
21st century developments in the understanding and control of molecular solids
Jonathan W.
Steed

Department of Chemistry, Durham University, University Science Laboratories, South Road, Durham, DH1 3LE, UK. E-mail: jon.steed@durham.ac.uk; Fax: +44(0)191 384 4737; Tel: +44 (0)191 334 2085
First published on 6th November 2018
Abstract
The first decades of the 21st Century have seen very significant advances in the understanding, prediction and control of molecular solids. Advances in crystallization techniques, polymorphism, co-crystals, co-amorphous materials, crystal engineering and instrumentation have all contributed to what is now an extremely active field. There are huge fundamental implications as well as applications particularly in the pharmaceutical, agrochemicals and energetic materials sectors. This highlight article surveys some of the key recent developments.
 Jonathan W. Steed | Jonathan W. Steed is Professor of Chemistry at Durham University, winner of the 2010 Corday Morgan Prize and a Royal Society Wolfson Research Merit Award holder. His research concerns the molecular solid state and the development of functional responsive gels. |
Crystals have always fascinated both scientists and artists alike. Their symmetry and clean geometric shapes seem to arise like magic from the Earth. As early as 1657 Scotsman, William Davidson (called ‘Davissone’) noted the regular Platonic shapes in crystalline solids and natural forms, and began the field of crystallography: “a new subject which, so far as I know, none before me has elaborated”, Fig. 1. Fast forward to 1832 when keen visual observation of different crystal shapes allowed Friedrich Wöhler and Justus von Liebig to describe the first incidence of polymorphism in a molecular crystal; different crystal packing arrangements of the molecule benzamide.1 While Davissone, Wöhler and von Liebig recognised that there was something special about these remarkable, self-assembled materials it was not until the advent of X-ray diffraction in the early 20th century that it became possible to study their internal structure and begin to understand how and why crystals form in the way they do. Work in the 1960s by microscopist Walter McCrone2 brought about a much greater understanding of the diversity of solid forms molecules could adopt but even so, chemists remained far from being able to predict crystal structure in advance. As late as 1988 Maddox described the ongoing inability to predict crystal structure from molecular structure as an “ongoing scandal”.3 Maddox was writing around the time I began my own research in molecular crystals. Growing single crystals for X-ray analysis was a black art, and the rare sample that was big enough to make it down to the diffractometer room endured an anxious wait to see if it was stable out of the mother liquor, could pass the photographic checks and survive long enough at room temperature to give a usable data set.
 | ||
| Fig. 1 The occurrence of regular Platonic shapes in crystalline solids and natural forms (from W. Davisonne, Les Elements de la Philosophie de l’Art du Feu, ou Chemie, Paris, 1657). | ||
Against this backdrop of metaphorical magic and luck, I wanted to encapsulate in this short highlight article just how far we have come in the last two decades in the understanding, control and even prediction of the structure and properties of molecular solids. That is not to say, of course, that the field is all sewn up, but rather it is something of an exciting time to be looking at crystalline and amorphous materials.
A key driver in the solid forms arena is the burgeoning interest and funding from the pharmaceutical industry in particular, and to a lesser extent the agrochemicals and energetic materials sectors. Because molecular solid form impacts directly on solid state properties that are of direct relevance to drug formulation, the field has seen considerable investment and consequent dramatic growth in research effort.4–6 In addition, intellectual property considerations have been a very significant driver. Triggered in part by the 1984 Hatch-Waxman act7 allowing rapid access to market of generic pharmaceuticals and the subsequent Zantac (ranitidine hydrochloride) litigation,8 it is now almost universal to actively seek to identify new solid forms in pharmaceutical development. The patent protection offered by the Form A crystal patent, for example, was a significant driver behind the $1Bn 2015 acquisition of the US rights to the painkiller tapentadol hydrochloride.9 Litigation needs have also driven the development of some novel separation approaches such as the use of density separation fluids10 or magnetic levitation11 to separate polymorphs of APIs from one another or from excipients.
Perhaps the most obvious shift over the past 20 years has been the intense focus on co-crystals and even more recently on amorphous and co-amorphous materials as a means to enhance drug dissolution and bioavailability.12–14Fig. 2 summarizes the position of co-crystals and co-amorphous solids within the landscape of molecular materials. The classification of crystalline multi-component systems has been hotly debated.15 If the components bear formal charges they are considered salts. Neutral multi-component systems are considered solvates (including hydrates) if one component played the role of solvent during the crystallization process. If not, the system is a co-crystal or molecular complex. Sometimes the artificial distinction of whether a component is solid at room temperature has been used to distinguish solvates from co-crystals, however this criterion is unsatisfactory since it leads to different classification of some materials in labs of different temperature. Co-crystals also often (although not always) exhibit a well-defined stoichiometry and typically possess rather different properties to non-stoichiometric inclusion compounds where some sites are unoccupied, or one component can be free to move in a channel. Multicomponent crystalline solids can also exist as solid solutions in which one component is randomly distributed in a crystal of another.
