
Recent applications of biphotonic processes in organic synthesis
Jorge
Castellanos-Soriano
 a,
Jorge C.
Herrera-Luna
a,
David
Díaz Díaz
bcd,
M. Consuelo
Jiménez
a and
Raúl
Pérez-Ruiz
*a
a,
Jorge C.
Herrera-Luna
a,
David
Díaz Díaz
bcd,
M. Consuelo
Jiménez
a and
Raúl
Pérez-Ruiz
*a
aDepartamento de Química, Universitat Politècnica de València (UPV), Camino de Vera s/n, 46022, Valencia, Spain. E-mail: raupreru@qim.upv.es
bInstitut für Organische Chemie, Universität Regensburg, Universitätsstr. 31, 93053 Regensburg, Germany
cDepartamento de Química Orgánica, Universidad de La Laguna, Avda. Astrofísico Francisco Sánchez 3, 38206 La Laguna, Tenerife, Spain
dInstituto de Bio-Orgánica Antonio González, Universidad de La Laguna, Avda. Astrofísico Francisco Sánchez 2, 38206 La Laguna, Tenerife, Spain
First published on 2nd June 2020
Abstract
Currently, evolution of chemical transformations by visible light irradiation is highly desirable from cost, safety, availability, and environmental friendliness points of view. Besides, activation of less reactive substrates under very mild conditions becomes one of the most challenging tasks in organic synthesis. However, the insufficient energy provided by one photon of visible light for their activation definitely makes necessary the development of new protocols together with the design of new photocatalytic systems to overcome this limitation. In this context, the implementation of biphotonic processes has been found to be a solution for these drawbacks. This new mechanistic paradigm which combines light upconversion processes with energy/electron transfers holds great potential for high energy demanding bond activations, expanding the accessible reactivity window. Here, we wish to highlight the recent applications of biphotonic processes in organic synthesis.
Sunlight strikes our planet every day with more energy than we consume in a whole year. The question arises why the chemistry community has not used sunlight as a synthetic tool for developing photochemical processes for almost one century.† The main reason lies in the inability of most organic compounds to absorb light in the visible range. Moreover, electronically excited states are often only available upon irradiation with shorter wavelengths of ultraviolet (UV) light. These high-energy photons can therefore cause uncontrolled photodecomposition processes, a factor that has limited the broad utilization of photochemical synthesis in the fabrication of complex organic molecules. For this purpose, many researchers have explored ways to efficiently employ visible light energy for the activation of organic molecules,1 for example, the design, synthesis and development of photocatalysts that can absorb visible light and mediate the desired chemical transformations by different oxidative and reductive potentials. Activation of molecules with visible light offers the possibility of reaction pathways which are otherwise impossible to perform with classical nonphotochemical strategies.2
Another fascinating aspect of photochemistry boosted by visible light is the use of photons as “traceless and green reagents”, rendering photochemical processes green and sustainable. In this context, the last decade has witnessed a fantastic growth in the field of organic photocatalysis using visible light as an energy source, emerging as a new and powerful tool for activating molecules.3
However, the scope of photocatalytic bond activations is limited not only by the energy of one visible photon but also by the energetic losses suffered by the photocatalysts such as intramolecular charge transfers or internal conversions.3h A plausible approach to expand the accessible reactivity window to less activated substrates is the use of photon upconversion (UC),4 which converts low-energy radiation into higher-energy radiation by either the two-photon absorption (TPA) mechanism or the triplet–triplet annihilation (TTA) mechanism.5 This technology has been successfully applied in diverse scientific fields ranging from energy to biology.6 Although the utilization of biphotonic processes‡ as a potential synthetic tool is still in its infancy, it has gained great momentum over the past five years and has triggered renewed interest in photochemistry in general. Development of the biphotonic technology for addressing critical bond activations or electron transfers in organic synthesis has been found to be advantageous while retaining the benefit of mild reaction conditions using lower-energy visible light (Scheme 1).
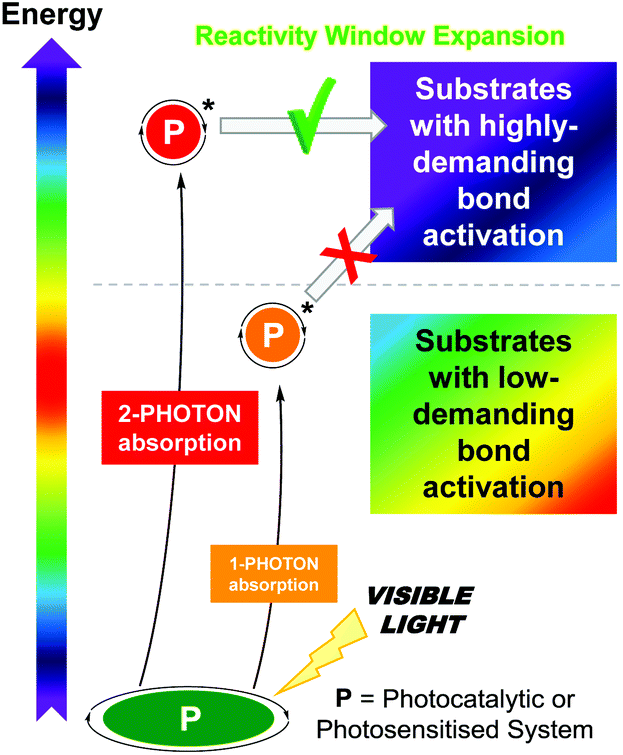 | ||
| Scheme 1 Conceptual scheme of this highlight article: the application of biphotonic processes expands the reactivity window for organic synthesis. | ||
In this highlight article, we therefore focus our attention on the existing examples of this emerging concept where several biphotonic processes such as TPA, TTA and consecutive photoinduced electron transfer (ConPET) have been successfully applied to chemical transformations.
