
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effect of iron re-deposition on the corrosion of impurity-containing magnesium
Daniel
Höche
*a,
Carsten
Blawert
a,
Sviatlana V.
Lamaka
a,
Nico
Scharnagl
a,
Chamini
Mendis
a and
Mikhail L.
Zheludkevich
ab
aMagIC – Magnesium Innovation Centre, Helmholtz-Zentrum Geesthacht, Max-Planck Str. 1, 21502 Geesthacht, Germany. E-mail: Daniel.hoeche@hzg.de
bDepartment of Materials and Ceramic Engineering/CICECO, University of Aviero, 3810-193 Aveiro, Portugal
First published on 26th November 2015
Abstract
This article provides a contribution towards the mechanistic understanding of surface phenomena observed during the corrosion of Mg-based substrates particularly in the low anodic polarization range. The concept considers the recent literature explaining cathodic hydrogen evolution from noble acting areas even during global anodic polarization. Heavy metal impurities in the ppm range or intermetallics are always present even in highly pure magnesium. Their potential effect was investigated here in more detail. The experimental results contribute to understanding the role of iron impurities in dark area formation and suggest a way for linking the observed phenomena to the recent literature. The shown enhanced cathodic activity of dark areas especially at the corrosion front and the superfluous hydrogen are linked to an iron re-deposition mechanism due to iron reduction. The proposed mechanism is based on the results obtained from innovative characterisation techniques using magnetic fields, diffraction experiments and transmission electron microscopy, which show the formation of iron rich zones, especially at the corrosion front offering “in statu nascendi” metallic Fe films acting as active cathodes for hydrogen reduction.
1. Introduction
The special behaviour of magnesium during anodic polarization is widely known and has been the subject of various scientific investigations.1,2 The strong hydrogen evolution reaction (HER) cannot be explained by the standard theory of anodic dissolution of metals. Already in the 1920s3 the phenomenon was known and also more than 30 years ago4–6 the issue was already of great interest. Recent discussions between Atrens, Kirkland and Birbilis7–9 reveal fundamental differences in the opinions on the reason for the explanation of the general Negative Difference Effect (NDE) mechanism. The NDE was also shown for aluminium alloys10,11 especially for Cu rich alloys containing the S-phase. Drazic et al.12 stated a direct relation to pitting driven dissolution mechanism. It is also generally known that the effect is stronger for less noble metallic alloys assuming comparable electrolyte exposure conditions. All these facts indicate that there can be a general mechanism behind the NDE.Several recently published studies try to explain the observed effects.13–16 Summaries can be found in the publications of Curioni et al.,17 Bender et al.,18 Atrens et al.19 or the very detailed work of Frankel et al.20 Williams, Frankel, Samaniego and Birbilis gave a strong feedback to the cathodic activity increase during anodic polarisation.1,7,21 Williams et al.1 have shown that there are persistent cathodes on the anodically polarized Mg surface. This indicates that superfluous hydrogen comes from a cathodic reaction which occurs at more noble inclusions present on an Mg surface. Birbilis et al.22 have also shown the enhanced catalytic response by measuring enhanced cathodic currents, and even the net polarity reversal of Mg surfaces held at an anodic potential. It is pointed out that the sites on the Mg surface enriched with noble metals (with higher exchange current density for hydrogen reduction, such as Cu, Al, Ni, Zn, Fe, and Mn) provide more favourable places for the cathodic reaction. Identifying the source of evolving hydrogen these studies do not clearly explain and validate how such an extensive amount of hydrogen can be cathodically generated on the noble inclusions, since they constitute only a minor fraction of the total surface area of the Mg-based substrates. In particular it is true for magnesium of relatively high purity where the concentration of impurities is at the ppm level.
Recent publication by Lebouil et al.23 promotes the so-called “filming” model. It is a description starting from initial impurity related reactions towards film formation (dark region of the Mg(OH)2/MgO bilayer) and its spatial extension. According to the model hydrogen gas is formed via persistent cathodic reaction on the bilayer. Thus, the NDE is not driven by anodic dissolution as also explained in the review of Thomas et al.24 The main question remaining is how this oxide/hydroxide layer which is not electronically conductive25 (it possesses only ionic conductivity) can act as an efficient cathode?
Thomas et al.24 stated that besides the mentioned filming effect impurities influence the HER: “…other metals may serve as preferred sites for high rates of HER, whilst their presence in the film could catalyse the HER, increasing the overall rate of the reaction forming Mg(OH)2”. This indicates a direct linking to the impurities. However the presence of such metallic inclusions within the oxide-hydroxide layer electrically decoupled from the Mg matrix should not contribute to the cathodic processes because of the negligible electron conductivity of oxides.
Fajardo et al.26 carried out galvanostatic experiments showing enhanced cathodic activity on ultra-highly pure Mg – UHP (0.1 ppm Fe measured using a spark source mass spectrometer). The surface appearance of low impurity level samples was surprisingly strongly affected after the tests, although they use 0.1 M NaCl test solution. The explanation is rather difficult since the cathodic reaction has to occur on α-Mg. It is commonly known that the Tafel reaction of H2 generation can occur on bare Mg. According to the theory for a Tafel-driven HER, two adsorbed protons (Had) are required. They have to arise via electrochemical adsorption (Volmer step) which for UHP Mg means:
| Mg + H2O + e− → Mg–Had + OH− |
As a consequence this implies that the HER depends on the amount of Had (adsorbed protons). Their amount depends on available electrons. If the system is globally polarized anodically a large number of electrons are generated which are consumed by HER (source: Mg2+ + 2e−). The availability of electrons is related to the very strong driving force for Mg oxidation. Thus, despite a certain polarisation resistance due to HER, the potential is driven so far negative of the equilibrium potential for proton reduction and the electron generation rate remains exceedingly high. As a result, there are sufficient electrons that can interact immediately. Such materials behaviour occurs if it is non-polarizable. As a result, the increase of anodic polarization and the increase of anodic reactions accelerate Had formation. This chemisorption on Mg is well known and was shown by Bird et al.27
However, chemical desorption via Tafel is “slow”. Thus the current increase is limited. The only possibility to get an increase of HER as measured must be a very fast process like electrochemical desorption. It is well known that this process (Heyrovsky) occurs as a rate determining step on 3d transition metals (Fe, Ni, and Cu) or even Pt as a pure transfer reaction.28 And it is also known that Heyrovsky reaction is the preferred mechanism in an alkaline environment. That is one reason why they are used for hydrogen production. It is very interesting that those conditions occur on Mg during corrosion.
The studies of Norskov et al.29 and Bird et al.27 of H2 on Mg(0001) showed the chemisorbed stage of Had on the surface. In this context we would like to refer to the study of Vegge et al.30 as well. The logical conclusion is that the Tafel process drives hydrogen formation reaction in the Mg primary phase.
It was shown by Pozzo et al.31 that transition metal dopants in magnesium can modify the catalytic activity. Their efficiency can be described via the Volcano plot with Ni, Fe and Rh sitting near the top of the volcano. As a consequence, the HER (Tafel-like) during anodic polarization of Mg could even increase at the 0.1 ppm Fe impurity level.
The occurrence of enhanced cathodic activity after cyclic polarization observed by Fajardo et al.26 seems to be related to a roughening effect since the strength of increase is limited. This does not exclude that dopants from Fe and Ni (0.1 ppm is sufficient) complement the catalytic properties of Mg leading to enhanced hydrogen kinetics. For high impurity levels a convolution of the mentioned effects and the major cathodic reactions on impurities drive the HER. This would also explain why the “strength of increase” increases with the impurity level until saturation due to the surface morphology and the electrolyte as observed in ref. 22 and 26.