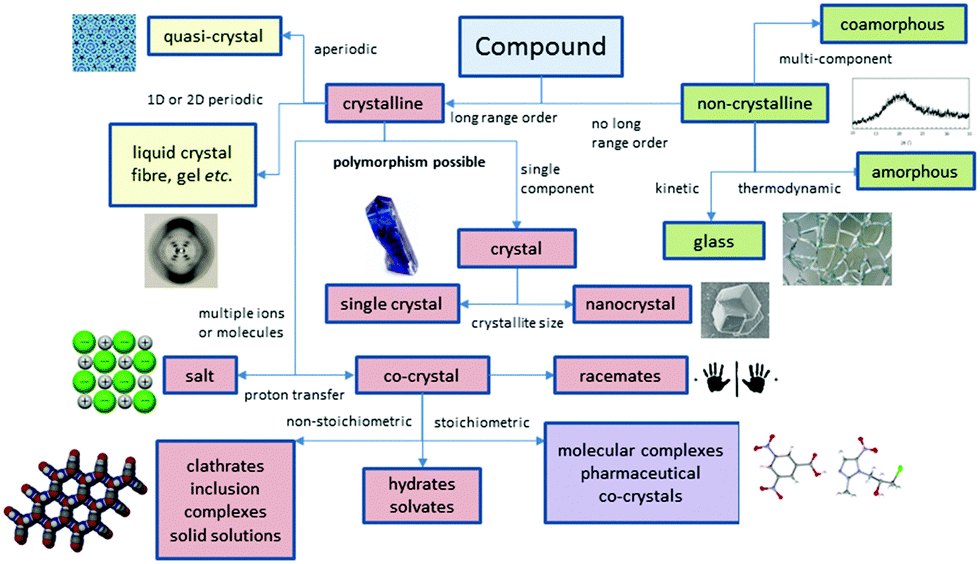 | ||
| Fig. 2 The scope of molecular materials (adapted from ref. 16). | ||
Very recently the regulatory status of co-crystals has been defined by the US FDA which states “[a] co-crystal with a pharmaceutically acceptable coformer … can be considered to be a pharmaceutical co-crystal and has a regulatory classification similar to that of a polymorph of the API”.17 There are now a number of FDA approved co-crystal drug products including Entresto® (sacubitril-valsartan), Lexapro® (escitalopram oxalate), and Depakote® (valproate sodium cocrystal with valproic acid).18 A particularly interesting recent result is the development of drug–drug cocrystals such as the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 cocrystal of tramadol and celecoxib.19 Amorphous materials and techniques used to study their local structure such as solid-state NMR spectroscopy18 and total scattering techniques20 have also seen considerable interest. The caveat is that amorphous materials are less thermodynamically stable than crystals and hence there is a need to find ways to stabilise them, particularly in the presence of moisture. In this context co-amorphous materials in the form of polymer blends with polymers such as polyvinylpyrrolidone are also increasingly common, but these have drawbacks such as the need for relatively large amounts of polymer. Small-molecule co-amorphous21–23 materials including co-amorphous drug–drug preparations24 are beginning to be explored as a high solubility alternative. As a result of the popularity of co-crystalline and co-amorphous materials there has been considerable progress in the identification of suitable co-formers and methods used to screen for new co-crystalline and co-amorphous forms, Fig. 3.25–30
:1 cocrystal of tramadol and celecoxib.19 Amorphous materials and techniques used to study their local structure such as solid-state NMR spectroscopy18 and total scattering techniques20 have also seen considerable interest. The caveat is that amorphous materials are less thermodynamically stable than crystals and hence there is a need to find ways to stabilise them, particularly in the presence of moisture. In this context co-amorphous materials in the form of polymer blends with polymers such as polyvinylpyrrolidone are also increasingly common, but these have drawbacks such as the need for relatively large amounts of polymer. Small-molecule co-amorphous21–23 materials including co-amorphous drug–drug preparations24 are beginning to be explored as a high solubility alternative. As a result of the popularity of co-crystalline and co-amorphous materials there has been considerable progress in the identification of suitable co-formers and methods used to screen for new co-crystalline and co-amorphous forms, Fig. 3.25–30
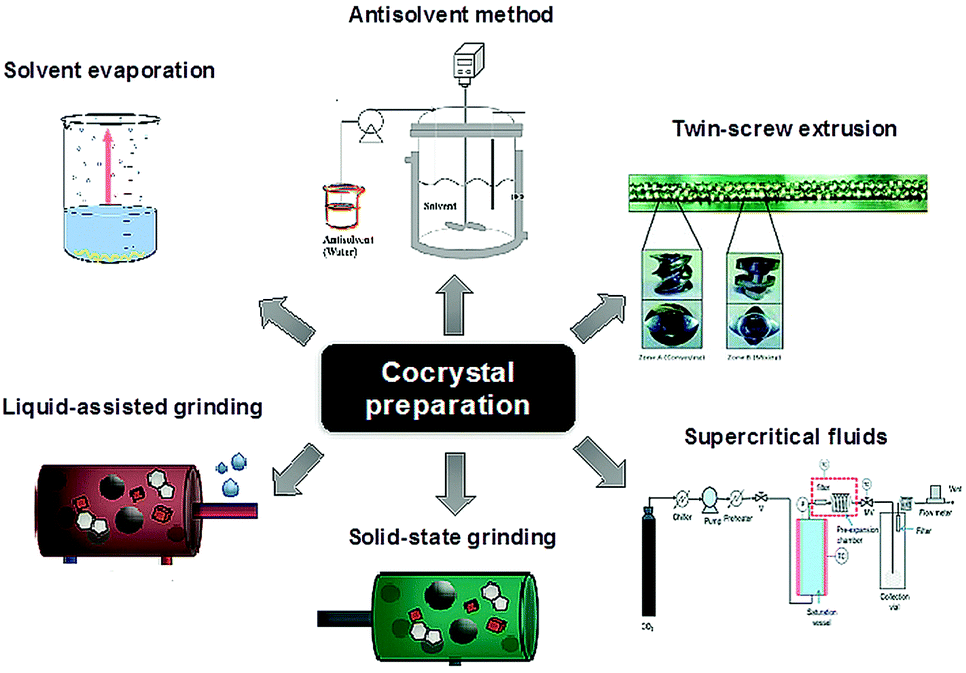 | ||
| Fig. 3 Methods for co-crystal preparation (reproduced with permission from ref. 18). | ||
Another key area is the significant advances in the way in which new molecular solids are prepared and characterised. In 2013 the Fujita group published an article in Nature entitled “X-ray analysis on the nanogram to microgram scale using porous complexes”.31 For the first time someone was proposing a solution to the age-old problem of compounds that are difficult to prepare as nice single crystals. Instead of trying to use artful techniques to induce the compound to crystallise by itself, the crystalline order is provided by pre-grown crystals of coordination networks containing guest binding cages. The target compound is then introduced into the cages by allowing it to diffuse into the network of this ‘crystalline sponge’. An ordered array of cages results in an ordered array of guest molecules suitable for single crystal diffraction, Fig. 4. While the method is not without its drawbacks such as low precision, and the need for the substrate to diffuse into the crystalline pores, it genuinely represents a step change in a very classical arena.
