
Photoredox-catalyzed indirect acyl radical generation from thioesters†
Alexander R.
Norman
,
Martina N.
Yousif
and
Christopher S. P.
McErlean
 *
*
School of Chemistry, University of Sydney, New South Wales 2006, Australia. E-mail: christopher.mcerlean@sydney.edu.au; Web: http://sydney.edu.au/science/chemistry/~mcerlean
First published on 5th October 2018
Abstract
A photoredox-catalyzed method for the indirect generation of acyl radicals from stable thioesters is described. The process is applicable to both aromatic and aliphatic substrates, and the resulting acyl radicals can undergo both intermolecular and intramolecular reactions. The mild reaction conditions allow for domino photoredox-catalyzed processes to occur. To illustrate the utility of the method, the total synthesis of a pharmaceutical agent is described.
Introduction
Acyl radicals have a long history as reactive intermediates in organic chemistry, however they are seldom utilised in modern synthesis.1–3 Traditionally, the generation of acyl radicals has relied upon the use of highly reactive acid derivatives, such as acyl chlorides, selenides and tellurides. Direct radical scission of the acyl–heteroatom bond in the presence of super-stoichiometric organo-tin reagents, or photolytically with high energy UV-light, reveals the desired acyl radical (Scheme 1A). The general incompatibility of these precursors with common reaction conditions, as well as the toxicity associated with both the substrates and reagents, greatly detracts from the appeal of these traditional methods and has spurred the development of alternative methods for acyl radical generation.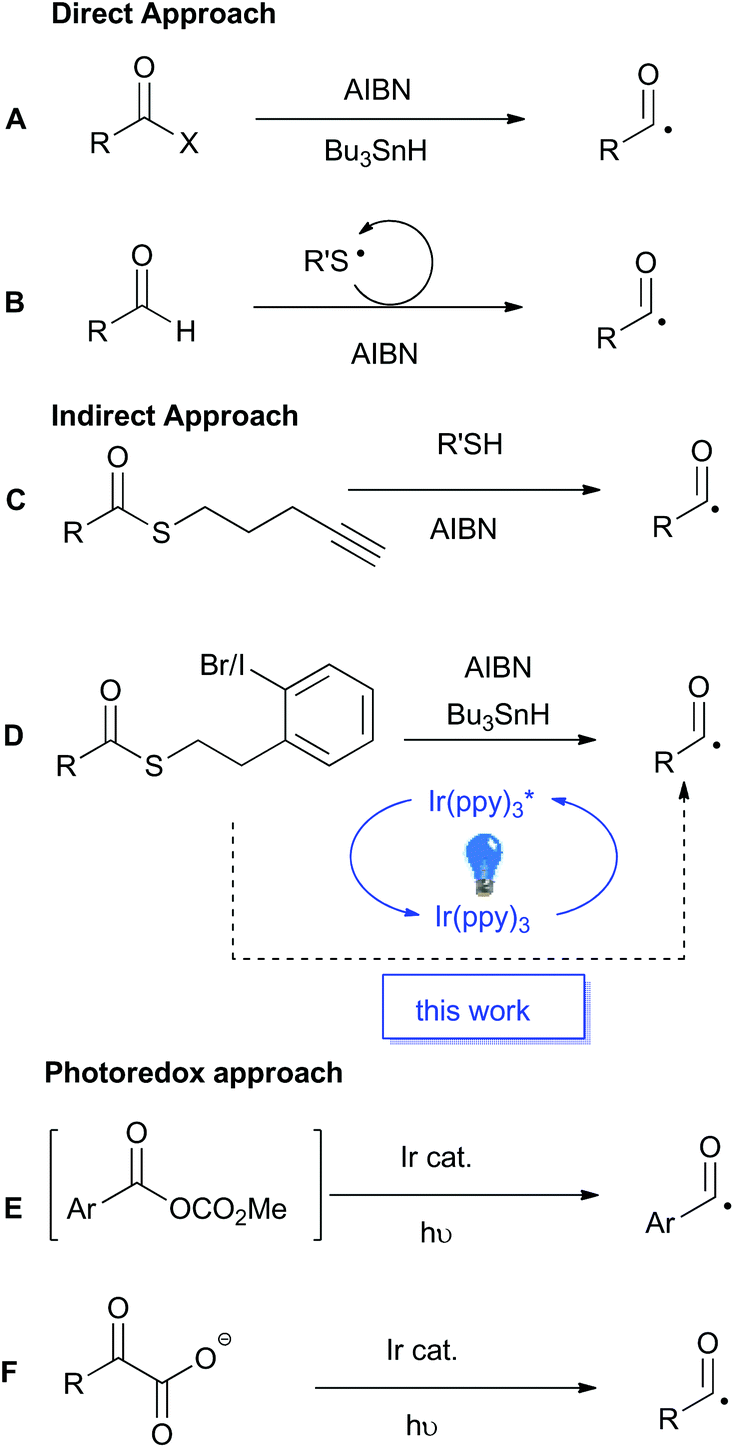 | ||
| Scheme 1 Previous approaches to acyl radical generation. | ||
As potent radicalophiles, sulfur-containing functional groups have played key roles in the development of alternative acyl radical generating methods. Tomioka and co-workers established an aerobic and thiol-catalysed protocol as a direct route from aldehyde precursors (Scheme 1B).4–6 In contrast, Benati, Spagnolo and co-workers described an efficient thiol–ene fragmentation reaction to liberate the desired acyl radical (Scheme 1C).7 This indirect approach, where the initial radical formation is distal to the acyl centre, permits the use of a more stable precursor, namely, a thioester. Similarly, Crich and co-workers were the first to disclose this indirect approach utilising a pendant aryl halide (Scheme 1D).8 Upon radical initiation, a stannyl radical abstracts the halogen to form an aryl radical which undergoes a rapid intramolecular cyclisation, releasing dihydrobenzothiophene and unveiling the desired acyl radical. Crich also reported a tin-free variant based on reduction of an aryl diazonium salt, but that reactive functional group is less well tolerated than the aryl halide.9
The principles of green chemistry that advocate for non-toxic reagents, catalytic processes, and energy minimisation,10 has driven the recent upsurge in development of photoredox catalyzed processes.11,12 In 2015 Wallentin and co-workers reported the photoredox catalyzed production of acyl radicals from in situ generated mixed carbonic anhydrides (Scheme 1E).13 The process worked well for aromatic substrates but was not viable for aliphatic substrates. In the same year MacMillan and co-workers reported the photoredox catalysed decarboxylation of α-keto acids as a highly efficient method for acyl radical generation (Scheme 1F).14 The protocol was applicable to both aromatic and aliphatic substrates, but the requisite α-keto acid precursors occur less frequently than ubiquitous carboxylic acids. MacMillan and co-workers recently reported a photocatalytic variant of Tomoika's method that utilized an aromatic thiol for the abstraction of the hydrogen atom from aldehydes.15 Surprisingly, the combination of sulfur-containing functional groups, such as thioesters, as latent acyl radicals with photoredox-catalysis has not yet been reported. As the Crich-type thioesters are available in a single step from carboxylic acid starting materials,8 we anticpated that the integration of those substrates into a mild photoredox catalyzed protocol would lead to a generally applicable and synthetically versatile method for acyl radical generation that would complement existing methods. The outcomes of our investigations into this integrated approach are reported below.
Optimization studies
Our studies began by identifying a suitable catalyst–thioester combination for the indirect transformation of haloaryl thioesters into the desired acyl radicals. The reported reduction potential of iodobenzene is −1.59 V in DMF (vs. SCE).16 The reduction potential of the photo-excited state of the commercially available fac-Ir(ppy)3 complex has been measured as −1.73 V (vs. SCE),17 which suggested that the iodoaryl thioester 1 (Table 1) should be reduced by that photocatalyst under the action of blue light. As shown in Table 1, this was indeed the case.|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entrya | Ir(ppy)3 loading (mol%) | Amine (equiv.) | Solventb | Irradiation time (h) | Conversion into 3c (%) | Conversion into 5c (%) | Unreacted 1c (%) |
| a Irradiated with a 4.5 W blue (465 nm) LED strip light. b Solvents were degassed. c Conversion by 1H NMR analysis of the crude reaction mixture. | |||||||
| 1 | 2.5 mol% | iPr2NEt (10) | CH3CN | 18 h | 40 | 6 | 54 |
| 2 | 2.5 mol% | NMM (10) | CH3CN | 18 h | 33 | 5 | 62 |
| 3 | 2.5 mol% | HCO2NH4 (10) | CH3CN | 18 h | 2 | — | 98 |
| 4 | 2.5 mol% | Hantzsch ester (2) | CH3CN | 18 h | — | — | 100 |
| 5 | 2.5 mol% | Bu3N (10) | CH3CN | 18 h | 80 | 20 | — |
| 6 | 2.5 mol% | Bu3N (10) | DMF | 18 h | 69 | 31 | Trace |
| 7 | 2.5 mol% | Bu3N (10) | MeOH | 18 h | 10 | — | 90 |
| 8 | 2.5 mol% | Bu3N (10) | PhMe | 18 h | <10 | — | >90 |
| 9 | 2.5 mol% | Bu3N (10) | CH2Cl2 | 18 h | 46 | 26 | 2–8 |
| 10 | 2.5 mol% | Bu3N (10) | THF | 18 h | 12 | — | 88 |
| 11 | 1 mol% | Bu3N (10) | CH3CN | 18 h | 78 | 22 | — |
| 12 | 0.1 mol% | Bu3N (10) | CH3CN | 18 h | 78 | 22 | — |
| 13 | 2.5 mol% | Bu3N (10) | CH3CN | 18 h | 78 | 22 | — |
| HCO2H (10) | |||||||
| 14 | 0 mol% | Bu3N (10) | CH3CN | 18 h | 0 | 0 | 100 |

A mixture of substrate 1, fac-Ir(ppy)3, and a series of amines were irradiated using a highly energy-efficient 4.5 W blue LED strip light. Reaction progress was monitored by 1H NMR analysis of the crude reaction mixture, with dihydrobenzothiophene (3) being diagnostic of acyl radical formation. In this way tributylamine was quickly identified as the most suitable sacrificial electron donor to complete the catalyst's redox cycle (entries 1–5).11,18 Polar aprotic solvents were well tolerated with the highest conversion being achieved in acetonitrile (entries 5–10). Given the expense of iridium-based catalysts, it was gratifying that the process suffered no loss of efficiency at catalyst loadings as low as 0.1 mol% (entries 11 and 12). And finally, the photoredox catalyzed nature of this process was confirmed by the lack of conversion in the absence of fac-Ir(ppy)3 (entry 14).
Under the reaction conditions, some proto-dehalogenated product 5 was observed. Beckwith and Boate have measured the rate constant for the formation of dihydrobenzothiophene (3) by intramolecular cyclisation of an aryl radical onto a sulfide to be 5 × 107 s−1 at 80 °C.19 Since Crich obtains uniformly high yields when using the tin-based reagents to generate the acyl radicals at 80 °C,20 we attribute the relative increase in proto-dehalogenation in this instance to the lower reaction temperature. Indeed, with a view to minimizing energy consumption, all of the photoredox catalyzed reactions in this work were performed at room temperature. Pleasingly, the presence of excess formic acid did not increase the amount of proto-dehalogenation (entry 13), even though it has previously been utilized as a hydrogen atom donor.21 We anticipated that the ability to perform the reaction in the presence of an acid would potentially facilitate further redox processes. The conditions shown in entry 13 were adopted for subsequent reactions.
Intramolecular cyclizations
With appropriate reaction conditions in hand, we began exploring the scope of the photoredox catalysed indirect acyl radical generation through a series of intramolecular cyclisation reactions. First, we explored the reaction of aromatic substrates to generate a series of chromanone and indanone derivatives 7b–18b (Scheme 2). Each of the allyl-containing aromatic substrates 7a–18a was generated by a N,N′-dicyclohexylcarbodiimide (DCC) mediated coupling of the carboxylic acid with the known thiol 6 (step 1) and was then subjected to the photoredox reaction conditions (step 2). The most apparent trend in reactivity correlates with the electron-withdrawing nature of the substituents on the aromatic ring. Compound 7a was smoothly transformed into the methylchromanone 7b in good yield. Likewise, the relatively electron-rich compounds 8a and 9a also cyclized in good yield to give chromanones 8b and 9b. In contrast, the relatively electron-poor substrate 10a was transformed into the bromochromanone 10b in low yield. The yield could be rescued to some degree by inclusion of an electron-donating group, as in compound 11a, which cyclized to give 11b in moderate overall yield. The highest yield was obtained for the most electron-rich substrate 12a, which cyclised to give the indanone 12b in 83% isolated yield. When substrates 13a–17a were subjected to the standard reaction conditions the desired chromanones 13b–17b were not isolated (see below). The identity of the heteroatom on the allyl tether could be altered to a nitrogen with no detrimental effect and compound 18a cyclized to give 18b in comparable yield to the oxygen analogue 7a.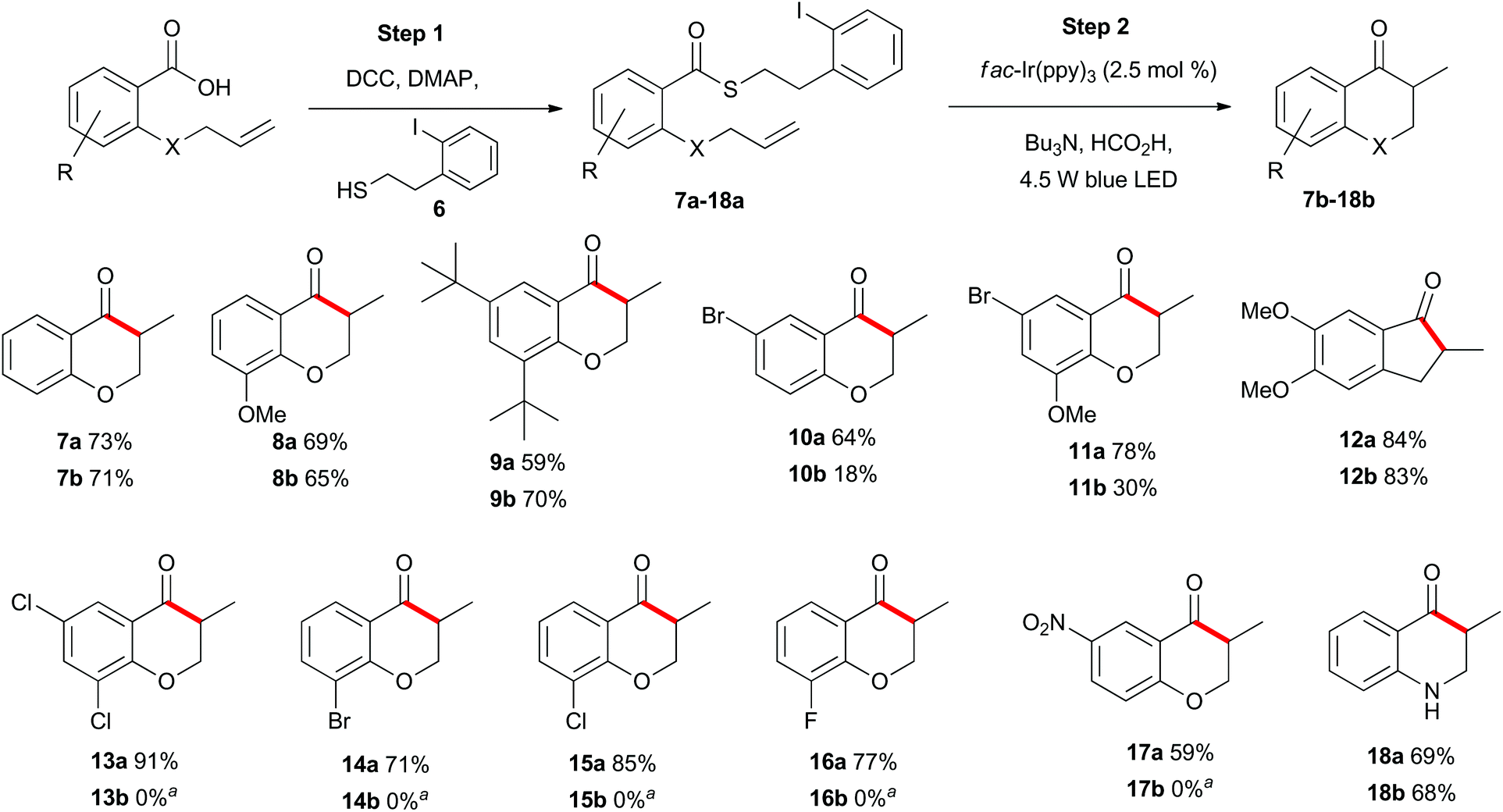 | ||
Scheme 2 Substrate scope of allyl-substituted aromatic compounds. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) See Schemes 6 and 11 for further details. See Schemes 6 and 11 for further details. | ||
Having examined the role of the substrate, we next investigated the scope of the alkene acceptor on aromatic substrates (Scheme 3). As already demonstrated, allyl units were well tolerated. The electron-deficient acrylate substrate 19a also cyclized to give 19b in good yield. The pendant cyclohexene unit of substrate 20a participated in the cyclization to give compound 20b. The propargyl containing substrate 21a was consumed under the standard reaction conditions, but surprisingly, product 21b could not be isolated from the reaction mixture. The highly electron-deficient styrenyl, malonyl, and vinylogous ester derivatives 22a, 23a and 24a proved to be incompatible with these standard reaction conditions (see below).
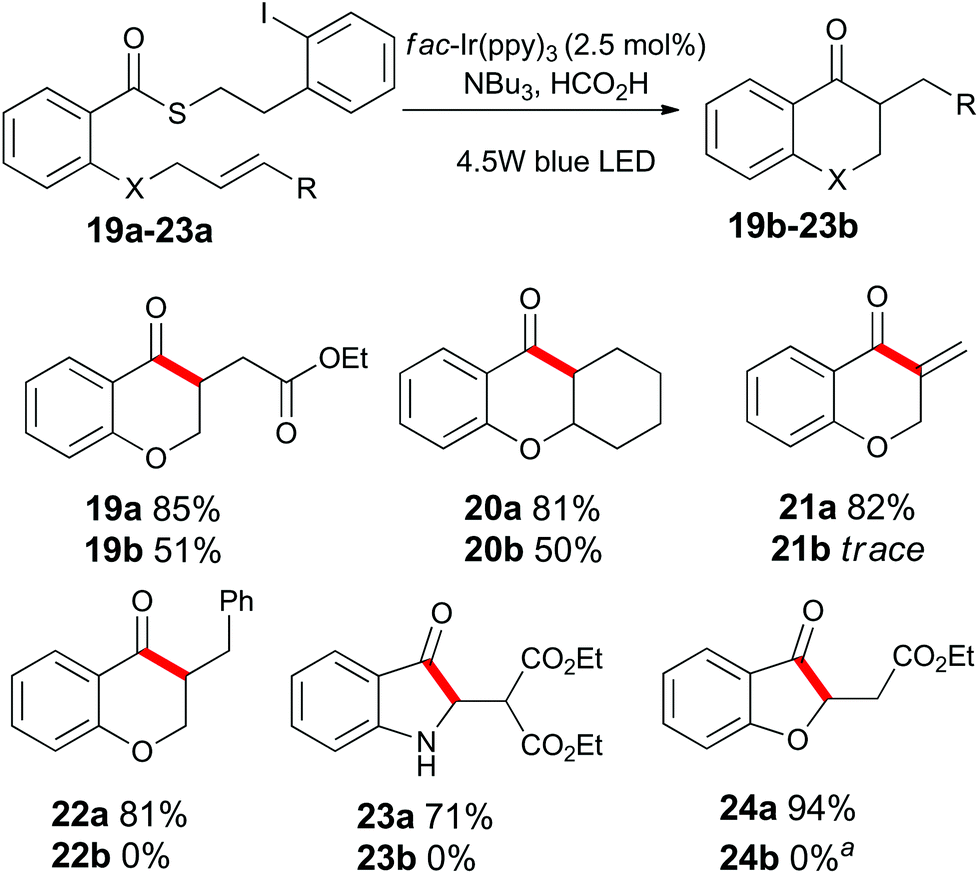 | ||
| Scheme 3 Substrate scope of alkene units on substituted aromatic compounds. aSee Scheme 7 for further details. | ||
As shown in Scheme 4, the heteroaromatic substrate 25a was transformed into the tricyclic compound 25b, and compound 26a which incorporated an all carbon tether and a vinylcyclopropyl unit, gave the expected ring-opened product 26b. This result served to confirm the radical nature of the process.
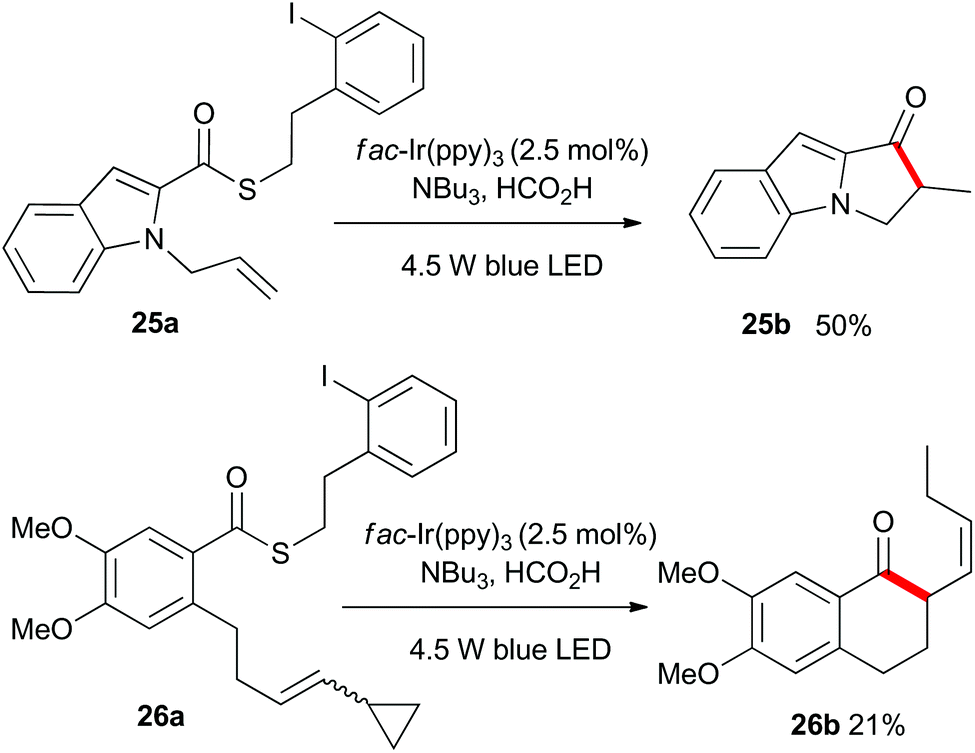 | ||
| Scheme 4 Heteroaromatic acyl radical cyclization and cyclization onto a vinylcyclopropyl unit. | ||
Finally, as shown in Scheme 5, we utilized substrate 27a which contains a latent aliphatic acyl radical and a tri-substituted alkene to further explore the scope of the reaction. Pleasingly, 27a cyclized quantitatively as monitored by 1H NMR analysis and the relatively volatile menthone 27b was isolated in 44% yield.
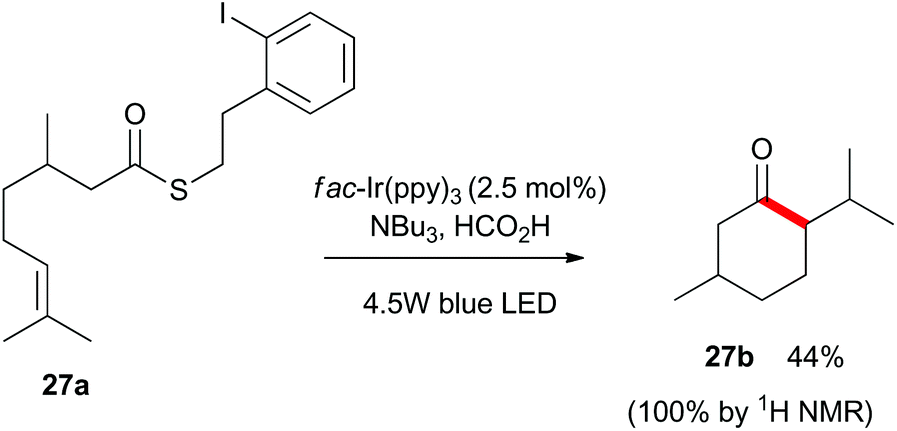 | ||
| Scheme 5 Aliphatic acyl radical cyclizing onto a tri-substituted alkene. | ||
Up to this point, the limitations on the photoredox-catalyzed indirect acyl radical generation correlated with electron deficient systems; whether it be electron deficient substrates such as 13a–17a or electron deficient acceptors 22a–24a. We rationalised this outcome on the basis of a preferential reaction between the photo-excited iridium catalyst and the electron-deficient unit. To overcome this shortcoming, we examined an alternative photo-induced method to reduce the aryl iodide and initiate the desired cascade. Minisci and coworkers utilized aryl radicals for the fast iodine abstraction from organic substrates containing relatively labile C(sp3)–I bonds.22 We examined the ability of several aryl radicals to abstract iodine from the less reactive C(sp2)–I bond of substrate 13a and initiate the indirect acyl radical generation sequence (Scheme 6). The required aryl radicals were generated from the corresponding diazonium salts 29a–29c using the Hantzsch ester (28) and ambient light. Li and Xu have previously reported the use of Hantzsch esters as mild photo-induced single electron reductants for radical processes.23,24 In this case, although conversions were modest (39% consumed starting material for the highest yielding example with 29a) the desired electron deficient chromanone 13b was isolated in a 53% yield based on recovered starting material. When compounds 13a, 17a and 29a were subjected to these reaction conditions, the previously inaccessible chromanones 14b and 17b were produced in moderate yields (Scheme 6). The corresponding tert-butyl Hantzsch ester (30) could be employed with equal efficiency. Similarly, when substrate 24a containing an electron-deficient alkene was subjected to these alternative photo-induced conditions, benzofuranone 24b was isolated in a 91% yield based on recovered starting material (Scheme 7).
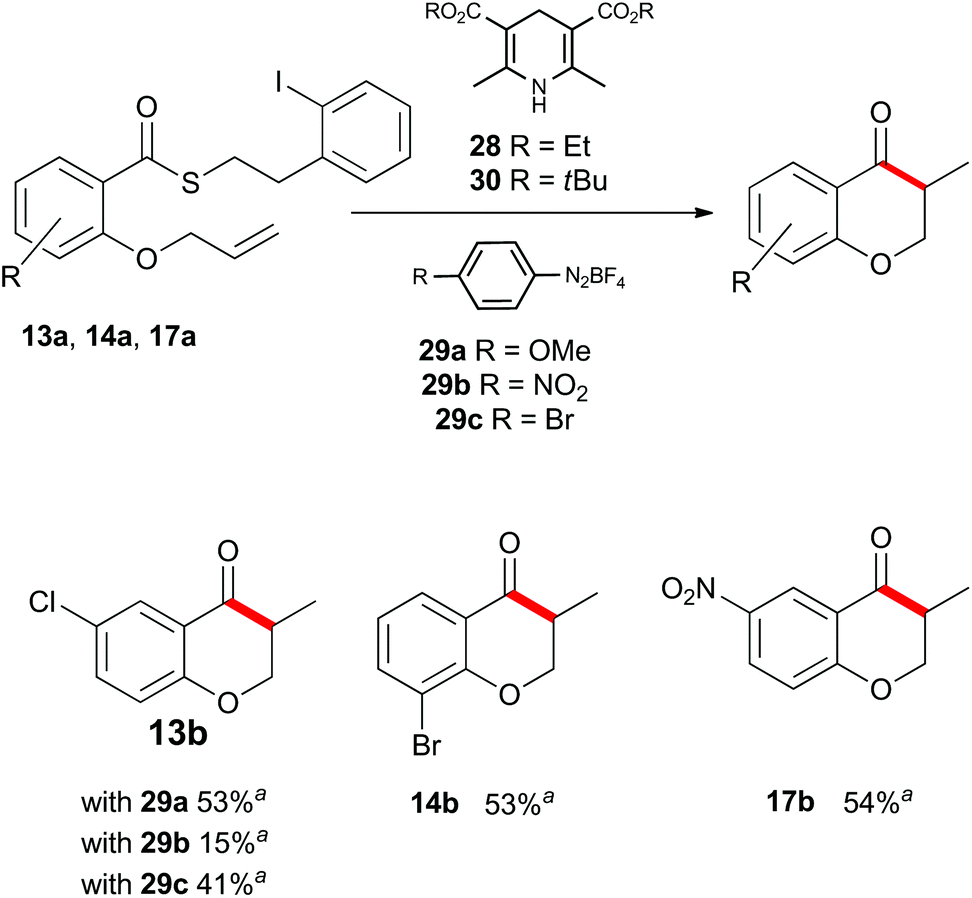 | ||
| Scheme 6 Indirect acyl radical generation and cyclization with electron-deficient substrates. aYield based on recovered starting material. | ||
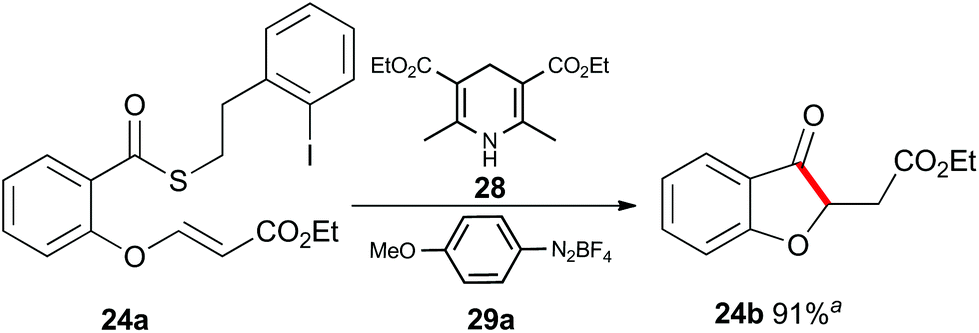 | ||
| Scheme 7 Indirect acyl radical generation and cyclization with electron-defficient alkenes. aYield based on recovered starting material. | ||
The use of the Hantzsch ester (28), a latent aryl radical (29a) and ambient light represents a complementary method that can be employed with electron deficient systems, albeit in a stoichiometric fashion.