The experiments of Curioni et al.32 showed that the highest current occurs close to the corrosion forefront. They also measured a decreasing cathodic current contribution of the dark film with increasing distance which contradicts to Fajardo's statement. Recent SVET studies by Williams et al.33 show the circular expansion of the mentioned dark area confirming the occurrence of the highest dissolution at the interface to unaffected magnesium and they clearly show cathodic current contribution over the entire dark film slightly decreasing with distance to the interface. This can be partially related to the ongoing densification of precipitated hydroxide on top of the dark zone. Brady et al.34 and their results measured by neutron scattering strongly support this aspect since they show a densification down to 40% nanoporosity. This value fits to modelling assumptions of Höche35 and to studies of Deslouis et al.36 Hydroxide formation itself is also related to pH conditions. Rossrucker et al.37 studied the pH dependence and showed increased HER with decreasing pH which is also related to the mentioned precipitation kinetics and densification.
Despite all differences the impurity effect remains. The occurrence of transition metal elements like iron on the corroded surface was shown by Cain et al.38 Also the latest results of Yang et al.39 clearly support the iron impact on HER. A possible morphology of the active zone is shown by Lynse et al.40 Interesting contribution was given by Salleh et al.41 Their results state that iron particles entrapped under corrosion products still can sustain HER at high rates which fits to the previously mentioned results and is in line with those of Brady et al.34 More important is the following statement of Salleh et al.:41 “Enhanced HER on Mg is an effect of the enhanced water self-dissociation upon magnesium, attributed to the presence/growth of the Mg(OH)2 layer on its surface”, considering the findings of Williams et al.42 by density functional theory – DFT calculations. These results suggest that water reduction can occur on Mg(OH)2, but do not explain where the required electrons come from. Deslouis et al.36 and especially Nelson et al.25 clearly showed the dielectric nature of Mg hydroxide which contradicts to this approach. Nevertheless, the question remains open: can contribution of this effect be stronger than the electrochemical driven water reduction especially on 3d transition metals at strongly negative potentials?
Discrepancies in dark area contribution strength can be explained by the different electrolyte used by the authors affecting the potential (electric) field distribution. This effect is clearly validated by Williams et al.33
Based on the results achieved by experiments with ferrofluids, by TEM studies and by grazing incidence X-ray diffraction (GIXRD), we suggest a mechanism of extensive hydrogen evolution related to re-deposited metallic Fe film at the corrosion forefront and subsequent cathodic Heyrovsky reaction of water reduction. Such reactions can occur at much higher rates than the one going via the conventional Volmer–Tafel route for hydrogen evolution. As a result, the extensive HER is a superposition of interacting effects whereas the impact of cathodic reactions on dark areas changes with corrosion duration depending on the local conductivity and the pH value. The cathodic performance of the dark film will be explained by the deposition of metallic films on the very surface as a result of reduction of iron oxides to Fe as shown by Le et al.,43 respectively. Armstrong et al.44 demonstrated an ongoing hydroxide precipitation on top as previously introduced by other authors.
The obstacles of experimental access to the in situ process occurring during dark film formation are well known.38,45 Thus, the validation of the surface morphology requires an unusual and innovative use of methods. Two promising candidates are magnetic domain visualisation according to the Bitter decoration method46 and in situ diffraction experiments47 allowing in situ phase formation studies during potentiodynamic polarisation. Apart from that, very interesting information can be received using Mössbauer spectroscopy.48
2. Experimental
2.1 Electrochemical tests
For complementary experiments different species of magnesium 99.98% (ppm: Fe-47, Ni-2, Cu-5), 99.95% (ppm: Fe-179, Ni-2, Cu-2), 99.95% (ppm: Fe-220, Ni-2, Cu-5) and 99.94% (ppm: Fe-65, Ni-2, Cu-3) were used. Bare materials were cut into pieces, ground, polished and rinsed with ethanol. Polishing was performed on a slow rotating disc via diamond paste (1 μm particles). For subsequent cleaning and rinsing a soft alkaline cleaning solution was used. Potentiodynamic cathodic polarization (sweep rate of 30 mV min−1) was performed in stirred 0.5 wt% NaCl solution at a pH value of 10.5 for the impurity elements to match typical conditions at the very surface during magnesium corrosion.The corrosion cell (333 ml) with a three electrode set-up consisted of an Ag/AgCl reference electrode, a Pt counter electrode and the specimen itself (1.54 cm2) as a working electrode. The electrolyte temperature was 21.5 ± 0.5 °C and the electrolyte being in equilibrium with the atmosphere was stirred during the potentiodynamic polarization measurements.
2.2 Hydrogen evolution
Additionally, for HER investigations on Mg-x ppm Fe (x = 47, 65, 179) an initial pH of 7 was used in a very slow stirred aqueous 5 wt% NaCl solution to accelerate H2 generation. HER was measured for different anodic potentiostatic polarization conditions without IR compensation by a modified test cell (WE – 1.54 cm2) connected to a hydrogen collecting burette. To minimize the loss of H2 due to adsorption at the cell wall the setup was initially flushed with hydrogen gas in order to minimize adhesion effects (secondary bubbles).2.3 Iron deposition simulation
In order to validate and to simulate the Fe impact on the enhanced catalytic activity magnesium 99.98% (47-ppm Fe) has been treated in iron containing salt solutions. The treatment was performed at open circuit potential in 0.5 M FeCl2 for 5 min and 30 min at pH 7 or 10.5. Immediately after the process the samples have been cathodically polarized as previously explained in Section 2.1, in order to study the corresponding hydrogen evolution rate.2.4 X-ray photoelectron spectroscopy (XPS)
X-ray photoelectron spectroscopy (XPS) experiments were carried out using a Kratos Axis Ultra DLD system working with a 15 kV X-ray gun using monochromatic Al-Kα radiation. The analyzed area size was set to 700 μm × 300 μm and the pass energy was set to 40 eV for the region scan.2.5 Transmission electron microscopy (TEM)
The specimen (corroded Mg-65 ppm Fe) for TEM investigation was prepared from regions adjacent to the dark zone area using focused ion beam (FIB) milling using a FEI Helios NanoLab 600 DualBeam FIB/SEM. The specimen surface was protected with a 300 nm Pt layer prior to FIB processing with Ga+ ions to protect the specimen from damage. The TEM samples were mounted on Cu grids for TEM investigations. Transmission electron microscopy was performed on a FEI CM200 Transmission electron microscope equipped with an EDAX energy dispersive X-ray (EDS) spectrometer operating at 200 kV.2.6 Magnetic field influenced corrosion and imaging by the Bitter decoration method
A water based ferrofluid containing up to 4% (Vol.) magnetite nanoparticles (av. d = 30 nm (EMG 705, FerroTec)) was used to visualize the arising magnetic structure using the Bitter decoration method.46 Dark area formation was studied on the Mg-220 ppm Fe sample sheet positioned on a strong neodymium–iron–boron magnet. The magnetic field interacts with the re-deposited iron by inducing a dipole moment, respectively, and magnetization which subsequently interacts with the ferrofluid. A drop of 0.5 wt% NaCl solution was placed on top of the Mg sample and the setup was placed under an optical microscope to observe the progress of dark area formation. After the corrosion front (dark area) was formed the ferrofluid was added to the drop of NaCl solution using a pipette (2 μl). During the procedure the Mg sample remained on the magnet. Since micro-convection within the droplet can influence the results, the whole setup was not moved afterwards. After the ferrofluid particles positioned themselves due to magnetic forces on the corroded Mg surface, the samples were rinsed with ethanol, dried at 60 °C and studied using SEM and EDX mapping and compared with a reference sample that is not positioned on the magnet. 22 corrosively active dark area spots were analysed: 15 tests on the neodymium–iron–boron magnet and 7 tests without placing the Mg sample in the strong magnetic field.In order to validate iron deposition, the corrosion conditions were simulated by the immersion of Mg test samples into iron salt containing solutions (as described in Section 2.3). The Bitter test has been repeated on a sample (Mg-47 ppm Fe) after 1 min in 0.5 wt% NaCl + 0.05 M FeCl2 solution at −1.2 V vs. OCP potentiostatic conditions. This cathodic potentiostatic treatment and the related hydrogen evolution are required to induce reduction reaction of iron-rich phases to metallic Fe. The used parameters were derived from the studies of Flis-Kabulska et al.,43,44,49 Zou et al.,50 Allenore et al.51,52 and Beverskog et al.53 For example Flis-Kabulska et al.49 showed reduction reactions and electrode activation due to Fe deposition at a cathodic overpotential of about −1.09 V vs. Hg/HgO in an alkaline electrolyte. A more detailed description will follow in Section 4.2.