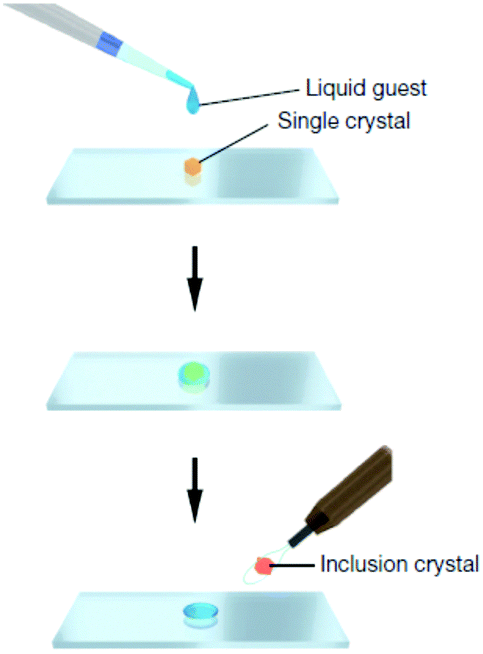 | ||
| Fig. 4 Preparation of a guest included network complex: a single piece of crystalline sponge is treated for 2 d (possibly under sealed conditions for volatile liquids) with a drop of a liquid guest and subjected to X-ray data collection (reproduced with permission from ref. 31). | ||
The crystalline sponge method is one of a host of recent, creative ways to prepare high quality crystals, new or different polymorphs and novel co-crystalline or amorphous forms. There are also recent developments in increasing throughput and control, influencing morphology or altering key properties such as dissolution rate, bioavailability, processability, stability and compressability.32 There has been considerable progress in the use of micro- or nanoconfinement for crystallization. Crystallization in microemulsion droplets has been shown to ‘leapfrog Ostwald's rule of stages’ and home in on the most thermodynamically stable solid form under a given set of conditions.33,34 In addition to molecular compounds such as pharmaceuticals,35 the method has also been applied nanomaterials such as nanographite.36 Nanoscale confinement in materials such as porous polymers and nanoporous glass is also a powerful new crystallization tool.37 As Ward points out in a recent review: “nanoscale confinement examines nucleation and phase transformations at length scales corresponding to the critical size, at which kinetics and thermodynamics of nucleation and growth intersect and dramatic departures in stability compared to bulk crystals can appear”.38 Confined crystallization has been shown to result in novel outcomes under conditions of both ‘rigid confinement’ in materials such as silica39 and ‘soft confinement’ in polymers.40 In both cases, solid-state NMR spectroscopy is a useful in situ probe of crystallization outcome. Confined crystallization has also been used on patterned surfaces41,42 and in gel microparticles to give polymorph selective crystallization, particularly for the highly polymorphic (and difficult to control) olanzapine precursor ROY.43 Our own work has focussed on small molecule organogels to limit convection and provide an active surface for crystal nucleation. This includes gels specifically tailored to have chemical functionality mimicking the ubiquitous ROY. The gels have variable effects, sometimes favouring the most thermodynamically stable polymorph as in the crystals of Form III carbamazepine (Fig. 5)44 and sometimes increasing nucleation rate of metastable polymorphs. Work in conjunction with gel structure calculation by Day suggests that an element of conformational biasing is involved with the observed selectivity.45 Related work by the Smith and Sanchez groups has shown that single46 and multicomponent gels involving dendrons and long chain aliphatic amines also results in effective pharmaceutical crystallization media.47 In addition, cationic surfactants in conjunction with nanocellulose have been used to gel-crystallize a novel solvate of sulfapyridine, the drug credited with saving Winston Churchill's life in 1943.48
 | ||
| Fig. 5 Crystals of Form III carbamazepine growing in a supramolecular organogel (reproduced with permission from ref. 44). | ||
A particularly notable development reported by Cristóbal Viedma in 2005 involves the grinding of crystals suspended in solution as a means to deracemization and ultimately the production of enantiomerically pure solids induced by nonlinear autocatalysis and recycling, a technique known as Viedma ripening.49,50 The technique has gained considerable prominence in the ensuing decade with recent work continuing to shed considerable mechanistic light on the role of abrasion and fracture of crystalline particles in attrition-enhanced deracemization processes.51
As well as solution-mediated crystallization strategies, the 21st century has seen an explosion in the application of mechanochemistry and liquid assisted grinding methods to the study of molecular solid form.52 While grinding often brings about amorphization, mechanochemistry can also proceed with recrystallization to give new polymorphs.53 It is also especially important in the discovery of novel co-crystals which may not form from solution, particularly in cases where the two components are of very different solubility.54 An interesting alternative is ‘vapour digestion’ – liquid assisted grinding without the grinding – in which exposure of pure components to solvent vapour results in co-crystal formation.55
Given the difficulty in obtaining single crystals for structural characterization from mechanochemical synthesis, creative techniques have evolved in order to study mechanochemical co-crystal formation such as terahertz time domain spectroscopy56 and milling apparatus compatible with in situ synchrotron X-ray diffraction.57 Rarer, non-mechanochemical solid–solid transformations have also become increasingly topical, particularly single-crystal-to-single-crystal transformations which can give mechanistic insight and also allow access to otherwise inaccessible polymorphs.58 In this context work by MacGillivray on topotactic transformations in co-crystals involving a template and photosensitive precursor can give rise to novel compounds such as ladderanes not readily accessible by conventional means.59 There was even a fascinating report in 2014 of a new form of ice, Ice XVI, generated by evacuation of neon from a structure II clathrate hydrate with retention of crystallinity.60 Ice XVI is the least dense pure solid form of water.
In conjunction with novel lab crystallization methods, there have also been significant advances in industrial techniques for preparing solid state materials and their application to co-crystals in particular. These include particularly advances in continuous crystallization,61 milling,62 twin screw extrusion63 and sonocrystallization.64
In addition to progress in the practical aspects of making crystals, there has also been a parallel growth in understanding the mechanism by which they form. For many years classical nucleation theory with its molecule-by-molecule growth of a critical sized nucleus was the dominant picture of crystal nucleation in most chemists minds.65 There has been an increasing appreciation of non-classical approaches, however. Two-step nucleation theory posits the evolution of a supersaturated system through a dense liquid phase that does not resemble the final crystal structure, followed by reorganisation of that dense aggregate cluster into an ordered structure (Fig. 6a).65 Non-classical mechanisms of crystal growth such as oriented attachment of smaller particles is also being increasingly recognised (Fig. 6b).66 Oriented attachment involves the spontaneous self-assembly of adjoining crystals with common crystallographic orientations and offers a powerful explanation of solid state chiral recognition phenomena.67