Intermolecular additions
Having demonstrated that the photoredox-catalyzed indirect acyl radical generation and subsequent reaction onto alkenes was viable for a range of substrates in an intramolecular sense, we next explored the intermolecular variant. As shown in Scheme 8, a range of aliphatic and aromatic thioesters 33a–38a participated in the intermolecular alkene addition with an excess of cyclohexene (31) or allyltrimethylsilane (32). As expected the overall yields of the intermolecular process were lower than the corresponding intramolecular cyclizations, with reactions involving the allylsilane 32 being consistently higher yielding. Indeed, Guindon and co-workers employed allyltrimethylsilane (32) as a replacement for allyltributylstannane in the allylation of β-alkoxy esters.25 This reagent was reported to be superior due to both reduced toxicity and increased reactivity. That previous work proceeded via an atom transfer radical addition (ATRA) mechanism and the eliminated species was likely R3SiX. In the present case (Scheme 8), there is no possibility to form a silyl halide and so the silicon unit is retained in the products.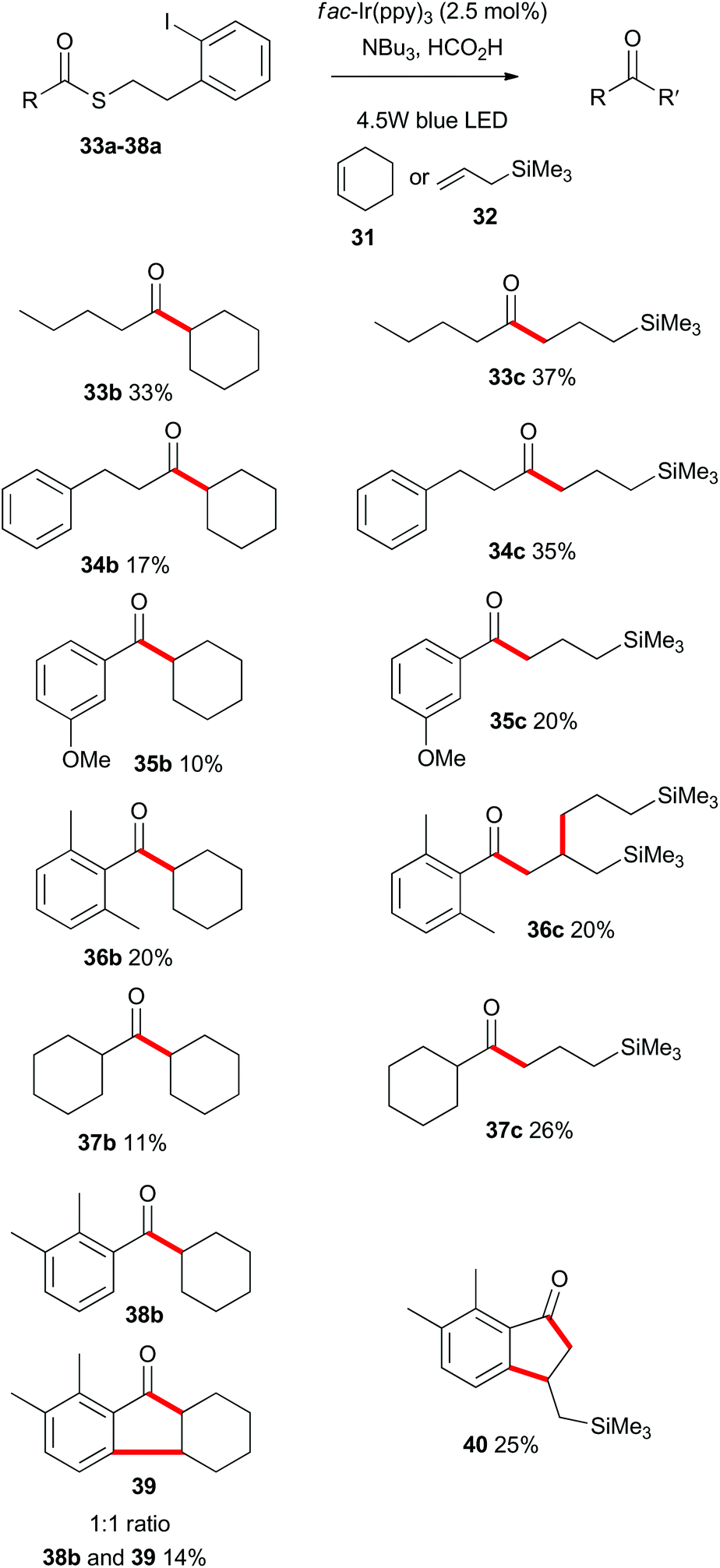 | ||
| Scheme 8 Scope of the intermolecular photoredox catalysed indirect acyl radical generation, alkene addition. | ||
Of particular interest are the reactions of substrates 36a and 38a. In those instances the radical intermediate generated by addition of the acyl radical onto an alkene, engaged in a secondary reaction. As such, 36a was transformed into 36c, and the ortho-substituted compound 38a was converted into hexahydrofluorenone 39 and indanone 40. These processes in which multiple bonds were formed under the same exceedingly mild photoredox-catalyzed reaction conditions, prompted us to explore other cascade processes.
Cascade reactions
To this point, we have shown that acyl radicals can be generated from aryl and alkyl thioesters in an indirect fashion under photoredox conditions, and that they undergo addition reactions to olefins in both an intra- and intermolecular fashion. The products of those radical addition reactions subsequently underwent hydrogen atom transfer (HAT) from the oxidized tributylaminiumyl radical to give a methyl or methylene group, as shown in the plausible mechanism depicted in Scheme 9. We next explored the opportunity to engage the intermediate radical (A) in a subsequent alkene addition reaction to give an intra–intermolecular cascade process.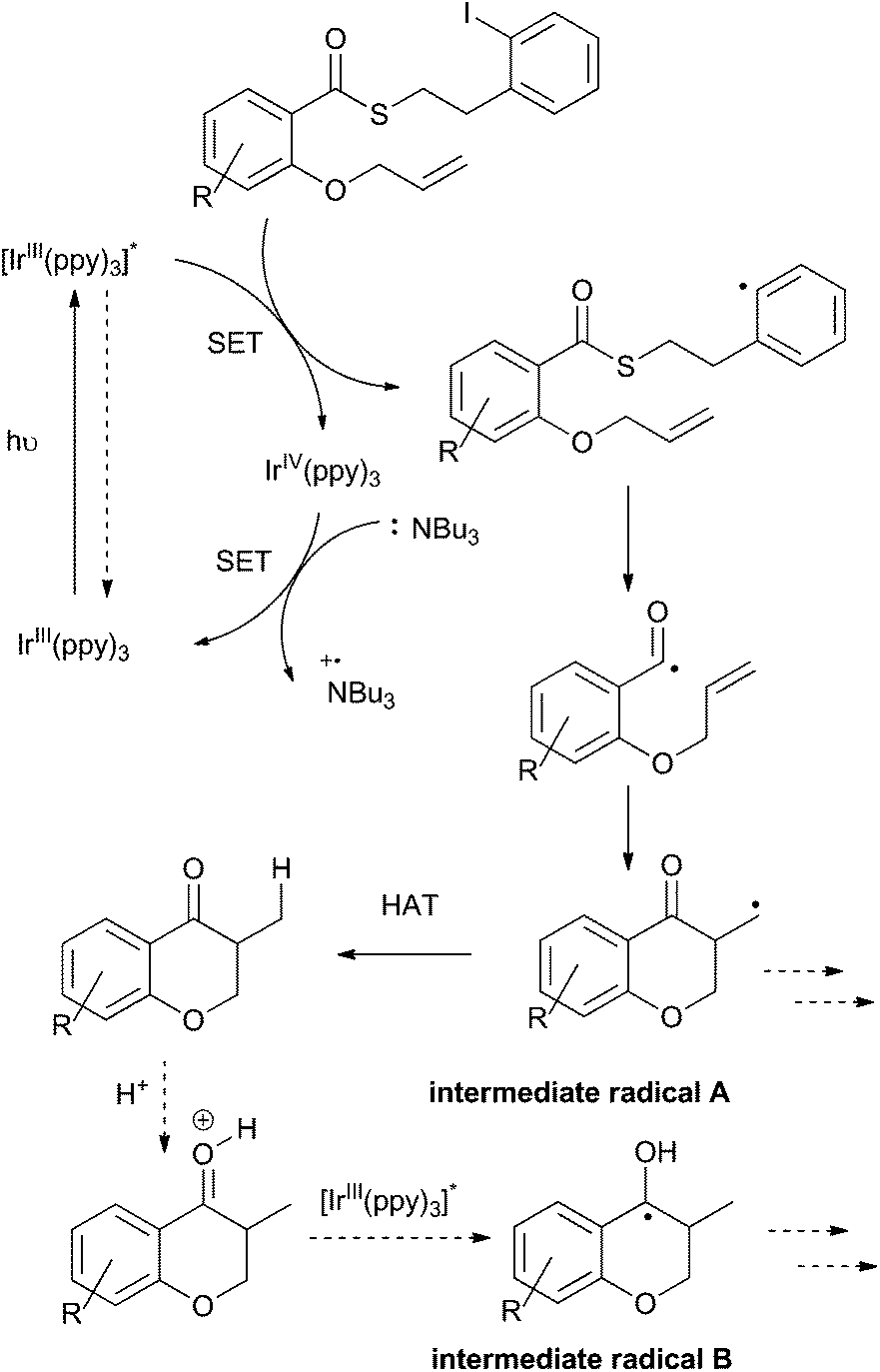 | ||
| Scheme 9 Plausible mechanism for the photoredox process. | ||
As shown in Scheme 10, irradiation of thioester 7a under the standard reaction conditions in the presence of excess allyltrimethylsilane (32) resulted in isolation of the chain-extended chromanone 41. We then attempted to perform a cascade radical allylation. Zard and co-workers developed the allyl transfer agent 42 for the production of allylated products under radical conditions.26,27 The fluoropyridyl unit of intermediate 43 is a potent radical acceptor and is reported to undergo facile β-scission under refluxing ethyl acetate or classical radical conditions to give the corresponding alkene 44 and pyridone 45. However, under the exceedingly mild reaction conditions employed for the photoredox-catalyzed process, no β-scission was observed and compound 46 was isolated in an almost 1:1 mixture with the double alkene addition product 47.
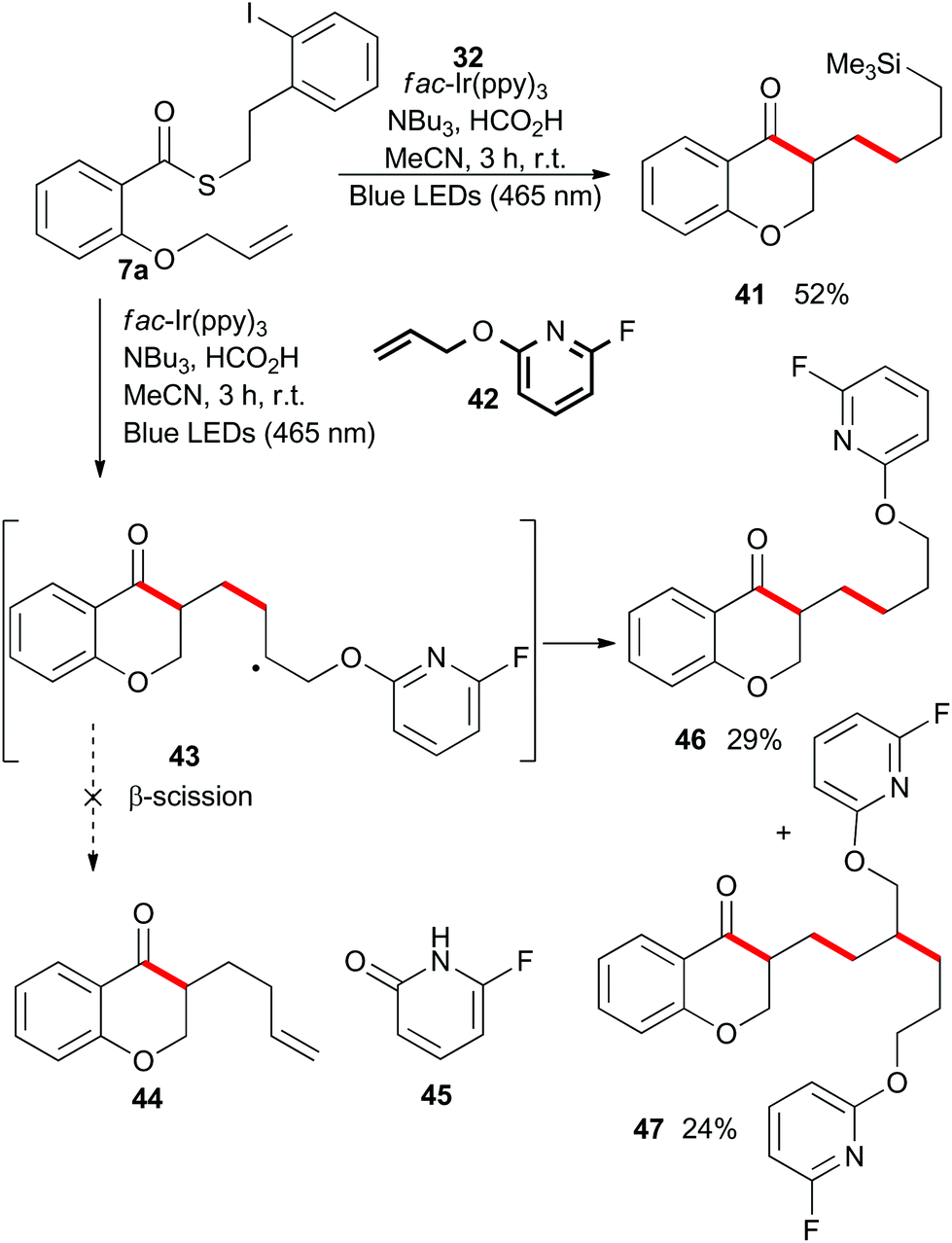 | ||
| Scheme 10 Intermolecular–intramolecular radical cascade reactions. | ||
Rueping and co-workers have previously shown that acetophenones undergo photoredox catalysed dimerisations in an acid-dependent manner.28 Our ability to conduct the photoredox-catalyzed indirect radical generation in the presence of formic acid suggested that product chromanones could be employed in a cascade reaction (Scheme 9).29–32 The reduction potential of unsubstituted chromanone has been measured at −1.9 V (vs. SCE),33,34 which lies outside the range of the photo-excited fac-Ir(ppy)3 complex. However, in the presence of formic acid, protonation would result in a significant change in redox behaviour.35,36 Reduction of the protonated species would give a benzylic radical (intermediate radical B) that could engage in a radical–radical dimerization reaction (colligation).
Gratifyingly, when the thioesters 7a, 15a, 16a and 20a were subjected to the standard reaction conditions, the dimeric products 7c, 15c, 16c, and 20c were isolated in good overall yield as a mixture of meso and racemic diastereomers (Scheme 11). The dimerization of the 3-halo thioesters (15a and 16a) was particularly facile. The ease of single electron reduction of protonated chromanones bearing electron withdrawing substituents provides an explanation for the intial troubles isolating certain cyclisation products described in Scheme 2.
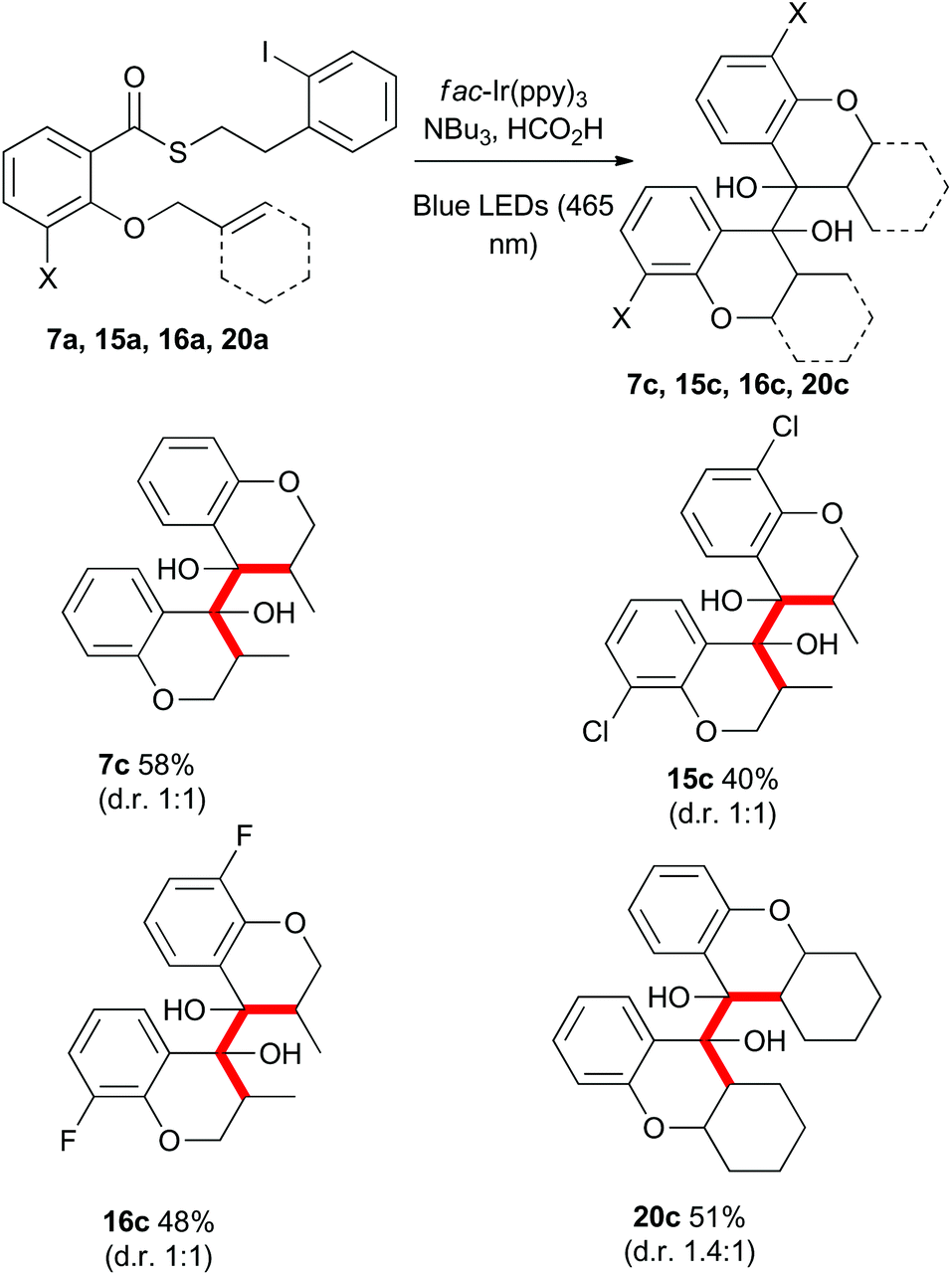 | ||
| Scheme 11 Radical cyclization–dimerization cascade reactions. | ||
Mechanistically, the formation of the dimeric compounds could occur either by colligation,37 or by addition of an initially formed radical onto an activated carbonyl unit. In situ1H NMR monitoring of the reaction showed that the starting materials were converted into the chromanone products before the dimeric species began to appear (Fig. 1), with no cross-reactivity between benzyl radicals and thioesters being observed. To further delineate the reaction pathway, two electronically different ketones were added to the mixture (Scheme 12). Acetophenone (48) is known to be reduced under photoredox conditions in an acid dependent manner,28 but pentanone (49) is not reduced. If the reaction proceeds by radical addition to carbonyl, then both compounds would participate in the reaction and mixed pinacol-type products would be observed. In the event, pinacol 50 was observed in the reaction conducted in the presence of acetophenone (48). In contrast, when the reaction was conducted in the presence of pentanone (49), only the dimeric compound 7c was observed. This strongly suggests that the dimerization proceeds via a colligation pathway.
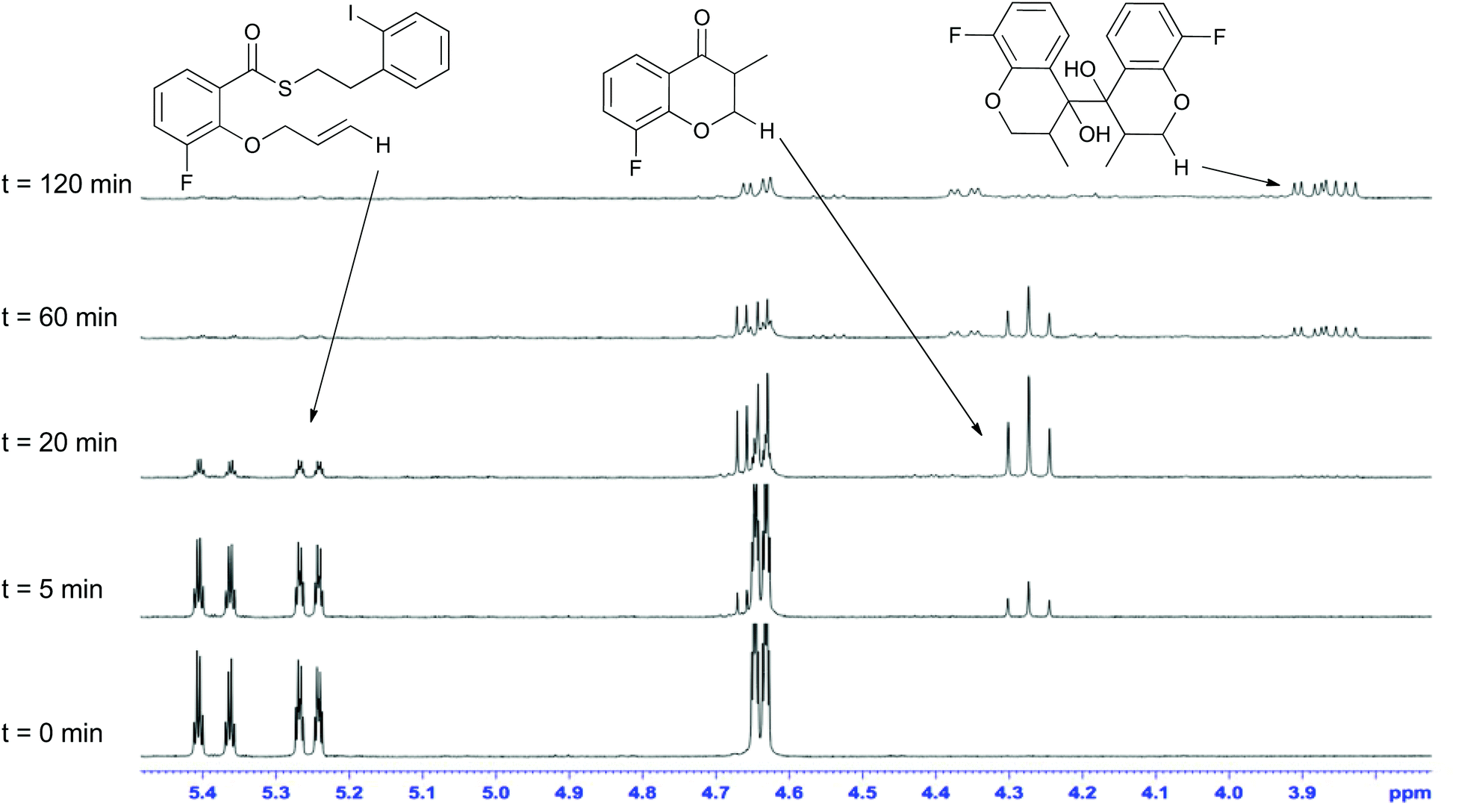 | ||
| Fig. 1 In situ 1H NMR analysis of the dimerization reaction mixture. | ||
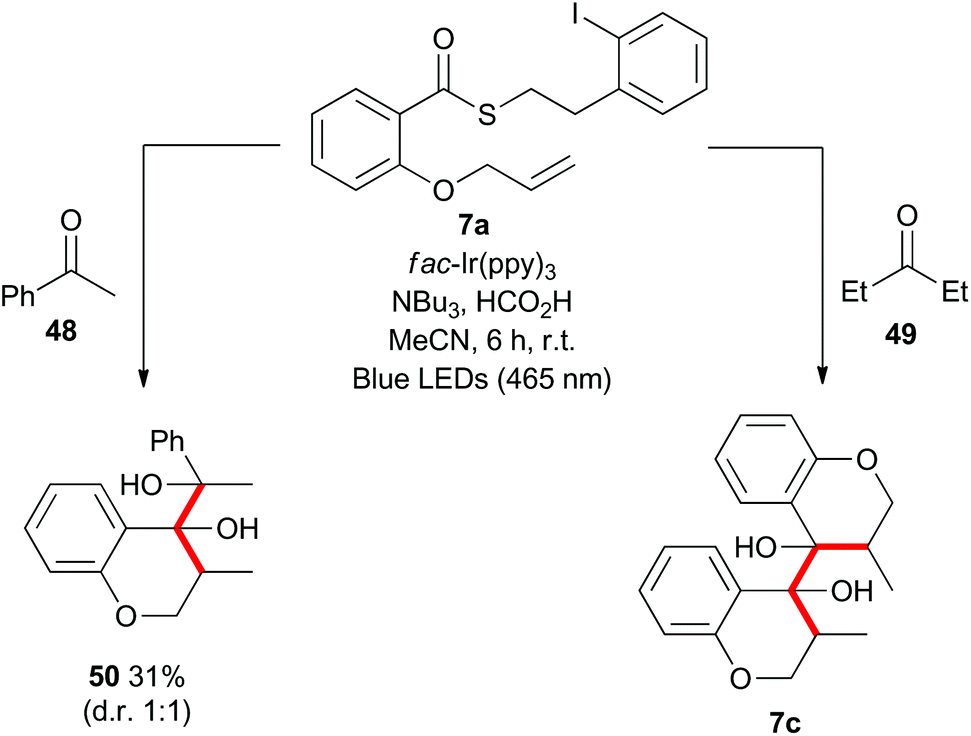 | ||
| Scheme 12 Radical cyclization–dimerization with different ketones. | ||
Benzylically stabilized radicals such as intermediate B in Scheme 9 are known to be resistant to alkene addition reactions. Indeed, when pre-formed chromanone 7b was subjected to the acidic photoredox reaction conditions in the presence of an excess of either allylmethoxyphenol 51 or allyltrimethylsilane 32, the only observed product was the dimer 7c (Scheme 13). We attempted to overcome the barrier to intermolecular radical addition by conducting the reaction in an intramolecular fashion (Scheme 14). As such, the alkene containing chromanone 53b was subjected to the acidic photoredox reaction conditions. Spectacularly, the desired tricyclic compound 54 was not observed, but rather the dimeric compound 53c was isolated in good yield.38
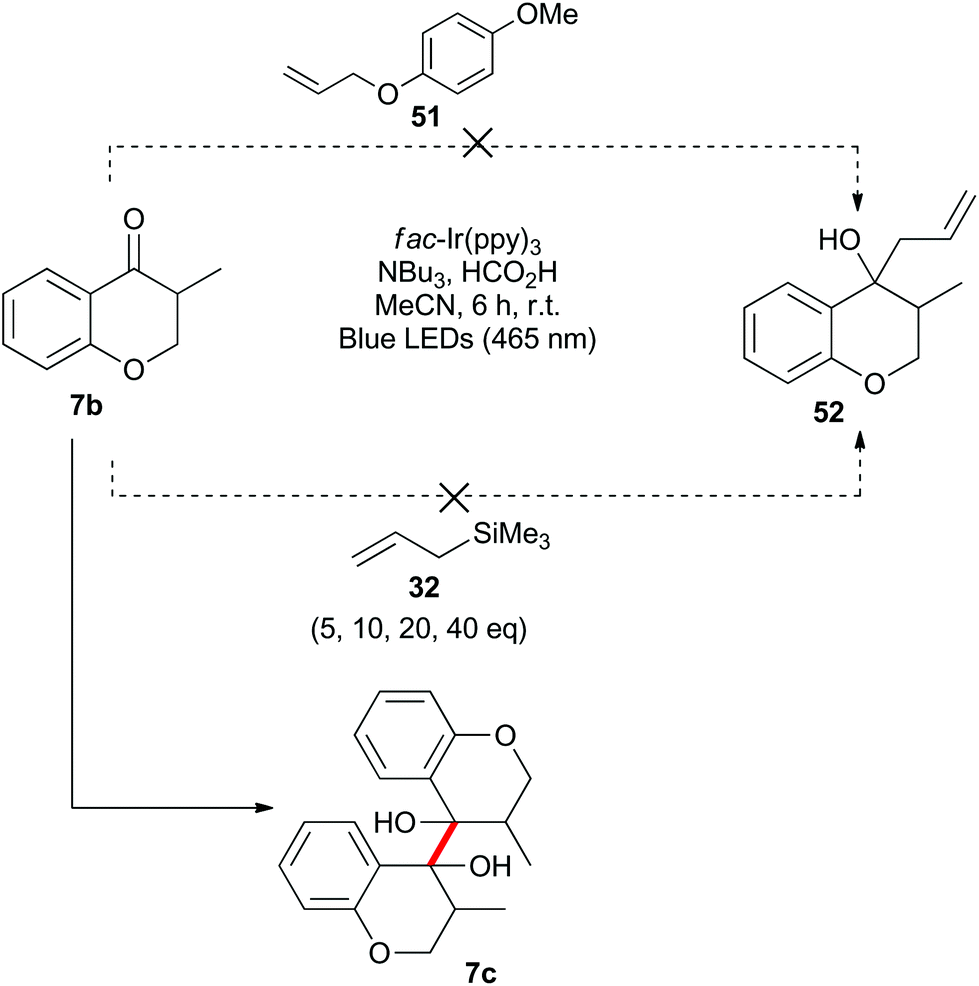 | ||
| Scheme 13 Attempted intermolecular benzylic radical addition reactions. | ||
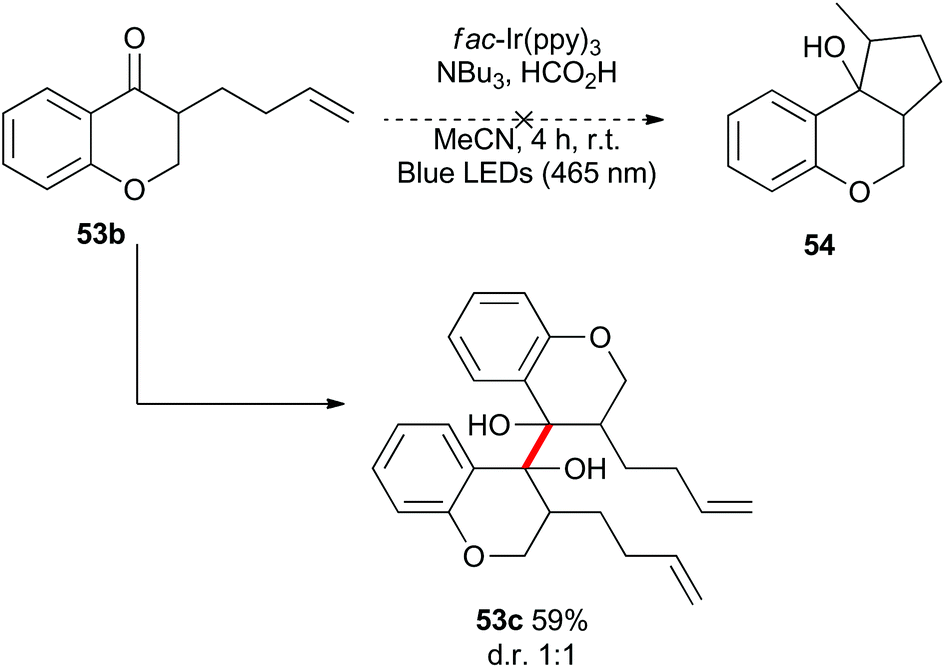 | ||
| Scheme 14 Attempted intramolecular benzylic radical addition reaction. | ||
Synthetic compatibility
The examples described above demonstrate that the iodoarene thioester can be easily generated from a range of carboxylic acids and that it can be chemoselectively engaged by the application of photoredox reaction conditions. For this indirect acyl radical generation to be generally applicable, the thioester functionality must tolerate a wide range of reaction conditions. Intuitively, we can expect that the thioester unit will be incompatible with strongly basic conditions, but the window of tolerance has not been well defined. As such, we subjected compound 55 to a range of reactions involving basic and nucleophilic reagents. As shown in Scheme 15, compound 55 was converted into acetal 56 by the action of catalytic Brønsted acid in an excess of diol. No esterification was observed. The thioester unit was similarly compatible with the Lewis acid TiCl4, and underwent Mukayami aldol reaction with silyl ether 57 to give compound 58. The thioester was also stable in the presence of the mildly basic phosphorane 59, during the transformation of 55 into 60. And finally, the thioester remained intact after exposure to aniline 61 and a hydride source, during the conversion of 55 into 62. Together, these results instilled confidence that the iodoarene thioester could be carried through a practicable synthetic sequence before undergoing reaction in a chemoselective fashion.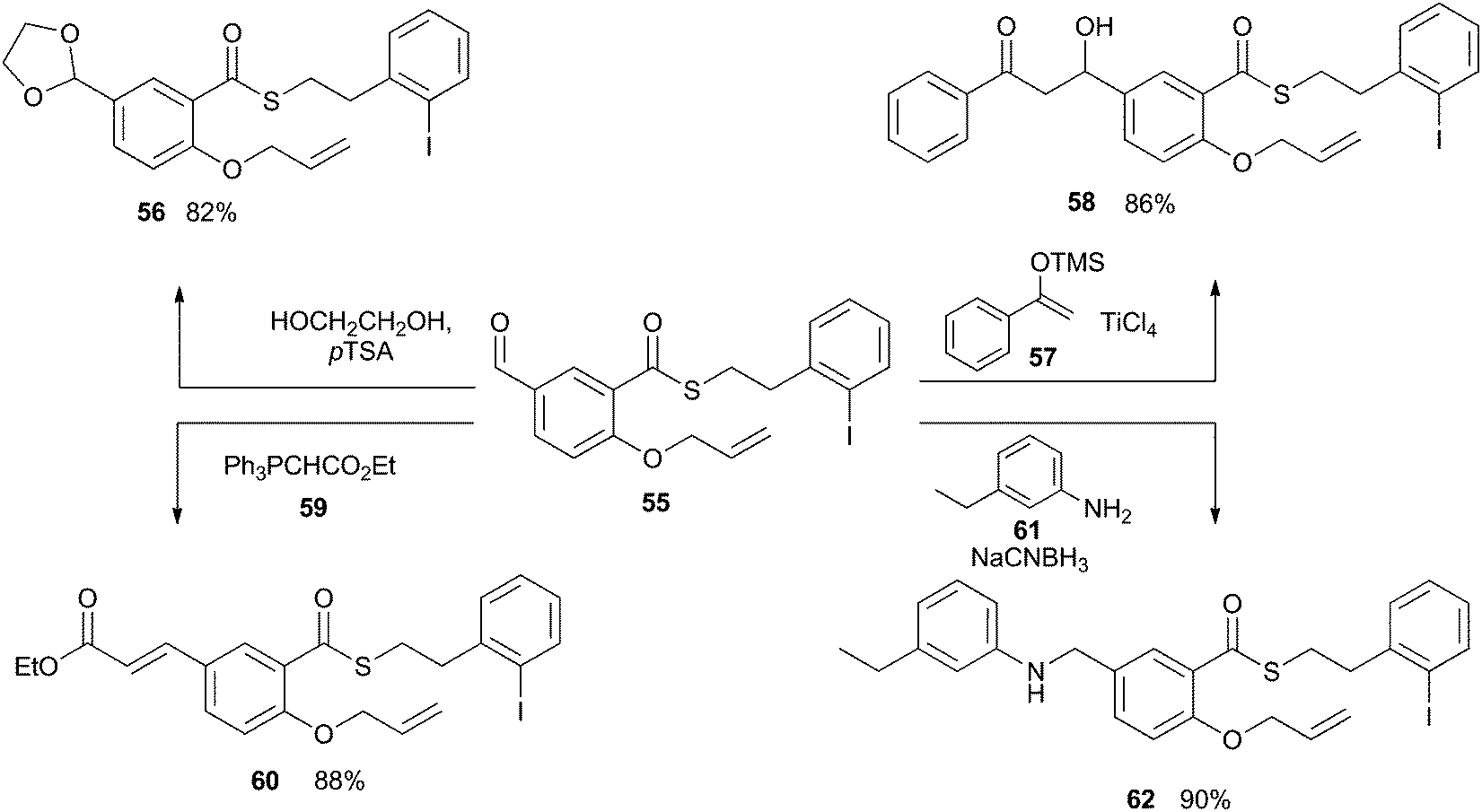 | ||
| Scheme 15 Synthetic compatibility of the thioester unit with common reaction conditions. | ||
Synthetic application
To highlight the synthetic potential of the indirect generation of acyl radicals under photoredox-catalyzed conditions, we undertook the synthesis of clinical agent donepezil (63) (Fig. 2). Donepezil (63) was disclosed by Sugimoto and co-workers in 1992 and approved by the FDA in 1996 for the treatment of mild–severe dementia associated with Alzheimer's disease.39 As a reversible inhibitior of acetylcholinesterase, donepezil (63) acts by increasing the concentration of acetylcholine in the central nervous system, thereby enhancing cholinergic function. Strictly a palliative treatment for the cognitive symptoms of the disease, donepezil (63) neither prevents nor slows the neurodegeneration associated with the Alzheimer's disease. Excluding the patent literature, there have been 11 previous syntheses of donepezil (63).39–49 As depicted in Fig. 2, most approaches follow Sugimoto's original strategy and utilize an indanone alkylation to unite the two halves of the molecule.39 The exceptions are Fillon's late-stage Freidel–Crafts alkylation,46 Rao's late stage oxidation,41 and Stambuli's palladium-catalyzed cascade construction of both the indanone and pyrrolidine connections.44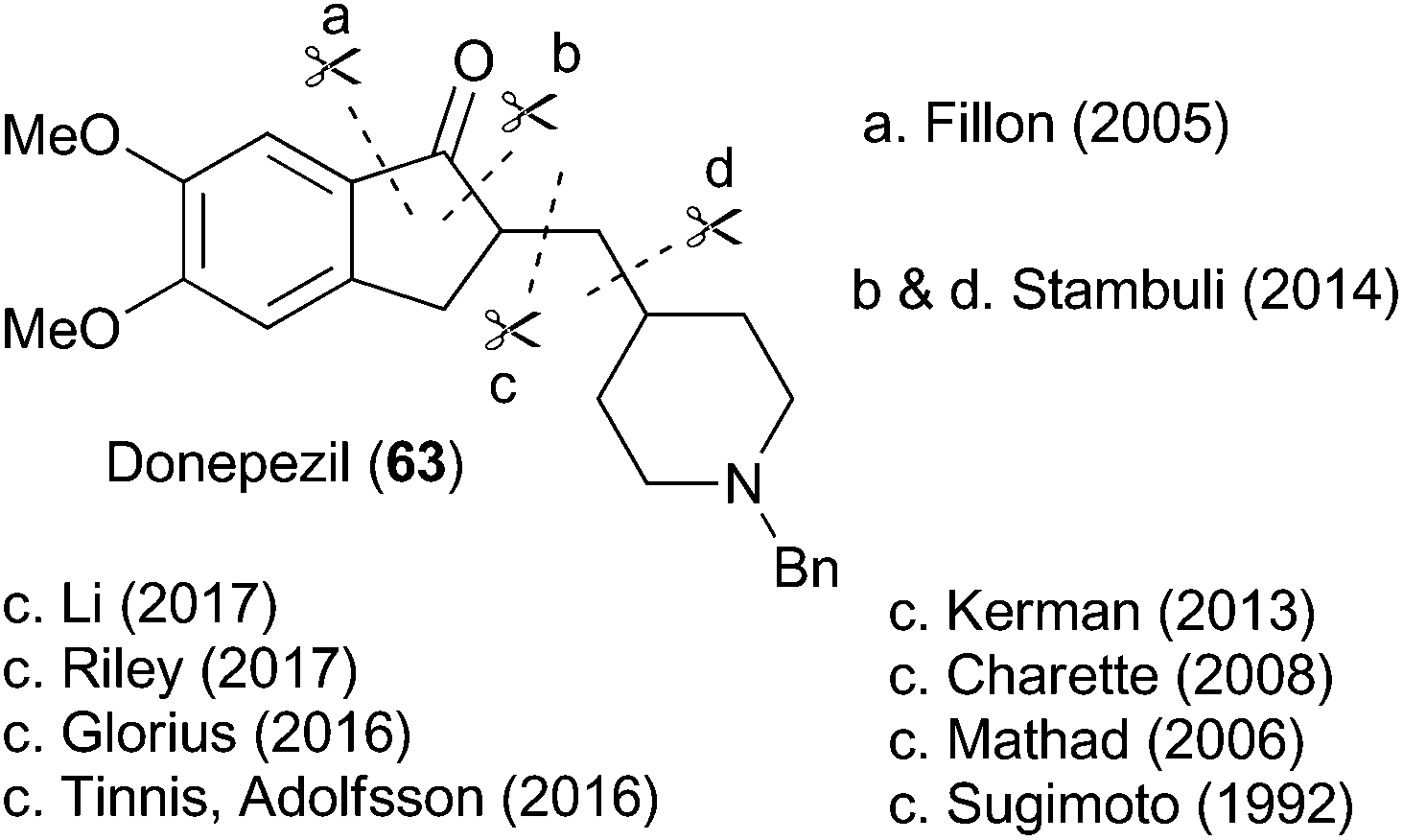 | ||
| Fig. 2 Previous approaches to Donepezil. | ||
Our synthesis is shown in Scheme 16. The commercially available iodobenzoic acid 64 was deprotonated and then subjected to a copper-mediated allylation to give 65. DCC-mediated coupling of 65 with thiol 6 installed the requisite thioester 12a. Separately, the commercially available pyrrolidinyl alcohol 66 was iodinated and subjected to an elimination reaction to give the vinyl pyrrolidine 68. Cross metathesis using Grubbs’ first generation catalyst united fragments 12a and 68 and delivered the cyclisation precursor 69. It is noteworthy that the thioester was stable under the metathesis conditions, providing further illustration of its synthetic utility. The key photoredox-catalyzed acyl radical formation and intramolecular alkene addition was performed in the standard way and delivered compound 70 in 51% yield. The Boc unit was replaced by a benzyl group to complete the 5 step synthesis of donepezil (63).
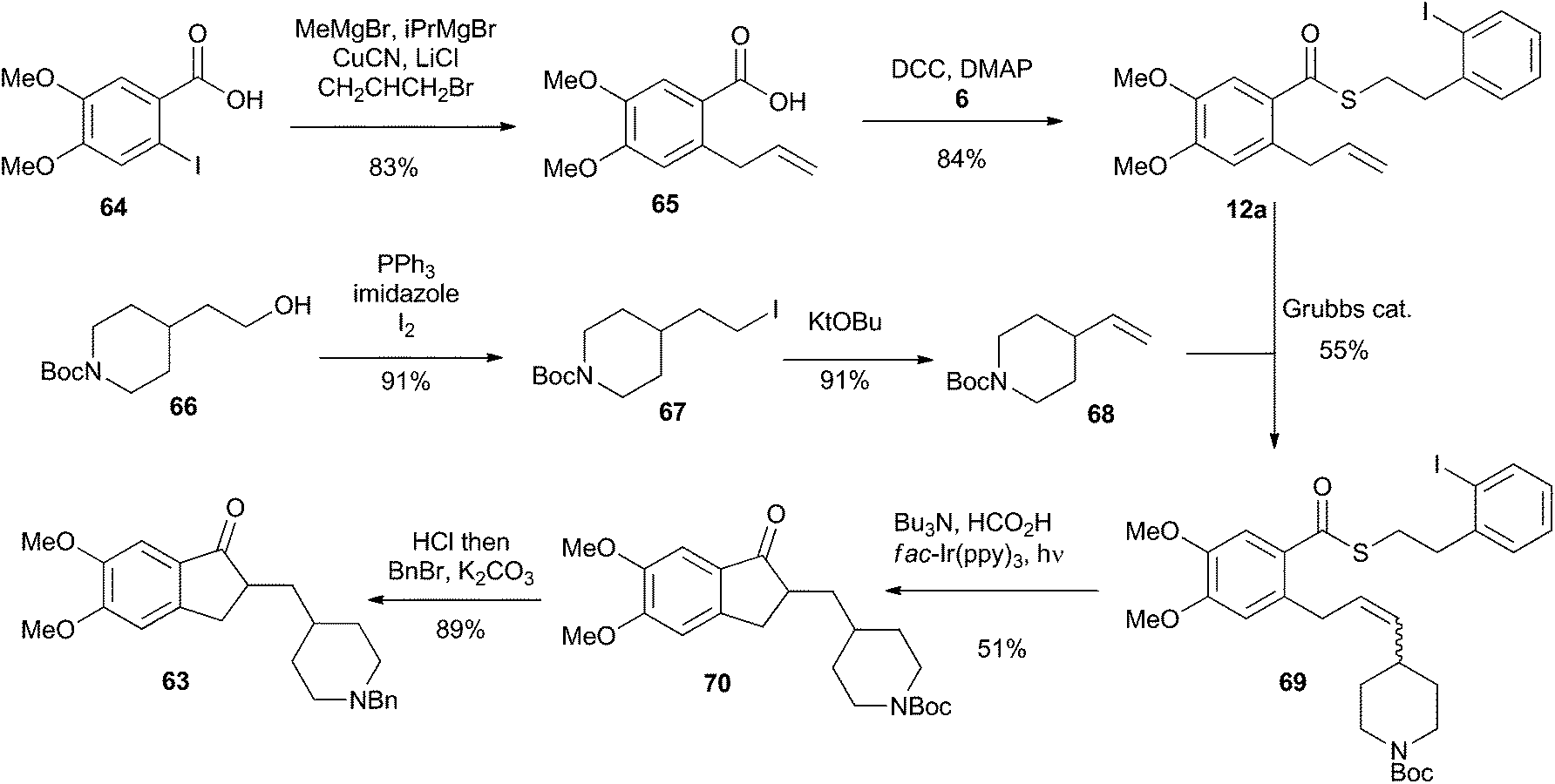 | ||
| Scheme 16 Synthesis of Donepezil utilizing an indirect acyl radical generation and intramolecular cyclization reaction. | ||