2.7 Grazing incidence X-ray diffraction (GIXRD)
Samples for GIXRD analysis were prepared as previously described in Section 2.1 by anodic polarization for 30 minutes. During the formation of the dark zone the samples were positioned (focus alignment) and analysed under exposed conditions. Measurements were performed on a Bruker D8 Advance (DaVinci design) equipped with a eulerian cradle and in grazing incidence geometry (Göbel mirror, 2° sollers, LynxEye detector-0D) at different incident angles to record depth information (4s counting time per step). Angles have been varied stepwise between 0.15° and 0.9°. Data analysis was carried out using common analysis software (BrukerEVA and PDF + 4 2014).3. Results
3.1 Potentiodynamic polarization and hydrogen evolution
Fig. 1 presents the cathodic polarization curves obtained on Mg samples with different amounts of Fe impurities. The measurements were performed after different times of 100 mV potentiostatic anodic polarization. It can be clearly seen that the increased level of Fe leads to higher cathodic currents because of more intensive hydrogen evolution on inclusions. However another important effect is also observed. The cathodic activity for the same type of Mg is increasing with time of anodic polarization. This effect can be related to the increase of effective cathode area during anodic polarization. The increase of cathodic active area during corrosion due to the enrichment of impurities was already shown in the literature.16,22,26 The measured potential is shifted towards the noble side due to increasing Fe containing areas related to the uncovered impurities and the proposed film deposition effect (any other noble compounds can interact as well). Continuously, the present surface conditions (surface fraction ratio of cathode (Fe) vs. anode (Mg)) change and a mixed potential occurs which is still cathodic to the impurity elements and close to the magnesium OCP. The achieved corrosion kinetics is determined by the impurity content and by the number of iron-rich particles getting detached from the substrate due to anodic polarization, as it will be described in Section 4. Furthermore, the maximum cathodic current is also dependent on the iron content. Fig. 1 clearly validates the direct correlation between the dark film area and the Fe content.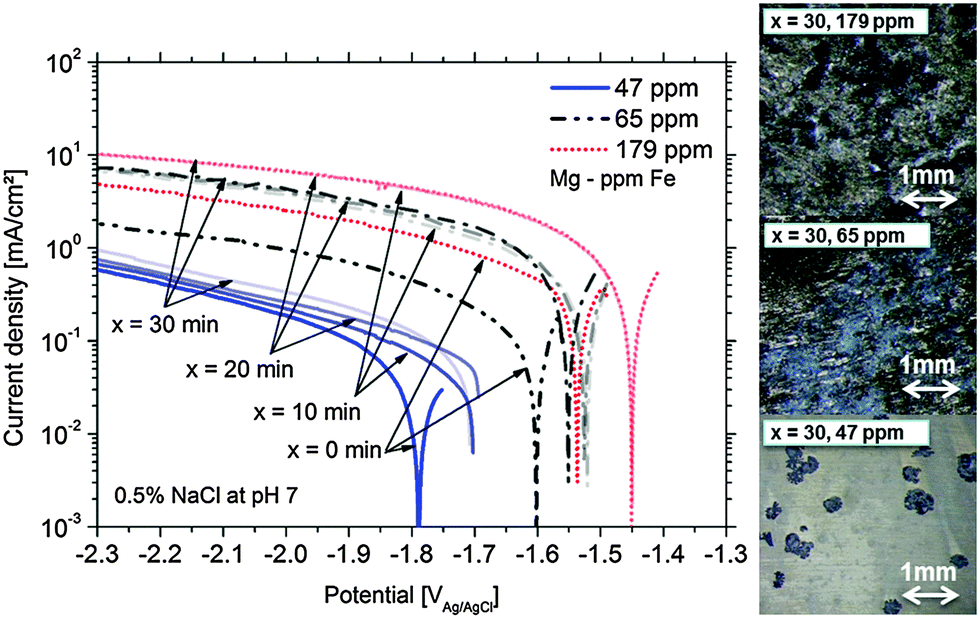 | ||
| Fig. 1 Enhanced cathodic activity after anodic potentiostatic polarization for x = 0, 10, 20, 30 minutes at +100 mV vs. OCP for pure Mg containing different Fe levels and their surface appearance after x = 30 min (initially all had a shiny surface). | ||
The maximum of cathodic current was measured even after 10 min at +100 mV vs. OCP of the 179 ppm Fe sample (sample surface was fully dark). According to the observations the maximum is determined by the rough “dark” area and the IR (ohmic) properties of the electrolyte.
For further verification, some hydrogen evolution rate tests were performed for magnesium samples with different impurity levels (Fig. 2). At low (uncorrected) anodic polarization the HER increases with the amount of impurities. A further increase to a certain amount of anodic polarization leads to a smaller increase of HER since IR-effects become relevant.