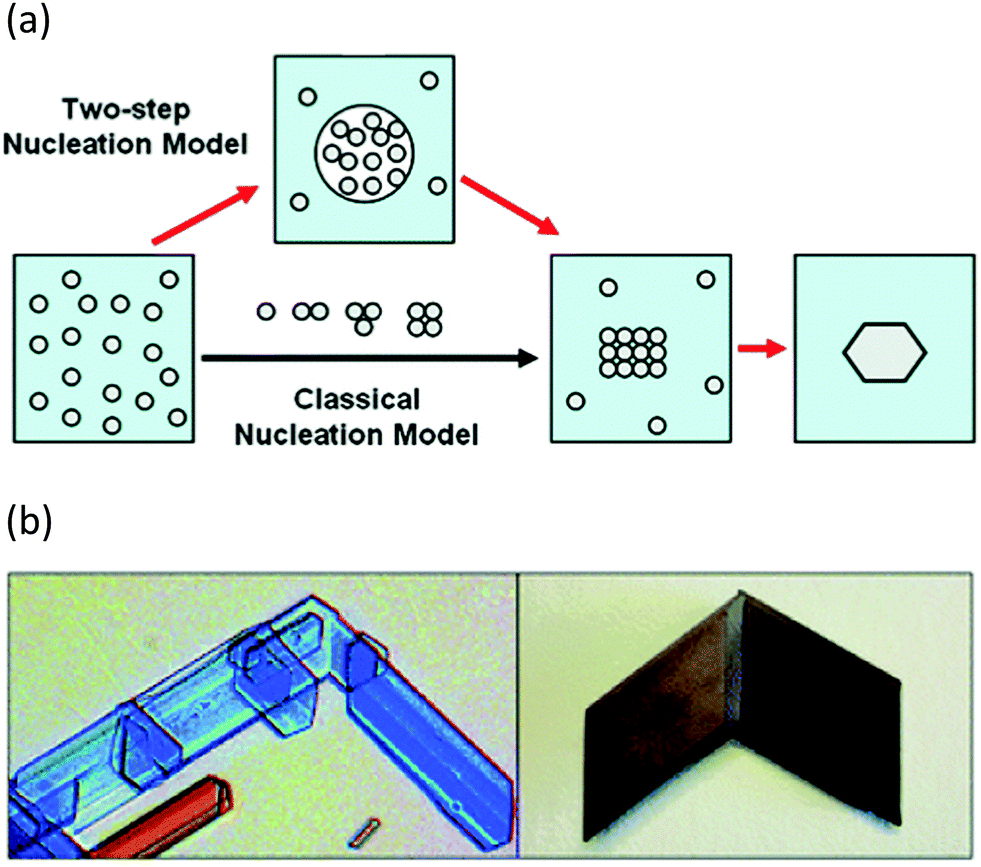 | ||
| Fig. 6 (a) Classical vs. non-classical nucleation models (reproduced with permission from ref. 65) (b) Oriented attachment in gypsum crystals (reproduced with permission from ref. 66). | ||
The past decade has seen tremendous progress in computational crystal structure prediction (CSP) methodologies. A harbinger of change was the striking success of hybrid DFT methods in correctly calculating all four trial structures, including that of a co-crystal, in the 2007 Cambridge blind tests.68 While subsequent blind tests have shown that many challenges remain, particularly with conformationally flexible molecules and multicomponent systems,69 a hybrid DFT approach based on Monte Carlo parallel tempering for structure generation and final ranking with DFT predicted all five experimental structures in the 2016 blind tests.70 Routine application of CSP remains limited by computational expense and the expertise needed to apply the methods. However, landmark work by the Price and Florence groups in 2011 resulted in the calculation of a new catermeric polymorph of the epilepsy medication carbamazepine, termed Form V. All of the previously known Forms I–IV are based on hydrogen bonded dimers and hence the fact that the catemeric Form V had not been experimentally observed despite its calculated stability must arise from the dominance of the dimers at the early nucleation stages. Price and Florence were able to overcome this barrier by experimentally growing the crystals of the predicted Form V on a template of the closely related dihydrocarbamazepine, Fig. 7. The approach is a general one and has very recently resulted in a new form of tolfenamic acid based on a computed prediction that identified a form of mefenamic acid as a suitable template.71 CSP approaches in combination with experimental TEM data also represent a powerful experimental approach to identification of new polymorphs present in tiny amounts, as in the case of a new form of theophylline present as a mixture with the well-known Form II.72
 | ||
| Fig. 7 Crystals of the computationally predicted catemeric carbamazepine Form V (I–iii) growing on a catermeric dihydrocarbamazepine Form II seed crystal (reproduced with permission from ref. 86). | ||
The fact that CSP methods apparently ‘overpredict’ the number of possible stable forms is an ongoing puzzle.73 However, wide-ranging exploration of the experimental space is beginning to show that some of these elusive calculated forms are real. In 2015 in silico polymorph screening revealed two potentially thermodynamically stable forms of the cardiovascular drug Dalcetrapib in addition to the known polymorphs.74 Subsequent investigation of the compound at high pressure allowed the experimental structure determination of one of the predicted forms. The experimental work showed that the new form is metastable at ambient pressure. The work effectively derisks the undesirable appearance of a more stable polymorph during late-stage development, as happened in the famous ritonavir case.75 Similarly application of magnetic fields during coronene crystallization also revealed a novel, albeit not predicted, polymorph of this well-known compound.76
In addition to the major advances in computational CSP, there has also been a great deal of progress in ‘soft’ predictions in terms of the identification of trends in crystal engineering and their practical use in preparing designed solid forms. The importance of the Cambridge Structural Database, which is now approaching one million entries,77 is hard to overstate in this context. Some of the earliest systematic observational predictions were made back in 1990 by Margaret Etter in the form of Etter's rules which focussed on hydrogen bonded systems in general and some specific systems such as nitroanilines, diaryl ureas, and some co-crystals in particular.78 In 1994 Brock and Dunitz made further systematic observations ‘towards a grammar of crystal packing’79 and in 1995 Desiraju published his powerful supramolecular synthon approach,80 which has been a substantial driver in a broad range of crystal engineering work ever since.81 Work by Aakeröy, for example, has shown that there is a hierarchy of supramolecular synthons that can be used to design multi-component cocrystals based on relative hydrogen bond acidity/basicity.82 There has also been growing interest in solid forms that do not conform to expected trends such as those with multiple molecules in the asymmetric unit (Z′ > 1).83,84 Interestingly, while much work in crystal engineering has been concerned with the ability to design and control the ways in which molecules pack to give a crystal, work by Hosseini has also focussed on the macroscopic engineering of crystals themselves in a “crystal welding” approach involving the isomorphous deposition of closely related crystals of different metal complexes.85
The past 20 years has also seen tremendous progress in molecular assembly in non-periodic structures. The field has been stimulated by the award of the 2011 Nobel Prize in chemistry to Shechtman for his work on quasicrystals.87 Within the field of molecular materials, developments on aperiodic ordered arrays of tetracarboxylic acid rhombus or Penrose tilings on surfaces have been enabled by the ability to directly image molecular arrays by STM.88,89
The search for understanding of crystal structure has also led to intense scrutiny of the interactions the hold crystals together. Not just strong and directional interactions that are subject to facile design such as hydrogen bonds, but also weaker or less obvious interactions that have previously gone unrecognised. The past two decades has seen an explosive growth in the understanding of halogen bonding in particular,90 with halogen and hydrogen bonds acting as potentially mutually orthogonal supramolecular synthons.91 A number of other less well recognized supramolecular interactions such as anion–π interactions,92,93 chalcogen bonding,94 pnictogen bonding,95 and π–hole interactions96 are also now being identified and finding application in crystal engineering.
