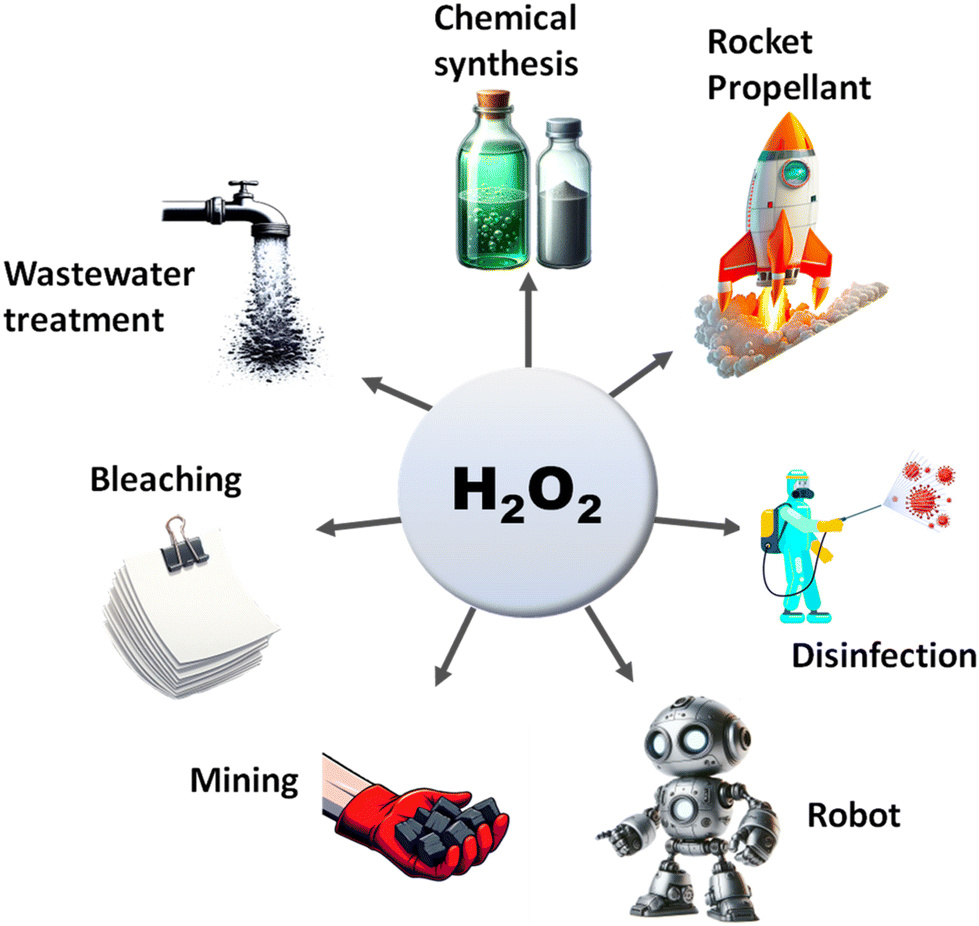 | ||
| Fig. 2 Diverse applications of hydrogen peroxide. | ||
A number of semiconductors, namely metal oxides,30 metal sulfides,35–37 metal phosphides,38,39 transition metal dichalcogenides,40 transition metal oxyhydroxides,41 metal–organic frameworks (MOFs),42–44 carbon-based semiconductors,45–47 metal-free organic semiconductors like g-C3N4,48–50 resins,51–58 covalent triazine frameworks (CTFs),59 covalent organic frameworks (COFs),60,61 covalent heptazine frameworks (CHFs)62 and Bi-containing semiconductors,63–65 have been extensively investigated for photocatalytic H2O2 production. Despite significant research, the field still faces challenges such as low photoconversion efficiency and rapid e−/h+ pair recombination, which limit its practical applications. In light of these challenges and considering the environmental benefits of reduced metal utilization, there is an urgent demand for the development of innovative, visible-light-responsive, metal-free photocatalysts to maximize the overall efficiency of solar energy utilization.
Among the plethora of semiconductor photocatalysts studied, graphitic carbon nitride (g-C3N4) has gained significant interest, particularly in the area of photocatalytic H2O2 production. This attention can be attributed to its advantageous electronic band structure, exceptional physicochemical stability, facile synthesis methods, cost-effectiveness, and high resistance to photocorrosion.66–72 Additionally, the specific alignment of its conduction and valence bands favours oxygen (O2) reduction while concurrently limiting the undesired oxidative breakdown of H2O2, which gives it an edge over oxide-based alternatives.73 This benefit is further amplified by its high selectivity in generating H2O2 through a mechanism involving the formation of superoxide radicals (˙O2−) and subsequent 1,4-endoperoxide intermediates on its melem units. These intermediates effectively inhibit O–O bond cleavage, optimizing H2O2 yield during oxygen reduction.74 In a pioneering study, Shiraishi and colleagues were the first to showcase the capability of a g-C3N4 structure to photocatalytically produce H2O2 in a water/alcohol mixture. Despite this breakthrough, the process exhibited low efficiency when no sacrificial agent was involved, primarily due to the speedy recombination of charge species and insufficient O2 adsorption on the catalyst's surface.74 In the case of unmodified g-C3N4, several obstacles impede its practical utility. These challenges encompass rapid charge carrier recombination, limited light absorption capacity, suboptimal electrical conductivity, low specific surface area, and slow water oxidation kinetics due to the weak oxidation ability of holes. To optimize the photoconversion efficiency of pure g-C3N4, a multitude of nanoengineering strategies have been effectively deployed. These include elemental substitutions with metals and non-metals, the introduction of defects, precise control over morphological attributes, surface functionalization techniques, and the construction of heterojunctions.69,75–77 These approaches aim to enlarge the light absorption spectrum and separation efficiency of photoinduced charge carriers. However, it is challenging to significantly enhance both the oxidation and reduction capabilities of g-C3N4 using a single modification approach. This is because the efficacy of g-C3N4 largely relies on the potentials of its VB and CB to facilitate specific half-reactions, namely OH−/˙OH and O2/˙O2− thermodynamically. Additionally, optimizing g-C3N4 for enhanced visible light absorption is often at odds with achieving strong redox potentials. Because enhanced light absorption typically requires a narrower band gap, while stronger redox abilities necessitate more negative potentials for the CB and more positive potentials for the VB, numerous techniques have been suggested for altering the properties of g-C3N4. However, when considering multiple objectives at once, only the formation of heterojunctions that combine g-C3N4 with other suitable semiconductor materials seems to comprehensively address all of the concerns previously mentioned.77–81
To date, a diverse array of heterojunctions involving g-C3N4 have been engineered (Fig. 3) based on the band positions and charge separation mechanism. These include type II, Z-scheme, and S-scheme heterojunctions.82–84 Among the variety of heterojunctions, establishing a Z-scheme or S-scheme heterojunction between g-C3N4 and another oxidative semiconductor presents a viable and logical solution for a singular modification strategy of g-C3N4. The crux of this approach lies in establishing a robust and closely-knit interface between g-C3N4 and the other semiconductor, one that features well-aligned CB and VB edges. When materials with different work functions come into contact, an internal electric field is created between them. This field can expedite the movement of photoexcited electron–hole pairs and curtail exciton recombination.85,86 Moreover, this well-structured composite not only augments the absorption of visible light but also assures significant oxidation and reduction abilities for photogenerated holes and electrons, respectively. Both of these properties are important for improving the photocatalytic performance.
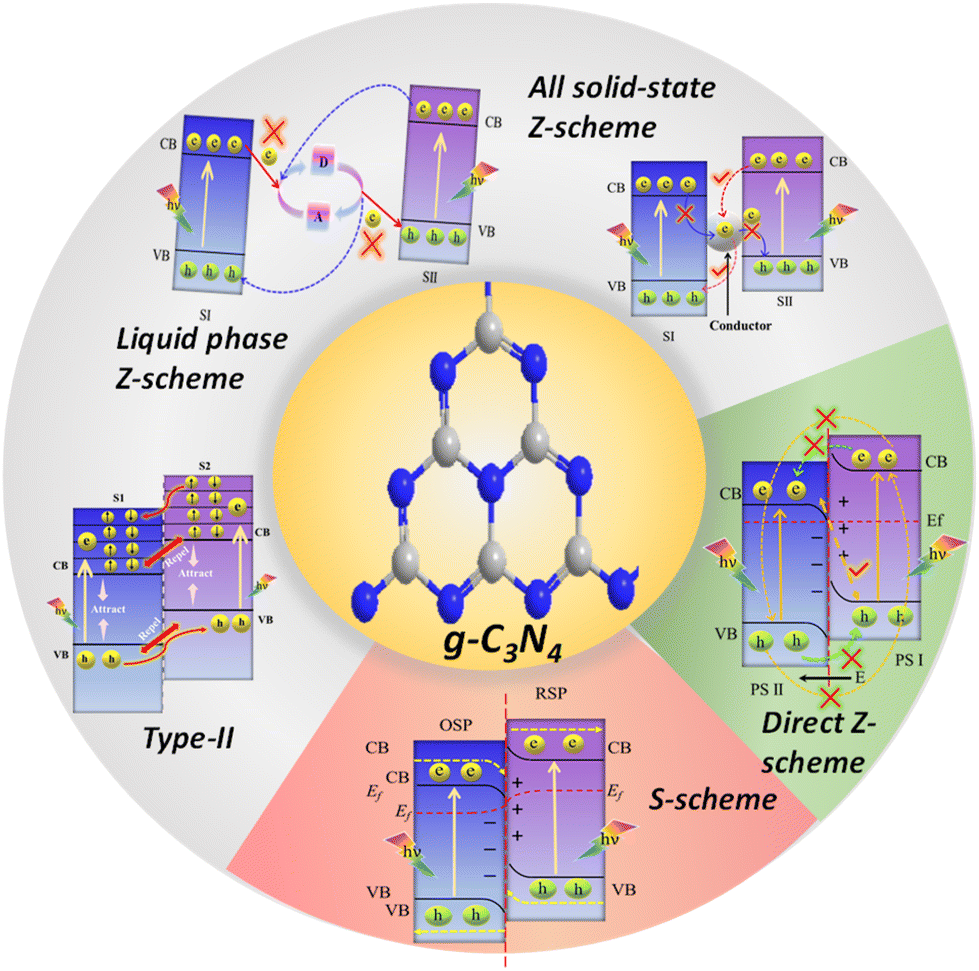 | ||
| Fig. 3 Illustration of charge transfer mechanisms in different g-C3N4-based heterojunctions. | ||
On the basis of the inimitable advantages of direct Z- and S-scheme heterojunctions in preserving the strong redox ability of the photocatalytic system, Z- and S-scheme-based photocatalysts were widely explored in photocatalytic H2O2 production. In particular, numerous articles using g-C3N4-based Z- and S-scheme heterojunctions for photocatalytic H2O2 production have emerged; therefore, an exhaustive overview of this topic is highly in demand. This work discusses a new breakthrough in g-C3N4-based Z-scheme and S-scheme heterojunctions towards H2O2 production under visible light illumination (Fig. 4). We exclusively focus on g-C3N4 as a photocatalyst with a detailed discussion of its properties and its Z-scheme and more relevant S-scheme heterojunctions for photocatalytic H2O2 applications (Fig. 1). In this review, first of all, the proposed background and upsurge in research on direct Z- and S-scheme heterojunctions are elaborately introduced. Secondly, the design principles and characterization methods of Z- and S-scheme heterojunctions have been systematically described. Moreover, the photocatalytic applications of current advancements in g-C3N4-based direct Z- and S-scheme heterojunctions have been described in detail, with respect to H2O2 generation, highlighting the important finding of the recently reported state of the art. The review also systematically describes the fundamentals of H2O2 evolution along with a brief description of the peroxide formation pathway over g-C3N4 and detection techniques. We have included a short narrative on different types of reaction conditions for optimising photocatalytic H2O2 generation efficiency. Finally, we try to summarize the future prospects of this emerging area of research towards green fuel generation along with the associated challenges and measures taken to address the issues encountered.
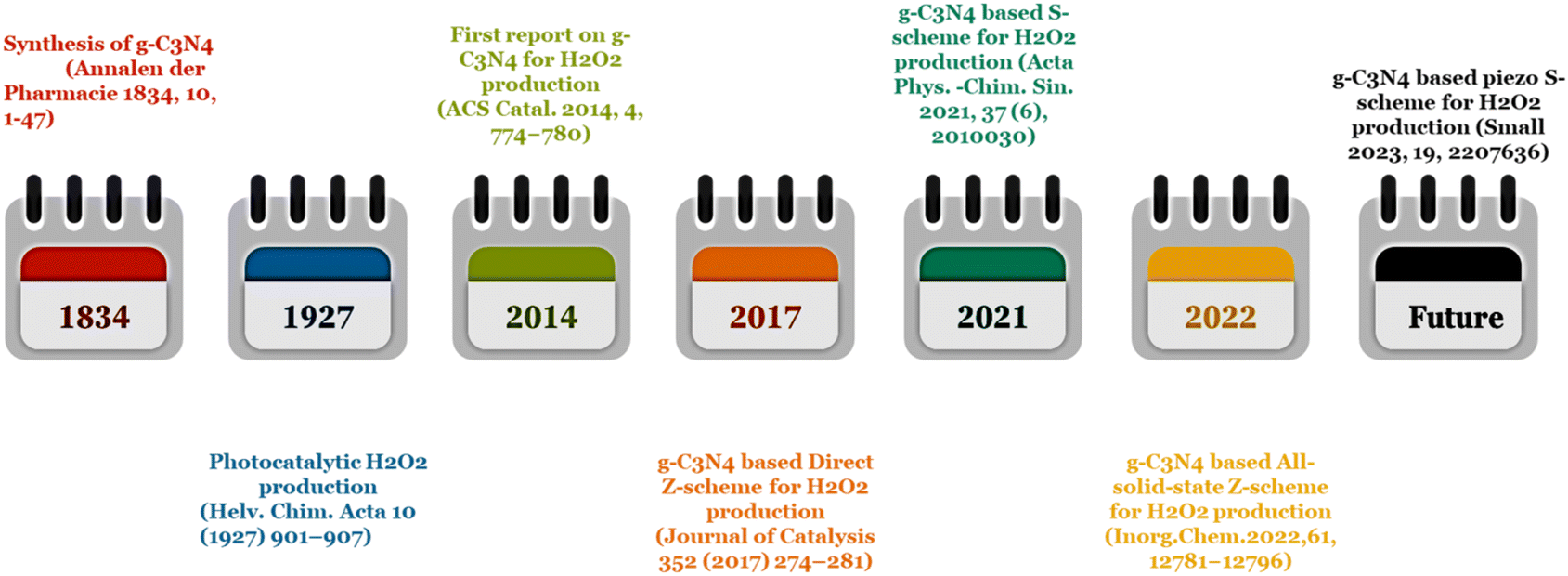 | ||
| Fig. 4 Evolutionary timeline of g-C3N4-based Z- and S-scheme heterojunctions in photocatalytic hydrogen peroxide production. | ||
2. Graphitic carbon nitride (g-C3N4): a sustainable photocatalyst
Graphitic carbon nitride (g-C3N4), the most stable allotrope composed of the abundant elements carbon and nitrogen, is a noteworthy metal-free conjugated organic n-type semiconductor. Since its discovery in 2009 by Wang et al., g-C3N4 has gained significant attention in the arena of photocatalysis, especially for its role in photocatalytic hydrogen production through water splitting.87 Its robust physicochemical stability, abundance on Earth, desirable electronic band structure, and straightforward preparation process make it an excellent photocatalyst.88,89 Interestingly, the early version of this material, known as “melon”, was first created by Berzelius in 1834 and later named by Liebig.90 Despite its long history, the application of g-C3N4 in heterogeneous catalysis only began around 2006, marking a new era in its utilisation. Carbon nitride is known for its diverse structural forms, including α-C3N4, β-C3N4, cubic-C3N4, pseudocubic-C3N4, and notably, graphitic-C3N4 (g-C3N4). The latter, g-C3N4, is particularly esteemed as the most stable allotrope, thanks to its outstanding chemical and thermal stability.91 Studies on the inner workings of g-C3N4 have unveiled its intricate structure, which is composed of tri-s-triazine/heptazine (C6N7) aromatic heterocycle rings, each featuring secondary nitrogen atoms (Fig. 5). Notably, the tri-s-triazine/heptazine (C6N7) components within g-C3N4 have been identified as more energetically stable compared to s-triazine (C3N3) units, with this stability manifesting as a 30 kJ mol−1 lower energy level.92 Each layer in the planar structure of g-C3N4 is an alternating arrangement of carbon and nitrogen. These rings are linked by planar tertiary amino groups, creating a two-dimensional, pi-conjugated, polymeric structure. This arrangement results in a delocalized π-electron cloud, which underlies the minimal basic nature of the tertiary amines present. Additionally, these layers are held together by weak van der Waals forces, with a notable gap of 0.326 nm between them. This gap facilitates exfoliation of the material into two-dimensional graphitic nanosheets. In each layer, the atoms are strategically organized in a honeycomb configuration and are strongly bonded through covalent bonds, endowing g-C3N4 with its highly stable physicochemical properties.93,94 Moreover, g-C3N4 stands out among its five structural phases by having the lowest band gap. This feature is primarily due to the presence of sp2-hybridized carbon and nitrogen atoms, which facilitate π-conjugated electronic structures.72 The electronic structure of g-C3N4 is remarkably efficient for visible light absorption, a feature linked to its moderate band gap energy, approximately estimated at 2.7 eV. Additionally, the material exhibits band edge potentials of +1.6 eV for the VB and −1.1 eV for the CB, when measured against the normal hydrogen electrode (NHE). These values align with an optical wavelength in the vicinity of 460 nm, which gives g-C3N4 its distinctive yellow colour, a direct result of its specific absorption profile within this wavelength range.95 Analysis of the HOMO and LUMO reveals that the VB of g-C3N4 primarily consists of 2p orbitals of nitrogen, while the CB contains 2p orbitals of both carbon and nitrogen.92 This structural arrangement endows g-C3N4 with a considerably negative CB, thereby enhancing the reducing power of electrons. This feature makes it highly effective for various catalytic reactions.92 Additionally, g-C3N4 exhibits exceptional thermal stability up to 600 °C in air. This resilience is attributed to its aromatic C–N heterocycles. Its chemical stability is equally impressive; it remains stable and does not dissolve in a range of substances, including water, acids, alkalis, and several organic solvents such as alcohols, diethyl ether, and toluene.69 This resistance to solvents renders g-C3N4 a versatile material, especially suitable for liquid-phase reactions in a variety of solutions. Beyond its chemical and physical properties, g-C3N4 is also recognized for its biocompatibility and environmentally friendly attributes, making it an attractive material for diverse applications. Despite extensive research, the practical application of pristine g-C3N4 faces several challenges. These include rapid recombination and inefficient separation of charge carriers due to the merging of N 2p and C 2p states in the CB, and structural irregularities or defects. Additionally, its limited electron mobility, stemming from poor conductivity, and suboptimal solar energy absorption or weak response to visible light further restricts its use. The material's low specific surface area (10–15 m2 g−1) results in fewer active sites, impacting its hydrophilicity, crystallinity, and surface properties, while its moderately positive VB energy limits its oxidation capability. The structure of g-C3N4 is comprised of two types of carbon atoms: sp2 hybridized carbon (N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) and nitrogen-bridging carbon (C–NH2), as well as three types of nitrogen atoms, namely pyridine nitrogen (C–NC), central nitrogen (N–(C)3), and amino nitrogen (C–N–H). Pyridine nitrogen acts as a site for oxidation reactions on the surface of g-C3N4, while carbon atoms, pyridine nitrogen, and central nitrogen serve as reduction centres during photocatalytic reactions.50 However, the bridge-connected nitrogen cannot be photoexcited, leading to the blocking of charge flow between triazine units and impeding interlayer charge carrier transfer due to weak van der Waals forces. This leads to the recombination of photoinduced charged species, diminishing the photocatalytic performance. Furthermore, the replacement of highly electronegative nitrogen in the structure by carbon contributes to high symmetry, slowing internal electron transport and e−/h+ pair recombination. Given that g-C3N4 is synthesized through the thermal condensation of nitrogen-rich molecules, and the composition of the precursor is adjustable, it is possible to design and construct electron transfer channels within its structure. These channels can break the structural symmetry, enhancing charge transport and separation. To address these issues sustainably, numerous modification approaches have been explored, such as controlling structural defects,76 doping with metals or non-metals,96 incorporating functional groups,71 altering its morphology and dimensionality,97 introducing metal constituents,91 employing exfoliation,89 and heterojunction formation.98 Among these strategies, designing heterojunction structures based on g-C3N4, using semiconductors with varying bandgaps, has become a focal point. These structures are advantageous for separating electron–hole pairs and leveraging the synergistic benefits of different components to enhance performance. Currently, there is growing excitement around the construction of Z- and S-scheme heterostructures using g-C3N4 as a component to balance its strong reducing ability in the CB, offset its weak ability to oxidize holes, and ultimately improve the charge separation efficiency.86
N) and nitrogen-bridging carbon (C–NH2), as well as three types of nitrogen atoms, namely pyridine nitrogen (C–NC), central nitrogen (N–(C)3), and amino nitrogen (C–N–H). Pyridine nitrogen acts as a site for oxidation reactions on the surface of g-C3N4, while carbon atoms, pyridine nitrogen, and central nitrogen serve as reduction centres during photocatalytic reactions.50 However, the bridge-connected nitrogen cannot be photoexcited, leading to the blocking of charge flow between triazine units and impeding interlayer charge carrier transfer due to weak van der Waals forces. This leads to the recombination of photoinduced charged species, diminishing the photocatalytic performance. Furthermore, the replacement of highly electronegative nitrogen in the structure by carbon contributes to high symmetry, slowing internal electron transport and e−/h+ pair recombination. Given that g-C3N4 is synthesized through the thermal condensation of nitrogen-rich molecules, and the composition of the precursor is adjustable, it is possible to design and construct electron transfer channels within its structure. These channels can break the structural symmetry, enhancing charge transport and separation. To address these issues sustainably, numerous modification approaches have been explored, such as controlling structural defects,76 doping with metals or non-metals,96 incorporating functional groups,71 altering its morphology and dimensionality,97 introducing metal constituents,91 employing exfoliation,89 and heterojunction formation.98 Among these strategies, designing heterojunction structures based on g-C3N4, using semiconductors with varying bandgaps, has become a focal point. These structures are advantageous for separating electron–hole pairs and leveraging the synergistic benefits of different components to enhance performance. Currently, there is growing excitement around the construction of Z- and S-scheme heterostructures using g-C3N4 as a component to balance its strong reducing ability in the CB, offset its weak ability to oxidize holes, and ultimately improve the charge separation efficiency.86
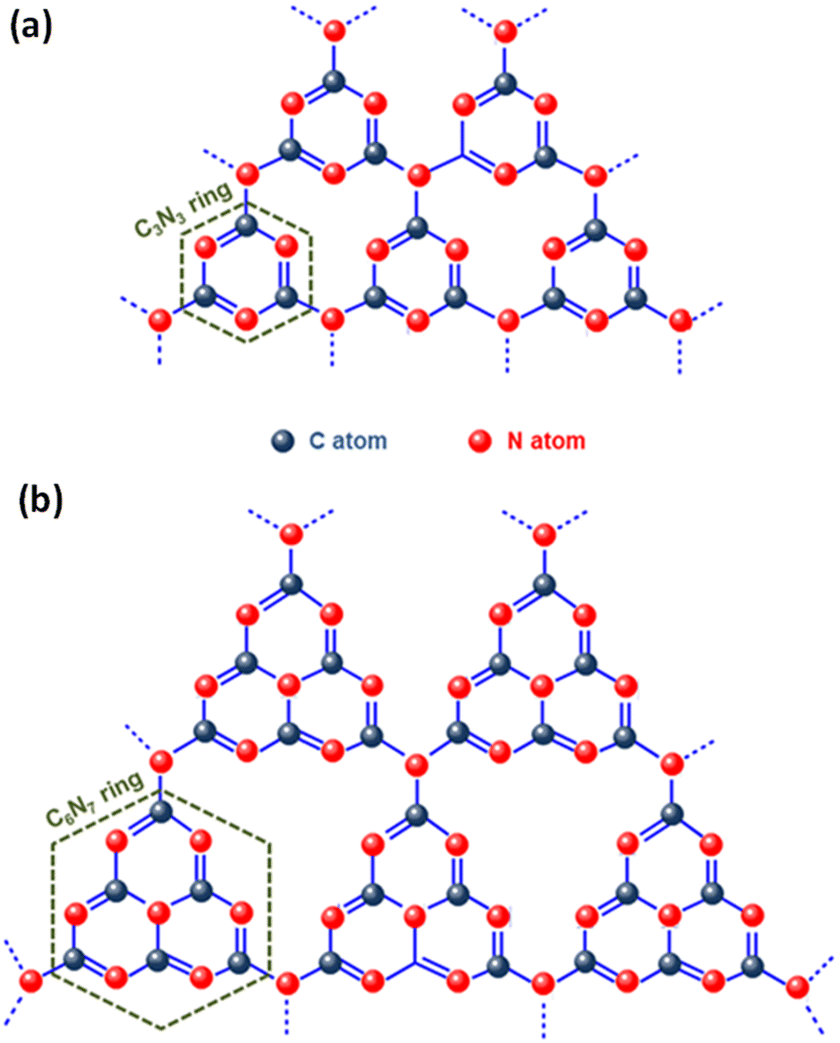 | ||
| Fig. 5 Structural representation of (a) s-triazine and (b) tri-s-triazine rings within g-C3N4. The figures are adapted with permission from ref. 88. Copyright 2020, Wiley-VCH. | ||
The planar structure of g-C3N4 makes it a suitable candidate for the construction of Z- and S-scheme photocatalytic systems. This is particularly due to its increased surface area, which can be easily achieved through a facile exfoliation process. This larger surface area offers a huge number of active sites for photocatalytic reactions, leading to enhanced catalytic activity. Additionally, g-C3N4 has garnered considerable interest as a reduction photocatalyst for its significant capabilities in environmental applications, specifically in various photocatalytic reduction processes. Researchers have been exploring the coupling of g-C3N4 with a wide range of oxidation semiconductor photocatalysts to create Z- and S-scheme nanocomposites. This area of study is crucial for several reasons. Firstly, it helps to maintain a higher redox capability, which is essential for efficient catalytic reactions. Secondly, it enhances the ability to harness solar radiation, making the photocatalytic system more energy-efficient.
3. Construction of heterojunctions
Heterojunctions, which arise from the fusion of two semiconductors with unique bandgaps and electron configurations, lead to the phenomenon of band bending at their interfaces. These interfaces can be differentiated into two distinct types based on their bandgap alignments and the relative positions of their valence bands (VB) and conduction bands (CB): type-II (staggered-gap) and the Z-scheme heterojunction.The type-II heterojunction is recognized for its advantageous alignment where the VB and CB of S2 are higher in energy than those of S1 (as depicted in Fig. 6). This promotes the separation of electrons and holes upon light illumination; electrons move from a higher to a lower energy state, with holes moving in the reverse direction, thereby spatially separating them, but at the expense of the redox potential of the system, adversely affecting photocatalytic processes. Furthermore, the movement of charge carriers would be progressively impeded as like charge carriers continue to build up in their populations. Further in-depth analysis of the charge transfer process revealed a decrease in redox potential, leading to slower redox reaction rates due to the relocation of electrons to the less reductive CB of S1 and the accumulation of holes in the less oxidative VB of S2. This situation also led to the dissipation of some amount of energy unproductively, without directly contributing to photocatalytic reactions.99 Additionally, the proposed charge separation process is impeded by electrostatic repulsion between like charges, such as electrons in the CBs of semiconductors and holes in their VBs. Furthermore, the CB of S1 contains a limited array of molecular orbitals with nearly continuous energy levels, each capable of holding just two electrons. Upon photoexcitation, these orbitals fill up with photoinduced electrons, preventing electrons from the CB of S2 from entering. This situation is akin to a hotel where rooms (orbitals) can only be reoccupied once the initial guests (electrons in S1) have vacated, leaving no vacancy for new guests (electrons from S2) until the former are moved elsewhere.86 This observation contributed significantly to the emergence of the Z-scheme family. The Z-scheme heterojunction approach has been recognized as a promising alternative to type-II heterojunctions in this context. Moreover, a novel charge transfer configuration, termed the S-scheme heterojunction, has also been recently introduced.100 This new paradigm is gradually establishing a foothold in the realm of photocatalysis, with Z-scheme and S-scheme heterojunctions already having a wide range of applications.
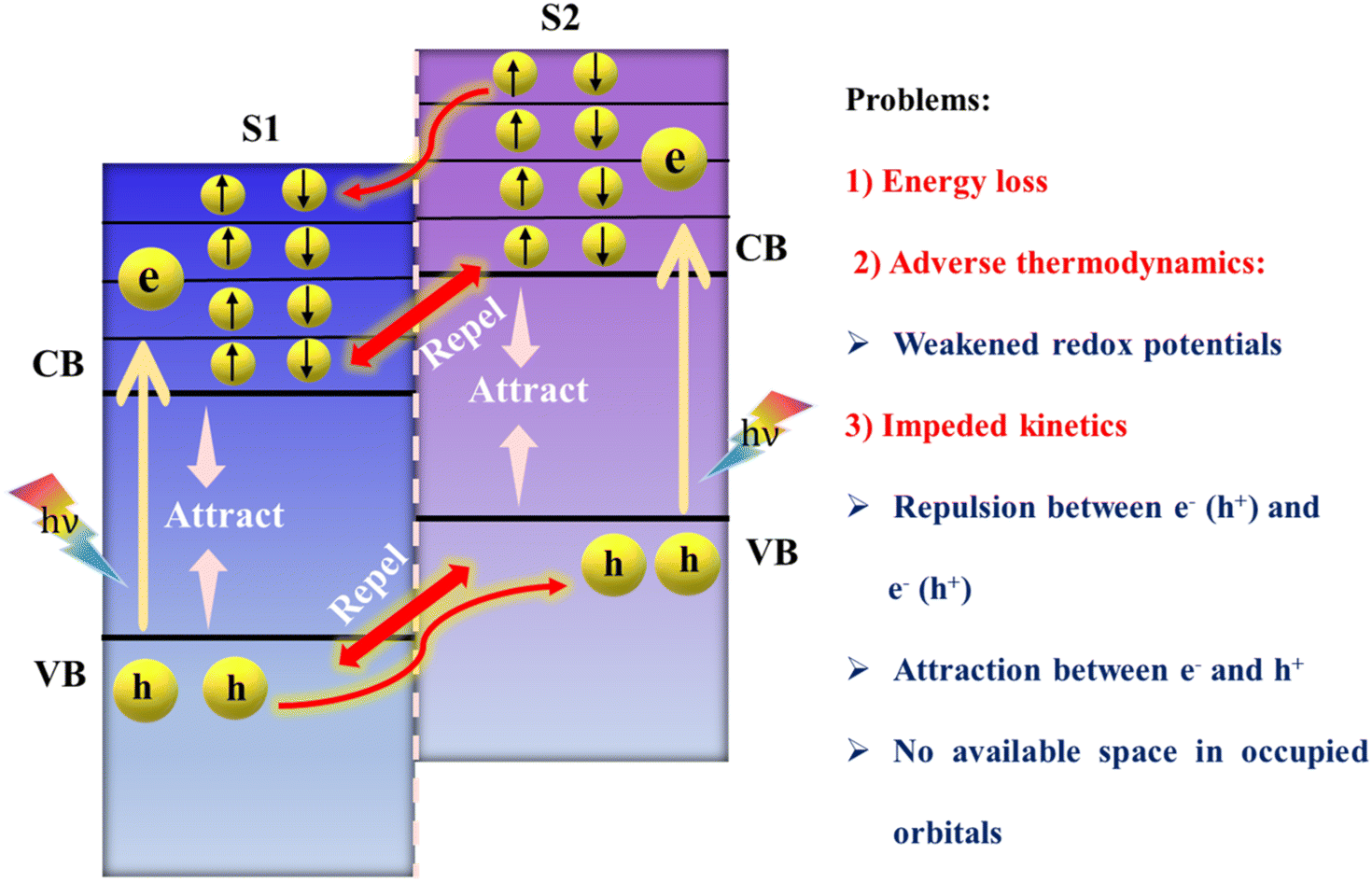 | ||
| Fig. 6 Suggested charge transfer pathway in conventional type-II heterojunctions and associated challenges. | ||
3.1. Z-Scheme-based heterojunctions
The Z-scheme concept emerges from the natural photosynthesis process performed in plants’ chlorophyll pigment to facilitate the thermodynamically uphill conversion of water and carbon dioxide into oxygen and carbohydrates (Fig. 7).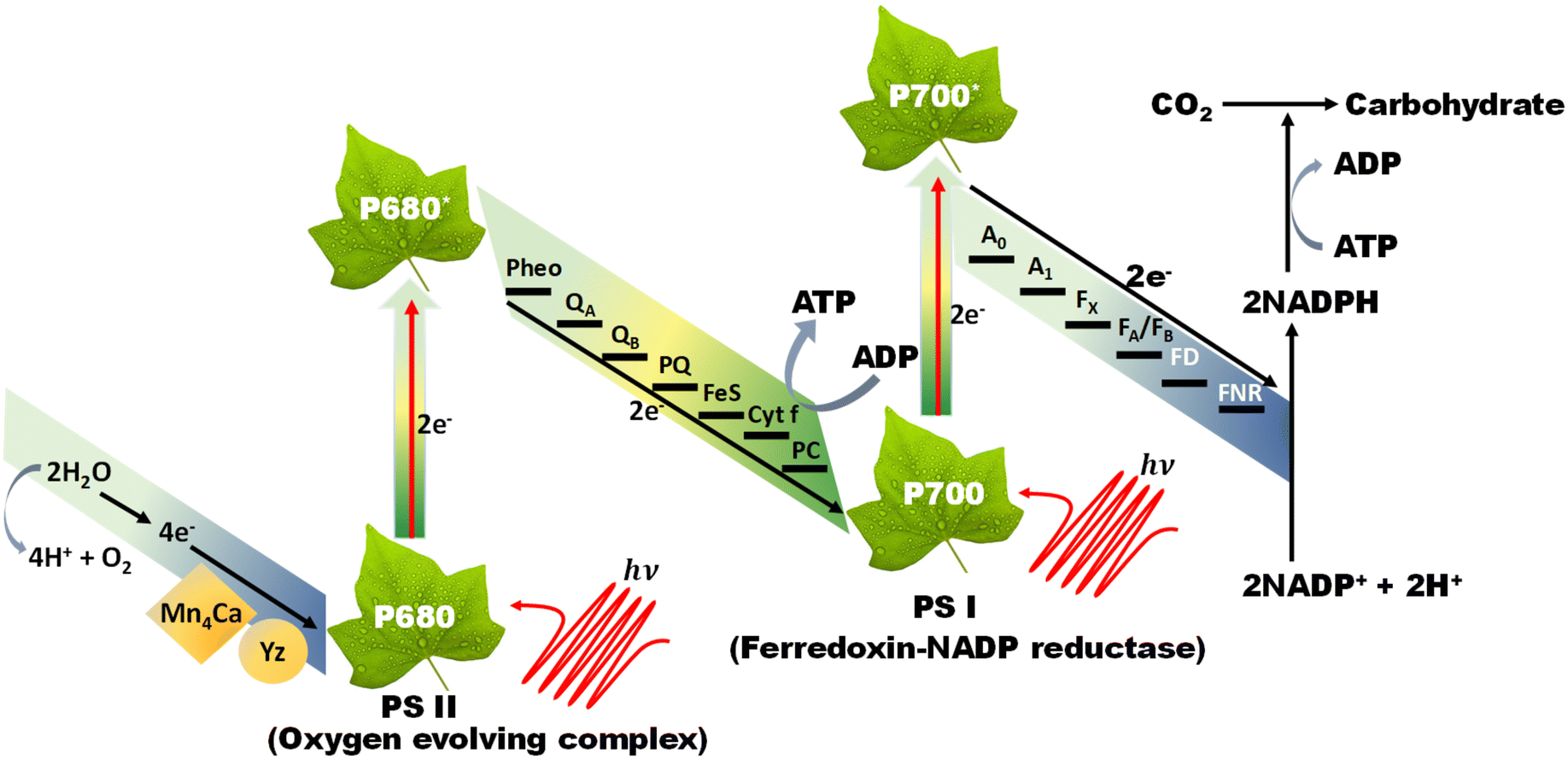 | ||
| Fig. 7 Diagrammatic representation of the natural photosynthesis process. | ||
This transformation is facilitated through a two-step photoexcitation process under solar energy irradiation, enabling plants to exploit solar energy efficiently with a quantum efficiency approaching 100%. This model represents the natural and intricate mechanisms by which plants harness solar energy to perform essential life-sustaining transformations.101 Basically, photosystem I (PS I) and photosystem II (PS II) are the two main components of chlorophyll, which are connected in series via an electron transfer chain. PS I and PS II harvest solar energy, corresponding to wavelengths of λ ≤ 700 nm and λ ≤ 680 nm, respectively, in the chlorophyll reaction centres and electrons are excited from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO). After that, the photoinduced electrons in the LUMO of PS II recombine with the HOMO of ferredoxin-NAPD reductase (PS I) via an electron transfer chain, which stimulates chemical osmotic potential that causes the transformation of adenosine diphosphate (ADP) into adenosine triphosphate (ATP). The photoexcited electrons accumulated in the HOMO of PS I drift to ferredoxin-NADP+ reductase through ferredoxin for the conversion of NADP+ into NADPH coenzyme in which the reduction of carbon dioxide to carbohydrate occurs. The photogenerated holes that are present in the HOMO of PS II act as an oxygen-evolving centre, where the accumulated photogenerated holes oxidize water molecules at the manganese–calcium oxide cluster. Both the above-discussed processes occur simultaneously at different regions of the chlorophyll pigment in plants. This type of charge transfer demonstrates the robust redox ability of PS I and PS II, which is different from the conventional type-II heterojunction. The electron flow path of the cascade profile resembles the letter ‘Z’ of the English alphabet, from which the name of the Z-scheme system is derived. Thus, excellent redox potential, as well as efficient charge carrier separation, can be attained in the Z-scheme system by the combination of reduction and oxidation semiconductors. Inspired by the unique features of the natural Z-scheme photosynthesis process, the biomimetic artificial Z-scheme photocatalysts synthesized have been extensively studied in the last 30 years, indicating extraordinary advancement in the field of research. The ground-breaking idea of the artificial Z-scheme model was first initiated by Bard in 1979 by imitating the natural photosynthesis process of plants by employing a shuttle redox pair as an electron transport pathway between two photosystems to maximize the redox potential of the photocatalytic heterojunction systems.102 The accumulation of photoinduced electrons in the CB of PS I make it an electron surplus region, which shows strong reduction potential and suppresses photodegradation from self-oxidation. Similarly, PS II serves as a hole-rich region by the accumulation of photoinduced holes in the VB of PS II, which show a strong oxidation ability and protect PS II from photoreduction. However, this statement reveals that PS II should be a semiconductor with a strong oxidation ability, while PS I should have a strong reduction ability to protect against the corresponding photooxidation and photoreduction and simultaneously promote hydrogen peroxide production. Hence, photocatalysts having poor resistance towards photooxidation cannot be employed as PS II. In summary, a photocatalyst with a smaller resistance to photoreduction is not appropriate for use as PS I.103
Furthermore, the number of photoinduced electron and hole pairs in the photocatalytic Z-scheme heterojunction system is just half of that in the conventional photocatalytic system because of the recombination of excitons via the electron mediator. However, the Z-scheme heterojunction can efficiently reduce bulk electron–hole recombination and increase the lifespan of photoinduced charge carriers in the isolated photosystem. Based on the condition of whether an additional charge mediator is introduced or not, and the type of the electron mediator taken for charge carrier transfer, the Z-scheme photocatalytic systems can be classified into two types: indirect Z-scheme systems and direct Z-scheme systems (PS-PS systems), respectively.104
3.1.1.1. Conventional Z-scheme systems. The conventional Z-scheme photocatalyst, the PS-A/D-PS system, involves an electron acceptor/donor (A/D) pair in the liquid phase and two photosystems (PS II and PS I), where this pair operates much like a Ferris wheel between the two photosystems, but without any physical contact between PS II and PS I (Fig. 8a).106 The direction of electron transport from the CB of PS II to the VB of PS I relies on the below-mentioned redox reactions of the A/D pair:
Forward reactions:
| Acceptor (A) + ne− (from the CB of PS II) → donor (D) |
| Donor (D) + nh+ (from the VB of PS I) → Acceptor (A) |
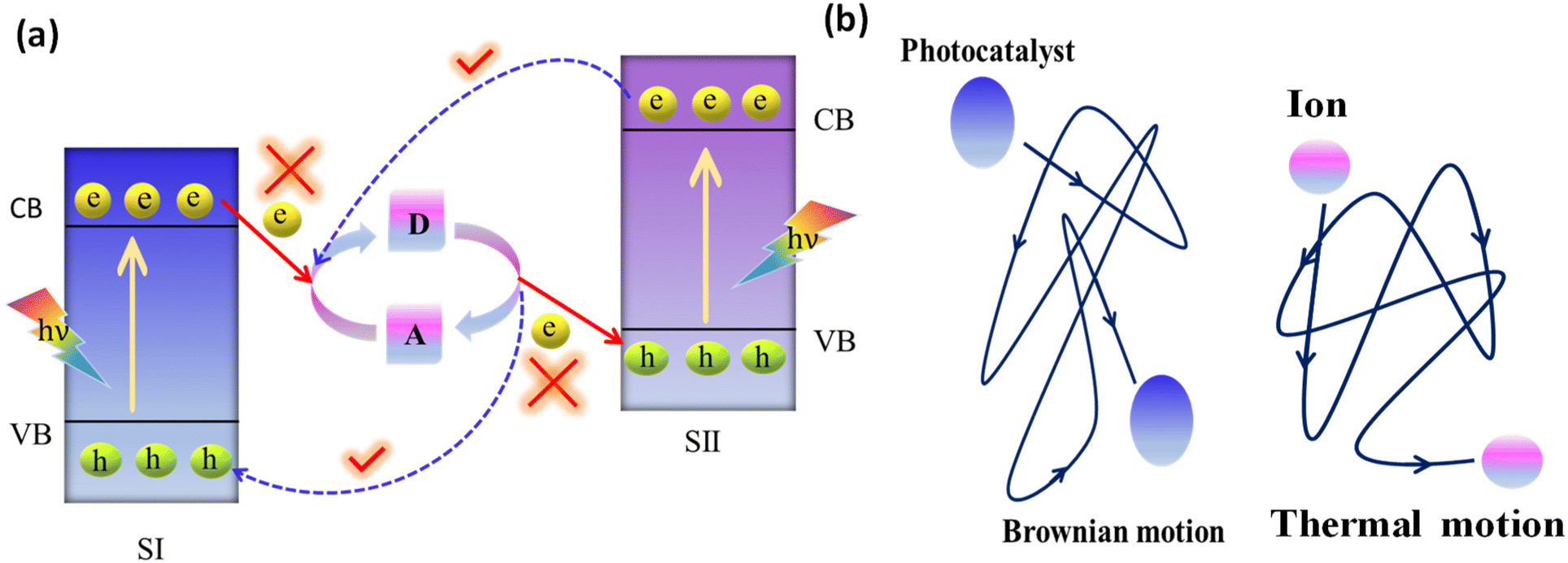 | ||
| Fig. 8 (a) Suggested (solid lines) and favored (dotted lines) charge transfer pathways in conventional liquid-phase Z-scheme heterojunctions. (b) Diagrammatic depiction of the Brownian movement of photocatalyst particles and thermal movement of redox couple ions in a solution. | ||
When the photocatalysts are exposed to photons having energy equal to or higher than the bandgap energy (Eg), photoexcitation occurs. Then the reduction of the electron acceptor (A) takes place by the transfer of photoexcited electrons from the CB of PS II to the electron donor (D), and simultaneously, the created electron donor (D) will revert back into its oxidized form (i.e., electron acceptor, A) by consuming the photoinduced holes from the VB of PS I. As a result, the photogenerated electrons from the CB of PS II are indirectly transported to recombine with the photogenerated holes from the VB of PS I with the assistance of the regeneration of the A/D redox pair. The net effect is that photoinduced electrons in the CB of PS I and photoinduced holes in the VB of PS II are efficiently separated (i.e., the system shows largely suppressed charge recombination) and accumulate in the highest possible CB and lowest possible VB, providing a considerably large redox potential for the photocatalytic surface redox reaction from the combination of the two photosystems.