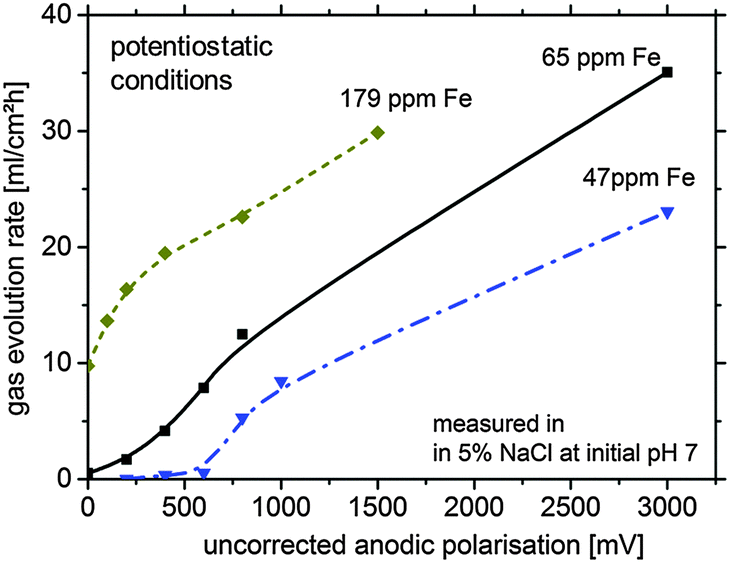 | ||
| Fig. 2 Typical hydrogen gas evolution rate under various potentiostatic conditions (not IR-corrected) for different magnesium systems in 5 wt% sodium chloride solution (lines are just for better visualisation; error >10%). The duration of hydrogen collection per point was 12 min for 179 ppm Fe, 30 min for 65 ppm Fe and 60 min for 47 ppm Fe. | ||
As shown in Fig. 2, especially for the 179 ppm Fe sample, the HER without external polarization is at a high level. This is related to the self-accelerating corrosion process due to the interaction of impurities, HER and the proposed re-deposition. Dissolution of Mg leads to uncovering of impurities at the surface making them more accessible for contact with the electrolyte. This mechanism contributes to the increased effective surface area of the cathodic sites leading to the increase in the HER rate and was recently suggested by Taheri et al.16
However, the large amount of hydrogen cannot be explained by impurities alone. One of the possible explanations is re-deposition of iron from dissolved iron rich impurities detached from the corroding matrix by a mechanism, which has been validated for aluminum alloy 2024 when copper is re-deposited on the aluminum surface around S-phase intermetallics (73). In the case of Fe re-deposition as a thin film, a large cathode can be formed from relatively small dissolved iron precipitates drastically increasing the surface area available for HER. This would also explain the unproportional difference between the measured HER of Fig. 2 (factor of approx. 10–100) and the impurity content (factor of approx. four). The sources of Fe ions in the solution are the small iron containing impurity particles freed from the Mg matrix in the course of dissolution. The free sub-micron scale iron rich particles, losing galvanic protection after the electrical contact with the matrix is lost, can be dissolved because of free corrosion in the aggressive electrolyte. They generate iron cations, and due to local conditions at the corrosion front, subsequent re-deposition takes place. This effect will be described in more detail in Section 4.
If this knowledge is taken into consideration, the whole hydrogen generation process has two major sources:
(1) Continuing water reduction at the existing particles of impurities partially embedded in the Mg matrix
(2) HER at growing areas of metallic re-deposited iron at the corrosion forefront (dark area growth).
The following part of this manuscript is focused on providing experimental evidence for the iron re-deposition process during Mg corrosion.
3.2 Magnesium – dark film simulation
At first, the experiments were carried out in order to show that the addition of Fe(II) to the electrolyte can increase the cathodic activity. The suggestion is that Fe2+ from solution can be reduced on the Mg surface to form metallic Fe film which will act as a large cathode. As shown in Fig. 3 for Mg immersed in different iron salt containing solutions (0.5 M FeCl2 at pHs 7 and 10.5 for 5 min and 30 min, respectively), an enhanced cathodic activity is observed when Fe(II) is present in the solution. The observed cathodic activity is similar to the measured activity of magnesium with 65 ppm Fe after dark area formation as shown in Fig. 3 as well. This increase of activity provides an additional contribution to the idea of formation of metallic Fe deposits on the magnesium surface creating additional sides for enhanced cathodic activity.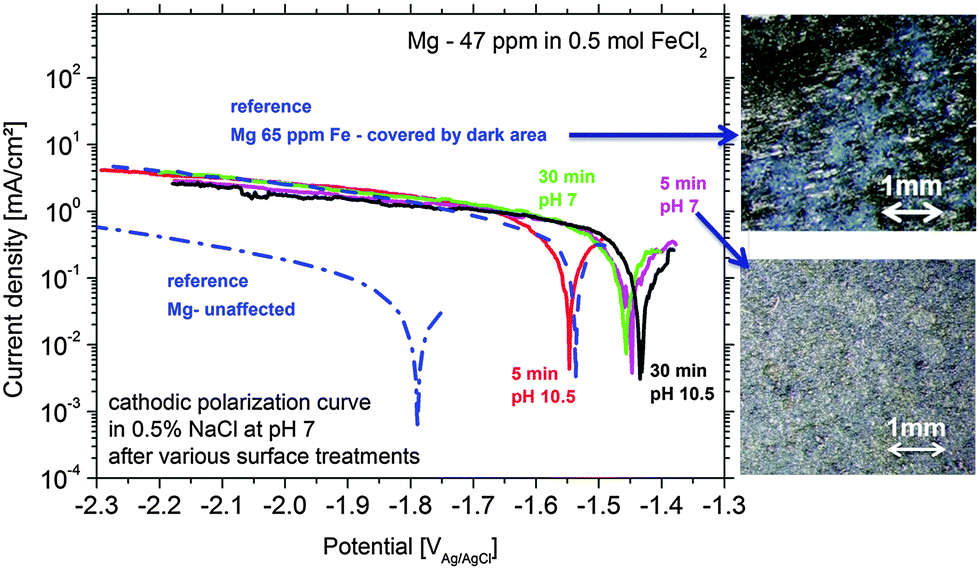 | ||
| Fig. 3 Increased cathodic activity of Mg-47 ppm Fe samples after immersion treatments in 0.5 M FeCl2 solution for 5 and 30 min at pH 7 and 10.5. For comparison the initial bare- and corroded Mg-65 ppm Fe with the fully corroded surface are shown. The dark surface appearance of two test samples is shown by optical micrographs (initially all had a shiny surface). | ||
The validation of metallic film formation during Mg corrosion in an aqueous environment is not a trivial task, especially from the point of view of experimental access to in situ conditions. X-ray photoelectron spectroscopy (XPS) was used here to obtain information about the chemical state at the very surface (first 5–10 nm) of corroded Mg samples. However the samples have to be dried before the measurement since the method requires high vacuum. As a consequence the samples were immediately dried after the tests in order to stop all electrochemical processes as fast as possible. Measurements on the Mg-179 ppm Fe sample do not reveal any iron peak due to the detection limit (∼10−3 to 10−2 at%54) and even more important, the continuing Mg(OH)2 formation after removing the electrolyte. Thus, the XPS study was also performed on a specimen after being immersed in the iron chloride solution and after cathodic polarization according to Fig. 3. As illustrated in Fig. 4, the signal of iron 2p3/2 of this test sample contains multiple excitations.
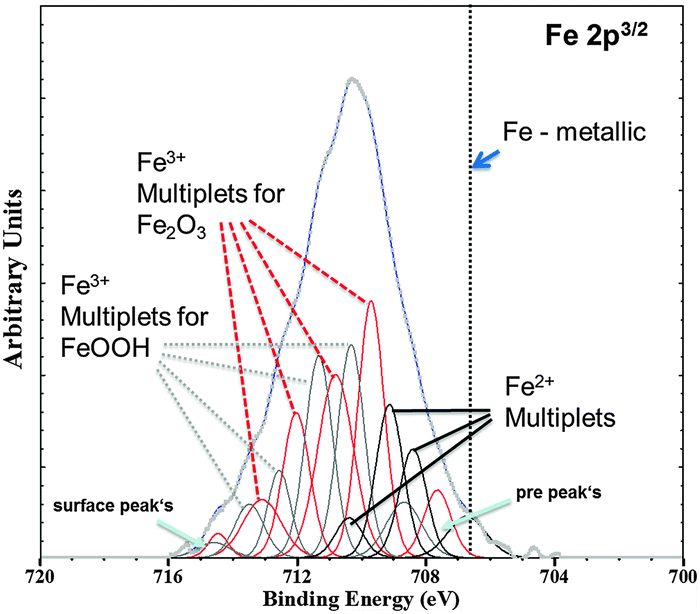 | ||
| Fig. 4 Fe 2p3/2 XPS spectra (Shirley background corrected) of the Mg-47 ppm Fe sample after immersion in 0.5 molar FeCl2 solution for 5 min at initial pH 7 validated deposition of the Fe rich phase. Multiplet fitting according to the study of Grosvenor et al.57 is implemented as well. Differentiation between α and γ modifications of Fe2O3 and FeOOH, respectively, is not indicated. | ||
According to the literature55–57 multiple peak-fitting of Fe compounds is critical due to convoluted signals related to spin–orbit coupling with unpaired 3d electrons and electrostatic interaction. The evaluation, as shown by Grosvenor et al.57 applying the Shirley background, matches very well and therefore was used in first approximation. The presence of Fe2O3 and FeOOH was detected, however without distinguishing between α and γ modification. The physical meaning of the pre- and surface peak in Fig. 4 is explained in Grosvenor's work as well. Fe2+ signals are contributed to remaining iron chloride and other oxides. Metallic Fe (expected at 706.6 eV, ref. 56) could not be detected in this case because of the very fast oxidation of the thin Fe film after terminating immersion. Thus, much experimental effort (especially in situ) is required to prove that metallic Fe is formed at the corrosion front.