The intense focus on the discovery and transformation of pharmaceutical solid forms in particular has led in some cases to the discovery of novel and unanticipated ways to bring about solid-state phase transformations. Application of CO2 gas pressure to the drugs clarithromycin and lansoprazole brings about facile conversion from unstable solvates to thermodynamically stable polymorphs.97 Despite the lack of the permanent pores that are associated with more rigid materials such as MOFs and zeolites, organic molecular solids have proven to be remarkably mobile and surprisingly permeable to fluid diffusion.98 As a result new applications for organic solids in gas storage and separation are being envisaged.99 A particular striking development in this context are the crystalline arrays of molecules with permanently engineered and predictable pores produced by the Cooper group.100 Remarkably this has also included liquids with permanent porosity.101
While of huge importance, the pharmaceutical sector has not been the only one to drive interest in organic solids. The solid state properties of agrochemicals and energetic materials are also of considerable interest and importance. As with pharmaceuticals, crystal engineering approaches and particular co-crystal formation can be used to modify or tune the behaviour of these substances. Work by the Aakeröy group, for example, has shown that co-crystallization approaches can be used to modulate the detonation properties of the energetic material ethylenedinitramine.102 In the agrochemicals sector co-crystal formation has been used to increase the melting point of the fungicides cyprodinil and dithianon.103
The 2016 Nobel Prize in chemistry has shone a spotlight onto the area of molecular machines.104–106 While much work on molecular machines is on isolated molecules either as a proof-of-principle in solution or confined on surfaces, crystalline arrays of molecular machines offer perhaps the most significant scope for a functional, high-density device. In this context work by the Garcia-Garibay group has focussed on what they term “amphidynamic crystals” including crystalline molecular motors and gyroscopes based on a wheel and axel type of approach.107 The growing control of crystal structure–property relationships has also given rise to the ability of engineer functional crystals – a field termed “crystal adaptronics” which has given rise to systems such as mechanically reconfigurable elastic, superelastic and ferroelastic molecular crystals with properties such as bending and shape memory.108,109
Finally, it is noteworthy that the contribution made by progress in instrumentation, particularly in X-ray crystallography has made the study of molecular solids, particularly under non-ambient conditions, increasingly accessible. The availability of high intensity sources such as the UK Diamond synchrotron with the I19 and I11 single crystal and high resolution powder diffraction beamlines110 makes structural characterisation of even marginal crystals and poorly crystallizing compounds such as gelators increasingly possible. Structure determination on tiny nanocrystals using electron diffraction applied to both biological samples111 and small molecules is also potentially revolutionary.112 Sample environments such as the very high pressures offered by the diamond anvil cell and high pressure gas cells, as well as ultra-low temperature down to 2 K are also making novel and dynamic structural experiments increasingly common.113
Conclusions
The first decades of the 21st century have seen a quiet revolution in the understanding, prediction, control and importance of the organic and molecular solid state, rivalling that of ‘harder’ materials such as metal–organic frameworks. With the increasing molecular weight and hydrophobicity of recently discovered drugs, the optimization of solid state properties is becoming ever more important. Coupled with the intellectual property opportunities new solid forms can offer, formulation challenges are providing a strong industrial driver for the continued expansion in the exploration of molecular solid state chemistry. Understanding and control of molecular solids even in aperiodic and non-crystalline systems is experiencing dramatic advances as a result of techniques such as total scattering, high resolution microscopy, high intensity X-ray sources and electron diffraction. Control of molecular 3D packing offers scope for the realisation of arrays of functional molecular systems such as molecular machines and advanced properties such as controlled porosity, ferroelasticity and shape memory. The near future promises exciting progress in the determination of the structure of amorphous or nanocrystalline materials, the reliable and convenient prediction of all accessible crystal structures and in the understanding of the crystal nucleation process.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
I thank the Royal Society for a Wolfson Research Merit Award and the Engineering and Physical Sciences Research Council for their support of our work in the crystallization arena. I am also very grateful to Prof. Len Barbour, Dr Katharina Edkins and Dr David Berry for constructive advice on the manuscript.Notes and references
- J. Thun, L. Seyfarth, J. Senker, R. E. Dinnebier and J. Breu, Angew. Chem., Int. Ed., 2007, 46, 6729–6731 CrossRef CAS PubMed.
- W. C. McCrone, in Physics and Chemistry of the OrganicSolid State, ed. D. Fox, M. M. Labes and A. Weissberger, Interscience, New York, 1965, vol. 11, pp. 726–767 Search PubMed.
- J. Maddox, Nature, 1988, 335, 201 CrossRef.
- J. Bernstein, Cryst. Growth Des., 2011, 11, 632–650 CrossRef CAS.
- A. Y. Lee, D. Erdemir and A. S. Myerson, Annu. Rev. Chem. Biomol. Eng., 2011, 2, 259–280 CrossRef CAS PubMed.
- G. Saurabh and C. Kaushal, J. Chem. Pharm. Res., 2011, 3, 6–17 Search PubMed.
- G. Boehm, L. Yao, L. Han and Q. Zheng, Acta Pharm. Sin. B, 2013, 3, 297–311 CrossRef.
- J. Bernstein, in International Union of Crystallography Monographs on Crystallography, Clarendon Press, Oxford, 2002, ch. 10, pp. 298–301 Search PubMed.
- https://www.prnewswire.com/news-releases/depomed-to-acquire-us-rights-to-nucynta-tapentadol-nucynta-er-tapentadol-extended-release-tablets-and-nucynta-tapentadol-oral-solution-from-janssen-pharmaceuticals-inc-for-105-billion-300021321.html, accessed 5th October 2018.
- J. L. Atwood, Cryst. Growth Des., 2015, 15, 2874–2877 CrossRef CAS.
- M. B. J. Atkinson, D. K. Bwambok, J. Chen, P. D. Chopade, M. M. Thuo, C. R. Mace, K. A. Mirica, A. A. Kumar, A. S. Myerson and G. M. Whitesides, Angew. Chem., Int. Ed., 2013, 52, 10208–10211 CrossRef CAS PubMed.
- D. J. Berry and J. W. Steed, Adv. Drug Delivery Rev., 2017, 117, 3–24 CrossRef CAS PubMed.
- N. Qiao, M. Li, W. Schlindwein, N. Malek, A. Davies and G. Trappitt, Int. J. Pharm., 2011, 419, 1–11 CrossRef CAS PubMed.
- H. G. Brittain, Cryst. Growth Des., 2012, 12, 1046–1054 CrossRef CAS.