Due to the free thermal motion of redox ion pairs in the liquid phase, rapid Brownian movement of photocatalyst nanoparticles and the absence of spatial constraints between redox ion pairs and surface-active sites of photocatalysts, thermodynamically favourable unwanted backward reactions occur (Fig. 8b).107 Particularly, the photoexcited electrons in PS I possess a higher reduction potential than those in PS II. So, the redox pair acceptors are more prone to receive electrons from PS I instead of PS II. In a similar way, photoinduced holes in PS II have a higher oxidation potential than those in PS I. Therefore, the donor of redox pairs is more willing to give electrons to PS II on behalf of PS I. Hence, the system cannot assure a strong redox potential, which has a negative impact on the photocatalytic reaction.99 The commonly utilised electron mediators for the PS-A/D-PS Z-scheme systems are IO3−/I−, Fe3+/Fe2+, [Co(bpy)3]3+/2+, [Co(phen)3]3+/2+, VO2+, and VO2+.108
Backward reactions:
| Acceptor (A) + ne− (from the CB of PS I) → donor (D) |
| Donor (D) + nh+ (from the VB of PS II) → Acceptor (A) |
Most of the redox pair mediators show light-shielding effects since the redox couples strongly absorb visible light and show colour, which reduces the supply of solar photons and their subsequent utilization. Redox couples are generally pH sensitive and difficult to keep in their long-term active state, which restricts the implementation of redox couples to operate PS-A/D-PS systems over a wide range of pH values. For example, simple ion redox cycles like iodate/iodine (IO3−/I−) can only operate effectively under more basic conditions of pH ≥ 9 due to the presence of active IO3−/I− ion pairs instead of inactive I3−/I− formation, available under lower pH conditions (eqn (1) and (2)). Similarly, Fe3+/Fe2+ redox pairs are only stable under more acidic conditions of pH less than 2.5 due to the formation of Fe(OH)3 precipitate through Fe3+ hydrolysis at high pH.108
| I3− + 2e− → 3I− E° vs. NHE = +0.536 V | (1) |
| IO3− + 6e− + 3H2O → I− + 6OH− E° vs. NHE = +0.670 V | (2) |
Achieving electron transport via an aqueous environment between photocatalyst nanoparticles and redox ion pairs presents a significant challenge, due to the absence of free electrons in water. Furthermore, the reactions of ionic redox couples are only possible under liquid phase conditions; this restricts their application to solid and gas phase photocatalysis. Such PS-A/D-PS systems also cannot be used for the degradation of pollutants in the solution because the latter probably hinders the forward redox reaction of the A/D pair, resulting in limited uses of the PS-A/D-PS systems in the field of photocatalytic water splitting.103 In order to address the above-discussed problems, the redox couple mediator-free all-solid-state Z-scheme has been proposed.
3.1.1.2. All-solid-state Z-scheme systems. In the solid-state Z-scheme mechanism, the A/D pair of a conventional liquid-phase Z-scheme system is exchanged for a solid-state conductive material (C) as the electron transfer bridge connecting the two semiconductors (Fig. 3), which are either noble metals (Au, Ag, Cu, Bi, Pd and Pt nanoparticles) or carbon-based materials like graphene, reduced graphene oxide (rGO), exfoliated graphite, graphene oxide, carbon dots (CDs), MXene, and carbon nanotubes (CNTs), broadening its application from liquid to gas ranging from H2O splitting to CO2 reduction. First, Tada et al. introduced the idea of solid-state Z-scheme systems by applying ‘Au’ as a solid-state conductive material in the development of the CdS/Au/TiO2 ternary system in 2006 for the photocatalytic water splitting reaction.109 Basically, a minimal interfacial Ohmic contact resistance is developed between a solid-state semiconductive material and two semiconductors (PS I and PS II system) at the junction interface; this generates an outstanding interparticle electron transfer capability by reducing the electron transfer path from the CB of PS II to the VB of PS I. Although the use of a solid conductive material extends the application scope, as well as improves the stability and recovery of the photocatalyst, it still has some drawbacks. Firstly, because of their strong reducing capabilities, the photoinduced electrons from the CB of PS II tend to recombine with the photoinduced holes from PS I, which possess high oxidizing capabilities, especially across solid conductors. Secondly, electron transfer through certain metal conductors, such as Au, having a higher work function (lower Fermi level, Ef) than those of PS I and PS II, faces challenges owing to the presence of dual Schottky barriers at the interfaces where the semiconductors meet the metal. Excited electrons of PS I and PS II naturally move towards a metal conductor having a larger work function. Additionally, to ensure the conductor particles are exactly positioned between two photocatalysts, it is challenging to accurately manage the synthesis process from the perspective of material design. The bulk conductor particles are most likely haphazardly distributed on the photocatalyst surface and exhibit point-to-point interfacial contact with the semiconductor, suggesting that they may serve as cocatalysts rather than charge-transfer shuttles.86,110 Appropriate preparation techniques are crucial for the creation of diverse PS-C-PS systems to make sure that the PS and the electron mediator are in close interfacial contact. PS-C-PS system development is constrained since the PS and solid-state conductive material must be integrated together as a single unit, in contrast to the flexibility of the PS-A/D-PS system. By applying a controlled synthesis procedure, the ideal geometrically precise positioning of the solid conductor sandwiched between two semiconductors is quite imprecise. The usage of noble metals as electron mediators is rare, expensive, photocorrosive, and displays a shielding effect, which greatly limits their viability for real applications.99 Challenges persist with regard to the dynamics of electron transfer within liquid Z-scheme configurations, as these mechanisms have yet to be fully resolved. To summarize, the challenge of electron transfer within liquid Z-scheme systems continues to be an open question.
 | ||
| Fig. 9 Comparison of charge transfer pathways in (a–c) direct Z-scheme versus (d–f) p–n junction. | ||
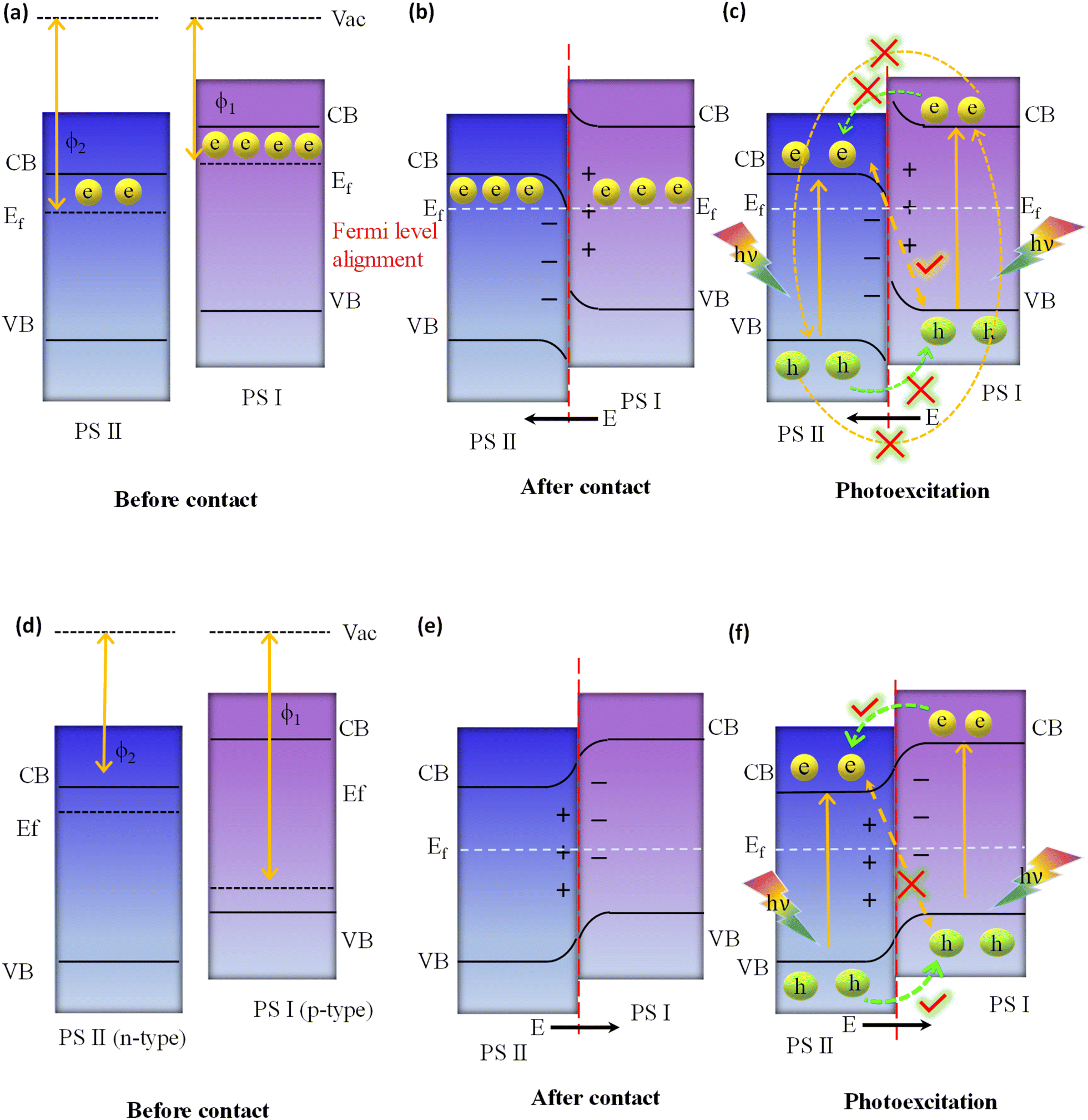
In the direct Z-scheme, the CB and VB potentials of PS I are positioned at a higher level along with a lower work function or higher Fermi level in comparison with PS II, as indicated in Fig. 9a. When PS I and PS II are in close proximity to one another under dark conditions, there will be a spontaneous flow of electrons from PS I to PS II at the interfacial surface. This process will continue until PS I and PS II have achieved equilibrium in their Fermi levels, as depicted in Fig. 9b. Consequently, PS I and PS II become oppositely charged, i.e., positive and negative, due to the loss and gain of electrons, respectively, at their interface, resulting in the establishment of an internal built-in electric field that is directed from PS I to PS II. At the same time, upward and downward band bending occurs at the interface region of PS I and PS II because of the donation and accumulation of electrons, respectively, which forms a potential barrier. Under light irradiation, the photogenerated electrons in the CB of PS II and holes in the VB of PS I are more prone to recombine at the contact interface under the influence of the three factors (i.e., internal built-in electric field, band edge bending, and coulombic attraction between electrons and holes) that act as the driving force for recombination (see Fig. 9c).116 In this instance, the Z-scheme charge transfer method is favoured. On the other hand, factors like the internal built-in electric field, inducing an extra potential barrier caused by band edge bending, and coulombic repulsion inhibit the type II like charge transfer (i.e., flow of photoinduced electrons from the CB of PS I to the CB of PS II, as well as photoinduced electrons from the VB of PS II to the VB of PS I), which are not suitable for Z-scheme charge transfer pathways as illustrated in Fig. 9c. Finally, weak charge carriers are removed via recombination, while the powerful photogenerated electrons and holes are conserved in the CB of PS I and the VB of PS II, respectively, to participate in the specific surface redox reaction. It should be highlighted that the electric field induced in this process also plays a significant role in suppressing the recombination of photoinduced electrons present in the CB of photosystem I (PS I) and photoinduced holes in the VB of photosystem II (PS II), as outlined in Fig. 9c. Conversely, the p–n heterojunction is a subclass of the type II system illustrated in Fig. 9d, substantially differing from the direct Z-scheme system depicted in Fig. 9a. The Fermi level of PS I, a p-type semiconductor, is situated lower than that of PS II, an n-type semiconductor. When the p-type and n-type semiconductors come into contact with each other under dark conditions, free electrons from the n-type can diffuse to the p-type semiconductor until equilibrium is achieved, leading to the formation of an interfacial built-in electric field directed from n to p, as shown in Fig. 9e. Consequently, a negative charge is created at the PS I interface, while PS II becomes positively charged due to electron transfer from PS II to PS I. This field facilitates electron movement from PS I (CB) to PS II (CB), while hole transfer occurs from PS II (VB) to PS I (VB) under light irradiation, i.e. a p–n junction charge carrier transfer pathway is realized (see Fig. 9f). In this scenario, due to the orientation of the existing built-in interfacial electric field, electron movement from PS II (CB) to recombine with holes in PS I (VB) is restricted, i.e. a direct Z-scheme charge carrier transfer pathway (as illustrated in Fig. 9f). Nonetheless, this mechanism of charge transfer is akin to the conventional type II system, sharing its inherent limitations. Therefore, within a p–n junction, the mode of photoinduced charge carrier flow is not conducive to the direct Z-scheme mode, as represented in Fig. 9f.116 Interestingly, in summary, the direct Z-scheme photocatalyst, the type II heterojunction and the p–n junction systems have the same band alignment. But, the ways of moving charges in the type II heterojunction and the p–n junction systems are different from those of the direct Z-scheme photocatalyst. Notably, the direct Z-scheme system is able to maintain the strong redox potential of the photoinduced charge carriers. In contrast, charge segregation of the type II heterojunction, as well as p–n junction photocatalysts, is facilitated by sacrificing their photogenerated charge carriers, which possess exceptional redox potential since electrons move from high to low reduction potential and holes move from high to low oxidation potential against the repulsive force of similar migrating charges. The primary causes of their differing charge transfer paths from those of direct Z-scheme systems are not having any IEFs in the type II heterojunction and creating the opposite direction of IEF in the p–n junction. Because of their similar band constructions, it is crucial to determine whether a manufactured photocatalytic system adopts a direct Z-scheme or type II heterojunction (or p–n junction).
Analogous to direct Z-scheme photocatalysts, all-solid-state photocatalysts similarly demonstrate bending at the edges of the VB and CB. This behaviour is primarily observed at the interface of the metal and the semiconductor in these all-solid-state photocatalysts. In situations where the metal demonstrates a larger work function than the semiconductor (ϕm > ϕs), there is a movement of free electrons from the semiconductor towards the metal. This directional transfer results in an upward bending of the semiconductor band edge, a phenomenon induced by the generated electric field. Conversely, if the metal possesses a smaller work function than the semiconductor (ϕm < ϕs), there will be a migration of free electrons in the opposite direction, that is, from the metal to the semiconductor, causing the semiconductor band edge to bend downward. For conventional Z-scheme photocatalysts, direct interfacial contact between two distinct photocatalysts does not exist. Consequently, there is no redistribution of free electrons between the two photocatalysts, and no internal electric field forms between them. In this context, the edges of the VB and CB of each component remain unaffected when conventional Z-scheme photocatalysts are constituted.
3.1.2.1. Theoretical requirements for the formation of a direct Z-scheme heterojunction. Direct Z-scheme heterojunction preparation has four fundamental prerequisites. First, the energy bands of two semiconductors should be aligned in a staggered pattern as that of a type II heterojunction. The second need is that a strong oxidation potential should be present in one semiconductor while a strong reduction potential is present in the other. Third, the reductive side photocatalyst should have higher CB and VB positions and a higher Fermi level (smaller work function) than the one used in the oxidative side. Fourth, photosystem II should be a semiconductor with a strong oxidation ability, while photosystem I should have a strong reduction ability to protect them from the corresponding photooxidation and photoreduction and simultaneously promote hydrogen peroxide production. Hence, a photocatalyst with weak resistance to photooxidation cannot be used as PS II. In the same way, a photocatalyst with weak resistance to photoreduction is not suitable as PS I. Therefore, for the system as a whole to attain the desired level of stability over time, PS I and PS II must have a high resistance to photoreduction and photooxidation processes, respectively.119 The direct Z-scheme effectively addresses the limitations found in the type-II heterojunction, conventional Z-scheme, and all-solid-state Z-scheme systems, but it does not completely answer certain scientific questions. One significant issue is the difficulty in establishing an interfacial built-in electric field (IEF) at identical Fermi levels when two semiconductors are juxtaposed. This is because the potential difference tends to disappear, subsequently impeding charge transfer at the interface.120 In response to these unresolved questions, Fu and colleagues proposed an alternative as a potential solution: the step-scheme (S-scheme) photocatalytic system.
To address the challenges and discrepancies observed in charge transfer within type-II heterojunctions, the traditional Z-scheme, and both all-solid-state and direct Z-scheme systems, a novel S-scheme heterojunction paradigm was introduced, building upon the principles of the direct Z-scheme heterojunction. Since Fu et al. (Yu's group) unveiled the concept of S-scheme photocatalytic systems in 2019,100 there has been growing interest in S-scheme heterojunctions. This approach offers a clear representation of electron transfer, resembling an upward movement on a staircase or appearing as an “N” shape on a microscopic scale. The development of S-scheme heterojunctions was, therefore, a natural progression in the field. The distinctive benefits of S-scheme heterojunctions, including efficient separation and transport of photogenerated carriers and a strong redox capacity, have catalyzed rapid advancements in the domain. To fully understand the S-scheme photocatalytic heterojunctions, one must delve into their formation and progression.
4. Fundamentals of S-scheme heterojunctions
4.1. Construction of S-scheme heterostructures
At its core, an S-scheme heterostructure is established between two intimately connected semiconductors that exhibit a staggered band alignment, akin to those seen in a type II configuration (see Fig. 11a). Yet, what differentiates the S-scheme heterostructure photocatalyst is its unique charge transfer mechanism. While type II is commonly employed, it is not without its thermodynamic and kinetic limitations, leading to a diminished redox capability. Drawing inspiration from the direct Z-scheme, the S-scheme heterojunction is comprised of an oxidation semiconductor photocatalyst (OSP) and a reduction semiconductor photocatalyst (RSP). A semiconductor photocatalyst characterized by a higher conduction band minimum (CBM), a higher valence band maximum (VBM), and an elevated Fermi level is designated as the reduction semiconductor photocatalyst (RSP). Conversely, the counterpart in this relationship is identified as the oxidation semiconductor photocatalyst (OSP). Common examples of OSPs and RSPs are depicted in Fig. 10. For the S-scheme heterojunction to form, the CB, VB, and Ef of the RSP should be positioned higher and should have a lower work function compared to those of the OSP.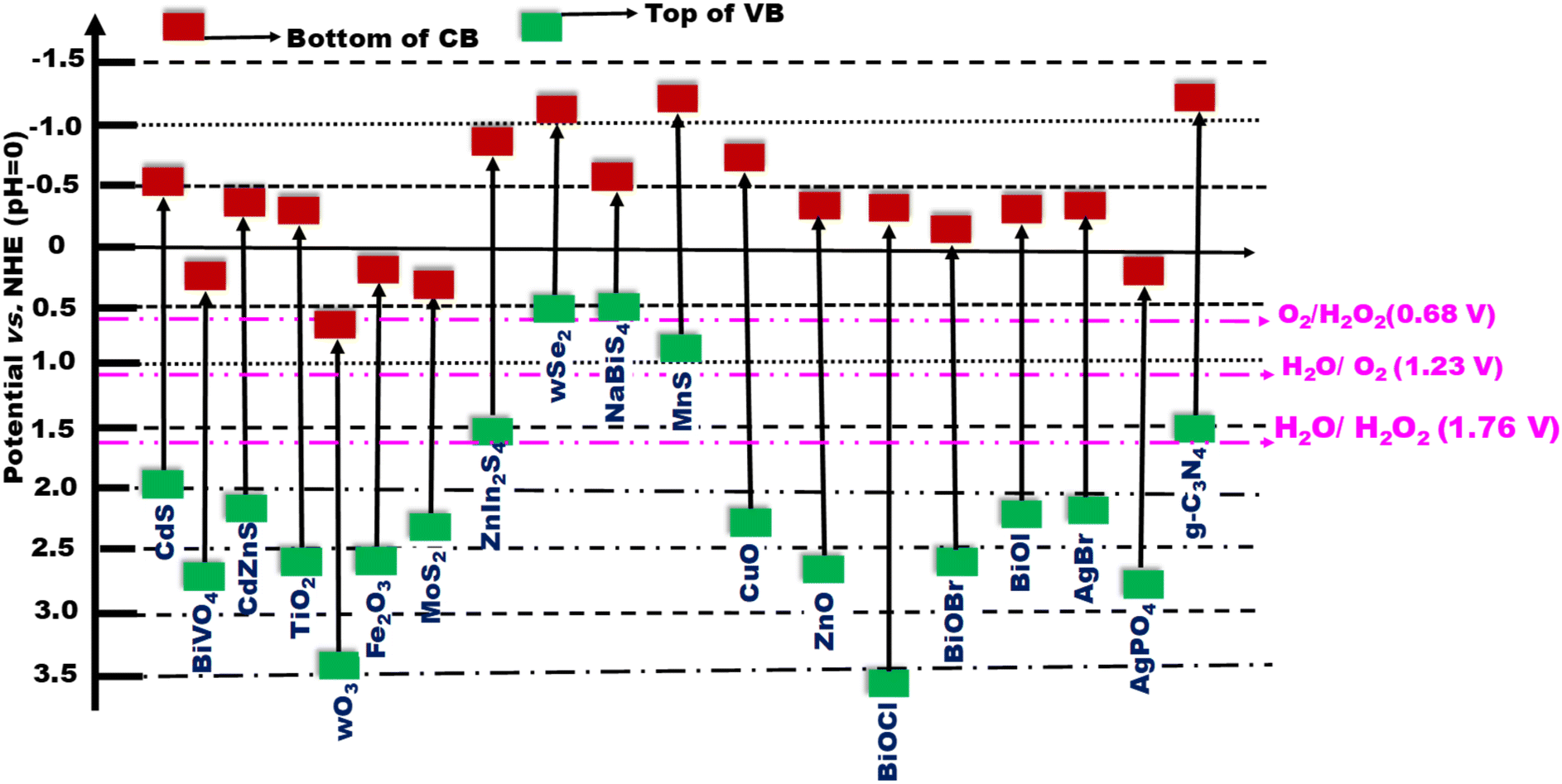 | ||
| Fig. 10 Band structures of various semiconductors frequently utilized to create Z- or S-scheme heterojunctions with g-C3N4. | ||
 | ||
| Fig. 11 (a–d) Charge transfer pathways in an S-scheme heterojunction. | ||
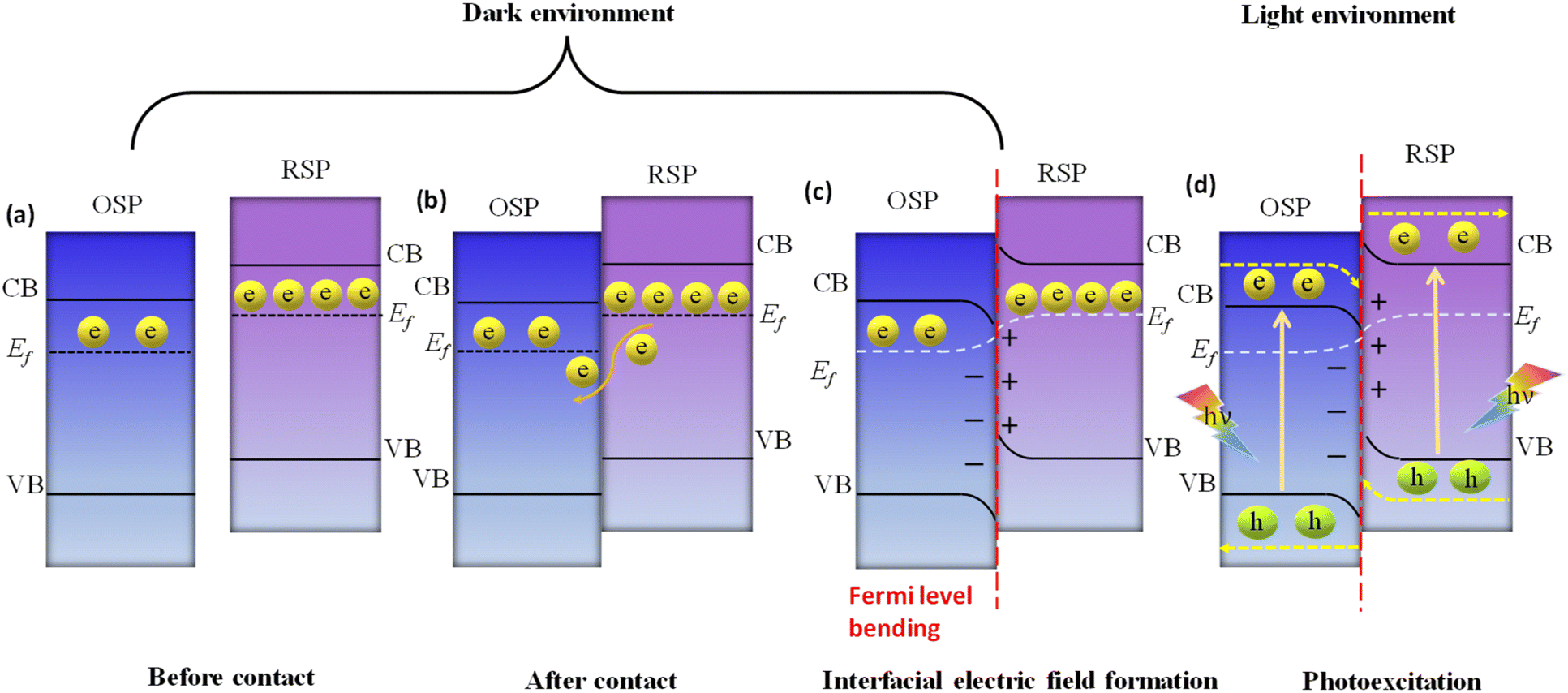
Here, we examine the development of the S-scheme heterojunction between two n-type semiconductors. Given that the RSP has a higher Ef than the OSP, as depicted in Fig. 11a, when the RSP and OSP come into contact under dark conditions, there will be a spontaneous migration of free electrons from the RSP to the OSP (Fig. 11b) via the interface to attain Fermi-level equilibrium only at the exact contact point and Fermi level bending within the interface zone developing depletion and accumulation layers of electrons at the interface in the RSP and OSP respectively, since the RSP side loses electrons and is positively charged and vice versa (Fig. 11c). There will be a gradual reduction and increment of the Fermi levels of the RSP and OSP, respectively, near the interface zone until Fermi level equilibrium is reached, as confirmed by the surface potential results. However, the Fermi level of the bulk remains as before.121
Upon close interaction, four areas emerge, each having its own unique electron density profile: the OSP bulk, the OSP interface, the RSP interface, and the RSP bulk (see Fig. 11d).24 Given that Ef is influenced by electron density, its value should differ across these four regions. An increase in electron density at the OSP interface (or a decrease at the RSP interface) leads to a corresponding elevation (or reduction) in Ef. Furthermore, the extent of the interface region is influenced by the disparity in the Fermi levels between the OSP and RSP. At the same time, upward and downward band edge bending occurs at the interface region of the RSP and OSP because of the donation and accumulation of electrons, respectively. Simultaneously, an interfacial electric field (IEF) is formed, which is directed from the RSP to the OSP. When exposed to light, electrons get promoted to the CB, while holes are formed in the VB. The photogenerated electrons in the CB of the OSP and holes in the VB of the RSP are more prone to recombine at the contact interface under the influence of five factors (i.e., interfacial built-in electric field, band edge bending, pinning of the Fermi level, the existence of a potential difference and coulombic attraction between electrons and holes), which act as the driving force for recombination. These combined influences diminish the potential to produce oxidative entities. Moreover, the probability of charge recombination within either the OSP or RSP is notably reduced in such a scenario. On the other hand, an interfacial built-in electric field, band edge bending, and coulombic repulsion inhibit double charge transfer like type-II. Meanwhile, the photogenerated electrons exhibit a greater potential in the CB of one semiconductor (RSP), whereas the photogenerated holes possess a higher potential in the VB of another semiconductor (OSP). Concurrently, electrons in the CB of the RSP and holes in the VB of the OSP migrate toward the surface of the photocatalyst due to the influence of the IEF, where catalytic reactions take place. This strategy leverages the interfacial electric field as a driving force, enabling charge carriers to be spatially separated during illumination while maintaining robust redox capabilities, providing a potent thermodynamic impetus for the production of H2O2 through photocatalysis. The S-scheme mechanism is facilitated by the synergistic interaction between two semiconductor materials, driven by an interfacial electric field. This field effectively limits the transmission distance of photogenerated charge carriers, thereby mitigating losses during their migration process. Consequently, S-scheme heterojunctions not only enhance the transfer and separation of carriers but also expedite redox reactions, thereby promoting efficient H2O2 production. Finally, weak charge carriers are removed via recombination, while the powerful photogenerated electrons and holes are conserved in the CB of the RSP and the VB of the OSP, respectively, to participate in the surface redox reaction. Furthermore, S-scheme heterojunctions spatially segregate active sites responsible for oxidation and reduction processes, leading to a substantial suppression of charge carrier recombination. Following the assembly of an S-scheme heterojunction, since the Fermi energy (Ef) within the bulk remains unaltered, photogenerated electrons maintain their original high reduction potential in the RSP, while photogenerated holes retain their initial high oxidation capacity in the OSP. Hence, the S-scheme system ensures charge separation and endows the system with a strong redox potential, which boosts the photocatalytic performance. Previously reported heterojunction systems have many contradictions based on their charge transfer mechanism, which can be solved by this principle.
4.2. Maintaining the driving force behind S-scheme transfer
Under ideal conditions, when semiconductors with distinct Fermi levels are in contact with each other, electron movement aligns these levels, as shown in Fig. 9a and b. According to this theoretical assumption, the interfacial electric field in the heterojunction should weaken and eventually vanish under dark conditions, leading to a deviation from S-scheme transfer post-light illumination due to the loss of its driving force.99,122 Contrary to this theoretical expectation, the sustained migration of photoinduced charge carriers via the S-scheme in such heterojunctions has been confirmed through numerous sophisticated characterization techniques, presenting a stark contradiction to the initial assumption. This discrepancy stems from the actual equilibrium condition of the Fermi level at the heterojunction, which significantly differs from the idealized predictions.123 In a perfect scenario, Fermi levels within a heterojunction would align without considering the impact of surface states. Yet, interfacial defects and the resulting high contact resistance in real-world heterojunctions lead to Fermi level pinning, as strongly supported by surface potential tests.124 Even when equilibrium is achieved, a persistent potential difference between the interface of two semiconductors is observed.125 Furthermore, the variance in electron density at the interface of the heterojunction causes the bending of Fermi levels, as illustrated in Fig. 11c.99 This occurs as electrons move from the Ef of the reduction photocatalyst to that of the oxidation photocatalyst, reducing electron density at the interface compared to the internal area and thus bending the Fermi levels. Such dynamics establish a dynamic equilibrium within the components of the heterojunction, ensuring a continuous driving force for the perseverance of S-scheme charge transfer.The driving force behind carrier separation within the interface of the heterojunction is a pivotal factor, traditionally considered solely due to the interfacial built-in electric field. Yet, recent research suggests that the diffusion effect contributes to charge separation, where the variance in electron concentration across the S-scheme heterojunction enables diffusion-controlled carrier separation to surpass that of the interfacial electric field of the heterojunction.126 Moreover, two critical aspects, namely, effective interfacial contact area and interfacial contact resistance, must be considered for optimizing charge separation. The effective interfacial contact area is crucial as it defines the extent of the driving force, with a larger area enhancing the carrier separation efficiency.127 On the other hand, the interfacial contact resistance significantly affects the rate at which charge carriers migrate; excessively high resistance can impede carrier movement. Therefore, improving lattice matching is essential for constructing S-scheme heterojunctions that facilitate efficient carrier migration.128
4.3. Design principles of S-scheme heterojunctions and theoretical requirements for implementing S-scheme charge transfer mechanisms
In general, to assemble an S-scheme heterostructure, two individual n-type semiconductors are chosen, which serve as the oxidation semiconductor photocatalyst (OSP) and the reduction semiconductor photocatalyst (RSP).129 However, as research and development progress, several questions arise: (i) Can p-type based semiconductors also be used in the creation of S-scheme photocatalysts? In this context, to answer the above question, several reports proposed that S-scheme photocatalysts could definitely be established by using p-type semiconductors.99,130 Which means both the RSP and OSP can be n-type or p-type semiconductors and vice versa. For constructing an S-scheme heterojunction, the major crucial point is that the RSP semiconductor has a higher CB and Fermi level than those of the OSP. Mainly, four feasible scenarios, (1) n–n heterojunction, (2) p–n heterojunction, (3) n–p heterojunction, and (4) p–p heterojunction, are valid options to meet the aforementioned condition. It is noteworthy that experimental verification has been obtained for all four forms of heterojunctions.131,132 Type-II and S-scheme charge transfers in semiconductor heterojunctions are often confused with each other because their formation mechanisms and the interleaving of semiconductor energy bands, along with electron transfer dynamics in the dark state, are very similar. In the n-type semiconductor heterojunction, when the CB of PS1 is more negative and its Fermi level is higher than those of PS2, electron transfer follows an S-scheme, not type-II, as electrons move from the higher Ef of PS1 to the lower Ef of PS2 due to the unbalanced state of Ef under dark conditions, creating an interfacial electric field (IEF) directed from PS1 to PS2. This setup makes type-II charge transfer impossible: the interfacial energy band of PS1 with a higher Ef bends upward, hindering electron movement towards the interface and favouring hole migration, while the interfacial band of PS2 with a lower Ef bends downward, facilitating the opposite. Consequently, this mismatch in band bending and electric field direction exclusively supports S-scheme electron transfer, as shown in Fig. 12a and b. This principle similarly applies to two p-type semiconductors that are in contact, where a higher Fermi level and more negative CB position of PS1 compared to PS2 also lead to S-scheme transfer, as illustrated in Fig. 12c and d.123 Additionally, when an n-type and a p-type semiconductor come into contact, the relationship between their Fermi levels and CB positions determines the charge transfer type: a higher Fermi level and more negative CB in the n-type results in S-scheme transfer (Fig. 12e and f), whereas a more negative CB and lower Fermi level in the p-type align with type-II transfer conditions, as the interfacial electric field direction aligns with the band bending state (Fig. 12g and h).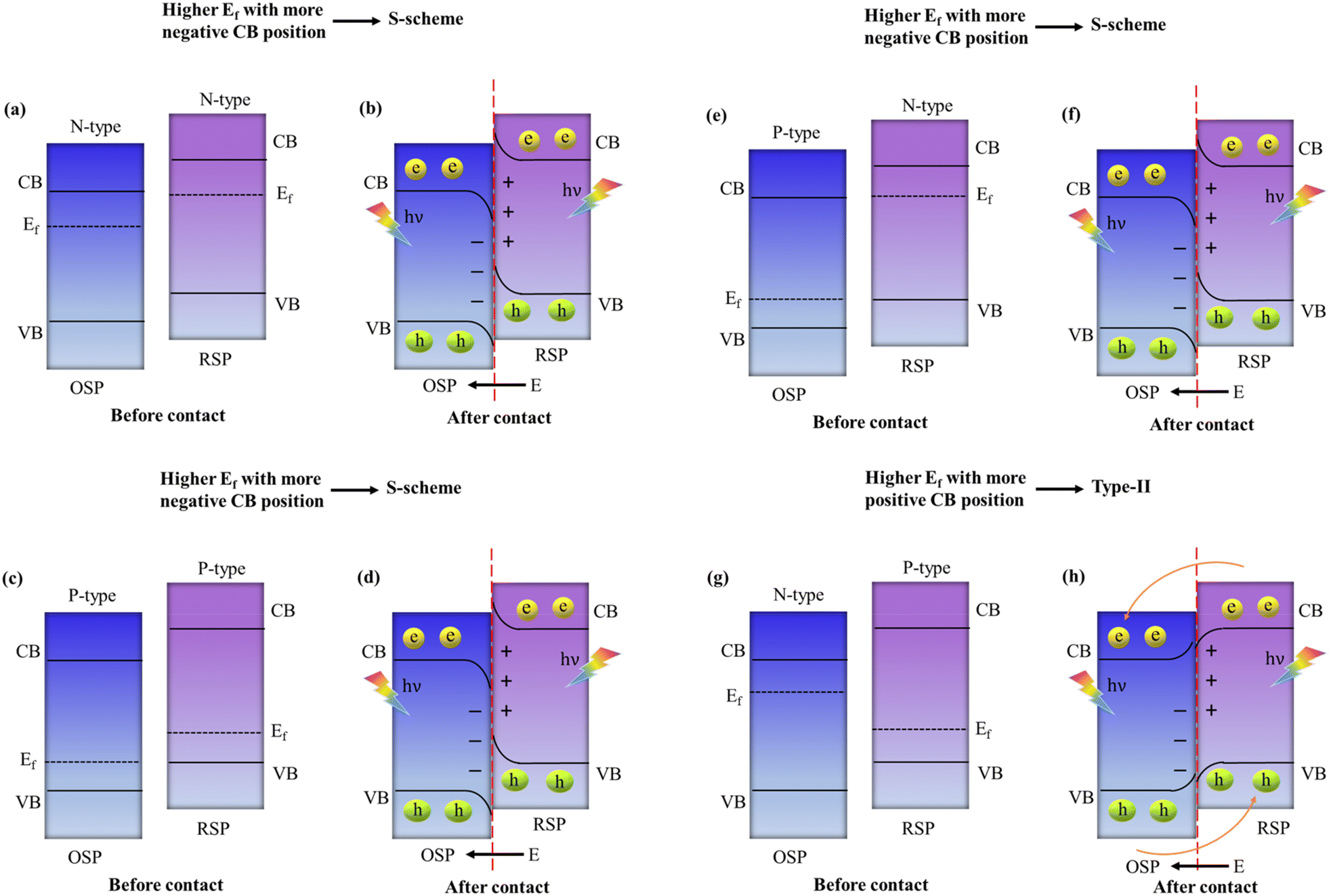 | ||
| Fig. 12 Criteria for initiating (a–f) S-scheme and (g and h) type-II charge transfer mechanisms. | ||
In summary, the electron transfer type between semiconductors that are in contact in a heterojunction hinges on the relative positions of their CB and Fermi levels. For two semiconductors that are in contact, S-scheme transfer occurs when the more negative CB of the semiconductor accompanies a higher Fermi level, whereas type-II transfer takes place when the more negative CB of the semiconductor accompanies a lower Fermi level. It is important to highlight that the charge transfer direction is determined by the positions of the energy bands of the constituent semiconductor within the heterojunction, and this is not influenced by the type of semiconductor involved.