Photosensitized two-photon absorption (TPA) reactions : The TPA mechanism involves an intramolecular simultaneous absorption of two photons via a virtual state,7 and relaxation would lead to an emission with a greater frequency than those of the absorbed photons. Despite TPA requiring the use of high intensity excitation by laser beams with concentrated peaks, numerous potential applications in biophotonics, optics and nano-micro fabrication have been reported.5 However, the involvement of TPA processes in organic transformations is quite rare, presumably because of difficulties in experimental designs. Nevertheless, some examples can be found in the literature focused mainly on photosensitized biphotonic irradiation for DNA damage. Miranda and co-workers8 demonstrated the photochemistry of an upper thymine-like triplet state (nπ* triplet). The proposed mechanism involved the biphotonic excitation of benzophenone by high-energy laser pulses and a successful intramolecular sensitization. To avoid energetic losses, a dyad containing both the photosensitizer and the acceptor linked by an amide bridge was synthesized (Scheme 2). Thus, the benzophenone Tn formed by biphotonic excitation transferred its energy to thymine-like T2 (nπ*), allowing the subsequent Norrish–Yang reaction to obtain the corresponding products. In this vein, the same research group has very recently published the intermolecular version of this reaction,9 using 2′-methoxyacetophenone as a photosensitizer and tert-butyluracil as a modified pyrimidine base; however, the strategy was now different. After the first UVA (355 nm) photon was absorbed by the sensitizer, triplet–triplet energy transfer (TTEnT) occurred, giving rise to the nucleobase triplet which was able to absorb the second photon (355 nm) reaching a Tn state. The viability of this strategy was supported by two model reactions: (i) the Norrish–Yang photocyclization of tert-butyluracil and (ii) the photohydration of its uracil analogue, lacking the tert-butyl substituent.
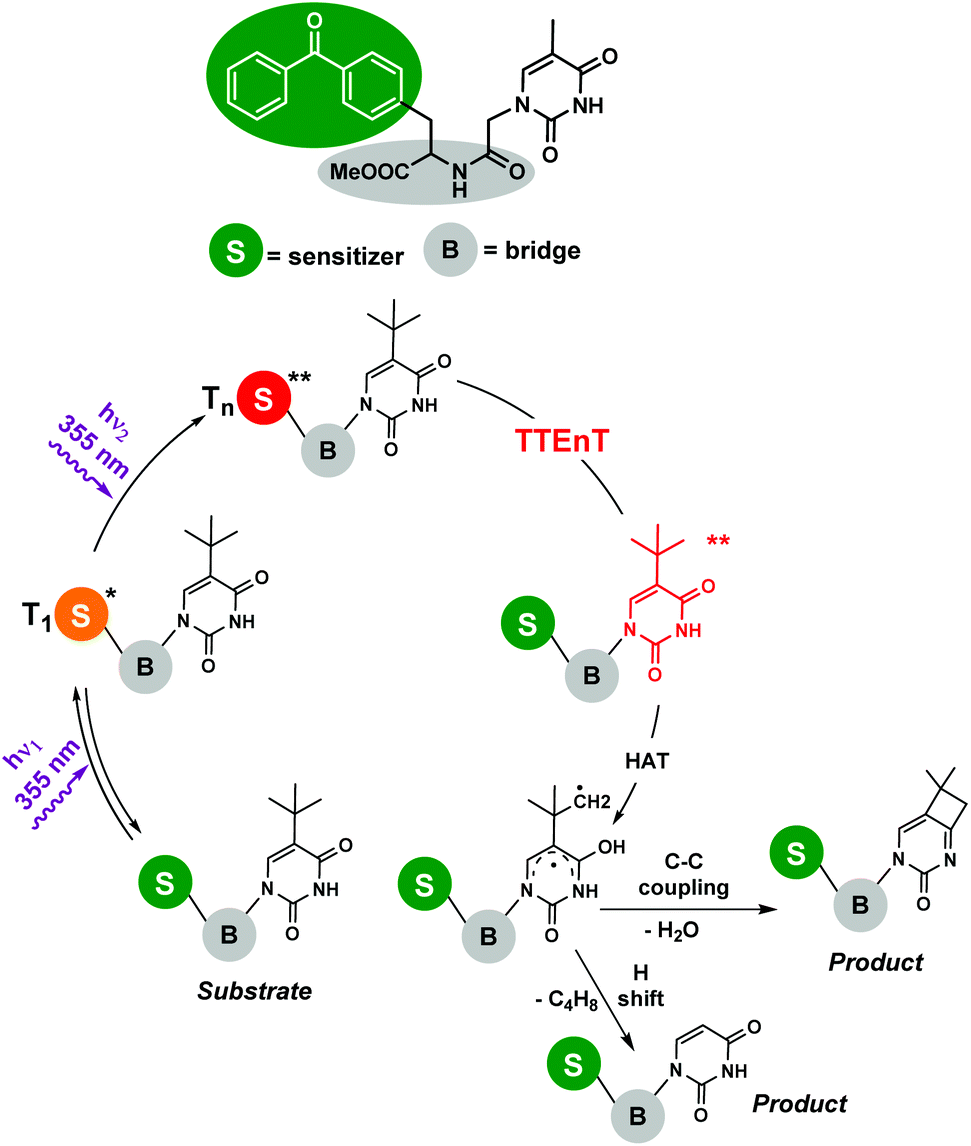 | ||
| Scheme 2 Photoreaction of the benzophenone-modified nucleobase dyad. Involvement of a TPA process upon laser flash photolysis (λexc = 355 nm). | ||
Carbazole-based compounds were also found to be effective biphotonic sensitizers for the DNA photodamage.10 Experimentally, the DNA photocleavage was successfully achieved in the presence of photosensitizers upon irradiation with not only visible light but also with 800 nm NIR light via a TPA process. The proposed reaction mechanism involved a hydrogen atom abstraction (HAT) by N-centred radicals (type I mechanism) under anaerobic conditions. Accordingly, subsequent computational studies on these findings evidenced the great efficiency of these compounds as sensitizers in the NIR region; however, the rationalization of the mechanism of action revealed the involvement of solvated electrons (e−(aq)) produced by a spontaneous photoionization that in turn were responsible for DNA strand cleavage.11
In this respect, Wenger and co-workers also observed that efficient two-photon excitation of a metal complex generated an e−(aq) upon ionization.12 This fact allowed them to control the reactivity over several photochemical reactions relying on the light intensity. Hence, the one-photon mechanism (low-powered intensity) permitted energy transfer from the photocatalyst to the substrates achieving debromination, cis–trans isomerization or HAT; in contrast, the use of high-powered light intensity (TPA) electron transfer from the e−(aq) took place, affording in this case dechlorinated, hydrogenated and dimer products, respectively (Scheme 3).