- S. Aitipamula, R. Banerjee, A. K. Bansal, K. Biradha, M. L. Cheney, A. R. Choudhury, G. R. Desiraju, A. G. Dikundwar, R. Dubey, N. Duggirala, P. P. Ghogale, S. Ghosh, P. K. Goswami, N. R. Goud, R. R. K. R. Jetti, P. Karpinski, P. Kaushik, D. Kumar, V. Kumar, B. Moulton, A. Mukherjee, G. Mukherjee, A. S. Myerson, V. Puri, A. Ramanan, T. Rajamannar, C. M. Reddy, N. Rodriguez-Hornedo, R. D. Rogers, T. N. G. Row, P. Sanphui, N. Shan, G. Shete, A. Singh, C. C. Sun, J. A. Swift, R. Thaimattam, T. S. Thakur, R. Kumar Thaper, S. P. Thomas, S. Tothadi, V. R. Vangala, N. Variankaval, P. Vishweshwar, D. R. Weyna and M. J. Zaworotko, Cryst. Growth Des., 2012, 12, 2147–2152 CrossRef CAS.
- J. W. Steed, Trends Pharm. Sci., 2013, 34, 185–193 CrossRef CAS PubMed.
- U.S. Department of Health and Human Services, Food and Drug Administration Center for Drug Evaluation and Research (CDER), Regulatory Classification of Pharmaceutical Co-Crystals Guidance for Industry, http://www.fda.gov/ucm/groups/fdagov-public/@fdagov-drugs-gen/documents/document/ucm516813.pdf, accessed November 2018.
- A. Karagianni, M. Malamatari and K. Kachrimanis, Pharmaceutics, 2018, 10, 18 CrossRef PubMed.
- M. Merlos, E. Portillo-Salido, A. Brenchat, B. Aubel, J. Buxens, A. Fisas, X. Codony, L. Romero, D. Zamanillo and J. M. Vela, Eur. J. Pharmacol., 2018, 833, 370–378 CrossRef CAS PubMed.
- S. Bates, G. Zografi, D. Engers, K. Morris, K. Crowley and A. Newman, Pharm. Res., 2006, 23, 2333–2349 CrossRef CAS PubMed.
- W. Wu, K. Löbmann, J. Schnitzkewitz, A. Knuhtsen, D. S. Pedersen, H. Grohganz and T. Rades, Int. J. Pharm., 2018, 549, 380–387 CrossRef CAS PubMed.
- S. J. Dengale, H. Grohganz, T. Rades and K. Löbmann, Adv. Drug Delivery Rev., 2016, 100, 116–125 CrossRef CAS PubMed.
- M. J. Goodwin, O. M. Musa, D. J. Berry and J. W. Steed, Cryst. Growth Des., 2018, 18, 701–709 CrossRef CAS.
- K. Löbmann, R. Laitinen, H. Grohganz, K. C. Gordon, C. Strachan and T. Rades, Mol. Pharmaceutics, 2011, 8, 1919–1928 CrossRef PubMed.
- N. Blagden, S. J. Coles and D. J. Berry, CrystEngComm, 2014, 16, 5753–5761 RSC.
- N. Blagden, D. J. Berry, A. Parkin, H. Javed, A. Ibrahim, P. T. Gavan, L. L. De Matos and C. C. Seaton, New J. Chem., 2008, 32, 1659–1672 RSC.
- S. L. Morissette, Ö. Almarsson, M. L. Peterson, J. F. Remenar, M. J. Read, A. V. Lemmo, S. Ellis, M. J. Cima and C. R. Gardner, Adv. Drug Delivery Rev., 2004, 56, 275–300 CrossRef CAS PubMed.
- K. Fucke, S. A. Myz, T. P. Shakhtshneider, E. V. Boldyreva and U. J. Griesser, New J. Chem., 2012, 36, 1969–1977 RSC.
- N. Qiao, M. Z. Li, W. Schlindwein, N. Malek, A. Davies and G. Trappitt, Int. J. Pharm., 2011, 419, 1–11 CrossRef CAS PubMed.
- D. Tan, L. Loots and T. Friscic, Chem. Commun., 2016, 52, 7760–7781 RSC.
- Y. Inokuma, S. Yoshioka, J. Ariyoshi, T. Arai, Y. Hitora, K. Takada, S. Matsunaga, K. Rissanen and M. Fujita, Nature, 2013, 495, 461–466 CrossRef CAS PubMed.
- B. Rodríguez-Spong, C. P. Price, A. Jayasankar, A. J. Matzger and N. r. Rodríguez-Hornedo, Adv. Drug Delivery Rev., 2004, 56, 241–274 CrossRef PubMed.
- C. Chen, O. Cook, C. E. Nicholson and S. J. Cooper, Cryst. Growth Des., 2011, 11, 2228–2237 CrossRef CAS.
- C. E. Nicholson, C. Chen, B. Mendis and S. J. Cooper, Cryst. Growth Des., 2011, 11, 363–366 CrossRef CAS.
- C. E. Nicholson and S. J. Cooper, Crystals, 2011, 1, 195–205 CrossRef CAS.
- N. J. Hargreaves and S. J. Cooper, Cryst. Growth Des., 2016, 16, 3133–3142 CrossRef CAS.
- J.-M. Ha, J. H. Wolf, M. A. Hillmyer and M. D. Ward, J. Am. Chem. Soc., 2004, 126, 3382–3383 CrossRef CAS PubMed.
- Q. Jiang and M. D. Ward, Chem. Soc. Rev., 2014, 43, 2066–2079 RSC.
- X. Yang, T. C. Ong, V. K. Michaelis, S. Heng, J. Huang, R. G. Griffin and A. S. Myerson, CrystEngComm, 2014, 16, 9345–9352 RSC.
- X. Yang, T.-C. Ong, V. K. Michaelis, S. Heng, R. G. Griffin and A. S. Myerson, CrystEngComm, 2015, 17, 6044–6052 RSC.
- A. Y. Lee, I. S. Lee, S. S. Dettet, J. Boerner and A. S. Myerson, J. Am. Chem. Soc., 2005, 127, 14982–14983 CrossRef CAS PubMed.
- A. Singh, I. S. Lee and A. S. Myerson, Cryst. Growth Des., 2008, 9, 1182–1185 CrossRef.
- Y. Diao, K. E. Whaley, M. E. Helgeson, M. A. Woldeyes, P. S. Doyle, A. S. Myerson, T. A. Hatton and B. L. Trout, J. Am. Chem. Soc., 2012, 134, 673–684 CrossRef CAS PubMed.
- J. A. Foster, M.-O. M. Piepenbrock, G. O. Lloyd, N. Clarke, J. A. K. Howard and J. W. Steed, Nat. Chem., 2010, 2, 1037–1043 CrossRef CAS PubMed.
- J. A. Foster, K. K. Damodaran, A. Maurin, G. M. Day, H. P. G. Thompson, G. J. Cameron, J. C. Bernal and J. W. Steed, Chem. Sci., 2017, 8, 78–84 RSC.
- F. Aparicio, E. Matesanz and L. Sánchez, Chem. Commun., 2012, 48, 5757–5759 RSC.