4.4. Tactics for Fermi level adjustment to facilitate type-II to S-scheme transfer conversion
Adjusting the Fermi levels between semiconductors can significantly influence the electron transfer mechanisms, facilitating a shift from type-II to S-scheme transfer. The direction of band bending at the interface is critical for determining the nature of electron transfer, whether it be type-II or S-scheme. By modifying the relative positions of Ef, one can change the band bending direction, thereby switching the electron transfer mechanism and enhancing carrier separation efficiency.133–135 This strategy of Fermi level adjustment is a key method for controlling carrier dynamics within heterojunctions. For example, Huang and colleagues successfully altered electron transfer in a C3N4/W18O49 system by adjusting the relative positions of Ef.136 Meanwhile, PS1 and PS2 are in contact, and an electric field is generated from PS2 to PS1 at their interface due to the lower Fermi level of PS1 than that of PS2. This field directs electrons to consumption at PS2 while accumulating at PS1, causing the interfacial energy band to bend downwards and upwards for PS1 and PS2, leading to a type-II electron transfer mechanism. Interestingly, the study found that adsorbing triethanolamine on PS1 raised its Fermi level above that of PS2. This adjustment reversed the band bending directions, upward for PS1 and downward for PS2, facilitating an S-scheme charge transfer. This discovery highlights a method for controlling the pathways of carrier transfer.123 In addition to molecular adsorption, non-metallic doping is another effective method for altering Fermi levels. For instance, Kim and co-workers enhanced the Fermi level in C3N4 by doping with boron. They observed that when boron-doped C3N4 was combined with ZnO, resulted in a reversed interfacial band bending state, providing a shift in the charge transfer mechanism.134 Moreover, the creation of interfacial chemical bonds can also adjust the Fermi levels and change the band bending directions, offering another avenue for control.135 While it is theoretically feasible to transition the electron transfer dynamics from type-II to S-scheme through Fermi level adjustments, in practice, the Fermi level in semiconductor heterojunctions may remain fixed due to significant contact resistance. Consequently, a comprehensive set of characterization tools is recommended to verify the potential for actual Fermi level modification.4.5. Enhancing the driving force behind S-scheme transfer: approaches and strategies
The interfacial electric field is key to driving S-scheme charge transfer effectively, with its strength being crucial for charge carrier separation efficiency.137–139 To boost this strength, various strategies have been devised, including the development of dual S-scheme heterojunctions.140 Moreover, these heterojunctions are also involved in three distinct photocatalyst systems to address the challenges such as less interfacial contact and the weak driving force seen in single component S-scheme heterojunctions. Under dark conditions, when oxidation-type (OSP) and reduction-type (RSP) photocatalysts are juxtaposed, energy band bending and the creation of an internal electric field occur at the interface, resulting in the accumulation of electron and hole pairs in the OSP and RSP, respectively. Upon illumination, the electric field drives the electrons from the CB of the OSP to recombine with holes present in the VB of the RSP, facilitating the accumulation of high redox potential electron and hole pairs in the CB of the RSP and the VB of the OSP, respectively. This dual arrangement creates two interfacial electric fields, providing excellent spatial charge carrier separation and transfer compared to that in single S-scheme based heterojunctions, thereby significantly boosting the photocatalytic efficiency.141 Moreover, precisely increasing the Fermi level gradient and widening the Fermi level gap serve as another potent method to amplify both the strength of the interfacial electric field as a driving force, and expanding the space–charge region, thereby strongly promoting more directed carrier transport.125 Additionally, creating interfacial chemical bonds further strengthens the built-in electric field and speeds up charge carrier transport, demonstrating a multifaceted approach to improving the efficacy of S-scheme electron transfer mechanisms.142–144 Moreover, it should be noted that, beyond semiconductor materials, organic compounds with comparable lowest unoccupied molecular orbital (LUMO) and highest occupied molecular orbital (HOMO) energy levels can also be integrated into S-scheme heterojunctions.145In summary, the initial concept of S-scheme heterojunctions has evolved from n–n type to encompass various possibilities. This expansion opens up new avenues for the design and manipulation of diverse S-scheme heterojunctions in future research and applications for effective charge carrier separation and enhanced photocatalytic activities. For clarity, Table 1 summarizes the specific differences between direct Z-scheme and S-scheme heterostructures. Table 2 details a comparison of different types of heterojunctions, focusing on their band positions and mechanisms for charge transfer and separation. Additionally, we discuss several characterisation methods to verify direct Z- or S-scheme charge dynamics and provide multiple instances of g-C3N4-based direct Z- or S-scheme heterojunction systems for photocatalytic H2O2 evolution.
| Entry | Direct Z-scheme | S-Scheme |
|---|---|---|
| 1 | When semiconductors with distinct Fermi levels are in contact with each other, the electron movement aligns these levels. According to this theoretical assumption, the interfacial electric field in the heterojunction should diminish over time and eventually vanish under dark conditions, meaning that carrier migration post-light irradiation would not follow the direct Z-scheme due to the loss of its driving force, which is clearly at odds with experimental observations from surface potential tests | Interfacial defects and the resulting high contact resistance in actual synthesized heterojunctions lead to Fermi level pinning, as strongly supported by surface potential tests. Even when equilibrium is achieved, a persistent potential difference between the interface of two semiconductors is observed. Furthermore, the variance in electron density at the interface of the heterojunction causes interfacial Fermi level bending. This occurs as electrons move from the Ef of the reduction photocatalyst to that of the oxidation photocatalyst, reducing electron density at the interface compared to the internal area and thus bending the Fermi levels at the interface. Such dynamics establish a dynamic equilibrium within the components of a heterojunction, ensuring the continuous driving force for the perseverance of S-scheme charge transfer. The Fermi level of the bulk remains unchanged, allowing photoinduced charge carriers to retain their original high redox potential in the respective semiconductor components |
| 2 | It is silent about the nature of the charge transfer direction | The charge carrier transfer direction is determined by the positions of the energy bands of the constituent semiconductors within the heterojunction, and is not influenced by the type of semiconductor involved |
| 3 | It does not address the intensity of interfacial band edge bending, which creates an additional potential barrier to inhibit type-II like charge transfer | The intensity of interfacial band edge bending is directly related to the Fermi level gap between two semiconductors. The literature reports that type-II charge transfers often occur when semiconductors with small Fermi level differences form an S-scheme heterojunction. In such cases, the reduction photocatalyst is preferentially excited, causing both type-II and S-scheme transfers to coexist. Conversely, a large Fermi level difference dominates the S-scheme migration path |
| 4 | It does not address the factors influencing the extent of the interfacial space–charge region and the strength of the interfacial electric field | The strategy of precisely increasing the Fermi level gradient and widening the Fermi level gap between OSP and RSP amplifies both the strength of the interfacial electric field as a driving force and expands the interfacial space–charge region, thereby strongly promoting more directed carrier transport |
| 5 | The direct Z-scheme does not address the switching of carrier transfer | By modifying the relative positions of the Fermi levels, one can change the interfacial band bending direction, thereby switching the electron transfer mechanism, which is a key method for controlling carrier dynamics within the heterojunctions |
| Entry | Type | Charge transfer mode and rate | Scope of implementation | Benefits | Main challenges |
|---|---|---|---|---|---|
| 1 | Type-II | Nil and moderate | — | Improve charge separation efficiency | • Rapid recombination |
| • Sluggish charge migration | |||||
| • Reduced redox potential | |||||
| • Charge-separation efficiency at the cost of reduced redox ability | |||||
| 2 | Traditional liquid phase Z-scheme | Redox ion pairs and slow | Liquid phase | Excellent light transmittance | • Strong pH dependency (harsh operational conditions): |
| (I) The IO3−/I− system operates effectively only at pH levels above 9 due to the formation of inactive I3− at lower pH values | |||||
| (II) Fe3+/Fe2+ remains stable only under acidic conditions with a pH below 2.5, as Fe(OH)3 precipitates at higher pH values | |||||
| • The shielding effect is due to light absorption by the redox mediator. For example, aqueous Fe3+ absorbs light strongly up to 464 nm | |||||
| • Reverse reaction arises from the back donation of charge carriers | |||||
| • Both PS I and II are needed, each with appropriate oxidation and reduction capabilities, to initiate redox reactions involving various A/D pairs | |||||
| • Limited to the liquid phase due to the ionic characteristics of redox pairs, hindering scalability potential | |||||
| 3 | All-solid-state Z-scheme | Conductor and moderate | Liquid and gas phases | Enhance conductivity, improve electron transfer, and establish excellent interfacial connections | • Expensive manufacturing and light-blocking properties |
| • While there is flexibility in designing the PS-A/D-PS system, creating the PS-C-PS system is more restrictive because both the PS and electron mediator must be integrated into a single unit | |||||
| • Expensive electron mediators, such as metals and nanocarbons, are necessary to regulate directional electron transfer | |||||
| • Appropriate preparation methods are vital for diverse PS-C-PS system development: | |||||
| (I) To guarantee close interfacial contact between the PS and electron mediator | |||||
| (II) To achieve a balance between the level of reduction in the nanocarbon mediator and the overall hydrophilicity of the composites | |||||
| 4 | Direct Z-scheme | Built-in interfacial electric field and fast | Liquid and gas phases | Strong redox power | • There is considerable uncertainty regarding the mechanism of electron transfer |
| • Prevent the recombination of electron–hole pairs | |||||
| • Quantum efficacy | |||||
| • PS-PS systems exhibit less efficient directional electron transfer compared to PS-C-PS systems due to the lack of an electron mediator | |||||
| • The creation of an interfacial electric field heavily relies on the characteristics of PS I and PS II, where PS I possesses a higher Fermi level than PS II | |||||
| • Different direct Z-scheme verification tests are necessary to differentiate from heterojunction type photocatalytic systems | |||||
| 5 | S-Scheme | Built-in interfacial electric field and fast | — | Adjustable built-in interfacial electric field intensity, stable interfacial carrier transportation, strong redox capability | • Mainly confined to powdered photocatalysts, not applicable to photochemistry and solar cells with an external circuit; insufficient management of reaction thermodynamics and dynamics in S-scheme photocatalysts |
5. Characterization techniques for direct Z- or S-scheme heterojunction photocatalysts
To date, three characterization techniques (i.e., ex-situ/in situ light illuminated XPS, surface potential measurement by in situ-KPFM and fs-TAS) have been developed to directly confirm the formation of direct Z and S-scheme heterojunctions.5.1. In situ/ex situ illuminated X-ray photoelectron spectroscopy (ISIXPS)
To confirm the pathway of photogenerated electron–hole transfer in S-scheme heterojunctions, the in situ light illuminated XPS (ISIXPS) technique was first suggested by Yu and colleagues and is recognized as a particularly effective and influential method. XPS, or X-ray photoelectron spectroscopy, is a sophisticated method for surface examination that offers insights into the elemental composition, content percentages, electronic states, and bonding patterns of different materials.146 This technique is useful for detecting alterations to the elemental binding energy when the chemical surroundings, i.e. electron density of an element, are changed. For instance, when there is a reduction in the electron density of a particular element, it results in a positive shift in binding energy, whereas a rise in electron density causes a negative shift.In S-scheme photocatalytic systems upon light irradiation, both the OSP and RSP of the heterojunction experience a reduction or increment in electron density because of the distinctive exciton migration dynamics of the S-scheme, which is caused by the movement of photoexcited electrons from the CB of the OSP into the VB of the RSP, influenced by an interfacial electric field (IEF). Consequently, this process leads to a lowered binding energy in the RSP and an increased binding energy in the OSP under illumination conditions. XPS characterization thus facilitates monitoring of the directional charge migration related to specific elements, providing a detailed analysis of the associated OSP and RSP semiconductor structures.107
During XPS analysis, a solid specimen, connected electrically to apparatus, will be placed in an ultrahigh vacuum environment, typically maintained below 10−9 Pa. Upon exposure of the sample surface to high-energy X-ray beams, such as 1486.6 eV for Al Kα X-rays, the electrons gain enough energy to be ejected from the surface into the vacuum, leading to atom ionization. This phenomenon is also known as the photoelectric effect (as illustrated in Fig. 13a).
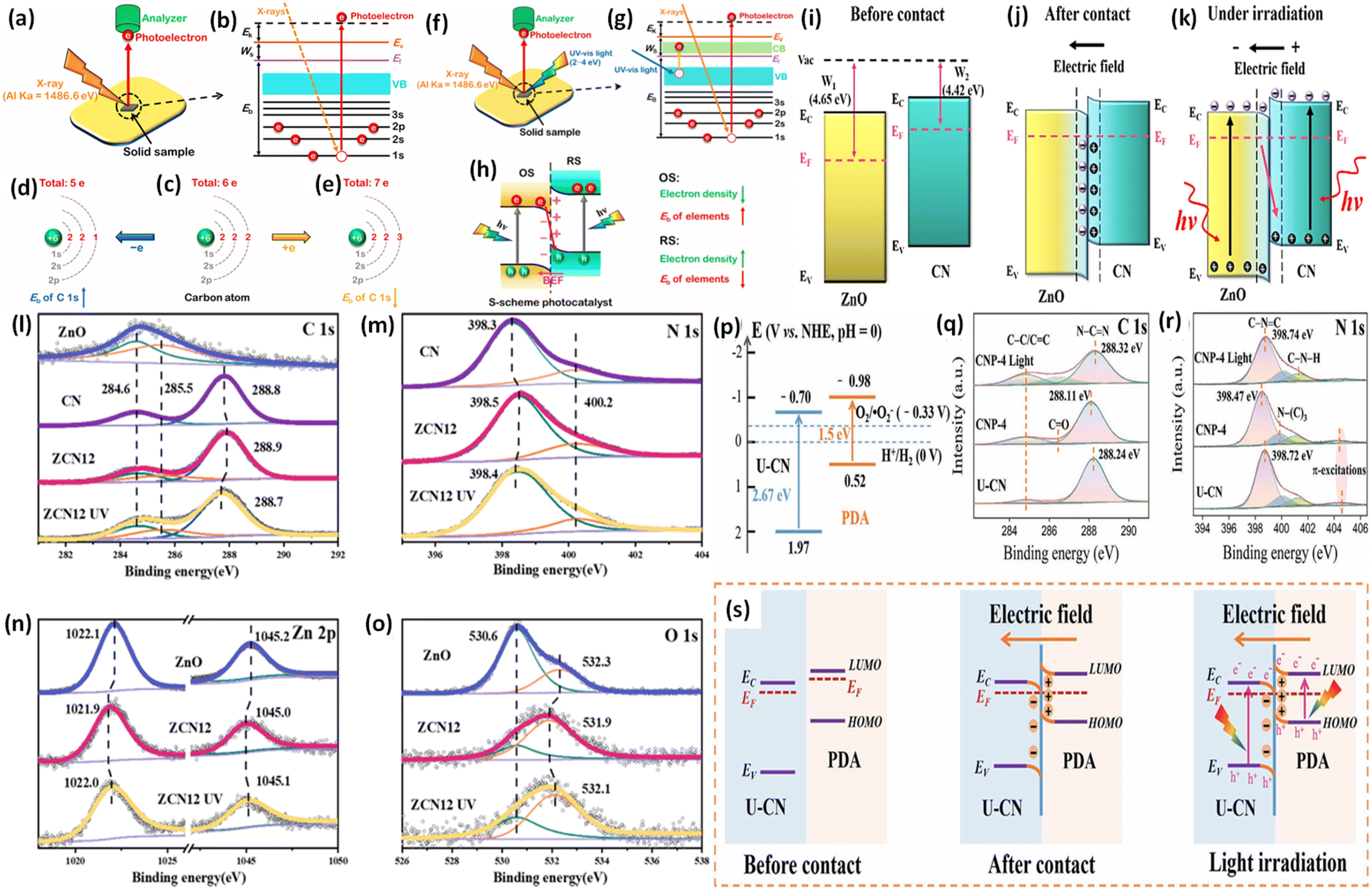 | ||
| Fig. 13 (a) Illustration of the XPS measurement principle. (b) Photoelectron production during XPS studies under X-ray irradiation. (c–e) Impact on the binding energy (Eb) of C 1s following electron loss or gain. (f) Detailed schematic of the working principle of ISIXPS analysis under dual irradiation with UV-visible light and X-rays. (g) Electron excitation processes during ISIXPS measurements under simultaneous UV-visible light and X-ray irradiation. (h) Variations in electron density and elemental binding energy (Eb) of an S-scheme heterojunction photocatalyst under photoexcitation. The figures are adapted with permission from ref. 149. Copyright 2022, ACS. (i–k) Light-induced S-scheme charge transfer process between ZnO and CN. Spectra of (l) C 1s, (m) N 1s, (n) Zn 2p, and (o) O 1s in ZnO, CN, and ZCN12 measured using high-resolution ISI-XPS in the dark and under UV light illumination. The figures are adapted with permission from ref. 151. Copyright 2021, ACS. (p) Arrangement of band structures of U-CN and PDA. In situ XPS measurements of (q) C 1s and (r) N 1s for U-CN and CNP-4. (s) The S-scheme charge transfer process occurring within the CNP-4 heterojunction. The figures are adapted with permission from ref. 152. Copyright 2023, Wiley-VCH. | ||
These ejected electrons, or photoelectrons, possessing varying kinetic energies (Ek), traverse through the XPS device's analysis slit. The ensuing electron current, indicative of the rate of photoelectron emission and contingent on Ek, is then documented (Fig. 13b).147 On a microscopic scale, atomic orbital energies are quantized, meaning every electron possesses a distinct energy level. Based on their proximity to the nucleus, ranging from near to far, the atomic orbitals can be categorized into distinct types like 1s, 2s, 2p, and 3s. The energy needed to knock out an electron from the material is termed its ionization or binding energy (Eb). Notably, electrons residing in inner orbitals (e.g., 1s) exhibit a higher Eb than those in outer orbitals (e.g., 2p). The inner-shell electrons can be excited by X-rays due to their extraordinarily powerful energy (for example, the energy (hν) of Al Kα X-rays is 1486.6 eV). With enough energy, the photoexcited electrons can overcome the barriers of the sample's work function (Ws) and binding energy (Eb), allowing them to eventually be ejected from the surface of the sample with Ek. As a result, Eb can be determined using the following eqn (3):148
| Eb = hv − Ek − Ws | (3) |
In XPS experiments, Eb of the inner-shell electrons is determined by measuring the kinetic energy (Ek) of emitted photoelectrons. By analyzing this Eb value, one can discern the elements present in the sample, as each element typically possesses a distinct Eb value. A shift or change in Eb of electrons in the inner shell, also called the chemical shift, occurs when an electron loses or gains an electron in the outer orbitals of the atom. For instance, a carbon atom contains two electrons in each of its 1s, 2s, and 2p orbitals, as depicted in Fig. 13c. The nucleus of carbon contains six protons and six neutrons, hence possessing six positive charges. Two fundamental forces bind the nuclei and electrons of atoms together: the coulombic force, which is caused by protons, and the nuclear force, which is influenced by protons as well as neutrons. As the carbon nucleus has a fixed number of protons and neutrons, the overall attractive force remains consistent. When a carbon atom loses a valence electron from the 2p orbital, its electron count is reduced to five (as shown in Fig. 13d). This loss enhances the force exerted on each remaining electron, resulting in an increase in Eb of the C 1s electron. On the other hand, if a carbon atom gains an electron in the 2p orbital, its electron count rises to seven, reducing the force per electron and thereby decreasing Eb of C 1s (illustrated in Fig. 13e). Electron acceptance or loss can be challenging in some situations. For instance, in covalent compounds, atomic orbitals hybridize to form new molecular orbitals, altering the electron distribution and density. Hence, Eb of C 1s will likewise change depending on the chemical environment in which the carbon atom is found. When the electron density around a carbon atom is reduced, Eb of C 1s is increased and vice versa. While these shifts in Eb might be subtle compared to direct electron acceptance or loss, they remain detectable through XPS techniques.149 ISIXPS emerges as a vital tool to confirm the electron transfer routes in S-scheme semiconductor materials. In this technique, a combination of high-energy X-rays and ultraviolet–visible (UV-vis) light is employed to irradiate the sample's surface, as depicted in Fig. 13f. The resulting excited photoelectrons with kinetic energy (Ek) then leave the sample surface, enter the vacuum, and are captured by the analyzer. To delve deeper into the role of UV-vis light during ISIXPS experiments, electron excitation processes are detailed in Fig. 13g. The inner-shell electrons can be excited by X-rays with very high energy (Al Kα = 1486.6 eV), allowing them to overcome the barriers of binding energy (Eb) and work function (Ws) to emerge as free photoelectrons carrying Ek ultimately. In contrast, the energy of UV-vis light (between 2 and 4 eV) is significantly lower compared to X-rays. As a result, it can only excite the valence electrons to move from the VB to the CB, while ensuring these excited electrons remain bound to the atom's nucleus, as shown in Fig. 13g. Therefore, following excitation of the outer-shell valence electrons, the analyzer in ISIXPS measurements detects Eb of electrons in the inner shell. As valence electrons move from the VB to the semiconductor's CB, photogenerated electrons are formed through the excitation process. These energized electrons can transfer between two semiconductors while residing in them. Like conventional XPS analysis, the loss of photogenerated electrons causes a reduction in electron density and an increase in elemental binding energy (Eb), while the acceptance of photogenerated electrons results in increased electron density and a decrease in Eb. As per the previous discussion, the proof of electron movement in S-scheme heterojunctions under dark conditions and during light exposure can be obtained by combining normal XPS and ISIXPS data analysis. The variation in Eb of an individual element can be utilized to approximate the alteration in electron density. This enables the determination of electron movement directions in S-scheme heterojunctions.150 Conventional XPS reveals the electron flow dynamics between the RSP and OSP upon intimate interaction. Given that the RSP possesses a higher Fermi level (Ef) than the OSP, electrons migrate from the RSP to OSP after coming into contact. Analyzing the RSP post-contact reveals a decline in its electron density compared to its pre-contact state, which marks an upsurge in Eb of its constituent elements. In contrast, the post-contact OSP showcases an enhanced electron density in the S-scheme photocatalytic system compared to its initial state, implying a consequent decrease in its Eb. Additionally, the migration pathway of photoexcited electrons in the S-scheme heterojunction can be verified using ISIXPS. When subjected to light, photoinduced electrons in the CB of the OSP are driven to move to the VB of the RSP by the interfacial built-in electric field (IEF), resulting in a decrease in the electron density of the OSP and an increase in the RSP as visualized in Fig. 13h. As a result, under light irradiation, the elemental Eb of the OSP increases, and that of the RSP decreases in the S-scheme photocatalyst.149
On the basis of XPS and ISIXPS mechanistic analyses, we present two case studies that utilize ISIXPS to unveil the charge transfer dynamics within S-scheme heterojunctions. For example, Liu's group prepared an S-scheme heterojunction between MOF-derived ZnO and g-C3N4 (CN) through a two-step calcination process.151 The CB and Fermi level (Ef) of CN are positioned higher than those in ZnO, making them suitable for the development of an S-scheme heterojunction. To validate charge carrier transfer in the ZnO/CN (ZCN-12) S-scheme heterojunction photocatalyst, both conventional XPS and ISIXPS tests are carried out. When ZnO and CN come into close contact, there will be spontaneous diffusion of electrons from CN to ZnO via their interfaces because Ef of CN is higher than that of ZnO to attain Fermi level equilibrium at the exact contact point, developing depletion and accumulation regions of electrons at the interface of CN and ZnO, respectively, since CN loses electrons and becomes positively charged and vice versa. At the same time, upward and downward band bending occurs at the interface region of CN and ZnO because of the donation and accumulation of electrons, respectively. Simultaneously, an interfacial electric field is formed, which is directed from CN to ZnO. Therefore, relative to CN, the C 1s and N 1s peak positions in the ZCN12 composite move to a higher Eb, implying a reduction in electron density within the CN component of ZCN12 (as seen in Fig. 13l and m). On the other hand, when set against ZnO, the Zn 2p and O 1s peak positions in the ZCN12 composite tend to a lower Eb (illustrated in Fig. 13n and o). This suggests increased electron density in ZnO, implying that ZnO receives electrons from CN. These shifts in electron density between CN and ZnO confirm electron migration from CN to ZnO after contact in the absence of light (Fig. 13i and j). During ISIXPS experiments, the sample surface is exposed to UV light with a wavelength of 365 nm. In the ZCN12 compound, there is a noticeable shift to a lower binding energy (Eb) for the C 1s and N 1s peaks (as seen in Fig. 13l and m), whereas the Zn 2p and O 1s peaks shift to a higher Eb (as illustrated in Fig. 13n and o). This suggests an increase in electron density in the CN regions and a decrease in the ZnO part. Such observations reinforce the idea of photogenerated electron migration from ZnO to CN when subjected to UV light, a process visually mapped out in Fig. 13k. Through the findings from both regular XPS and ISIXPS, the electron movement patterns in the ZnO/g-C3N4 S-scheme heterojunction are clearly demonstrated. Similarly, Zhang et al. successfully developed a 2D/2D close contact S-scheme heterojunction system, C3N4/PDA (CNP), through the in situ self-polymerization method, showing an excellent H2O2 production efficiency.152 The band diagrams for U-CN and PDA are illustrated in Fig. 13p. When compared to individual U-CN, the observed C 1s and N 1s peaks in CNP-4 move to a lower Eb after contact in the absence of light (as depicted in Fig. 13q and r). No significant variations are evident post-PDA deposition, implying that the primary peaks in CNP-4 are derived from U-CN. These findings indicate an increased electron density in U-CN and a reduced one in PDA within CNP-4. This validates electron movement from PDA to U-CN when they come into contact without light exposure (see Fig. 13s). When exposed to light, the C 1s and N 1s peaks of CNP-4 exhibit a shift toward an elevated Eb, as seen in Fig. 13q and r. This shift suggests a reduction in electron density within U-CN and a corresponding increase in PDA, signifying the migration of photogenerated electrons from U-CN towards PDA, a process supported by the IEF as demonstrated in Fig. 13s. Through the combined insights from XPS and ISIXPS studies, the pathway of electron transfer in the C3N4/PDA (CNP) S-scheme heterojunction is elucidated both in the absence of light and upon photoactivation. To sum up, XPS and ISIXPS stand out as highly sensitive tools capable of identifying minor shifts in elemental Eb resulting from variations in electron density, in both the absence and presence of light. These shifts in elemental Eb effectively represent the interactions and electron movements between two components. Additionally, XPS and ISIXPS serve as widely applicable techniques for tracking electron transfer within heterojunction photocatalysts.
5.2. Surface potential
Atomic force microscopy (AFM) emerged in 1985, pioneered by scientists at IBM.153 This cutting-edge technique is renowned for delivering detailed images and insights into surface characteristics and material properties on the nanoscale. Essentially, AFM operates by using a probe to “sense” or “interact” with a material's surface, enabling evaluations of its morphology, roughness, and composition. An advanced extension of AFM is Kelvin probe force microscopy (KPFM). This technique broadens the horizons of AFM by measuring the surface potential or work function of catalysts.154 The above can be achieved when employing a conductive tip in AFM followed by applying a minor amount of alternating current voltage (VAC) inside the tip as well as the material under observation (Fig. 14a). This process helps in measuring the contact potential difference. Furthermore, a bias voltage (VDC) enables the measurement of the electrostatic force through VCPC with the conductive tip. KPFM provides insights into the complex electrical characteristics and charge orientation of the material's surface.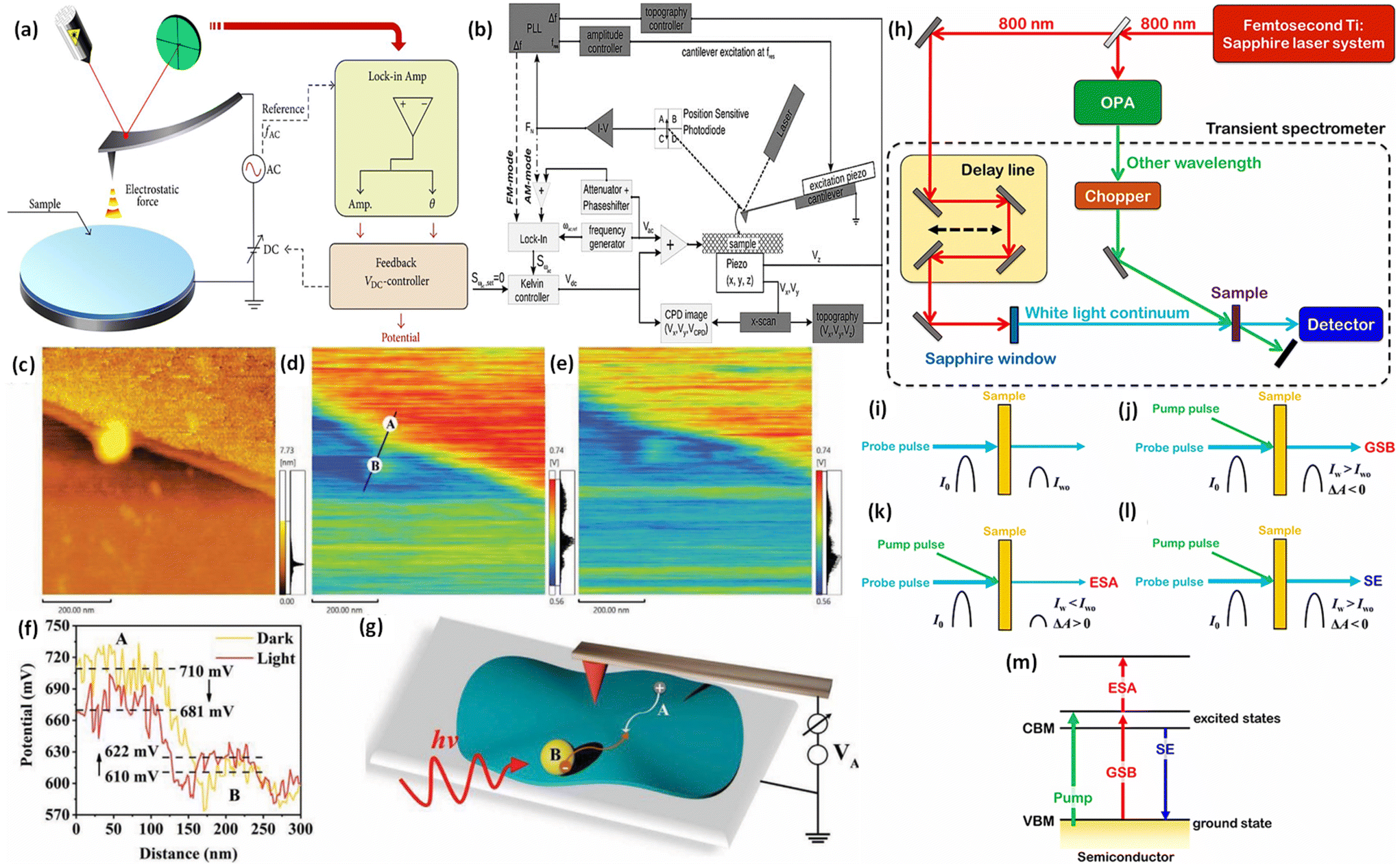 | ||
| Fig. 14 (a) In situ AFM-KPFM principles and operation. (b) A typical beam deflection setup for AFM. The figures are adapted with permission from ref. 156. Copyright 2024, Elsevier. (c) AFM picture from the CP photocatalytic system. Surface potential variation in CP in the dark (d) and under light illumination (e). (f) The surface potential measured through line scanning. (g) Schematic representation of in situ light irradiated KPFM. The figures are adapted with permission from ref. 157. Copyright 2021, Wiley-VCH. (h) Schematic illustration of the operation of the fs-TAS instrument. The intensity of the probe pulse subsequent to traversing the specimen (i) in the absence of the pump pulse and (j–l) with it. Three distinct signals, GSB, ESA, and SE, can be discerned through the utilisation of pump pulse excitation. (m) Correlations between relaxation and electron transition pathways in the semiconductor. The figures are adapted with permission from ref. 161. Copyright 2023, RSC. | ||
Merging the functionalities of AFM and KPFM in a singular device provides dual advantages: it captures both the physical topography and the electrical attributes at the nanoscale. Such a combination becomes pivotal when delving into surface electronic characteristics, including aspects like charge deviation distribution, surface potential fluctuation, and work function difference. This integrated AFM-KPFM approach enables a richer exploration of how surface structures are interlinked with their local electronic attributes, as shown in Fig. 14b. The localized surface potential, represented as φ = φ (x, y), is intimately connected with the electronic distribution over the material. One is able to derive this potential through counterbalancing the electrostatic force across the tip as well as the specimen. By adjusting the bias voltage, one can identify the surface potential at particular points (local surface potential) over the specimen.155Fig. 14b illustrates a typical depiction of beam deflection AFM and demonstrates the ability of AFM-KPFM to offset electrostatic forces by applying a bias voltage to the specimen. Utilizing in situ atomic force microscopy in combination with Kelvin probe force microscopy (AFM-KPFM) in potential mode offers a way to assess the charge dynamics, specifically examining the movement of photoexcited electrons from the oxidation semiconductor to the reduction semiconductor of a heterojunction photocatalyst when exposed to light.156 Such a transfer is evident from the enhanced surface potential on the oxidation semiconductor post-light exposure, supporting the existence of the S-scheme heterojunction. KPFM imaging was utilized to visualize the separation of excitons within an S-scheme heterojunction photocatalyst. The utilization of in situ-KPFM has emerged as a significant method to investigate charge separation as well as transfer mechanisms during light irradiation. This approach can determine the distribution of surface potential at heterogeneous interfaces. Typically, when a semiconductor receives electrons, its Fermi level rises, leading to a subsequent reduction in surface potential. Conversely, when it loses electrons, the Fermi level goes down, resulting in an increase in surface potential. During the S-scheme charge carrier migration mechanism, electrons shift from the OSP to the RSP under light excitation. This induces electron gain in the RSP and loss in the OSP. As a result, it is simple to determine the direction of electron transfer by monitoring surface potential variations. In this domain, Cheng and colleagues unveiled the spatial dynamics of charge separation and movement in a CdS/polymer (CP) photocatalyst using in situ-KPFM, as depicted in Fig. 14(c–g).157 In Fig. 14d and f, in the absence of light, the potential variation on the surface of the conjugated polymer PT (point A) as well as CdS (point B) was approximately 100 mV. This suggests an interfacial electric field (IEF) moving from PT (point A) to CdS (point B), acting as the driving force for electron movement. When exposed to light, there was a noticeable decline in the surface potential at point A (PT), whereas the surface potential at point B (CdS) increased, indicating the transition of photoexcited electrons from CdS towards the polymer.
5.3. Transient absorption spectroscopy (TAS)
Transient absorption spectroscopy (TAS) is a valuable tool for investigating ultrafast electron dynamics within photoexcited semiconductors, offering temporal resolution ranging from femtoseconds to picoseconds. The femtosecond transient absorption spectroscopy (fs-TAS) method is frequently employed as a “pump–probe” technique, offering a potent means to investigate the electronic state configuration and charge dynamics in semiconductor materials. More specifically, it offers a dynamic representation of carrier occupation across semiconductor energy levels over time.158 Furthermore, fs-TAS unravels the intricate relaxation pathways through which photoexcited electron transfer occurs from the high energy CB to the low energy VB of a photocatalyst.159 By analyzing fs-TAS data, the charge dynamics of heterojunction photocatalysts can be revealed, allowing us to determine the cause of enhanced catalytic efficiency.160 In practical fs-TAS experiments, a monochromatic laser known as a “pump pulse” of high energy is used to excite a semiconductor photocatalyst. Subsequently, a white pulse called a “probe pulse” of low energy is employed to estimate the number of photoinduced charge carriers in excited CB and ground state VB energy levels. The time delay between the laser and the white pulse is adjusted using an optical delay line. By using an optical delay line, the time delay between the laser and the white pulse can be monitored. The relaxation process is explained by analyzing the fluctuation between the number of photoexcited charge carriers and the delay time.161 The fs-TAS setup consists of three key elements: a Ti-sapphire laser system that produces an 800 nm femtosecond beam, an optical parametric amplifier (OPA), and a transient spectrometer (Fig. 14h). A beam splitter divides this beam into two distinct beams with different intensities. Following this, the beam with higher intensity is modulated by a chopper and utilized to generate a shorter wavelength pulse through the OPA. This pump pulse is then used to excite electrons of the semiconductor photocatalyst from the VB to the CB. On the other hand, the beam with lower intensity passes through an optical delay line and interacts with sapphire, producing a continuous white pulse. This white pulse serves as a probe pulse, which is directed at the sample. A critical step is the precise alignment of the pump and probe pulses at the same point on the sample. As the probe pulse passes through the sample and reaches the detector, the pump pulse is blocked by a stopper after it has passed through the sample.The optical delay line enables the adjustment of the path difference between the probe and pump pulses, creating a controlled delay time between them. This delay enables the two pulses to arrive at the sample at different moments. By assessing the absorption of the probe pulse in both the presence and absence of pump pulse illumination, while varying the delay time between them, differential optical absorption spectra of the sample can be obtained. Changes in the signal intensity of the probe pulse as it passes through the sample indicate variations in the number of excited carriers.162 Therefore, examining these signals offers information about the relaxation dynamics of excitons as they move from a higher energy state to a lower one. The fs-TAS spectrum is a particular form of differential optical absorption spectrum obtained from transmittance measurements. Generally, the detector evaluates the transmittance (T) of the sample, and the absorbance (A) of the sample can be calculated as follows:163
A = −log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) T = log(I0/I) T = log(I0/I) |
In this context, I0 and I denote the intensity of the probe pulse before and after it travels through the sample, respectively. Additionally, the change in absorption of the probe pulse (ΔA) for the sample is defined as follows:
| ΔA = Aw − Awo = log(I0/Iw) − log(I0/Iwo) = log(Iwo/Iw) |
In this case, Aw and Awo represent the absorption of the probe pulse by the sample with and without excitation by the pump pulse, respectively. Iw and Iwo denote the intensities of the probe pulse after traversing the sample with and without excitation by the pump pulse, respectively. It is crucial to note that ΔA changes with both the wavelength of the probe pulse and the time delay between the pump and probe pulses. As the delay time is adjusted by the optical delay line, ΔA is recorded at various wavelengths.164
Consequently, a three-dimensional image function, ΔA (λ, t), depicting the relationship between wavelength and delay time is obtained. This three-dimensional image enables an examination of how ΔA varies with wavelength at a constant delay time and how it changes with delay time at a specific wavelength, reflecting the variation in the number of excited state charge carriers over delay time at that wavelength. Following this, we explore the relationship between ΔA and the carrier kinetics of the sample. In the absence of a pump pulse, when only the probe pulse illuminates the semiconductor photocatalyst, the semiconductor absorbs part of the probe pulse. The intensity of absorption depends on the number of ground state electrons in the VB and the semiconductor's absorption coefficient, as shown in Fig. 14i. When the pump pulse is introduced to the semiconductor sample, ground state electrons in the VB are excited and transition to the CB of the semiconductor. This leads to a decrease in the number of electrons in the VB and an increase in the CB. When the semiconductor sample is then illuminated by the probe pulse, the absorption of the probe pulse is reduced (Iw > Iwo) due to the decreased number of ground state electrons in the VB, resulting in a negative ΔA signal, commonly known as ground state bleaching (GSB), as depicted in Fig. 14j. Additionally, some of the electrons in the excited state absorb the probe pulse that was not absorbed by the ground state electrons, moving to higher energy excited states. As a result, the absorption of the probe pulse by the sample increases (Iw < Iwo), leading to a positive ΔA signal, referred to as excited state absorption (ESA) or photoinduced absorption (PIA), as illustrated in Fig. 14k. Moreover, the semiconductor sample in the excited state is inherently unstable and returns to the ground state, through either stimulated or spontaneous radiation. During this transition, the semiconductor sample emits fluorescence, leading to an increased intensity of light detected by the detector (Iw > Iwo). This process results in a negative ΔA signal known as stimulated emission (SE), as shown in Fig. 14l.165
Three distinct ΔA signals can be identified in fs-TAS measurements, each associated with specific electron transition and relaxation processes within semiconductors, as outlined in Fig. 14m. Based on the previously discussed ΔA signals, two main types of fs-TAS spectra can be identified. In a photoactivated semiconductor, when an excited electron moves from the energy levels of the CB to those of the VB, it loses energy through interactions with surrounding particles. This lost energy is predominantly released as heat, resulting in a difference between the energy of the excitation light and the emitted light, commonly known as fluorescence. As a result, the luminescence spectrum, also referred to as the fluorescence spectrum, shifts to lower energy levels, a phenomenon known as the Stokes shift.
The extent of the Stokes shift varies due to different physicochemical properties and energy level structures among various semiconductors.166 In the fs-TAS spectrum, the ground state bleaching (GSB) signal aligns with the steady-state absorption spectrum, while the stimulated emission (SE) signal matches the fluorescence spectrum. When the Stokes shift is minor, the wavelengths of the GSB and SE peaks overlap to some extent, resulting in the final fs-TAS spectrum being a composite of the GSB, SE, and excited state absorption (ESA) peaks, as shown in Fig. 15a. On the other hand, when there is a significant Stokes shift, the wavelength of the GSB peak does not overlap with that of the SE peak, leading to the differentiation of three distinct peaks in the fs-TAS spectrum, as illustrated in Fig. 15b. Heterojunction photocatalysts exhibit markedly higher photocatalytic efficiency compared to single photocatalysts, largely due to the spatial separation of charge carriers. To understand the mechanism behind this enhanced performance in heterojunctions, it is crucial to study the dynamics of photoinduced carriers within these photocatalysts, with a particular focus on the ultrafast interfacial electron transfer process. The application of fs-TAS measurements enables the observation of charge transfer processes at picosecond (ps) and femtosecond (fs) timescales, making it an effective tool for investigating heterojunction photocatalysts. Significant studies have been carried out to explore the kinetics of interfacial electron transfer in S-scheme photocatalysts using fs-TAS.
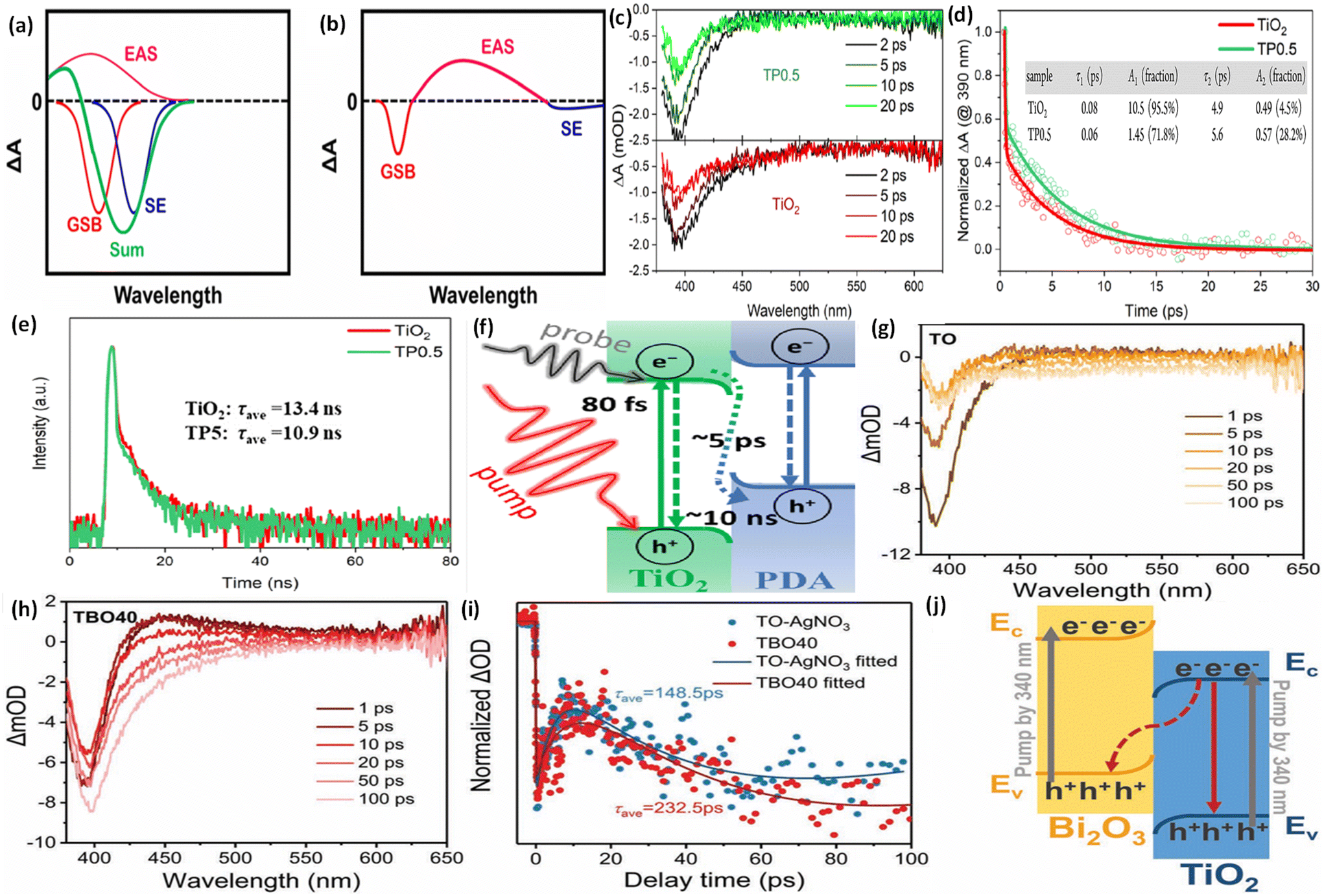 | ||
| Fig. 15 Representative fs-TAS spectra for (a) a minor Stokes shift and (b) a major Stokes shift. The figures are adapted with permission from ref. 161. Copyright 2023, RSC. (c) fs-TAS. (d) GSB signal decay profiles for fs-TAS in TiO2 and TP0.5. (e) TRPL spectra for TiO2 and TP0.5. (f) Diagram showing the kinetics of electron transfer in TP0.5. The figures are adapted with permission from ref. 150. Copyright 2022, ACS. TAS of TO (g) and TBO40 (h) photocatalyst obtained using 340 nm excitation. (i) Fitted transient absorption decay curves for TO-AgNO3 and TBO40 at 395 nm, captured within the 100 ps. (j) Diagrammatic representation of S-scheme charge transfer in the TBO40 photocatalyst. The figures are adapted with permission from ref. 168. Copyright 2022, Wiley-VCH. | ||
In this regard, Wang and co-workers studied the dynamics of photoexcited charge carriers in photocatalysts with a TiO2/polydopamine (TiO2/PDA) S-scheme heterojunction employing fs-TAS.167 In their research, the fs-TAS spectra for both pure TiO2 and the most efficient TiO2/PDA sample (referred to as TP0.5) showed ground-state bleach (GSB) peaks around 375 nm, as depicted in Fig. 15c. The analysis of the GSB recovery kinetics (Fig. 15d) indicated a quick decay of the GSB signal within just 30 ps. This rapid decay (τ1) on the femtosecond timescale was attributed to electron trapping in the TiO2 material, while slower decay (τ2) was associated with electron transfer kinetics. Notably, TP0.5 had a larger proportion of τ2 compared to pure TiO2, indicating a novel charge transfer mechanism in TP0.5. This mechanism was attributed to interfacial charge transport within the TiO2/PDA S-scheme heterojunction, with charge transfer from the CB of TiO2 to the VB of PDA occurring within approximately 5 ps. Moreover, the average lifetime (τave) value of TP0.5 in the time-resolved photoluminescence (TRPL) spectra indicated the band-to-band recombination of electrons and holes within TiO2 and was estimated to be around 10 ns (Fig. 15e). In conclusion, the interfacial charge transfer process took place much faster than charge recombination, thus aiding in the effective separation of excitons within the TiO2/PDA S-scheme heterojunction photocatalysts, as illustrated in Fig. 15f. This study provides insights into the mechanism behind the enhanced performance of S-scheme heterojunction photocatalysts by elucidating the kinetics of interfacial electron transfer. Similarly, He and colleagues carried out an extensive investigation using fs-TAS to thoroughly explore electron transfer processes within the TiO2/Bi2O3 S-scheme heterojunction.168 Their study included fs-TAS measurements on two samples: pure TiO2 (denoted as TO) and TiO2/Bi2O3 with optimized photocatalytic performance (referred to as TBO40). In the fs-TAS spectra of both TO and TBO40, as depicted in Fig. 15g and h, notable features were seen, including a GSB peak at approximately 395 nm and a broad ESA peak extending from 425 to 550 nm. To determine whether these spectral signals were attributed to electrons or holes, control experiments were performed using either an electron scavenger (such as AgNO3 and benzoquinone) or a hole scavenger (like lactic acid and ethanol). Specifically, the fs-TAS spectra of TO were examined in the presence of an electron scavenger (AgNO3) or a hole scavenger (furfuryl alcohol). The addition of these electron or hole scavengers resulted in noticeable changes to both GSB and ESA signals, indicating that the dynamics of photoexcited charge carriers played a role in both of these signals. By analyzing the fitting results of GSB recovery kinetics within the first 100 ps, as shown in Fig. 15i, it was found that the average lifetimes (τave) of holes increased from 148.5 ps for TO-AgNO3 to 232.5 ps for TBO40. This enhancement in hole lifetimes suggested the superior electron-accepting ability of Bi2O3 within the heterojunction. As a result, this indicated an efficient interfacial charge transfer process from the CB of TiO2 to the VB of Bi2O3, as depicted in Fig. 15j.