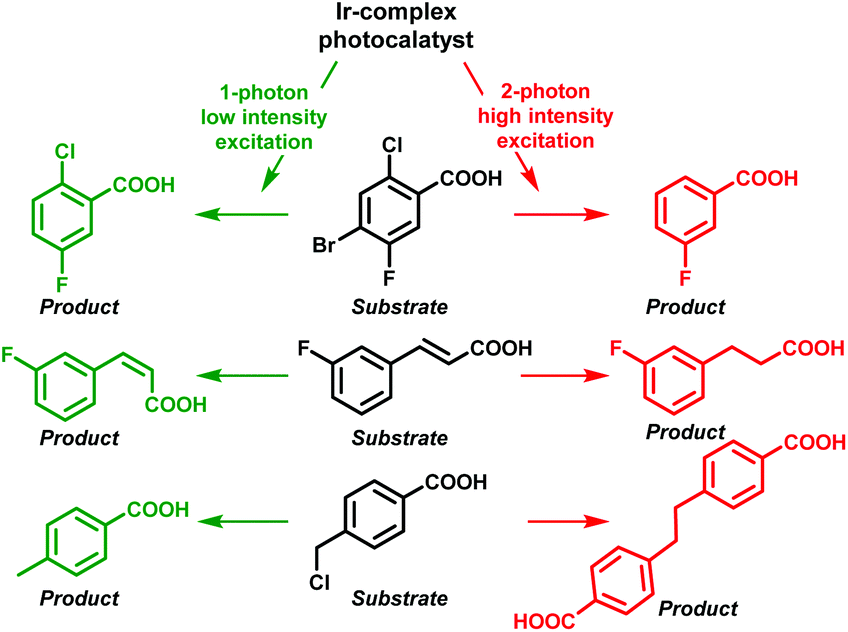 | ||
| Scheme 3 Control over the reactivity relying on the one- or two-photon mechanism. | ||
Regarding the challenging dechlorination reactions, Yamaji and co-workers carried out two-colour two-laser photolysis on several chlorinated diketones.13 The results revealed the formation of halogen-free diketones as the sole products by a Cl elimination mechanism in which the upper triplet Tn (n ≥ 2) state was involved. Finally, chemical reactions involving quinoline-based photoremovable protecting groups using TPA processes have been recently proven as a powerful tool for physiological studies.14 Thus, photolysis at longer wavelengths (740 nm light from a Ti:sapphire laser) allowed the efficient release of homopiperonylic acid in a high yield under simulated physiological conditions.
Photocatalytic reactions by triplet–triplet annihilation (TTA) : Photon upconversion based on TTA between bimolecular organic systems has become one of the most attractive wavelength conversion technologies. Various combinations of compounds showing TTA properties can be found in the literature.15Scheme 4 shows the photochemical events associated with this synchronized biphotonic process which includes intersystem crossing (ISC), TTEnT, TTA and upconverted fluorescence (hν2).
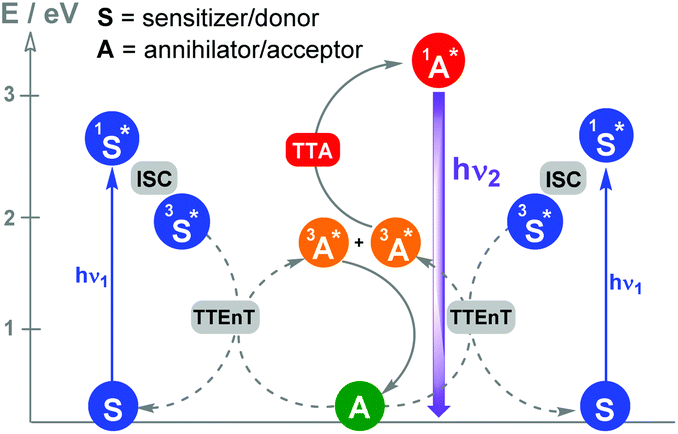 | ||
| Scheme 4 Photon upconversion technology based on TTA. After the absorption of low-energy photons (hν1), a sensitizer triplet is produced by intersystem crossing (ISC) from its singlet. Subsequently, TTEnT from sensitizer triplets to the acceptor (Dexter mechanism) occurs. When two acceptor molecules in their triplet states are capable of colliding during their lifetimes, a higher singlet energy level is formed by TTA and, consequently, generates delayed upconverted fluorescence (hν2). | ||
This transformation of two low-energy photons (2 × hν1) into one higher-energy photon (hν2) has been already productively applied in diverse scientific areas of research;16 however, organic synthetic procedures using TTA processes have only recently been developed. For the first time, TTA was embedded in chemical reactions involving electron transfer activation of challenging organic molecules.17 Thus, we pioneered this elegant concept investigating the photophysical and photochemical aspects of combining TTA events of two simple organic dyes (diketone and oxazole) with reductive activations of aryl bromides.17a This framework involved detailed spectroscopic, theoretical, and synthetic studies and ultimately led to the development of a new set of photocatalytic redox hydrodebrominations. Concomitantly with this work, we also contributed with the first intragel aerobic photoreduction of aryl halides catalysed by platinum(II) octaethyl-porphyrin (PtOEP) and 9,10-diphenylanthracene (DPA) as the TTA system in supramolecular gel networks.17b Thus, the gel network provided a stable microenvironment for the challenging multi-step process under aerobic conditions, at room temperature and without additional additives. These results demonstrated that low weight molecular (LWM) gelators could be used as confined reaction media or micro/nanoreactors, providing the background for more demanding photophysical processes.
We further developed this methodology for more complex processes such as C–C coupling reactions. In particular, photocatalytic aromatic functionalization of N-methyl pyrrole was achieved using a metal-free TTA system based on diiodoBOPHY-like derivative (BOPHY) and DPA.18 The reaction reported good to high product yields and the mechanistic aspects of this procedure were elucidated by means of combining product analysis, spectroscopic data and computational studies (Scheme 5). It is worth mentioning that an economical blue laser pointer (λexc = 445 nm ± 10, 2 W) was employed that facilitated the management of the optimal conditions instead of using the laser flash photolysis technique.