Conclusions
Acyl radicals are reactive intermediates infrequently employed in organic synthesis. In an effort to increase their utility and generality, we have developed a photoredox-catalyzed process to access these useful intermediates in an indirect manner from thioesters. Ubiquitous carboxylic acids can be converted into relatively robust Crich-type thioesters in a single step. These aryliodide containing thioesters are stable to many common reaction conditions, including moderately basic and nucleophilic conditions. The latent acyl radical can be revealed at room temperature using the commercially available fac-Ir(ppy)3 complex and low energy blue light. Altering the photocatalyst system to an aryldiazonium salt and the Hantzsch ester enables substrates with otherwise competing reactivity to be employed in the process. The liberated acyl radical can undergo reaction onto a variety of electronically and sterically different alkenes in both an inter- and intramolecular fashion. These include chain elongation with alkenes containing functional groups capable of β-scission reactions (no β-scission was observed), and pinacol couplings for the generation of dimeric and cross-coupled diols.The photoredox-catalyzed indirect acyl radical generation has been employed for the total synthesis of the clinically used pharmaceutical, donepezil. Application of the described procedure enabled a strategically new disconnection to be envisaged for this popular synthetic target. And importantly, the short synthesis of donepezil demonstrates that this methodology is applicable to target-based synthesis.
In conclusion, we report a mild method for the generation of acyl radicals that is complementary to existing methods. The scope of the protocol both in terms of subtrates that can be employed and reactions that can be conducted, gives us confidence that this new protocol will help facilitate the increased use of acyl radicals for key bond formation during target-based synthesis.
Experimental section
General experimental
All reactions were performed under an inert atmosphere (nitrogen or argon) in oven dried glassware, unless otherwise stated. Toluene, acetonitrile, methanol, diethyl ether, dichloromethane, tetrahydrofuran and dimethylformamide were purified using an Innovative Technology, Inc., PureSolv™ solvent purification system. Tributylamine was dried over potassium hydroxide then fractionally distilled. All other solvents and reagents were used as received from commercial sources.Melting points were determined using a Stanford Research Systems Optimelt automated melting point system and are uncorrected. Infrared spectra were acquired on a Bruker ALPHA FT-IR as thin films or neat. 1H and 13C NMR spectra were recorded in deuterochloroform on a Bruker AVANCE III 500, a Bruker AVANCE III 400, a Bruker AVANCE 300, or a Bruker AVANCE 200 spectrometer (1H frequencies 500, 400, 300, 200 MHz; 13C frequencies 125, 100, 75 and 50 MHz respectively). 1H chemical shifts are expressed as parts per million (ppm) with residual chloroform (δ 7.26) as an internal reference and are reported as chemical shift (δH); relative integral; multiplicity (s = singlet, br = broad, d = doublet, t = triplet, dd = doublet of doublets, ddd = doublet of doublet of doublets, m = multiplet); and coupling constants (J) reported in Hz. 13C NMR chemical shifts are expressed as parts per million (ppm) with residual chloroform (δ 77.16) as internal reference and are reported as chemical shift (δC); multiplicity (assigned from DEPT or HSQC experiments). High resolution mass spectra were recorded on a Bruker Apex II Fourier Transform Ion Cyclotron Resonance mass spectrometer with a 7.0 T magnet, fitted with an off-axis Analytica electrospray source. Column chromatography was performed using 40–60 μm (230–400 mesh) silica gel using commercial solvents. Analytical thin layer chromatography was performed using preconditioned plates (Merck TLC silica gel 60 F254 on aluminium) and visualised using UV light (254 nm and 365 nm), ethanolic anisaldehyde, vanillin or potassium permanganate solution.
Experimental procedures
![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) ), 7.30–7.25 (2 H, m, 2 × Ar–), 6.91 (1 H, ddd, J = 7.9, 6.8, 2.3 Hz, Ar–), 3.85 (2 H, t, J = 6.8 Hz, C2OH), 3.01 (2 H, t, J = 6.8 Hz, C2Ar), 1.64 (1 H, brs, O); 13C NMR (126 MHz, CDCl3) δ: 141.1 (C), 139.7 (CH), 130.3 (CH), 128.4 (CH), 128.3 (CH), 100.8 (C), 62.3 (CH2), 43.7 (CH2); LRMS (APCI) m/z 385 (67), 357 (67), 231 ([M − OH]+, 100).
), 7.30–7.23 (2 H, m, 2 × Ar–), 6.90 (1 H, ddd, J = 7.9, 6.60, 2.4 Hz, Ar–), 3.12–3.08 (2 H, m, SC2), 2.99–2.95 (2 H, m, ArC2), 2.33 (3 H, s, C3); 13C NMR (101 MHz, CDCl3) δ: 195.6 (C), 142.6 (C), 139.7 (CH), 130.0 (CH), 128.5 (2 × CH), 100.5 (C), 40.5 (CH2), 30.7 (CH3), 29.2 (CH2); νmax/cm−1 3055, 2926, 1689, 1465, 1436, 1132, 1105, 1047; LRMS (ESI+) m/z 645 (37), 385 (47), 329 ([M + Na]+, 100).
), 7.30–7.25 (2 H, m, 2 × Ar–), 6.91 (1 H, ddd, J = 7.9, 6.8, 2.3 Hz, Ar–), 3.85 (2 H, t, J = 6.8 Hz, C2OH), 3.01 (2 H, t, J = 6.8 Hz, C2Ar), 1.64 (1 H, brs, O); 13C NMR (126 MHz, CDCl3) δ: 141.1 (C), 139.7 (CH), 130.3 (CH), 128.4 (CH), 128.3 (CH), 100.8 (C), 62.3 (CH2), 43.7 (CH2); LRMS (APCI) m/z 385 (67), 357 (67), 231 ([M − OH]+, 100).
), 7.30–7.23 (2 H, m, 2 × Ar–), 6.90 (1 H, ddd, J = 7.9, 6.60, 2.4 Hz, Ar–), 3.12–3.08 (2 H, m, SC2), 2.99–2.95 (2 H, m, ArC2), 2.33 (3 H, s, C3); 13C NMR (101 MHz, CDCl3) δ: 195.6 (C), 142.6 (C), 139.7 (CH), 130.0 (CH), 128.5 (2 × CH), 100.5 (C), 40.5 (CH2), 30.7 (CH3), 29.2 (CH2); νmax/cm−1 3055, 2926, 1689, 1465, 1436, 1132, 1105, 1047; LRMS (ESI+) m/z 645 (37), 385 (47), 329 ([M + Na]+, 100).
Intramolecular cyclizations
), 7.79 (1 H, dd, J = 7.7, 1.8 Hz, Ar–), 7.44 (1 H, ddd, J = 8.4, 7.4, 1.8 Hz, Ar–), 7.33 (1 H, dd, J = 7.6, 1.8 Hz, Ar–), 7.28 (1 H, td, J = 7.4, 1.3 Hz, Ar–), 6.99 (1 H, td, J = 7.6, 1.0 Hz, Ar–), 6.95 (1 H, dd, J = 8.4, 1.0 Hz, Ar–), 6.90 (1 H, ddd, J = 7.9, 7.2, 1.8 Hz, Ar–), 6.10 (1 H, ddt, J = 17.3, 10.6, 5.1 Hz, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 5.49 (1 H, dq, J = 17.3, 1.6 Hz, CHCH), 5.31 (1 H, dq, J = 10.5, 1.4 Hz, CHCH), 4.68 (2 H, dt, J = 5.1, 1.6 Hz, OC2), 3.28–3.24 (2 H, m, SC2), 3.11–3.08 (2H, m, SCH2C2); 13C NMR (101 MHz, CDCl3) δ: 190.9 (C), 157.0 (C), 143.1 (C), 139.6 (CH), 133.5 (CH), 132.7 (CH), 130.2 (CH), 129.9 (CH), 128.5 (CH), 128.4 (CH), 127.5 (C), 120.7 (CH), 118.1 (CH2), 113.6 (CH), 100.5 (C), 69.9 (CH2), 40.5 (CH2), 29.6 (CH2); νmax/cm−1 3071, 2925, 2865, 1673, 1633, 1595, 1482, 1446, 1284, 1194, 1010; LRMS (ESI+) m/z 447 ([M + Na]+, 100).
), 7.47–7.44 (1 H, m, Ar–), 7.02–6.96 (1 H, m, Ar–), 6.97–6.93 (1 H, m, Ar–), 4.50 (1 H, dd, J = 11.3, 5.1 Hz, OCH), 4.15 (1H, t, J = 11.3 Hz, OCH), 2.89–2.82 (1 H, m, CCH3), 1.22 (3 H, d, J = 7.0 Hz, C3); 13C NMR (126 MHz, CDCl3) δ: 194.9 (C), 161.9 (C), 135.8 (CH), 127.5 (CH), 121.5 (CH), 120.7 (C), 117.9 (CH), 72.4 (CH2), 40.9 (CH), 10.8 (CH3); νmax/cm−1 2973, 2934, 2876, 1690, 1606, 1478, 1387, 1325, 1296, 1212, 1148, 1129; LRMS (ESI+) m/z 523 (98), 347 (73), 199 (100), 185 ([M + Na]+, 18).
), 7.35–7.27 (3 H, m, 3 × Ar–), 7.11–7.03 (2 H, m, 2 × Ar–), 6.92 (1 H, ddd, J = 7.9, 7.1, 1.9 Hz, Ar–), 6.13 (1 H, ddt, J = 17.2, 10.4, 6.0 Hz, CCHH), 5.36 (1 H, dq, J = 17.2, 1.6 Hz, CHCH), 5.22 (1 H, dq, J = 10.4, 1.4 Hz, CHCH), 4.61 (2 H, dt, J = 6.0, 1.3 Hz, OC2), 3.88 (3 H, s, OC3), 3.29–3.25 (2 H, m, SC2), 3.11–3.07 (2 H, m, SCH2C2); 13C NMR (101 MHz, CDCl3) δ: 191.3 (C), 153.5 (C), 146.4 (C), 143.0 (C), 139.7 (CH), 134.1 (CH), 133.1 (C), 130.3 (CH), 128.6 (CH), 128.5 (CH), 124.0 (CH), 120.6 (CH), 118.2 (CH2), 116.0 (CH), 100.5 (C), 74.9 (CH2), 56.3 (CH3), 40.5 (CH2), 29.7 (CH2); νmax/cm−1 2937, 1674, 1639, 1580, 1470, 1438, 1266, 1232, 1010; LRMS (ESI+) m/z 1383 (36), 931 (100), 477 ([M + Na]+, 58), 436 (14); HRMS (ESI+) calculated for [C19H19IO3SNa] 476.99918, found 476.99903.
), 7.04 (1 H, dd, J = 8.0, 1.5 Hz, Ar–), 6.95 (1 H, t, J = 7.9 Hz, Ar–), 4.60 (1 H, dd, J = 11.3, 5.0 Hz, OCH), 4.22 (1 H, t, J = 11.1 Hz, OCH), 3.91 (3 H, s, OC3), 2.90–2.84 (1 H, m, CCH3), 1.23 (3 H, d, J = 7.0 Hz, C3); 13C NMR (101 MHz, CDCl3) δ: 194.8 (C), 151.8 (C), 148.9 (C), 121.3 (C), 121.0 (CH), 118.7 (CH), 116.5 (CH), 72.9 (CH2), 56.4 (CH3), 40.7 (CH), 10.9 (CH3); νmax/cm−1 2963, 2932, 1688, 1606, 1492, 1452, 1301, 1270, 1214, 983, 730; LRMS (ESI+) m/z 413 (100), 393 (82), 358 (35), 273 (28), 240 (36), 215 ([M + Na]+, 19), 186 (65).
), 7.49 (1 H, d, J = 2.5 Hz, Ar–), 7.43 (1 H, d, J = 2.5 Hz, Ar–), 7.36–7.28 (2 H, m, Ar–), 6.95–6.90 (1 H, m, Ar–), 6.07 (1 H, ddt, J = 17.3, 10.5, 5.3 Hz, CCH2), 5.42 (1 H, dq, J = 17.3, 1.7 Hz, CHCH), 5.25 (1 H, dq, J = 10.5, 1.5 Hz, CHCH), 4.38 (2 H, dt, J = 5.3, 1.5 Hz, OC2), 3.30–3.26 (2 H, m, SC2), 3.12–3.08 (2 H, m, SCH2C2), 1.41 (9 H, s, 3 × C3), 1.32 (9 H, s, J = 3 × C3); 13C NMR (101 MHz, CDCl3) δ: 193.7 (C), 153.7 (C), 145.5 (C), 143.0 (C), 142.9 (C), 139.7 (CH), 133.8 (CH), 132.5 (C), 130.2 (CH), 128.60 (CH), 128.56 (CH), 128.1 (CH), 124.4 (CH), 117.4 (CH2), 100.5 (C), 75.7 (CH2), 40.7 (CH2), 35.6 (C), 34.7 (C), 31.5 (3 × CH3), 30.9 (3 × CH3), 29.7 (CH2); νmax/cm−1 2960, 2869, 1675, 1466, 1438, 1362, 1230, 1203, 1169, 988, 928, 829, 746; LRMS (ESI+) m/z 559 ([M + Na]+, 100); HRMS (ESI+) calculated for [C26H33O2ISNa] 559.11382, found 559.11420.
), 7.53 (1 H, d, J 2.5 Hz, Ar–), 4.52 (1 H, dd, J = 11.1, 5.0 Hz, OCH), 4.12 (1 H, dd, J = 10.9, 10.9 Hz, OCH), 2.86–2.79 (1 H, m, OCH2C), 1.40 (9 H, s, 3 × CC3), 1.31 (9 H, s, 3 × CC3), 1.22 (3 H, d, J = 7.0 Hz, CHCH3); 13C NMR (101 MHz, CDCl3) δ: 196.0 (C), 159.0 (C), 143.3 (C), 138.3 (C), 130.5 (CH), 121.5 (CH), 120.7 (C), 71.8 (CH2), 40.8 (CH), 35.2 (C), 34.6 (C), 31.5 (3 × CH3), 29.8 (3 × CH3), 11.1 (CH3); νmax/cm−1 2958, 2870, 1688, 1604, 1478, 1444, 1244; LRMS (ESI+) m/z 685 (32) 611 (23), 571 (100), 355 (24), 297 ([M + Na]+, 37).
), 7.83 (1 H, dd, J = 7.9, 1.2 Hz, Ar–), 7.52 (1 H, dd, J = 8.8, 2.6 Hz, Ar–), 7.34 (1 H, dd, J = 7.6, 2.0 Hz, Ar–), 7.30 (1 H, td, J = 7.3, 1.2 Hz, Ar–), 6.92 (1 H, ddd, J = 7.9, 7.0, 2.1 Hz, Ar–), 6.07 (1 H, ddt, J = 17.3, 10.5, 5.2 Hz, CCHH), 5.47 (1 H, dq, J = 17.3, 1.6 Hz, CHCH), 5.32 (1 H, dq, J = 10.6, 1.4 Hz, CHCH), 4.65 (2 H, dt, J = 5.1, 1.6 Hz, OC2), 3.28–3.24 (2 H, m, SC2), 3.10–3.07 (2 H, m, SCH2C2); 13C NMR (101 MHz, CDCl3) δ: 189.7 (C), 156.1 (C), 142.9 (C), 139.7 (CH), 136.0 (CH), 132.4 (CH), 132.3 (CH), 130.2 (CH), 129.0 (C), 128.6 (CH), 128.5 (CH), 118.5 (CH2), 115.5 (CH), 113.1 (C), 100.5 (C), 70.2 (CH2), 40.4 (CH2), 29.8 (CH2); νmax/cm−1 2921, 1676, 1633, 1479, 1274, 1177, 1129, 1011; LRMS (ESI+) m/z 527 ([M + Na]+, 100), 525 ([M + Na]+, 100); HRMS (ESI+) calculated for [C18H16O2BrISNa] 524.89913 and 526.89704, found 524.89970 and 524.89772.
), 7.53 (1 H, dd, J = 8.8, 2.5 Hz, Ar–), 6.87 (1 H, d, J = 8.8 Hz, Ar–), 4.50 (1 H, dd, J = 11.4, 5.1 Hz, OCH), 4.14 (1 H, t, J = 11.2 Hz, OCH), 2.85 (1 H, dqd, J = 11.0, 7.0, 5.1 Hz, CCH3), 1.22 (1 H, d, J = 7.0 Hz, CHC3); 13C NMR (101 MHz, CDCl3) δ: 193.7 (C), 160.8 (C), 138.5 (CH), 129.9 (CH), 122.0 (C), 120.0 (CH), 114.2 (C), 72.4 (CH2), 40.6 (CH), 10.7 (CH3); νmax/cm−1 2966, 2932, 2879, 1687, 1599, 1477, 1457, 1420, 1316, 1297, 1183, 1021; LRMS (EI+) m/z 242 ([M]+, 45), 240 ([M]+, 46), 200 (90), 198 (100), 172 (32), 170 (28).