Similarly, Cheng and colleagues used fs-TAS in their research to explore the dynamics of photogenerated carriers in a CdS/PDB S-scheme heterojunction.169 Initially, two-dimensional mapping TA spectra were obtained to detect TA signals across femtosecond to nanosecond timescales, as shown in Fig. 16(a–d). Notably, an additional peak at 710 nm was observed in the CdS/PDB composite compared to pure CdS. This specific wavelength corresponded to an energy level of 1.75 eV, aligning with the energy difference between the CBM of CdS and the HOMO of PDB, as depicted in Fig. 16e. Subsequently, decay kinetic curve fitting was conducted to determine fluorescence lifetimes, as illustrated in Fig. 16f and g. The lifetimes τ2 and τ3 of CdS/PDB were significantly shorter than those of pure CdS. Additionally, a new lifetime, labeled as τ4, was introduced into the fitting process to achieve an accurate fit. These results provided strong evidence for the transfer of photoexcited electrons from the CB of CdS to the HOMO of PDB, as illustrated in Fig. 16h. The studies mentioned above have mainly offered indirect insights into interfacial electron transfer within S-scheme heterojunction photocatalysts, inferred from the analysis of electron transfer kinetics obtained by fitting the recovery kinetics of GSB. However, it should be noted that the recombination of electrons in the CB of the OSP and holes in the VB of the RSP can lead to fluorescence emission as well as a negative ΔA signal in fs-TAS spectra, a signal that is often overlooked. Future research could, therefore, focus on detecting and analyzing this negative ΔA signal, which arises from the interfacial charge transfer processes within S-scheme heterojunction photocatalysts. Such studies could provide a more detailed understanding of these intricate electron transfer dynamics.
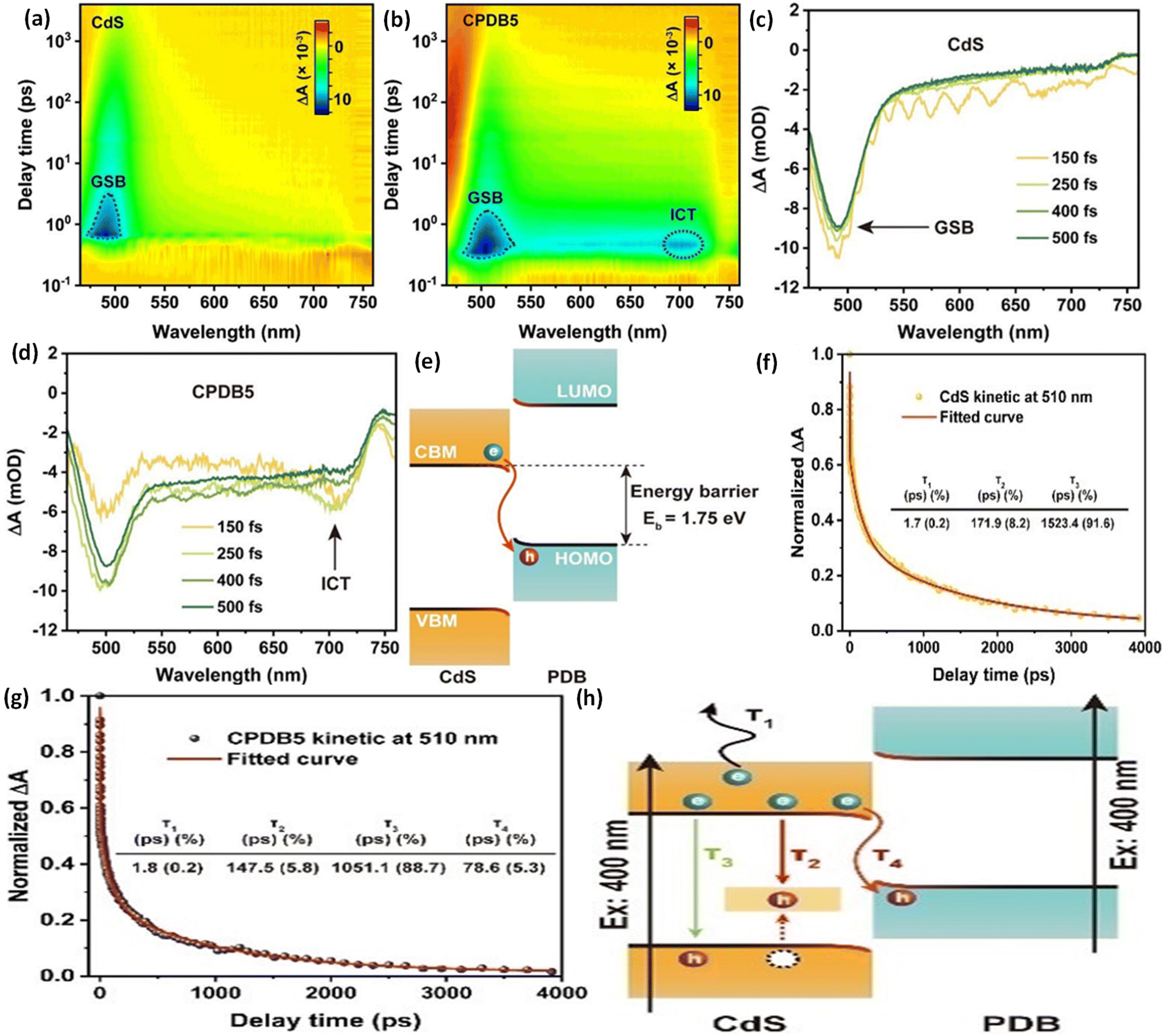 | ||
| Fig. 16 2D mapping of TA spectra for (a) pristine CdS and (b) CPDB5 photocatalyst. TAS signals of (c) CdS and (d) CPDB photocatalyst over fs–ns timescales. (e) Depiction of the energy gap between the CBM of CdS and the HOMO level of PDB. Normalized decay kinetics of (f) CdS and (g) CPDB5 measured at a wavelength of 510 nm. (h) Decay routes for photoinduced electrons within CdS and CPDB photocatalysts. The figures are adapted with permission from ref. 169. Copyright 2023, Wiley-VCH. | ||
The results from fs-TAS indicate that the improved photocatalytic performance of heterojunction photocatalysts is due to rapid interfacial charge transfer and the effective use of photogenerated carriers in redox reactions at the right time.
5.4. DFT calculations
Density functional theory (DFT) serves as an indispensable computational tool for investigating the electronic structure and charge dynamics in many-electron systems, including Z/S-scheme heterojunctions commonly found in photocatalytic reactions. By modeling electron interactions at atomic and molecular levels, DFT provides insights into electronic properties. It generates essential information, including band structure, lattice parameters, and energies related to binding and adsorption, which are crucial for addressing challenges like photocatalyst stability, substrate adsorption strength, etc.170 In addition to offering theoretical guidance for regulating carrier behavior, DFT calculations have been particularly useful in indirectly confirming the charge flow dynamics in S-scheme photocatalytic systems, as evidenced in recent publications. However, it is important to note that DFT is a theoretical method and may not account for all complexities encountered under actual testing conditions.171 By delving into the electronic arrangement of the materials using DFT calculations, one is able to obtain a clearer understanding of charge transfer dynamics, enabling more effective regulation of electronic structures and more accurate prediction of material properties. One is able to gain a clearer understanding of the kinetics of charge transfer. The interfacial carrier mobility pathway as well as the interfacial electric field (IEF) direction in Z- or S-scheme photocatalysts can be confirmed through DFT simulations along with metrics like the variation in average planar electron density, denoted as Δρ(z), and the mapping of differential charge density. Simulating the interfacial electric field serves as a valuable approach for investigating the carrier transfer mechanism. The interfacial directional flow of electrons and holes within a heterojunction can be hypothesized through examining the interfacial electric field within the components of a heterojunction.172Generally, DFT is utilized to ascertain the work function (ϕ) of each component within a photocatalytic system. The Fermi energy level for each semiconductor can subsequently be verified using the formula ϕ = Evac − Ef, where ϕ signifies the work function and Evac stands for the vacuum energy. Through the optimization of average potential distributions for each component, the work function (ϕ) is determined. Electron migration from one component of the photocatalytic system to another can be determined by computing the work functions of the two semiconductors. A three-dimensional differential charge density map shows the build-up of charge at the interface, with yellow and green denoting electron accumulation and depletion, respectively. The fluctuation in average electron density along the Z-axis serves as a guide for discerning the routes of electron transfer among the components of the photocatalytic system.171,172
In this regard, Sun et al. confirmed the routes of charge transfer in the Sv-ZIS@3DA-CN S-scheme heterojunction through DFT analyses.173 Clear differences in charge density at the interface are evident from DFT simulations, with the 3DA-CN side mainly appearing green and the Sv-ZIS side predominantly yellow, symbolizing an electron depletion zone and an accumulation zone, respectively (Fig. 17b and d). The distribution of electronic charge surrounding S atoms in Sv-ZIS is notably more localized than that surrounding C atoms in 3DA-CN, indicating an intense bond between S and C atoms. In the average planar electron density difference curve, the 3DA-CN side exhibits positive charge, indicative of negative electron density, while the Sv-ZIS side displays negative charge, indicative of positive electron density (Fig. 17c). Consequently, both the differential charge density map and the average planar electron density curve demonstrate that electrons are transferred from 3DA-CN to Sv-ZIS (Fig. 17b–d). Fig. 17e illustrates the density of states (DOS) surrounding the Fermi levels of 3DA-CN, Sv-ZIS, and Sv-ZIS/CN composite. Notably, there is significant electron density near the Fermi level of Sv-ZIS/CN, indicating electron redistribution between 3DA-CN and Sv-ZIS, suggestive of a strong interaction between them. As a result, an interfacial built-in electric field (IEF) is formed and directed from 3DA-CN towards Sv-ZIS in the 3DA-CN@Sv-ZIS S-scheme photocatalytic system. The optimized geometric simulated structure yielded work function (Φ) values of 4.83 eV and 4.67 eV for Sv-ZIS and 3DA-CN, respectively, as depicted in Fig. 17a, f and g. Notably, the Sv-ZIS system has a higher work function and lower Fermi level compared to 3DA-CN. Consequently, upon interfacial close contact, spontaneous electronic migration occurs from the 3DA-CN component towards the Sv-ZIS component. This movement continued as long as the Fermi level became sufficiently flat to impact on band bending.
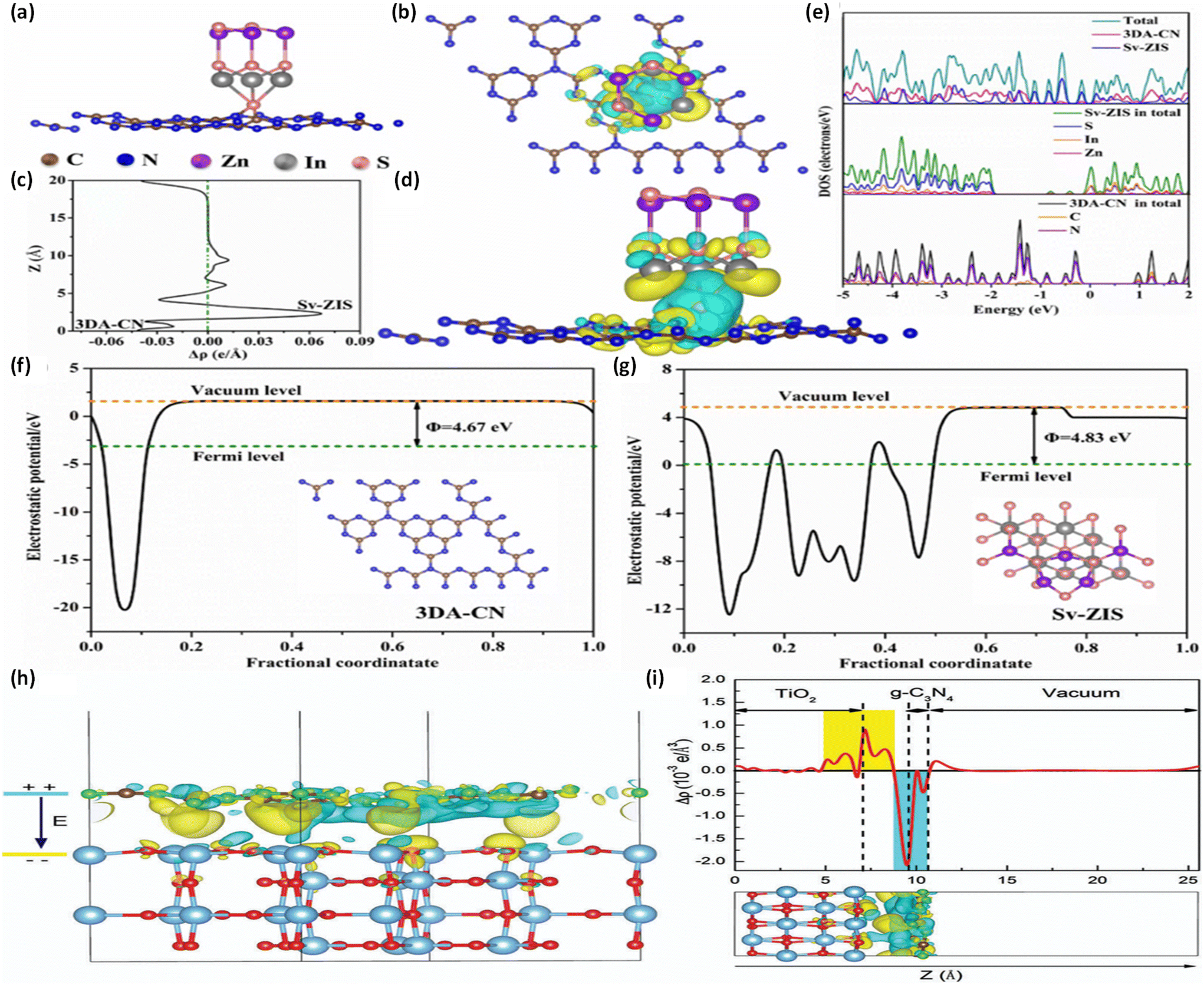 | ||
| Fig. 17 (a) Lateral perspective of the Sv-ZIS/CN heterojunction following geometrical optimization. (b) Top view of the charge density difference. (c) Planar-averaged difference in electron density. (d) Side view of the charge density difference. (e) DOS for the Sv-ZIS/CN composite. Work functions for (f) 3DA-CN and (g) Sv-ZIS photocatalysts. The figures are adapted with permission from ref. 173. Copyright 2022, RSC. (h) Variation in charge density (0.0004 e Å−3) across the g-C3N4/TiO2 interface. (i) Planar-averaged difference in electron density (Δρ(z)) for the g-C3N4/TiO2 interface, with yellow regions showing electron accumulation, while cyan areas denote electron depletion. The figures are adapted with permission from ref. 118. Copyright 2016, RSC. | ||
One useful tool for studying the carrier transfer mechanism is internal electric field simulation. This approach was used to examine the interfacial electric field as well as the interfacial charge transfer direction across TiO2 and g-C3N4.118 The values of work function for TiO2 along with g-C3N4 photocatalysts are 6.43 eV and 5.32 eV, respectively, implying electron migration from g-C3N4 to TiO2. A 3D visualization of the interfacial charge density difference (as shown in Fig. 17h) displays where charges accumulate (yellow) and are depleted (cyan). Additionally, a variation in average charge density along the Z-axis was observed (Fig. 17i), further confirming electron migration from g-C3N4 towards TiO2 leading to the creation of holes on the g-C3N4 side.
In their research, Zhu and co-workers analyzed the charge transfer dynamics in different materials by computing their work functions.174 The study indicated that the work functions for g-C3N4, SnS2, and O-C3N4 were 4.3, 6.0, and 3.6 eV, respectively, as depicted in Fig. 18a. This indicates that the Fermi level positions of g-C3N4 and O-C3N4 are more negative than that of SnS2. Fig. 18b visually represents the charge density difference, where accumulation and depletion regions of charge are highlighted in yellow and cyan colour. Within the g-C3N4/SnS2 photocatalyst, the charge is primarily localized on g-C3N4 (blue) and on SnS2 (yellow), implying charge transfer from g-C3N4 towards SnS2. A similar electron transfer is observed in the O-C3N4/SnS2 system. Furthermore, the strengths of the interfacial electric field (IEF) within g-C3N4/SnS2 as well as O-C3N4/SnS2 S-scheme heterojunctions were found to be 1.32 × 109 V m−1 and 5.31 × 109 V m−1, respectively, as shown in Fig. 18c and d. These values suggest a stronger IEF along with efficient electron flow in the oxygen-doped system, aligning with the experimental observations that demonstrate superior photocatalytic activity in O-C3N4/SnS2 compared to the g-C3N4/SnS2 system. In conclusion, this study offered not only a theoretical understanding of the S-scheme mechanism within the g-C3N4/SnS2 heterojunction but also introduced a new method for altering the strength of the IEF in S-scheme photocatalysts by incorporating nonmetal atoms.
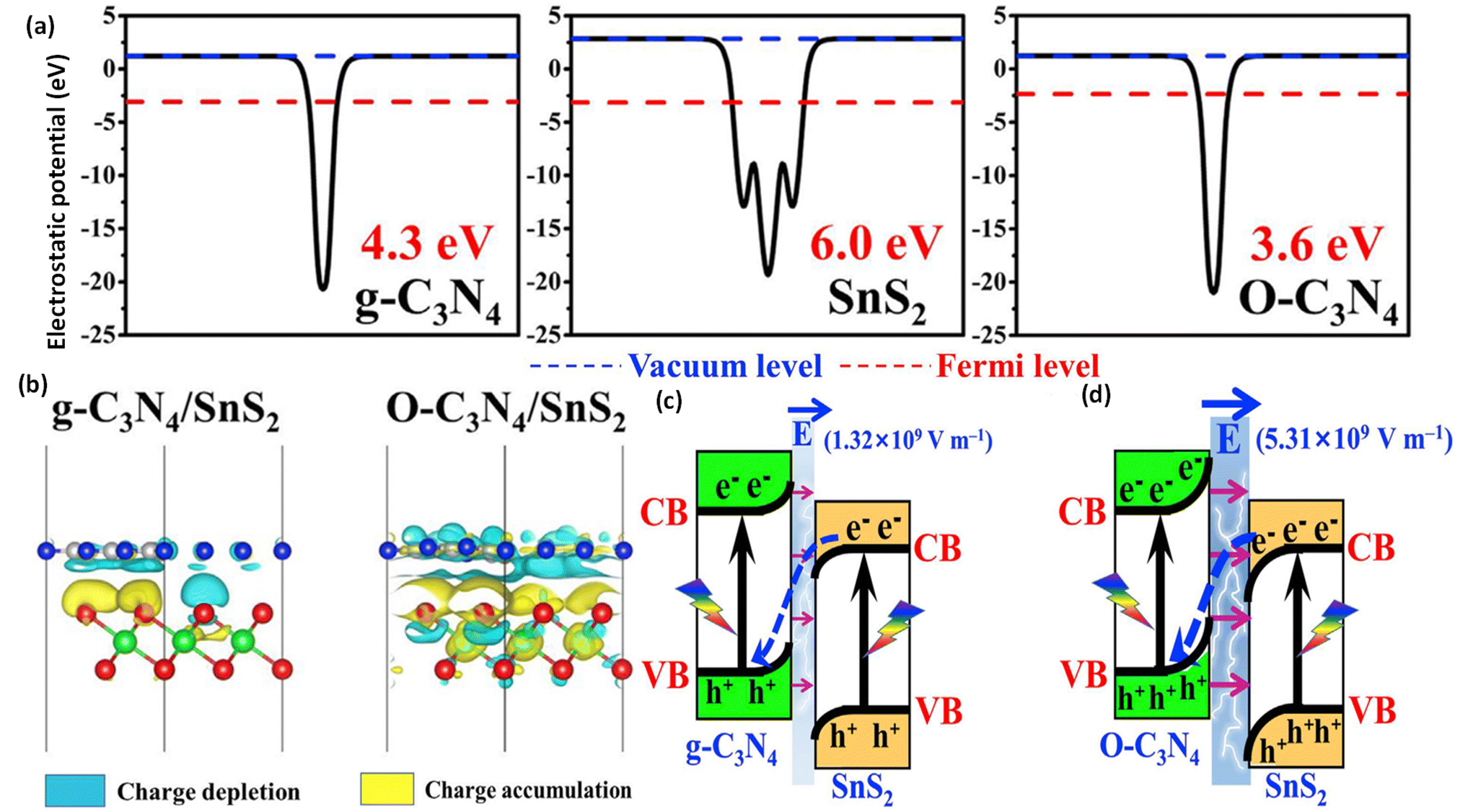 | ||
| Fig. 18 (a) Work function values for g-C3N4, SnS2, and O-C3N4 photocatalysts. (b) Charge density variations within the composite photocatalysts. Illustration of the S-scheme charge transfer routes for (c) g-C3N4/SnS2 and (d) O-C3N4/SnS2 heterojunctions. The figures are adapted with permission from ref. 174. Copyright 2021, Elsevier. | ||
Hence, DFT analyses serve as a foundational tool for determining both IEF orientation and the charge carrier transfer pathway.
5.5. Reactive free radical trapping testing by EPR
It is well known that the photocatalytic process can produce a variety of reactive free radicals. Among these, holes must have a strong oxidative power in order to produce hydroxyl radicals, while electrons with strong reductive capabilities are the source of superoxide radicals. Within an S-scheme photocatalyst, the excitons with a weaker redox potential tend to recombine, leaving behind potent charge carriers of robust redox potential. Hence, evaluating the formation of hydroxyl (˙OH) and superoxide (˙O2−) radicals through EPR during the photocatalytic reaction is an effective strategy to authenticate the more relevant S-scheme charge migration pathway.78 In this regard, Fu and colleagues presented the EPR profiles for DMPO-˙OH and DMPO-˙O2− for WO3, g-C3N4, and 15%WO3/g-C3N4 photocatalysts.100 In Fig. 19b, g-C3N4 showcased an absence of DMPO-˙OH signals, because its photoinduced holes had a weak capacity for oxidation (2.14 V, in relation to NHE at pH = 0), as shown in Fig. 19a. However, both WO3 and 15%WO3/g-C3N4 composite showed a pronounced DMPO-˙OH response, proving that photogenerated holes of WO3 did not migrate towards the VB of g-C3N4, but instead resided over the VB of WO3. Conversely, in Fig. 19c, pronounced DMPO-˙O2− responses were evident in g-C3N4 along with the 15%WO3/g-C3N4 heterojunction, indicating that their photoexcited electrons possessed enough reduction power for the reduction of O2 to ˙O2−. According to these EPR data, the photoinduced holes and electrons remained in the VB of WO3 and in the CB of g-C3N4, subsequently proving to be a more relevant S-scheme charge migration pathway than the type-II mechanism.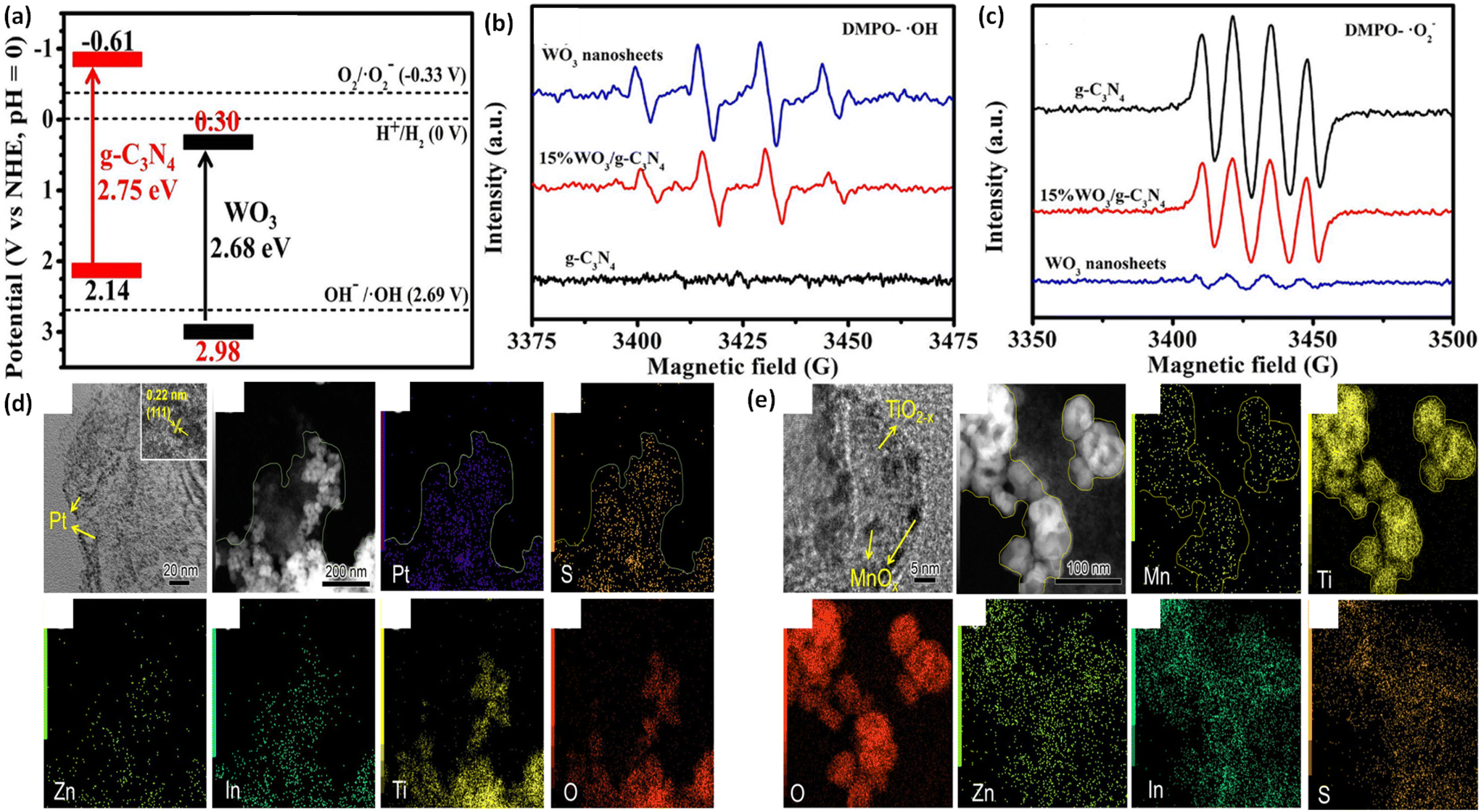 | ||
| Fig. 19 (a) Band arrangements of g-C3N4 and WO3 nanosheets. EPR spectra showing (b) DMPO-˙OH in an aqueous solution and (c) DMPO-˙O2− in a methanol dispersion, with g-C3N4, WO3 nanosheets, and 15%WO3/g-C3N4 photocatalysts. The figures are adapted with permission from ref. 100. Copyright 2019, Elsevier. (d) TEM, STEM, and EDX spectroscopy element mapping images of the Pt photodeposited O-ZIS/TiO2−x heterojunction. (e) TEM, STEM, and EDX element mapping images of the MnOx photodeposited O-ZIS/TiO2−x photocatalyst. The figures are adapted with permission from ref. 175. Copyright 2022, Elsevier. | ||
5.6. Selective photodeposition of noble metal cocatalysts
In photocatalytic reactions, noble metals like Au and Pt, with large work functions, typically serve as electron reservoirs. These metals are commonly deposited selectively on the electron-dense area of the photocatalyst, which are the reduction sites, through the process known as photodeposition. Hence, the targeted photoreduction of these noble metals represents an efficient approach to determine the surface characteristics of a particular semiconductor of any heterojunction system and identify its electron-rich zone or reduction site as well as electron-deficient zone or oxidation site. Within the S-scheme heterojunction, upon irradiation, the OSP side undergoes electron loss, while the RSP side gains electrons. Consequently, a high electron density develops on the RSP side, which causes noble metals to deposit on the RSP surface selectively. In this regard, Liu and colleagues developed an S-scheme O-ZnIn2S4/TiO2−x heterojunction. They observed that upon exposure to light, Pt nanoparticles selectively deposited themselves onto the O-ZnIn2S4 nanosheet surfaces, whereas MnOx nanoparticles favored the TiO2−x surface, as illustrated in Fig. 19d and e.175 Such observations provide strong evidence that O-ZnIn2S4 and TiO2−x serve as the reductive and oxidative sites in photocatalytic processes being rich in electrons and holes, respectively. Consequently, it has been demonstrated that the O-ZnIn2S4/TiO2−x heterojunction exhibits an S-scheme charge transfer mechanism.5.7. Ultraviolet photoelectron spectroscopy (UPS) analysis
UPS is a less commonly utilized method than XPS for determining the valence band density of states on the surface of the materials.176 However, its working principles are quite similar to those of XPS. Specifically, UPS involves the ejection of photoelectrons when the material is irradiated with deep ultraviolet (UV) light. This process causes the dislocation of electrons out of the VB instead of the inner-shell electrons. Fig. 20a shows that UV light excites electrons from the VB to surpass the vacuum level (Evac), creating a VB reflection above Evac. These electrons, originating from any point within the VB, must reach the surface to escape and be detected. Electrons that retain energy while escaping are known as secondary electrons, ranging from Ef to above Evac, as shown in Fig. 20b, where Ef is applicable for the photoelectron spectrometer as well as the sample. Electrons are released towards the vacuum, allowing the spectrometer to measure both the VB and secondary electrons surpassing the vacuum level, which is illustrated in Fig. 20c. The resulting UPS spectrum mainly features secondary electrons.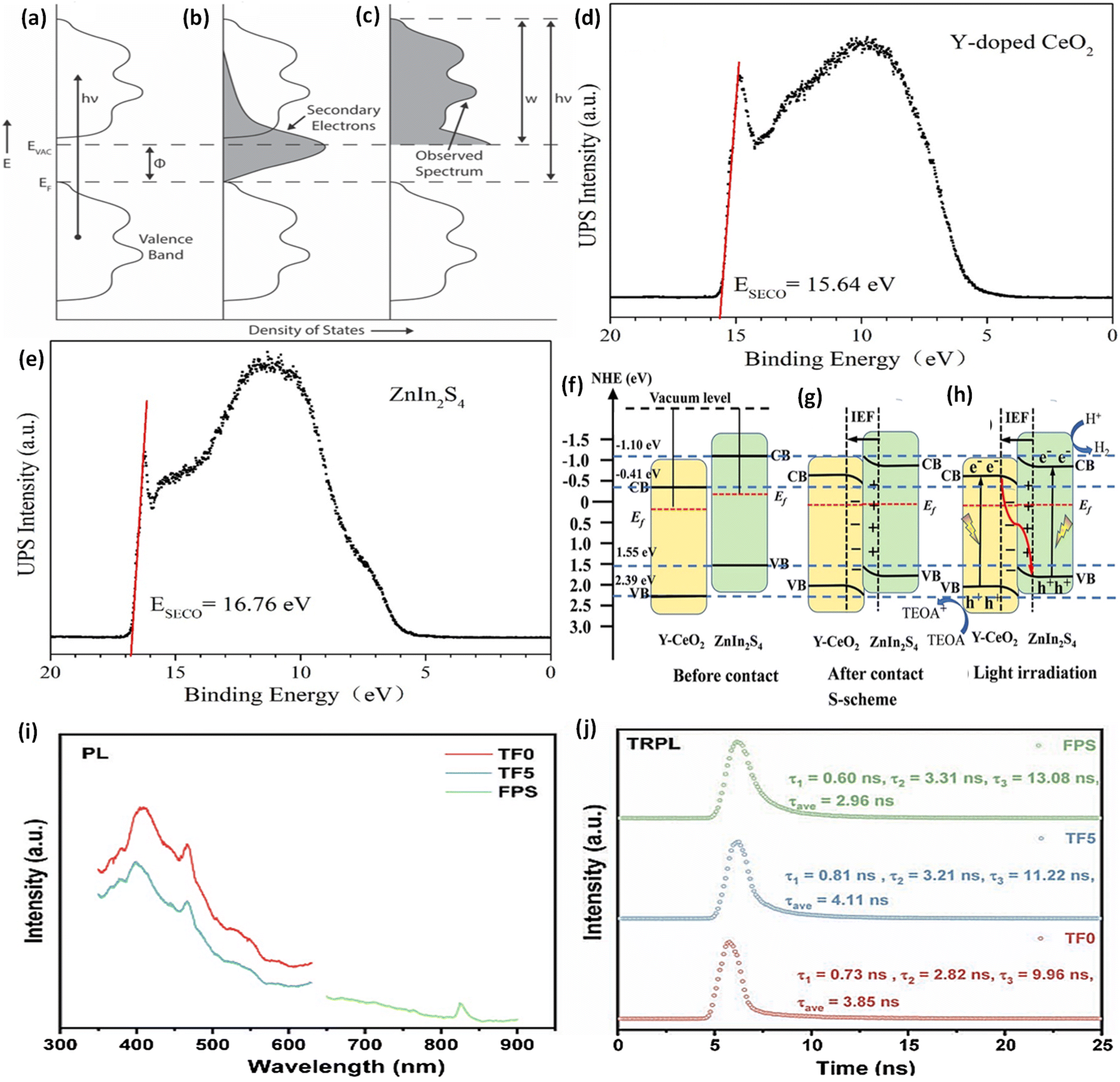 | ||
| Fig. 20 Schematic representation of a UPS spectrum. (a) Deep UV light excites electrons from the VB to above the vacuum level (Evac) in a metal sample. (b) Secondary electrons. (c) The UPS spectrum comprises elastically reached photoelectrons and secondary electrons above Evac. The sum of the spectrum width (w) and work function (Φ) equals the photon energy. The figures are adapted with permission from ref. 176. Copyright 2023, Elsevier. (d) and (e) UPS spectra for Y-doped CeO2 and ZnIn2S4. The S-scheme heterojunction charge carrier transport process between Y-doped CeO2 and ZnIn2S4: (f) prior to contact, (g) post-contact, (h) during light exposure. The figures are adapted with permission from ref. 178. Copyright 2022, Elsevier. The PL spectra (i) and the TRPL spectra (j) of samples TF0, TF5, and FPS. The figures are adapted with permission from ref. 182. Copyright 2022, Wiley-VCH. | ||
Analysis of this spectrum in relation to Ef enables the plotting of the secondary electron cutoff (SECO). Alignment of Ef within the spectrometer and the surface of the specimen facilitates determination of the work function (Φ) of the respective material, a critical parameter representing the minimum energy needed to remove electrons from the Ef of the material, thus reflecting the challenge in electron mobility out of the material as follows:177
| Φ = hν − SECO |
Acquiring a UPS spectrum was straightforward, but deriving meaningful data could be challenging due to specimen preparation and surface charge accumulation concerns.156 Whitten and co-workers proposed a specific protocol for UPS analysis to address these challenges.176 To explore the charge transfer pathway in the Y-CeO2@ZnIn2S4 heterojunction, UPS analysis was utilized. This technique, specifically applied to Y-CeO2 and ZnIn2S4, facilitated the calculation of work functions (Φ) by subtracting the incident UV light energy (21.22 eV) from the energy of secondary electron cutoff (ESECO), as demonstrated in Fig. 20d and e.178 The results indicated a greater work function value for Y-CeO2 (5.58 eV) than ZnIn2S4 (4.46 eV), suggesting a lower Fermi level (Ef) for the former. This inconsistency in Ef was further investigated using ESR spectra, VB-XPS, and UV-visible DRS. These investigations led to a proposed mechanism, illustrated in Fig. 20(f–h), where spontaneous interfacial electron transfer occurred from ZnIn2S4 to Y-CeO2 to attain Fermi level equilibrium. This electron movement initiates an interfacial electric field (IEF), preventing further electron transfer. Y-doping in CeO2 causes the band edge to shift downward, while ZnIn2S4 inclusion develops an upward band bending near the interface. When exposed to light, electrons move from the VB creating holes in the CB, leading to the recombination of photoexcited electrons and holes from the CB of Y-CeO2 and the VB of ZnIn2S4, respectively. The study highlights the efficacy of UPS in determining Fermi levels and supporting the S-scheme carrier dynamics.
5.8. Steady-state PL spectroscopy
The application of steady-state photoluminescence (PL) spectroscopy is prevalent due to its non-destructive, rapid, and easier sample-preparation nature. This technique is essential for assessing optoelectronic characteristics across a variety of samples, such as heterostructures,179 defect analysis,180 and evaluating solar cell efficiency.181 The mechanism underlying PL is demonstrated in Fig. 21a, revealing distinct optical behaviors in organic versus inorganic semiconductors. Inorganic semiconductors, characterized by tightly bound atoms within a crystal lattice, exhibit delocalized electronic states, forming a broad VB and CB. In contrast, organic materials are better understood through organic semiconductor theory. This review focuses primarily on PL emissions from inorganic semiconductors. When a direct band gap semiconductor absorbs a photon, the energy of which exceeds the band gap (Eg), an electron from the VBM moves to the CBM, resulting in a vacancy in the VBM as shown in Fig. 21a. The photoexcited electron then moves to the lower energy state of CB, dissipating its excess energy as thermal energy. Ultimately, it transitions back to the VBM, emitting photons in the process. In inorganic PL, two primary emission types are recognized: intrinsic and extrinsic emissions. Intrinsic (inter band) emissions involve the direct return of the electron to the VB, whereas extrinsic (sub-band-gap) emissions arise as the photoexcited electron becomes captured at defect or impurity sites inside the band gap, indicating the defect and impurity characteristics of the parent system.156 The freshly generated photons must undergo depletion of energy to successfully depart the surface of the catalyst (Fig. 21b). Without this loss, photons may reflect back, preventing their escape. Fig. 21c illustrates three key extrinsic radiation mechanisms. One trapping level is involved in extrinsic emissions of both type I and type II, with the energy gap that separates the inter band and sub-band-gap emission lines precisely mirroring the trap energy position inside Eg. However, type III extrinsic radiation demands multiple trap levels for both holes and electrons. For type III, it is impractical to identify the trap energy position by examining the energy gap that separates the inter band and sub-band-gap emission lines. Nonetheless, in scenarios where the emissions from defects or impurities involve two trap energy positions of type III, one being a deep trapping level (situated at a considerable distance from the band edge) and the other a shallow trapping level (close to opposite band edge), the emissions are characterized by pronounced thermal quenching. At elevated temperatures, carriers stuck in the shallow condition can undergo thermal activation, moving towards the closest band edge, which consequently results in a reduction of emission intensity. The concept also holds true for both type I and type II once the trap zone is near a band edge. The standard configuration for steady-state PL spectroscopy is depicted in Fig. 21d, featuring an appropriate light source for sample excitation, a spectrometer to gather emission spectra, a highly responsive detection device for detecting the scattered radiation, plus an electronic device for optical–electrical data conversion. Within PL spectroscopy, the responses of optoelectronic elements change with light frequency, leading to a spectrum detected by the detector of the system that differs from the sample's original emission spectrum, as depicted in Fig. 21e. Such a difference emphasises the importance of considering the PL spectroscopy system's overall spectral response. An adjustment variable, derived from correlating the system's anticipated and observed spectra, is employed to adjust subsequent measurements, ensuring precise steady PL spectra analysis, despite system-specific deviations.181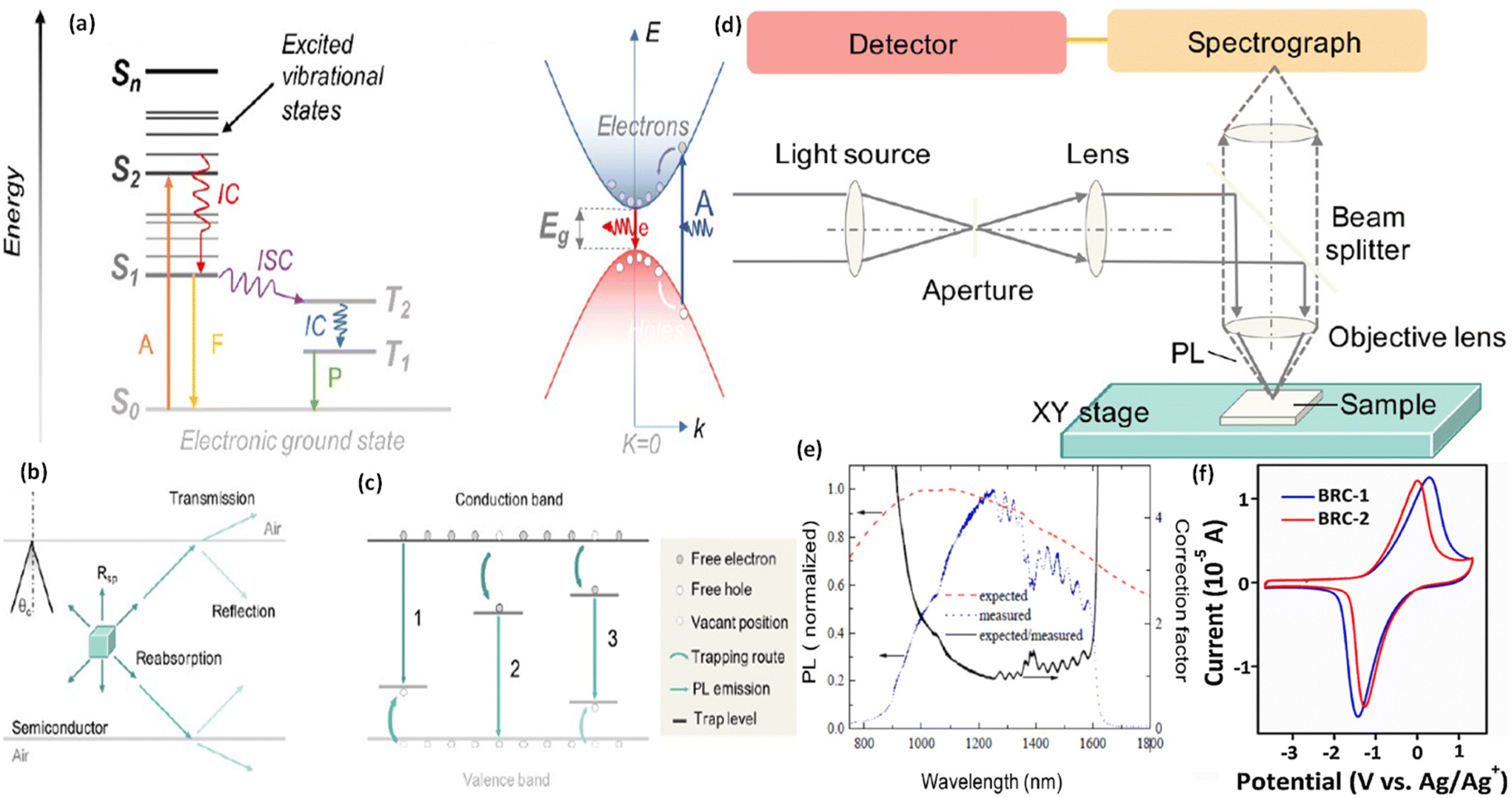 | ||
| Fig. 21 (a) PL phenomena in organic and inorganic materials with direct bandgaps. Abbreviations: IC (internal conversion), P (phosphorescence), A (absorption), F (fluorescence), ISC (intersystem crossing), e (emission), Eg (bandgap). E and k represent the kinetic energy and wave vector of electrons or holes. Schematic illustration depicting (b) loss mechanisms of spontaneously generated photons within a semiconductor and (c) potential sub-bandgap luminescence pathways arising from defects and impurities. (d) Micro-PL spectroscopy system with reflection geometry and confocal optics. (e) Normalized spectra of a tungsten–halogen light source, showing interference oscillation from 1200 to 1600 nm. The figures are adapted with permission from ref. 181. Copyright 2020, ACS. (f) Cyclic voltammetry curve of BRC-1 and BRC-2 photocatalysts, referenced against the Ag/Ag+ electrode. The figures are adapted with permission from ref. 183. Copyright 2017, Elsevier. | ||
In this context, the FePS3/TiO2 (TiO2/FPS) heterojunction system is utilized as a case study.182 Both PL and time-resolved photoluminescence (TRPL) techniques were employed to elucidate exciton dynamics. Fig. 20(i) presents the PL spectra of samples (TF0 and TF5), showing two prominent peaks around 408 nm followed by 467 nm, equating to energy values of 3.04 eV and 2.66 eV, respectively. The PL peak near 408 nm indicates band-to-band emission activity, whereas the PL peak close to 468 nm is likely to be due to band edge-free excitons. The PL intensity of TiO2 decreases noticeably, when TiO2 nanoparticles are deposited onto FPS nanosheets, suggesting a lower rate of exciton recombination of the TiO2/FPS composite (TF5) than that of pure TiO2 (TF0). Besides, an emission peak of 826 nm is visible in the PL spectra of the FPS composite, relating to the inter band emission of the composite photocatalyst. Further understanding can be gained by examining the exciton lifespans through TRPL analysis, as illustrated in Fig. 20(j). The fitted and average exciton lifetimes of the TF5 composite are significantly higher than those in TF0, suggesting efficient charge separation and migration. Likewise, the TRPL spectra and associated exciton lifetimes for FPS nanosheets are shown in Fig. 20(j). FPS demonstrates a shorter average exciton lifespan than those of TiO2 and TF5, suggesting a more rapid recombination of excitons. These observations are in line with the minimal photocatalytic performance observed in FPS, when compared to TF0 and TF5, highlighting the enhanced photocatalytic efficiency of the TiO2/FPS photocatalyst.