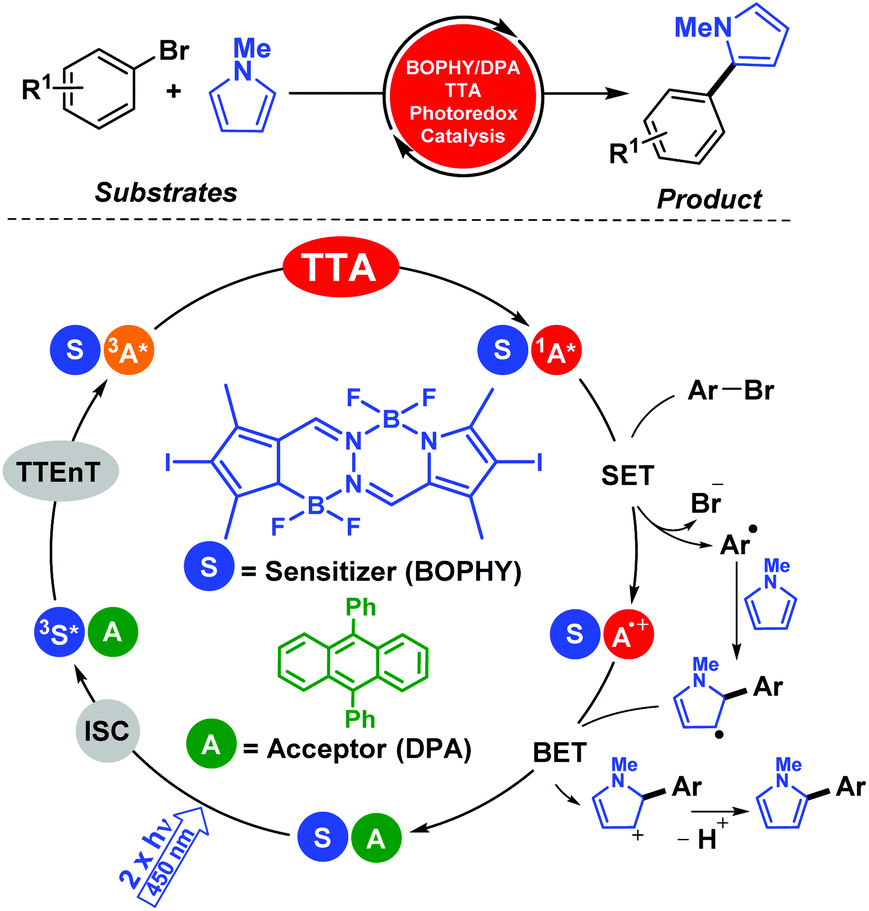 | ||
| Scheme 5 Aromatic functionalization of N-methyl pyrrole photocatalyzed by the TTA technology. | ||
Among biological applications, near-infrared (NIR) radiation was also used for several organic transformations by utilizing the photophysical process of TTA.19,20 Orange or blue light was accessed from low-energy NIR light (λexc = 730 nm) by pairwise combination of the TTA couple.19 For instance, the delayed emission released from the NIR-to-orange upconversion system (Pd-complex/diketopyrrolopyrrole derivative as a sensitizer/annihilator) was absorbed by a photocatalyst (eosin Y or Rose Bengal) that subsequently photocatalyzed low energy demanding reactions such as dehalogenation of α-bromoacetophenone, amine oxidation or C–N bond activation. Besides, the combination of a NIR absorbing sensitizer Pt-complex with tetratertbutylperylene as a blue-emitter was adapted to the prototypical [Ru(bpy)3]2+-catalysed reaction, the intramolecular [2 + 2] cyclization of enones, obtaining moderate yields (48%). This TTA system was also employed directly for catalysing the cyclization of dienyl azides to pyrroles (80% yield) or the polymerization of methyl methacrylate. On the other hand, a photochromic reaction involving a C–N bond breaking was successfully achieved by TTA, where NIR light irradiation (λexc = 635 nm) was also implemented.20
Despite suitable TTA systems in aqueous media being barely known,21 monodechlorination of trichloroacetate in air-saturated aqueous solution was successfully achieved by means of the TTA technology.21d This procedure constituted a promising green strategy for further photochemical transformations in homogeneous aqueous solution.
Consecutive photoinduced electron transfer (ConPET) reactions : Plants produce their own energy autotrophically from water, carbon dioxide and visible light. This combination, the renowned photosynthesis, follows a two-step photon excitation Z-scheme mechanism where accumulative photon absorption is involved. Surprisingly, the application of the Z-scheme photosynthesis concept to activate substrates with high energy demanding bonds for organic synthesis is very recent (Scheme 6).
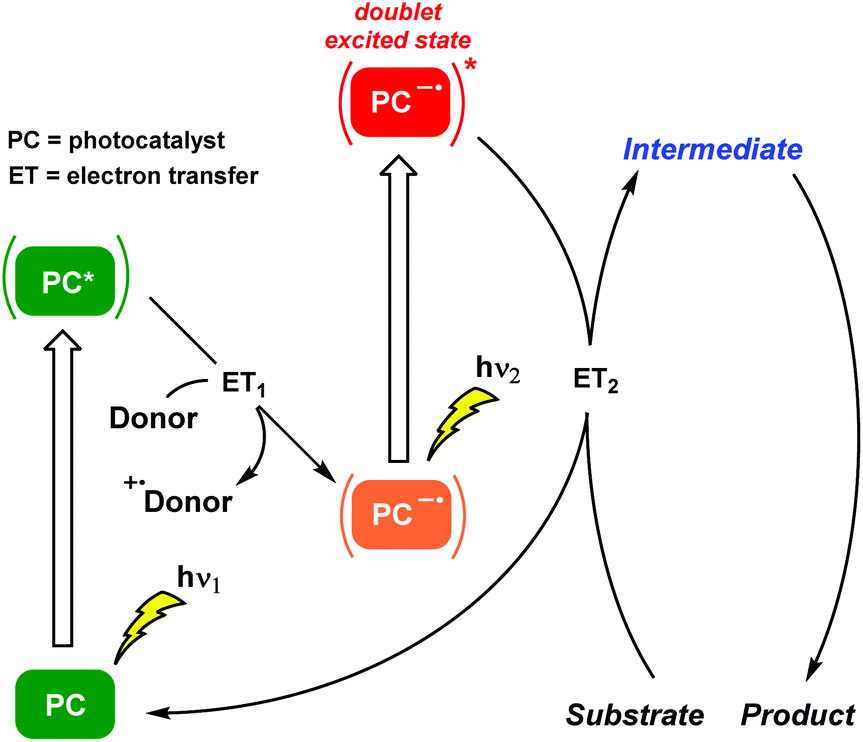 | ||
| Scheme 6 The ConPET mechanism, which mimics the Z-scheme, adapted to organic transformations. Selective excitation of the PC led to its excited state which is quenched by an appropriate donor generating the PC radical anion. Then, excitation of the PC radical anion results in the formation of an excited PC radical anion (doublet excited state), a highly reducing agent. | ||
The kick-off work engaging these two terms is attributed to König and co-workers.22 Photogeneration of radical anions of aryl halides (Br, Cl) was achieved by a ConPET process involving a doublet excited state. An organic perylene diimide (PDI)-based photocatalyst was capable of accumulating the energy of two visible light photons in order to reduce stable aryl halides, generating the corresponding aryl radicals which were trapped by hydrogen atom donors or by specific nucleophiles to form C–C bonds. Thus, this outstanding chemical model mimicked the Z scheme in biological photosynthesis and permitted the photocatalytic conversion of less reactive chemical bonds in organic synthesis. Subsequently, the same PDI-based photocatalyst was incorporated into the metal–organic polymer. This heterogeneous approach facilitated the ConPET process for the visible light-driven dehalogenation of aryl halides reducing markedly the irradiation times.23 Regarding chromophore-diimide derivatives, a heterogeneous strategy based on the synergistic effects of ConPET and HAT processes was applied to aryl halide photoreduction. A new polyoxometalate-incorporated naphthalenediimide (NDI)-based metal–organic framework (MOF) was designed and, after visible light irradiation, the host–guest MOF could stably generate NDI˙− which was the critical intermediate for obtaining the photoreduced products.24 Strategically, diimide-like photocatalysts required a priori tedious synthesis, in addition to low solubility in organic solvents and sometimes the use of elevated temperatures. Moreover, alternative investigations related to the PDI system might suggest other mechanistic interpretations.25
Various organic, inexpensive, and soluble dyes such as 1,8-dihydroxyanthraquinone (Aq-OH), rhodamine 6G (Rh-6G) or 9,10-dicyanoanthracene (DCA) were found to be suitable photocatalysts for ConPET processes. For instance, Aq-OH transformed into its coloured radical anion or semiquinone anion in the presence of triethylamine upon the irradiation of one visible light photon. Then, excitation of these species by visible light led to an electron transfer process, activating aryl halides that reacted in dehalogenation or C–C bond-forming reactions.26