), 7.41 (1 H, d, J = 2.3 Hz, Ar–), 7.33–7.25 (2 H, m, Ar–), 7.13 (1 H, d, J = 2.3 Hz, Ar–), 6.91 (1 H, ddd, J = 7.9, 6.8, 2.3 Hz, Ar–), 6.08 (1 H, ddt, J = 17.2, 10.3, 6.0 Hz, CCH2), 5.34 (1 H, dq, J = 17.2, 1.6 Hz, CHCH), 5.22 (1 H, dq, J = 10.3, 1.3 Hz, CHCH), 4.58 (2 H, dt, J = 6.0, 1.3 Hz, OC2), 3.86 (3 H, s, OCH3), 3.28–3.24 (2 H, m, SC2), 3.10–3.06 (2 H, m, SCH2C2); 13C NMR (101 MHz, CDCl3) δ: 190.0 (C), 154.1 (C), 145.6 (C), 142.7 (C), 139.7 (CH), 133.9 (C), 133.6 (CH), 130.2 (CH), 128.6 (CH), 128.5 (CH), 123.0 (CH), 119.0 (CH), 118.6 (CH2), 116.4 (C), 100.5 (C), 74.9 (CH2), 56.5 (CH3), 40.3 (CH2), 29.7 (CH2); νmax/cm−1 3080, 2936, 2863, 1675, 1639, 1569, 1468, 1441, 1419, 1403, 1303, 1255, 1221, 974; LRMS (ESI+) m/z 555 ([M + Na]+, 94), 557 ([M + Na]+, 100); HRMS (ESI+) calculated for [C19H18O3SIBrNa] 554.90969 and 556.90761, found 554.91019 and 556.90817.
), 7.11 (1 H, d, J = 2.3 Hz, Ar–), 4.60 (1 H, dd, J = 11.4, 5.0 Hz, OCH), 4.20 (1 H, t, J = 11.1 Hz, OCH), 3.90 (3 H, s, OC3), 2.87 (1 H, dqd, J = 10.9, 7.0, 5.0 Hz, CCH3), 1.22 (3 H, d, J = 7.0 Hz, CHC3); 13C NMR (101 MHz, CDCl3) δ: 193.5 (C), 151.0 (C), 149.8 (C), 122.0 (C), 121.0 (CH), 119.5 (CH), 113.6 (C), 72.9 (CH2), 56.7 (CH3), 40.6 (CH), 10.8 (CH3); νmax/cm−1 2967, 2933, 1694, 1486, 1578, 1486, 1460, 1440, 1294, 1263, 1248; LRMS (ESI+) m/z 271 ([M + H]+, 100), 273 ([M + H]+, 100); HRMS (ESI+) calculated for [C11H11O3BrNa] 292.97838 and 294.97633, found 292.97873 and 294.97661.
), 7.34 (1 H, s, Ar–), 7.34 (1 H, dd, J = 7.6, 1.9 Hz, Ar–), 7.30 (1 H, td, J = 7.4, 1.3 Hz, Ar–), 6.92 (1 H, ddd, J = 7.8, 7.2, 1.9 Hz, Ar–), 6.74 (1 H, s, Ar–), 5.97 (1 H, ddd, J = 16.9, 10.3, 6.4 Hz, CCH2), 5.06–5.01 (2 H, m, CHC2), 3.92 (6 H, s, 2 × OC3), 3.60 (2 H, dt, J = 6.4, 1.5 Hz, C2CHCH2), 3.29–3.25 (2 H, s, SC2), 3.11–3.07 (2 H, m, SCH2C2); 13C NMR (126 MHz, CDCl3) δ: 192.5 (C), 151.9 (C), 146.9 (C), 142.9 (C), 139.7 (CH), 137.4 (CH), 133.1 (C), 130.3 (CH), 129.5 (C), 128.6 (CH), 128.5 (CH), 115.9 (CH2), 113.5 (CH), 112.1 (CH), 100.6 (C), 56.3 (CH3), 56.1 (CH3), 40.7 (CH2), 37.6 (CH2), 29.8 (CH2); νmax/cm−1 2933, 2844, 1659, 1516, 1266, 1193, 1113; LRMS (ESI+) m/z 491 ([M + Na]+, 100); HRMS (ESI+) calculated for [C20H21IO3SNa] 491.01483, found 491.01437.
), 6.85 (1 H, s, Ar–), 3.95 (3 H, s, OC3), 3.89 (3 H, s, OC3), 3.29 (1 H, dd, J = 16.6, 7.3 Hz, CHCH), 2.71–2.59 (2 H, m, CHC), 1.28 (3 H, d, J = 7.4 Hz, C3); 13C NMR (101 MHz, CDCl3) δ: 208.3 (C), 155.6 (C), 149.6 (C), 148.8 (C), 129.1 (C), 107.5 (CH), 104.6 (CH), 56.3 (CH3), 56.2 (CH3), 42.3 (CH), 34.9 (CH2), 16.7 (CH3); νmax/cm−1 2964, 2931, 2872, 2839, 1684, 1590, 1499, 1457, 1437, 1364, 1319, 1295, 1258, 1239, 1214, 1180, 1124, 1043, 1002; LRMS (ESI+) m/z 435 (100), 229 ([M + Na]+, 74).
), 7.57 (1 H, d, J = 2.6 Hz, Ar–), 7.52 (1 H, d, J = 2.6 Hz, Ar–), 7.32–7.30 (2 H, m, Ar–), 6.95–6.91 (1 H, m, Ar–), 6.09 (1 H, ddt, J = 17.2, 10.3, 5.9 Hz, CCH2), 5.41 (1 H, dq, J = 17.2, 1.5 Hz, CHCH), 5.28 (1 H, dq, J = 10.3, 1.3 Hz, CHCH), 4.56 (2 H, dt, J = 5.9, 1.3 Hz, OC2), 3.32–3.28 (2 H, m, SC2), 3.12–3.07 (2 H, m, SCH2C2); 13C NMR (101 MHz, CDCl3) δ: 189.6 (C), 151.6 (C), 142.5 (C), 139.8 (CH), 134.9 (C), 133.4 (CH), 132.8 (CH), 130.5 (C), 130.2 (CH), 129.7 (C), 128.7 (CH), 128.6 (CH), 127.6 (CH), 119.3 (CH2), 100.5 (C), 76.0 (CH2), 40.3 (CH2), 29.9 (CH2); νmax/cm−1 3070, 2926, 1673, 1643, 1462, 1437, 1418, 1211, 1174; LRMS (ESI+) m/z 515 ([M + Na]+, 100), 413 (42), 393 (46), 360 (16), 331 (11); HRMS (ESI+) calculated for [C18H15Cl2IO2SNa] 514.91067 and 516.90770, found 514.91072 and 516.90775.
), 7.70 (1 H, dd, J = 8.0, 1.6 Hz, Ar–), 7.64 (1 H, dd, J = 7.8, 1.7 Hz, Ar–), 7.39–7.30 (2 H, m, Ar–), 7.05 (1 H, t, J = 7.8 Hz, Ar–), 6.93 (1 H, ddd, J = 7.9, 6.7, 2.3 Hz, Ar–), 6.14 (1 H, ddt, J = 17.2, 10.4, 5.9 Hz, CCH2), 5.43 (1 H, dq, J = 17.2, 1.6 Hz, CHCH), 5.28 (1 H, dq, J = 10.4, 1.3 Hz, CHCH), 4.57 (2 H, dt, J = 5.9, 1.3 Hz, OC2), 3.31–3.27 (2 H, m, SC2), 3.12–3.08 (2 H, m, SCH2C2); 13C NMR (100 MHz, CDCl3) δ: 190.8 (C), 153.8 (C), 142.7 (C), 139.8 (CH), 137.1 (CH), 134.4 (C), 133.1 (CH), 130.2 (CH), 128.59 (2 × CH), 128.56 (CH), 125.3 (CH), 119.2 (C), 118.9 (CH2), 100.5 (C), 76.0 (CH2), 40.4 (CH2), 29.8 (CH2); νmax/cm−1 3064, 2927, 1677, 1640, 1437, 1417, 1229, 1179, 1134, 1010; LRMS (ESI+)m/z 525 ([M + Na]+, 100), 527 ([M + Na]+, 82); HRMS (ESI+) calculated for [C18H16O2SIBrNa] 524.89913 and 526.89704, found 524.89920 and 526.89719.
), 7.61 (1 H, dd, J = 7.8, 1.7 Hz, Ar–), 7.53 (1 H, dd, J = 7.9, 1.7 Hz, Ar–), 7.34–7.30 (2 H, m, Ar–), 7.11 (1 H, t, J = 7.9 Hz, Ar–), 6.93 (1 H, ddd, J = 8.0, 6.8, 2.2 Hz, Ar–), 6.13 (1 H, ddt, J = 17.2, 10.3, 5.9 Hz, CCH2), 5.43 (1 H, dq, J = 17.2, 1.5 Hz, CHCH), 5.28 (1 H, dq, J = 10.3, 1.3 Hz, CHCH), 4.59 (2 H, dt, J = 5.9, 1.3 Hz, OC2), 3.32–3.28 (2 H, m, SC2), 3.12–3.08 (2 H, m, SCH2C2); 13C NMR (101 MHz, CDCl3) δ: 190.7 (C), 152.8 (C), 142.7 (C), 139.7 (CH), 134.3 (C), 134.0 (CH), 133.1 (CH), 130.2 (CH), 129.6 (C), 128.6 (2 × CH), 127.7 (CH), 124.7 (CH), 118.8 (CH2), 100.5 (C), 75.8 (CH2), 40.3 (CH2), 29.8 (CH2); νmax/cm−1 3065, 2928, 1665, 1641, 1440, 1227, 1180, 1145, 978, 939, 748, 733; LRMS (ESI+) m/z 483 ([M + Na]+, 36), 481 ([M + Na]+, 100); HRMS (ESI+) calculated for [C18H16O2SI35ClNa] 480.94964, found 480.95016; calculated for [C18H16O2SI37ClNa] 482.94666, found 482.94720.
), 7.50 (1 H, dd, J = 7.9, 1.4 Hz, Ar–), 7.35–7.29 (2 H, m, 2 × Ar–), 7.23 (1 H, ddd, J = 11.1, 8.2, 1.6 Hz, Ar–), 7.08 (1 H, td, J = 8.0, 4.7 Hz, Ar–), 6.94–6.91 (1 H, m, Ar–), 6.13–6.07 (1 H, m, CCH2), 5.39 (1 H, dq, J = 17.2, 1.5 Hz, CHCH), 5.26 (1 H, dq, J = 10.4, 1.2 Hz, CHCH), 4.69–4.67 (2 H, m, OC2), 3.30–3.27 (2 H, m, SC2), 3.12–3.08 (2 H, m, SCH2C2); 13C NMR (101 MHz, CDCl3) δ: 190.3 (1 C, d, J = 3.3 Hz, C), 156.0 (1 C, d, J = 248.9 Hz, C), 144.9 (1 C, d, J = 12.4 Hz, C), 142.8 (C), 139.7 (CH), 133.6 (1 C, d, J = 1.8 Hz, C), 133.2 (CH), 130.2 (CH), 128.6 (CH), 128.5 (CH), 124.4 (1 C, d, J = 1.4 Hz, CH), 123.7 (1 C, d, J = 7.7 Hz, CH), 120.4 (1 C, d, J = 19.7 Hz, CH), 119.1 (CH2), 100.5 (C), 75.7 (1 C, d, J = 5.8 Hz, CH), 40.4 (CH2), 29.7 (CH2); 19F NMR (376 MHz, CDCl3) δ: −127.80; νmax/cm−1 2928, 1676, 1639, 1468, 1264, 1227, 1009; LRMS (ESI+) m/z 465 ([M + Na]+, 100); HRMS (ESI+) calculated for [C18H16O2FSNa] 464.97919, found 464.97946.
), 8.33 (1 H, dd, J = 9.2, 2.9 Hz, Ar–), 7.84 (1 H, dd, J = 8.0, 1.0 Hz, Ar–), 7.35–7.29 (2 H, m, 2 × Ar–), 7.05 (1 H, d, J = 9.2 Hz, Ar–), 6.94 (1 H, ddd, J = 7.9, 6.8, 2.2 Hz, Ar–), 6.08 (1 H, ddt, J = 17.3, 10.6, 5.2 Hz, CHCHH), 5.50 (1 H, dq, J = 17.3, 1.6 Hz, CHCH), 5.38 (1 H, dq, J = 10.6, 1.3 Hz, CHCH), 4.78 (1 H, dt, J = 5.1, 1.5 Hz, OC2), 3.33–3.29 (2 H, m, SC2), 3.13–3.09 (2 H, m, SCH2C2); 13C NMR (101 MHz, CDCl3) δ: 189.2 (C), 161.2 (C), 142.6 (C), 141.2 (C), 139.8 (CH), 131.3 (CH), 130.2 (CH), 128.7 (CH), 128.64 (CH), 128.63 (CH), 127.8 (C), 125.9 (CH), 119.3 (CH2), 113.3 (CH), 100.5 (C), 70.5 (CH2), 40.3 (CH2), 29.9 (CH2); νmax/cm−1 3084, 1637, 1608, 1563, 1342, 1276, 1086; LRMS (ESI+) m/z 492 ([M + Na]+, 100); HRMS (ESI+) calculated for [C18H16NO4SINa] 491.97369, found 491.97428.
), 7.95 (1 H, dd, J = 8.1, 1.6 Hz, Ar–), 7.84 (1 H, dd, J = 7.9, 1.3 Hz, Ar–), 7.37–7.29 (3 H, m, 3 × Ar–), 6.93 (1 H, ddd, J = 7.8, 7.3, 1.9 Hz, Ar–), 6.69 (1 H, dd, J = 8.6, 0.8 Hz, Ar–), 6.62 (1 H, ddd, J = 8.1, 7.0, 1.1 Hz, Ar–), 5.96 (1 H, ddt, J = 17.2, 10.3, 5.1 Hz, CCHH), 5.31 (1 H, dq, J = 17.2, 1.7 Hz, CHCH), 5.21 (1 H, dq, J = 10.3, 1.6 Hz, CHCH), 3.89 (2 H, tt, J = 5.4, 1.8 Hz, NC2), 3.27–3.24 (2 H, m, SC2), 3.10–3.07 (2 H, m, SCH2C2); 13C NMR (126 MHz, CDCl3) δ: 193.1 (C), 149.0 (C), 143.0 (C), 139.7 (CH), 135.0 (CH), 134.4 (CH), 130.9 (CH), 130.2 (CH), 128.54 (CH), 128.46 (CH), 118.0 (C), 116.4 (CH2), 115.0 (CH), 112.1 (CH), 100.6 (C), 45.3 (CH2), 40.9 (CH2), 29.0 (CH2); νmax/cm−1 3476, 3359, 3059, 2925, 1694, 1630, 1582, 1516, 1447, 1197, 1162, 1011; LRMS (ESI+) m/z 522 (42), 424 ([M + H]+, 100); HRMS (ESI+) calculated for [C18H18INOSNa] 446.00460, found 446.00472.
), 7.28 (1 H, ddd, J = 8.3, 6.9, 1.5 Hz, Ar–), 6.73 (1 H, ddd, J = 8.1, 7.1, 1.1 Hz, Ar–), 6.65 (1 H, dd, J = 8.3, 1.3 Hz, Ar–), 4.44 (1 H, brs, N), 3.55 (1 H, dd, J = 11.7, 5.3 Hz, NHCH), 3.27 (1 H, t, J = 11.6 Hz, NHCH), 2.74–2.64 (1 H, m, CCH3), 1.22 (3 H, d, J = 6.9 Hz, C3); 13C NMR (101 MHz, CDCl3) δ: 196.6 (C), 151.8 (C), 134.9 (CH), 128.0 (CH), 118.9 (C), 118.0 (CH), 115.7 (CH), 48.9 (CH2), 41.2 (CH), 12.7 (CH3); νmax/cm−1 3348, 2964, 2930, 2872, 2815, 1656, 1611, 1512, 1365, 1343, 1237, 1155; LRMS (ESI+) m/z 393 (84), 345 (100), 184 ([M + Na]+, 71); HRMS (ESI+) calculated for [C10H11NONa] 184.07329, found 184.07325.
), 7.77 (1 H, dd, J = 7.8, 1.8 Hz, Ar–), 7.43 (1 H, ddd, J = 8.4, 7.4, 1.8 Hz, Ar–), 7.35 (1 H, dd, J = 7.6, 1.8 Hz, Ar–), 7.29 (1 H, td, J = 7.4, 1.3 Hz, Ar–), 7.09 (1 H, dt, J = 15.7, 4.0 Hz, CH2CCH), 7.03 (1 H, td, J = 7.6, 1.0 Hz, Ar–), 6.93–6.88 (2 H, m, 2 × Ar–), 6.32 (1 H, dt, J = 15.7, 2.1 Hz, CH2CHC), 4.80 (2 H, dd, J = 4.0, 2.1 Hz, ArOC2), 4.21 (2 H, q, J = 7.1 Hz, CO2C2), 3.30–3.25 (2 H, m, SC2), 3.13–3.07 (2 H, m, SCH2C2), 1.28 (3 H, t, J = 7.1 Hz, C3); 13C NMR (101 MHz, CDCl3) δ: 190.7 (C), 166.1 (C), 156.1 (C), 142.9 (C), 141.6 (CH), 139.6 (CH), 133.6 (CH), 130.2 (CH), 129.9 (CH), 128.5 (CH), 128.4 (CH), 127.8 (C), 122.7 (CH), 121.3 (CH), 113.3 (CH), 100.5 (C), 67.6 (CH2), 60.7 (CH2), 40.5 (CH2), 29.6 (CH2), 14.3 (CH3); νmax/cm−1 2980, 2935, 1718, 1669, 1637, 1596, 1484, 1443, 1288, 1180, 1039; LRMS (ESI+) m/z 514 (100), 497 ([M + H]+, 40); HRMS (ESI+) calculated for [C21H21O4ISNa] 519.00974, found 519.00972.
), 7.50–7.45 (1 H, m, Ar–), 7.04–7.00 (1 H, m, Ar–), 6.97 (1 H, dd, J = 8.4, 1.2 Hz, Ar–), 4.60 (1 H, dd, J = 11.2, 5.3 Hz, ArOCH), 4.30 (1 H, dd, 11.9, 11.2 Hz, ArOCH), 4.19 (2 H, qd, J = 7.2, 2.0 Hz, CO2C2), 3.33 (1 H, dddd, J = 11.9, 8.3, 5.1, 5.1 Hz, ArCOC), 2.93 (1 H, dd, J = 16.9, 4.8 Hz, CHCO2Et), 2.41 (1 H, dd, J = 16.9, 8.2 Hz, CHCO2Et), 1.28 (3 H, t, J = 7.2 Hz, C3); 13C NMR (101 MHz, CDCl3) δ: 192.7 (C), 171.5 (C), 161.9 (C), 136.1 (CH), 127.5 (CH), 121.7 (CH), 120.7 (C), 118.0 (CH), 70.4 (CH2), 61.1 (CH2), 42.7 (CH), 30.5 (CH2), 14.3 (CH3); νmax/cm−1 2980, 2927, 1730, 1688, 1605, 1479, 1298, 1213, 1175, 1128, 1035, 1012; LRMS (ESI+) m/z 682 (29), 491 (24), 420 (100), 403 (43), 257 (49), 234 (M+, 33).
), 7.43 (1 H, ddd, J = 8.4, 7.3, 1.8 Hz, Ar–), 7.35 (1 H, dd, J = 7.6, 1.8 Hz, Ar–), 7.29 (1 H, td, J = 7.4, 1.3 Hz, Ar–), 7.02 (1 H, d, J = 9.0 Hz, Ar–), 7.00–6.96 (1 H, m, Ar–), 6.93–6.89 (1 H, m, Ar–), 6.00–5.91 (2 H, m, CC), 4.94–4.88 (1 H, m, OC), 3.26–3.22 (2 H, m, SC2), 3.10–3.06 (2 H, m, SCH2C2), 2.22–2.11 (1 H, m, OCHCH), 2.06–1.95 (4 H, m, OCHCHC2CH), 1.68–1.65 (1 H, m, OCHCHHCH2CH); 13C NMR (101 MHz, CDCl3) δ: 190.9 (C), 156.8 (C), 143.2 (C), 139.6 (CH), 133.5 (CH), 132.6 (CH), 130.2 (CH), 130.0 (CH), 128.4 (CH), 128.3 (CH), 128.0 (C), 125.8 (CH), 120.4 (CH), 114.5 (CH), 100.5 (C), 72.7 (CH), 40.5 (CH2), 29.6 (CH2), 28.5 (CH2), 25.2 (CH2), 19.2 (CH2); νmax/cm−1 3029, 2932, 2865, 1676, 1631, 1593, 1478, 1447, 1282, 1239, 1190, 1009; LRMS (ESI+) m/z 619 (100), 487 ([M + Na]+, 78), 321 (47); HRMS (ESI+) calculated for [C21H21O2ISNa] 487.01991, found 487.02033.
), 7.47 (1 H, ddd, J = 8.3, 7.2, 1.8 Hz, Ar–), 7.01–6.96 (2 H, m, 2 × Ar–), 4.60–4.57 (1 H, m, OC), 2.57–2.53 (1 H, m, COC), 2.13–2.09 (1 H, m, C), 1.79–1.58 (6 H, m, 6 × C), 1.47–1.37 (1 H, m, C); 13C NMR (101 MHz, CDCl3) δ: 195.9 (C), 161.4 (C), 136.0 (CH), 127.5 (CH), 121.2 (CH), 119.8 (C), 118.0 (CH), 76.3 (CH), 48.1 (CH), 29.5 (CH2), 24.13 (CH2), 24.06 (CH2), 20.6 (CH2); νmax/cm−1 2934, 2860, 1685, 1605, 1461, 1306, 1229, 1150, 1121; LRMS (APCI) m/z 389 (30), 203 ([M + H]+, 100).
), 7.79 (1 H, dd, J = 7.8, 1.8 Hz, Ar–), 7.48 (1 H, ddd, J = 8.4, 7.3, 1.8 Hz, Ar–), 7.36 (1 H, dd, J = 7.6, 1.9 Hz, Ar–), 7.30 (1 H, td, J = 7.4, 1.3 Hz, Ar–), 7.15 (1 H, dd, J = 8.4, 0.9 Hz, Ar–), 7.06 (1 H, td, J = 7.6, 0.9 Hz, Ar–), 6.94–6.90 (1 H, m, Ar–), 4.82 (2 H, d, J = 2.4 Hz, OC2), 3.29–3.25 (2 H, m, SC2), 3.12–3.08 (2 H, m, SCH2C2), 2.55 (1 H, t, J = 2.4 Hz, CH2CC); 13C NMR (101 MHz, CDCl3) δ: 190.8 (C), 155.8 (C), 143.0 (C), 139.7 (CH), 133.5 (CH), 130.3 (CH), 130.0 (CH), 128.6 (CH), 128.5 (CH), 128.1 (C), 121.6 (CH), 114.2 (CH), 100.5 (C), 78.1 (C), 76.4 (CH), 56.8 (CH2), 40.5 (CH2), 29.7 (CH2); νmax/cm−1 3291, 3059, 2923, 1669, 1634, 1596, 1482, 1447, 1286, 1260, 1011; LRMS (ESI+) m/z 445 ([M + Na]+, 100); HRMS (ESI+) calculated for [C18H15O2SINa] 444.97296, found 444.97346.
), 7.46 (1 H, ddd, J = 8.4, 7.4, 1.8 Hz, Ar–), 7.44–7.41 (2 H, m, 2 × Ar–), 7.36–7.28 (3 H, m, 3 × Ar–), 7.28–7.24 (2 H, m, 2 × Ar–), 7.05–7.00 (2 H, m, 2 × Ar–), 6.90 (1 H, ddd, J = 7.8, 7.4, 1.8 Hz, Ar–), 6.80 (1 H, dt, J = 16.0, 1.6 Hz, CPh), 6.45 (1 H, dt, J = 16.0, 5.7 Hz, CCHPh), 4.85 (2 H, dd, J = 5.6, 1.6 Hz, OC2), 3.30–3.26 (2 H, m, SC2), 3.12–3.08 (2 H, m, SCH2C2); 13C NMR (101 MHz, CDCl3) δ: 190.9 (C), 157.1 (C), 143.1 (C), 139.6 (CH), 136.5 (C), 133.6 (CH), 133.3 (CH), 130.2 (CH), 129.9 (CH), 128.7 (2 × CH), 128.5 (CH), 128.4 (CH), 128.1 (CH), 127.6 (C), 126.8 (2 × CH), 124.0 (CH), 120.9 (CH), 113.8 (CH), 100.6 (C), 69.9 (CH2), 40.5 (CH2), 29.7 (CH2); νmax/cm−1 3057, 3025, 2924, 2863, 1671, 1633, 1594, 1482, 1446, 1285, 1241, 1193, 1162, 1111; LRMS (ESI+) m/z 523 ([M + Na]+, 100); HRMS (ESI+) calculated for [C24H21O2ISNa] 523.01991, found 523.02024.
), 8.55 (1 H, d, J = 13.5 Hz, CC), 8.07 (1 H, dd, J = 8.0, 1.4 Hz, Ar–), 7.83 (1 H, dd, J = 7.8, 1.0 Hz, Ar–), 7.60–7.55 (1 H, m, Ar–), 7.40 (1 H, d, J = 8.1 Hz, Ar–), 7.34–7.28 (2 H, m, Ar–), 7.16 (1 H, ddd, J = 7.3, 7.1, 0.9 Hz, Ar–), 6.93 (1 H, ddd, J = 7.8, 6.7, 2.3 Hz, Ar–), 4.43 (2 H, q, J = 7.1 Hz, C2CH3), 4.27 (2 H, q, J = 7.1 Hz, C2CH3), 3.40–3.36 (2 H, m, SC2), 3.14–3.10 (2 H, m, C2Ar), 1.41 (3 H, t, J = 7.1 Hz, C3), 1.34 (3 H, t, J = 7.1 Hz, C3); 13C NMR (101 MHz, CDCl3) δ: 193.4 (C), 167.0 (C), 166.2 (C), 149.7 (CH), 142.6 (C), 139.8 (CH), 139.3 (C), 134.7 (CH), 130.7 (CH), 130.2 (CH), 128.62 (CH), 128.56 (CH), 124.5 (C), 123.5 (CH), 115.9 (CH), 100.6 (C), 97.3 (C), 60.7 (CH2), 60.5 (CH2), 40.3 (CH2), 29.6 (CH2), 14.62 (CH3), 14.56 (CH3); νmax/cm−1 2978, 2928, 1716, 1693, 1656, 1610, 1589, 1225, 1191, 1170; LRMS (ESI+) m/z 576 ([M + Na]+, 100); HRMS (ESI+) calculated for [C23H24NO5SINa] 576.03121, found 576.03198.
), 7.83 (1 H, dd, J = 7.8, 1.3 Hz, Ar–), 7.74 (1 H, d, J = 12.3 Hz, OCCH), 7.55 (1 H, ddd, J = 8.2, 7.5, 1.7 Hz, Ar–), 7.33–7.28 (3 H, m, 3 × Ar–), 7.14 (1 H, dd, J = 8.2, 0.9 Hz, Ar–), 6.93 (1 H, ddd, J = 7.9, 6.7, 2.3 Hz, Ar–), 5.57 (1 H, d, J = 12.2 Hz, OCHC), 4.20 (2 H, q, J = 7.1 Hz, OC2), 3.30–3.26 (2 H, m, SC2), 3.11–3.07 (2 H, m, SCH2C2), 1.28 (3 H, t, J = 7.1 Hz, C3); 13C NMR (101 MHz, CDCl3) δ: 189.8 (C), 166.9 (C), 158.9 (CH), 153.2 (C), 142.6 (C), 139.7 (CH), 133.8 (CH), 130.2 (CH), 130.0 (CH), 129.5 (C), 128.6 (2 × CH), 125.4 (CH), 119.8 (CH), 103.4 (CH), 100.5 (C), 60.3 (CH2), 40.3 (CH2), 29.7 (CH2), 14.4 (CH3); νmax/cm−1 3056, 2978, 1712, 1646, 1479, 1446, 1225, 1199, 1123, 1046; LRMS (ESI+) m/z 505 ([M + Na]+, 100); HRMS (ESI+) calculated for [C20H19IO4SNa] 504.99409, found 504.99422.
), 7.70 (1 H, dt, J = 8.1, 1.1 Hz, Ar–), 7.46 (1 H, s, Ar–), 7.37–7.31 (4 H, m, Ar–), 7.20–7.16 (1 H, m, Ar–), 6.93 (1 H, ddd, 8.0, 7.0, 2.1 Hz, Ar–), 6.00 (1 H, ddt, J = 17.1, 10.3, 5.0 Hz, CCH2), 5.17 (2 H, dt, J = 5.0, 1.7 Hz, NC2), 5.12 (1 H, dq, J = 10.3, 1.5 Hz, CHCH), 4.92 (1 H, dq, J = 17.1, 1.6 Hz, CHCH), 3.33–3.29 (2 H, m, SC2), 3.13–3.10 (2 H, m, SCH2C2); 13C NMR (101 MHz, CDCl3) δ: 184.4 (C), 142.8 (C), 139.7 (CH), 139.5 (C), 133.83 (C), 133.77 (CH), 130.3 (CH), 128.5 (2 × CH), 126.2 (C), 126.0 (CH) 123.0 (CH), 121.2 (CH), 116.4 (CH2), 110.9 (CH), 110.8 (CH), 100.6 (C), 47.2 (CH2), 40.8 (CH2), 28.9 (CH2); νmax/cm−1 3058, 2926, 1640, 1510, 1454, 1154, 1131, 1010; HRMS (ESI+) calculated for [C20H18NOISNa] 470.00460, found 470.00477.
), 7.42 (1 H, dddd, J = 8.4, 1.0, 1.0, 1.0 Hz, Ar–), 7.36 (1 H, ddd, J = 8.4, 6.8, 1.1 Hz, Ar–), 7.19 (1 H, ddd, J = 8.2, 6.8, 1.2 Hz, Ar–), 7.02 (1 H, d, J = 0.9 Hz, Ar–), 4.68 (1 H, dd, J = 10.8, 8.0 Hz, NCH), 3.98 (1 H, dd, J = 10.8, 4.8 Hz, NCH), 3.32–3.27 (1 H, m, NCH2C), 1.46 (3 H, d, J = 7.5 Hz, CH3); 13C NMR (101 MHz, CDCl3) δ: 196.2 (C), 135.3 (C), 135.2 (C), 132.3 (C), 125.2 (CH), 124.3 (CH), 121.6 (CH), 110.6 (CH), 99.4 (CH), 48.0 (CH2), 45.6 (CH), 15.7 (CH3); νmax/cm−1 3077, 2972, 2882, 1711, 1539, 1348, 1232, 1166; LRMS (ESI+) m/z 238 (77), 234 (30), 214 (32), 200 (40), 186 ([M + H]+, 100).