5.9. Cyclic voltammetry (CV)
Cyclic voltammetry (CV) is another technique that, although not yet validated for the S-scheme charge transfer pathway, has proved to be successful in Z-scheme heterojunctions. In this method, the cyclic voltammogram provides the oxidation and reduction capability of the semiconductor, indicated by the peak and trough points, respectively. When the reduction–oxidation potential of a heterojunction system is higher than that of the pristine semiconductor, it suggests an enhancement in the redox power of the photocatalytic system, thereby indicating the creation of a Z-scheme heterostructure. This is in contrast to the type II double charge transfer mechanism, which is expected to reduce the redox potential of the system (Fig. 21f).183 In this regard, Ding et al. verified the formation of a direct Z-scheme heterojunction within the BiO1−xBr/Bi2O2CO3 photocatalytic system.183 To effectively apply this method, one should compare the CV plot of both the composite photocatalyst and the pristine semiconductor to realise an enhanced redox power in the heterojunction system. Hence, this examination can also serve to validate the direct Z- or more relevant S-scheme interfacial charge transfer within the heterojunctions. For clarity, Table 3 summarizes the specific characterization techniques employed to confirm the charge transfer pathway in direct Z- or S-scheme heterojunctions.| Entry | Characterization method | Operational concept | Overview of procedures | Information shared | Process involved in charge dynamics |
|---|---|---|---|---|---|
| 1 | Ex situ XPS | Variations in electron density | Detailed energy shifts indicate either an increase or decrease in electron density, offering insights into the direction of electrons when determining band positions through methods such as the Mulliken electronegativity principle, XPS, or M–S plot | Loss/gain of electrons | Electron transfer from RSP to OSP |
| 2 | In situ irradiated XPS | Loss/acceptance of photogenerated electrons causes a reduction/an increase in electron density and an increase/a decrease in elemental binding energy (Eb) | A combination of high-energy X-rays and ultraviolet-visible (UV-vis) light is employed to irradiate the sample's surface. When subjected to light, photoinduced electrons in the CB of OSP are driven by the interfacial built-in electric field (IEF) to move to the VB of the RSP, resulting in a decrease in the electron density of the OSP and an increase in the RSP. As a result, under light irradiation, the elemental Eb of the OSP increases, and that of the RSP decreases within the heterojunction photocatalyst | Loss/gain of photoinduced electrons | Photoinduced electron transfer from the OSP to the RSP |
| 3 | In situ irradiated KPFM | Surface potential distribution of the heterogenous interface | Typically, when a semiconductor receives electrons, its Fermi level rises, leading to a subsequent reduction in surface potential. Conversely, when it loses electrons, the Fermi level goes down, resulting in an increase in surface potential | Loss/gain of photoinduced electrons | Photoinduced electron transfer from the OSP to the RSP |
| 4 | fs-TAS | Detecting the ultrafast interfacial charge transfer signal at the interface of oxidation and reduction photocatalysts | The peak at a specific wavelength in TA spectra is associated with the energy released from the recombination of electrons in the OSP and holes in the RSP | TA spectra signal shows the interfacial transfer of photogenerated electrons from the OSP to the RSP | Recombination of electrons in the OSP and holes in the RSP |
| 5 | DFT | Theoretical machine learning tool designed to forecast band structure and behavior | Various parameters are configured to anticipate the behavior of semiconductor band structures and ascertain their electronic arrangement, aiding in the prediction of interfacial charge transfer | Work function, loss or gain of electrons | Dissimilar Fermi levels between the RSP and the OSP, electron transfer from the RSP to the OSP |
| 6 | EPR | Records signals during trapping experiments | DMPO signals, produced by radicals, are captured by the instrument with or without light irradiation for comparison purposes, particularly when evaluating pristine semiconductors against various synthesized composites | Production of ˙O2− and ˙OH radicals | Electrons confined within the CB of the RSP and holes retained in the VB of the OSP |
| 7 | Metal photodeposition | Mz+ + ze− → M0 | Photoreduction of metals occurs where electrons accumulate between the components of the heterojunction and gives the possible mechanism that occurs for such accumulation. Identify the electron-rich zone or reduction site as well as electron-deficient zone or oxidation site of a heterojunction | Loss/gain of photogenerated electrons | Photogenerated electrons retained within the CB of the RSP and holes confined in the VB of the OSP |
| 8 | UPS | Determination of Ef, CB, and VB positions | The UPS spectra can be linearly approximated to derive the band structures of semiconductor photocatalysts, enabling the design of charge transfer mechanisms with the assistance of the radicals identified during trapping experiments | Work function | Dissimilar Fermi levels between the RSP and the OSP |
| 10 | TRPL | Band-to-band (intrinsic) and sub-band-gap (extrinsic) emissions | The fitted and average exciton lifetimes in the composite are significantly higher than those in the pristine sample, suggesting efficient charge separation and migration. A shorter average exciton lifespan suggests a more rapid recombination of excitons | Lifetimes of charge carriers | Lifetime decay kinetics |
| 11 | CV | The curve shows reduction and oxidation potentials of a material | Contrasting the CV curves of pristine semiconductors with those of the composite will indicate whether the redox capability in the composite is enhanced as a result of direct Z- or S-scheme, given that type II typically diminishes the redox capability | Redox power of the photocatalytic system | Electrons confined within the CB of the RSP and holes retained in the VB of the OSP |
6. Fundamentals and reaction mechanisms of photocatalytic H2O2 production over g-C3N4
The process of producing H2O2 through light-driven methods using g-C3N4 as a semiconductor photocatalyst encompasses a series of intricate and ongoing reactions. When g-C3N4 is exposed to light energy that is sufficiently higher than the band gap energy, electrons within the VB become energized and move to the CB, leaving behind positively charged holes (h+) in the VB (Fig. 22a). These photogenerated electrons and holes migrate from the inner part of g-C3N4 to its surface. However, during this migration, there is a tendency for these carriers to recombine, leading to the conversion of energy, which is either emitted as light or generates heat. The carriers that evade recombination reach the catalyst's surface, and some engage in surface reduction or oxidation reactions with oxygen and water molecules, respectively, while others inevitably experience recombination (Fig. 22b). Two significant challenges in H2O2 production are the limited ability of the photocatalyst to harness light effectively and the undesirable recombination of charge carriers, which is often attributed to the disorganized flow of charges after their separation. Moreover, the dynamics of how reactant molecules adhere and become activated on the catalyst's surface play a vital role in this process. Additionally, understanding the specific mechanisms of H2O2 formation, along with the thermodynamic energy involved, is crucial for enhancing the efficiency of photocatalytic H2O2 generation. According to ongoing research developments, the pathways for the photocatalytic production of H2O2 involve the oxygen reduction reaction (ORR) and the water oxidation reaction (WOR).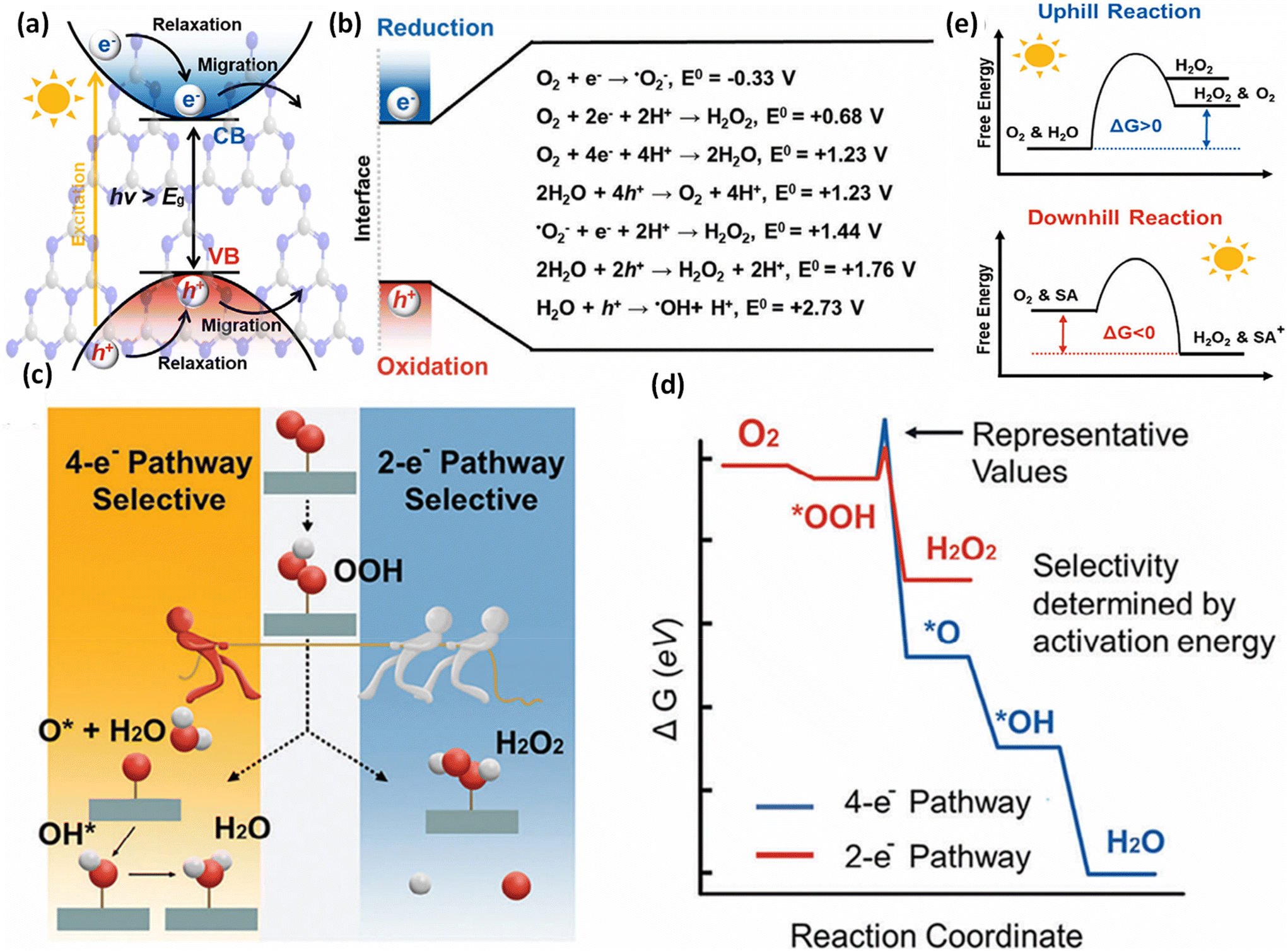 | ||
| Fig. 22 Illustrations of various stages of (a) photoexcitation and transfer of charge carriers. (b) Surface redox reaction processes relevant to solar H2O2 formation. (c and d) The competition between two-electron and four-electron routes in the oxygen reduction reaction, represented by a free energy diagram. (e) The concept of uphill and downhill processes in this production process. The figures are reproduced with permission from ref. 189. Copyright 2024, Wiley-VCH. | ||
6.1. Oxygen reduction reaction (ORR) pathway
The photocatalytic oxygen reduction reaction (ORR) follows/encompasses three distinct reaction pathways facilitated by one-, two-, and four-electron transfers, as outlined in eqn (4), (7) and (8). Typically, the formation of H2O2 through photocatalysis is predominantly realized by the proton-coupled two-electron ORR, where protons are chiefly sourced from water or organic electron donors.184 This two-electron ORR can transpire through an indirect stepwise/sequential single electron transfer pathway, as depicted in eqn (4) and (5), or alternatively, through a concerted two-electron pathway, as illustrated in eqn (7). Given that the ORR processes are significantly influenced by the energy position of the CB edge, O2 is reduced by photoexcited electrons in the CB. In the indirect, sequential one-electron-transfer ORR, O2 is first reduced to ˙O2−, as depicted in eqn (4), which is subsequently followed by the formation of H2O2, as illustrated in eqn (5). Given that the redox potential of O2/˙O2− is set at −0.33 V versus NHE, it is a thermodynamic prerequisite for a semiconductor engaging in a sequential one-electron-transfer ORR to have a CB position that is more negative than −0.33 V versus NHE.Importantly, this specific CB position not only satisfies the conditions for the one-electron-transfer ORR but also aligns with the thermodynamic prerequisites for the concerted two-electron-transfer ORR.185 When compared to the direct two-electron-transfer route, the indirect stepwise single-electron-transfer ORR is kinetically desirable due to only one electron being required in each step.186 Nevertheless, to uphold thermodynamic viability, it necessitates a larger bandgap, consequentially restricting the light absorption capacity. Moreover, complications arise as ˙O2− has the potential to interact with holes, producing singlet oxygen (1O2), as shown in eqn (6), which subsequently decreases the selectivity for H2O2.30 Therefore, to prevent such side reactions and inefficiencies due to the presence of unpredictable reactions at more negative potentials, the direct two-electron-transfer ORR is recommended as it holds promising implications for the enhanced production and selectivity of H2O2. Conversely, the semiconductor with CB positions falling between the O2/˙O2− redox potential (−0.33 V vs. NHE) and O2/H2O2 redox potential (+0.68 V vs. NHE) can produce H2O2 directly through the two-electron-transfer pathway, as shown in eqn (7). Hence, the concerted two-electron pathway proves to be more favorable thermodynamically for the photoinduced generation of H2O2 in an ORR because it requires less energy in the CB level. However, the synthesis of H2O2via the two-electron-transfer ORR pathway presents a formidable challenge, chiefly due to unsatisfactory selectivity, determined by the activation energy, stemming from the competing four-electron-transfer pathway, which enjoys a more favorable thermodynamic barrier (Fig. 22c and d). Precisely, the redox potential of O2/H2O (+1.23 V vs. NHE) is more positive compared to O2/H2O2 (+0.68 V vs. NHE), establishing H2O as the prevalent product. Particularly, semiconductors with a CB more positive than +0.68 V versus NHE will find H2O2 synthesis thermodynamically impeded/prohibited, favoring the four-electron-transfer process to yield H2O instead.
| O2 + e− → ˙O2− (E° = −0.33 V vs. NHE) | (4) |
| ˙O2− + e− + 2H+ → H2O2 (E° = +1.44 V vs. NHE) | (5) |
| ˙O2− + h+ → 1O2 (E° = +0.34 V vs. NHE) | (6) |
| O2 + 2e− + 2H+ → H2O2 (E° = +0.68 V vs. NHE) | (7) |
| O2 + 4e− + 4H+ → 2H2O (E° = +1.23 V vs. NHE) | (8) |
6.2. Water oxidation reaction (WOR) pathway
Contrary to the ORR, the WOR is initiated by photoinduced holes within the VB of g-C3N4. Similar to the ORR, the WOR can proceed through three distinct reaction pathways, involving one-, two-, and four-electron transfers, represented by eqn (10), (9) and (12), respectively. Remarkably, the redox potential for the 2e− transfer pathway is +1.76 V versus NHE (eqn (9)), which is higher than +1.23 V versus NHE for the 4e− transfer pathway (eqn (12)). This signifies that the formation of H2O2 is thermodynamically less favorable compared to the oxygen evolution reaction (OER). Consequently, this thermodynamic preference results in the predominant generation of O2 and poses a challenge for H2O2 production during the WOR. The production of H2O2 through water oxidation can occur via a concerted 2e− transfer WOR or an alternative indirect, stepwise single e− transfer WOR pathway. In the indirect route, when the VB edge surpasses +2.73 V versus NHE, the single electron transfer WOR has the potential to generate ˙OH, as illustrated by eqn (10), and subsequently, a pair of ˙OH can merge to produce H2O2, in accordance with eqn (11). However, the realization of this process is entangled with difficulties attributed to the high standard potential (+2.73 V vs NHE), sluggish reaction rates, and diminished selectivity over the OER process.187 The pronounced oxidation potential of the H2O/˙OH couple restricts the formation of ˙OH, which also plays a pivotal role in the decomposition of H2O2.185 Relative to the indirect step, the direct concerted 2e− transfer WOR, represented by eqn (9), demonstrates superior thermodynamic favorability for the synthesis of H2O2. This necessitates the VB edge position of the photocatalyst to be located within the +1.76 and +2.73 V range versus NHE. However, this photoinduced WOR route, indicated by eqn (9), invariably intertwines with competing reactions, such as the single-electron WOR generating ˙OH (eqn (10)) and the four-electron reaction resulting in O2 (eqn (12)). While the process involving the formation of H2O2 through the pairing of hydroxyl radicals induced by holes is theoretically feasible (as outlined in eqn (11)), the actual yield of H2O2 is constrained by the necessity of having a high concentration of these hydroxyl radicals. This requirement significantly limits the production efficiency of H2O2. Additionally, the light-driven two-electron WOR path, which is essential in this process, requires a high oxidation potential. This high potential, unfortunately, leads to a situation where the newly formed H2O2 is prone to rapid decomposition, further complicating the effective generation of H2O2. The photocatalytic WOR for synthesizing H2O2 typically does not occur in isolation, mainly due to its inherent limitations. However, an innovative approach involves coupling the two-electron WOR pathway with the hydrogen evolution reaction (HER). This coupling results in a unique intermediate photocatalytic process that simultaneously produces H2 and H2O2. Furthermore, H2O2 synthesis can be effectively achieved through a dual-pathway strategy, which integrates both the four-electron WOR and the two-electron WOR.184 In this synergistic process, the four-electron WOR pathway supplies oxygen for the ORR pathway, which in turn facilitates the generation of H2O2.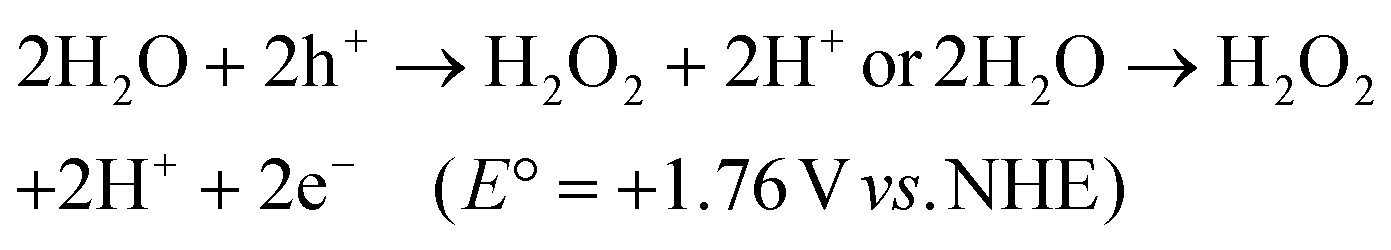 | (9) |
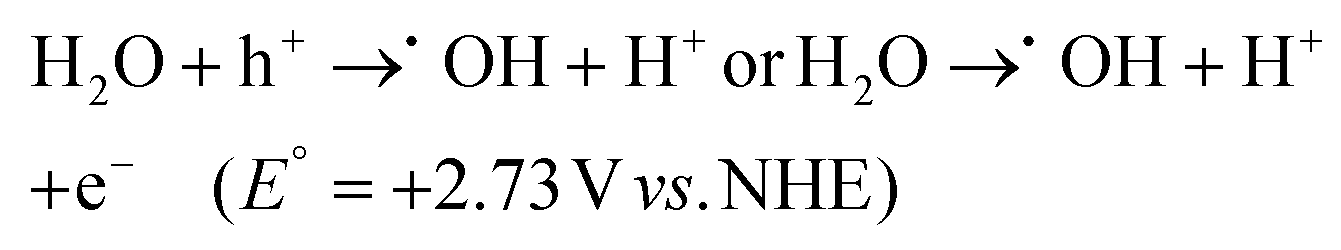 | (10) |
| ˙OH + ˙OH → H2O2 (E° = +1.14 V vs. NHE) | (11) |
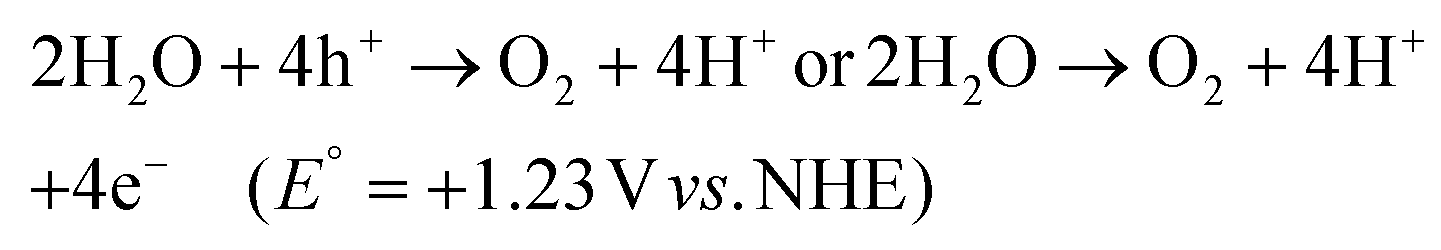 | (12) |
6.3. Integrated dual-channel pathway
H2O2 can be produced through a novel approach by combining the ORR and the WOR. For instance, merging the ORR pathway with the WOR pathway can yield H2O2via dual pathways from both O2 and H2O, enabling 100% atom utilization efficiency as all atoms of O2 and H2O contribute to forming H2O2 (eqn (13)), integrating the two-electron ORR and the two-electron WOR, which is a combination of eqn (7) and (9).188 This integrated approach harnesses the full energy of photoproduced electrons and holes, thus optimizing the photocatalysis process. Additionally, it enables H2O2 formation without utilizing sacrificial agents. Another way to synthesize H2O2 involves combining the four-electron WOR process with the two-electron ORR process, utilizing H2O as an electron donor. This process is outlined using water and oxygen as the sole reactants. Thermodynamic considerations reveal a challenge in the direct synthesis of H2O2 through two-electron water oxidation driven by photogenerated holes, as detailed in eqn (9). This difficulty arises because the oxidation of water to oxygen (O2) in a four-electron process, which occurs at a potential of +1.23 V versus NHE, is more thermodynamically favorable than the two-electron process leading to H2O2, which happens at +1.76 V versus NHE. It is important to note, however, that this alternative route described in eqn (14), combining eqn (7) and (12), generates excess O2, resulting in less than 100% atom utilization efficiency. In the production of H2O2, as outlined in eqn (13) and (14), the water oxidation half-reaction is thermodynamically challenging, requiring an uphill effort with a notable gain in free energy, specifically 204 kJ mol−1 and 117 kJ mol−1 respectively. This process of generating H2O2 from O2 and H2O, without the use of sacrificial agents, aligns well with the principles of a photosynthesis process, as characterized by a positive free energy change (ΔG > 0) (Fig. 22e).189 In this scenario, solar energy is effectively harnessed and stored in the form of H2O2 through photosynthesis. Conversely, when employing a hole scavenger, like organic alcohols, in the oxidation half-reaction (seen in eqn (15)), the production of H2O2 shifts to a photocatalytic process. This is because it is a thermodynamically favorable or “downhill” reaction, where the free energy change is negative (ΔG < 0) (Fig. 22e).189 In such photocatalytic processes, while solar light speeds up the reaction, it does not result in the storage of solar energy.189 It is important to note that the terms “photosynthesis” and “photocatalytic” are often used interchangeably to describe the H2O2 generation reaction. However, a clear understanding and differentiation of these terms is essential for accurate scientific communication, especially considering their distinct thermodynamic properties and the role of solar energy in each process.| 2H2O + O2 → 2H2O2 (ΔG = 204 kJ mol−1) | (13) |
| H2O + O2 → H2O2 + ½O2 (ΔG = 117 kJ mol−1) | (14) |
| R–CH2OH + 2h+ → RCHO + 2H+ | (15) |
Electrochemical investigations have shown that the CB of g-C3N4 is situated at approximately −1.13 V versus NHE. This level is adequately negative, surpassing the required potentials for both O2/H2O2 (0.68 V vs. NHE) and O2/˙O2− (−0.33 V vs. NHE) conversions, endowing it with the necessary reduction potential to generate H2O2. However, the VB of g-C3N4, at around 1.51 V vs. NHE, does not meet the 1.76 V vs. NHE threshold needed for H2O2 production through the WOR. Such findings show that g-C3N4 possesses a reduction potential that is adequate for H2O2 synthesis via the ORR under the influence of visible light.49 In the context of overall photocatalytic H2O2 generation, the OER plays a pivotal role, as illustrated in eqn (12). The VB potential of g-C3N4, estimated to be around 1.51 V vs. NHE, is sufficiently high to facilitate water oxidation, yielding oxygen (1.23 V vs. NHE) and thereby driving the photocatalytic OER. Nonetheless, the overpotential associated with the OER often acts as a bottleneck in oxygen evolution, thereby impeding the overall efficiency of the photocatalytic production of H2O2. Thus, enhancing the OER reaction kinetics is vital for boosting overall H2O2 production efficiency.
Despite the adequacy of the CB and VB of g-C3N4 for initiating the reaction, the photocatalytic efficiency remains limited. The efficiency of surface reactions on photocatalysts is largely influenced by the nature of the surface atoms present, the microstructure, and the density of active sites. Consequently, for effective photocatalytic H2O2 generation, it is crucial to modify g-C3N4 to better support the adsorption and activation of O2 and H2O molecules.
7. Dynamics of hydrogen peroxide formation and breakdown
Hydrogen peroxide is inherently unstable, readily decomposing when exposed to ultraviolet (UV) light, under alkaline conditions, or at elevated temperatures. Additionally, the failure to promptly remove H2O2 from the surface of the catalyst can lead to further reactions with electrons or holes, thus diminishing the efficiency of photocatalytic H2O2 production. During the one-step reduction of H2O2, ˙OH radicals are formed, whereas h+ can interact with H2O2 to produce ˙O2− (see eqn (16) and (17)).184,185,188| H2O2 + e− → ˙OH + OH− | (16) |
| H2O2 + h+ → ˙O2− + 2H+ | (17) |
The kinetics of H2O2 photoproduction is characteristically reversible, attributed to the simultaneous occurrence of its formation and decomposition, which hampers satisfactory yields of H2O2. This photocatalytic production is a balance between the formation and decomposition processes of H2O2. The kinetics of H2O2 formation and decomposition are characterized by zeroth- and first-order processes, respectively, with the overall behavior described by empirical eqn (18) and (19).
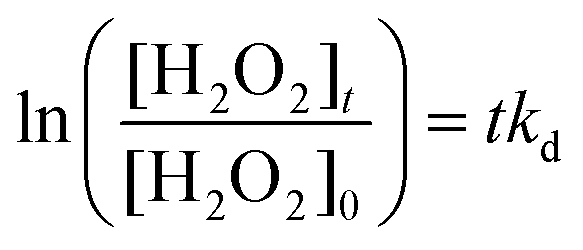 | (18) |
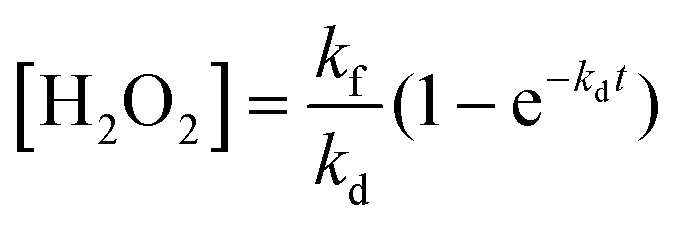 | (19) |
Here, kf and kd represent the rate constants for the formation (μmol L−1 min−1) and decomposition (min−1) of H2O2, respectively, where t denotes time (min) and [H2O2]t (μmol L−1) refers to the concentration of H2O2 at a given time t. Consequently, a higher kf coupled with a lower kd indicates increased activity towards H2O2 generation, highlighting the efficiency of the process. The complexity of H2O2 decomposition is influenced by various factors, including the intensity of light and the surface properties of the photocatalyst, where reducing light energy by employing visible light or decreasing the adsorption capacity of the photocatalyst for H2O2 can affect the process.30
8. Photocatalytic generation of H2O2 by g-C3N4
Shiraishi et al. initially explored the mechanism behind the photocatalytic generation of H2O2 by g-C3N4, highlighting the crucial function of O2.74 Through employing 5,5-dimethyl-1-pyrroline N-oxide (DMPO), a reagent for spin-trapping, their study demonstrated the limited occurrence of one-electron O2 reduction to form the ˙OOH radical on g-C3N4. This conclusion was supported by electron spin resonance (ESR) analysis, as shown in Fig. 23a, where there was the presence of a negligible adduct signal, when g-C3N4 served as the photocatalyst. This minimal signal infers that the electron prefers to quickly reduce the superoxo radical (transitioning from d to f) rather than engaging in one-electron O2 reduction (transitioning from d to e). Analysis of the Raman spectra, as presented in Fig. 23b, reveals that isotopic labeling leads to the appearance of new broadband signals at 891 cm−1 in the EtOH/H2O/16O2 system and at 861 cm−1 in the EtOH/water/18O2 system, signifying the generation of 1,4-endoperoxide species on the tri-s-triazine unit of the photoexcited g-C3N4 surface, via a two-electron oxygen reduction pathway (illustrated in Fig. 23c, from d to f). The virtually nonexistent ˙OOH radical formation (referenced in Fig. 23c; from d to e) likely results from the rapid reduction of the superoxo radical (transitioning from d to f), facilitated by the stabilization of endoperoxide species at the C1 and N4 positions of the triazine ring. Consequently, this mechanism effectively hinders/inhibits one-electron O2 reduction (d → e → a), favoring instead a preferential two-electron reduction pathway. Such a process explains the exceptionally high selectivity of g-C3N4 (around 90%) for H2O2 production.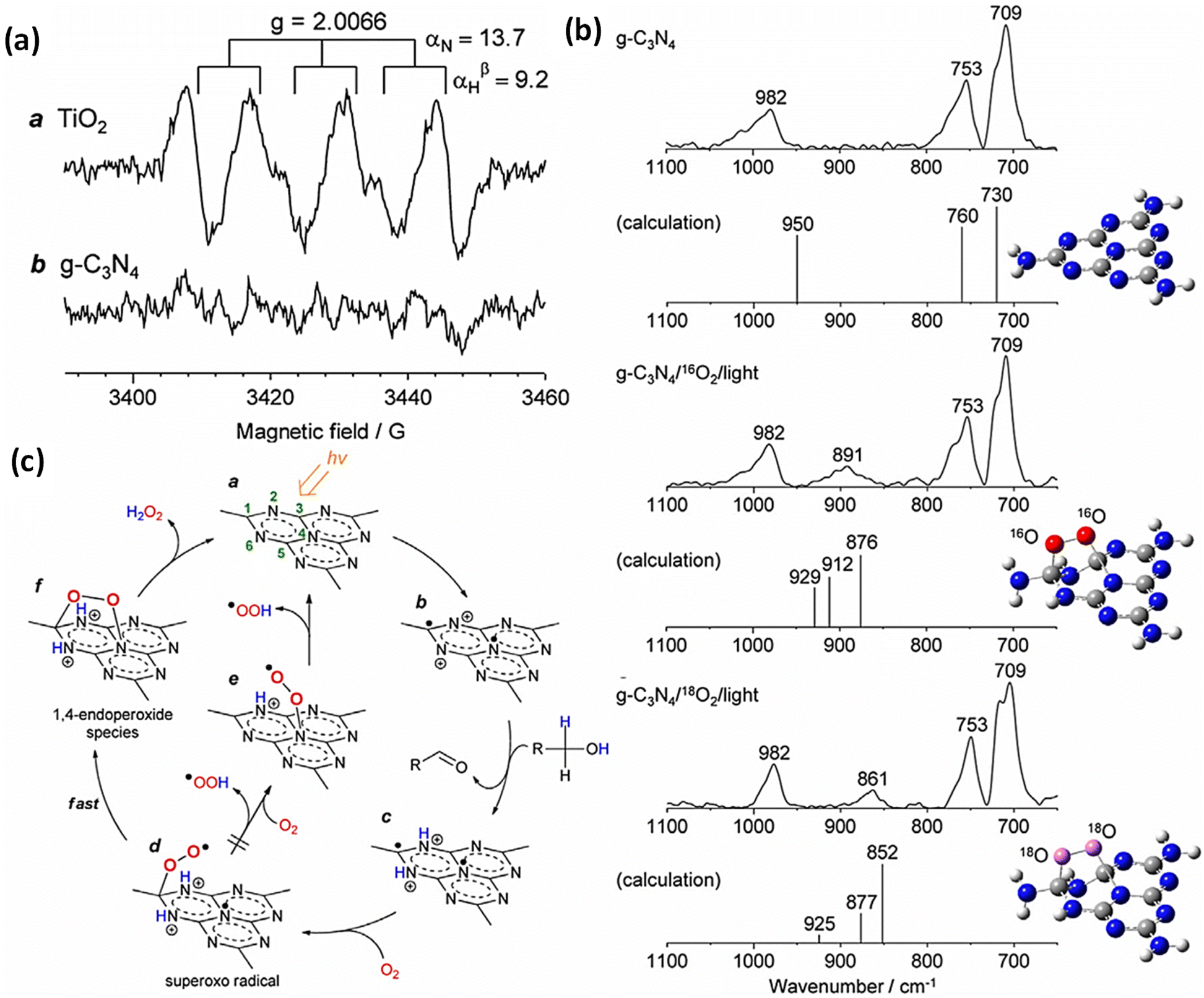 | ||
| Fig. 23 (a) ESR spectra of solutions resulting from the photoillumination of TiO2 and g-C3N4 in an ethanol/water/O2 system. (b) Raman spectra and calculated Raman shifts for g-C3N4 following photoreaction with 16O2 and 18O2 in an ethanol/water mixture. (c) The mechanism for the selective formation of H2O2 on g-C3N4 under visible light illumination. The figures are reproduced with permission from ref. 66. Copyright 2023, Wiley-VCH. | ||
Drawing from the aforementioned information, Shiraishi et al. introduced the mechanism for the photocatalytic production of H2O2 by g-C3N4, as shown in Fig. 23c. Initially, illumination of g-C3N4 generates photogenerated charges (a). These charges then separate, with electrons localizing at the C1 and N4 sites of the triazine ring and holes at the N2 and N6 sites (b). The α- and β-hydrogens of the alcohol remove these holes, generating an aldehyde (b to c), while oxygen captures the electrons from either the C1 or N4 site to form a superoxo radical (c to d). Subsequently, this radical is quickly reduced by another electron at the para position, leading to the formation of the 1,4-endoperoxide species (d to f). The cycle is completed with the protonation of this species, producing H2O2 (f to a), thereby finishing the photocatalytic process.
9. Performance evaluation method: apparent quantum yields (AQYs) and solar-to-chemical conversion (SCC) efficiencies
The apparent quantum yield (AQY) and solar-to-chemical conversion (SCC) efficiency serve as pivotal parameters for assessing the photocatalytic performances of photocatalysts, specifically in the domain of H2O2 production. The AQY is essentially determined by the ratio of the number of electrons transferred for H2O2 production to the number of incident photons at a given wavelength, and is calculated according to eqn (20)–(22):42,190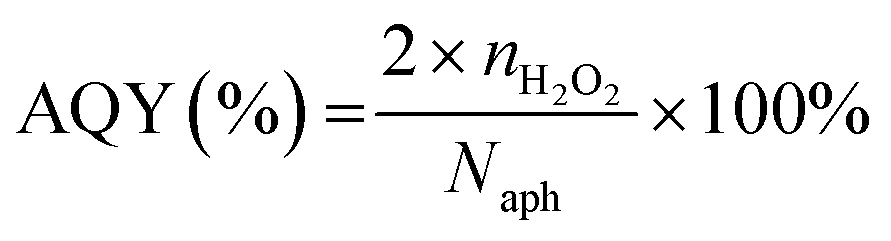 | (20) |
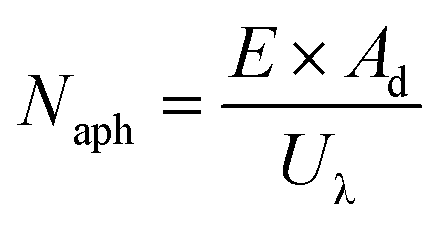 | (21) |
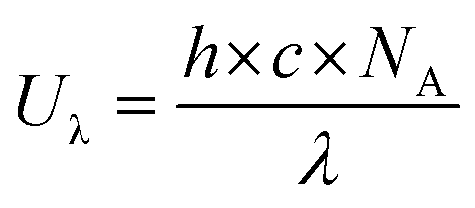 | (22) |
In the above formulas, nH2O2 represents the molar amount of H2O2 produced, Naph is the number of incident photons entering the reaction vessel, and E signifies the measured difference in light intensity transmitted before and after being absorbed by the photocatalyst (mW cm−2). Additionally, Ad denotes the area of the light collector part of the radiometer (cm2), Uλ is the molar photon energy of the given wavelength λ (J molphoton−1), h = 6.626 × 10−34 J s−1 is the Planck constant, c = 3 × 108 m s−1 represents the speed of light in a vacuum, NA = 6.022 × 1023 mol−1 is the Avogadro number, and λ specifies the given wavelength of incident photons (nm). AQY measurements, pivotal for understanding photocatalytic H2O2 production performance, are conventionally executed in a borosilicate glass bottle, utilizing an air mass (AM) 1.5 solar simulator in conjunction with wavelength-dependent band-pass filters, facilitating a meticulous and detailed evaluation of photocatalytic efficiency under varying conditions.
Moreover, solar-to-chemical conversion (SCC) efficiency also plays an integral role in the comprehensive evaluation of photocatalysts, providing a nuanced understanding of the conversion efficiency of solar energy into chemical energy, enriching the overall interpretative framework for analyzing photocatalytic performances and their optimization. The SCC efficiency is a critical metric and is instrumental in evaluating the transformation of captured solar energy into H2O2. The methodology for measuring SCC efficiency aligns closely with the conditions employed for AQY measurements, with the noteworthy exception being the incorporation of a λ > 420 nm cutoff filter. This filter is indispensable for preventing the consequential decomposition of synthesized H2O2 by ultraviolet light, ensuring the integrity of the measurement.
The mathematical representation for calculating SCC efficiency is given by eqn (23):185,190
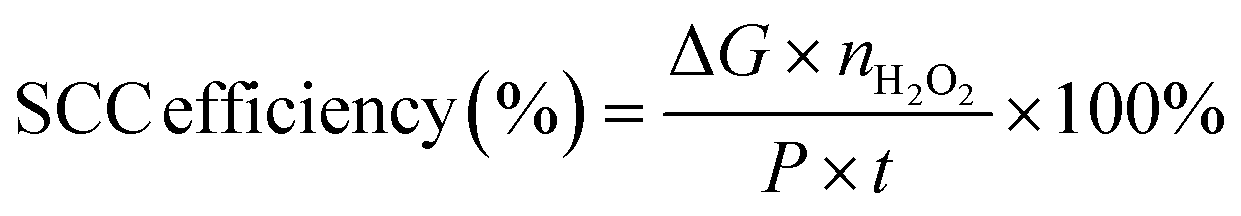 | (23) |
Here, ΔG = 117 kJ mol−1 symbolizes the Gibbs free energy required for the generation of H2O2 from H2O and 1/2O2, P represents the power of the incident photons, denoted in watts (W), and t delineates the illumination time, measured in seconds (s). The number of incident photons, coupled with the average intensity of irradiation, can be meticulously quantified by a radiometer, rendering a more accurate depiction of the solar energy harnessed for H2O2 production.