To date, the organic photocatalyst Rh-6G has been most widely used for organic synthesis.27 The first work in this series described the selective aromatic functionalisation of arenes and heteroarenes by controlling the bond activation through light-controlled regulation of redox potentials.27a Subsequently, a series of synthetic applications photocatalyzed by Rh-6G through the ConPET process were reported. Thus, the synthesis of pyrrolo-[1,2-a]quinolines and ullazines was achieved in one pot in moderate to good yields (35–75%).27b Heteroaromatic biaryls were synthesized in moderate to excellent yields (41–91%) and, remarkably, this reaction worked for a broad range of brominated electron-rich heteroarenes and chlorinated heteroarenes bearing electron-withdrawing groups.27c A photo-Arbuzov reaction for the construction of aryl phosphonates with consistent yields was also achieved by this methodology through C–P bond formation.27d Moreover, this Rh-6G-ConPET protocol was found to be an alternative strategy of the C–H arylation procedures using halogenated nucleobases under UV-light irradiation.27e Combination of ConPET (Rh-6G) with metal catalysis (lanthanide ions) permitted the activation of even chlorobenzenes in order to form C–C and C–P bonds.27f Finally, it was demonstrated that gel networks could act as the reaction media for aerobic photocatalyzed C–C coupling reactions by Rh-6G through the ConPET mechanism, providing similar results to those obtained under inert atmosphere conditions.27g
Spectroscopic investigations obtained some insights into the mechanistic aspects of this ConPET process using Rh-6G. Hence, fluorescence spectroscopy showed that the Rh-6G radical anion could be generated from the singlet or triplet excited state of Rh-6G, relying on the sacrificial donor and varying its concentration.27h Very recently, the fate of the excited Rh-6G radical anion has been studied by femtosecond spectroscopy.27i The lifetime of the excited Rh-6G radical anion (350 fs) was found to be too short for diffusion-controlled electron transfer to the substrate. Therefore, the authors postulated that the excited Rh-6G radical anion generated e−(aq) as an additional step in the photocatalytic cycle, which was now responsible for the aryl halide reductions.
In this context, the excited DCA radical anion lifetime previously ranged in the ns scale and it displayed an exceptionally high reducing potential of ca. −3 V.28 Therefore, DCA possessed all prime qualifications for its use as a strongly reducing photocatalyst. Indeed, photocatalytic aromatic substitutions of poor reactive aryl halides by DCA were found to be operated via the ConPET mechanism.29 Thus the resulting excited DCA radical anion readily effected C–C, C–P, C–S, and C–B bond formations. The detailed information of the reaction mechanism of this biphotonic catalytic process was supported by product analysis, spectroscopic measurements, and computational studies (Scheme 7).
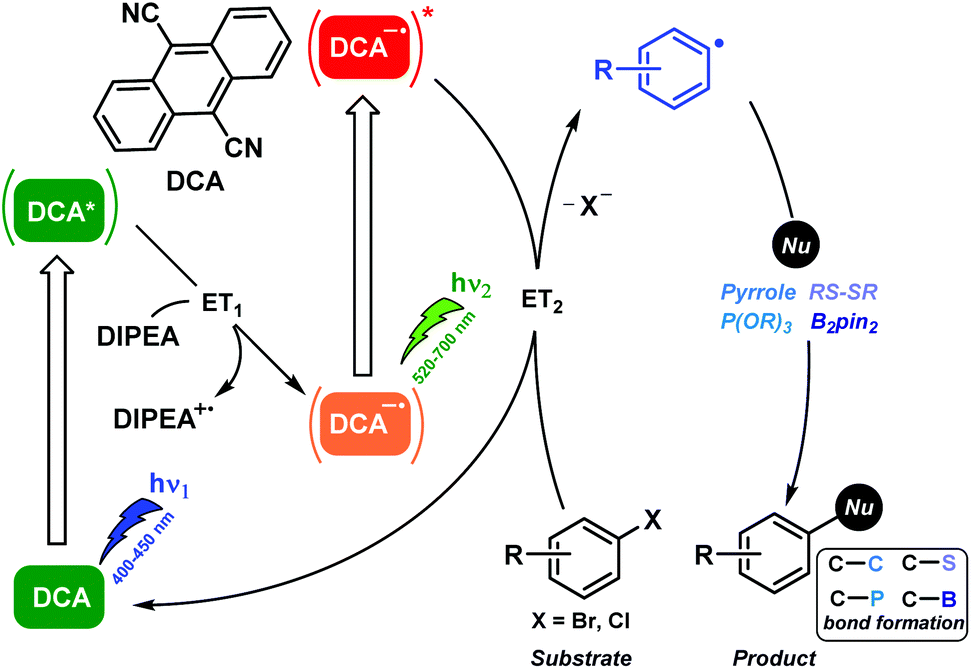 | ||
| Scheme 7 Aromatic substitutions of aryl halides photocatalyzed by DCA through the ConPET mechanism using cold white LEDs (410–700 nm). Fluorescence studies revealed the quantitative formation of the DCA radical anion that absorbs green light, giving rise to its doublet excited state; this species was capable of injecting one electron to the aryl halides. | ||
In summary, the biphotonic technology has emerged as a powerful tool for addressing important bond activations or electron transfer processes in organic synthesis under mild conditions and using lower-energy visible light. These biphotonic processes include TPA, TTA and ConPET reactions, which have been used to trigger, for instance, reductive activations of aryl bromides, C–C or C–heteroatom coupling reactions, dehalogenation of aryl halides and functionalisation of arenes and heteroarenes. The success of this approach lies on a judicious selection of the photocatalytic system to achieve the required photoregulation of the involved redox potentials. In the coming years, we foresee a significant increase in the use of this technology to assist modern organic synthesis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Generalitat Valenciana (CIDEGENT/2018/044) and the Spanish Government (CTQ2016-78875-P, BES-2017-080215 and BEAGAL18/00166) is gratefully acknowledged.Notes and references
- (a) G. D. Scholes, G. R. Fleming, A. Olaya-Castro and R. van Grondelle, Lessons from nature about solar light harvesting, Nat. Chem., 2011, 3, 763 CrossRef CAS PubMed; (b) G. D. Scholes, T. Mirkovic, D. B. Turner, F. Fassioli and A. Buchleitner, Solar light harvesting by energy transfer: from ecology to coherence, Energy Environ. Sci., 2012, 5, 9374 RSC; (c) B. Demmig-Adams, J. J. Stewart, T. A. Burch and W. W. Adams, Insights from placing photosynthetic light harvesting into context, J. Phys. Chem. Lett., 2014, 5, 2880 CrossRef CAS PubMed.