3.3 TEM studies
The observed effects are related to phenomena occurring at a nanometer scale. Via TEM studies shown in Fig. 5 it was possible to take a cross-sectional micrograph (screenshot) of the re-deposition situation at the corrosion front. Since measurements were performed under dry (vacuum) conditions MgO was observed on top instead of Mg(OH)2. This is related to the dehydration/decomposition of the nanocrystalline Mg(OH)2 forming MgO58–60 on the corroded sample under dry conditions (like in a vacuum).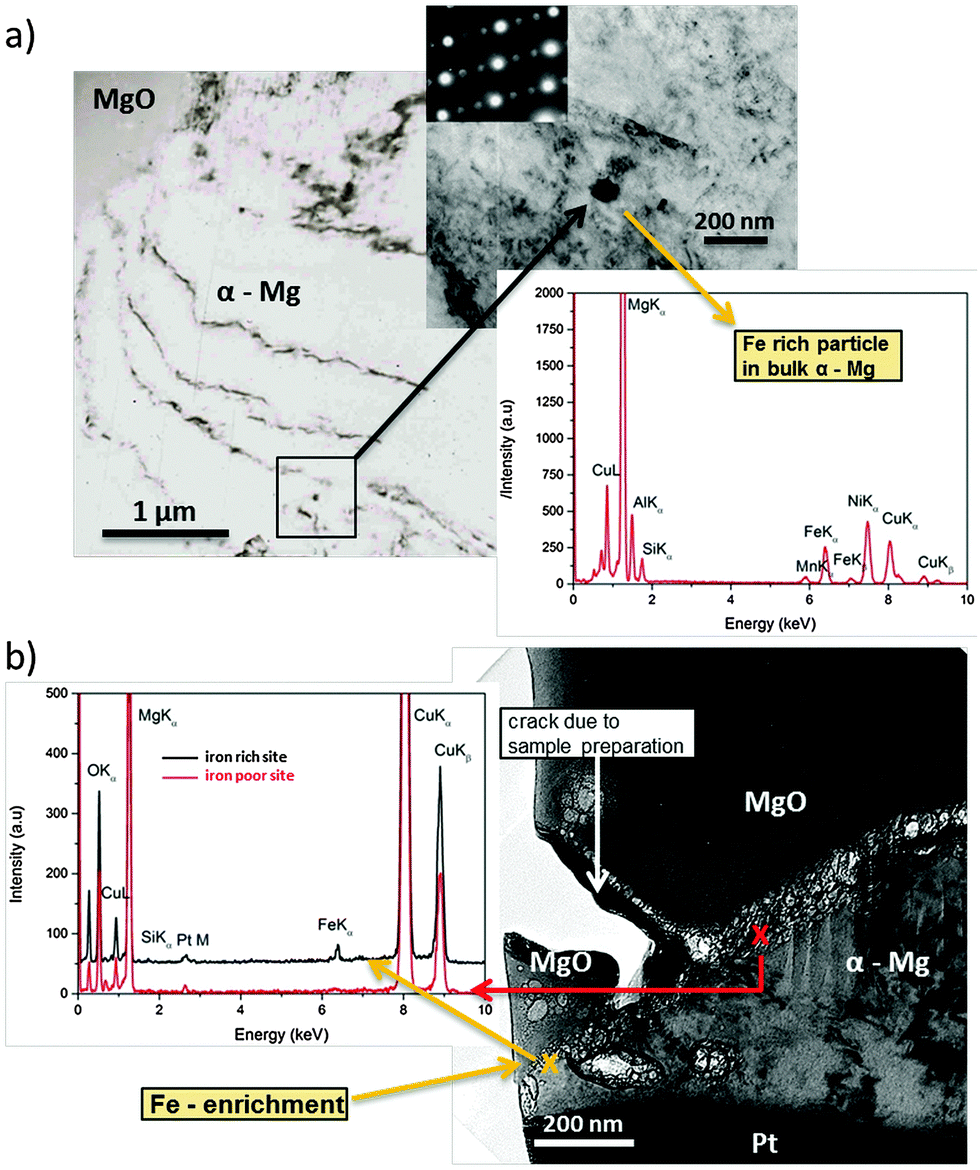 | ||
| Fig. 5 TEM micrographs on a cross-section of corroded (dark area zone) Mg-65 ppm Fe. (a) Fe-rich particle containing Si and Mn embedded into magnesium. Iron diffraction pattern indicates a ferrite structure. (b) Iron enrichment allocated at the corrosion front. Si and Mn were not observed there. (Cu, Ni and Pt are related to experimental artefacts.) | ||
Fig. 5a shows a typical Fe rich particle (∼80 nm diameter) containing Si and Mn related to the metallurgical history of the sample. It has a ferrite crystalline structure and is fully embedded. Fig. 5b delivers clear indication for the re-deposition mechanism. At the foremost position of the corrosion front the EDS analysis showed a significant iron signal whereas Si and Mn (as it should be for particles) did not occur (Si very weak). Thus, the detected iron originates from re-deposition rather than iron-rich particles. Additionally, the position is close to voids in an area with previously removed particles due to corrosion related detachment from the matrix. It cannot be directly proven that one of the particles was detached but, since the EDS shows iron as introduced by the re-reposition mechanism, it is most probable. The fact that, at a similar interface position between MgO and α-Mg without involved iron-rich particles, no EDS, Fe signal occurs, once again affirming the proposed mechanism.
3.4 Magnetic domains
Since ferromagnetic iron is involved, there should be an interaction of an external magnetic field (neodymium–iron–boron magnet) with the dark area. Consequently, the induced magnetization alters the magnetic properties. It is also known that iron oxides (excluding FeO),61 oxy-hydrides and hydroxides62–64 behave as ferromagnetic materials. Due to the interaction of the dipole moments of iron and iron-rich phases with the external magnetic field, the coverage by nano-magnets of water-based ferrofluid should be influenced. The alignment according to the magnet field vector![[B with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/b_char_0042_20d1.gif) allows correlation with iron distribution. After optimization of solution compositions, droplet sizes, magnet positions, and minimization of ferrofluid agglomeration, several tests were performed and compared.
allows correlation with iron distribution. After optimization of solution compositions, droplet sizes, magnet positions, and minimization of ferrofluid agglomeration, several tests were performed and compared.