In essence, SCC efficiency provides a meticulous evaluation of the capability to convert captured solar energy into H2O2, extending a nuanced understanding of the operational efficacy of photocatalysts in harnessing solar energy for chemical transformations, thereby enriching the analysis and optimization potential of photocatalytic processes.
10. Quantification method for photogenerated H2O2
Accurate measurement of photogenerated H2O2 is crucial for advancing the field of photocatalytic hydrogen peroxide production since the concentrations achieved through photocatalytic methods generally range from the micromolar to millimolar levels. Presently, the primary methods for this quantification are spectrophotometric analysis and chemical titration techniques. In spectrophotometry, the concentration of H2O2 is typically ascertained by employing redox reactions involving H2O2 and specific colour-forming agents. These reactions transform colourless H2O2 into substances with distinct colours, which are then quantified via UV-visible or fluorescence spectrophotometry supported by pre-established calibration curves. One classical approach employs iodometry, where H2O2 oxidizes iodide ions to form triiodide ions, displaying a unique absorbance peak around 350–353 nm (eqn (24)).30 To facilitate this redox reaction, a surplus of potassium iodide (KI) and a catalytic amount ammonium molybdate ((NH4)2MoO4) are introduced into the initial colourless medium, where the stoichiometric reduction of H2O2 occurs.| 3I− + 2H+ + H2O2 → I3− + 2H2O | (24) |
A variety of indicators have been investigated for their efficacy, including the catalysis of potassium iodide by potassium hydrogen phthalate, the oxidation of N,N-diethyl-p-phenylenediamine (DPD) facilitated by peroxidase (POD) and other reactions such as the oxidation of VO3− salts, the acid-catalyzed oxidation of o-tolidine, and the oxidation of oxo(5,10,15,20-tetra(4-pyridyl)porphyrinato) titanium(IV) (TiO(tpypH4)4+). Additionally, the reduction of Cu(II) in the presence of 2,9-dimethyl-1,10-phenanthroline (DMP), the reduction of cerium(IV) salts, and the horseradish peroxidase (HRP)-catalyzed dimerization of p-hydroxyphenylacetic acid (POHPAA) have been studied. However, the reliability of these colorimetric indicators can be compromised in the presence of specific organic electron donors. For instance, p-benzoquinone has been found to interfere with DPD and VO3−, leading to inaccuracies in H2O2 quantification. Chemical titration, specifically using potassium permanganate (KMnO4), is another commonly employed method for detecting H2O2. This technique capitalizes on the reductive decolorization of KMnO4 by H2O2 (eqn (25)). The reaction can be summarized as follows:
 | (25) |
However, this titration method is susceptible to interference from aromatic compounds and suffers from the issue of over-oxidation of a variety of species including H2O2 due to the robust oxidizing nature of KMnO4, thus affecting the accuracy of H2O2 quantification. Thus, spectrophotometric techniques appear to offer more targeted and reliable results than titration methods, primarily because the resulting products are identifiable by their specific wavelengths. Despite the range of available H2O2 detection methods, there is a pressing need for a standardized, accurate method for quantifying H2O2 concentrations in photocatalytic systems.
11. Advancements in g-C3N4-based direct Z-scheme and S-scheme catalysts for photocatalytic hydrogen peroxide generation
In this section, we provide an overview of recent studies on g-C3N4-based direct Z-scheme and more relevant S-scheme heterostructures aimed at generating hydrogen peroxide (H2O2) through photocatalysis. We provide a concise overview of the results from different research groups and include Tables 4 and 5, which outline the reaction conditions and efficiencies documented in each study. The development of a heterostructure based on g-C3N4, either in the form of a direct Z-scheme or the increasingly significant S-scheme, when combined with oxidation-type photocatalysts, enables the overall photosynthesis of H2O2 by facilitating the simultaneous production of H2O oxidation and O2 reduction products by utilizing the spatially separated oxidation and reduction components.191 This system simultaneously enhances the photocatalytic activity by broadening light absorption, improving stability, and boosting charge carrier and redox active site separation and modulating their transportation, while also prolonging carrier lifetimes and optimizing redox potential. It also aligns with the optical responses of adjacent components, induces band bending, and creates an IEF with preferred band alignment, promoting the segregation of useful photoinduced charge carriers, concurrently addressing the aforementioned challenges of g-C3N4. This topic is currently at the forefront of research in artificial photocatalysis. Our goal is to provide an understanding of how to create efficient g-C3N4-based photocatalysts for the generation of H2O2.| Entry | Photocatalyst | Electron donor | Concentration of photocatalyst | Irradiation conditions | H2O2 production | AQY | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | g-C3N4/perylene imides | None | 50 mg catalyst in 50 mL of distilled water | 300 W Xe lamp with a 420 nm cutoff filter Vis (λ > 420 nm) | 120 μmol (2 h) | 3.2% (λ = 420 nm) | 195 |
| 2 | ZnPPc/NB-g-C3N4 | None | Catalyst (10 mg) in H2O (20 mL) | Xe lamp through a UV cutoff filter (800 nm > λ > 400 nm) | 114 μmol g−1 h−1 | N/A | 196 |
| 3 | Ti3C2/g-C3N4/BiOCl | IPA | 50 mg catalyst in 50 mL mixed aqueous solution containing 2.5 mL isopropanol (IPA, 5 vol%) | 300 W Xe lamp, simulated sunlight | 1275 μM (60 min) | NA | 205 |
| 4 | Bi4O5Br2/g-C3N4 | None | 50 mg catalyst in 50 mL water | 300 W Xe lamp visible-light (λ > 420 nm) | 124 μM (60 min) | 11.8% (420 nm) | 50 |
| 5 | α-Fe2O3/CQD@g-C3N4 | None | 5.0 mg catalyst in 10 mL of water | 300 W Xe-lamp (420 nm cutoff) | 1.16 μmol L−1 min−1 | 17.80% at 420 nm | 207 |
| 6 | TiO2/B-doped g-C3N4 | EtOH | 20 mg catalyst in 20 mL of DI water containing 1 mL of EtOH | 250 W visible light | 110 μmol h−1 | NA | 214 |
| 7 | K+-doped g-C3N4 (KCN)/WO3 | EtOH | 0.2 g catalyst in 200 mL of deionized water, and 5 mL of ethanol | 300 W xenon lamp | 1.33 mmol L−1 in 4 h | NA | 210 |
| 8 | Cu2(OH)PO4/g-C3N4 | None | 0.2 g catalyst in deionized water (200 mL) | Solar simulator (300 W Xe lamp and 200 W infrared light) | 7.2 mmol L−1 | NA | 198 |
| 9 | Cu2(OH)2CO3/g-C3N4 | None | 0.2 g catalyst in deionized water (200 mL) | Simulated solar light source | 8.9 mmol L−1 (18 h) | NA | 199 |
| 10 | RF resin/g-C3N4 | None | 0.05 g of catalyst in 30 mL pure water (1.67 g L−1) | 300 W xenon lamp with a 420 nm cutoff filter | 72.8 μmol (12 h) | NA | 193 |
| 11 | Cd0.6Zn0.4 S/g-C3N4 | Ethanol | 10 mg catalyst in 100 mL deionized water containing 10% ethanol | 300 W Xe lamp with a 420 nm cutoff filter | 1098.5 μmol g−1 h−1 | NA | 11 |
| 12 | rGO-decorated W18O49@g-C3N4 | None | 10 mg of the photocatalyst in 50 mL of ultrapure water | 300 W Xe lamp with a 400 nm cutoff filter visible light source (400 < λ < 780 nm) | (71, 58.5, and 9 μmol g−1 h−1) under simulated solar, visible (>400 nm), and NIR light (>800 nm) | 5.8% at 380 nm | 208 |
| 13 | W18O49/crystalline g-C3N4 | IPA | Water/IPA | Full spectrum | 5550 μM g−1 h−1 | NA | 211 |
| 14 | S vacancy (MoS2−v)/carbon nitride nanotube (TCN) | IPA | 50 mg of the photocatalyst in 90 mL of water and 10 mL of the IPA mixture | 300 W Xe lamp (λ > 420 nm) | 1879 μmol g−1 h−1 | NA | 202 |
| 15 | NixPy/C/N-ZnO@B-doped g-C3N4 | EtOH | 20 mg catalyst in the solution containing 19 mL of deionised water and 1 mL of ethanol | UV-visible light irradiation (250 W Hg lamp) | 2873.46 μmol h−1 g−1 | NA | 209 |
| 16 | O-doped g-C3N4/ZnIn2S4-Zn | IPA | 10 mg catalyst in 50 mL of mixed solution containing 10% of isopropyl alcohol. | 300 W Xe lamp (λ > 420 nm) | 168.67 μmol L−1 in 80 minutes | 18.61% at 400 nm | 200 |
| Entry | Photocatalyst | Electron donor | Concentration of photocatalyst | Irradiation conditions | H2O2 production | AQY | Ref. |
|---|---|---|---|---|---|---|---|
| 17 | ZnO/g-C3N4 | EtOH | 20 mg catalyst in 45 mL of water and 5 mL of the ethanol mixture | 300 W xenon lamp (λ > 350 nm) | 1544 μmol L−1 | NA | 152 |
| 18 | C3N4/PDA | EtOH | Photocatalyst (20 mg) in an ethanol solution (20%) | 300 W xenon arc lamp (simulated sunlight illumination) | 3801.25 μmol g−1 h−1 | 2.22% at 400 nm | 153 |
| 19 | S-pCN/WO2.72 | None | 50 mg catalyst in 100 mL of deionized water | 300 W Xe arc lamp (λ > 420 nm) | 87 μM in 3 h | 1.5% at 420 nm | 212 |
| 20 | PDI-Ala/S-C3N4 | None | 50 mg photocatalyst in 50 mL distilled water | 300 W xenon lamp (420 nm < λ < 780 nm) | 28.3 μmol h−1 g−1 | NA | 146 |
| 21 | PDINH@PCN | IPA | 50 mg catalyst in 50 mL water/IPA with a volume ratio of 9:1 |
300 W xenon lamp | 922.4 μmol h−1 g−1 | NA | 194 |
| 22 | Sv-ZnIn2S4@g-C3N4 | IPA | Catalyst (20 mg) in solution (45 mL H2O and 5 mL isopropanol) | 300 W xenon lamp (λ ≥ 420 nm) | 1310.18 μM | NA | 174 |
| 23 | Sulfur-doped g-C3N4/TiO2 | None | 10 mg catalyst in 50 mL of deionized water | 300 W Xe lamp | 2128 μmol h−1 g−1 | 0.61% at 365 nm | 213 |
| 24 | Zn-TCPP/g-C3N4 | EtOH | Catalyst (30 mg) in 50 mL of ethanol aqueous solution (10 vol%) | 300 W Xe lamp | 532.7 μmol L−1 in 90 min | 7.01% at 365 nm | 197 |
| 25 | g-C3N4/HMoP | EtOH | 0.1 g catalyst in 10 mL of ethanol and 190 mL of deionized water | 300 W xenon lamp (λ > 420 nm) | Simulated sunlight (137.1 μmol L−1 h−1), visible light (113.1 μmol L−1 h−1) | NA | 215 |
| 26 | CuInS2/PCN | IPA | 20 mg of photocatalyst in 5% isopropanol aqueous solution (50 mL) | Visible light | 1247.6 μmol L−1 h−1 | 16.0% at 420 nm | 201 |
| 27 | g-C3N4/α-MnS | None | 50 mg catalyst in 50 mL of deionized water | 300 W Xe lamp with visible light cutoff filter | 209.4 μM h−1 | 8.5% at 450 nm | 203 |
| 28 | α-MnO2@B/O-g-C3N4/d-Ti3C2 | EtOH | 0.02 g catalysts in18 mL of DI water and 2 mL of ethanol | 250 W Xe lamp | 2846.4 μmol h−1 g−1 | NA | 206 |
| 29 | K+/I−-CN/CdSe-D | None | Catalyst (10 mg) in water (50 mL) | Visible light irradiation (λ > 400 nm). | 2240.23 μmol h−1 g−1 | NA | 204 |
In research conducted by Shiraishi et al., it was found that resorcinol–formaldehyde (RF) resins could function as a metal-free photocatalyst for generating H2O2 in the absence of a sacrificial agent.192 The SCC efficiency of the resin synthesized at 250 °C achieved a global high of 0.5%. Studies have shown that RF resins created at low hydrothermal temperatures (below 200 °C) display weak photocatalytic activity. This is attributed to the lack of quinone units (CO group), which act as electron acceptors (A), thereby impeding the production of H2O2. The formation of the quinone group during hydrothermal synthesis at high temperatures (above 250 °C) requires auto-pressurization, which is a crucial step in the process. To overcome the issues associated with high-temperature synthesis, the group of Su introduced g-C3N4 into RF resins using a low-cost, solvent-free ball milling mechanochemical method. This innovative approach effectively lowered the temperature required for quinone group formation from 250 °C to 180 °C, making the process more accessible (Fig. 24a).193 This Z-scheme structure led to an extended visible light absorption range beyond 800 nm, resulting in the production of 72.8 μmol of H2O2 within 12 hours when exposed to visible light without the need for sacrificial agents. This represents a 2.37-fold enhancement in comparison with the performance of the unmodified RF resin (Fig. 24b). This innovative approach offers fresh perspectives on the preparation of Z-scheme photocatalysts and the formation of quinone groups in RF resin, contributing to advancements in generating H2O2 within sacrificial agent free systems using visible light, and presenting possibilities for widespread applications. In a different study, Li and colleagues developed a metal-free, entirely organic S-scheme heterojunction, PDINH/PCN, by depositing perylene-3,4,9,10-tetracarboxylic diimide (PDINH) onto porous carbon nitride (PCN) through a crystallization process.194 This structure includes PDINH nanosheets, which are formed through interactions involving hydrogen bonding and π–π stacking, combined with PCN nanosheets. The PDINH/PCN composite achieved a peak rate of H2O2 production at 922.4 μmol h−1 g−1, significantly surpassing those of individual PCN and PDINH, as evidenced by ESR spectra showing strong ˙O2− and ˙OH radical signals. This indicates that H2O2 synthesis occurs through ORR and WOR dual pathways, utilizing the CB electrons of PCN and the HOMO holes of PDINH in the S-scheme heterojunction for these processes.
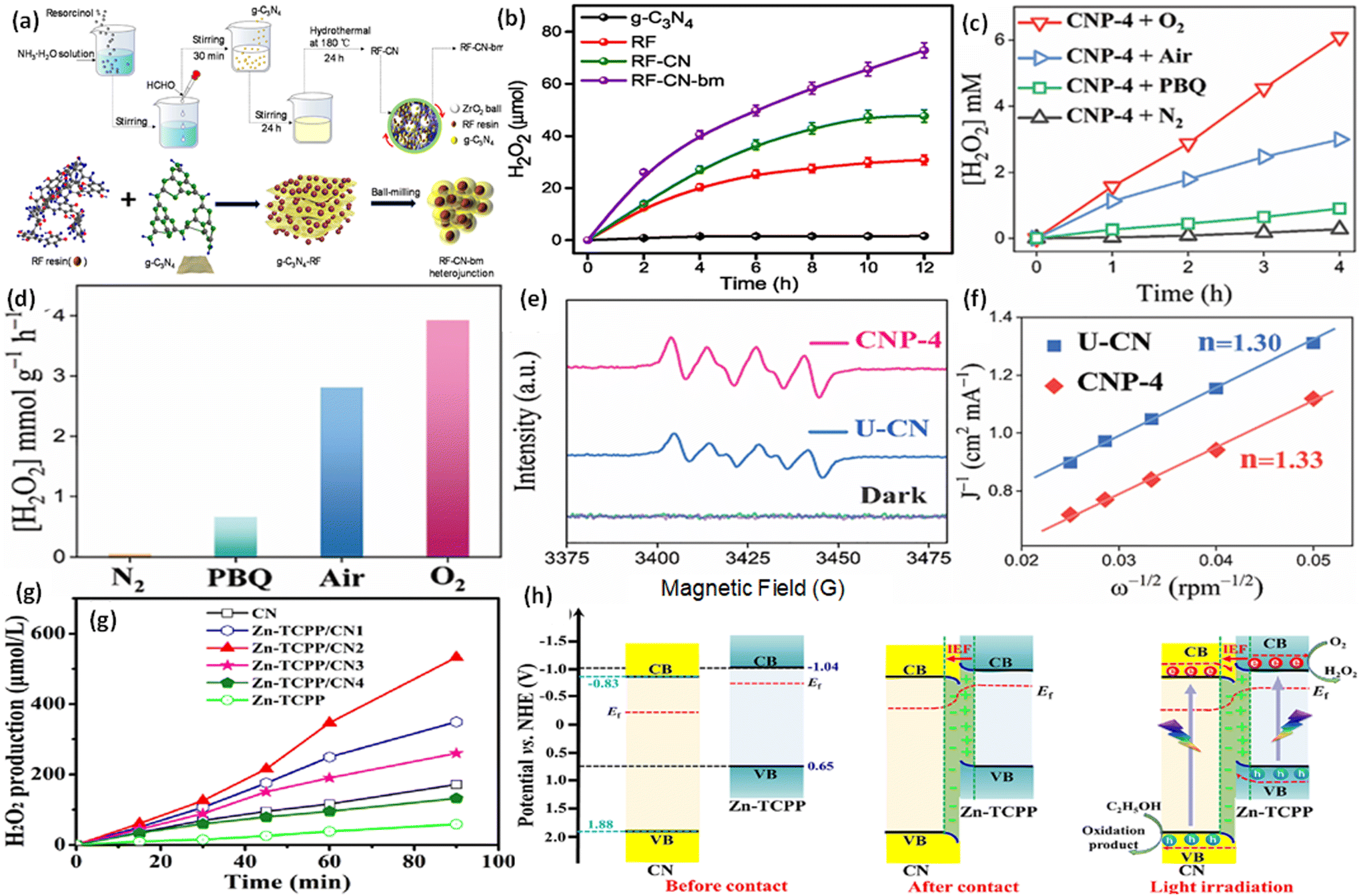 | ||
| Fig. 24 (a) Schematic depiction of RF-CN-bm preparation. (b) H2O2 generation comparison for g-C3N4, RF, RF-CN, and RF-CN-bm. The figures are adapted with permission from ref. 193. Copyright 2023, Elsevier. (c) H2O2 formation by the CNP-4 heterojunction under varying conditions. (d) Histogram showing H2O2 yield after 1 h of illumination. (e) ESR spectra of the DMPO-˙O2− adduct for U-CN and CNP-4 in the absence and presence of light irradiation. (f) Koutecky–Levich plots from RDE analysis of U-CN and CNP-4 at a fixed potential of −1.0 V versus NHE. The figures are adapted with permission from ref. 152. Copyright 2023, Wiley-VCH. (g) H2O2 formation over time when exposed to simulated sunlight. (h) Schematic of the photocatalytic mechanisms in the Zn-TCPP/CN S-scheme heterojunction for H2O2 generation. The figures are adapted with permission from ref. 197. Copyright 2023, Elsevier. | ||
Organic/inorganic hybrid heterostructures are emerging as effective solutions for H2O2 photosynthesis, typically formed through weak noncovalent interactions like electrostatic interactions, van der Waals forces, and hydrogen bonds. However, efficient charge transfer at the organic–inorganic interface remains a challenge. Using specifically tailored polymer structures, which can covalently bond, may overcome this issue by reducing interfacial charge transfer resistance and enhancing charge migration. Metal-free Z-scheme photocatalysts are particularly promising in this regard, given the wide range of suitable organic moieties for heterostructure creation. For instance, the Z-scheme photocatalyst PIx-NCN, created by covalently attaching polyimides (PIs) to g-C3N4 nanosheets, demonstrates efficient stepwise single-electron ORR and WOR processes for H2O2 production.195 Notably, the optimized PI5.0-NCN variant shows a threefold increase in H2O2 production rate to 60 μmol h−1 compared to pristine NCN. Zhang and co-workers developed a novel S-scheme photocatalyst named C3N4/PDA (CNP) by combining ultrathin g-C3N4 (U-CN) with in situ self-polymerized polydopamine (PDA).152 This photocatalyst significantly enhances H2O2 production, reaching 3801.25 μmol g−1 h−1, when exposed to light, surpassing the rates of U-CN and PDA by approximately 2 and 11 times, respectively. The S-scheme structure of CNP facilitates improved light utilization, charge carrier segregation, and transmission, resulting in its enhanced efficiency in producing H2O2 through photocatalysis. The production process, inhibited under ˙O2− scavenging conditions, involves an indirect stepwise single electron O2 reduction pathway, as confirmed by ESR spectroscopy and rotating disk electrode (RDE) characterization studies (Fig. 24(c–f)). These findings indicate that H2O2 is derived from ˙O2− intermediates. Li and colleagues devised a new S-scheme heterojunction, termed PDI-Ala/S-C3N4, which was entirely organic.145 This was achieved by affixing N,N′-bis(propionic acid)-perylene-3,4,9,10-tetracarboxylic diimide (PDI-Ala) onto sulfur-doped g-C3N4 using an in situ self-assembly technique. The effectiveness of this heterojunction is attributed to the aromatic nature of g-C3N4 and the highly conjugated π bonds of PDI-Ala. This enables strong binding through interactions such as π–π stacking and C–N bonds. The resulting composite exhibited a H2O2 formation rate of 28.3 μmol h−1 g−1, outperforming PDI-Ala and S-C3N4 by 2.9 and 1.6 times, respectively. This improved performance is credited to its unique conjugated structure, interfacial electric field, and the formation of new chemical linkages, all of which enhance charge carrier migration and segregation within the system. Additionally, Ye and colleagues developed a Z-scheme photocatalyst through the in situ polymerization of zinc polyphthalocyanine (ZnPPc) onto boron-doped, nitrogen-deficient g-C3N4 nanosheets (NBCN).196 DFT analysis showed electron transfer from nitrogen atoms of NBCN to the phthalocyanine ring and zinc atoms of ZnPPc, as confirmed by in situ XPS under varying light conditions, which indicated that zinc acted as an electron acceptor. UV-vis DRS demonstrated that the presence of ZnPPc on NBCN broadened the visible light absorption, with a red-shifted Q band at 710 nm, indicating enhanced solar spectrum utilization. This heterojunction facilitated O2 evolution from the WOR process in NBCN and two-electron exchange for the ORR in ZnPPc, resulting in a remarkable rate of photocatalytic H2O2 generation of 114 μmol g−1 h−1 in pure water without sacrificial agents. Furthermore, Xia and colleagues engineered an S-scheme heterojunction between Zn-TCPP/CN for photocatalytic H2O2 synthesis.197 This was achieved by attaching zinc porphyrin (Zn-TCPP) photosensitizer onto g-C3N4 (CN) via a –CONH– bridging bond through the process of calcination. The optimal Zn-TCPP/CN2 composite achieved a H2O2 production rate of 532.7 μmol L−1 in 90 min, surpassing those of pristine CN and Zn-TCPP by 3.1 and 9.0 times, respectively (Fig. 24g). In situ irradiated XPS and KPFM analyses offered insights into the charge dynamics in the S-scheme between Zn-TCPP and CN, improving carrier separation and redox capabilities, consequently enhancing the photocatalytic performance (Fig. 24h). Moreover, theoretical calculations demonstrated a strong interplay between Zn-TCPP and CN, promoting electron dispersion and the segregation of charge carriers.
Bismuth oxyhalides, Bi-rich materials recognized for their outstanding mass transfer properties, stability under light exposure, and responsiveness to visible light, effectively catalyze water oxidation owing to their high VB potential. In this context, Zhang and colleagues created a direct Z-scheme photocatalyst, Bi4O5Br2/g-C3N4, by affixing Bi4O5Br2 nanorods onto g-C3N4 nanosheets through a water-assisted self-assembly technique.64 This innovative heterostructure demonstrated an impressive rate of H2O2 production, reaching 124 μM in 60 min in pure water, which was nearly 25 times higher than that achieved with g-C3N4 alone. The notable improvement in H2O2 generation is attributed to the face-to-face attachment of Bi4O5Br2 nanorods and g-C3N4 nanosheets in the Z-scheme heterojunction photocatalyst. This arrangement enables effective transfer of charge carriers and supplies robust redox electrons for O2 reduction as well as holes for H2O oxidation.
Recently, transition metal sulfides and metal selenides have emerged as promising materials for photocatalytic H2O2 generation. Their narrow bandgaps, broad spectral sensitivity, and adjustable energy band levels are key factors contributing to their efficacy in this application. The VB of specific metal sulfides falls within a suitable range, creating a significant thermodynamic driving force for the WOR. Moreover, the CB of certain metal sulfides is more negatively positioned than the reduction potential of O2, providing an ample potential for the ORR. Hence, it is advisable to investigate a broader range of transition metal sulfides for applications in solar-driven H2O2 generation systems. In this context, Yu and co-workers synthesized a Z-scheme photocatalyst by attaching Cd0.6Zn0.4S nanoparticles to ultrathin g-C3N4, employing a direct synthesis approach to create a distinctive point-to-face arrangement.11 This morphology prevents agglomeration due to the plentiful surface binding sites provided by UCN and enables fast interfacial charge migration by shortening the carrier travel path. In situ light illuminated KPFM, DFT calculations, and XPS studies revealed a strong interfacial electric field between Cd0.6Zn0.4S and g-C3N4, leading to an accelerated Z-scheme exciton migration and segregation route. The Cd0.6Zn0.4S/g-C3N4 composite demonstrates greater surface potential and electric field than its individual parts (Fig. 25(a–d)), enhancing the redox potential and reducing carrier recombination. This leads to a remarkable photocatalytic H2O2 production rate of 1098.5 μmol g−1 h−1 (Fig. 25e). Additionally, the free energy diagram also supports the higher O2 reducing ability of the composite (Fig. 25f).
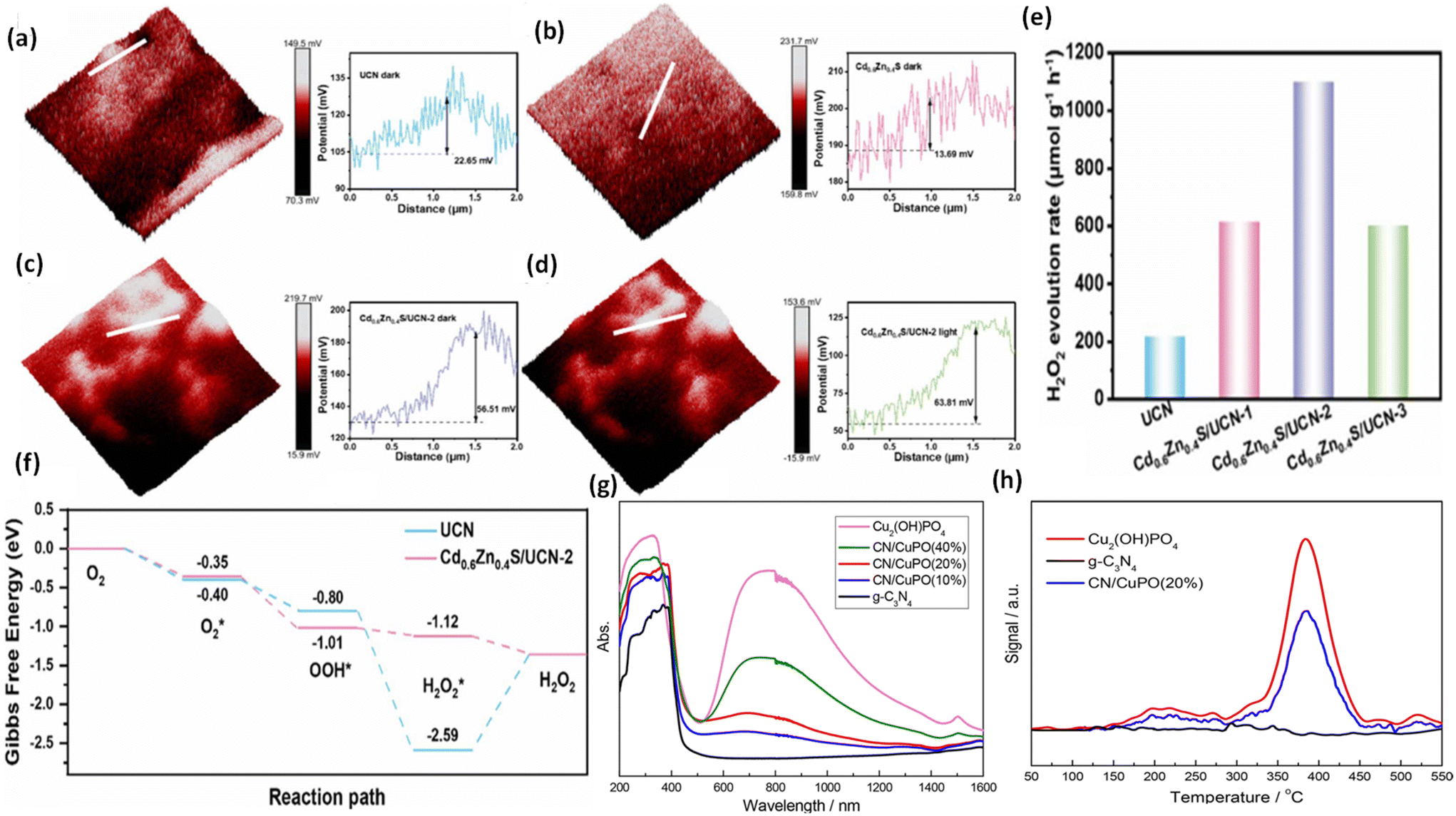 | ||
| Fig. 25 3D surface charge potential distribution and corresponding line-scanning profiles of (a) UCN, (b) Cd0.6Zn0.4S, and (c) Cd0.6Zn0.4S/UCN-2 composite in the dark, and (d) the Cd0.6Zn0.4S/UCN-2 heterojunction photocatalyst under illumination. (e) Production rate comparison of UCN and Cd0.6Zn0.4S/UCN composites using visible light. (f) Free energy diagrams illustrating O2 conversion to H2O2 by UCN and Cd0.6Zn0.4S/UCN-2 systems. The figures are adapted with permission from ref. 11. Copyright 2023, RSC. (g) UV–vis spectra comparison. (h) O2-TPD analysis comparison of g-C3N4, Cu2(OH)PO4, and CN/CuPO(20%) samples. The figures are adapted with permission from ref. 198. Copyright 2018, ACS. | ||
Moreover, combining conventional narrow bandgap semiconductors with g-C3N4 to form heterostructures has emerged as a successful approach for creating photocatalysts that are responsive to near-infrared (NIR) light. This enhances solar absorption in the NIR region and improves the separation of charges. In this context, Wang and colleagues created a Z-scheme photocatalyst, Cu2(OH)PO4/g-C3N4, capable of a full-spectrum response for producing H2O2, which was achieved by using an in situ hydrothermal method.198 Cu2(OH)PO4, with strong NIR absorption (800–1200 nm), as shown in Fig. 25g, enhances the overall solar absorption of the composite. Additionally, the [PO4]3− content of Cu2(OH)PO4 improves O2 adsorption, crucial for photocatalytic H2O2 production, as indicated by the O2-TPD signal (Fig. 25h). The optimized CN/CuPO (20%) photocatalyst achieves a 7.2 mmol L−1 H2O2 concentration under simulated sunlight without sacrificial agents, outperforming neat g-C3N4 and Cu2(OH)PO4 by 13 and 31.3 times, respectively. Likewise, Li and collaborators prepared a Cu2(OH)2CO3/g-C3N4 composite capable of responding to the full spectrum of light.199 They achieved this using a self-sacrificial method, wherein partial decomposition of g-C3N4 resulted in a Cu2(OH)2CO3/g-C3N4 composite with a highly effective combined interface. This composite exhibited significant absorption in the near-infrared (NIR) range (800–1200 nm) and achieved a H2O2 concentration of 8.9 mmol L−1 when exposed to simulated sunlight. This performance surpasses those of pristine g-C3N4 and Cu2(OH)2CO3 by more than 16 and 26.9 times, respectively. This highlights the importance of integrating NIR-based semiconductors into Z-scheme photocatalysts for enhanced H2O2 production.
In another report, Liu et al. developed a 2D/2D Z-scheme photocatalyst in the O-doped g-CN/ZnInS-Zn (ZIS-Z/OCN) system with defect engineering for improved photoinduced ROS production.200 ZIS-Z/OCN, featuring a better interfacial electric field (IEF), reduced charge transport distances, and zinc vacancies, efficiently minimizes hot charge-carrier recombination and speeds up charge movement at the hetero-interface. This leads to exceptional molecular oxygen activation under visible light, surpassing those of pristine ZIS-Z and OCN. Consequently, the 40ZIS-Z/OCN sample produces 168.67 μmol L−1 of H2O2 during 80 min of visible light exposure. Furthermore, Sun et al. developed a unique photocatalyst by merging agaric-like microporous g-C3N4 with sulfur vacancy-modified, flower-like ZnIn2S4, forming an S-scheme heterojunction (g-C3N4/Sv-ZnIn2S4) that showed a H2O2 production efficiency of 1310.18 μM, which was four times higher than that of pristine g-C3N4 (Fig. 26a).173 The efficiency of the catalyst is enhanced by its strategic morphology, sulfur vacancies, and the design of interfacial S–C bonds, along with an interfacial built-in electric field that guides the S-scheme electron transfer. H2O2 generation follows an indirect two-step single-electron transfer route. The charge migration path is evidenced by EPR spectra: under light, the g-C3N4/Sv-ZnIn2S4 composites show stronger DMPO-˙O2− and DMPO-˙OH signals compared to their individual components, indicating enhanced ˙O2− and ˙OH radical production and effective charge migration facilitated by the S–C bonds and S-scheme heterojunction, thus boosting H2O2 production (Fig. 26b and c). Jiang and co-workers developed an iron-free photo-Fenton-like (PFL) reaction system using a direct Z-scheme heterostructure composed of sulfur vacancy modified MoS2 with carbon nitride nanotubes (MoS2−v/TCN), offering a breakthrough for generating hydrogen peroxide over a wide pH range and addressing the traditional limitations of Fenton oxidation (Fig. 27a).201 The band structure of MoS2−v/TCN facilitates the rapid segregation of photoexcited electrons and holes. DFT results reveal that the Z-scheme charge dynamics lowers the energy barrier for *tOOH and *HO2− formation, while S vacancies on MoS2−v improve photoinduced electron utilization by MoS2−v/TCN (Fig. 27b and c). This results in a high performance and stable H2O2 generation (254.8–269.9 μmol g−1 h−1 in pure water, 1879 μmol g−1 h−1 in 10% isopropanol) over the pH range of 3–9. This approach provides an innovative strategy for on-site generation and activation of H2O2 in Fenton oxidation reactions. In related research by Zhang et al., an S-scheme heterojunction, CuInS2/PCN, was developed through an in situ low-temperature hydrothermal method.202 The catalytic efficiency of the optimized sample, 3CuInS2/PCN, showed a H2O2 production rate of 1247.6 μmol L−1 h−1, substantially exceeding the performance of the separate components. It was 11.6 times higher than that of PCN (107.4 μmol L−1 h−1) and 16.0 times higher than that of CuInS2 (78.0 μmol L−1 h−1) (Fig. 27d). Furthermore, the apparent quantum yield for H2O2 generation using 3CuInS2/PCN achieved 16.0% at a wavelength of 420 nm.
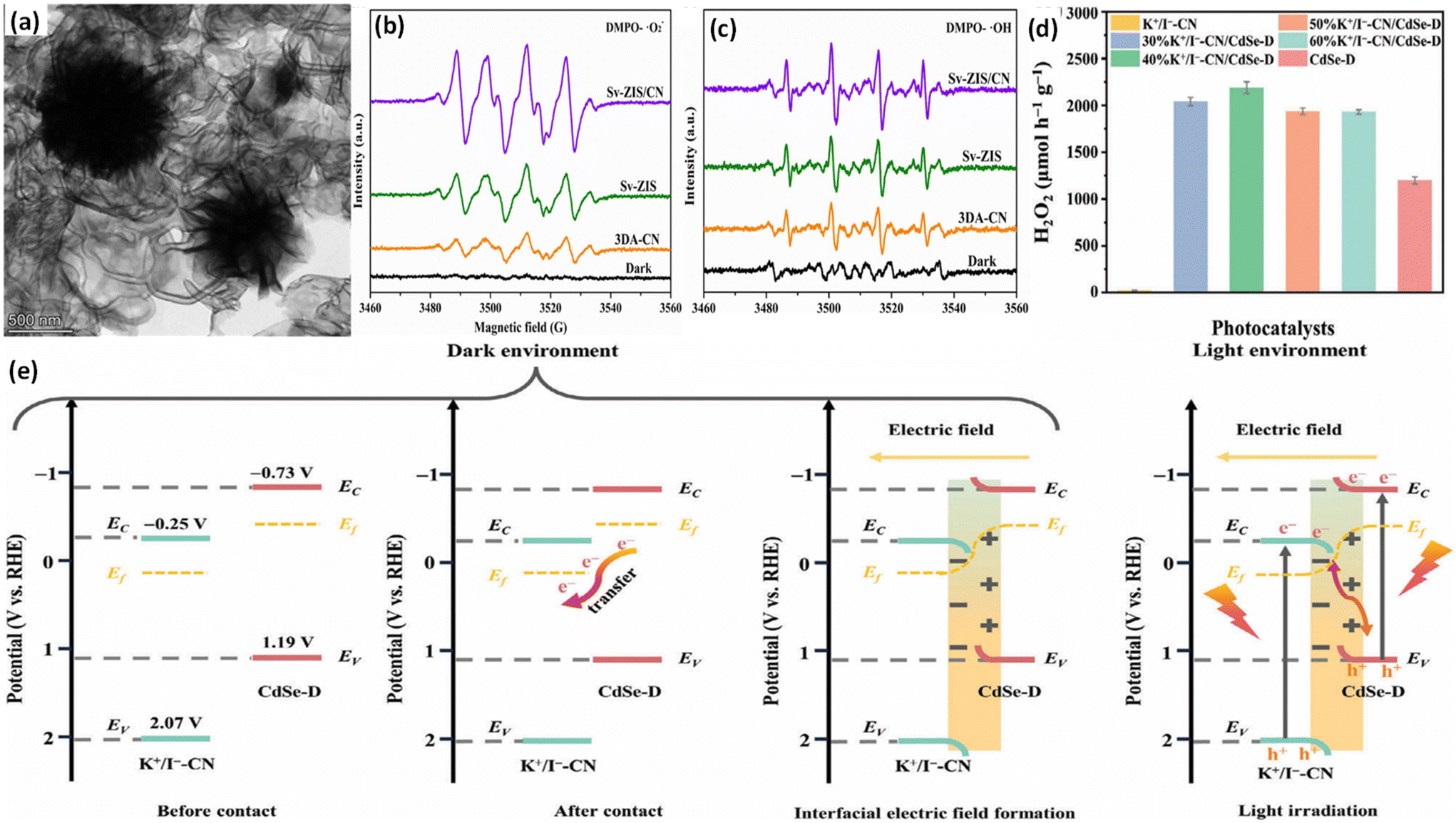 | ||
| Fig. 26 (a) TEM images showing the Sv-ZIS/CN composite. ESR spectra of (b) DMPO-˙O2− and (c) DMPO-˙OH for Sv-ZIS/CN, Sv-ZIS, and 3DA-CN samples. The figures are adapted with permission from ref. 173. Copyright 2022, RSC. (d) H2O2 production rates over the catalysts under visible light (λ > 400 nm) illumination. (e) Schematic depicting the charge transfer mechanism in the K+/I−-CN/CdSe-D S-scheme heterojunction. The figures are adapted with permission from ref. 204. Copyright 2024, Wiley-VCH. | ||
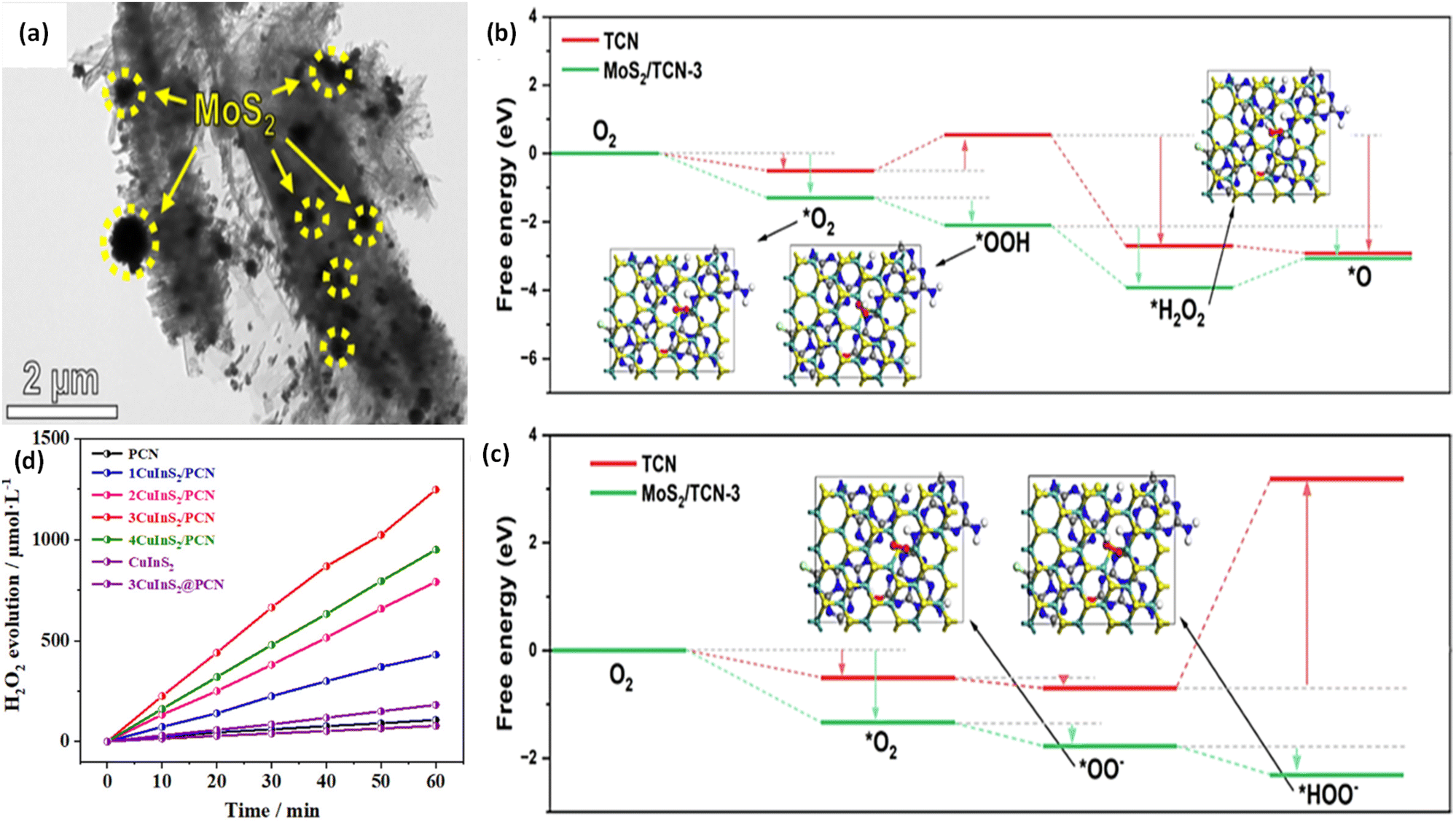 | ||
| Fig. 27 (a) SEM images of MoS2−v/TCN-3. Free energy change graphs for the *HO2− 2e− ORR pathway under (b) acidic and (c) alkaline conditions. The figures are adapted with permission from ref. 201. Copyright 2023, Elsevier. (d) Photocatalytic H2O2 generation by PCN, CuInS2, 3CuInS2@PCN, and xCuInS2/PCN samples. The figures are adapted with permission from ref. 202. Copyright 2023, Elsevier. | ||
Additionally, Wang et al. developed an S-scheme p–n heterojunction photocatalyst, PCN/MnS, by combining p-type α-MnS with n-type protonated g-C3N4 (PCN) through a one-step synthesis method.203 This composite develops a nanosphere morphology of α-MnS with lamellar-derived protonated g-C3N4 layers on its surface, forming a heterojunction that improves the surface area, light absorption capacity, and charge migration. This structure significantly boosts the photocatalytic production rate of H2O2 to 209.4 μM h−1, outperforming pure α-MnS by 1.5 times in air without needing O2 gas bubbling or sacrificial agents. Moreover, the group of He synthesized a K+/I−-CN/CdSe-D S-scheme photocatalyst by incorporating potassium and iodide ions intercalated into carbon nitride together with diethylenetriamine modified cadmium selenide through a dual approach involving molten salt and microwave-assisted hydrothermal techniques.204 The incorporation of I− and K+ ions into C3N4 improves light harvesting and reduces the distance required for charge transfer between layers. Additionally, the formation of interfacial C–Se bonds effectively regulates the charge migration process. The 40%K+/I−-CN/CdSe-D composite demonstrated an outstanding H2O2 formation rate of 2240.23 μmol h−1 g−1 in the absence of a sacrificial donor through a stepwise single-electron ORR route (Fig. 26d). This method of combined interfacial chemical bonding with ionic intercalation and an S-scheme heterojunction offers a novel approach for reducing the recombination of photoexcited charge carriers and improving the efficiency of H2O2 photosynthesis (Fig. 26e).