- (a) V. Balzani, G. Bergamini and P. Ceroni, Light: A very peculiar reactant and product, Angew. Chem., Int. Ed., 2015, 54, 11320 CrossRef CAS PubMed; (b) N. Hoffmann, Photochemical reactions as key steps in organic synthesis, Chem. Rev., 2008, 108, 1052 CrossRef CAS PubMed; (c) N. J. Turro, V. Ramamurthy and J. C. Scaiano, Modern Molecular Photochemistry of Organic Molecules, University Science Books, Sausalito, CA, 2010, p. 1084 Search PubMed; (d) A. G. Griesbeck, M. Oelgemöller and F. Ghetti, CRC Handbook of Organic Photochemistry and Photobiology, CRC Press, Boca Raton, FL, 3rd edn, 2012, p. 1694 Search PubMed; (e) A. Albini and M. Fagnoni, Handbook of Synthetic Photochemistry, Wiley-VCH, Weinheim, 2009, p. 463 CrossRef; (f) M. Montaldi, A. Credi, L. Prodi and T. M. Gandolfi, CRC Handbook of Photochemistry, CRC Press, Boca Raton, FL, 3rd edn, 2006, p. 664 CrossRef; (g) A. G. Griesbeck and J. Mattay, Synthetic Organic Photochemistry, Marcel Dekker, New York, 2005, p. 648 Search PubMed.
- (a) A. Hossain, A. Bhattacharyya and O. Reiser, Copper's rapid ascent in visible-light photoredox catalysis, Science, 2019, 364, 450 CrossRef PubMed; (b) Q. Q. Zhou, Y. Q. Zou, L. Q. Lu and W. J. Xiao, Visible-light-induced organic photochemical reactions through energy-transfer pathways, Angew. Chem., Int. Ed., 2019, 58, 1586 CrossRef CAS PubMed; (c) F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Energy transfer catalysis mediated by visible light: Principles, Applications, Chem. Soc. Rev., 2018, 47, 7190 RSC; (d) J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, The merger of transition metal and photocatalysis, Nat. Rev. Chem., 2017, 1, 0052 CrossRef CAS; (e) N. A. Romero and D. A. Nicewicz, Organic photoredox catalysis, Chem. Rev., 2016, 116, 10075 CrossRef CAS PubMed; (f) K. L. Skubi, T. R. Blum and T. P. Yoon, Dual catalysis strategies in photochemical synthesis, Chem. Rev., 2016, 116, 10035 CrossRef CAS PubMed; (g) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (h) D. M. Schultz and T. P. Yoon, Solar synthesis: Prospects in visible light photocatalysis, Science, 2014, 343, 1239176 CrossRef PubMed.
- (a) M. Zhang, Y. Lin, T. J. Mullen, W.-F. Lin, L.-D. Sun, C.-H. Yan, T. E. Patten, D. Wang and G.-y. Liu, Improving hematite's solar water splitting efficiency by incorporating rare-earth upconversion nanomaterials, J. Phys. Chem. Lett., 2012, 3, 3188 CrossRef CAS PubMed; (b) F. Gonell, M. Haro, R. S. Sánchez, P. Negro, I. Mora-Seró, J. Bisquert, B. Julián-López and S. Gimenez, Photon up-conversion with lanthanide-doped oxide particles for solar H2 generation, J. Phys. Chem. C, 2014, 118, 11279 CrossRef CAS.
- C. Ye, L. Zhou, X. Wang and Z. Liang, Photon upconversion: from two-photon absorption (TPA) to triplet–triplet annihilation (TTA), Phys. Chem. Chem. Phys., 2016, 18, 10818 RSC , and references therein.
- For selected reviews: (a) Q.-C. Sun, Y. C. Ding, D. M. Sagar and P. Nagpal, Photon upconversion towards applications in energy conversion and bioimaging, Prog. Surf. Sci., 2017, 92, 281 CrossRef CAS; (b) L. Frazer, J. K. Gallaher and T. W. Schmidt, ACS Energy Lett., 2017, 2, 1346 CrossRef CAS; (c) A. Gulzar, J. Xu, P. Yang, F. He and L. Xu, Upconversion processes: versatile biological applications and biosafety, Nanoscale, 2017, 9, 12248 RSC.
- K. D. Nanda and A. I. Krylov, Visualizing the contributions of virtual states to two-photon absorption cross sections by natural transition orbitals of response transition density matrices, J. Phys. Chem. Lett., 2017, 8, 3256 CrossRef CAS PubMed.
- V. Vendrell-Criado, G. M. Rodríguez-Muñiz, M. Yamaji, V. Lhiaubet-Vallet, M. C. Cuquerella and M. A. Miranda, Two-photon chemistry from upper triplet states of thymine, J. Am. Chem. Soc., 2013, 135, 16714 CrossRef CAS PubMed.
- O. R. Alzueta, J. Cadet, M. C. Cuquerella and M. A. Miranda, Photosensitised biphotonic chemistry of pyrimidine derivatives, Org. Biomol. Chem., 2020, 18, 2227 RSC.