Fig. 6 shows the SEM images and corresponding Mg, O and Fe – EDS elemental mapping obtained after short exposure of Mg (220 ppm Fe) to a drop of 0.5 wt% NaCl solution. The shape of the dark area can barely be recognized in Fe mapping in Fig. 6a (partially indicated by the white line) when the immersion and addition of the ferrofluid were performed in the absence of the external magnet since the magnetization of dark zones is very low. In contrast, in Fig. 6b when electrolyte exposure and addition of the ferrofluid to the droplet were performed on a sample positioned on the neodymium–iron–boron magnet the Bitter decoration method was more efficient. The arising dark areas show magnetic-force-related interaction because the magnetite nanoparticles of the ferrofluid clearly reproduce the shape of the dark area and align themselves around it (while the external magnet serves as a trigger for the weak magnetic field of the dark areas due to induced magnetization). Note that in Fig. 6b the ferrofluid is mostly assembled around the dark zones and weaker in the middle, suggesting that there is a higher content of Fe-rich phases concentrated at the periphery of the corrosion front of the dark area.
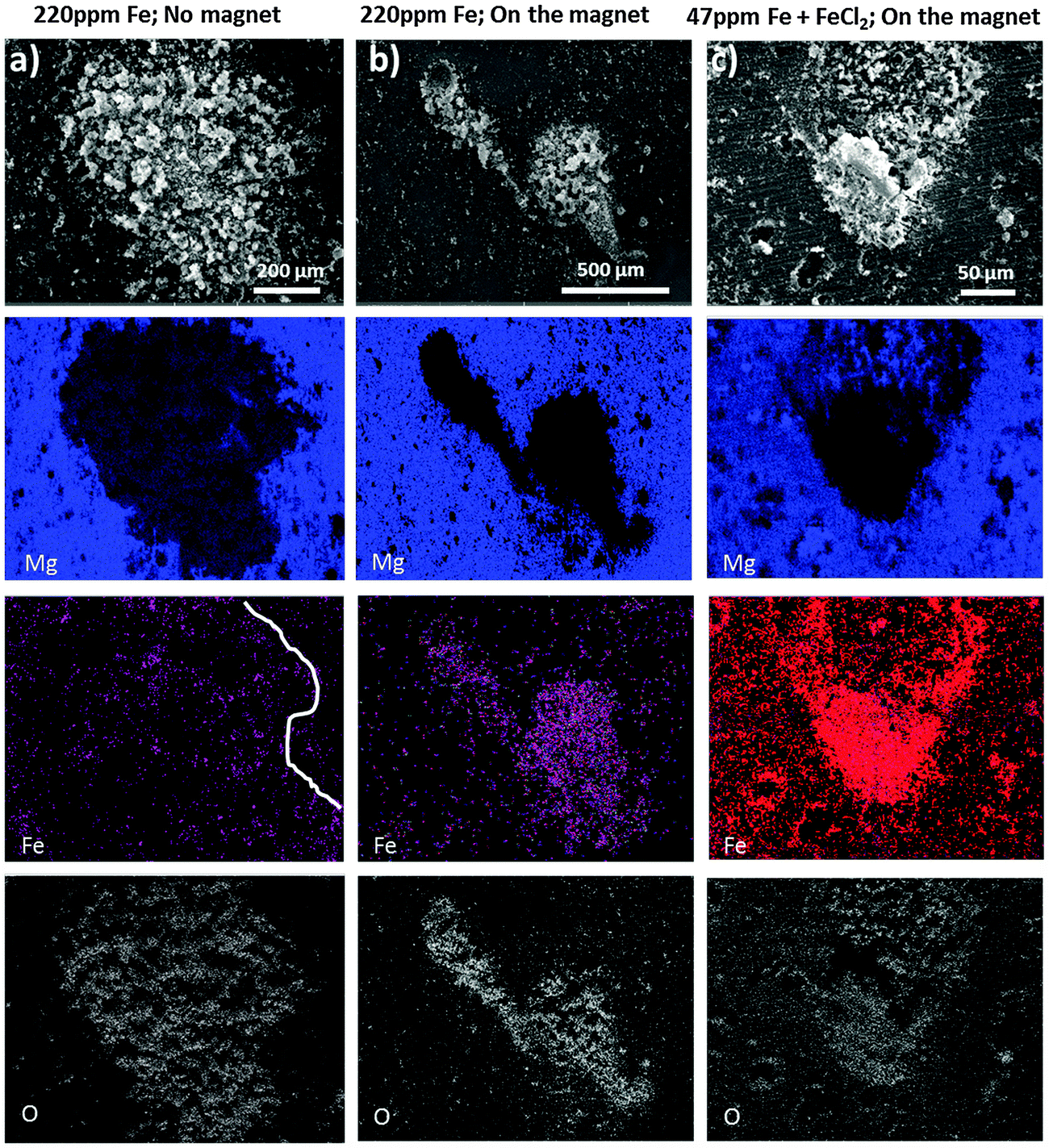 | ||
| Fig. 6 SEM-EDS elemental mapping of the Mg surface (containing either 220 ppm Fe (a) and (b) or 47 ppm Fe (c) after short exposure to 0.5% NaCl and subsequently added ferrofluid). Additionally, the sample in (c) was polarized in 0.5% NaCl containing 0.05 M FeCl2 before positioning on the magnet and adding the ferrofluid. | ||
Certain morphological features, like the geometrical shape of the dark area in Fig. 6a, can be recognized at the top right corner, which indicates that both magnetic and capillary effects could attract magnetic nanoparticles to the black area of the sample that was not in contact with the magnet. A comparison with Fig. 6b suggests that with the magnet the pattern formation is dominated by the magnetic-field-induced interaction.
The observed magnetic field related effect can also be validated on samples used for the dark area simulation via iron salt treatments. Even though there is a higher amount of iron deposited on the entire surface, there is a much stronger magnetic field at the dark area revealed by the enrichment and alignment of ferrofluid as shown in Fig. 6c. Note that in these experiments, the external magnet only served as an amplifier/trigger, increasing the weak magnetic field of the black areas. If there does not present ferromagnetic material at the dark areas, the external magnet would not have anything to magnetize and no specific interaction of the dark area with the ferrofluid would occur. Moreover, no alignment of the ferrofluid was observed at the non-corroded surface away from the corroded areas.
The specimen which was corroded without the magnet (Fig. 7a) does not show the pattern formation due to low magnetization. In contrast the specimen, which was corroded on the magnet in (Fig. 7b), clearly shows that the formed black area interacts by magnetic forces because the magnetite nanoparticles of the ferrofluid are aligned in recognizable branches/pattern according to Bitter around the dark area. Note that this pattern could not have been formed exclusively by the magnetic field of the underlying magnet. If this would be the case, the ferrofluid enriched and aligned in the bottom right corner of Fig. 7b should not remain on its position, but would move to the strongest magnetic field located lower on the sample and indicated by the vertical line in Fig. 7d. It is assumed that the underlying magnet acts as a trigger for the existing weak magnetic field. Due to its magnetization effect the interaction of Fe-rich dark areas with the ferrofluid was amplified. Also note that the Bitter-like patterns of the ferrofluid are not observed at long distances to the black area. Their aligned agglomerations are selectively found closely around the iron-rich dark areas.
 | ||
| Fig. 7 Corrosion forefront (a) without magnetic structures, (b) and (c) with pattern formation according to Bitter decoration revealing the selective alignment of ferrofluid (marked by lines) around the black area formed under different conditions, and (d) magnetic field structure. | ||
The validation of this behaviour is shown in Fig. 7c. It was observed that similar pattern formation occurs in the same way for the iron deposition simulation case applying FeCl2 treatment with subsequent sufficient cathodic polarization here (−1.2 V vs. OCP) in order to enable Fe re-deposition. All results clearly indicate the magnetic effect of dark areas and the existence of ferromagnetic material in dark areas, formed on corroding bare magnesium.