- J. Buendía, E. Matesanz, D. K. Smith and L. Sánchez, CrystEngComm, 2015, 17, 8146–8152 RSC.
- C. Ruiz-Palomero, S. R. Kennedy, M. L. Soriano, C. D. Jones, M. Valcarcel and J. W. Steed, Chem. Commun., 2016, 52, 7782–7785 RSC.
- L.-C. Sögütoglu, R. R. E. Steendam, H. Meekes, E. Vlieg and F. P. J. T. Rutjes, Chem. Soc. Rev., 2015, 44, 6723–6732 RSC.
- C. Viedma, Phys. Rev. Lett., 2005, 94, 065504 CrossRef PubMed.
- C. Xiouras, A. A. Fytopoulos, J. H. Ter Horst, A. G. Boudouvis, T. Van Gerven and G. D. Stefanidis, Cryst. Growth Des., 2018, 18, 3051–3061 CrossRef CAS.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friscic, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- D. Hasa and W. Jones, Adv. Drug Delivery Rev., 2017, 117, 147–161 CrossRef CAS PubMed.
- T. Friščić and W. Jones, Cryst. Growth Des., 2009, 9, 1621–1637 CrossRef.
- D. Braga, S. L. Giaffreda, F. Grepioni, M. R. Chierotti, R. Gobetto, G. Palladino and M. Polito, CrystEngComm, 2007, 9, 879–881 RSC.
- K. L. Nguyen, T. Friščić, G. M. Day, L. F. Gladden and W. Jones, Nat. Mater., 2007, 6, 206–209 CrossRef CAS PubMed.
- T. Friscic, I. Halasz, P. J. Beldon, A. M. Belenguer, F. Adams, S. A. J. Kimber, V. Honkimaki and R. E. Dinnebier, Nat. Chem., 2013, 5, 66–73 CrossRef CAS PubMed.
- A. Chaudhary, A. Mohammad and S. M. Mobin, Cryst. Growth Des., 2017, 17, 2893–2910 CrossRef CAS.
- X. C. Gao, T. Friščić and L. R. MacGillivray, Angew. Chem., Int. Ed., 2004, 43, 232–236 CrossRef CAS PubMed.
- A. Falenty, T. C. Hansen and W. F. Kuhs, Nature, 2014, 516, 231 CrossRef CAS PubMed.
- T. McGlone, N. E. B. Briggs, C. A. Clark, C. J. Brown, J. Sefcik and A. J. Florence, Org. Process Res. Dev., 2015, 19, 1186–1202 CrossRef CAS.
- S. Korde, S. Pagire, H. Pan, C. Seaton, A. Kelly, Y. Chen, Q. Wang, P. Coates and A. Paradkar, Cryst. Growth Des., 2018, 18, 2297–2304 CrossRef CAS.
- D. E. Crawford, C. K. G. Miskimmin, A. B. Albadarin, G. Walker and S. L. James, Green Chem., 2017, 19, 1507–1518 RSC.
- M. D. L. de Castro and F. Priego-Capote, Ultrason. Sonochem., 2007, 14, 717–724 CrossRef PubMed.
- D. Erdemir, A. Y. Lee and A. S. Myerson, Acc. Chem. Res., 2009, 42, 621–629 CrossRef CAS PubMed.
- C. Viedma, L. A. Cuccia, A. McTaggart, B. Kahr, A. T. Martin, J. M. McBride and P. Cintas, Chem. Commun., 2016, 52, 11673–11676 RSC.
- C. Viedma, J. M. McBride, B. Kahr and P. Cintas, Angew. Chem., Int. Ed., 2013, 52, 10545–10548 CrossRef CAS PubMed.
- M. A. Neumann, F. J. J. Leusen and J. Kendrick, Angew. Chem., Int. Ed., 2008, 47, 2427–2430 CrossRef CAS PubMed.
- S. L. Price, Chem. Soc. Rev., 2014, 43, 2098–2111 RSC.
- A. M. Reilly, R. I. Cooper, C. S. Adjiman, S. Bhattacharya, A. D. Boese, J. G. Brandenburg, P. J. Bygrave, R. Bylsma, J. E. Campbell, R. Car, D. H. Case, R. Chadha, J. C. Cole, K. Cosburn, H. M. Cuppen, F. Curtis, G. M. Day, R. A. DiStasio Jr, A. Dzyabchenko, B. P. van Eijck, D. M. Elking, J. A. van den Ende, J. C. Facelli, M. B. Ferraro, L. Fusti-Molnar, C.-A. Gatsiou, T. S. Gee, R. de Gelder, L. M. Ghiringhelli, H. Goto, S. Grimme, R. Guo, D. W. M. Hofmann, J. Hoja, R. K. Hylton, L. Iuzzolino, W. Jankiewicz, D. T. de Jong, J. Kendrick, N. J. J. de Klerk, H.-Y. Ko, L. N. Kuleshova, X. Li, S. Lohani, F. J. J. Leusen, A. M. Lund, J. Lv, Y. Ma, N. Marom, A. E. Masunov, P. McCabe, D. P. McMahon, H. Meekes, M. P. Metz, A. J. Misquitta, S. Mohamed, B. Monserrat, R. J. Needs, M. A. Neumann, J. Nyman, S. Obata, H. Oberhofer, A. R. Oganov, A. M. Orendt, G. I. Pagola, C. C. Pantelides, C. J. Pickard, R. Podeszwa, L. S. Price, S. L. Price, A. Pulido, M. G. Read, K. Reuter, E. Schneider, C. Schober, G. P. Shields, P. Singh, I. J. Sugden, K. Szalewicz, C. R. Taylor, A. Tkatchenko, M. E. Tuckerman, F. Vacarro, M. Vasileiadis, A. Vazquez-Mayagoitia, L. Vogt, Y. Wang, R. E. Watson, G. A. de Wijs, J. Yang, Q. Zhu and C. R. Groom, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 439–459 CrossRef CAS PubMed.
- D. H. Case, V. K. Srirambhatla, R. Guo, R. E. Watson, L. S. Price, H. Polyzois, J. K. Cockcroft, A. J. Florence, D. A. Tocher and S. L. Price, Cryst. Growth Des., 2018, 18, 5322–5331 CrossRef CAS.
- M. D. Eddleston, K. E. Hejczyk, E. G. Bithell, G. M. Day and W. Jones, Chem. – Eur. J., 2013, 19, 7883–7888 CrossRef CAS PubMed.
- S. L. Price, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2013, 69, 313–328 CrossRef CAS PubMed.
- M. A. Neumann, J. van de Streek, F. P. A. Fabbiani, P. Hidber and O. Grassmann, Nat. Commun., 2015, 6, 7793 CrossRef CAS PubMed.