:1 diasteteromeric mixture of 26a (670 mg, 1.28 mmol, 82%) as a colourless oil. Rf: 0.19 (10% diethyl ether in hexanes); 1H NMR (400 MHz, CDCl3) δ: 7.84–7.82 (1 H + 0.6 × 1 H, m, Ar–), 7.35–7.28 (3 H + 0.6 × 3 H, m, 3 × Ar–), 6.94–6.90 (1 H + 0.6 × 1 H, m, Ar–), 6.75 (1 H, s, Ar–), 6.70 (0.6 × 1 H, s, Ar–), 5.54 (0.6 × 1 H, dt, J = 15.0, 7.0 Hz, CH2CCH), 5.35 (1 H, dtd, 10.7, 7.4, 0.8 Hz, CH2CCH), 4.99 (0.6 × 1 H, ddt, J = 15.2, 8.5, 1.3 Hz, CH2CHC), 4.78–4.73 (1 H, m, CH2CHC), 3.92 (3 H, s, ArOC3), 3.91 (0.6 × 3 H, s, ArOC3), 3.91 (3 H, s, ArOC3), 3.91 (0.6 × 3 H, s, ArOC3), 3.29–3.25 (2 H + 0.6 × 2 H, m, SC2), 3.11–3.07 (2 H + 0.6 × 2 H, m, SCH2C2), 2.93–2.89 (2 H, m, ArC2), 2.87–2.83 (0.6 × 2 H, m, ArC2), 2.48–2.46 (2 H, m, C2CHCH), 2.27–2.25 (0.6 × 2 H, m, C2CHCH), 1.56–1.47 (1 H, m, CHCHC), 1.38–1.29 (1 H, m, CHCHC), 0.68–0.63 (2 H + 0.6 × 2 H, m, 2 × CH(CH)2), 0.31–0.26 (2 H + 0.6 × 2 H, m, 2 × CH(CH)2); 13C NMR (101 MHz, CDCl3) δ: 192.39 (C), 192.35 (C), 151.64 (C), 151.60 (C), 146.63 (C), 146.59 (C), 142.84 (C), 142.83 (C), 139.7 (CH + CH), 135.49 (C), 135.46 (C), 134.9 (CH), 134.6 (CH), 130.18 (CH), 130.17 (CH), 129.4 (C), 129.3 (C), 128.51 (CH + CH), 128.49 (CH + CH), 127.3 (CH), 127.1 (CH), 113.74 (CH), 113.69 (CH), 112.14 (CH), 112.08 (CH), 100.63 (C), 100.60 (C), 56.24 (CH3), 56.22 (CH3), 56.08 (CH3), 56.06 (CH3), 40.70 (CH2), 40.67 (CH2), 34.6 (CH2), 33.8 (CH2), 33.7 (CH2), 29.8 (CH2), 29.7 (CH2 + CH2), 13.6 (CH), 9.7 (CH), 7.0 (2 × CH2), 6.5 (2 × CH2); νmax/cm−1 3001, 2934, 1661, 1516, 1464, 1265, 1193, 1111; LRMS (ESI+) m/z 545 ([M + Na]+, 100); HRMS (ESI+) calculated for [C24H27O3SINa] 545.06178, found 545.06249.
), 6.66 (1 H, s, Ar–), 5.65 (1 H, dtd, J = 10.8, 7.2, 1.0 Hz, CHCCH2), 5.50 (1 H, ddt, J = 10.8, 8.8, 1.6 Hz, CCHCH2), 3.93 (3 H, s, OC3), 3.91 (3 H, s, OC3), 3.44 (1 H, dddd, J = 10.9, 8.8, 4.6, 1.0 Hz, CCHCH), 3.04–2.92 (2 H, m, ArC2), 2.21–1.97 (4 H, m, CHC2 + C2CH3), 1.03 (3 H, t, J = 7.5 Hz, CH2C3); 13C NMR (101 MHz, CDCl3) δ: 197.6 (C), 153.6 (C), 148.1 (C), 138.8 (C), 135.1 (CH), 126.1 (CH), 125.8 (C), 110.3 (CH), 109.1 (CH), 56.20 (CH3), 56.16 (CH3), 46.2 (CH), 30.8 (CH2), 28.3 (CH2), 21.3 (CH2), 14.4 (CH3); νmax/cm−1 2959, 2932, 2867, 1670, 1599, 1511, 1265, 1146; LRMS (ESI+) m/z 543 (100), 283 ([M + Na]+, 80); HRMS (ESI+) calculated for [C16H20O3Na] 283.13047, found 283.13072.
), 7.29–7.27 (2 H, m, Ar–), 6.93–6.89 (1 H, m, Ar–), 5.10–5.07 (1 H, m, CC), 3.14–3.10 (2 H, m, SC2), 3.01–2.97 (2 H, m, SCH2C2), 2.56 (1 H, dd, J = 14.5, 5.9 Hz, CHCO), 2.37 (1 H, dd, J = 14.5, 8.2 Hz, CHCO), 2.07–1.95 (3 H, m, C2CH2C), 1.70 (3 H, d, J = 1.1 Hz, (3C)2C), 1.60 (3 H, s, (3C)2C), 1.39–1.32 (1 H, m, CH2CHCH), 1.27–1.20 (1 H, m, CH2CHCH), 0.94 (3 H, d, J = 6.7 Hz, 3CCH); 13C NMR (101 MHz, CDCl3) δ: 199.0 (C), 142.8 (C), 139.7 (CH), 131.8 (C), 130.2 (CH), 128.53 (CH), 128.51 (CH), 124.3 (CH), 100.5 (C), 51.5 (CH2), 40.8 (CH2), 36.8 (CH2), 30.9 (CH), 29.0 (CH2), 25.9 (CH3), 25.5 (CH2), 19.6 (CH3), 17.8 (CH3); νmax/cm−1 2963, 2926, 1688, 1465, 1436, 1010; LRMS (ESI+) m/z 471 (100), 455 (66), 439 ([M + Na]+, 93); HRMS (ESI+) calculated for [C18H25OISNa] 439.05630, found 439.05665.
HCO), 2.31–2.28 (1 H, m, CHCO), 2.19–1.66 (13 H, m, 2 × CHCO, 2 × CCH2CO, 2 × CH3CHCH, 2 × CH3CHCH2CH, CH3CHCH2CH, 2 × CCO, 2 × (CH3)2C), 1.51–1.32 (3 H, m, 2 × CH3CHCH, CH3CHCH2CH), 1.00 (3 H, d, J = 6.4 Hz, CHC3), 0.98 (3 H, d, J = 6.7 Hz, CHC3), 0.93 (3 H, d, J = 6.6 Hz, H3CCHC3), 0.91 (3 H, d, J = 6.9 Hz, H3CCHC3), 0.85 (3 H, d, J = 6.8 Hz, H3CCHC3), 0.84 (3 H, d, J = 6.6 Hz, H3CCHC3); 13C NMR (126 MHz, CDCl3) δ: 214.7 (C), 212.6 (C), 57.3 (CH), 56.1 (CH), 51.0 (CH2), 48.2 (CH2), 35.6 (CH), 34.5 (CH), 34.1 (CH2), 29.6 (CH2), 28.0 (CH2), 27.1 (CH2), 27.0 (CH), 26.0 (CH), 22.4 (CH3), 21.6 (CH3), 21.4 (CH3), 21.0 (CH3), 20.0 (CH3), 18.8 (CH3); νmax/cm−1 2955, 2926, 2871, 1708, 1456, 1368, 1203, 1116.
CH2), 5.49 (1 H, dq, J = 17.3, 1.6 Hz, CHCH), 5.31 (1 H, dq, J = 10.5, 1.4 Hz, CHCH), 4.68 (2 H, dt, J = 5.1, 1.6 Hz, OC2), 3.28–3.24 (2 H, m, SC2), 3.11–3.08 (2H, m, SCH2C2); 13C NMR (101 MHz, CDCl3) δ: 190.9 (C), 157.0 (C), 143.1 (C), 139.6 (CH), 133.5 (CH), 132.7 (CH), 130.2 (CH), 129.9 (CH), 128.5 (CH), 128.4 (CH), 127.5 (C), 120.7 (CH), 118.1 (CH2), 113.6 (CH), 100.5 (C), 69.9 (CH2), 40.5 (CH2), 29.6 (CH2); νmax/cm−1 3071, 2925, 2865, 1673, 1633, 1595, 1482, 1446, 1284, 1194, 1010; LRMS (ESI+) m/z 447 ([M + Na]+, 100).
), 7.47–7.44 (1 H, m, Ar–), 7.02–6.96 (1 H, m, Ar–), 6.97–6.93 (1 H, m, Ar–), 4.50 (1 H, dd, J = 11.3, 5.1 Hz, OCH), 4.15 (1H, t, J = 11.3 Hz, OCH), 2.89–2.82 (1 H, m, CCH3), 1.22 (3 H, d, J = 7.0 Hz, C3); 13C NMR (126 MHz, CDCl3) δ: 194.9 (C), 161.9 (C), 135.8 (CH), 127.5 (CH), 121.5 (CH), 120.7 (C), 117.9 (CH), 72.4 (CH2), 40.9 (CH), 10.8 (CH3); νmax/cm−1 2973, 2934, 2876, 1690, 1606, 1478, 1387, 1325, 1296, 1212, 1148, 1129; LRMS (ESI+) m/z 523 (98), 347 (73), 199 (100), 185 ([M + Na]+, 18).
), 7.35–7.27 (3 H, m, 3 × Ar–), 7.11–7.03 (2 H, m, 2 × Ar–), 6.92 (1 H, ddd, J = 7.9, 7.1, 1.9 Hz, Ar–), 6.13 (1 H, ddt, J = 17.2, 10.4, 6.0 Hz, CCHH), 5.36 (1 H, dq, J = 17.2, 1.6 Hz, CHCH), 5.22 (1 H, dq, J = 10.4, 1.4 Hz, CHCH), 4.61 (2 H, dt, J = 6.0, 1.3 Hz, OC2), 3.88 (3 H, s, OC3), 3.29–3.25 (2 H, m, SC2), 3.11–3.07 (2 H, m, SCH2C2); 13C NMR (101 MHz, CDCl3) δ: 191.3 (C), 153.5 (C), 146.4 (C), 143.0 (C), 139.7 (CH), 134.1 (CH), 133.1 (C), 130.3 (CH), 128.6 (CH), 128.5 (CH), 124.0 (CH), 120.6 (CH), 118.2 (CH2), 116.0 (CH), 100.5 (C), 74.9 (CH2), 56.3 (CH3), 40.5 (CH2), 29.7 (CH2); νmax/cm−1 2937, 1674, 1639, 1580, 1470, 1438, 1266, 1232, 1010; LRMS (ESI+) m/z 1383 (36), 931 (100), 477 ([M + Na]+, 58), 436 (14); HRMS (ESI+) calculated for [C19H19IO3SNa] 476.99918, found 476.99903.
), 7.04 (1 H, dd, J = 8.0, 1.5 Hz, Ar–), 6.95 (1 H, t, J = 7.9 Hz, Ar–), 4.60 (1 H, dd, J = 11.3, 5.0 Hz, OCH), 4.22 (1 H, t, J = 11.1 Hz, OCH), 3.91 (3 H, s, OC3), 2.90–2.84 (1 H, m, CCH3), 1.23 (3 H, d, J = 7.0 Hz, C3); 13C NMR (101 MHz, CDCl3) δ: 194.8 (C), 151.8 (C), 148.9 (C), 121.3 (C), 121.0 (CH), 118.7 (CH), 116.5 (CH), 72.9 (CH2), 56.4 (CH3), 40.7 (CH), 10.9 (CH3); νmax/cm−1 2963, 2932, 1688, 1606, 1492, 1452, 1301, 1270, 1214, 983, 730; LRMS (ESI+) m/z 413 (100), 393 (82), 358 (35), 273 (28), 240 (36), 215 ([M + Na]+, 19), 186 (65).
), 7.49 (1 H, d, J = 2.5 Hz, Ar–), 7.43 (1 H, d, J = 2.5 Hz, Ar–), 7.36–7.28 (2 H, m, Ar–), 6.95–6.90 (1 H, m, Ar–), 6.07 (1 H, ddt, J = 17.3, 10.5, 5.3 Hz, CCH2), 5.42 (1 H, dq, J = 17.3, 1.7 Hz, CHCH), 5.25 (1 H, dq, J = 10.5, 1.5 Hz, CHCH), 4.38 (2 H, dt, J = 5.3, 1.5 Hz, OC2), 3.30–3.26 (2 H, m, SC2), 3.12–3.08 (2 H, m, SCH2C2), 1.41 (9 H, s, 3 × C3), 1.32 (9 H, s, J = 3 × C3); 13C NMR (101 MHz, CDCl3) δ: 193.7 (C), 153.7 (C), 145.5 (C), 143.0 (C), 142.9 (C), 139.7 (CH), 133.8 (CH), 132.5 (C), 130.2 (CH), 128.60 (CH), 128.56 (CH), 128.1 (CH), 124.4 (CH), 117.4 (CH2), 100.5 (C), 75.7 (CH2), 40.7 (CH2), 35.6 (C), 34.7 (C), 31.5 (3 × CH3), 30.9 (3 × CH3), 29.7 (CH2); νmax/cm−1 2960, 2869, 1675, 1466, 1438, 1362, 1230, 1203, 1169, 988, 928, 829, 746; LRMS (ESI+) m/z 559 ([M + Na]+, 100); HRMS (ESI+) calculated for [C26H33O2ISNa] 559.11382, found 559.11420.
), 7.53 (1 H, d, J 2.5 Hz, Ar–), 4.52 (1 H, dd, J = 11.1, 5.0 Hz, OCH), 4.12 (1 H, dd, J = 10.9, 10.9 Hz, OCH), 2.86–2.79 (1 H, m, OCH2C), 1.40 (9 H, s, 3 × CC3), 1.31 (9 H, s, 3 × CC3), 1.22 (3 H, d, J = 7.0 Hz, CHCH3); 13C NMR (101 MHz, CDCl3) δ: 196.0 (C), 159.0 (C), 143.3 (C), 138.3 (C), 130.5 (CH), 121.5 (CH), 120.7 (C), 71.8 (CH2), 40.8 (CH), 35.2 (C), 34.6 (C), 31.5 (3 × CH3), 29.8 (3 × CH3), 11.1 (CH3); νmax/cm−1 2958, 2870, 1688, 1604, 1478, 1444, 1244; LRMS (ESI+) m/z 685 (32) 611 (23), 571 (100), 355 (24), 297 ([M + Na]+, 37).
), 7.83 (1 H, dd, J = 7.9, 1.2 Hz, Ar–), 7.52 (1 H, dd, J = 8.8, 2.6 Hz, Ar–), 7.34 (1 H, dd, J = 7.6, 2.0 Hz, Ar–), 7.30 (1 H, td, J = 7.3, 1.2 Hz, Ar–), 6.92 (1 H, ddd, J = 7.9, 7.0, 2.1 Hz, Ar–), 6.07 (1 H, ddt, J = 17.3, 10.5, 5.2 Hz, CCHH), 5.47 (1 H, dq, J = 17.3, 1.6 Hz, CHCH), 5.32 (1 H, dq, J = 10.6, 1.4 Hz, CHCH), 4.65 (2 H, dt, J = 5.1, 1.6 Hz, OC2), 3.28–3.24 (2 H, m, SC2), 3.10–3.07 (2 H, m, SCH2C2); 13C NMR (101 MHz, CDCl3) δ: 189.7 (C), 156.1 (C), 142.9 (C), 139.7 (CH), 136.0 (CH), 132.4 (CH), 132.3 (CH), 130.2 (CH), 129.0 (C), 128.6 (CH), 128.5 (CH), 118.5 (CH2), 115.5 (CH), 113.1 (C), 100.5 (C), 70.2 (CH2), 40.4 (CH2), 29.8 (CH2); νmax/cm−1 2921, 1676, 1633, 1479, 1274, 1177, 1129, 1011; LRMS (ESI+) m/z 527 ([M + Na]+, 100), 525 ([M + Na]+, 100); HRMS (ESI+) calculated for [C18H16O2BrISNa] 524.89913 and 526.89704, found 524.89970 and 524.89772.
), 7.53 (1 H, dd, J = 8.8, 2.5 Hz, Ar–), 6.87 (1 H, d, J = 8.8 Hz, Ar–), 4.50 (1 H, dd, J = 11.4, 5.1 Hz, OCH), 4.14 (1 H, t, J = 11.2 Hz, OCH), 2.85 (1 H, dqd, J = 11.0, 7.0, 5.1 Hz, CCH3), 1.22 (1 H, d, J = 7.0 Hz, CHC3); 13C NMR (101 MHz, CDCl3) δ: 193.7 (C), 160.8 (C), 138.5 (CH), 129.9 (CH), 122.0 (C), 120.0 (CH), 114.2 (C), 72.4 (CH2), 40.6 (CH), 10.7 (CH3); νmax/cm−1 2966, 2932, 2879, 1687, 1599, 1477, 1457, 1420, 1316, 1297, 1183, 1021; LRMS (EI+) m/z 242 ([M]+, 45), 240 ([M]+, 46), 200 (90), 198 (100), 172 (32), 170 (28).
), 7.41 (1 H, d, J = 2.3 Hz, Ar–), 7.33–7.25 (2 H, m, Ar–), 7.13 (1 H, d, J = 2.3 Hz, Ar–), 6.91 (1 H, ddd, J = 7.9, 6.8, 2.3 Hz, Ar–), 6.08 (1 H, ddt, J = 17.2, 10.3, 6.0 Hz, CCH2), 5.34 (1 H, dq, J = 17.2, 1.6 Hz, CHCH), 5.22 (1 H, dq, J = 10.3, 1.3 Hz, CHCH), 4.58 (2 H, dt, J = 6.0, 1.3 Hz, OC2), 3.86 (3 H, s, OCH3), 3.28–3.24 (2 H, m, SC2), 3.10–3.06 (2 H, m, SCH2C2); 13C NMR (101 MHz, CDCl3) δ: 190.0 (C), 154.1 (C), 145.6 (C), 142.7 (C), 139.7 (CH), 133.9 (C), 133.6 (CH), 130.2 (CH), 128.6 (CH), 128.5 (CH), 123.0 (CH), 119.0 (CH), 118.6 (CH2), 116.4 (C), 100.5 (C), 74.9 (CH2), 56.5 (CH3), 40.3 (CH2), 29.7 (CH2); νmax/cm−1 3080, 2936, 2863, 1675, 1639, 1569, 1468, 1441, 1419, 1403, 1303, 1255, 1221, 974; LRMS (ESI+) m/z 555 ([M + Na]+, 94), 557 ([M + Na]+, 100); HRMS (ESI+) calculated for [C19H18O3SIBrNa] 554.90969 and 556.90761, found 554.91019 and 556.90817.
), 7.11 (1 H, d, J = 2.3 Hz, Ar–), 4.60 (1 H, dd, J = 11.4, 5.0 Hz, OCH), 4.20 (1 H, t, J = 11.1 Hz, OCH), 3.90 (3 H, s, OC3), 2.87 (1 H, dqd, J = 10.9, 7.0, 5.0 Hz, CCH3), 1.22 (3 H, d, J = 7.0 Hz, CHC3); 13C NMR (101 MHz, CDCl3) δ: 193.5 (C), 151.0 (C), 149.8 (C), 122.0 (C), 121.0 (CH), 119.5 (CH), 113.6 (C), 72.9 (CH2), 56.7 (CH3), 40.6 (CH), 10.8 (CH3); νmax/cm−1 2967, 2933, 1694, 1486, 1578, 1486, 1460, 1440, 1294, 1263, 1248; LRMS (ESI+) m/z 271 ([M + H]+, 100), 273 ([M + H]+, 100); HRMS (ESI+) calculated for [C11H11O3BrNa] 292.97838 and 294.97633, found 292.97873 and 294.97661.
), 7.34 (1 H, s, Ar–), 7.34 (1 H, dd, J = 7.6, 1.9 Hz, Ar–), 7.30 (1 H, td, J = 7.4, 1.3 Hz, Ar–), 6.92 (1 H, ddd, J = 7.8, 7.2, 1.9 Hz, Ar–), 6.74 (1 H, s, Ar–), 5.97 (1 H, ddd, J = 16.9, 10.3, 6.4 Hz, CCH2), 5.06–5.01 (2 H, m, CHC2), 3.92 (6 H, s, 2 × OC3), 3.60 (2 H, dt, J = 6.4, 1.5 Hz, C2CHCH2), 3.29–3.25 (2 H, s, SC2), 3.11–3.07 (2 H, m, SCH2C2); 13C NMR (126 MHz, CDCl3) δ: 192.5 (C), 151.9 (C), 146.9 (C), 142.9 (C), 139.7 (CH), 137.4 (CH), 133.1 (C), 130.3 (CH), 129.5 (C), 128.6 (CH), 128.5 (CH), 115.9 (CH2), 113.5 (CH), 112.1 (CH), 100.6 (C), 56.3 (CH3), 56.1 (CH3), 40.7 (CH2), 37.6 (CH2), 29.8 (CH2); νmax/cm−1 2933, 2844, 1659, 1516, 1266, 1193, 1113; LRMS (ESI+) m/z 491 ([M + Na]+, 100); HRMS (ESI+) calculated for [C20H21IO3SNa] 491.01483, found 491.01437.
), 6.85 (1 H, s, Ar–), 3.95 (3 H, s, OC3), 3.89 (3 H, s, OC3), 3.29 (1 H, dd, J = 16.6, 7.3 Hz, CHCH), 2.71–2.59 (2 H, m, CHC), 1.28 (3 H, d, J = 7.4 Hz, C3); 13C NMR (101 MHz, CDCl3) δ: 208.3 (C), 155.6 (C), 149.6 (C), 148.8 (C), 129.1 (C), 107.5 (CH), 104.6 (CH), 56.3 (CH3), 56.2 (CH3), 42.3 (CH), 34.9 (CH2), 16.7 (CH3); νmax/cm−1 2964, 2931, 2872, 2839, 1684, 1590, 1499, 1457, 1437, 1364, 1319, 1295, 1258, 1239, 1214, 1180, 1124, 1043, 1002; LRMS (ESI+) m/z 435 (100), 229 ([M + Na]+, 74).
), 7.57 (1 H, d, J = 2.6 Hz, Ar–), 7.52 (1 H, d, J = 2.6 Hz, Ar–), 7.32–7.30 (2 H, m, Ar–), 6.95–6.91 (1 H, m, Ar–), 6.09 (1 H, ddt, J = 17.2, 10.3, 5.9 Hz, CCH2), 5.41 (1 H, dq, J = 17.2, 1.5 Hz, CHCH), 5.28 (1 H, dq, J = 10.3, 1.3 Hz, CHCH), 4.56 (2 H, dt, J = 5.9, 1.3 Hz, OC2), 3.32–3.28 (2 H, m, SC2), 3.12–3.07 (2 H, m, SCH2C2); 13C NMR (101 MHz, CDCl3) δ: 189.6 (C), 151.6 (C), 142.5 (C), 139.8 (CH), 134.9 (C), 133.4 (CH), 132.8 (CH), 130.5 (C), 130.2 (CH), 129.7 (C), 128.7 (CH), 128.6 (CH), 127.6 (CH), 119.3 (CH2), 100.5 (C), 76.0 (CH2), 40.3 (CH2), 29.9 (CH2); νmax/cm−1 3070, 2926, 1673, 1643, 1462, 1437, 1418, 1211, 1174; LRMS (ESI+) m/z 515 ([M + Na]+, 100), 413 (42), 393 (46), 360 (16), 331 (11); HRMS (ESI+) calculated for [C18H15Cl2IO2SNa] 514.91067 and 516.90770, found 514.91072 and 516.90775.
), 7.70 (1 H, dd, J = 8.0, 1.6 Hz, Ar–), 7.64 (1 H, dd, J = 7.8, 1.7 Hz, Ar–), 7.39–7.30 (2 H, m, Ar–), 7.05 (1 H, t, J = 7.8 Hz, Ar–), 6.93 (1 H, ddd, J = 7.9, 6.7, 2.3 Hz, Ar–), 6.14 (1 H, ddt, J = 17.2, 10.4, 5.9 Hz, CCH2), 5.43 (1 H, dq, J = 17.2, 1.6 Hz, CHCH), 5.28 (1 H, dq, J = 10.4, 1.3 Hz, CHCH), 4.57 (2 H, dt, J = 5.9, 1.3 Hz, OC2), 3.31–3.27 (2 H, m, SC2), 3.12–3.08 (2 H, m, SCH2C2); 13C NMR (100 MHz, CDCl3) δ: 190.8 (C), 153.8 (C), 142.7 (C), 139.8 (CH), 137.1 (CH), 134.4 (C), 133.1 (CH), 130.2 (CH), 128.59 (2 × CH), 128.56 (CH), 125.3 (CH), 119.2 (C), 118.9 (CH2), 100.5 (C), 76.0 (CH2), 40.4 (CH2), 29.8 (CH2); νmax/cm−1 3064, 2927, 1677, 1640, 1437, 1417, 1229, 1179, 1134, 1010; LRMS (ESI+)m/z 525 ([M + Na]+, 100), 527 ([M + Na]+, 82); HRMS (ESI+) calculated for [C18H16O2SIBrNa] 524.89913 and 526.89704, found 524.89920 and 526.89719.
), 7.61 (1 H, dd, J = 7.8, 1.7 Hz, Ar–), 7.53 (1 H, dd, J = 7.9, 1.7 Hz, Ar–), 7.34–7.30 (2 H, m, Ar–), 7.11 (1 H, t, J = 7.9 Hz, Ar–), 6.93 (1 H, ddd, J = 8.0, 6.8, 2.2 Hz, Ar–), 6.13 (1 H, ddt, J = 17.2, 10.3, 5.9 Hz, CCH2), 5.43 (1 H, dq, J = 17.2, 1.5 Hz, CHCH), 5.28 (1 H, dq, J = 10.3, 1.3 Hz, CHCH), 4.59 (2 H, dt, J = 5.9, 1.3 Hz, OC2), 3.32–3.28 (2 H, m, SC2), 3.12–3.08 (2 H, m, SCH2C2); 13C NMR (101 MHz, CDCl3) δ: 190.7 (C), 152.8 (C), 142.7 (C), 139.7 (CH), 134.3 (C), 134.0 (CH), 133.1 (CH), 130.2 (CH), 129.6 (C), 128.6 (2 × CH), 127.7 (CH), 124.7 (CH), 118.8 (CH2), 100.5 (C), 75.8 (CH2), 40.3 (CH2), 29.8 (CH2); νmax/cm−1 3065, 2928, 1665, 1641, 1440, 1227, 1180, 1145, 978, 939, 748, 733; LRMS (ESI+) m/z 483 ([M + Na]+, 36), 481 ([M + Na]+, 100); HRMS (ESI+) calculated for [C18H16O2SI35ClNa] 480.94964, found 480.95016; calculated for [C18H16O2SI37ClNa] 482.94666, found 482.94720.
), 7.50 (1 H, dd, J = 7.9, 1.4 Hz, Ar–), 7.35–7.29 (2 H, m, 2 × Ar–), 7.23 (1 H, ddd, J = 11.1, 8.2, 1.6 Hz, Ar–), 7.08 (1 H, td, J = 8.0, 4.7 Hz, Ar–), 6.94–6.91 (1 H, m, Ar–), 6.13–6.07 (1 H, m, CCH2), 5.39 (1 H, dq, J = 17.2, 1.5 Hz, CHCH), 5.26 (1 H, dq, J = 10.4, 1.2 Hz, CHCH), 4.69–4.67 (2 H, m, OC2), 3.30–3.27 (2 H, m, SC2), 3.12–3.08 (2 H, m, SCH2C2); 13C NMR (101 MHz, CDCl3) δ: 190.3 (1 C, d, J = 3.3 Hz, C), 156.0 (1 C, d, J = 248.9 Hz, C), 144.9 (1 C, d, J = 12.4 Hz, C), 142.8 (C), 139.7 (CH), 133.6 (1 C, d, J = 1.8 Hz, C), 133.2 (CH), 130.2 (CH), 128.6 (CH), 128.5 (CH), 124.4 (1 C, d, J = 1.4 Hz, CH), 123.7 (1 C, d, J = 7.7 Hz, CH), 120.4 (1 C, d, J = 19.7 Hz, CH), 119.1 (CH2), 100.5 (C), 75.7 (1 C, d, J = 5.8 Hz, CH), 40.4 (CH2), 29.7 (CH2); 19F NMR (376 MHz, CDCl3) δ: −127.80; νmax/cm−1 2928, 1676, 1639, 1468, 1264, 1227, 1009; LRMS (ESI+) m/z 465 ([M + Na]+, 100); HRMS (ESI+) calculated for [C18H16O2FSNa] 464.97919, found 464.97946.
), 8.33 (1 H, dd, J = 9.2, 2.9 Hz, Ar–), 7.84 (1 H, dd, J = 8.0, 1.0 Hz, Ar–), 7.35–7.29 (2 H, m, 2 × Ar–), 7.05 (1 H, d, J = 9.2 Hz, Ar–), 6.94 (1 H, ddd, J = 7.9, 6.8, 2.2 Hz, Ar–), 6.08 (1 H, ddt, J = 17.3, 10.6, 5.2 Hz, CHCHH), 5.50 (1 H, dq, J = 17.3, 1.6 Hz, CHCH), 5.38 (1 H, dq, J = 10.6, 1.3 Hz, CHCH), 4.78 (1 H, dt, J = 5.1, 1.5 Hz, OC2), 3.33–3.29 (2 H, m, SC2), 3.13–3.09 (2 H, m, SCH2C2); 13C NMR (101 MHz, CDCl3) δ: 189.2 (C), 161.2 (C), 142.6 (C), 141.2 (C), 139.8 (CH), 131.3 (CH), 130.2 (CH), 128.7 (CH), 128.64 (CH), 128.63 (CH), 127.8 (C), 125.9 (CH), 119.3 (CH2), 113.3 (CH), 100.5 (C), 70.5 (CH2), 40.3 (CH2), 29.9 (CH2); νmax/cm−1 3084, 1637, 1608, 1563, 1342, 1276, 1086; LRMS (ESI+) m/z 492 ([M + Na]+, 100); HRMS (ESI+) calculated for [C18H16NO4SINa] 491.97369, found 491.97428.
), 7.95 (1 H, dd, J = 8.1, 1.6 Hz, Ar–), 7.84 (1 H, dd, J = 7.9, 1.3 Hz, Ar–), 7.37–7.29 (3 H, m, 3 × Ar–), 6.93 (1 H, ddd, J = 7.8, 7.3, 1.9 Hz, Ar–), 6.69 (1 H, dd, J = 8.6, 0.8 Hz, Ar–), 6.62 (1 H, ddd, J = 8.1, 7.0, 1.1 Hz, Ar–), 5.96 (1 H, ddt, J = 17.2, 10.3, 5.1 Hz, CCHH), 5.31 (1 H, dq, J = 17.2, 1.7 Hz, CHCH), 5.21 (1 H, dq, J = 10.3, 1.6 Hz, CHCH), 3.89 (2 H, tt, J = 5.4, 1.8 Hz, NC2), 3.27–3.24 (2 H, m, SC2), 3.10–3.07 (2 H, m, SCH2C2); 13C NMR (126 MHz, CDCl3) δ: 193.1 (C), 149.0 (C), 143.0 (C), 139.7 (CH), 135.0 (CH), 134.4 (CH), 130.9 (CH), 130.2 (CH), 128.54 (CH), 128.46 (CH), 118.0 (C), 116.4 (CH2), 115.0 (CH), 112.1 (CH), 100.6 (C), 45.3 (CH2), 40.9 (CH2), 29.0 (CH2); νmax/cm−1 3476, 3359, 3059, 2925, 1694, 1630, 1582, 1516, 1447, 1197, 1162, 1011; LRMS (ESI+) m/z 522 (42), 424 ([M + H]+, 100); HRMS (ESI+) calculated for [C18H18INOSNa] 446.00460, found 446.00472.