3.5 In situ phase formation aspects
GIXRD diffraction patterns are measured in situ, directly during the growth of the dark areas. The results in Fig. 8a show the expected strong Mg(OH)2 signal. The shape of the hydroxide peaks is broadened due to crystal size effects (nanocrystallinity). In order to get the information of Fe rich phases the pattern has to be analyzed carefully.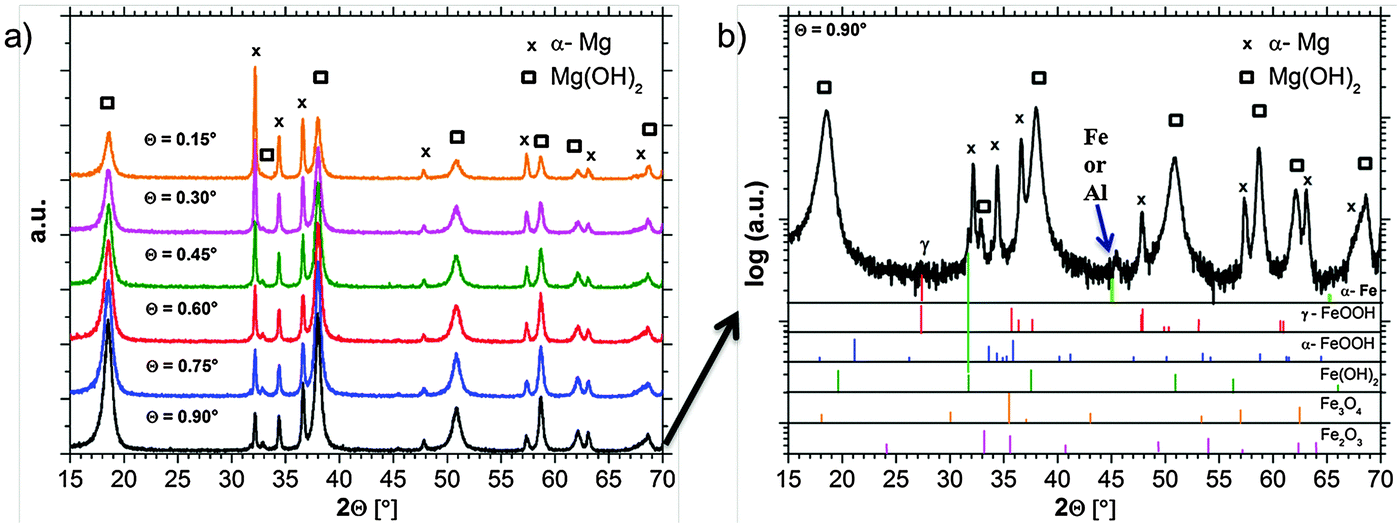 | ||
| Fig. 8 (a) GIXRD pattern (measured in situ) after the formation of the dark area on Mg-65 ppm Fe in 0.5% NaCl solution showing the growth of Mg(OH)2. (b) GIXRD diffraction patterns measured at an incident angle of 0.90° (logarithmic scale) including the indication of probable phases. | ||
Besides the expected α-Mg and hydroxide pattern, weak signals in the background occur, which can be related to iron-rich phases. Due to the phase formation conditions which lead to a very thin (nanocrystalline) film and due to the low concentration of Fe, only weak and broadened signals occur. Fig. 8b explicitly shows a signal which is related to Fe(OH)2 at 2θ = 31.8°. Furthermore, there are indications for lepidocrocite (γ-FeOOH) formation at 2θ = 27.5°. Other phases like magnetite (Fe3O4), hematite (Fe2O3), or goethite (α-FeOOH) cannot be assigned without doubts due to the peak overlap and the detection limit. The peak at 2θ = 45° raises questions since it fits to ferrite (α-Fe) [(011) = 44.68°; (002) = 65.03°] and aluminum (sample holder) [(111) = 38.47°; (200) = 44.74°; (220) = 65.13°]. Aluminum cannot be excluded since the strongest reference peak at 2θ = 38.47° is overlapped by the Mg(OH)2 signal.
The observation that the strongest iron-related signal occurs at 0.9° incident beam angle points out that iron-rich phases are located below magnesium hydroxide. This fits very well not only to recent research results in Section 3.4 based on the ferrofluid tests, but also to the studies of Curioni et al.32 and Yang et al.39
4. Discussion – mechanistic understanding of hydrogen generation
4.1 Implementation of the re-deposition mechanism
During the corrosion of bare Mg, containing a sufficient content of impurities like iron-rich particles, the introduced phenomena occur. Immersion into an aggressive/corrosive environment for a critical period of time changes the surface morphology due to anodic dissolution, release of impurities and the increase of the dark area fraction which is cathodically active (as shown in previously published studies17,23,24 and indicated in Fig. 9). The contribution of reactions at dark areas to the global hydrogen evolution rate becomes stronger with progressing corrosion. This is related to uncovered impurity surfaces and the increase of active film area as shown by other authors16 and stated by Yang et al.39 The evolving dark surface area acts as the cathode.16,17,24,33 After coverage of the whole surface by the dark regions, the impact of the cathodically active dark film becomes dominant and the relative cathodic current contribution of the single impurities decreases. The results presented here explain the formation and the growth mechanism of dark areas. There is experimental evidence that the increase of the cathodic area is related to the re-deposition of dissolved iron species at the Mg surface.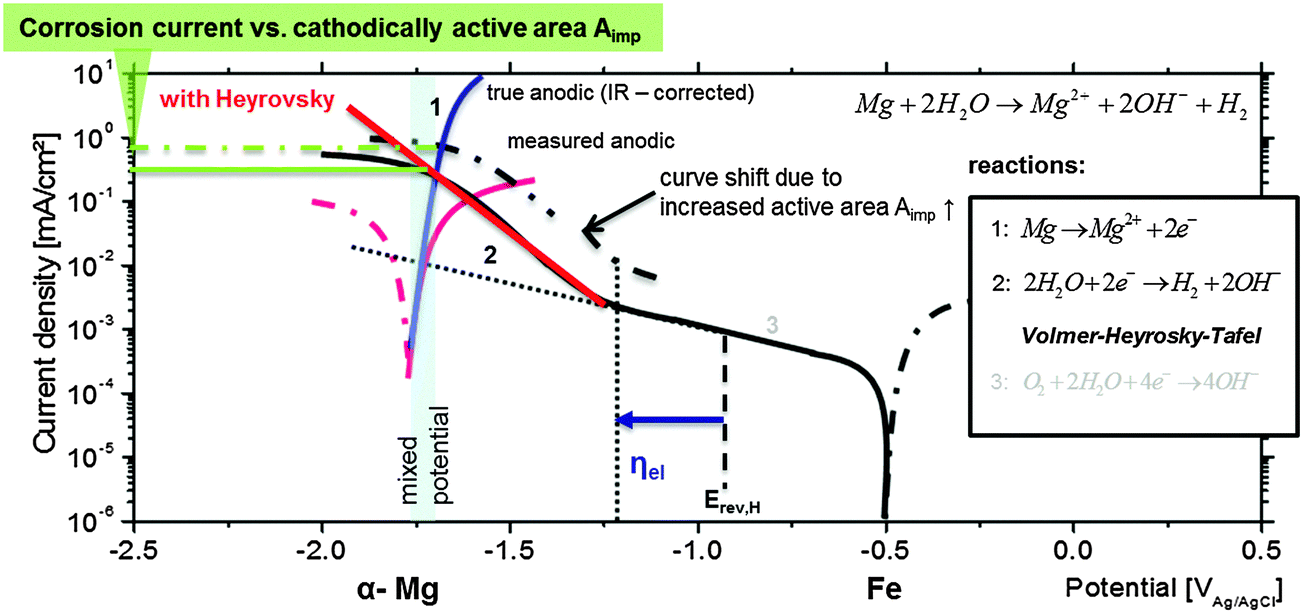 | ||
| Fig. 9 Applied mixed potential theory, corrosion current density as a result of occurring reactions, the overpotential ηelvs. Erev,H, the amount of cathodically active area Aimp, and its relation to single polarization curves. The RHE potential is indicated as well. | ||
The potential electrodeposition of Fe from alkaline electrolytes on different metallic substrates (Au, Ni) was already demonstrated by Yuan et al.,65,66 Allanore et al.,51,52,67 Flis-Kabulska et al.,49,68 and Fray.69 According to Allanore et al.52 the re-deposition can also occur from solutions containing hematite particles.