- J. Bauer, S. Spanton, R. Henry, J. Quick, W. Dziki, W. Porter and J. Morris, Pharm. Res., 2001, 18, 859–866 CrossRef CAS.
- J. Potticary, L. R. Terry, C. Bell, A. N. Papanikolopoulos, P. C. M. Christianen, H. Engelkamp, A. M. Collins, C. Fontanesi, G. Kociok-Köhn, S. Crampin, E. Da Como and S. R. Hall, Nat. Commun., 2016, 7, 11555 CrossRef CAS PubMed.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- M. C. Etter, Acc. Chem. Res., 1990, 23, 120–126 CrossRef CAS.
- C. P. Brock and J. D. Dunitz, Chem. Mater., 1994, 6, 1118–1127 CrossRef CAS.
- G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 2311–2327 CrossRef CAS.
- J. D. Dunitz and A. Gavezzotti, Cryst. Growth Des., 2012, 12, 5873–5877 CrossRef CAS.
- C. B. Aakeröy, J. Desper and B. M. T. Scott, Chem. Commun., 2006, 1445–1447 RSC.
- C. P. Brock, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 807–821 CrossRef CAS PubMed.
- K. M. Steed and J. W. Steed, Chem. Rev., 2015, 115, 2895–2933 CrossRef CAS PubMed.
- C. R. R. Adolf, S. Ferlay, N. Kyritsakas and M. W. Hosseini, J. Am. Chem. Soc., 2015, 137, 15390–15393 CrossRef CAS PubMed.
- J.-B. Arlin, L. S. Price, S. L. Price and A. J. Florence, Chem. Commun., 2011, 47, 7074–7076 RSC.
- D. Shechtman, Angew. Chem., Int. Ed., 2011, 50, 10489 CrossRef.
- K. S. Mali, N. Pearce, S. De Feyter and N. R. Champness, Chem. Soc. Rev., 2017, 46, 2520–2542 RSC.
- A. Stannard, J. C. Russell, M. O. Blunt, C. Salesiotis, M. d. C. Giménez-López, N. Taleb, M. Schröder, N. R. Champness, J. P. Garrahan and P. H. Beton, Nat. Chem., 2011, 4, 112 CrossRef PubMed.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- C. B. Aakeroy, P. D. Chopade and J. Desper, Cryst. Growth Des., 2011, 11, 5333–5336 CrossRef CAS.
- A. Bauza, T. J. Mooibroek and A. Frontera, ChemPhysChem, 2015, 16, 2496–2517 CrossRef CAS PubMed.
- M. Giese, M. Albrecht and K. Rissanen, Chem. Commun., 2016, 52, 1778–1795 RSC.
- A. C. Legon, Phys. Chem. Chem. Phys., 2017, 19, 14884–14896 RSC.
- A. Bauza, T. J. Mooibroek and A. Frontera, ChemPhysChem, 2016, 17, 1608–1614 CrossRef CAS PubMed.
- A. Bauza, A. Frontera and T. J. Mooibroek, Cryst. Growth Des., 2016, 16, 5520–5524 CrossRef CAS.
- J. Tian, S. J. Dalgarno and J. L. Atwood, J. Am. Chem. Soc., 2011, 133, 1399–1404 CrossRef CAS PubMed.
- P. K. Thallapally, L. Dobrzanska, T. R. Gingrich, T. B. Wirsig, L. J. Barbour and J. L. Atwood, Angew. Chem., Int. Ed., 2006, 45, 6506–6509 CrossRef CAS PubMed.
- S. J. Dalgarno, P. K. Thallapally, L. J. Barbour and J. L. Atwood, Chem. Soc. Rev., 2007, 36, 236–245 RSC.
- A. Pulido, L. Chen, T. Kaczorowski, D. Holden, M. A. Little, S. Y. Chong, B. J. Slater, D. P. McMahon, B. Bonillo, C. J. Stackhouse, A. Stephenson, C. M. Kane, R. Clowes, T. Hasell, A. I. Cooper and G. M. Day, Nature, 2017, 543, 657–664 CrossRef CAS PubMed.
- N. Giri, M. G. Del Pópolo, G. Melaugh, R. L. Greenaway, K. Rätzke, T. Koschine, L. Pison, M. F. C. Gomes, A. I. Cooper and S. L. James, Nature, 2015, 527, 216–220 CrossRef CAS PubMed.
- C. B. Aakeroy, T. K. Wijethunga and J. Desper, Chem. – Eur. J., 2015, 21, 11029–11037 CrossRef PubMed.
- C. Sowa, R. E. Gold, T. Chiodo and R. Vogel, WO 2013030777, 2013.
- J. P. Sauvage, Angew. Chem., Int. Ed., 2017, 56, 11080–11093 CrossRef CAS PubMed.
- B. L. Feringa, Angew. Chem., Int. Ed., 2017, 56, 11059–11078 CrossRef PubMed.
- J. F. Stoddart, Angew. Chem., Int. Ed., 2017, 56, 11094–11125 CrossRef CAS PubMed.
- C. S. Vogelsberg and M. A. Garcia-Garibay, Chem. Soc. Rev., 2012, 41, 1892–1910 RSC.
- E. Ahmed, D. P. Karothu and P. Naumov, Angew. Chem., Int. Ed., 2018, 57, 8837–8846 CrossRef CAS PubMed.
- E. R. Engel and S. Takamizawa, Angew. Chem., Int. Ed., 2018, 57, 11888–11892 CrossRef CAS PubMed.
- S. P. Thompson, J. E. Parker, J. Potter, T. P. Hill, A. Birt, T. M. Cobb, F. Yuan and C. C. Tang, Rev. Sci. Instrum., 2009, 80, 075107 CrossRef CAS PubMed.
- M. T. B. Clabbers, E. van Genderen, W. Wan, E. L. Wiegers, T. Gruene and J. P. Abrahams, Acta Crystallogr., Sect. D: Struct. Biol., 2017, 73, 738–748 CrossRef CAS PubMed.
- T. Gruene, J. T. C. Wennmacher, C. Zaubitzer, J. J. Holstein, J. Heidler, A. Fecteau-Lefebvre, S. De Carlo, E. Müller, K. N. Goldie, I. Regeni, T. Li, G. Santiso-Quinones, G. Steinfeld, S. Handschin, E. van Genderen, J. A. van Bokhoven, G. H. Clever and R. Pantelic, Angew. Chem., Int. Ed., 2018 DOI:10.1002/anie.201811318.
- R. Lee, J. A. K. Howard, M. R. Probert and J. W. Steed, Chem. Soc. Rev., 2014, 43, 4300–4311 RSC.
| This journal is © The Royal Society of Chemistry 2018 |