
Cross-dehydrogenative coupling involving benzylic and allylic C–H bonds
Irene
Bosque,  *a
Rafael
Chinchilla,
a
Jose C.
Gonzalez-Gomez,
a
David
Guijarro
a
and
Francisco
Alonso
*a
*a
Rafael
Chinchilla,
a
Jose C.
Gonzalez-Gomez,
a
David
Guijarro
a
and
Francisco
Alonso
*a
*a
Rafael
Chinchilla,
a
Jose C.
Gonzalez-Gomez,
a
David
Guijarro
a
and
Francisco
Alonso
*a
Abstract
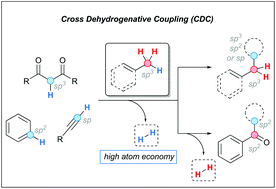
In recent years, the cross-dehydrogenative coupling (CDC) has demonstrated to be a powerful synthetic tool to form C–C bonds from two C–H bonds. This high atom-economic transformation is an attractive alternative to the classical substitution and coupling approaches for the benzylation and allylation of carbon centers, which rely on the reaction of adequately functionalized starting materials. This review emphasizes the utility of the CDC through the coupling of benzylic and allylic C–H bonds with C(sp)–H, C(sp2)–H and C(sp3)–H bonds.
 Please wait while we load your content...
Please wait while we load your content...