), 7.28 (1 H, ddd, J = 8.3, 6.9, 1.5 Hz, Ar–), 6.73 (1 H, ddd, J = 8.1, 7.1, 1.1 Hz, Ar–), 6.65 (1 H, dd, J = 8.3, 1.3 Hz, Ar–), 4.44 (1 H, brs, N), 3.55 (1 H, dd, J = 11.7, 5.3 Hz, NHCH), 3.27 (1 H, t, J = 11.6 Hz, NHCH), 2.74–2.64 (1 H, m, CCH3), 1.22 (3 H, d, J = 6.9 Hz, C3); 13C NMR (101 MHz, CDCl3) δ: 196.6 (C), 151.8 (C), 134.9 (CH), 128.0 (CH), 118.9 (C), 118.0 (CH), 115.7 (CH), 48.9 (CH2), 41.2 (CH), 12.7 (CH3); νmax/cm−1 3348, 2964, 2930, 2872, 2815, 1656, 1611, 1512, 1365, 1343, 1237, 1155; LRMS (ESI+) m/z 393 (84), 345 (100), 184 ([M + Na]+, 71); HRMS (ESI+) calculated for [C10H11NONa] 184.07329, found 184.07325.
), 7.77 (1 H, dd, J = 7.8, 1.8 Hz, Ar–), 7.43 (1 H, ddd, J = 8.4, 7.4, 1.8 Hz, Ar–), 7.35 (1 H, dd, J = 7.6, 1.8 Hz, Ar–), 7.29 (1 H, td, J = 7.4, 1.3 Hz, Ar–), 7.09 (1 H, dt, J = 15.7, 4.0 Hz, CH2CCH), 7.03 (1 H, td, J = 7.6, 1.0 Hz, Ar–), 6.93–6.88 (2 H, m, 2 × Ar–), 6.32 (1 H, dt, J = 15.7, 2.1 Hz, CH2CHC), 4.80 (2 H, dd, J = 4.0, 2.1 Hz, ArOC2), 4.21 (2 H, q, J = 7.1 Hz, CO2C2), 3.30–3.25 (2 H, m, SC2), 3.13–3.07 (2 H, m, SCH2C2), 1.28 (3 H, t, J = 7.1 Hz, C3); 13C NMR (101 MHz, CDCl3) δ: 190.7 (C), 166.1 (C), 156.1 (C), 142.9 (C), 141.6 (CH), 139.6 (CH), 133.6 (CH), 130.2 (CH), 129.9 (CH), 128.5 (CH), 128.4 (CH), 127.8 (C), 122.7 (CH), 121.3 (CH), 113.3 (CH), 100.5 (C), 67.6 (CH2), 60.7 (CH2), 40.5 (CH2), 29.6 (CH2), 14.3 (CH3); νmax/cm−1 2980, 2935, 1718, 1669, 1637, 1596, 1484, 1443, 1288, 1180, 1039; LRMS (ESI+) m/z 514 (100), 497 ([M + H]+, 40); HRMS (ESI+) calculated for [C21H21O4ISNa] 519.00974, found 519.00972.
), 7.50–7.45 (1 H, m, Ar–), 7.04–7.00 (1 H, m, Ar–), 6.97 (1 H, dd, J = 8.4, 1.2 Hz, Ar–), 4.60 (1 H, dd, J = 11.2, 5.3 Hz, ArOCH), 4.30 (1 H, dd, 11.9, 11.2 Hz, ArOCH), 4.19 (2 H, qd, J = 7.2, 2.0 Hz, CO2C2), 3.33 (1 H, dddd, J = 11.9, 8.3, 5.1, 5.1 Hz, ArCOC), 2.93 (1 H, dd, J = 16.9, 4.8 Hz, CHCO2Et), 2.41 (1 H, dd, J = 16.9, 8.2 Hz, CHCO2Et), 1.28 (3 H, t, J = 7.2 Hz, C3); 13C NMR (101 MHz, CDCl3) δ: 192.7 (C), 171.5 (C), 161.9 (C), 136.1 (CH), 127.5 (CH), 121.7 (CH), 120.7 (C), 118.0 (CH), 70.4 (CH2), 61.1 (CH2), 42.7 (CH), 30.5 (CH2), 14.3 (CH3); νmax/cm−1 2980, 2927, 1730, 1688, 1605, 1479, 1298, 1213, 1175, 1128, 1035, 1012; LRMS (ESI+) m/z 682 (29), 491 (24), 420 (100), 403 (43), 257 (49), 234 (M+, 33).
), 7.43 (1 H, ddd, J = 8.4, 7.3, 1.8 Hz, Ar–), 7.35 (1 H, dd, J = 7.6, 1.8 Hz, Ar–), 7.29 (1 H, td, J = 7.4, 1.3 Hz, Ar–), 7.02 (1 H, d, J = 9.0 Hz, Ar–), 7.00–6.96 (1 H, m, Ar–), 6.93–6.89 (1 H, m, Ar–), 6.00–5.91 (2 H, m, CC), 4.94–4.88 (1 H, m, OC), 3.26–3.22 (2 H, m, SC2), 3.10–3.06 (2 H, m, SCH2C2), 2.22–2.11 (1 H, m, OCHCH), 2.06–1.95 (4 H, m, OCHCHC2CH), 1.68–1.65 (1 H, m, OCHCHHCH2CH); 13C NMR (101 MHz, CDCl3) δ: 190.9 (C), 156.8 (C), 143.2 (C), 139.6 (CH), 133.5 (CH), 132.6 (CH), 130.2 (CH), 130.0 (CH), 128.4 (CH), 128.3 (CH), 128.0 (C), 125.8 (CH), 120.4 (CH), 114.5 (CH), 100.5 (C), 72.7 (CH), 40.5 (CH2), 29.6 (CH2), 28.5 (CH2), 25.2 (CH2), 19.2 (CH2); νmax/cm−1 3029, 2932, 2865, 1676, 1631, 1593, 1478, 1447, 1282, 1239, 1190, 1009; LRMS (ESI+) m/z 619 (100), 487 ([M + Na]+, 78), 321 (47); HRMS (ESI+) calculated for [C21H21O2ISNa] 487.01991, found 487.02033.
), 7.47 (1 H, ddd, J = 8.3, 7.2, 1.8 Hz, Ar–), 7.01–6.96 (2 H, m, 2 × Ar–), 4.60–4.57 (1 H, m, OC), 2.57–2.53 (1 H, m, COC), 2.13–2.09 (1 H, m, C), 1.79–1.58 (6 H, m, 6 × C), 1.47–1.37 (1 H, m, C); 13C NMR (101 MHz, CDCl3) δ: 195.9 (C), 161.4 (C), 136.0 (CH), 127.5 (CH), 121.2 (CH), 119.8 (C), 118.0 (CH), 76.3 (CH), 48.1 (CH), 29.5 (CH2), 24.13 (CH2), 24.06 (CH2), 20.6 (CH2); νmax/cm−1 2934, 2860, 1685, 1605, 1461, 1306, 1229, 1150, 1121; LRMS (APCI) m/z 389 (30), 203 ([M + H]+, 100).
), 7.79 (1 H, dd, J = 7.8, 1.8 Hz, Ar–), 7.48 (1 H, ddd, J = 8.4, 7.3, 1.8 Hz, Ar–), 7.36 (1 H, dd, J = 7.6, 1.9 Hz, Ar–), 7.30 (1 H, td, J = 7.4, 1.3 Hz, Ar–), 7.15 (1 H, dd, J = 8.4, 0.9 Hz, Ar–), 7.06 (1 H, td, J = 7.6, 0.9 Hz, Ar–), 6.94–6.90 (1 H, m, Ar–), 4.82 (2 H, d, J = 2.4 Hz, OC2), 3.29–3.25 (2 H, m, SC2), 3.12–3.08 (2 H, m, SCH2C2), 2.55 (1 H, t, J = 2.4 Hz, CH2CC); 13C NMR (101 MHz, CDCl3) δ: 190.8 (C), 155.8 (C), 143.0 (C), 139.7 (CH), 133.5 (CH), 130.3 (CH), 130.0 (CH), 128.6 (CH), 128.5 (CH), 128.1 (C), 121.6 (CH), 114.2 (CH), 100.5 (C), 78.1 (C), 76.4 (CH), 56.8 (CH2), 40.5 (CH2), 29.7 (CH2); νmax/cm−1 3291, 3059, 2923, 1669, 1634, 1596, 1482, 1447, 1286, 1260, 1011; LRMS (ESI+) m/z 445 ([M + Na]+, 100); HRMS (ESI+) calculated for [C18H15O2SINa] 444.97296, found 444.97346.
), 7.46 (1 H, ddd, J = 8.4, 7.4, 1.8 Hz, Ar–), 7.44–7.41 (2 H, m, 2 × Ar–), 7.36–7.28 (3 H, m, 3 × Ar–), 7.28–7.24 (2 H, m, 2 × Ar–), 7.05–7.00 (2 H, m, 2 × Ar–), 6.90 (1 H, ddd, J = 7.8, 7.4, 1.8 Hz, Ar–), 6.80 (1 H, dt, J = 16.0, 1.6 Hz, CPh), 6.45 (1 H, dt, J = 16.0, 5.7 Hz, CCHPh), 4.85 (2 H, dd, J = 5.6, 1.6 Hz, OC2), 3.30–3.26 (2 H, m, SC2), 3.12–3.08 (2 H, m, SCH2C2); 13C NMR (101 MHz, CDCl3) δ: 190.9 (C), 157.1 (C), 143.1 (C), 139.6 (CH), 136.5 (C), 133.6 (CH), 133.3 (CH), 130.2 (CH), 129.9 (CH), 128.7 (2 × CH), 128.5 (CH), 128.4 (CH), 128.1 (CH), 127.6 (C), 126.8 (2 × CH), 124.0 (CH), 120.9 (CH), 113.8 (CH), 100.6 (C), 69.9 (CH2), 40.5 (CH2), 29.7 (CH2); νmax/cm−1 3057, 3025, 2924, 2863, 1671, 1633, 1594, 1482, 1446, 1285, 1241, 1193, 1162, 1111; LRMS (ESI+) m/z 523 ([M + Na]+, 100); HRMS (ESI+) calculated for [C24H21O2ISNa] 523.01991, found 523.02024.
), 8.55 (1 H, d, J = 13.5 Hz, CC), 8.07 (1 H, dd, J = 8.0, 1.4 Hz, Ar–), 7.83 (1 H, dd, J = 7.8, 1.0 Hz, Ar–), 7.60–7.55 (1 H, m, Ar–), 7.40 (1 H, d, J = 8.1 Hz, Ar–), 7.34–7.28 (2 H, m, Ar–), 7.16 (1 H, ddd, J = 7.3, 7.1, 0.9 Hz, Ar–), 6.93 (1 H, ddd, J = 7.8, 6.7, 2.3 Hz, Ar–), 4.43 (2 H, q, J = 7.1 Hz, C2CH3), 4.27 (2 H, q, J = 7.1 Hz, C2CH3), 3.40–3.36 (2 H, m, SC2), 3.14–3.10 (2 H, m, C2Ar), 1.41 (3 H, t, J = 7.1 Hz, C3), 1.34 (3 H, t, J = 7.1 Hz, C3); 13C NMR (101 MHz, CDCl3) δ: 193.4 (C), 167.0 (C), 166.2 (C), 149.7 (CH), 142.6 (C), 139.8 (CH), 139.3 (C), 134.7 (CH), 130.7 (CH), 130.2 (CH), 128.62 (CH), 128.56 (CH), 124.5 (C), 123.5 (CH), 115.9 (CH), 100.6 (C), 97.3 (C), 60.7 (CH2), 60.5 (CH2), 40.3 (CH2), 29.6 (CH2), 14.62 (CH3), 14.56 (CH3); νmax/cm−1 2978, 2928, 1716, 1693, 1656, 1610, 1589, 1225, 1191, 1170; LRMS (ESI+) m/z 576 ([M + Na]+, 100); HRMS (ESI+) calculated for [C23H24NO5SINa] 576.03121, found 576.03198.
), 7.83 (1 H, dd, J = 7.8, 1.3 Hz, Ar–), 7.74 (1 H, d, J = 12.3 Hz, OCCH), 7.55 (1 H, ddd, J = 8.2, 7.5, 1.7 Hz, Ar–), 7.33–7.28 (3 H, m, 3 × Ar–), 7.14 (1 H, dd, J = 8.2, 0.9 Hz, Ar–), 6.93 (1 H, ddd, J = 7.9, 6.7, 2.3 Hz, Ar–), 5.57 (1 H, d, J = 12.2 Hz, OCHC), 4.20 (2 H, q, J = 7.1 Hz, OC2), 3.30–3.26 (2 H, m, SC2), 3.11–3.07 (2 H, m, SCH2C2), 1.28 (3 H, t, J = 7.1 Hz, C3); 13C NMR (101 MHz, CDCl3) δ: 189.8 (C), 166.9 (C), 158.9 (CH), 153.2 (C), 142.6 (C), 139.7 (CH), 133.8 (CH), 130.2 (CH), 130.0 (CH), 129.5 (C), 128.6 (2 × CH), 125.4 (CH), 119.8 (CH), 103.4 (CH), 100.5 (C), 60.3 (CH2), 40.3 (CH2), 29.7 (CH2), 14.4 (CH3); νmax/cm−1 3056, 2978, 1712, 1646, 1479, 1446, 1225, 1199, 1123, 1046; LRMS (ESI+) m/z 505 ([M + Na]+, 100); HRMS (ESI+) calculated for [C20H19IO4SNa] 504.99409, found 504.99422.
), 7.70 (1 H, dt, J = 8.1, 1.1 Hz, Ar–), 7.46 (1 H, s, Ar–), 7.37–7.31 (4 H, m, Ar–), 7.20–7.16 (1 H, m, Ar–), 6.93 (1 H, ddd, 8.0, 7.0, 2.1 Hz, Ar–), 6.00 (1 H, ddt, J = 17.1, 10.3, 5.0 Hz, CCH2), 5.17 (2 H, dt, J = 5.0, 1.7 Hz, NC2), 5.12 (1 H, dq, J = 10.3, 1.5 Hz, CHCH), 4.92 (1 H, dq, J = 17.1, 1.6 Hz, CHCH), 3.33–3.29 (2 H, m, SC2), 3.13–3.10 (2 H, m, SCH2C2); 13C NMR (101 MHz, CDCl3) δ: 184.4 (C), 142.8 (C), 139.7 (CH), 139.5 (C), 133.83 (C), 133.77 (CH), 130.3 (CH), 128.5 (2 × CH), 126.2 (C), 126.0 (CH) 123.0 (CH), 121.2 (CH), 116.4 (CH2), 110.9 (CH), 110.8 (CH), 100.6 (C), 47.2 (CH2), 40.8 (CH2), 28.9 (CH2); νmax/cm−1 3058, 2926, 1640, 1510, 1454, 1154, 1131, 1010; HRMS (ESI+) calculated for [C20H18NOISNa] 470.00460, found 470.00477.
), 7.42 (1 H, dddd, J = 8.4, 1.0, 1.0, 1.0 Hz, Ar–), 7.36 (1 H, ddd, J = 8.4, 6.8, 1.1 Hz, Ar–), 7.19 (1 H, ddd, J = 8.2, 6.8, 1.2 Hz, Ar–), 7.02 (1 H, d, J = 0.9 Hz, Ar–), 4.68 (1 H, dd, J = 10.8, 8.0 Hz, NCH), 3.98 (1 H, dd, J = 10.8, 4.8 Hz, NCH), 3.32–3.27 (1 H, m, NCH2C), 1.46 (3 H, d, J = 7.5 Hz, CH3); 13C NMR (101 MHz, CDCl3) δ: 196.2 (C), 135.3 (C), 135.2 (C), 132.3 (C), 125.2 (CH), 124.3 (CH), 121.6 (CH), 110.6 (CH), 99.4 (CH), 48.0 (CH2), 45.6 (CH), 15.7 (CH3); νmax/cm−1 3077, 2972, 2882, 1711, 1539, 1348, 1232, 1166; LRMS (ESI+) m/z 238 (77), 234 (30), 214 (32), 200 (40), 186 ([M + H]+, 100).
:1 diasteteromeric mixture of 26a (670 mg, 1.28 mmol, 82%) as a colourless oil. Rf: 0.19 (10% diethyl ether in hexanes); 1H NMR (400 MHz, CDCl3) δ: 7.84–7.82 (1 H + 0.6 × 1 H, m, Ar–), 7.35–7.28 (3 H + 0.6 × 3 H, m, 3 × Ar–), 6.94–6.90 (1 H + 0.6 × 1 H, m, Ar–), 6.75 (1 H, s, Ar–), 6.70 (0.6 × 1 H, s, Ar–), 5.54 (0.6 × 1 H, dt, J = 15.0, 7.0 Hz, CH2CCH), 5.35 (1 H, dtd, 10.7, 7.4, 0.8 Hz, CH2CCH), 4.99 (0.6 × 1 H, ddt, J = 15.2, 8.5, 1.3 Hz, CH2CHC), 4.78–4.73 (1 H, m, CH2CHC), 3.92 (3 H, s, ArOC3), 3.91 (0.6 × 3 H, s, ArOC3), 3.91 (3 H, s, ArOC3), 3.91 (0.6 × 3 H, s, ArOC3), 3.29–3.25 (2 H + 0.6 × 2 H, m, SC2), 3.11–3.07 (2 H + 0.6 × 2 H, m, SCH2C2), 2.93–2.89 (2 H, m, ArC2), 2.87–2.83 (0.6 × 2 H, m, ArC2), 2.48–2.46 (2 H, m, C2CHCH), 2.27–2.25 (0.6 × 2 H, m, C2CHCH), 1.56–1.47 (1 H, m, CHCHC), 1.38–1.29 (1 H, m, CHCHC), 0.68–0.63 (2 H + 0.6 × 2 H, m, 2 × CH(CH)2), 0.31–0.26 (2 H + 0.6 × 2 H, m, 2 × CH(CH)2); 13C NMR (101 MHz, CDCl3) δ: 192.39 (C), 192.35 (C), 151.64 (C), 151.60 (C), 146.63 (C), 146.59 (C), 142.84 (C), 142.83 (C), 139.7 (CH + CH), 135.49 (C), 135.46 (C), 134.9 (CH), 134.6 (CH), 130.18 (CH), 130.17 (CH), 129.4 (C), 129.3 (C), 128.51 (CH + CH), 128.49 (CH + CH), 127.3 (CH), 127.1 (CH), 113.74 (CH), 113.69 (CH), 112.14 (CH), 112.08 (CH), 100.63 (C), 100.60 (C), 56.24 (CH3), 56.22 (CH3), 56.08 (CH3), 56.06 (CH3), 40.70 (CH2), 40.67 (CH2), 34.6 (CH2), 33.8 (CH2), 33.7 (CH2), 29.8 (CH2), 29.7 (CH2 + CH2), 13.6 (CH), 9.7 (CH), 7.0 (2 × CH2), 6.5 (2 × CH2); νmax/cm−1 3001, 2934, 1661, 1516, 1464, 1265, 1193, 1111; LRMS (ESI+) m/z 545 ([M + Na]+, 100); HRMS (ESI+) calculated for [C24H27O3SINa] 545.06178, found 545.06249.
), 6.66 (1 H, s, Ar–), 5.65 (1 H, dtd, J = 10.8, 7.2, 1.0 Hz, CHCCH2), 5.50 (1 H, ddt, J = 10.8, 8.8, 1.6 Hz, CCHCH2), 3.93 (3 H, s, OC3), 3.91 (3 H, s, OC3), 3.44 (1 H, dddd, J = 10.9, 8.8, 4.6, 1.0 Hz, CCHCH), 3.04–2.92 (2 H, m, ArC2), 2.21–1.97 (4 H, m, CHC2 + C2CH3), 1.03 (3 H, t, J = 7.5 Hz, CH2C3); 13C NMR (101 MHz, CDCl3) δ: 197.6 (C), 153.6 (C), 148.1 (C), 138.8 (C), 135.1 (CH), 126.1 (CH), 125.8 (C), 110.3 (CH), 109.1 (CH), 56.20 (CH3), 56.16 (CH3), 46.2 (CH), 30.8 (CH2), 28.3 (CH2), 21.3 (CH2), 14.4 (CH3); νmax/cm−1 2959, 2932, 2867, 1670, 1599, 1511, 1265, 1146; LRMS (ESI+) m/z 543 (100), 283 ([M + Na]+, 80); HRMS (ESI+) calculated for [C16H20O3Na] 283.13047, found 283.13072.
), 7.29–7.27 (2 H, m, Ar–), 6.93–6.89 (1 H, m, Ar–), 5.10–5.07 (1 H, m, CC), 3.14–3.10 (2 H, m, SC2), 3.01–2.97 (2 H, m, SCH2C2), 2.56 (1 H, dd, J = 14.5, 5.9 Hz, CHCO), 2.37 (1 H, dd, J = 14.5, 8.2 Hz, CHCO), 2.07–1.95 (3 H, m, C2CH2C), 1.70 (3 H, d, J = 1.1 Hz, (3C)2C), 1.60 (3 H, s, (3C)2C), 1.39–1.32 (1 H, m, CH2CHCH), 1.27–1.20 (1 H, m, CH2CHCH), 0.94 (3 H, d, J = 6.7 Hz, 3CCH); 13C NMR (101 MHz, CDCl3) δ: 199.0 (C), 142.8 (C), 139.7 (CH), 131.8 (C), 130.2 (CH), 128.53 (CH), 128.51 (CH), 124.3 (CH), 100.5 (C), 51.5 (CH2), 40.8 (CH2), 36.8 (CH2), 30.9 (CH), 29.0 (CH2), 25.9 (CH3), 25.5 (CH2), 19.6 (CH3), 17.8 (CH3); νmax/cm−1 2963, 2926, 1688, 1465, 1436, 1010; LRMS (ESI+) m/z 471 (100), 455 (66), 439 ([M + Na]+, 93); HRMS (ESI+) calculated for [C18H25OISNa] 439.05630, found 439.05665.
HCO), 2.31–2.28 (1 H, m, CHCO), 2.19–1.66 (13 H, m, 2 × CHCO, 2 × CCH2CO, 2 × CH3CHCH, 2 × CH3CHCH2CH, CH3CHCH2CH, 2 × CCO, 2 × (CH3)2C), 1.51–1.32 (3 H, m, 2 × CH3CHCH, CH3CHCH2CH), 1.00 (3 H, d, J = 6.4 Hz, CHC3), 0.98 (3 H, d, J = 6.7 Hz, CHC3), 0.93 (3 H, d, J = 6.6 Hz, H3CCHC3), 0.91 (3 H, d, J = 6.9 Hz, H3CCHC3), 0.85 (3 H, d, J = 6.8 Hz, H3CCHC3), 0.84 (3 H, d, J = 6.6 Hz, H3CCHC3); 13C NMR (126 MHz, CDCl3) δ: 214.7 (C), 212.6 (C), 57.3 (CH), 56.1 (CH), 51.0 (CH2), 48.2 (CH2), 35.6 (CH), 34.5 (CH), 34.1 (CH2), 29.6 (CH2), 28.0 (CH2), 27.1 (CH2), 27.0 (CH), 26.0 (CH), 22.4 (CH3), 21.6 (CH3), 21.4 (CH3), 21.0 (CH3), 20.0 (CH3), 18.8 (CH3); νmax/cm−1 2955, 2926, 2871, 1708, 1456, 1368, 1203, 1116.
Alternative photo-induced cyclizations
), 7.35–7.34 (2 H, m, 2 × Ar–), 4.06 (3 H, s, OC3); 13C NMR (126 MHz, CD3CN) δ: 171.2 (C), 136.7 (2 × CH), 118.8 (2 × CH), 102.5 (C), 58.4 (CH3); 19F NMR (471 MHz, CD3CN) δ: −151.7; 11B NMR (160 MHz, CD3CN) δ: −1.15; νmax/cm−1 3120, 2252, 1583, 1569, 1291, 1051, 1002; LRMS (ESI+) m/z 357 (100), 135 (M+, 56).
), 8.87–8.84 (2 H, m, 2 × Ar–); 13C NMR (126 MHz, (CD3)2CO) δ: 155.1 (C), 135.9 (2 × CH), 127.4 (2 × CH), 122.5 (C); 19F NMR (471 MHz, (CD3)2CO) δ: −150.7; 11B NMR (160 MHz, (CD3)2CO) δ: −0.96; νmax/cm−1 3119, 3106, 2307, 1539, 1359, 1317, 1040; LRMS (ESI+) m/z 387 (45), 150 (M+, 100).
), 8.32–8.29 (2 H, m, 2 × Ar–); 13C NMR (101 MHz, (CD3)2CO) δ: 138.4 (C), 135.9 (2 × CH), 134.9 (2 × CH), 115.5 (C); 19F NMR (471 MHz, (CD3)2CO) δ: −150.8; 11B NMR (160 MHz, (CD3)2CO) δ: −0.94; νmax/cm−1 3103, 2287, 1556, 1041, 1009; LRMS (ESI−) m/z 455 (100), 185 (M+, 79) 183 (M+, 79), 157 ([M–N2]+, 79), 155 ([M–N2]+, 79).
:1, 3 × 15 mL). The organic extracts were washed with brine (20 mL), dried (Na2SO4) and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (1–5% diethyl ether in hexanes) to give 13b (19 mg, 0.082 mmol, 20%) as a dark yellow solid and recovered starting material (122 mg, 0.25 mmol, 61%). Rf: 0.38 (10% diethyl ether in hexanes); m.p. 84–86 °C: 1H NMR (400 MHz, CDCl3) δ: 7.77 (1 H, d, J = 2.6 Hz, Ar–), 7.53 (1 H, d, J = 2.6 Hz, Ar–), 4.63 (1 H, dd, J = 11.5, 5.1 Hz, OCH), 4.23 (1 H, t, J = 11.3 Hz, OCH), 2.89 (1 H, dqd, J = 11.2, 7.0, 5.1 Hz, CCH3), 1.23 (3 H, d, J = 7.0 Hz, C3); 13C NMR (101 MHz, CDCl3) δ: 192.9 (C), 156.0 (C), 135.4 (CH), 126.8 (C), 125.6 (CH), 123.8 (C), 122.2 (C), 72.9 (CH2), 40.5 (CH), 10.6 (CH3); νmax/cm−1 3074, 2989, 2932, 2879, 1694, 1591, 1469, 1435, 1372, 1291, 1245, 1205, 1183, 1012; HRMS (ESI+) calculated for [C10H9Cl2O2] 230.99741 and 232.99446, found 230.99739 and 232.99445.
:1, 3 × 15 mL). The organic extracts were washed with brine (20 mL), dried (Na2SO4) and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (2–5% diethyl ether in hexanes) to give 14b (9.0 mg, 0.037 mmol, 18%) as a pale yellow oil and recovered starting material (67 mg, 0.13 mmol, 66%). Rf: 0.33 (10% diethyl ether in hexanes); 1H NMR (400 MHz, CDCl3) δ: 7.86 (1 H, dd, J = 7.9, 1.6 Hz, Ar–), 7.72 (1 H, dd, J = 7.7, 1.6 Hz, Ar–), 6.92 (1 H, t, J = 7.8 Hz, Ar–), 4.64 (1 H, dd, J = 11.4, 5.1 Hz, OCH), 4.24 (1 H, t, J = 11.3 Hz, OCH), 2.90 (1 H, dqd, J = 11.1, 7.0, 5.1 Hz, CCH3), 1.23 (3 H, d, J = 7.0 Hz, C3); 13C NMR (101 MHz, CDCl3) δ: 194.0 (C), 158.2 (C), 139.1 (CH), 126.9 (CH), 122.2 (CH), 121.9 (C), 111.5 (C), 72.9 (CH2), 40.5 (CH), 10.7 (CH3); νmax/cm−1 2975, 2932, 2875, 1693, 1594, 1468, 1439, 1288, 1259, 1221, 1071; LRMS (ESI+) m/z 283 (59), 243 ([M + Na]+, 100), 241 ([M + Na]+, 100); HRMS (ESI+) calculated for [C10H10O2Br] 240.98587 and 242.98382, found 240.98591 and 242.98384.
:1, 3 × 15 mL). The organic extracts were washed with brine (20 mL), dried (Na2SO4) and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (60–80% dichloromethane in hexanes) to give 17b (6.0 mg, 0.029 mmol, 18%) as a white solid and recovered starting material (49 mg, 0.10 mmol, 66%). Rf: 0.23 (20% diethyl ether in hexanes); m.p. 123–125 °C; 1H NMR (400 MHz, CDCl3) δ: 8.79 (1 H, d, J = 2.9 Hz, Ar–), 8.32 (1 H, dd, J = 9.2, 2.9 Hz, Ar–), 7.10 (1 H, d, J = 2.9 Hz, Ar–), 4.64 (1 H, dd, J = 11.5, 5.3 Hz, OCH), 4.26 (1 H, t, J = 11.4 Hz, OCH), 2.95 (1 H, dqd, J = 11.4, 7.0, 5.2 Hz, CCH3), 1.26 (3 H, t, J = 7.0 Hz, C3); 13C NMR (101 MHz, CDCl3) δ: 192.7 (C), 165.8 (C), 142.4 (C), 130.2 (CH), 124.1 (CH), 120.3 (C), 119.3 (CH), 72.8 (CH2), 40.6 (CH), 10.5 (CH3); νmax/cm−1 3089, 2926, 2889, 2852, 1699, 1616, 1519, 1484, 1437, 1343, 1296, 1263, 1133, 1008; LRMS (ESI+) m/z 413 (59), 362 (62), 301 (79), 230 ([M + Na]+, 100); HRMS (ESI+) calculated for [C10H10NO4] 208.06043, found 208.06048.
:1, 3 × 15 mL). The organic extracts were washed with brine (20 mL), dried (Na2SO4) and concentrated in vacuo. The residue was purified by flash chromatography on silica gel (50–100% dichloromethane in hexanes – 1% diethyl ether in dichloromethane) to give 24b (15 mg, 0.072 mmol, 36%) as a yellow oil and recovered starting material (60 mg, 0.12 mmol, 61%). Rf: 0.25 (20% diethyl ether in hexanes); 1H NMR (400 MHz, CDCl3) δ: 7.68 (1 H, ddd, J = 7.7, 1.4, 0.6 Hz, Ar–), 7.62 (1 H, ddd, J = 8.4, 7.1, 1.5 Hz, Ar–), 7.14–7.08 (2 H, m, 2 × Ar–), 4.88 (1 H, dd, J = 7.6, 3.8 Hz, OCCH2), 4.19–4.11 (2 H, m, OC2), 3.07 (1 H, dd, J = 17.0, 3.8 Hz, OCHCH), 2.82 (1 H, dd, J = 17.0, 7.6 Hz, OCHCH), 1.18 (3 H, t, J = 7.1 Hz, C3); 13C NMR (101 MHz, CDCl3) δ: 200.5 (C), 172.7 (C), 169.5 (C), 138.2 (CH), 124.4 (CH), 122.3 (CH), 121.1 (C), 113.7 (CH), 81.2 (CH), 61.4 (CH2), 36.2 (CH2), 14.1 (CH3); νmax/cm−1 2981, 2926, 1718, 1650, 1614, 1464, 1326, 1192, 1026.