- Y.-C. Zheng, M.-L. Zheng, K. Li, S. Chen, Z.-S. Zhao, X.-S. Wang and X.-M. Duan, Novel carbazole-based two-photon photosensitizer for efficient DNA photocleavage in anaerobic condition using near-infrared light, RSC Adv., 2015, 5, 770 RSC.
- H. Gattuso, E. Dumont, M. Marazziab and A. Monari, Two-photon-absorption DNA sensitization via solvated electron production: unraveling photochemical pathways by molecular modeling and simulation, Phys. Chem. Chem. Phys., 2016, 18, 18598 RSC.
- C. Kerzig and O. S. Wenger, Reactivity control of a photocatalytic system by changing the light intensity, Chem. Sci., 2019, 10, 11023 RSC.
- M. Yamaji, Y. Suwa, R. Shimokawa, C. Paris and M. A. Miranda, Photochemical reactions of halogenated aromatic 1,3-diketones in solution studied by steady state, one- and two-color laser flash photolyses, Photochem. Photobiol. Sci., 2015, 14, 1673 RSC.
- A.-L. K. Hennig, D. Deodato, N. Asad, C. Herbivo and T. M. Dore, Two-Photon Excitable Photoremovable Protecting Groups Based on the Quinoline Scaffold for Use in Biology, J. Org. Chem., 2020, 85, 726 CrossRef CAS PubMed.
- A. S. Gertsen, M. Koerstz and K. V. Mikkelsen, Benchmarking triplet–triplet annihilation photon upconversion schemes, Phys. Chem. Chem. Phys., 2018, 20, 12182 RSC.
- (a) M. Barawi, F. Fresno, R. Pérez-Ruiz and V. A. de la Peña O'Shea, Photoelectrochemical hydrogen evolution driven by visible-to-ultraviolet photon upconversion, ACS Appl. Energy Mater., 2019, 2, 207 CrossRef CAS; (b) N. Yanai and N. Kimizuka, New triplet sensitization routes for photon upconversion: thermally activated delayed fluorescence molecules, inorganic nanocrystals, and singlet-to-triplet absorption, Acc. Chem. Res., 2017, 50, 2487 CrossRef CAS PubMed; (c) T. F. Schulze and T. W. Schmidt, Photochemical upconversion: present status and prospects for its application to solar energy conversion, Energy Environ. Sci., 2015, 8, 103 RSC; (d) J. Zhou, Q. Liu, W. Feng, Y. Sun and F. Li, Upconversion luminescent materials: advances and applications, Chem. Rev., 2015, 115, 395 CrossRef CAS PubMed; (e) G. Chen, H. Qiu, P. N. Prasad and X. Chen, Upconversion nanoparticles: design, nanochemistry, and applications in theranostics, Chem. Rev., 2014, 114, 5161 CrossRef CAS PubMed; (f) F. N. Castellano; and T. J. Schmidt, Photochemical upconversion: the primacy of kinetics, J. Phys. Chem. Lett., 2014, 5, 4062 CrossRef PubMed; (g) C. E. McCusker and F. N. Castellano, Orange-to-blue and red-to-green photon upconversion with a broadband absorbing copper(I) MLCT sensitizer, Chem. Commun., 2013, 49, 3537 RSC; (h) K. Börjesson, D. Dzebo, B. Albinsson and K. Moth-Poulsen, Photon upconversion facilitated molecular solar energy storage, J. Mater. Chem. A, 2013, 1, 8521 RSC; (i) S. Guo, W. Wu, H. Guo and J. Zhao, Room-temperature long-lived triplet excited states of naphthalenediimides and their applications as organic triplet photosensitizers for photooxidation and triplet–triplet annihilation upconversions, J. Org. Chem., 2012, 77, 3933 CrossRef CAS PubMed; (j) T. Gallavardin, C. Armagnat, O. Maury, P. L. Baldeck, M. Lindgren, C. Monnereau and C. Andraud, An improved singlet oxygen sensitizer with two-photon absorption and emission in the biological transparency window as a result of ground state symmetry-breaking, Chem. Commun., 2012, 48, 1689 RSC; (k) R. S. Khnayzer, J. Blumhoff, J. A. Harrington, A. Haefele, F. Denga and F. N. Castellano, Upconversion-powered photoelectrochemistry, Chem. Commun., 2012, 48, 209 RSC; (l) J. Z. Zhao, S. M. Ji and H. M. Guo, Triplet–triplet annihilation based upconversion: from triplet sensi-tizers and triplet acceptors to upconversion quantum yields, RSC Adv., 2011, 1, 937 RSC.
- (a) M. Majek, U. Faltermeier, B. Dick, R. Pérez-Ruiz and A. Jacobi von Wangelin, Application of visible-to-uv photon upconversion to photoredox catalysis: the activation of aryl bromides, Chem. – Eur. J., 2015, 21, 15496 CrossRef CAS PubMed; (b) M. Haering, R. Pérez-Ruiz, A. Jacobi von Wangelin and D. Díaz Díaz, Intragel photoreduction of aryl halides by green-to-blue upconversion under aerobic conditions, Chem. Commun., 2015, 51, 16848 RSC.
- C. G. López-Calixto, M. Liras, V. A. de la Peña O'Shea and R. Pérez-Ruiz, Synchronized biphotonic process triggering c-c coupling catalytic reactions, Appl. Catal., B, 2018, 237, 18 CrossRef.
- B. D. Ravetz, A. B. Pun, E. M. Churchill, D. N. Congreve, T. Rovis and L. M. Campos, Photoredox catalysis using infrared light via triplet fusion upconversion, Nature, 2019, 565, 343 CrossRef CAS PubMed.
- A. Tokunaga, L. M. Uriarte, K. Mutoh, E. Fron, J. Hofkens, M. Sliwa and J. Abe, Photochromic reaction by red light via triplet fusion upconversion, J. Am. Chem. Soc., 2019, 141, 17744 CrossRef CAS PubMed.
- (a) K. A. El Roz and F. N. Castellano, Photochemical upconversion in water, Chem. Commun., 2017, 53, 11705 RSC; (b) W. Xu, W. Liang, W. Wu, C. Fan, M. Rao, D. Su, Z. Zhong and C. Yang, Supramolecular assembly-improved triplet–triplet annihilation upconversion in aqueous solution, Chem. – Eur. J., 2018, 24, 16677 CrossRef CAS PubMed; (c) H. Kouno, Y. Sasaki, N. Yanai and N. Kimizuka, Supramolecular crowding can avoid oxygen quenching of photon upconversion in water, Chem. – Eur. J., 2019, 25, 6124 CrossRef CAS PubMed; (d) C. Kerzig and O. S. Wenger, Sensitized triplet-triplet annihilation upconversion in water and its application to photochemical transformations, Chem. Sci., 2018, 57, 6670 RSC.