The incorporation of cocatalysts onto g-C3N4 is a commonly employed method to improve its photocatalytic performance for generating H2O2. Cocatalysts can capture photoinduced electrons from g-C3N4 and provide catalytic centers to promote surface redox reactions. Additionally, certain cocatalysts can improve the light absorption capabilities of g-C3N4. MXene, CQD, rGO, and metallic transition metal compounds such as metal phosphides are frequently used as cocatalysts. They can create Schottky junctions with g-C3N4, serving as efficient electron sinks because of their high work functions, which substantially decrease inherent recombination of electron–hole pairs. Ti3C2 MXene, a layered two-dimensional transition metal carbide known for its superior electrical conductivity, forms a Schottky junction when combined with n-type semiconductors. In this regard, Yang and team members developed a Ti2C3/g-C3N4/BiOCl (TC/g-CN/BOC) composite consisting of both a Z-scheme and Schottky junction with significantly enhanced H2O2 production efficiency.205 This composite, under sunlight exposure, enables the transfer of photoexcited electrons from the CB of BiOCl to the VB of g-C3N4 to merge with holes, encouraging oxidation in the VB of BiOCl. Concurrently, electrons from the CB of g-C3N4 transfer towards Ti3C2, aiding in the reduction of O2 to H2O2. This TC/g-CN/BOC composite shows impressive photocatalytic H2O2 production, reaching 1275 μM in 60 min under simulated sunlight. Similarly, Parida et al. developed a Schottky functionalized direct Z-scheme ternary heterojunction photocatalyst, α-MnO2@B/O-g-C3N4/d-Ti3C2, employing a simple two-step synthesis method for photocatalytic H2O2 production.206 The 5-MX/MBOCN variant, with 10% ethanol, achieved an optimal photocatalytic H2O2 formation rate of 2846.4 μmol h−1 g−1, which was 2.5 times higher than its binary MBOCN counterpart. The enhanced performance is owing to the combination of direct Z-scheme charge migration and the incorporation of a metallic delaminated MXene cocatalyst in the Schottky junction, providing a novel approach for optimizing charge migration and boosting H2O2 photosynthesis efficiency. Furthermore, Chen et al. developed a ternary system, α-Fe2O3/CQD@g-C3N4, using a solvothermal process.207 The α-Fe2O3/CQD@g-C3N4 hybrid experiences boosted e−/h+ separation efficiency due to the established Z-scheme heterojunction along with the electron capturing ability of carbon quantum dots (CQDs), which in turn improved the H2O2 production rate. The photocatalyst attains a H2O2 production rate of 1.16 μmol L−1 min−1 when exposed to visible light, outperforming pure g-C3N4 by 19 times. Additionally, the inclusion of Fe2O3 shifts H2O2 formation from a sequential single-electron indirect ORR to a direct two-electron ORR, providing active sites for H2O2 formation via the 2e− WOR route, owing to its VB position being more positive than the H2O2/H2O electrode potential. Similarly, Li et al. synthesized a ternary Z-scheme composite, rGO-decorated W18O49@g-C3N4 (r-CNW), by employing a straightforward in situ hydrothermal technique for H2O2 production, in which rGO acted as both a charge transfer facilitator and a co-catalyst for the oxygen reduction reaction.208 The optimal r-CNW-2 sample showed significantly improved H2O2 generation rates (71, 58.5, and 9 μmol g−1 h−1) under simulated solar, visible (>400 nm), and NIR light (>800 nm), which were 1.3 and 2 times greater than those of g-C3N4 (Fig. 28a). This improvement in performance, especially under NIR light, is credited to the transfer of LSPR hot electrons to the CB of g-C3N4, which boosts the oxygen reduction reaction. The Parida group created a ternary nanohybrid CNZ/BCN-NixPy−x using ZIF-8 derived C,N-codoped ZnO (CNZ) and B-doped g-C3N4 (BCN) decorated with NixPy nanoparticles through a facile calcination–phosphidation strategy.209 The optimized CNZ/BCN-NixPy−2 photocatalyst notably achieved a high H2O2 production rate of 2873.46 μmol h−1 g−1 when exposed to light, nearly twice the performance of the binary CNZ/BCN (1:1) composite (Fig. 28b). This improvement is attributed to the complex intimate contact of the CNZ and BCN Z-scheme heterojunction, along with NixPy cocatalyst integration, which improves photogenerated charge separation, mobilizes photoexcitons, increases surface reactive sites, and enhances photon absorption (Fig. 28c). Oxygen vacancies also play a crucial role, acting as active sites for H2O2 generation, increasing photoexciton transmission, and reducing electron–hole recombination.
 | ||
| Fig. 28 (a) H2O2 production by g-C3N4 and r-CNW-2 under simulated solar, visible, and NIR light illumination for 1 h. The figures are adapted with permission from ref. 208. Copyright 2023, Elsevier. (b) H2O2 generation rates of CNZ/BCN(1:1) and CNZ/BCN-NixPy–x samples. (c) Diagram of photocatalytic H2O2 and H2 formation mechanisms by the CNZ/BCN-NixPy−2 composite. The figures are adapted with permission from ref. 209. Copyright 2023, ACS. (d) Diagrams of the S-scheme heterojunction between WO2.72 and 0.5S-pCN before and after contact, showing IEF formation and band bending. The figures are adapted with permission from ref. 212. Copyright 2021, Elsevier. (e) N2 adsorption–desorption isotherms and pore size distribution profiles. (f) H2O2 photocatalysis by samples under UV-vis light. The figures are adapted with permission from ref. 151. Copyright 2021, ACS. In situ DRIFTS spectra of SCN/T9 during H2O2 generation at (g) 600–4000 cm−1, (h) 600–900 cm−1, and (i) 1300–1550 cm−1. The figures are adapted with permission from ref. 213. Copyright 2023, Elsevier. | ||
Shi et al. developed a Z-scheme heterojunction composite, KCN/WO3, by thermally copolymerizing K+-doped g-C3N4 (KCN) with WO3 nanoparticles.210 The K+ doping of the g-C3N4 layers reduced electronic localization and expanded the π conjugated system, facilitating electron transmission. The process of K+ doping and WO3 coupling realised an enhanced charge segregation in g-C3N4. The KCN10/WO3(10%) composite, leveraging the low CB of KCN10 and the high VB of WO3, significantly increased H2O2 photocatalytic production to 1.33 mmol L−1 in 4 h without O2 bubbling, a 27-fold improvement over undoped g-C3N4. W18O49, a sub-stoichiometric tungsten oxide with a tunable bandgap and broad light absorption from visible to NIR, is favored for g-C3N4 modification due to its abundant oxygen vacancies and LSPR effect, similar to noble metals. These vacancies lead to collective charge oscillations, enhancing the LSPR effect. Based on this concept, Zhang and co-workers developed a W18O49@g-C3N4 two-dimensional layered Z-scheme heterostructure, using in situ solvothermal growth of W18O49 nanobelts over the surface of g-C3N4 nanosheets.211 The heterostructure composite using crystalline g-C3N4 (W18O49-CCN) exhibited higher crystallinity and an improved interface compared to its amorphous counterpart (W18O49-ACN), enhancing the photogenerated charge carrier transport efficiency. This resulted in extended light absorption from Vis to NIR and significantly higher charge separation/transport efficiency in W18O49-CCN. W18O49-CCN achieved a H2O2 evolution rate of 5550 μM g−1 h−1 under the full spectrum, which was about 2.4 times that of W18O49-ACN. This performance is attributed to the synergistic effect of the Z-scheme structure and improved interfacial structure in W18O49-CCN. Similarly, Li and colleagues synthesized a 2D S-scheme photocatalyst by combining sulfur-doped porous g-C3N4 (S-pCN) and tungsten oxide (WO2.72) semiconductors via a solvent evaporation-induced self-assembly.212 This novel composite, showcasing optimal contact and excellent photocatalytic performance, achieved a H2O2 yield of 87 μM in 3 h under illumination without sacrificial agents in distilled water. This efficiency is due to the oxygen vacancies and the tunable localized surface plasmon resonance (LSPR) effect of WO2.72, alongside the S-scheme mechanism (Fig. 28d).
Liu et al. created a ZnO/g-C3N4 (ZCN) S-scheme heterojunction photocatalyst featuring a hierarchical porous structure, specifically designed for H2O2 production, through calcining the urea-impregnated ZIF-8 MOF.151 The formation of macropores in ZCN12 was observed in FESEM images, resulting from ZIF-8 micropore expansion and rupture due to the release of NH3 gas during calcination, while N2 physisorption confirmed the presence of micropores and mesopores, as shown in Fig. 28e. This unique hierarchical porous structure of ZCN12 not only enhances mass transport and light absorption but also expands its surface area, offering additional active sites on the surface for photocatalytic H2O2 generation. The in situ development of g-C3N4 on ZnO forms a tightly integrated S-scheme heterojunction, leading to the composite's exceptional H2O2 production rate of 1544 μmol L−1 h−1. This rate is 3.4 and 5 times greater than those of g-C3N4 and ZnO individually, respectively (Fig. 28f). The improvement in H2O2 production is ascribed to both the S-scheme charge transfer mechanism along with the sponge-like architecture of the photocatalyst.
Jiang et al. successfully engineered a 3DOM SCN/T S-scheme composite by electrostatically combining TiO2 nanoparticles with sulfur-doped g-C3N4 in a 3D ordered macroporous (3DOM) architecture, achieving a high yield of 2128 μmol h−1 g−1 in H2O2 production from pure water.213 This performance is credited to the combined effects of the 3DOM structure, which enhances surface reactive sites, amplifies light absorption (via multiple scattering and the slow photon effect), and facilitates mass transfer, together with the S-scheme heterojunction that enhances exciton separation and redox efficiency. Additionally, this system effectively combines the strong oxidation properties of TiO2 with the strong reduction capabilities of SCN, enabling efficient ORR and WOR processes for H2O2 production. In situ DRIFTS analysis after visible-light irradiation revealed enhanced absorption peaks at 805, 877, 995, 1100–1250, and 1330–1514 cm−1, corresponding to tri-triazine units, adsorbed H2O2, adsorbed ˙O2− and ˙OOH species, and C–N stretching in aromatic rings (Fig. 28h and i). Conversely, water absorption bands at 3500 and 1573 cm−1 decreased, indicating its consumption as a proton carrier in O2 reduction (Fig. 28g), with no ˙OH bands being detected. This suggests that the triazine structure in the 3DOM SCN/T S-scheme heterojunctions predominantly facilitate the O2 reduction pathway in photocatalytic H2O2 formation.
Again, the Parida group developed a Z-scheme heterojunction by attaching mixed-phase, macroporous TiO2 (n-type) nanoparticles onto B-doped g-C3N4 (BCN) (p-type) nanosheets, using sonication and calcination methods.214 Their optimal photocatalyst, TBCN-8, demonstrated the highest efficacy, producing 110 μmol h−1 of H2O2. Liu et al. developed an S-scheme heterojunction named CN-HMoP by embedding polyoxometalate (HMoP) nanoparticles into g-C3N4 nanosheets using a calcination–post hydrothermal method.215 This process involves the formation of Mo–N bonds between the Mo atoms of HMoP and the amino groups and N atoms of the triazine rings of g-C3N4. These bonds facilitate the layer-by-layer insertion of HMoP nanoparticles into the g-C3N4 lamellae, leading to enhanced charge transfer and reduced carrier transfer resistance by exfoliating g-C3N4 nanosheets. Furthermore, the resultant S-scheme heterostructure not only facilitates the effective spatial separation of exciton pairs but also maintains high redox potentials, contributing to its superior photocatalytic performance. Under simulated sunlight and visible light, H2O2 generation rates of CN-HMoP were 137.1 and 113.1 μmol L−1 h−1, respectively, significantly outperforming those of pristine CN by 17.7 and 16.8 times.
12. Summary, challenges and future prospects
Hydrogen peroxide (H2O2), widely used in chemical synthesis, wastewater treatment, metal extraction, and textile bleaching, is gaining global importance for its eco-friendliness. The photocatalytic production of H2O2, utilizing water, oxygen, and solar energy, emerges as a sustainable and cost-effective method, offering a greener and more efficient solution compared to the traditional, polluting anthraquinone process. This approach aligns with carbon-neutral objectives and is ideal for local, decentralized H2O2 production. g-C3N4, a metal-free polymer semiconductor, is promising for photocatalysis due to its attributes like high activity, non-toxicity, stability, effective electronic structure, visible light absorption, readily available precursors, and affordability. Its tri-s-triazine-based structure efficiently drives two-electron transfer to O2 under visible light, forming superoxo radicals and 1,4-endoperoxide, thus enhancing H2O2 production through the ORR pathway. However, the OER is crucial but hindered by its high overpotential, limiting overall H2O2 production. Addressing challenges like improving OER kinetics, increasing H2O2 selectivity, managing the competition between the 4-electron ORR and HER, and enhancing the driving force are crucial for optimizing H2O2 production efficiency using g-C3N4. Despite its potential, the photocatalytic performance of g-C3N4 in H2O2 production is impeded by several factors: rapid recombination of photoinduced charges, a narrow optical response range, weak oxidation capability, and low H2O2 yield and selectivity. Additionally, its practical application is hindered by a low specific surface area, poor electronic conductivity, and the need for sacrificial agents to consume photoinduced holes, all of which restrict its effectiveness for H2O2 generation. Thus, the construction of a g-C3N4-based direct Z- or S-scheme heterostructure with oxidation-type photocatalysts enables the overall photosynthesis of H2O2 by facilitating the simultaneous production of H2O oxidation and O2 reduction products by utilizing the spatially separated oxidation and reduction components. This system simultaneously enhances photocatalytic activity by broadening light absorption, improving stability, and boosting charge carrier and redox active site separation, while also prolonging carrier lifetimes and optimizing redox potential. It also aligns with the optical responses of adjacent components, induces band bending, and creates an IEF with preferred band alignment, facilitating the spatial separation of valuable photoinduced carriers, concurrently addressing the mentioned challenges of g-C3N4. Oxidation semiconductors, designed to respond to visible light, should have a more positive VB oxidation potential than that of H2O, which is essential for the realization of the WOR pathway. Typically, inorganic metal oxides and oxyhalides exhibit excellent stability, potent oxidation capabilities, and effective affinity for surface hydroxyl (*OH) species, aiding the selective formation of H2O2 through the WOR. Thus, combining g-C3N4 with oxide/oxyhalide photocatalysts can capitalize on their robust redox potential for enhanced photocatalytic performance. The efficiency of photocatalytic H2O2 production significantly relies on several factors: the capacity to harvest light, the rates of charge generation and recombination, the efficiency of charge separation and transfer to the surface of the catalyst, the degree of oxygen adsorption, and the rate of surface redox reactions. Recent research indicates the mechanism of the ORR based on the initial configuration of O2 adsorption: end-on mode adsorption leads to a stepwise ORR pathway due to the differing reactivities of the oxygen atoms, typical of inorganic photocatalysts, in which O2 binds to metal sites or oxygen vacancies through one of the oxygen atoms. Conversely, g-C3N4 favors a bridging adsorption mode, with both oxygen atoms attached to the surface via π–π interactions, enabling the simultaneous occurrence of both sequential single-electron and concerted two-electron ORR pathways when combined with oxide/oxyhalide photocatalysts.In recent years, significant progress has been made in photocatalytic H2O2 production using g-C3N4-based direct Z- or S-scheme photocatalysts. However, despite these advancements, the technology remains far from practical, scaled-up application and is still in its infancy, facing numerous challenges that hinder commercial viability and practical applications. This review offers a comprehensive analysis of the current state of the art and suggests further research directions to bridge the gap towards industrial application. The focus for future work should be on enhancing performance and developing high-performance photocatalysts to facilitate the transition to economically viable and industrially feasible photocatalytic H2O2 production. Our insights are intended to both guide and inspire the development of high-performance g-C3N4-based direct Z- or S-scheme photocatalysts, highlighting the potential for innovative next-generation photocatalyst development in this promising yet challenging field.
(1) In photocatalytic H2O2 generation, reaction routes are crucial for selectivity and yield. The 2e− WOR is inefficient due to a high oxidative potential, while the lower potential of the 4e− WOR not only produces O2 for the 2e− oxygen reduction reaction (ORR), but also facilitates ORR kinetics through intelligently designed interfaces and active sites. Identifying these sites and understanding electron transfer mechanisms are vital. Recent studies show that adjusting the conduction band edge negatively in g-C3N4 semiconductors enhances the one-electron ORR, leading to superoxide radical formation, crucial for H2O2 generation via the two-step ORR process. The adsorption of these radicals on the surface of the catalyst is key for improving the selectivity and efficiency of H2O2 production. Additionally, the choice of electron donor significantly affects the ORR routes in these systems, although the mechanisms behind this are not fully understood. Further research is essential to understand the active site nature, reaction pathways, and their role in high catalytic performance for effective photochemical H2O2 generation and the design of novel catalytic systems.
(2) Mesoporous structures in catalysts enhance rapid mass diffusion, aiding in displacing H2O2 from the surface and preventing its decomposition, while simplifying the separation process. Focusing on visible light absorption, rather than UV light, further decreases H2O2 decomposition. Utilizing heterogeneous reaction systems, such as two- or three-phase systems, helps detach H2O2 from catalysts, minimizing decomposition and improving oxygen solubility and diffusion. Surface functionalization to increase hydrophobicity plays a crucial role in creating a barrier between the catalyst and H2O2, thus preventing excess reduction of H2O2 and boosting efficient H2O2 production under visible light. This hydrophobic modification also enhances oxygen diffusion to active sites while preventing H2O2 adsorption during the ORR process.
(3) The DFT method is used to calculate the oxygen adsorption energy and photoinduced carrier distribution, aiding in a deeper understanding of photocatalytic reaction pathways. Techniques like transient-photoinduced current (TPC), transient-photoinduced voltage (TPV), and in situ X-ray absorption spectroscopy (XAS) offer atom-level insights into H2O2 photoproduction. Analyzing O2 adsorption behaviors, surface effects on O2 adsorption and activation, intermediate states in H2O2 generation, and the influence of active species is essential. In situ IR and Raman spectroscopy verify conformational changes to the catalyst and intermediates in reactions. However, existing gas–solid in situ methods inadequately represent the liquid–solid reaction environment in photocatalytic H2O2 synthesis, necessitating liquid–solid in situ spectral technology for an accurate understanding of H2O and O2 interactions. Theoretical calculations like band structures, charge density mappings, adsorption energies, and reaction pathways serve as supplementary evidence to support the reliability of the photocatalytic synthesis mechanism. Techniques including in situ environmental transmission electron microscopy (ETEM), in situ Fourier transform infrared (FTIR) spectroscopy, and ultrafast transient absorption (TA) spectroscopy contribute to microscale observations of photocatalyst structures and reactivity, interface reactions, intermediate formation, adsorption/desorption processes and the kinetics of electron transfer during photocatalysis.
(4) The development of standardized protocols for measuring photocatalytic H2O2 production is crucial. Consistent standards are essential for accurately comparing the efficacy of different research findings in this field.
(5) Photocatalytic hydrogen peroxide (H2O2) production often uses sacrificial agents and powdered catalysts in water, posing recyclability challenges. Embedding these catalysts on films or glass improves recyclability but reduces H2O2 yield due to a smaller surface area. The key challenge is to balance high yield with catalyst recycling, requiring innovative designs. Charge carrier separation is the key to producing H2O2 efficiently. However, employing organic scavengers to improve this results in byproducts and decomposition problems, which complicate H2O2 purification and reduce its scalability. Thus, there is a growing need for scavenger-free photocatalysts that work efficiently under ambient conditions, utilizing photoexcited holes and electrons for the ORR and WOR. Moreover, developing self-sustaining O2 systems for solar-to-chemical conversion is more economically viable than relying on external oxygen sources.
(6) An innovative approach for enhancing solar energy efficiency involves a photosynthesis system for H2O2 that also conducts other oxidation reactions to produce valuable chemicals. Currently, most reactions use low-value sacrificial agents like isopropanol (IPA), which prevent carrier recombination but contaminate H2O2, increasing purification costs. IPA, which is converted into isopropyl aldehyde, could be replaced with compounds like benzyl alcohol or lactic acid, which have oxidized products with higher commercial value, improving h+ use and adding value to the process.
(7) Focusing on utilizing seawater and wastewater for H2O2 production offers a sustainable and cost-effective approach, addressing the issue of freshwater scarcity. The abundance of seawater makes it an ideal alternative for H2O2 generation. Using g-C3N4-based photocatalysts for this purpose could notably reduce costs and boost the feasibility of photocatalytic H2O2 evolution technologies.
(8) The photoproduction of H2O2 is influenced by factors like reactor design, light intensity, reaction conditions, temperature, catalyst dosages, and variations in ions and pH levels. Despite their importance, these critical factors have been underexplored in recent research. Hence, a thorough understanding of their impact on catalytic efficiency is crucial for enhancing H2O2 production for industrial applications.
(9) Enhancing Z- and S-scheme heterojunction performance requires using appropriate preparation techniques to ensure robust connections between reductive and oxidative photocatalysts and minimizing interfacial charge carrier resistance. The templated hydrothermal approach merges the RSP and OSP effectively and involves preparing one type of photocatalyst as a substrate, onto which the other type of precursor electrostatically attaches and is eventually converted into a bonded photocatalyst on the substrate under supercritical hydrothermal conditions. For stable Z- and S-scheme heterojunctions with polymeric photocatalysts, on-surface thermal polymerization is commonly used.
(10) Over time, g-C3N4-based heterojunction photocatalysts can lose their effectiveness due to reduced component interactions, affecting charge transfer efficiency and performance. Enhancing interactions and stability in g-C3N4 systems for real-world applications requires further research. Cost reduction in H2O2 production can be achieved through efficient synthesis and separation processes, but there are challenges to address, such as long-term stability and the need for additional purification due to the powdered nature of the materials. The production of H2O2, which requires a constant oxygen flow and sacrificial agents, could be simplified and made more cost-effective by using oxygen from the air and water from abundant sources like seawater or wastewater, eliminating the need for sacrificial agents.
(11) The Z- or S-scheme-based heterojunctions, with their strong interfacial electric fields, effectively encourage the anisotropic separation of e− and h+, reducing recombination in bulk and surface areas. However, optimizing this field is challenging due to charge carrier and defect saturation. A suggested and effective strategy to counter this involves enhancing the interfacial electric field (IEF) and charge carrier separation through piezoelectric effects, using piezoelectric and ferroelectric polarization fields, particularly in semiconductors with non-centrosymmetric polar structures. For g-C3N4-based heterostructures, developing piezoelectric and ferroelectric polarization through specific modifications can improve carrier transfer efficiency and suppress recombination, making the research and development of these piezoelectric heterojunctions a promising field.
In conclusion, this research opens new avenues for designing cost-effective, highly selective photocatalysts with enhanced activity and stability, which could revolutionize the field of photocatalytic H2O2 production and have far-reaching implications for renewable energy, environmental remediation, and green chemistry.
Author contributions
Subrat Kumar Sahoo: thought design, concept analysis, visualization, writing – original draft, and revising the manuscript. Lopamudra Acharya, Lijarani Biswal and Priyanka Priyadarshini: writing – reviewing, editing and finalising. Kulamani Parida: supervision, reviewing and revising the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful to Siksha ‘O’ Anusandhan (Deemed to be University) management for their constant support and motivation.References
- S. Chu, Y. Cui and N. Liu, The path towards sustainable energy, Nat. Mater., 2017, 16, 16–22 CrossRef PubMed.
- J. Gong, C. Li and M. R. Wasielewski, Advances in solar energy conversion, Chem. Soc. Rev., 2019, 48, 1862–1864 RSC.
- C. F. Shih, T. Zhang, J. Li and C. Bai, Powering the future with liquid sunshine, Joule, 2018, 2(10), 1925–1949 CrossRef CAS.
- N. S. Lewis and D. G. Nocera, Powering the planet: Chemical challenges in solar energy utilization, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Electrochemical photolysis of water at a semiconductor electrode, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- B. Zhang and L. Sun, Artificial photosynthesis: opportunities and challenges of molecular catalysts, Chem. Soc. Rev., 2019, 48, 2216–2264 RSC.
- T. Hisatomi and K. Domen, Reaction systems for solar hydrogen production via water splitting with particulate semiconductor photocatalysts, Nat. Catal., 2019, 2, 387–399 CrossRef CAS.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Solar water splitting cells, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- A. Kudo and Y. Miseki, Heterogeneous photocatalyst materials for water splitting, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- Y. Shiraishi, T. Takii, T. Hagi, S. Mori, Y. Kofuji, Y. Kitagawa, S. Tanaka, S. Ichikawa and T. Hirai, Resorcinol–formaldehyde resins as metal-free semiconductor photocatalysts for solar-to-hydrogen peroxide energy conversion, Nat. Mater., 2019, 18, 985–993 CrossRef CAS PubMed.
- W. Yu, Z. Zhu, C. Hu, S. Lin, Y. Wang, C. Wang, N. Tian, Y. Zhang and H. Huang, Point-to-face Z-scheme junction Cd0.6Zn0.4S/g C3N4 with a robust internal electric field for high-efficiency H2O2 production, J. Mater. Chem. A, 2023, 11, 6384–6393 RSC.
- C. Xia, Y. Xia, P. Zhu, L. Fan and H. Wang, Direct electrosynthesis of pure aqueous H2O2 solutions up to 20% by weight using a solid electrolyte, Science, 2019, 366, 226–231 CrossRef CAS PubMed.
- K. Mase, M. Yoneda, Y. Yamada and S. Fukuzumi, Seawater usable for production and consumption of hydrogen peroxide as a solar fuel, Nat. Commun., 2016, 7, 11470 CrossRef CAS PubMed.
- R. N. Gurram, M. Al-Shannag, N. J. Lecher, S. M. Duncan, E. L. Singsaas and M. Alkasrawi, Bioconversion of paper mill sludge to bioethanol in the presence of accelerants or hydrogen peroxide pretreatment, Bioresour. Technol., 2015, 192, 529–539 CrossRef CAS PubMed.
- K. P. Bryliakov, Catalytic asymmetric oxygenations with the environmentally benign oxidants H2O2 and O2, Chem. Rev., 2017, 117, 11406–11459 CrossRef CAS PubMed.
- J. Xu, X. Zheng, Z. Feng, Z. Lu, Z. Zhang, W. Huang and Y. Cui, Organic wastewater treatment by a single-atom catalyst and electrolytically produced H2O2, Nat. Sustainability, 2021, 4, 233–241 CrossRef PubMed.
- S. R. Jennings, D. J. Dollhopf and W. P. Inskeep, Acid production from sulfide minerals using hydrogen peroxide weathering, J. Appl. Geochem., 2000, 15, 235–243 CrossRef CAS.
- M. Venugopal and A. V. Saramma, Characterization of alkaline protease from Vibrio fluvialis strain VM10 isolated from a mangrove sediment sample and its application as a laundry detergent additive, Process Biochem., 2006, 41, 1239–1243 CrossRef CAS.
- Y. Yang, Y. Xue, H. Zhang and H. Chang, Flexible H2O2 microfluidic fuel cell using graphene/Prussian blue catalyst for high performance, Chem. Eng. J., 2019, 369, 813–817 CrossRef CAS.
- Research and Markets, Global Hydrogen Peroxide Market Report 2024, https://www.researchandmarkets.com/report/hydrogen-peroxide#rela3-5741554.
- V. C. C. Cheng, S. C. Wong, G. S. W. Kwan, W. T. Hui and K. Y. Yuen, Disinfection of N95 respirators by ionized hydrogen peroxide during pandemic coronavirus disease 2019 (COVID-19) due to SARS-CoV-2, J. Hosp. Infect., 2020, 105, 358 CrossRef CAS PubMed.
- Y. Wu, H. Che, B. Liu and Y. Ao, Promising Materials for Photocatalysis–Self–Fenton System: Properties, Modifications, and Applications, Small Struct., 2023, 4, 2200371 CrossRef CAS.
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. Fierro, Hydrogen peroxide synthesis: an outlook beyond the anthraquinone process, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS PubMed.
- S. Yang, A. Verdaguer-Casadevall, L. Arnarson, L. Silvioli, V. Colic, R. Frydendal, J. Rossmeisl, I. Chorkendorff and I. E. Stephens, Toward the decentralized electrochemical production of H2O2: a focus on the catalysis, ACS Catal., 2018, 8, 4064–4081 CrossRef CAS.
- Z. Lu, G. Chen, S. Siahrostami, Z. Chen, K. Liu, J. Xie, L. Liao, T. Wu, D. Lin, Y. Liu, T. F. Jaramillo, J. K. Nørskov and Y. Cui, High-efficiency oxygen reduction to hydrogen peroxide catalysed by oxidized carbon materials, Nat. Catal., 2018, 1, 156–162 CrossRef CAS.
- E. Jung, H. Shin, B. H. Lee, V. Efremov, S. Lee, H. S. Lee, J. Kim, W. H. Antink, S. Park, K. S. Lee, S. P. Cho, J. S. Yoo, Y. E. Sung and T. Hyeon, Atomic-level tuning of Co–N–C catalyst for high-performance electrochemical H2O2 production, Nat. Mater., 2020, 19, 436–442 CrossRef CAS PubMed.
- Q. Chang, P. Zhang, A. H. B. Mostaghimi, X. Zhao, S. R. Denny, J. H. Lee, H. Gao, Y. Zhang, H. L. Xin, S. Siahrostami, J. G. Chen and Z. Chen, Promoting H2O2 production via 2-electron oxygen reduction by coordinating partially oxidized Pd with defect carbon, Nat. Commun., 2020, 11, 2178 CrossRef CAS PubMed.
- C. Xia, S. Back, S. Ringe, K. Jiang, F. Chen, X. Sun, S. Siahrostami, K. Chan and H. Wang, Confined local oxygen gas promotes electrochemical water oxidation to hydrogen peroxide, Nat. Catal., 2020, 3, 125–134 CrossRef CAS.
- Q. Wu, J. Cao, X. Wang, Y. Liu, Y. Zhao, H. Wang, Y. Liu, H. Huang, F. Liao, M. Shao and Z. Kang, A metal-free photocatalyst for highly efficient hydrogen peroxide photoproduction in real seawater, Nat. Commun., 2021, 12, 483 CrossRef CAS PubMed.
- L. Wang, J. Zhang, Y. Zhang, H. Yu, Y. Qu and J. Yu, Inorganic metal–oxide photocatalyst for H2O2 production, Small, 2022, 18, 2104561 CrossRef CAS PubMed.
- C. Chu, Q. Zhu, Z. Pan, S. Gupta, D. Huang, Y. Du, S. Weon, Y. Wu, C. Muhich, E. Stavitski, K. Domen and J. H. Kim, Spatially separating redox centers on 2D carbon nitride with cobalt single atom for photocatalytic H2O2 production, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 6376–6382 CrossRef CAS PubMed.
- Z. Teng, N. Yang, H. Lv, S. Wang, M. Hu, C. Wang, D. Wang and G. Wang, Edge-functionalized g-C3N4 nanosheets as a highly efficient metal-free photocatalyst for safe drinking water, Chem, 2019, 5, 664–680 CAS.
- Z. Wei, M. Liu, Z. Zhang, W. Yao, H. Tan and Y. Zhu, Efficient visible-light-driven selective oxygen reduction to hydrogen peroxide by oxygen-enriched graphitic carbon nitride polymers, Energy Environ. Sci., 2018, 11, 2581–2589 RSC.
- E. Baur and C. Neuweiler, Photolytic formation of hydrogen peroxide, Helv. Chim. Acta, 1927, 10, 901–907 CrossRef CAS.
- Y. Li, Y. Zhao, H. Nie, K. Wei, J. Cao, H. Huang, M. Shao, Y. Liu and Z. Kang, Interface photo charge kinetics regulation by carbon dots for efficient hydrogen peroxide production, J. Mater. Chem. A, 2021, 9, 515–522 RSC.
- S. M. Ghoreishian, K. S. Ranjith, B. Park, S.-K. Hwang, R. Hosseini, R. Behjatmanesh-Ardakani, S. M. Pourmortazavi, H. U. Lee, B. Son, S. Mirsadeghi, Y.-K. Han and Y. S. Huh, Full-Spectrum Responsive Bi2S3@CdS S-Scheme Heterostructure with Intimated Ultrathin RGO toward Photocatalytic Cr(VI) Reduction and H2O2 Production: Experimental and DFT Studies, Chem. Eng. J., 2021, 419, 129530 CrossRef CAS.
- A. Wang, H. Liang, F. Chen, X. Tian, S. Yin, S. Jing and P. Tsiakaras, Facile synthesis of C3N4/NiIn2S4 heterostructure with novel solar steam evaporation efficiency and photocatalytic H2O2 production performance, Appl. Catal., B, 2022, 310, 121336 CrossRef CAS.
- F. Xue, Y. Si, M. Wang, M. Liu and L. Guo, Toward efficient photocatalytic pure water splitting for simultaneous H2 and H2O2 production, Nano Energy, 2019, 62, 823–831 CrossRef CAS.
- F. Xue, Y. Si, C. Cheng, W. Fu, X. Chen, S. Shen, L. Wang and M. Liu, Electron transfer via homogeneous phosphorus bridges enabling boosted photocatalytic generation of H2 and H2O2 from pure water with stoichiometric ratio, Nano Energy, 2022, 103, 107799 CrossRef CAS.
- W. Wang, W. Gu, G. Li, H. Xie, P. K. Wong and T. An, Few-layered tungsten selenide as a co-catalyst for visible-light-driven photocatalytic production of hydrogen peroxide for bacterial inactivation, Environ. Sci. Nano, 2020, 7, 3877–3887 RSC.
- W. Shi, W. Sun, Y. Liu, K. Zhang, H. Sun, X. Lin and F. Guo, A self-sufficient photo-Fenton system with coupling in situ production H2O2 of ultrathin porous g-C3N4 nanosheets and amorphous FeOOH quantum dots, J. Hazard. Mater., 2022, 436, 129141 CrossRef CAS PubMed.
- X. Y. Ji, Y. Y. Wang and J. Tao, Metal–organic frameworks for photocatalytic oxygen reduction reaction to hydrogen peroxide, Mater. Chem. Front., 2023, 7, 5120–5139 RSC.
- J. Sun, C. Jeet, M. Deng, A. Laemont, X. Feng, Y. Y. Liu and P. Van Der Voort, Metal-Organic Frameworks and Covalent Organic Frameworks as Photocatalysts for H2O2 Production from Oxygen and Water, J. Mater. Chem. A, 2023, 11, 21516–21540 RSC.
- J. Qiu, D. Dai and J. Yao, Tailoring metal–organic frameworks for photocatalytic H2O2 production, Coord. Chem. Rev., 2024, 501, 215597 CrossRef CAS.
- T. He, J. Wu, Y. Li, K. Wei, Y. Liu, H. Huang, Y. Liu and Z. Kang, A step-by-step design for dual channel metal-free photocatalysts towards high yield H2O2 photo-production from air and water, Chem. Eng. J., 2023, 451, 138551 CrossRef CAS.
- Y. Fu, C. A. Liu, M. Zhang, C. Zhu, H. Li, H. Wang, Y. Song, H. Huang, Y. Liu and Z. Kang, Photocatalytic H2O2 and H2 Generation from Living Chlorella vulgaris and Carbon Micro Particle Comodified g–C3N4, Adv. Energy Mater., 2018, 8, 1802525 CrossRef.
- X. Hu, X. Zeng, Y. Liu, J. Lu and X. Zhang, Carbon-based materials for photo-and electrocatalytic synthesis of hydrogen peroxide, Nanoscale, 2020, 12, 16008–16027 RSC.
- L. Xie, X. Wang, Z. Zhang, Y. Ma, T. Du, R. Wang and J. Wang, Photosynthesis of Hydrogen Peroxide Based on g-C3N4: The Road of a Cost–Effective Clean Fuel Production, Small, 2023, 19, 2301007 CrossRef CAS PubMed.
- C. Liang, X. M. Wang, W. Liu, H. Y. Liu, D. W. Huang, Y. Z. Zhang and C. G. Niu, Functionalized graphitic carbon nitride based catalysts in solar-to-chemical conversion for hydrogen peroxide production, Chem. Eng. J., 2023, 466, 142931 CrossRef CAS.
- Y. Li, Z. He, L. Liu, Y. Jiang, W. J. Ong, Y. Duan, W. Ho and F. Dong, Inside-and-out modification of graphitic carbon nitride (g-C3N4) photocatalysts via defect engineering for energy and environmental science, Nano Energy, 2023, 105, 108032 CrossRef CAS.
- Y. Shiraishi, M. Matsumoto, S. Ichikawa, S. Tanaka and T. Hirai, Polythiophene-doped resorcinol–formaldehyde resin photocatalysts for solar-to-hydrogen peroxide energy conversion, J. Am. Chem. Soc., 2021, 143, 12590–12599 CrossRef CAS PubMed.
- Q. Tian, L. Jing, S. Ye, J. Liu, R. Chen, C. A. H. Price, F. Fan and J. Liu, Nanospatial charge modulation of monodispersed polymeric microsphere photocatalysts for exceptional hydrogen peroxide production, Small, 2021, 17, 2103224 CrossRef CAS PubMed.
- X. Li, Q. Zheng, X. Wang, Q. Zheng, Y. Zhang, Y. Cong and S. W. Lv, Introduction of electron-deficient unit in resorcinol-formaldehyde resin to construct donor-acceptor conjugated polymer for enhancing photocatalytic H2O2 production, J. Mater. Chem. A, 2024, 12, 8420–8428 RSC.
- C. Zhao, X. Wang, Y. Yin, W. Tian, G. Zeng, H. Li, S. Ye, L. Wu and J. Liu, Molecular Level Modulation of Anthraquinone–containing Resorcinol–formaldehyde Resin Photocatalysts for H2O2 Production with Exceeding 1.2% Efficiency, Angew. Chem., Int. Ed., 2023, 62, e202218318 CrossRef CAS PubMed.
- X. Wang, X. Yang, C. Zhao, Y. Pi, X. Li, Z. Jia, S. Zhou, J. Zhao, L. Wu and J. Liu, Ambient Preparation of Benzoxazine–based Phenolic Resins Enables Long–term Sustainable Photosynthesis of Hydrogen Peroxide, Angew. Chem., 2023, 135, e202302829 CrossRef.
- Y. Xu, X. Hu, Y. Chen, S. Lin, C. Wang, F. Gou, X. Yang, W. Zheng and D. K. Ma, 3−Hydroxythiophenol–Formaldehyde Resin Microspheres Modulated by Sulfhydryl Groups for Highly Efficient Photocatalytic Synthesis of H2O2, Adv. Sci., 2023, 2304948 CrossRef PubMed.
- Q. Tian, X. K. Zeng, C. Zhao, L. Y. Jing, X. W. Zhang and J. Liu, Exceptional photocatalytic hydrogen peroxide production from sandwich–structured graphene interlayered phenolic resins nanosheets with mesoporous channels, Adv. Funct. Mater., 2023, 33, 2213173 CrossRef CAS.
- Y. Xiang, Z. Xia, W. Hu, C. Tong and C. Lü, Sustainable photocatalytic synthesis of hydrogen peroxide from catechol-formaldehyde resin microspheres modulated by nitrogen-doped carbon dots, Green Chem., 2024, 26, 1478–1487 RSC.
- X. Xu, Y. Sui, W. Chen, W. Huang, X. Li, Y. Li, D. Liu, S. Gao, W. Wu, C. Pan, H. Zhong and M. Wen, The photocatalytic H2O2 production by metal-free photocatalysts under visible-light irradiation, Appl. Catal., B, 2023, 123271 Search PubMed.
- Z. Yong and T. Ma, Solar–to–H2O2 Catalyzed by Covalent Organic Frameworks, Angew. Chem., 2023, 135, e202308980 CrossRef.
- Y. B. Dong, K. H. Xie, G. Wang, F. Zhao, M. C. Wang, H. Y. Zhang, H. R. Ma, Z. Z. Chen, L. Jiang, Y. Geng and Y. Geng, Covalent organic framework based photocatalysts for efficient visible-light driven hydrogen peroxide production, Inorg. Chem. Front., 2024, 11, 1322–1338 RSC.
- H. Cheng, H. Lv, J. Cheng, L. Wang, X. Wu and H. Xu, Rational Design of Covalent Heptazine Frameworks with Spatially Separated Redox Centers for High-Efficiency Photocatalytic Hydrogen Peroxide Production, Adv. Mater., 2022, 34, 2107480 CrossRef CAS PubMed.
- S. M. Ghoreishian, K. S. Ranjith, M. Ghasemi, B. Park, S. K. Hwang, N. Irannejad, M. Norouzi, S. Y. Park, R. B. Ardakani, S. M. Pourmortazavi, S. Mirsadeghi, Y. K. Han and Y. S. Huh, Engineering the photocatalytic performance of B-C3N4@ Bi2S3 hybrid heterostructures for full–spectrum–driven Cr(VI) reduction and in situ H2O2 generation: Experimental and DFT studies, Chem. Eng. J., 2023, 452, 139435 CrossRef CAS.
- X. Zhao, Y. You, S. Huang, Y. Wu, Y. Ma, G. Zhang and Z. Zhang, Z–scheme photocatalytic production of hydrogen peroxide over Bi4O5Br2/g-C3N4 heterostructure under visible light, Appl. Catal., B, 2020, 278, 119251 CrossRef CAS.