Intermolecular additions
084, 1602, 1585, 1327, 1258, 1157, 1018; 1H NMR (400 MHz; CDCl3) δ: 7.19–7.17 (3 H, m), 2.93 (1 H, ttt, J = 11.2, 3.2), 2.30 (3 H, s), 2.22 (3 H, s), 1.90–1.84 (2 H, m), 1.83–1.77 (2 H, m), 1.45–1.40 (2 H, m), 1.32–1.28 (4 H, m); 13C NMR (101 MHz; CDCl3) δ: 210.7 (C), 141.1 (C), 138.1 (C), 134.4 (C), 131.6 (CH), 125.3 (CH), 124.2 (CH), 50.2 (CH), 29.8 (CH2), 28.7 (2 × CH2), 26.1 (CH2), 25.9 (CH2), 20.4 (CH3), 16.6 (CH3); m/z HRMS (ESI): calcd for C15H20ONa ([M + Na]+): 239.14064; found: 239.14086; and compound 39 (2 mg, 7%) as a yellow oil. νmax/cm−1: 2926, 2853, 17084, 1602, 1585, 1327, 1258, 1157, 1018; 1H-NMR (400 MHz; CDCl3) δ: 7.31 (1 H, d, J = 7.7), 7.16 (1 H, d, J = 7.6), 3.29–3.23 (1 H, m), 2.71 (1 H, td, J = 6.9, 4.4), 2.59 (3 H, s), 2.30 (3 H, s), 2.16–2.04 (3 H, m), 1.74–1.65 (2 H, m), 1.58–1.48 (2 H, m), 1.26–1.24 (1 H, m); 13C NMR (101 MHz; CDCl3) δ: 209.1 (C), 157.0 (C), 137.5 (C), 136.3 (C), 135.3 (CH), 133.1 (C), 121.8 (CH), 49.5 (CH), 38.0 (CH), 32.1 (CH2), 23.4 (CH2), 23.1 (CH2), 22.7 (CH2), 19.2 (CH3), 13.9 (CH3); m/z HRMS (ESI): calcd for C15H18ONa ([M + Na]+): 237.12499; found: 237.12521.
Cascade intramolecular cyclization – intermolecular addition
), 7.46 (1 H, ddd, J = 8.3, 7.1, 1.8 Hz, Ar–), 7.01 (1 H, ddd, J = 7.8, 7.1, 1.1 Hz, Ar–), 6.95 (1 H, d, J = 8.3 Hz, Ar–), 4.52 (1 H, dd, J = 11.4, 4.4 Hz, OCH), 4.28 (1 H, dd, J = 11.4, 8.4 Hz, OCH), 2.66–2.63 (1 H, m, OCH2C), 1.91–1.84 (1 H, m, CHCH2CHCH2), 1.57–1.28 (5 H, m, CHC2CHC2), 0.55–0.47 (2 H, m, C2Si(CH3)3) −0.03 (9 H, s, Si(C3)3); 13C NMR (126 MHz, CDCl3) δ: 194.8 (C), 161.6 (C), 135.8 (CH), 127.6 (CH), 121.5 (CH), 120.8 (C), 117.8 (CH), 70.6 (CH2), 46.1 (CH), 31.0 (CH2), 26.2 (CH2), 24.1 (CH2), 16.6 (CH2), −1.6 (3 × CH3); νmax/cm−1 2951, 2922, 2858, 1691, 1606, 1478, 1246; LRMS (ESI+) m/z 435 (100), 344 (45), 299 ([M + Na]+, 40), 254 (44), 186 (54), 130 (50); HRMS (ESI+) calculated for [C16H24O2SiNa] 299.14378, found 299.14386.
), 6.63 (1 H, dd, J = 8.2, 1.5 Hz, Ar–), 6.46 (1 H, dd, J = 7.8, 2.5 Hz, Ar–), 6.07 (1 H, ddt, J = 17.2, 10.5, 1.5 Hz, CH2C), 5.40 (1 H, dq, J = 17.2, 1.6 Hz CHCH), 5.27 (1 H, dq, J = 10.4, 1.4 Hz, CHCH), 4.80 (2 H, dt, J = 5.6, 1.4 Hz, OC2); 13C NMR (101 MHz, CDCl3) δ: 162.7 (1 C, d, J = 13.6 Hz, C), 162.3 (1 C, d, J = 240.4 Hz, C), 142.7 (1 C, d, J = 8.0 Hz, CH), 133.0 (CH), 118.1 (CH2), 107.4 (1 C, d, J = 5.1 Hz, CH), 100.2 (1 C, d, J = 35.5 Hz, CH), 67.3 (CH2); 19F NMR (376 MHz, CDCl3) δ: −70.25; νmax/cm−1 2952, 2923, 1614, 1576, 1467, 1440, 1422, 1317, 1230, 1030; LRMS (ESI+) m/z 441 (67), 301 (100), 214 (33), 176 ([M + Na]+, 39).
), 7.62 (1 H, q, J = 8.2 Hz, Ar–), 7.46 (1 H, ddd, J = 8.4, 7.2, 1.8 Hz, Ar–), 7.02 (1 H, ddd, J = 7.9, 7.1, 1.0 Hz, Ar–), 6.96 (1 H, dd, J = 8.3, 0.8 Hz, Ar–), 6.58 (1 H, dd, J = 7.9, 1.4 Hz, Ar–), 6.44 (1 H, dd, J = 7.8, 2.5 Hz, Ar–), 4.53 (1 H, dd, J = 11.4, 4.4 Hz, OCHCH), 4.29 (1 H, dd, J = 11.4, 8.5 Hz, OCHCH), 4.28 (2 H, t, J = 6.6 Hz, OC2CH2), 2.72–2.64 (1 H, m, OCHHC), 2.03–1.89 (1 H, m, COCHCH), 1.87–1.77 (2 H, m, OCH2C2), 1.69–1.52 (3 H, m, COCHCHC2); 13C NMR (100 MHz, CDCl3) δ: 194.5 (C), 163.2 (1 C, d, J = 13.7 Hz, C), 162.4 (1 C, d, J = 240.1 Hz, C), 161.6 (1 C, d, J = 8.1 Hz, CH), 135.9 (CH), 127.6 (CH), 121.5 (CH), 120.8 (C), 117.8 (CH), 107.3 (1 C, d, J = 5.1 Hz, CH), 99.9 (1 C, d, J = 35.6 Hz, CH), 70.6 (CH2), 66.4 (CH2), 46.1 (CH), 29.0 (CH2), 26.3 (CH2), 23.8 (CH2); 19F NMR (376 MHz, CDCl3) δ: −70.21; νmax/cm−1 2925, 2857, 1689, 1606, 1574, 1478, 1453, 1439, 1322, 1229, 1016; LRMS (ESI+) m/z 653 (41), 531 (13), 338 ([M + Na]+, 100); HRMS (ESI+) calculated for [C18H18FNO3Na] 338.11629, found 338.11674; and 47 (18.0 mg, 0.038 mmol, 24%) as an inseparable mixture of diastereomers isolated as a pale yellow oil. Rf: 0.09 (10% diethyl ether in hexanes); 1H NMR (400 MHz, CDCl3) δ: 7.89–7.87 (1 H, m, Ar–H), 7.63–7.59 (2 H, m, 2 × Ar–H), 7.48–7.44 (1 H, m, Ar–H), 7.02–6.99 (1 H, m, Ar–H), 6.96–6.94 (1 H, m, Ar–H), 6.59–6.55 (2 H, m, 2 × Ar–H), 6.45–6.41 (2 H, m, 2 × Ar–H), 4.53–4.50 (1 H, m, OCHHCHC(O)), 4.31–4.19 (5 H, m, OCH2CHCH2CH2CH2 + OCHHCHC(O)), 2.68–2.61 (1 H, m, C(O)CH), 1.98–1.78 (4 H, m, C(O)CHCHHCH2CHCH2CH2), 1.59–1.56 (5 H, m, C(O)CHCHHCH2CHCH2); 13C NMR (100 MHz, CDCl3) δ: 194.5, 163.57, 163.56, 163.38, 163.32, 163.25, 163.2, 161.6, 161.2, 142.7, 142.62, 142.60, 142.5, 135.9, 127.6, 121.5, 120.7, 117.8, 107.32, 107.31, 107.27, 107.26, 100.2, 100.1, 99.8, 99.7, 70.6, 68.9, 68.82, 68.76, 46.4, 46.2, 37.7, 37.6, 29.1, 28.9, 27.9, 27.7, 26.34, 26.30, 23.9, 23.8; 19F NMR (376 MHz, CDCl3) δ: −70.18, −70.23; νmax/cm−1: 2920, 2843, 1684, 1621, 1580, 1490, 1470, 1450, 1210, 1012; LRMS (ESI+) m/z 491 ([M + Na]+, 100); HRMS (ESI+) calculated for [C26H27N2O4F2] 469.19334, found 469.19324.
Cascade intramolecular cyclization – colligation
), 7.26–7.22 (2 H, m, 2 × Ar–), 6.96–6.91 (2 H, m, 2 × Ar–), 6.87 (2 H, dd, J = 8.2, 1.1 Hz, 2 × Ar–), 4.41 (2 H, dd, J = 10.9, 4.1 Hz, 2 × OCH), 3.68 (2 H, dd, J = 10.9, 7.0 Hz, 2 × OCH), 2.62 (2 H, s, 2 × O), 2.36 (2 H, pd, J = 6.9, 4.2 Hz, 2 × OCHHC), 0.57 (6 H, d, J = 6.9 Hz, 2 × C3); 13C NMR (101 MHz, CDCl3) δ: 156.1 (2 × C), 129.5 (2 × CH), 129.1 (2 × CH), 125.6 (2 × C), 120.4 (2 × CH), 116.6 (2 × CH), 76.2 (2 × C), 70.4 (2 × CH2), 34.4 (2 × CH), 12.9 (2 × CH3); νmax/cm−1 3473, 2960, 2925, 2855, 1606, 1580, 1486, 1448, 1221, 1046, 1023; LRMS (ESI−)m/z 397 (71), 361 (33), 325 ([M − H]−, 100); HRMS (ESI+) calculated for [C20H22O4Na] 349.14103, found 349.14158. Diastereomer 2 (7.5 mg, 0.023 mmol, 29%) a colourless oil. Rf: 0.29 (40% diethyl ether in hexanes); 1H NMR (400 MHz, CDCl3) δ: 7.19 (2 H, ddd, J = 8.2, 7.2, 1.7 Hz, 2 × Ar–), 7.07 (2 H, dd, J = 7.9, 1.7 Hz, 2 × Ar–), 6.86–6.82 (2 H, m, 2 × Ar–), 6.76 (2 H, dd, J = 8.2, 1.3 Hz, 2 × Ar–), 3.94 (2 H, dd, J = 11.2, 3.7 Hz, 2 × OCH), 3.71 (2 H, dd, J = 11.2, 4.8 Hz, 2 × OCH), 2.70–2.66 (2 H, m, 2 × OCHHC), 2.51 (2 H, s, 2 × O), 1.08 (6 H, d, J = 7.0 Hz, 2 × C3); 13C NMR (101 MHz, CDCl3) δ: 155.5 (2 × C), 129.6 (2 × CH), 129.3 (2 × CH), 124.4 (2 × C), 120.6 (2 × CH), 116.6 (2 × CH), 77.7 (2 × C), 70.4 (2 × CH2), 33.8 (2 × CH), 14.8 (2 × CH3); νmax/cm−1 3485, 2962, 2927, 2874, 1607, 1580, 1487, 1450, 1310, 1225, 1047, 1025; LRMS (ESI−)m/z 361 (100), 325 ([M − H]−, 30); HRMS (ESI+) calculated for [C20H22O4Na] 349.14103, found 349.14156.
), 7.06 (2 H, dd, J = 7.9, 1.5 Hz, 2 × Ar–), 6.82 (2 H, t, J = 7.9 Hz, 2 × Ar–), 4.67 (2 H, dd, J = 11.0, 3.9 Hz, 2 × OCH), 3.92 (2 H, dd, J = 11.0, 5.5 Hz, 2 × OCH), 2.44–2.38 (4 H, m, 2 × (CCH3 and O)), 0.71 (6 H, d, J = 6.9 Hz, 2 × C3); 13C NMR (101 MHz, CDCl3) δ: 151.4 (2 × C), 130.2 (2 × CH), 127.5 (2 × CH), 126.6 (2 × C), 121.3 (2 × C), 119.9 (2 × CH), 76.5 (2 × C), 70.7 (2 × CH2), 33.5 (2 × CH), 13.5 (2 × CH3); νmax/cm−1 3495, 2962, 2928, 2856, 1474, 1444, 1289, 1245, 1125, 1081, 1027; HRMS (ESI+) calculated for [C10H11O235Cl35ClNa] 417.06309, found 417.06300, calculated for [C10H11O235Cl37ClNa] 419.06014, found 419.06013; Diastereomer 2 (6.5 mg, 0.020 mmol, 20%) a pale yellow oil. Rf: 0.19 (40% diethyl ether in hexanes); 1H NMR (400 MHz, CDCl3) δ: 7.31 (2 H, dd, J = 7.8, 1.6 Hz, 2 × Ar–), 6.99 (2 H, dd, J = 8.0, 1.6 Hz, 2 × Ar–), 6.80 (2 H, t, J = 7.9 Hz, 2 × Ar–), 4.15 (2 H, dd, J = 11.3, 3.8 Hz, 2 × OCH), 3.89 (2 H, dd, J = 11.3, 4.5 Hz, 2 × OCH), 2.70–2.66 (2 H, m, 2 × CCH3), 2.47 (2 H, s, 2 × O), 1.07 (6 H, d, J = 7.0 Hz, 2 × C3); 13C NMR (101 MHz, CDCl3) δ: 151.2 (2 × C), 130.3 (2 × CH), 127.8 (2 × CH), 125.7 (2 × C), 121.5 (2 × C), 120.5 (2 × CH), 77.8 (2 × C), 71.0 (2 × CH2), 33.6 (2 × CH), 14.8 (2 × CH3); νmax/cm−1 3520, 2963, 2931, 1597, 1476, 1444, 1245, 1080; HRMS (ESI+) calculated for [C10H11O235Cl35ClNa] 417.06309, found 417.06314, calculated for [C10H11O235Cl37ClNa] 419.06014, found 419.06019.
), 6.86 (2 H, dt, J = 8.1, 1.5 Hz, 2 × Ar–), 6.78 (2 H, td, J = 8.0, 5.0 Hz, 2 × Ar–), 4.13 (2 H, dd, J = 11.3, 3.7 Hz, 2 × OCH), 3.89 (2 H, dd, J = 11.3, 4.3 Hz, 2 × OCH), 2.74–2.65 (2 H, m, 2 × CCH3), 2.49 (2 H, s, 2 × O), 1.09 (6 H, d, J = 7.0 Hz, 2 × C3); 13C NMR (101 MHz, CDCl3) δ: 151.3 (2 C, d, J = 245.3 Hz, 2 × C), 144.0 (2 C, d, J = 11.2 Hz, 2 × C), 126.5 (2 C, d, J = 1.0 Hz, 2 × C), 124.2 (2 C, d, J = 3.6 Hz, 2 × CH), 119.7 (2 C, d, J = 7.2 Hz, 2 × CH), 115.9 (2 C, d, J = 17.9 Hz, 2 × CH), 77.4 (2 × C), 70.9 (2 × CH2), 33.5 (2 × CH), 14.8 (2 × CH3); 19F NMR (376 MHz, CDCl3) δ: −136.60; νmax/cm−1 3503, 2958, 2924, 2854, 1484, 1258, 1224; HRMS (ESI+) calculated for [C20H20O4FNa] 385.12219, found 385.12287; Diastereomer 2 a pale yellow oil (7 mg, 0.020 mmol, 24%). Rf: 0.17 (40% diethyl ether in hexanes); 1H NMR (400 MHz, CDCl3) δ: 7.07 (2 H, ddd, J = 10.7, 8.0, 1.5 Hz, 2 × Ar–), 6.99 (2 H, dt, J = 8.1, 1.4 Hz, 2 × Ar–), 6.83 (2 H, td, J = 8.0, 5.0 Hz, 2 × Ar–), 4.59 (2 H, dd, J = 11.0, 3.9 Hz, 2 × OCH), 3.86 (2 H, dd, J = 11.0, 5.8 Hz, 2 × OCH), 2.53 (2 H, s, 2 × O), 2.45–2.36 (2 H, m, 2 × CCH3), 0.69 (6 H, d, J = 6.9 Hz, 2 × C3); 13C NMR (101 MHz, CDCl3) δ: 151.2 (2 C, d, J = 245.5 Hz, 2 × C), 144.2 (2 C, d, J = 11.1 Hz, 2 × C), 127.7 (2 × C), 124.0 (2 C, d, J = 3.6 Hz, 2 × CH), 119.3 (2 C, d, J = 7.0 Hz, 2 × CH), 115.9 (2 C, d, J = 17.9 Hz, 2 × CH), 76.2 (2 C, d, J = 2.6 Hz, 2 × C), 70.6 (2 × CH2), 33.9 (2 × CH), 13.3 (2 × CH3); 19F NMR (376 MHz, CDCl3) δ: −136.78; νmax/cm−1 3510, 2959, 2925, 2854, 1483, 1455, 1258, 1220; HRMS (ESI+) calculated for [C20H20O4FNa] 385.12219, found 385.12268.
), 6.88 (2 H, dd, J = 8.2, 0.9 Hz, 2 × Ar–), 6.81–6.78 (2 H, m, 2 × Ar–), 6.65 (2 H, dd, J = 7.8, 1.5 Hz, 2 × Ar–), 5.09 (2 H, brs, 2 × OC), 2.23 (2 H, ddd, J = 12.3, 4.1, 2.6 Hz, 2 × CC), 2.18–2.15 (2 H, m, 2 × OCHCH), 1.80 (2 H, s, 2 × O), 1.76–1.73 (2 H, m, 2 × CCHCHHCH), 1.70–1.65 (2 H, m, 2 × CCHCH), 1.61–1.55 (6 H, m, 2 × OCHCHC2), 1.36–1.29 (2 H, m, 2 × CCHCHHCH), 0.89 (2 H, qd, J = 12.9, 3.7 Hz, 2 × CCHCH); 13C NMR (126 MHz, CDCl3) δ: 155.6 (2 × C), 129.7 (2 × CH), 128.3 (2 × CH), 122.7 (2 × C), 118.6 (2 × CH), 115.6 (2 × CH), 77.3 (2 × C), 71.1 (2 × CH), 38.4 (2 × CH), 31.2 (2 × CH2), 25.1 (2 × CH2), 23.0 (2 × CH2), 19.9 (2 × CH2); νmax/cm−1 3558, 2932, 2857, 1605, 1579, 1481, 1451, 1232; LRMS (ESI+) m/z 653 (55), 543 (26), 441 (35), 429 ([M + Na]+, 26), 393 (100), 283 (35); HRMS (ESI+) calculated for [C26H30O4Na] 429.20363, found 429.20442; Diastereomer 2 (7 mg, 0.0168 mmol, 21%) a colourless oil. Rf: 0.34 (30% diethyl ether in hexanes); 1H NMR (400 MHz, CDCl3) δ: 7.18–7.15 (2 H, m, 2 × Ar–), 6.80–6.73 (6 H, m, 6 × Ar–), 4.33 (2 H, brs, 2 × OC), 2.77 (2 H, s, 2 × O), 2.42–2.38 (2 H, m, 2 × CC), 2.00–1.97 (2 H, m, 2 × OCHCH), 1.83–1.75 (4 H, m, 2 × CCHCHCH), 1.59–1.48 (6 H, m, 2 × OCHCHC2), 1.35–1.33 (2 H, m, 2 × CCHCHHCH), 1.07 (2 H, qd, J = 12.8, 3.4 Hz, CCHCH); 13C NMR (101 MHz, CDCl3) δ: 155.2 (2 × C), 129.5 (2 × CH), 128.8 (2 × CH), 123.7 (2 × C), 120.0 (2 × CH), 116.2 (2 × CH), 77.9 (2 × C), 72.1 (2 × CH), 39.2 (2 × CH), 31.0 (2 × CH2), 25.4 (2 × CH2), 23.0 (2 × CH2), 19.7 (2 × CH2); νmax/cm−1 3484, 2932, 2860, 1606, 1580, 1483, 1453, 1232; HRMS (ESI+) calculated for [C26H30O4Na] 429.20363, found 429.20435.
), 7.63–7.60 (2 H, m, 2 × Ar–), 7.36–7.33 (2 H, m, 2 × Ar–), 7.30–7.22 (3 H, m, 3 × Ar–), 6.99–6.95 (1 H, m, Ar–), 6.82 (1 H, dd, J = 8.2, 1.2 Hz, Ar–), 4.84 (1 H, dd, J = 10.9, 3.3 Hz, OCH), 3.81 (1 H, dd, J = 10.9, 2.3 Hz, OCH), 2.00 (1 H, s, O), 1.84 (1 H, qdd, J = 7.1, 3.3, 2.3 Hz, OCHHC), 1.74 (3 H, s, CC3), 1.56 (1 H, s, O), 0.84 (3 H, d, J = 7.1 Hz, CHC3); 13C NMR (125 MHz, CDCl3) δ: 155.3 (C), 144.4 (C), 129.6 (CH), 128.7 (CH), 128.0 (2 × CH), 127.1 (CH), 126.6 (2 × CH), 123.1 (C), 119.8 (CH), 116.3 (CH), 79.7 (C), 76.3 (C), 70.0 (CH2), 33.3 (CH), 27.2 (CH3), 14.8 (CH3). Diastereomer 2 (7.5 mg, 0.026 mmol, 16%). Rf: 0.31 (2% diethyl ether in dichloromethane); 1H NMR (500 MHz, CDCl3) δ: 7.56 (1 H, dd, J = 7.9, 1.7 Hz, Ar–), 7.26–7.18 (26 H, m, Ar–, 2 × Ar–, 10 × Ar– and 10 × Ar–), 7.13–7.12 (2 H, m, 2 × Ar–), 7.07 (2 H, dd, J = 7.9, 1.7 Hz, 2 × Ar–), 6.94 (1 H, ddd, J = 7.9, 7.2, 1.3 Hz, Ar–), 6.84 (2 H, ddd, J = 7.9, 7.2, 1.3 Hz, 2 × Ar–), 6.76 (2 H, dd, J = 8.2, 1.3 Hz, 2 × Ar–), 6.74 (1 H, dd, J = 8.2, 1.4 Hz, Ar–), 3.93 (2 H, dd, J = 11.2, 3.7 Hz, 2 × OCH), 3.71 (2 H, dd, J = 11.2, 4.8 Hz, 2 × OCH), 3.53 (1 H, dd, J = 11.5, 3.4 Hz, OCH), 3.19 (1 H, dd, J = 11.5, 3.3 Hz, OCH), 2.70–2.65 (2 H, m, 2 × CCH3), 2.70 (1 H, s, O), 2.68 (1 H, s, O), 2.57 (2 H, s, 2 × O), 2.53 (2 H, s, 2 × O), 2.47 (1 H, qt, J = 7.1, 3.4 Hz, C), 2.27 (2 H, s, 2 × O), 1.69 (3 H, s, CC3), 1.59 (6 H, s, 2 × C3), 1.51 (6 H, s, 2 × C3), 1.08 (6 H, d, J = 7.0 Hz, 2 × C3), 0.92 (3 H, d, J = 7.2 Hz, CHC3); 13C NMR (101 MHz, CDCl3) δ: 155.5 (2 × C), 155.4 (C), 144.4 (C), 143.9 (2 × C), 143.6 (2 × C), 129.6 (2 × CH), 129.5 (CH), 129.3 (2 × CH), 129.0 (CH), 128.1 (CH), 127.5 (4 × CH), 127.43 (4 × CH), 127.37 (CH), 127.3 (CH and 4 × CH), 127.2 (2 × CH), 127.1 (4 × CH), 127.0 (2 × CH), 126.4 (2 × CH), 124.4 (2 × C), 123.4 (C), 120.6 (2 × CH), 120.1 (CH), 116.6 (2 × CH), 116.4 (CH), 79.8 (C), 79.0 (2 × C), 78.7 (2 × C), 77.6 (2 × C), 75.8 (C), 70.4 (2 × CH2), 70.0 (CH2), 33.7 (2 × CH), 32.3 (CH), 27.2 (CH3), 25.3 (2 × CH3), 25.1 (2 × CH3), 14.78 (2 × CH3), 14.77 (CH3).
), 7.49 (1 H, ddd, J = 8.4, 7.1, 1.8 Hz, Ar–), 7.06 (1 H, ddd, J = 8.0, 7.1, 1.0 Hz, Ar–), 6.98 (1 H, dd, J = 8.3, 1.0 Hz, Ar–), 6.31 (1 H, t, J = 1.2 Hz, CCH), 5.58 (1 H, td, J = 1.7, 1.0 Hz, CCH), 5.01 (2 H, t, J = 1.5 Hz, OCH2); 13C NMR (101 MHz, CDCl3) δ: 182.1 (C), 162.1 (C), 139.0 (C), 136.1 (CH), 128.1 (CH), 122.5 (CH2), 122.1 (CH), 122.0 (C), 118.2 (CH), 71.3 (CH2); νmax/cm−1 2983, 2855, 1685, 1608, 1475, 1465, 1322, 1212, 1148, 1111; LRMS (ESI+) m/z 497 (27), 479 (40), 355 (33), 321 (100), 237 (46), 161 ([M + H]+, 12).
), 7.46 (1 H, ddd, J = 8.4, 7.1, 1.8 Hz, Ar–), 7.01 (1 H, ddd, J = 7.9, 7.1, 1.0 Hz, Ar–), 6.95 (1 H, dd, J = 8.4, 1.0 Hz, Ar–), 5.81 (1 H, ddt, J = 17.1, 10.2, 6.6 Hz, CCHH), 5.08 (1 H, dq, 17.1, 1.7 Hz, CHCH), 5.01 (1 H, dq, J = 10.2, 1.5 Hz, CHCH), 4.52 (1 H, dd, J = 11.5, 4.5 Hz, OCH), 4.27 (1 H, dd, J = 11.5, 8.7 Hz, OCH), 2.71–2.69 (1 H, m, OCHHC), 2.24–2.18 (2 H, m, C2CHCH2), 2.05–2.02 (1 H, m, OCH2CHCH), 1.60–1.57 (1 H, m, OCH2CHCH); 13C NMR (101 MHz, CDCl3) δ: 194.5 (C), 161.6 (C), 137.6 (CH), 135.9 (CH), 127.6 (CH), 121.5 (CH), 120.8 (C), 117.8 (CH), 115.8 (CH2), 70.5 (CH2), 45.3 (CH), 31.1 (CH2), 25.6 (CH2); νmax/cm−1 3075, 2978, 2923, 2871, 1690, 1606, 1479, 1325, 1298, 1215; LRMS (ESI+) m/z 499 (23), 457 (38), 427 (100), 225 ([M + Na]+, 35); HRMS (ESI+) calculated for [(C13H14O2)2Na] 427.18798, found 427.18771.
:1 mixture of diastereomers. Diastereomer 1, a yellow solid. Rf: 0.22 (20% diethyl ether in hexanes); m.p. 76–78 °C; 1H NMR (400 MHz, CDCl3) δ: 7.18 (2 H, ddd, J = 8.2, 7.2, 1.7 Hz, 2 × Ar–), 7.00 (2 H, dd, J = 7.9, 1.6 Hz, 2 × Ar–), 6.82 (2 H, ddd, J = 8.0, 7.2, 1.3 Hz, 2 × Ar–), 6.75 (2 H, dd, J = 8.2, 1.3 Hz, 2 × Ar–), 5.77 (2 H, dddd, J = 17.2, 10.2, 7.1, 6.2 Hz, 2 × CCHH), 5.01 (2 H, dq, J = 17.2, 1.7 Hz, 2 × CHCH), 4.96 (2 H, ddt, J = 10.2, 2.1, 1.1 Hz, 2 × CHCH), 3.98 (2 H, dd, J = 11.6, 3.3 Hz, 2 × OCH), 3.88 (2 H, dd, J = 11.6, 4.2 Hz, 2 × OCH), 2.64 (2 H, s, 2 × O), 2.47–2.45 (2 H, m, 2 × OCHHC), 2.23–2.21 (2 H, m, 2 × CHCHCH2), 2.09–2.06 (2 H, m, 2 × CHCHCH2), 1.86–1.80 (2 H, m, 2 × OCH2CHCH), 1.37–1.26 (2 H, m, 2 × OCH2CHCH); 13C NMR (101 MHz, CDCl3) δ: 155.5 (2 × C), 138.4 (2 × CH), 129.6 (2 × CH), 128.9 (2 × CH), 124.8 (2 × C), 120.6 (2 × CH), 116.6 (2 × CH), 115.3 (2 × CH2), 77.7 (2 × C), 67.0 (2 × CH2), 38.0 (2 × CH), 31.9 (2 × CH2), 26.8 (2 × CH2); νmax/cm−1 3482, 3074, 2925, 2866, 1639, 1606, 1579, 1487, 1450, 1306, 1280, 1225, 1019; LRMS (ESI+) m/z 429 ([M + Na]+, 100), 393 (53); HRMS (ESI+) calculated for [C26H30O4Na] 429.20363, found 429.20443. Diastereomer 2, a yellow solid. Rf: 0.27 (20% diethyl ether in hexanes); m.p. 133–135 °C; 1H NMR (400 MHz, CDCl3) δ: 7.24 (2 H, ddd, J = 8.2, 7.2, 1.7 Hz, 2 × Ar–), 7.13 (2 H, dd, J = 7.7, 1.5 Hz, 2 × Ar–), 6.92–6.86 (4 H, m, 4 × Ar–), 5.59 (2 H, ddt, J = 17.0, 10.2, 6.7 Hz, 2 × CCHH), 4.95–4.88 (4 H, m, 2 × CHC2), 4.61 (2 H, dd, J = 11.1, 3.6 Hz, 2 × OCH), 3.92 (2 H, dd, J = 11.2, 5.3 Hz, 2 × OCH), 2.48 (2 H, s, 2 × O), 2.19–2.15 (2 H, m, 2 × OCHHC), 2.12–2.01 (2 H, m, 2 × CHCHCH2), 1.89–1.78 (2 H, m, 2 × CHCHCH2), 1.36–1.29 (2 H, m, 2 × OCH2CHCH), 1.08–1.03 (2 H, m, 2 × OCH2CHCH); 13C NMR (101 MHz, CDCl3) δ: 155.9 (2 × C), 138.3 (2 × CH), 129.6 (2 × CH), 128.6 (2 × CH), 124.9 (2 × C), 120.0 (2 × CH), 116.5 (2 × CH), 115.1 (2 × CH2), 76.5 (2 × C), 67.0 (2 × CH2), 38.2 (2 × CH), 32.0 (2 × CH2), 26.4 (2 × CH2); νmax/cm−1 3465, 3074, 2925, 2861, 1486, 1449, 1280, 1220, 1044, 1020; LRMS (ESI+) m/z 429 ([M + Na]+, 76), 393 (100); HRMS (ESI+) calculated for [C26H30O4Na] 429.20363, found 429.20446.