- I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Reduction of aryl halides by consecutive visible light-induced electron transfer processes, Science, 2014, 346, 725 CrossRef CAS PubMed.
- L. Zeng, T. Liu, C. He, D. Shi, F. Zhang and C. Duan, Organized aggregation makes insoluble perylene diimide efficient for the reduction of aryl halides via consecutive visible light-induced electron-transfer processes, J. Am. Chem. Soc., 2016, 138, 3958 CrossRef CAS PubMed.
- J. He, J. Li, Q. Han, C. Si, G. Niu, M. Li, J. Wang and J. Niu, Photoactive metal–organic framework for the reduction of aryl halides by the synergistic effect of consecutive photoinduced electron-transfer and hydrogen-atom-transfer processes, ACS Appl. Mater. Interfaces, 2020, 12, 2199 CrossRef CAS PubMed.
- M. Marchini, A. Gualandi, L. Mengozzi, P. Franchi, M. Lucarini, P. G. Cozzi, V. Balzani and P. Ceroni, Mechanistic insights into two-photon-driven photocatalysis in organic synthesis, Phys. Chem. Chem. Phys., 2018, 20, 8071 RSC.
- J. I. Bardagi, I. Ghosh, M. Schmalzbauer, T. Ghosh and B. König, Anthraquinones as photoredox catalysts for the reductive activation of aryl halides, Eur. J. Org. Chem., 2018, 34 CrossRef CAS.
- (a) I. Ghosh and B. König, Chromoselective photocatalysis: controlled bond activation through light-color regulation of redox potentials, Angew. Chem., Int. Ed., 2016, 55, 7676 CrossRef CAS PubMed; (b) A. Das, I. Ghosh and B. König, Synthesis of pyrrolo[1,2-a]quinolines and ullazines by visible light mediated one- and twofold annulation of N-arylpyrroles with arylalkynes, Chem. Commun., 2016, 52, 8695 RSC; (c) L. Marzo, I. Ghosh, F. Esteban and B. König, Metal-free photocatalyzed cross coupling of bromoheteroarenes with pyrroles, ACS Catal., 2016, 6, 6780 CrossRef CAS; (d) R. S. Shaikh, S. J. S. Düsel and B. König, Visible-light photo-arbuzov reaction of aryl bromides and trialkyl phosphites yielding aryl phosphonates, ACS Catal., 2016, 6, 8410 CrossRef CAS; (e) A. Graml, I. Ghosh and B. König, Synthesis of arylated nucleobases by visible light photoredox catalysis, J. Org. Chem., 2017, 82, 3552 CrossRef CAS PubMed; (f) A. U. Meyer, T. Slanina, A. Heckel and B. König, Lanthanide ions coupled with photoinduced electron transfer generate strong reduction potentials from visible light, Chem. – Eur. J., 2017, 23, 7900 CrossRef CAS PubMed; (g) M. Haring, A. Abramov, K. Okumura, I. Ghosh, B. König, N. Yanai, N. Kimizuka and D. Díaz Díaz, Air-sensitive photoredox catalysis performed under aerobic conditions in gel networks, J. Org. Chem., 2018, 83, 7928 CrossRef PubMed; (h) J. M. Haimerl, I. Ghosh, B. König, J. M. Lupton and J. Vogelsang, Chemical photocatalysis with rhodamine 6g: investigation of photoreduction by simultaneous fluorescence correlation spectroscopy and fluorescence lifetime measurements, J. Phys. Chem. B, 2018, 122, 10728 CrossRef CAS PubMed; (i) F. Brandl, S. Bergwinkl, C. Allacher and B. Dick, Consecutive photoinduced electron transfer (conPET): the mechanism of the photocatalyst rhodamine-6g, Chem. – Eur. J., 2020 DOI:10.1002/chem.201905167.
- (a) J. Eriksen, H. Lund, A. I. Nyvad, T. Yamato, R. H. Mitchell, T. W. Dingle, R. V. Williams and R. Mahedevan, Electron-transfer fluorescence quenching of radical ions, Acta Chem. Scand., 1983, 37, 459 CrossRef; (b) M. Fujita, A. Ishida, T. Majima and S. Takamuku, Lifetimes of radical anions of dicyanoanthracene, phenazine, and anthraquinone in the excited state from the selective electron-transfer quenching, J. Phys. Chem., 1996, 100, 5382 CrossRef CAS; (c) T. Shida, Electronic Absorption Spectra of Radical Ions, Elsevier, Amsterdam, 1988, pp. 246 Search PubMed.
- M. Neumeier, D. Sampedro, M. Majek, V. A. de la Peña O'Shea, A. Jacobi von Wangelin and R. Pérez-Ruiz, Dichromatic photocatalytic substitutions of aryl halides with a small organic dye, Chem. – Eur. J., 2018, 24, 105 CrossRef CAS PubMed.
- G. Ciamician, Science, 1912, 36, 385 CrossRef CAS PubMed.
- D. Gust, T. A. Moore and A. L. Moore, Mimicking bacterial photosynthesis, Pure Appl. Chem., 1998, 70, 2189 CAS.
- S.E. Braslavsky, Glossary of terms used in photochemistry, 3rd edition (IUPAC Recommendations 2006), Pure Appl. Chem., 2007, 79(3), 293 CAS.
Footnotes |
| † More than one century ago, the Italian chemist G. Ciamician published a Science article (“The Photochemistry of the Future”) where he recognized that sunlight could be utilized as a promoter of organic reactions (see ref. 30). Inspired by the ability of plants to make use of solar energy, he was the first scientist to investigate photochemical reactions in a systematic way. His predictions on the advantages of utilizing solar energy to convert it into fuels have led to him being considered a pioneer of modern photochemistry (in 1998, this paper was cited for the first time in a JRC journal: see for instance ref. 31, source: SciFinder®). |
| ‡ Based on ref. 32: The biphotonic process is defined as “resulting from two-photon excitation”. The two-photon process is defined as “a photophysical or photochemical event triggered by a two-photon excitation”. And the two-photon excitation is defined as “excitation resulting from successive or simultaneous absorption of two photons by an atom or molecular entity. This term is used for successive absorption only if some of the excitation energy of the first photon remains in the atom or molecular entity before absorption of the second photon. The simultaneous two-photon absorption can also be called biphotonic excitation”. |
| This journal is © the Partner Organisations 2020 |