- Y. Zhou, H. Dong, Z. Xu, Q. Zha, M. Zhu and Y. Meng, Carbon modification facilitates piezocatalytic H2O2 production over BiOCl nanosheets: Correlation between piezoresponse and surface reaction, Appl. Catal., B, 2024, 343, 123504 CrossRef CAS.
- L. Sun, P. Li, Z. Shen, Y. Pang, X. Ma, D. Qu, L. An and Z. Sun, A Minireview: The Mechanism of H2O2 Photoproduction by Graphitic Carbon Nitride, Adv. Energy Sustainability Res., 2023, 4, 2300090 CrossRef CAS.
- S. Chandrappa, S. J. Galbao, A. Furube and D. H. Murthy, Extending the Optical Absorption Limit of Graphitic Carbon Nitride Photocatalysts: A Review, ACS Appl. Nano Mater., 2023, 6, 19551–19572 CrossRef CAS.
- W. J. Ong, L. L. Tan, Y. H. Ng, S. T. Yong and S. P. Chai, Graphitic carbon nitride (g-C3N4)-based photocatalysts for artificial photosynthesis and environmental remediation: are we a step closer to achieving sustainability?, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- M. Majdoub, Z. Anfar and A. Amedlous, Emerging chemical functionalization of g-C3N4: covalent/noncovalent modifications and applications, ACS Nano, 2020, 14, 12390–12469 CrossRef CAS PubMed.
- F. He, Y. Lu, Y. Wu, S. Wang, Y. Zhang, P. Dong, Y. Wang, C. Zhao, S. Wang, J. Zhang and S. Wang, Rejoint of Carbon Nitride Fragments into Multi–Interfacial Order–Disorder Homojunction for Robust Photo–Driven Generation of H2O2, Adv. Mater., 2024, 36, 2307490 CrossRef CAS PubMed.
- N. Wang, L. Cheng, Y. Liao and Q. Xiang, Effect of Functional Group Modifications on the Photocatalytic Performance of g–C3N4, Small, 2023, 19, 2300109 CrossRef CAS PubMed.
- P. Kumar, G. Singh, X. Guan, J. Lee, R. Bahadur, K. Ramadass, P. Kumar, M. G. Kibria, D. Vidyasagar, J. Yi and A. Vinu, Multifunctional carbon nitride nanoarchitectures for catalysis, Chem. Soc. Rev., 2023, 52, 7602–7664 RSC.
- J. Yi, C. Bahrini, C. Schoemaecker, C. Fittschen and W. Choi, Photocatalytic decomposition of H2O2 on different TiO2 surfaces along with the concurrent generation of HO2 radicals monitored using cavity ring down spectroscopy, J. Phys. Chem. C, 2012, 116, 10090–10097 CrossRef CAS.
- Y. Shiraishi, S. Kanazawa, Y. Sugano, D. Tsukamoto, H. Sakamoto, S. Ichikawa and T. Hirai, Highly selective production of hydrogen peroxide on graphitic carbon nitride (g-C3N4) photocatalyst activated by visible light, ACS Catal., 2014, 4, 774–780 CrossRef CAS.
- C. Tang, M. Cheng, C. Lai, L. Li, X. Yang, L. Du, G. Zhang, G. Wang and L. Yang, Recent progress in the applications of non-metal modified graphitic carbon nitride in photocatalysis, Coord. Chem. Rev., 2023, 474, 214846 CrossRef CAS.
- X. Yu, S. F. Ng, L. K. Putri, L. L. Tan, A. R. Mohamed and W. J. Ong, Point–defect engineering: leveraging imperfections in graphitic carbon nitride (g–C3N4) photocatalysts toward artificial photosynthesis, Small, 2021, 17, 2006851 CrossRef CAS PubMed.
- B. Zhu, B. Cheng, J. Fan, W. Ho and J. Yu, g–C3N4−based 2D/2D composite heterojunction photocatalyst, Small Struct., 2021, 2, 2100086 CrossRef CAS.
- M. Sayed, B. Zhu, P. Kuang, X. Liu, B. Cheng, A. A. A. Ghamdi, S. Wageh, L. Zhang and J. Yu, EPR investigation on electron transfer of 2D/3D g–C3N4/ZnO S–scheme heterojunction for enhanced CO2 photoreduction, Adv. Sustainable Syst., 2022, 6, 2100264 CrossRef CAS.
- Z. Zhao, K. Dai, J. Zhang and G. Dawson, In situ preparation of Mn0.2Cd0.8S–diethylenetriamine/porous g–C3N4 S–scheme heterojunction with enhanced photocatalytic hydrogen production, Adv. Sustainable Syst., 2023, 7, 2100498 CrossRef CAS.
- D. Liu, L. Jiang, D. Chen, Z. Hao, B. Deng, Y. Sun and H. Liu, Twin S-Scheme g-C3N4/CuFe2O4/ZnIn2S4 Heterojunction with a Self-Supporting Three-Phase System for Photocatalytic CO2 Reduction: Mechanism Insight and DFT Calculations, ACS Catal., 2024, 14, 5326–5343 CrossRef CAS.
- J. Fu, J. Yu, C. Jiang and B. Cheng, g–C3N4−Based heterostructured photocatalysts, Adv. Energy Mater., 2018, 8, 1701503 CrossRef.
- Q. Wang, X. Wang, Z. Yu, X. Jiang, J. Chen, L. Tao, M. Wang and Y. Shen, Artificial photosynthesis of ethanol using type-II g-C3N4/ZnTe heterojunction in photoelectrochemical CO2 reduction system, Nano Energy, 2019, 60, 827–835 CrossRef CAS.
- R. Shen, L. Zhang, N. Li, Z. Lou, T. Ma, P. Zhang, Y. Li and X. Li, W–N bonds precisely boost Z-scheme interfacial charge transfer in g-C3N4/WO3 heterojunctions for enhanced photocatalytic H2 evolution, ACS Catal., 2022, 12, 9994–10003 CrossRef CAS.
- J. Zhou, B. Gao, D. Wu, C. Tian, H. Ran, W. Chen, Q. Huang, W. Zhang, F. Qi, N. Zhang, Y. Pu, J. Qiu, Z. Hu, J. Du, Z. Liu, Y. Leng and X. Tang, Enhanced Photocatalytic Activity of Lead–Free Cs2TeBr6/g–C3N4 Heterojunction Photocatalyst and Its Mechanism, Adv. Funct. Mater., 2024, 34, 2308411 CrossRef CAS.
- J. Low, C. Jiang, B. Cheng, S. Wageh, A. A. Al-Ghamdi and J. Yu, A review of direct Z–scheme photocatalysts, Small Methods, 2017, 1, 1700080 CrossRef.
- B. Zhu, J. Sun, Y. Zhao, L. Zhang and J. Yu, Construction of 2D S–Scheme Heterojunction Photocatalyst, Adv. Mater., 2024, 36, 2310600 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. Carlsson, K. Domen and M. Antonietti, A metal-free polymeric photocatalyst for hydrogen production from water under visible light, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- L. Wang, W. Si, Y. Tong, F. Hou, D. Pergolesi, J. Hou, T. Lippert, S. X. Dou and J. Liang, Graphitic carbon nitride (g–C3N4)–based nanosized heteroarrays: promising materials for photoelectrochemical water splitting, Carbon Energy, 2020, 2, 223–250 CrossRef CAS.
- L. Acharya, S. P. Pattnaik, A. Behera, R. Acharya and K. M. Parida, Exfoliated Boron Nitride (e-BN) Tailored Exfoliated Graphitic Carbon Nitride (e-CN): An Improved Visible Light Mediated Photocatalytic Approach towards TCH Degradation and H2 Evolution, Inorg. Chem., 2021, 60, 5021–5033 CrossRef CAS PubMed.
- D. Ma, Z. Zhang, Y. Zou, J. Chen and J. W. Shi, The progress of g-C3N4 in photocatalytic H2 evolution: From fabrication to modification, Coord. Chem. Rev., 2024, 500, 215489 CrossRef CAS.
- S. Patanaik, D. P. Sahoo and K. M. Parida, An overview on Ag modified g-C3N4 based nanostructured materials for energy and environmental applications, Renewable Sustainable Energy Rev., 2018, 82, 1297–1312 CrossRef.
- B. P. Mishra and K. M. Parida, Orienting Z scheme charge transfer in graphitic carbon nitride based systems for photocatalytic energy and environmental applications, J. Mater. Chem. A, 2021, 9, 10039–10080 RSC.
- L. Biswal, S. Nayak and K. Parida, Recent progress on strategies for the preparation of 2D/2D MXene/gC3N4 nanocomposites for photocatalytic energy and environmental applications, Catal. Sci. Technol., 2021, 11, 1222–1248 RSC.
- S. Patnaik, D. P. Sahoo and K. M. Parida, Recent advances in anion doped g-C3N4 photocatalysts: A review, Carbon, 2021, 172, 682–711 CrossRef CAS.
- D. Jiang, S. Hu, Y. Qu, X. Tian, H. Du, C. Zhu, Z. Li, L. Yin, Y. Yuan and G. Liu, Infrared Irradiation–Lattice Vibration Coupling–Initiated N→ Π* Electronic Transition in Carbon Nitride Nanosheets for Increased Photocatalysis, Adv. Funct. Mater., 2024, 34, 2311803 CrossRef CAS.
- Y. Sun, V. Kumar and K. H. Kim, The assessment of graphitic carbon nitride (g-C3N4) materials for hydrogen evolution reaction: Effect of metallic and non-metallic modifications, Sep. Purif. Technol., 2023, 305, 122413 CrossRef CAS.
- A. Hayat, M. Sohail, A. El Jery, K. M. Al-Zaydi, K. F. Alshammari, J. Khan and M. Z. Ansari, Different dimensionalities, morphological advancements and engineering of g–C3N4−based nanomaterials for energy conversion and storage, Chem. Rec., 2023, 23, e202200171 CrossRef CAS PubMed.
- B. P. Mishra, L. Biswal, S. Das, L. Acharya and K. M. Parida, Architecture and kinetic studies of photocatalytic H2O2 generation and H2 evolution through regulation of spatial charge transfer via Z-scheme path over A(001) facet engineering TiO2@Mxene/B-g-C3N4 Ternary hybrid, Langmuir, 2023, 39, 957–971 CrossRef CAS PubMed.
- L. Zhang, J. Zhang, H. Yu and J. Yu, Emerging S–scheme photocatalyst, Adv. Mater., 2022, 34, 2107668 CrossRef CAS PubMed.
- J. Fu, Q. Xu, J. Low, C. Jiang and J. Yu, Ultrathin 2D/2D WO3/g-C3N4 step-scheme H2-production photocatalyst, Appl. Catal., B, 2019, 243, 556–565 CrossRef CAS.
- Y. Tachibana, L. Vayssieres and J. R. Durrant, Artificial photosynthesis for solar water-splitting, Nat. Photonics, 2012, 6, 511–518 CrossRef CAS.
- A. J. Bard, Photoelectrochemistry and heterogeneous photo-catalysis at semiconductors, J. Photochem., 1979, 10, 59–75 CrossRef CAS.
- P. Zhou, J. Yu and M. Jaroniec, All–solid–state Z–scheme photocatalytic systems, Adv. Mater., 2014, 22, 4920–4935 CrossRef PubMed.
- H. Li, W. Tu, Y. Zhou and Z. Zou, Z–Scheme photocatalytic systems for promoting photocatalytic performance: recent progress and future challenges, Adv. Sci., 2016, 3(11), 1500389 CrossRef PubMed.
- W. Zhang, A. R. Mohamed and W. J. Ong, Z–Scheme photocatalytic systems for carbon dioxide reduction: where are we now?, Angew. Chem., Int. Ed., 2020, 59, 22894–22915 CrossRef CAS PubMed.
- M. Jourshabani, B. K. Lee and Z. Shariatinia, From traditional strategies to Z-scheme configuration in graphitic carbon nitride photocatalysts: recent progress and future challenges, Appl. Catal., B, 2020, 276, 119157 CrossRef CAS.
- A. Meng, B. Zhu, Y. Zhong, S. Zhou, P. Han and Y. Su, S-scheme heterojunction photocatalysts for CO2 conversion: Design, characterization and categories, Energy Rev., 2023, 2, 100052 CrossRef.
- B. J. Ng, L. K. Putri, X. Y. Kong, Y. W. Teh, P. Pasbakhsh and S. P. Chai, Z–scheme photocatalytic systems for solar water splitting, Adv. Sci., 2020, 7, 1903171 CrossRef CAS PubMed.
- H. Tada, T. Mitsui, T. Kiyonaga, T. Akita and K. Tanaka, All-solid-state Z-scheme in CdS–Au–TiO2 three-component nanojunction system, Nat. Mater., 2006, 5, 782–786 CrossRef CAS PubMed.
- Q. Xu, L. Zhang, B. Cheng, J. Fan and J. Yu, S-scheme heterojunction photocatalyst, Chem, 2020, 6, 1543–1559 CAS.
- W. Jiang, D. Qu, L. An, X. Gao, Y. Wen, X. Wang and Z. Sun, Deliberate construction of direct Z-scheme photocatalysts through photodeposition, J. Mater. Chem. A, 2019, 7, 18348–18356 RSC.
- M. Grätzel, Photoelectrochemical cells, Nature, 2001, 414, 338–344 CrossRef PubMed.
- X. Wang, G. Liu, Z. G. Chen, F. Li, L. Wang, G. Q. Lu and H. M. Cheng, Enhanced photocatalytic hydrogen evolution by prolonging the lifetime of carriers in ZnO/CdS heterostructures, ChemComm, 2009, 23, 3452–3454 RSC.
- G. Liu, L. Wang, H. G. Yang, H. M. Cheng and G. Q. M. Lu, Titania-based photocatalysts—crystal growth, doping and heterostructuring, J. Mater. Chem., 2010, 20, 831–843 RSC.
- J. Yu, S. Wang, J. Low and W. Xiao, Enhanced photocatalytic performance of direct Z-scheme g-C3N4−TiO2 photocatalysts for the decomposition of formaldehyde in air, Phys. Chem. Chem. Phys., 2013, 15, 16883–16890 RSC.
- Q. Xu, L. Zhang, J. Yu, S. Wageh, A. A. Al-Ghamdi and M. Jaroniec, Direct Z-scheme photocatalysts: Principles, synthesis, and applications, Mater. Today, 2018, 21, 1042–1063 CrossRef CAS.
- S. Bai, J. Jiang, Q. Zhang and Y. Xiong, Steering charge kinetics in photocatalysis: intersection of materials syntheses, characterization techniques and theoretical simulations, Chem. Soc. Rev., 2015, 44, 2893–2939 RSC.
- J. Liu, B. Cheng and J. Yu, A new understanding of the photocatalytic mechanism of the direct Z-scheme g-C3N4/TiO2 heterostructure, Phys. Chem. Chem. Phys., 2016, 18, 31175–31183 RSC.
- X. Liu, Q. Zhang and D. Ma, Advances in 2D/2D Z–scheme heterojunctions for photocatalytic applications, Sol. RRL, 2021, 5, 2000397 CrossRef CAS.
- Q. Xu, S. Wageh, A. A. Al-Ghamdi and X. Li, Design principle of S-scheme heterojunction photocatalyst, J. Mater. Sci. Technol., 2022, 124, 171–173 CrossRef.
- Z. Zhang and J. T. Yates Jr, Band bending in semiconductors: chemical and physical consequences at surfaces and interfaces, Chem. Rev., 2012, 112, 5520–5551 CrossRef CAS PubMed.
- S. Mansingh, K. K. Das, N. Priyadarshini, D. P. Sahoo, D. Prusty, J. Sahu, U. A. Mohanty and K. Parida, Minireview elaborating S-scheme charge dynamic photocatalysts: Journey from Z to S, mechanism of charge flow, characterization proof, and H2O2 evolution, Energy Fuels, 2023, 37, 9873–9894 CrossRef CAS.
- F. Li, Z. Fang, Z. Xu and Q. Xiang, The confusion about S-scheme electron transfer: critical understanding and a new perspective, Energy Environ. Sci., 2024, 17, 497–509 RSC.
- X. C. Liu, M. S. Choi, E. Hwang, W. J. Yoo and J. Sun, Fermi level pinning dependent 2D semiconductor devices: challenges and prospects, Adv. Mater., 2022, 34, 2108425 CrossRef CAS PubMed.
- X. Y. Yue, L. Cheng, J. J. Fan and Q. J. Xiang, 2D/2D BiVO4/CsPbBr3 S-scheme heterojunction for photocatalytic CO2 reduction: Insights into structure regulation and Fermi level modulation, Appl. Catal., B, 2022, 304, 120979 CrossRef CAS.
- H. J. Ben, Y. Liu, X. Liu, X. F. Liu, C. C. Ling, C. A. Liang and L. Z. Zhang, Diffusion–controlled Z–scheme–steered charge separation across PDI/BIOI heterointerface for ultraviolet, visible, and infrared light–driven photocatalysis, Adv. Funct. Mater., 2021, 31, 2102315 CrossRef CAS.
- W. K. Jo, S. Kumar, S. Eslava and S. Tonda, Construction of Bi2WO6/RGO/g-C3N4 2D/2D/2D hybrid Z-scheme heterojunctions with large interfacial contact area for efficient charge separation and high-performance photoreduction of CO2 and H2O into solar fuels, Appl. Catal., B, 2018, 239, 586–598 CrossRef CAS.
- A. Tomasi, B. Paviet-Salomon, Q. Jeangros, J. Haschke, G. Christmann, L. Barraud, A. Descoeudres, J. P. Seif, S. Nicolay, M. Despeisse, S. De Wolf and C. Ballif, Simple processing of back-contacted silicon heterojunction solar cells using selective-area crystalline growth, Nat. Energy, 2017, 2, 1–8 Search PubMed.
- D. Ren, W. Zhang, Y. Ding, R. Shen, Z. Jiang, X. Lu and X. Li, In situ fabrication of robust cocatalyst–free CdS/g–C3N4 2D–2D step–scheme heterojunctions for highly active H2 evolution, Sol. RRL, 2020, 4, 1900423 CrossRef CAS.
- Q. Xu, S. Wageh, A. Al-Ghamdi and X. Li, Design principle of S-scheme heterojunction photocatalyst, J. Mater. Sci. Technol., 2022, 124, 171–173 CrossRef.
- Z. Dong, J. Zhou, Z. Zhang, Y. Jiang, R. Zhou and C. Yao, Construction of a p–n Type S-Scheme Heterojunction by Incorporating CsPbBr3 Nanocrystals into Mesoporous Cu2O Microspheres for Efficient CO2 Photoreduction, ACS Appl. Energy Mater., 2022, 5, 10076–10085 CrossRef CAS.
- Z. Wang, B. Cheng, L. Zhang, J. Yu and H. Tan, BiOBr/NiO S–scheme heterojunction photocatalyst for CO2 photoreduction, Sol. RRL, 2022, 6, 2100587 CrossRef CAS.
- Z. F. Huang, J. J. Song, X. Wang, L. Pan, K. Li, X. W. Zhang, L. Wang and J. J. Zou, Switching charge transfer of C3N4/W18O49 from type-II to Z-scheme by interfacial band bending for highly efficient photocatalytic hydrogen evolution, Nano Energy, 2017, 40, 308–316 CrossRef CAS.
- D. Kim and K. Yong, Boron doping induced charge transfer switching of a C3N4/ZnO photocatalyst from Z-scheme to type II to enhance photocatalytic hydrogen production, Appl. Catal., B, 2021, 282, 119538 CrossRef CAS.
- W. Xu, W. Tian, L. Meng, F. Cao and L. Li, Interfacial chemical bond–modulated Z–scheme charge transfer for efficient photoelectrochemical water splitting, Adv. Energy Mater., 2021, 11, 2003500 CrossRef CAS.
- Z. F. Huang, J. J. Song, X. Wang, L. Pan, K. Li, X. W. Zhang, L. Wang and J. J. Zou, Switching charge transfer of C3N4/W18O49 from type-II to Z-scheme by interfacial band bending for highly efficient photocatalytic hydrogen evolution, Nano Energy, 2017, 40, 308–316 CrossRef CAS.
- X. Yue, J. Fan and Q. Xiang, Internal electric field on steering charge migration: modulations, determinations and energy–related applications, Adv. Funct. Mater., 2022, 32, 2110258 CrossRef CAS.
- X. Lu, Y. Shi, D. Tang, X. Lu, Z. Wang, N. Sakai, Y. Ebina, T. Taniguchi, R. Ma, T. Sasaki and C. Yan, Accelerated ionic and charge transfer through atomic interfacial electric fields for superior sodium storage, ACS Nano, 2022, 16, 4775–4785 CrossRef CAS PubMed.
- A. Deng, Y. Sun, Z. Gao, S. Yang, Y. Liu, H. He, J. Zhang, S. Liu, H. Sun and S. Wang, Internal electric field in carbon nitride-based heterojunctions for photocatalysis, Nano Energy, 2023, 108, 108228 CrossRef CAS.
- M. E. Malefane, P. J. Mafa, M. Managa, T. T. I. Nkambule and A. T. Kuvarega, Understanding the principles and applications of dual Z-scheme heterojunctions: how far can we go?, J. Phys. Chem. Lett., 2023, 14, 1029–1045 CrossRef CAS PubMed.
- X. Ruan, C. Huang, H. Cheng, Z. Zhang, Y. Cui, Z. Li, T. Xie, K. Ba, H. Zhang, L. Zhang, X. Zhao, J. Leng, S. Jin, W. Zhang, W. Zheng, S. K. Ravi, Z. Jiang, X. Cui and J. Yu, A twin S–scheme artificial photosynthetic system with self–assembled heterojunctions yields superior photocatalytic hydrogen evolution rate, Adv. Mater., 2023, 35, 2209141 CrossRef CAS PubMed.
- X. Wang, X. Wang, J. Huang, S. Li, A. Meng and Z. Li, Interfacial chemical bond and internal electric field modulated Z-scheme Sv-ZnIn2S4/MoSe2 photocatalyst for efficient hydrogen evolution, Nat. Commun., 2021, 12, 4112 CrossRef CAS PubMed.
- S. Li, L. Meng, W. Tian and L. Li, Engineering Interfacial Band Bending over ZnIn2S4/SnS2 by Interface Chemical Bond for Efficient Solar–Driven Photoelectrochemical Water Splitting, Adv. Energy Mater., 2022, 12, 2200629 CrossRef CAS.
- T. Han, X. Cao, K. Sun, Q. Peng, C. Ye, A. Huang, W.-C. Cheong, Z. Chen, R. Lin, D. Zhao, X. Tan, Z. Zhuang, C. Chen, D. Wang and Y. Li, Anion-exchange-mediated internal electric field for boosting photogenerated carrier separation and utilization, Nat. Commun., 2021, 12, 4952 CrossRef CAS PubMed.
- X. Li, J. Liu, J. Huang, C. He, Z. Feng, Z. Chen, L. Wan and F. Deng, All organic S-scheme heterojunction PDI-Ala/S-C3N4 photocatalyst with enhanced photocatalytic performance, Acta Phys.-Chim. Sin., 2021, 37, 2010030 Search PubMed.
- C. Corcoran, H. Tavassol, M. Rigsby, P. Bagus and A. Wieckowski, Application of XPS to Study Electrocatalysts for Fuel Cells, J. Power Sources, 2010, 195, 7856–7879 CrossRef CAS.
- P. Bagus, E. Ilton and C. Nelin, The Interpretation of XPS Spectra: Insights into Materials Properties, Surf. Sci. Rep., 2013, 68, 273–304 CrossRef CAS.
- G. Greczynski and L. Hultman, X-ray Photoelectron Spectroscopy: Towards Reliable Binding Energy Referencing, Prog. Mater. Sci., 2020, 107, 100591 CrossRef CAS.
- J. Zhang, L. Zhang, W. Wang and J. Yu, In situ irradiated X-ray photoelectron spectroscopy investigation on electron transfer mechanism in S-scheme photocatalyst, J. Phys. Chem. Lett., 2022, 13, 8462–8469 CrossRef CAS PubMed.
- L. Wang, J. Zhang, H. Yu, I. H. Patir, Y. Li, S. Wageh, A. A. Al Ghamdi and J. Yu, Dynamics of Photogenerated Charge Carriers in Inorganic/Organic S-Scheme Heterojunctions, J. Phys. Chem. Lett., 2022, 13, 4695–4700 CrossRef CAS PubMed.
- B. Liu, C. Bie, Y. Zhang, L. Wang, Y. Li and J. Yu, Hierarchically porous ZnO/g-C3N4 S-scheme heterojunction photocatalyst for efficient H2O2 production, Langmuir, 2021, 37, 14114–14124 CrossRef CAS PubMed.
- X. Zhang, J. Yu, W. Macyk, S. Wageh, A. A. Al-Ghamdi and L. Wang, C3N4/PDA S–Scheme Heterojunction with Enhanced Photocatalytic H2O2 Production Performance and Its Mechanism, Adv. Sustainable Syst., 2023, 7, 2200113 CrossRef CAS.
- G. Binnig, C. F. Quate and C. Gerber, Atomic force microscope, Phys. Rev. Lett., 1986, 56, 930 CrossRef PubMed.
- T. Hackl, G. Schitter and P. Mesquida, AC Kelvin probe force microscopy enables charge mapping in water, ACS Nano, 2022, 16, 17982–17990 CrossRef CAS PubMed.
- U. Zerweck, C. Loppacher, T. Otto, S. Grafström and L. M. Eng, Accuracy and resolution limits of Kelvin probe force microscopy, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 125424 CrossRef.
- F. Li, G. Zhu, J. Jiang, L. Yang, F. Deng and X. Li, A review of updated S-scheme heterojunction photocatalysts, J. Mater. Sci. Technol., 2024, 177, 142–180 CrossRef.
- C. Cheng, B. He, J. Fan, B. Cheng, S. Cao and J. Yu, An inorganic/organic S–scheme heterojunction H2−production photocatalyst and its charge transfer mechanism, Adv. Mater., 2021, 33, 2100317 CrossRef CAS PubMed.
- J. Y. Xu, X. Tong, P. Yu, G. E. Wenya, T. McGrath, M. J. Fong, J. Wu and Z. M. Wang, Ultrafast dynamics of charge transfer and photochemical reactions in solar energy conversion, Adv. Sci., 2018, 5, 1800221 CrossRef PubMed.
- H. Gogoi, S. S. Pathak, S. Dasgupta, L. S. Panchakarla, S. Nath and A. Datta, Exciton Dynamics in Colloidal CdS Quantum Dots with Intense and Stokes Shifted Photoluminescence in a Single Decay Channel, J. Phys. Chem. Lett., 2022, 13, 6770–6776 CrossRef CAS PubMed.
- L. Cheng, X. Yue, J. Fan and Q. Xiang, Site–specific electron–driving observations of CO2−to–CH4 photoreduction on co–doped CeO2/crystalline carbon nitride S–scheme heterojunctions, Adv. Mater., 2022, 34, 2200929 CrossRef CAS PubMed.
- J. Zhang, B. Zhu, L. Zhang and J. Yu, Femtosecond transient absorption spectroscopy investigation into the electron transfer mechanism in photocatalysis, Chem. Commun., 2023, 59, 688–699 RSC.
- J. Ma, T. J. Miao and J. Tang, Charge carrier dynamics and reaction intermediates in heterogeneous photocatalysis by time-resolved spectroscopies, Chem. Soc. Rev., 2022, 51, 5777–5794 RSC.
- K. E. Knowles, M. D. Koch and J. L. Shelton, Three applications of ultrafast transient absorption spectroscopy of semiconductor thin films: spectroelectrochemistry, microscopy, and identification of thermal contributions, J. Mater. Chem. C, 2018, 6, 11853–11867 RSC.
- C. Ruckebusch, M. Sliwa, P. D. Pernot, A. De Juan and R. Tauler, Comprehensive data analysis of femtosecond transient absorption spectra: A review, J. Photochem. Photobiol., C, 2012, 13, 1–27 CrossRef CAS.
- X. Liu, P. Zeng, S. Chen, T. A. Smith and M. Liu, Charge Transfer Dynamics at the Interface of CsPbX3 Perovskite Nanocrystal–Acceptor Complexes: A Femtosecond Transient Absorption Spectroscopy Study, Laser Photonics Rev., 2022, 16, 2200280 CrossRef CAS.
- S. Chouksey, S. Sankaranarayanan, V. Pendem, P. K. Saha, S. Ganguly and D. Saha, Strong size dependency on the carrier and photon dynamics in InGaN/GaN single nanowalls determined using photoluminescence and ultrafast transient absorption spectroscopy, Nano Lett., 2017, 17, 4596–4603 CrossRef CAS PubMed.
- L. Wang, J. Zhang, H. Yu, I. H. Patir, Y. Li, S. Wageh, A. A. A. Ghamdi and J. Yu, Dynamics of photogenerated charge carriers in inorganic/organic S-scheme heterojunctions, J. Phys. Chem. Lett., 2022, 13, 4695–4700 CrossRef CAS PubMed.
- B. He, Z. Wang, P. Xiao, T. Chen, J. Yu and L. Zhang, Cooperative Coupling of H2O2 Production and Organic Synthesis over a Floatable Polystyrene–Sphere–Supported TiO2/Bi2O3 S–Scheme Photocatalyst, Adv. Mater., 2022, 34, 2203225 CrossRef CAS PubMed.
- C. Cheng, J. Zhang, B. Zhu, G. Liang, L. Zhang and J. Yu, Verifying the Charge–Transfer Mechanism in S–Scheme Heterojunctions Using Femtosecond Transient Absorption Spectroscopy, Angew. Chem., Int. Ed., 2023, 62, e202218688 CrossRef CAS PubMed.
- J. Wang, Z. Wang, K. Dai and J. Zhang, Review on inorganic-organic S-scheme photocatalysts, J. Mater. Sci. Technol., 2023, 165, 187–218 CrossRef CAS.
- J. Lu, S. Gu, H. Li, Y. Wang, M. Guo and G. Zhou, Review on multi-dimensional assembled S-scheme heterojunction photocatalysts, J. Mater. Sci. Technol., 2023, 160, 214–239 CrossRef CAS.
- F. Li, L. Cheng, J. Fan and Q. Xiang, Steering the behavior of photogenerated carriers in semiconductor photocatalysts: a new insight and perspective, J. Mater. Chem. A, 2021, 9, 23765–23782 RSC.
- L. Sun, X. Liu, X. Jiang, Y. Feng, X. Ding, N. Jiang and J. Wang, An internal electric field and interfacial S–C bonds jointly accelerate S-scheme charge transfer achieving efficient sunlight-driven photocatalysis, J. Mater. Chem. A, 2022, 10, 25279–25294 RSC.
- B. Zhu, H. Tan, J. Fan, B. Cheng, J. Yu and W. Ho, Tuning the strength of built-in electric field in 2D/2D g-C3N4/SnS2 and g-C3N4/ZrS2 S-scheme heterojunctions by nonmetal doping, J. Materiomics, 2021, 7, 988–997 CrossRef.
- J. Liu, J. Wan, L. Liu, W. Yang, J. Low, X. Gao and F. Fu, Synergistic effect of oxygen defect and doping engineering on S-scheme O-ZnIn2S4/TiO2-x heterojunction for effective photocatalytic hydrogen production by water reduction coupled with oxidative dehydrogenation, J. Chem. Eng., 2022, 430, 133125 CrossRef CAS.
- J. E. Whitten, Ultraviolet photoelectron spectroscopy: Practical aspects and best practices, Appl. Surf. Sci. Adv., 2023, 13, 100384 CrossRef.
- Y. Luo, Y. Tang, T. F. Chung, C. L. Tai, C. Y. Chen, J. R. Yang and D. Y. Li, Electron work function: an indicative parameter towards a novel material design methodology, Sci. Rep., 2021, 11, 11565 CrossRef CAS PubMed.
- X. Lu, L. Quan, H. Hou, J. Qian, Z. Liu and Q. Zhang, Fabrication of 1D/2D Y-doped CeO2/ZnIn2S4 S-scheme photocatalyst for enhanced photocatalytic H2 evolution, J. Alloys Compd., 2022, 925, 166552 CrossRef CAS.
- H. P. Komsa and A. V. Krasheninnikov, Electronic structures and optical properties of realistic transition metal dichalcogenide heterostructures from first principles, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 085318 CrossRef.
- T. Y. Jeong, H. Kim, S. J. Choi, K. Watanabe, T. Taniguchi, K. J. Yee, Y. S. Kim and S. Jung, Spectroscopic studies of atomic defects and bandgap renormalization in semiconducting monolayer transition metal dichalcogenides, Nat. Commun., 2019, 10, 3825 CrossRef PubMed.
- M. Tebyetekerwa, J. Zhang, Z. Xu, T. N. Truong, Z. Yin, Y. Lu, S. Ramakrishna, D. Macdonald and H. T. Nguyen, Mechanisms and applications of steady-state photoluminescence spectroscopy in two-dimensional transition-metal dichalcogenides, ACS Nano, 2020, 14, 14579–14604 CrossRef CAS PubMed.
- B. Xia, B. He, J. Zhang, L. Li, Y. Zhang, J. Yu, J. Ran and S. Z. Qiao, TiO2/FePS3 S–scheme heterojunction for greatly raised photocatalytic hydrogen evolution, Adv. Energy Mater., 2022, 12, 2201449 CrossRef CAS.
- J. Ding, Z. Dai, F. Qin, H. Zhao, S. Zhao and R. Chen, Z-scheme BiO1-xBr/Bi2O2CO3 photocatalyst with rich oxygen vacancy as electron mediator for highly efficient degradation of antibiotics, Appl. Catal., B, 2017, 205, 281–291 CrossRef CAS.
- W. Yu, C. Hu, L. Bai, N. Tian, Y. Zhang and H. Huang, Photocatalytic hydrogen peroxide evolution: What is the most effective strategy?, Nano Energy, 2022, 104, 107906 CrossRef CAS.
- S. Qu, H. Wu and Y. H. Ng, Clean Production of Hydrogen Peroxide: A Heterogeneous Solar–Driven Redox Process, Adv. Energy Mater., 2023, 13, 2301047 CrossRef CAS.
- H. Cheng, J. Cheng, L. Wang and H. Xu, Reaction pathways toward sustainable photosynthesis of hydrogen peroxide by polymer photocatalysts, Chem. Mater., 2022, 34, 4259–4273 CrossRef CAS.
- L. Wang, J. Sun, B. Cheng, R. He and J. Yu, S-scheme heterojunction photocatalysts for H2O2 production, J. Phys. Chem. Lett., 2023, 14, 4803–4814 CrossRef CAS PubMed.
- X. Zeng, Y. Liu, X. Hu and X. Zhang, Photoredox catalysis over semiconductors for light-driven hydrogen peroxide production, Green Chem., 2021, 23, 1466–1494 RSC.
- J. Yang, X. Zeng, M. Tebyetekerwa, Z. Wang, C. Bie, X. Sun, I. Marriam and X. Zhang, Engineering 2D Photocatalysts for Solar Hydrogen Peroxide Production, Adv. Energy Mater., 2024, 2400740 CrossRef.
- Y. Sun, L. Han and P. Strasser, A comparative perspective of electrochemical and photochemical approaches for catalytic H2O2 production, Chem. Soc. Rev., 2020, 49, 6605–6631 RSC.
- H. Hou, X. Zeng and X. Zhang, Production of hydrogen peroxide by photocatalytic processes, Angew. Chem., Int. Ed., 2020, 59, 17356–17376 CrossRef CAS PubMed.
- Y. Shiraishi, T. Takii, T. Hagi, S. Mori, Y. Kofuji, Y. Kitagawa, S. Tanaka, S. Ichikawa and T. Hirai, Resorcinol-formaldehyde resins as metal-free semiconductor photocatalysts for solar-to-hydrogen peroxide energy conversion, Nat. Mater., 2019, 18, 985–993 CrossRef CAS PubMed.
- P. Su, J. Zhang, Y. Zhou, Z. Wei, S. Zhao, B. Yang, X. Zhao and J. Chen, Efficient photocatalytic production of hydrogen peroxide by Z-scheme resorcinol-formaldehyde resin/g-C3N4 heterostructure under visible light, J. Chem. Eng., 2023, 454, 140504 CrossRef CAS.
- X. Li, D. Chen, N. Li, Q. Xu, H. Li and J. Lu, Efficient photocatalytic hydrogen peroxide production induced by the strong internal electric field of all-organic S-scheme heterojunction, J. Colloid Interface Sci., 2023, 633, 691–702 CrossRef CAS PubMed.
- L. Yang, G. Dong, D. L. Jacobs, Y. Wang, L. Zang and C. Wang, Two-channel photocatalytic production of H2O2 over g-C3N4 nanosheets modified with perylene imides, J. Catal., 2017, 352, 274–281 CrossRef CAS.
- Y. X. Ye, J. Pan, F. Xie, L. Gong, S. Huang, Z. Ke, F. Zhu, J. Xu and G. Ouyang, Highly efficient photosynthesis of hydrogen peroxide in ambient conditions, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, 2103964118 CrossRef PubMed.
- Y. Xia, B. Zhu, X. Qin, W. Ho and J. Yu, Zinc porphyrin/g-C3N4 S-scheme photocatalyst for efficient H2O2 production, Chem. Eng. J., 2023, 467, 143528 CrossRef CAS.
- X. Wang, Z. Han, L. Yu, C. Liu, Y. Liu and G. Wu, Synthesis of full-spectrum-response Cu2(OH)PO4/g-C3N4 photocatalyst with outstanding photocatalytic H2O2 production performance via a “two channel route”, ACS Sustainable Chem. Eng., 2018, 6, 14542–14553 CrossRef CAS.
- Z. Li, N. Xiong and G. Gu, Fabrication of a full-spectrum-response Cu2(OH)2 CO3/gC3N4 heterojunction catalyst with outstanding photocatalytic H2O2 production performance via a self-sacrificial method, Dalton Trans., 2019, 48, 182–189 RSC.
- H. Y. Liu, C. G. Niu, D. W. Huang, C. Liang, H. Guo, Y. Y. Yang and L. Li, Unravelling the role of reactive oxygen species in ultrathin Z-scheme heterojunction with surface zinc vacancies for photocatalytic H2O2 generation and CTC degradation, Chem. Eng. J., 2023, 465, 143007 CrossRef CAS.
- G. Jiang, X. You, B. An, B. Zhu, F. Liu, X. Duan, Y. Wang and R. Zhao, Constructing direct Z-scheme heterojunctions of defective MoS2-v on carbon nitride nanotubes for high-performance hydrogen peroxide production and iron-free photo-Fenton-like reactions over a wide pH range, Appl. Surf. Sci., 2023, 618, 156656 CrossRef CAS.
- Y. Zhang, X. Chen, Y. Ye and J. Chen, Photocatalytic oxygen reduction reaction over copper-indium-sulfide modified polymeric carbon nitride S-scheme heterojunction photocatalyst, J. Catal., 2023, 419, 9–18 CrossRef CAS.
- Y. Wang, Y. He, Y. Chi, P. Yin, L. Wei, W. Liu, X. Wang, H. Zhang and H. Song, Construction of S-scheme pn heterojunction between protonated g-C3N4 and α-MnS nanosphere for photocatalytic H2O2 production and in situ degradation of oxytetracycline, J. Environ. Chem. Eng., 2023, 11, 109968 CrossRef CAS.
- H. He, Z. Wang, J. Zhang, C. Shao, K. Dai and K. Fan, Interface Chemical Bond Enhanced Ions Intercalated Carbon Nitride/CdSe–Diethylenetriamine S–Scheme Heterojunction for Photocatalytic H2O2 Synthesis in Pure Water, Adv. Funct. Mater., 2024, 2315426 CrossRef CAS.
- Q. Yang, R. Li, S. Wei and R. Yang, Schottky functionalized Z-scheme heterojunction photocatalyst Ti2C3/g-C3N4/BiOCl: Efficient photocatalytic H2O2 production via two-channel pathway, Appl. Surf. Sci., 2022, 572, 151525 CrossRef CAS.
- B. P. Mishra, S. Das, L. Biswal, L. Acharya, J. Sahu and K. Parida, MXene Schottky Functionalized Z-scheme Ternary Heterostructure for Enhanced Photocatalytic H2O2 Production and H2 Evolution, J. Phys. Chem. C, 2024, 128, 1921–1935 CrossRef CAS.
- X. Chen, W. Zhang, L. Zhang, L. Feng, C. Zhang, J. Jiang, T. Yan and H. Wang, Sacrificial agent-free photocatalytic H2O2 evolution via two-electron oxygen reduction using a ternary α-Fe2O3/CQD@gC3N4 photocatalyst with broad-spectrum response, J. Mater. Chem., 2020, 8, 18816–18825 RSC.
- X. Li, F. Ye, H. Zhang, M. Ahmad, Z. Zeng, S. Wang, S. Wang, D. Gao and Q. Zhang, Ternary rGO decorated W18O49@g-C3N4 composite as a full-spectrum-responded Z-scheme photocatalyst for efficient photocatalytic H2O2 production and water disinfection, J. Environ. Chem. Eng., 2023, 11, 110329 CrossRef CAS.
- P. Behera, A. Ray, S. P. Tripathy, S. Subudhi, L. Acharya and K. Parida, NixPy Cocatalyst-Loaded MOF-Derived C/N–ZnO@B-Doped g-C3N4-Based Z-Scheme Nanohybrid: A Combinatorically Enhanced Ternary Photocatalyst towards Hydrogen Peroxide and Hydrogen Production, ACS Appl. Eng. Mater., 2023, 1, 2876–2891 CrossRef CAS.
- J. Shi, Z. Liu, Y. Luo, C. Guo, Y. Li, T. Yang, C. Ju, H. Wang, X. Li and Z. Fan, Enhanced photocatalytic production of hydrogen peroxide via two-channel pathway using modified graphitic carbon nitride photocatalyst: Doping K+ and combining WO3, Colloids Surf., A, 2021, 623, 126758 CrossRef CAS.
- X. Zhang, H. S. Chen and P. Yang, W18O49/crystalline g-C3N4 layered heterostructures with full solar energy harvesting towards efficient H2O2 generation and NO conversion, Nano Energy, 2024, 120, 109160 CrossRef CAS.
- X. Li, B. Kang, F. Dong, Z. Zhang, X. Luo, L. Han, J. Huang, Z. Feng, Z. Chen, J. Xu, B. Peng and Z. L. Wang, Enhanced photocatalytic degradation and H2/H2O2 production performance of S-pCN/WO2.72 S-scheme heterojunction with appropriate surface oxygen vacancies, Nano Energy, 2021, 81, 105671 CrossRef CAS.
- Z. Jiang, Q. Long, B. Cheng, R. He and L. Wang, 3D ordered macroporous sulfur-doped g-C3N4/TiO2 S-scheme photocatalysts for efficient H2O2 production in pure water, J. Mater. Sci. Technol., 2023, 162, 1–10 CrossRef CAS.
- A. Behera, P. Babu and K. Parida, Growth of macroporous TiO2 on B-doped gC3N4 nanosheets: a Z-scheme photocatalyst for H2O2 production and phenol oxidation under visible light, Inorg. Chem. Front., 2021, 8, 1489–1499 RSC.
- X. Liu, Y. Li, K. Lin and Y. Jiang, Construction of a novel heteropoly molybdophosphate/graphitized carbon nitride s-scheme heterostructure with enhanced photocatalytic H2O2 evolution activity, J. Colloid Interface Sci., 2024, 654, 1228–1239 CrossRef CAS.
| This journal is © the Partner Organisations 2024 |