Thioester stability studies
), 9.89 (1 H, s, CO), 8.37 (1 H, d, J = 2.0 Hz, Ar–), 7.99 (1 H, dd, J = 8.7, 1.9 Hz, Ar–), 7.85–7.83 (1 H, m, Ar–), 7.32–7.30 (2 H, m, 2 × Ar–), 7.10 (1 H, d, J = 8.7 Hz, Ar–), 6.95 (1 H, ddd, J = 8.0, 5.3, 3.7 Hz, Ar–), 3.38–3.35 (2 H, m, SC2), 3.14–3.11 (2 H, m, SCH2C2); 13C NMR (101 MHz, CDCl3) δ: 197.3 (C), 189.7 (CH), 164.4 (C), 142.1 (C), 139.9 (CH), 135.9 (CH), 132.5 (CH), 130.2 (CH), 128.84 (CH), 128.79 (C), 128.72 (CH), 120.0 (C), 119.5 (CH), 100.5 (C), 40.3 (CH2), 29.2 (CH2); νmax/cm−1 3055, 2926, 2828, 1693, 1625, 1584, 1480, 1197, 1147; LRMS (ESI−)v m/z 471 (384), 411 ([M − H]−, 100); HRMS (ESI+) calculated for [C16H14O3SI] 412.97028, found 412.96977.
O), 8.28 (1 H, d, J = 2.0 Hz, Ar–), 8.00 (1 H, dd, J = 8.7, 1.9 Hz, Ar–), 7.84 (1 H, d, J = 7.9 Hz, Ar–), 7.35 (1 H, dd, J = 7.7, 1.6 Hz, Ar–), 7.31 (1 H, t, J = 7.4 Hz, Ar–), 7.09 (1 H, d, J = 8.7 Hz, Ar–), 6.93 (1 H, td, J = 7.5, 1.6 Hz, Ar–), 6.13–6.05 (1 H, m, CCHH); 5.52–5.48 (1 H, m, CHCH), 5.38–5.35 (1 H, m, CHCH), 4.77 (2 H, dt, J = 5.2, 1.6 Hz, OC2), 3.31–3.28 (2 H, m, SC2), 3.12–3.09 (2 H, m, SCH2C2); 13C NMR (101 MHz, CDCl3) δ: 190.1 (CH), 190.0 (C), 161.3 (C), 142.8 (C), 139.7 (CH), 134.1 (CH), 132.7 (CH), 131.7 (CH), 130.2 (CH), 129.5 (C), 128.6 (CH), 128.5 (CH), 127.9 (C), 118.9 (CH2), 113.6 (CH), 100.5 (C), 70.1 (CH2), 40.4 (CH2), 29.8 (CH2); νmax/cm−1 2924, 1694, 1634, 1596, 1493, 1422, 1276, 1263, 1231, 1164, 1115, 1008; LRMS (ESI+) m/z 475 ([M + Na]+, 100); HRMS (ESI+) calculated for [C19H17IO3SNa] 474.98353, found 474.98317.
), 7.82 (1 H, dd, J = 7.9, 1.3 Hz, Ar–), 7.56 (1 H, dd, J = 8.6, 2.4 Hz, Ar–), 7.35 (1 H, dd, J = 7.6, 1.8 Hz, Ar–), 7.30 (1 H, td, J = 7.4, 1.3 Hz, Ar–), 6.97 (1 H, d, J = 8.6 Hz, Ar–), 6.92 (1 H, td, J = 7.6, 1.8 Hz, Ar–), 6.08 (1 H, ddt, J = 17.3, 10.6, 5.2 Hz, CCHH), 5.78 (1 H, s, ArC), 5.47 (1 H, dq, J = 17.3, 1.6 Hz, CHCH), 5.31 (1 H, dq, J = 10.6, 1.4 Hz, CHCH), 4.68 (2 H, dt, J = 5.1, 1.6 Hz, C2CH), 4.15–4.12 (2 H, m, OCHCHO), 4.04–4.01 (2 H, m, OCHCHO), 3.28–3.24 (2 H, m, SC2), 3.11–3.07 (2 H, m, SCH2C2); 13C NMR (126 MHz, CDCl3) δ: 190.6 (C), 157.7 (C), 143.1 (C), 139.7 (CH), 132.6 (CH), 131.8 (CH), 130.5 (C), 130.3 (CH), 128.6 (CH), 128.5 (2 × CH), 127.3 (C), 118.3 (CH2), 113.5 (CH), 103.2 (CH), 100.5 (C), 70.0 (CH2), 65.5 (2 × CH2), 40.6 (CH2), 29.6 (CH2); νmax/cm−1 2923, 2886, 1692, 1672, 1634, 1596, 1495, 1465, 1274, 1165, 1119, 1008; HRMS (ESI+) calculated for [C21H21IO4SNa] 519.00974, found 519.00979.
), 7.83 (1 H, dd, J = 7.9, 1.2 Hz, Ar–), 7.63 (1 H, d, J = 16.0 Hz, ArCCH), 7.59 (1 H, dd, J = 8.7, 2.3 Hz, Ar–), 7.35 (1 H, dd, J = 7.6, 1.9 Hz, Ar–), 7.30 (1 H, td, J = 7.4, 1.3 Hz, Ar–), 6.97 (1 H, d, J = 8.7 Hz, Ar–), 6.93 (1 H, td, J = 7.5, 1.9 Hz, Ar–), 6.37 (1 H, d, J = 16.0 Hz ArCHC), 6.08 (1 H, ddt, J = 17.3, 10.6, 5.2 Hz, CCHH), 5.48 (1 H, dq, J = 17.3, 1.6 Hz, CHCH), 5.33 (1 H, dq, J = 10.6, 1.4 Hz, CHCH), 4.70 (2 H, dt, J = 5.1, 1.6 Hz, ArOC2), 4.26 (2 H, q, J = 7.1 Hz, CO2C2), 3.30–3.26 (2 H, m, SC2), 3.12–3.08 (2 H, m, SCH2C2), 1.34 (3 H, t, J = 7.1 Hz, C3); 13C NMR (126 MHz, CDCl3) δ: 190.4 (C), 167.1 (C), 158.2 (C), 143.1 (CH), 142.9 (C), 139.7 (CH), 133.0 (CH), 132.2 (CH), 130.2 (CH), 129.6 (CH), 128.6 (CH), 128.5 (CH), 127.9 (C), 127.3 (C), 118.5 (CH2), 117.6 (CH), 113.8 (CH), 100.5 (C), 70.0 (CH2), 60.6 (CH2), 40.5 (CH2), 29.7 (CH2), 14.5 (CH3); νmax/cm−1 2979, 2929, 1708, 1636, 1598, 1495, 1260, 1166, 1120, 1010; LRMS (ESI+) m/z 545 ([M + Na]+, 100); HRMS (ESI+) calculated for [C23H23IO4SNa] 545.02539, found 545.02578.
), 7.36–7.26 (3 H, m, 3 × Ar–), 4.93 (1 H, d, J = 1.7 Hz, CCH), 4.45 (1 H, d, J = 1.7 Hz, CCH), 0.29 (9 H, s, 3 × C3); 13C NMR (101 MHz, CDCl3) δ: 155.8 (C), 137.7 (C), 128.3 (CH), 128.2 (2 × CH), 125.4 (2 × CH), 91.2 (CH2), 0.22 (3 × CH3); νmax/cm−1 2960, 1316, 1302, 1286, 1252, 1027, 1010; LRMS (ESI+) m/z 295 (73), 193 ([M + H]+, 100).
), 7.84 (1 H, d, J = 2.4 Hz, Ar–), 7.82 (1 H, dd, J = 7.9, 1.2 Hz, Ar–), 7.60–7.55 (2 H, m, 2 × Ar–), 7.49–7.45 (2 H, m, 2 × Ar–), 7.35 (1 H, dd, J = 7.6, 1.8 Hz, Ar–), 7.29 (1 H, td, J = 7.4, 1.2 Hz, Ar–), 6.98 (1 H, d, J = 8.6 Hz, Ar–), 6.91 (1 H, ddd, J = 7.8, 7.3, 1.8 Hz, Ar–), 6.10 (1 H, ddt, J = 17.3, 10.6, 5.2 Hz, CCHH), 5.48 (1 H, dq, J = 17.3, 1.7 Hz, CHCH), 5.34–5.30 (2 H, m, CHCH + COH), 4.68 (2 H, dt, J = 5.1, 1.6 Hz, OC2), 3.66 (1 H, brs, O), 3.38–3.35 (2 H, m, C2CHOH), 3.28–3.24 (2 H, m, SC2), 3.11–3.07 (2 H, m, SCH2C2); 13C NMR (101 MHz, CDCl3) δ: 200.2 (C), 190.9 (C), 156.6 (C), 143.1 (C), 139.7 (CH), 136.6 (C), 135.4 (C), 133.9 (CH), 132.7 (CH), 131.1 (CH), 130.2 (CH), 128.9 (2 × CH), 128.6 (CH), 128.4 (CH), 128.3 (2 × CH), 127.4 (C), 127.3 (CH), 118.2 (CH2), 113.8 (CH), 100.5 (C), 70.1 (CH2), 69.4 (CH), 47.2 (CH2), 40.5 (CH2), 29.7 (CH2); νmax/cm−1 3487, 3059, 2924, 1676, 1634, 1494, 1285, 1254, 1211, 1120, 1010; LRMS (ESI+) m/z 595 ([M + Na]+, 100); HRMS (ESI+) calculated for [C27H25IO4SNa] 595.04104, found 595.04148.
), 7.80 (1 H, d, J = 2.4 Hz, Ar–), 7.46 (1 H, dd, J = 8.5, 2.4 Hz, Ar–), 7.35 (1 H, dd, J = 7.6, 1.8 Hz, Ar–), 7.29 (1 H, td, J = 7.4, 1.2 Hz, Ar–), 7.10 (1 H, t, J = 7.7 Hz, Ar–), 6.94 (1 H, d, J = 8.6 Hz, Ar–), 6.94–6.92 (1 H, m, Ar–), 6.59 (1 H, brd, J = 7.5 Hz, Ar–), 6.49 (1 H, brt, J = 2.0 Hz, Ar–), 6.46 (1 H, ddd, J = 8.0, 2.5, 0.8 Hz, Ar–), 6.10 (1 H, ddt, J = 17.3, 10.6, 5.1 Hz, CCHH), 5.48 (1 H, dq, J = 17.2, 1.6 Hz, CHCH), 5.32 (1 H, dq, J = 10.6, 1.5 Hz, CHCH), 4.67 (2 H, dt, J = 5.1, 1.6 Hz, OC2), 4.29 (2 H, s, NC2), 4.01 (1 H, brs, N), 3.28–3.25 (2 H, m, SC2), 3.11–3.08 (2 H, m, SCH2C2), 2.57 (2 H, q, J = 7.6 Hz ArC2), 1.21 (3 H, t, J = 7.6 Hz, C3); 13C NMR (101 MHz, CDCl3) δ: 190.9 (C), 156.3 (C), 148.1 (C), 145.6 (C), 143.1 (C), 139.7 (CH), 132.8 (CH), 132.7 (CH), 132.0 (C), 130.2 (CH), 129.4 (CH), 129.0 (CH), 128.6 (CH), 128.5 (CH), 127.5 (C), 118.2 (CH2), 117.7 (CH), 113.9 (CH), 112.8 (CH), 110.4 (CH), 100.5 (C), 70.1 (CH2), 47.7 (CH2), 40.6 (CH2), 29.7 (CH2), 29.1 (CH2), 15.6 (CH3); νmax/cm−1 3408, 2962, 2928, 1667, 1633, 1604, 1493, 1284, 1268, 1164, 1122, 1010; LRMS (ESI+) m/z 580 ([M + Na]+, 100); HRMS (ESI+) calculated for [C27H28INO2SNa] 580.07776, found 580.07835.
Synthesis of donepezil
), 6.76 (1 H, s, Ar–), 6.04 (1 H, ddt, J = 17.7, 9.5, 6.5 Hz, CCH2), 5.07–5.02 (2 H, m, CHC2), 3.94 (3 H, s, OC3), 3.92 (3 H, s, OC3), 3.82 (2 H, dt, J = 6.5, 1.5 Hz, C2); 13C NMR (126 MHz, CDCl3) δ: 172.1 (C), 153.0 (C), 147.0 (C), 138.0 (C), 137.7 (CH), 119.7 (C), 115.7 (CH2), 114.3 (CH), 113.6 (CH), 56.2 (CH3), 56.1 (CH3), 38.6 (CH2); νmax/cm−1 3003, 2957, 1688, 1606, 1574, 1522, 1266, 1222, 1207, 1167; LRMS (ESI−) m/z 465 (42), 430 (37), 221 ([M − H]−, 100).
H), 3.21 (2 H, t, J = 7.2 Hz, C2I), 2.69 (2 H, brt, J = 12.5 Hz, 2 × NCH), 1.77 (2 H, q, J = 7.0 Hz, C2CH2I), 1.66–1.56 (3 H, m, 2 × NCH2CH and C), 1.44 (9 H, s, 3 × C3), 1.09 (2 H, qd, J = 12.2, 4.3 Hz, 2 × NCH2CH); 13C NMR (101 MHz, CDCl3) δ: 154.9 (C), 79.5 (C), 43.9 (2 × CH2), 40.0 (CH2), 36.8 (CH), 31.3 (2 × CH2), 28.6 (3 × CH3), 4.0 (CH2); νmax/cm−1 2973, 2925, 2848, 1687, 1447, 1364, 1277, 1223, 1161, 1111; LRMS (ESI+) m/z 701 (55), 362 ([M + Na]+, 100).
CHH), 4.99 (1 H, dt, J = 17.3, 1.5 Hz, CHCH), 4.94 (1 H, dt, J = 10.4, 1.4 Hz, CHCH), 4.07 (2 H, brd, J = 8.3 Hz, 2 × NCH), 2.72 (2 H, brt, J = 12.3 Hz, 2 × NCH), 2.13–2.05 (1 H, m, CCHCH2), 1.67 (2 H, brd, J = 13.1 Hz, 2 × NCH2CH), 1.44 (9 H, s, 3 × C3), 1.26 (2 H, qd, J = 12.3, 4.1 Hz, 2 × NCH2CH); 13C NMR (101 MHz, CDCl3) δ: 155.0 (C), 142.7 (CH), 113.1 (CH2), 79.4 (C), 43.8 (2 × CH2), 39.8 (CH), 31.5 (2 × CH2), 28.6 (3 × CH3); νmax/cm−1 2976, 2931, 2850, 1689, 1417, 1365, 1231, 1156; LRMS (ESI+) m/z 445 (72), 234 ([M + Na]+, 100); HRMS (ESI+) calculated for [C12H21NO2Na] 234.14645, found 234.14665.
), 7.33–7.27 (3 H + 0.15 × 3 H, m, 3 × Ar–), 6.91 (1 H + 0.15 × 1 H, ddd, J = 7.9, 7.0, 2.1 Hz, Ar–), 6.74 (0.15 × 1 H, s, Ar–), 6.70 (1 H, s, Ar–), 5.54 (1 H, dtd, J = 15.4, 6.5, 1.1 Hz, CH2CCH), 5.51–5.29 (0.15 × 2 H, m, CH2CC), 5.39 (1 H, ddt, J = 15.4, 6.5, 1.3 Hz, CH2CHC), 4.04 (2 H + 0.15 × 2 H, brs, 2 × NCH), 3.90 (3 H + 0.15 × 3 H, s, OC3), 3.89 (3 H + 0.15 × 3 H, s, OC3), 3.63 (0.15 × 2 H, dd, J = 7.1, 1.4 Hz, C2CHCH), 3.52 (2 H, d, J = 6.5 Hz, C2CHCH), 3.26–3.22 (2 H + 0.15 × 2 H, m, SC2), 3.09–3.05 (2 H + 0.15 × 2 H, m, SCH2C2), 2.80–2.66 (2 H + 0.15 × 2 H, m, 2 × NCH), 2.09–2.05 (1 H + 0.15 × 1 H, CHCHC), 1.64–1.61 (2 H + 0.15 × 2 H, m, 2 × NCHHCH), 1.45 (0.15 × 9 H, s, C(C3)3), 1.43 (9 H, s, C(C3)3), 1.28–1.19 (2 H + 0.15 × 2 H, m, 2 × NCHHCH); 13C NMR (101 MHz, CDCl3) δ: 192.5 (C + C), 154.93 (C), 154.91 (C), 151.9 (C), 151.8 (C), 146.79 (C), 146.76 (C), 142.78 (C), 142.75 (C), 139.7 (CH + CH), 135.8 (CH), 135.3 (CH), 134.0 (C), 133.7 (C), 130.1 (CH + CH), 129.4 (C), 129.3 (C), 128.51 (CH + CH), 128.49 (CH + CH), 127.5 (CH), 127.3 (CH), 113.4 (CH), 112.9 (CH), 112.3 (CH), 112.1 (CH), 100.6 (C + C), 79.3 (C + C), 56.2 (CH3 + CH3), 56.0 (CH3 + CH3), 43.9 (2 × (CH2 + CH2)), 40.6 ((CH2 + CH2), 38.9 (CH), 36.3 (CH2), 34.7 (CH), 32.0 (2 × (CH2 + CH2)), 31.1 (CH2), 29.7 (CH2 + CH2), 28.6 (3 × (CH3 + CH3)); νmax/cm−1 2972, 2931, 1689, 1516, 1465, 1424, 1267, 1166, 1112; LRMS (APCI) m/z 674 ([M + Na]+, 14), 615 (12), 543 (37), 319 (42), 295 (100); HRMS (APCI) calculated for [C30H38INO5SNa] 674.14076, found 674.14129.
), 6.84 (1 H, s, Ar–), 4.12–4.06 (2 H, m, 2 × NCH), 3.94 (3 H, s, OC3), 3.89 (3 H, s, OC3), 3.24 (1 H, dd, J = 17.6, 8.3 Hz, ArCH), 2.71–2.66 (4 H, ArCHC + 2 × NCH), 1.92–1.85 (1 H, m, CHCHCH), 1.73–1.63 (3 H, m, 2 × NCH2CH + NCH2CH2C), 1.44 (9 H, s, C(C3)3), 1.36–1.16 (3 H, m, 2 × NCH2CH + CHCHCH); 13C NMR (101 MHz, CDCl3) δ: 207.6 (C), 155.7 (C), 155.0 (C), 149.6 (C), 148.8 (C), 129.4 (C), 107.5 (CH), 104.5 (CH), 79.4 (C), 56.3 (CH3), 56.2 (CH3), 45.3 (CH), 44.2 (2 × CH2), 38.8 (CH2), 34.7 (CH), 33.4 (CH2), 32.9 (2 × CH2), 28.6 (3 × CH3); νmax/cm−1 2925, 2847, 1689, 1501, 1422, 1313, 1266, 1157; LRMS (ESI+) m/z 499 (26), 412 ([M + Na]+, 100).
), 7.16 (1 H, s, Ar–), 6.85 (1 H, s, Ar–), 3.96 (3 H, s, OC3), 3.90 (3 H, s, OC3), 3.56 (2 H, s, C2Ph), 3.23 (1 H, dd, J = 17.5, 8.1 Hz, ArCH), 2.94 (2 H, brs, 2 × NCH), 2.71–2.67 (2 H, m, ArCHC), 2.03 (2 H, brs, 2 × NCH), 1.93–1.88 (1 H, m, CHCHCH), 1.75–1.69 (2 H, m, 2 × NCH2CH), 1.42–1.30 (3 H, m, 2 × NCH2CH + CHCHCH); 13C NMR (101 MHz, CDCl3) δ: 207.9 (C), 155.6 (C), 149.6 (C), 148.9 (C), 138.9 (C – only observed through HMBC), 129.55 (C), 129.46 (2 × CH), 128.4 (2 × CH), 127.3 (CH), 107.5 (CH), 104.5 (CH), 63.4 (CH2), 56.4 (CH3), 56.2 (CH3), 53.8 (2 × CH2), 45.5 (CH), 38.8 (CH2), 34.4 (CH), 33.6 (CH2), 31.7 (2 × CH2); νmax/cm−1 2921, 2845, 1693, 1591, 1500, 1439, 1312, 1264, 1121, 1073; LRMS (ESI+) m/z 402 ([M + Na]+, 25), 380 ([M + H]+, 100).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
ARN is grateful to the University of Sydney for the Henry Bertie and Florence Mabel Gritton Scholarship, a Faculty of Science Postgraduate Research Excellence Award, and a John A. Lamberton Research Scholarship. MNY acknowledges receipt of an Australian Government RTP Scholarship.Notes and references
- C. Chatgilialoglu, D. Crich, M. Komatsu and I. Ryu, Chem. Rev., 1999, 99, 1991–2070 CrossRef CAS PubMed.
- D. L. Boger and R. J. Mathvink, J. Org. Chem., 1992, 57, 1429–1443 CrossRef CAS.
- D. Crich, C. Chen, J.-T. Hwang, H. Yuan, A. Papadatos and R. I. Walter, J. Am. Chem. Soc., 1994, 116, 8937–8951 CrossRef CAS.
- K. Yoshikai, T. Hayama, K. Nishimura, K.-I. Yamada and K. Tomioka, J. Org. Chem., 2005, 70, 681–683 CrossRef CAS PubMed.
- L. Benati, G. Calestani, R. Leardini, M. Minozzi, D. Nanni, P. Spagnolo and S. Strazzari, Org. Lett., 2003, 5, 1313–1316 CrossRef CAS PubMed.
- V. Chudasama, R. J. Fitzmaurice and S. Caddick, Nat. Chem., 2010, 2, 592–596 CrossRef CAS PubMed.
- L. Benati, G. Calestani, R. Leardini, M. Minozzi, D. Nanni, P. Spagnolo and S. Strazzari, Org. Lett., 2003, 5, 1313–1316 CrossRef CAS PubMed.
- D. Crich and Q. Yao, J. Org. Chem., 1996, 61, 3566–3570 CrossRef CAS.
- D. Crich and X. Hao, J. Org. Chem., 1997, 62, 5982–5988 CrossRef CAS.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- M. Reckenthäler and A. G. Griesbeck, Adv. Synth. Catal., 2013, 355, 2727–2744 CrossRef.
- G. Bergonzini, C. Cassani and C.-J. Wallentin, Angew. Chem., Int. Ed., 2015, 54, 14066–14069 CrossRef CAS PubMed.
- L. Chu, J. M. Lipshultz and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2015, 54, 7929–7933 CrossRef CAS PubMed.
- X. Zhang and D. W. C. MacMillan, J. Am. Chem. Soc., 2017, 139, 11353–11356 CrossRef CAS PubMed.
- A. J. Fry and R. L. Krieger, J. Org. Chem., 1976, 41, 54–57 CrossRef CAS.
- L. Flamigni, A. Barbieri, C. Sabatini, B. Ventura and F. Barigelletti, in Photochemistry and Photophysics of Coordination Compounds II, ed. V. Balzani and S. Campagna, Springer Berlin Heidelberg, Berlin, Heidelberg, 2007, pp. 143–203 Search PubMed.
- J. Hu, J. Wang, T. H. Nguyen and N. Zheng, Beilstein J. Org. Chem., 2013, 9, 1977–2001 CrossRef PubMed.
- A. L. J. Beckwith and D. R. Boate, J. Chem. Soc., Chem. Commun., 1986, 189–190 RSC.
- D. Crich, Helv. Chim. Acta, 2006, 89, 2167–2182 CrossRef CAS.
- J. D. Nguyen, E. M. D'Amato, J. M. R. Narayanam and C. R. J. Stephenson, Nat. Chem., 2012, 4, 854–859 CrossRef CAS PubMed.
- F. Minisci, E. Vismara, F. Fontana, G. Morini, M. Serravalle and C. Giordano, J. Org. Chem., 1986, 51, 4411–4416 CrossRef CAS.
- W. Chen, H. Tao, W. Huang, G. Wang, S. Li, X. Cheng and G. Li, Chem. – Eur. J., 2016, 22, 9546–9550 CrossRef CAS PubMed.
- W. Huang and X. Cheng, Synlett, 2017, 148–158 CAS.
- Y. Guindon, B. Guérin, C. Chabot and W. Ogilvie, J. Am. Chem. Soc., 1996, 118, 12528–12535 CrossRef CAS.
- N. Charrier, B. Quiclet-Sire and S. Z. Zard, J. Am. Chem. Soc., 2008, 130, 8898–8899 CrossRef CAS PubMed.
- L. Debien, B. Quiclet-Sire and S. Z. Zard, Acc. Chem. Res., 2015, 48, 1237–1253 CrossRef CAS PubMed.
- M. Nakajima, E. Fava, S. Loescher, Z. Jiang and M. Rueping, Angew. Chem., Int. Ed., 2015, 54, 8828–8832 CrossRef CAS PubMed.
- S. Borra, D. Chandrasekhar, S. Adhikary, S. Rasala, S. Gokulnath and R. A. Maurya, J. Org. Chem., 2017, 82, 2249–2256 CrossRef CAS PubMed.
- D. Chandrasekhar, S. Borra, J. B. Nanubolu and R. A. Maurya, Org. Lett., 2016, 18, 2974–2977 CrossRef CAS PubMed.
- A. Hossain, S. K. Pagire and O. Reiser, Synlett, 2017, 1707–1714 CAS.
- P. Bonilla, Y. P. Rey, C. M. Holden and P. Melchiorre, Angew. Chem., Int. Ed., 2018, 57, 12819–12823 CrossRef CAS PubMed.
- F. Fournier, J. Berthelot and Y. L. Pascal, Tetrahedron, 1984, 40, 339–347 CrossRef CAS.
- F. Fournier and M. Fournier, Can. J. Chem., 1986, 64, 881–890 CrossRef CAS.
- T. Schuster, M. Kurz and M. W. Göbel, J. Org. Chem., 2000, 65, 1697–1701 CrossRef CAS PubMed.
- T. Schuster, M. Bauch, G. Dürner and M. W. Göbel, Org. Lett., 2000, 2, 179–181 CrossRef CAS PubMed.
- W. Ding, L.-Q. Lu, J. Liu, D. Liu, H.-T. Song and W.-J. Xiao, J. Org. Chem., 2016, 81, 7237–7243 CrossRef CAS PubMed.
- The same mechanism may operate for the dimerization of α-ketoesters. J. Zhu, Y. Yuan, S. Wang and Z.-J. Yao, ACS Omega, 2017, 2, 4665–4677 CrossRef CAS.
- H. Sugimoto, Y. Iimura, Y. Yamanishi and K. Yamatsu, Bioorg. Med. Chem. Lett., 1992, 2, 871–876 CrossRef CAS.
- C. Schlepphorst, B. Maji and F. Glorius, ACS Catal., 2016, 6, 4184–4188 CrossRef CAS.
- X. Ma, Z. Li, F. Liu, S. Cao and H. Rao, Adv. Synth. Catal., 2014, 356, 1741–1746 CrossRef CAS.
- A. J. Veloso, P. M. Nagy, B. Zhang, D. Dhar, A. Liang, T. Ibrahim, S. Mikhaylichenko, I. Aubert and K. Kerman, Anal. Chim. Acta, 2013, 774, 73–78 CrossRef CAS PubMed.
- F. Tinnis, A. Volkov, T. Slagbrand and H. Adolfsson, Angew. Chem., Int. Ed., 2016, 55, 4562–4566 CrossRef CAS PubMed.
- K. C. Miles, C. Le and J. P. Stambuli, Chem. – Eur. J., 2014, 20, 11336–11339 CrossRef CAS PubMed.
- D. G. van Greunen, W. Cordier, M. Nell, C. van der Westhuyzen, V. Steenkamp, J.-L. Panayides and D. L. Riley, Eur. J. Med. Chem., 2017, 127, 671–690 CrossRef CAS PubMed.
- E. Fillion, D. Fishlock, A. Wilsily and J. M. Goll, J. Org. Chem., 2005, 70, 1316–1327 CrossRef CAS PubMed.
- P. Liu, R. Liang, L. Lu, Z. Yu and F. Li, J. Org. Chem., 2017, 82, 1943–1950 CrossRef CAS PubMed.
- G. Barbe and A. B. Charette, J. Am. Chem. Soc., 2008, 130, 18–19 CrossRef CAS PubMed.
- C. Elati, N. Kolla, S. R. Chalamala, P. Vankawala, V. Sundaram, H. Vurimidi and V. Mathad, Synth. Commun., 2006, 36, 169–174 CrossRef CAS.
- N. Sakai, K. Kawana, R. Ikeda, Y. Nakaike and T. Konakahara, Eur. J. Org. Chem., 2011, 3178–3183 CrossRef CAS.
- K. Hirano, A. T. Biju, I. Piel and F. Glorius, J. Am. Chem. Soc., 2009, 131, 14190–14191 CrossRef CAS PubMed.
- E. Fillion, D. Fishlock, A. Wilsily and J. M. Goll, J. Org. Chem., 2005, 70, 1316–1327 CrossRef CAS PubMed.
- J. R. Struble, J. Kaeobamrung and J. W. Bode, Org. Lett., 2008, 10, 957–960 CrossRef CAS PubMed.
- S. Hintz, J. Mattay, R. van Eldik and W.-F. Fu, Eur. J. Org. Chem., 1998, 1583–1596 CrossRef CAS.
- M. L. Bennasar, T. Roca and F. Ferrando, Org. Lett., 2004, 6, 759–762 CrossRef CAS PubMed.
- H. Bonin, D. Delbrayelle, P. Demonchaux and E. Gras, Chem. Commun., 2010, 46, 2677–2679 RSC.
- H. Santoso, M. I. Casana and C. D. Donner, Org. Biomol. Chem., 2014, 12, 171–176 RSC.
- D. T. Genna and G. H. Posner, Org. Lett., 2011, 13, 5358–5361 CrossRef CAS PubMed.
- O. Kose and S. Saito, Org. Biomol. Chem., 2010, 8, 896–900 RSC.
- Y.-S. Hon, T.-R. Hsu, C.-Y. Chen, Y.-H. Lin, F.-J. Chang, C.-H. Hsieh and P.-H. Szu, Tetrahedron, 2003, 59, 1509–1520 CrossRef CAS.
- K. Prantz and J. Mulzer, Chem. – Eur. J., 2010, 16, 485–506 CrossRef CAS PubMed.
- Z. Li, R. Yazaki and T. Ohshima, Org. Lett., 2016, 18, 3350–3353 CrossRef CAS PubMed.
- R. J. Armstrong, W. Niwetmarin and V. K. Aggarwal, Org. Lett., 2017, 19, 2762–2765 CrossRef CAS PubMed.
- J. Renou, J. Dias, G. Mercey, T. Verdelet, C. Rousseau, A.-J. Gastellier, M. Arboleas, M. Touvrey-Loiodice, R. Baati, L. Jean, F. Nachon and P.-Y. Renard, RSC Adv., 2016, 6, 17929–17940 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra for all synthesized compounds. See DOI: 10.1039/c8qo00867a |
| This journal is © the Partner Organisations 2018 |