A similar process is suggested by Flis-Kabulska et al.49,68 for enhanced electrocatalytic activity and reactivation of electrodes during electrolysis (with iron cathodes). They state that metallic reactive pure iron (“in statu nascendi”) is deposited after the reduction of Fe(OH)42− at the site where hydrogen gas bubbles are formed, which fits to reactions shown by Le et al. and Armstrong et al.43,44 In another work70 they also show exactly the same slope for the pseudo-Tafel line of the hydrogen formation of 170 mV dec−1 on active iron films as measured in Fig. 10 in this paper. Possible reduction reactions can be found in the study of Misawa et al.71 or Stratmann et al.72 Thus, one can also expect that similar iron deposition processes can occur at the Mg surface during corrosion. In the case of magnesium corrosion, the iron source is the impurities, iron particles or iron rich intermetallics which are normally cathodically protected. However, during the corrosion process they lose electrical contact with Mg, as shown by Taheri et al.,16 and thus, start to dissolve via a self-corrosion mechanism rather rapidly. Dissolved iron can be reduced, and as a result very thin metallic iron layers or patches are re-deposited close to the hydroxide layer. This film is sufficient to force the cathodic behavior as proposed and indicated previously in Fig. 1.
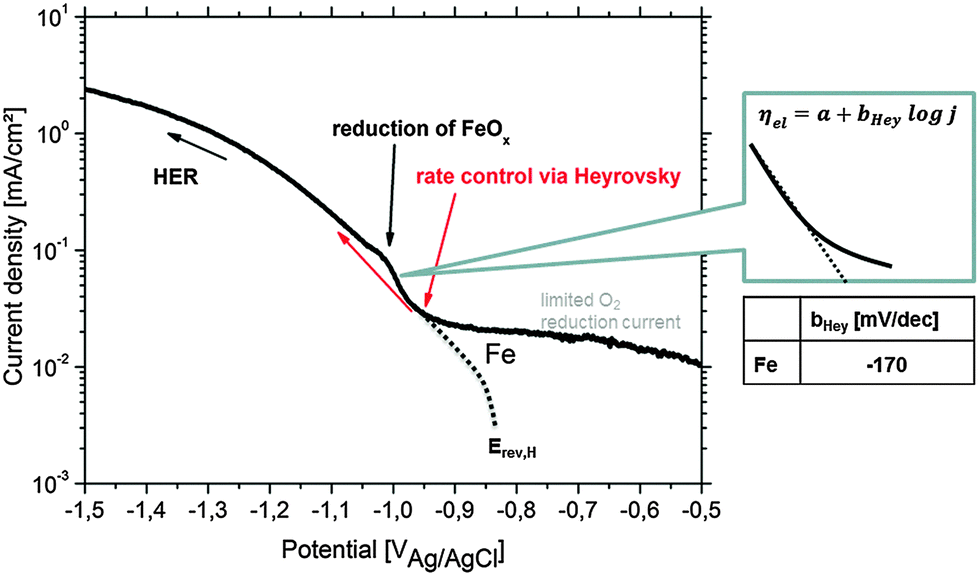 | ||
| Fig. 10 Cathodic polarization curve on Fe in 90% deaerated alkaline 0.5 wt% NaCl solution (pH = 10.5) simulating the conditions during Mg corrosion. The pseudo-Tafel slope bHey in the Heyrovsky range follows the relation 2 × 2.303RT/F ≤ bHey ≤ 3 × 2.303RT/F.83Erev,H and the (virtual) hydrogen reduction related curve are adapted from the study of Chialvo et al.78 Contribution to the current due to iron oxide reduction is labeled. | ||
It has to be kept in mind that the dark appearance is also partially caused by the ongoing roughening effect which has to be considered as well. Based on the literature and experimental results presented in this paper, the process for dark area formation and at least the reason for the negative difference effect are proposed as follows:
(I) Initial cathodic process on Fe rich particles and anodic dissolution of Mg.
(II) Detachment of iron particles from the metallic substrate by undermining or hydrogen bubbles as proven in ref. 16, 39 and 40.
(III) Chemical dissolution of detached iron-rich particles with the formation of Fe(II), Fe(III) or α-Fe2O3 compounds (similar to ref. 39)
(IV) Chemical reduction (e.g. by adsorbed atomic hydrogen) of dissolved iron species to metallic Fe according to ref. 43, 44, and 49 (probably close to bubbles in combination with localized pH increase)
(V) Electrochemical reduction of Fe species (similar to processes on other substrates51,52,67)
(VI) Deposition of thin metallic Fe film or patches mainly at the moving corrosion forefront due to the most suitable conditions combined with H2 generation
(VII) Accelerated HER on the freshly deposited iron due to the increased reactivity of pure metal (“in statu nascendi”) according to the Heyrovsky pathway as explained in Section 4.2
A similar re-deposition mechanism is also proven for S-phase (Al2MgCu) containing aluminum alloys by Buchheit et al.73 and later by Yasakau et al.74 and El Warraky et al.75 Due to the fast dissolution of Mg and then Al, the S-phase becomes porous and enriched with copper. The detachment of copper nanoparticles and their dissolution followed by re-deposition on the aluminium matrix covered with Al2O3 in the vicinity of the de-alloyed S-phase enlarge the cathode and propel further corrosion. In ref. 75 it was shown that a concentration of only 2 ppm CuCl2 diluted in 0.5 M NaCl is sufficient to cover the entire surface of Al of their test samples with Cu. This is a clear hint that this kind of dissolution and re-deposition mechanism demonstrated for Al–Cu might be also feasible for Mg–Fe. The related NDE on aluminum is weaker since local cathodic polarization on the active surface area is not as high as for magnesium. In the appendix an approximation of critical Fe content for the Mg–Fe system is given. Briefly, around 20 Fe intermetallic inclusions (fully dissolved) with an average diameter of 1 μm are theoretically enough to cover 1 cm2 of the Mg surface with a continuous Fe monolayer.
4.2 Heyrovsky pathway
The achieved results evidence that Fe impurities dissolved in the Mg matrix can be re-deposited on the surface, forming a thin iron-based film which acts as an effective cathode for the HER. However the observed high rate of HER, especially at slightly anodic polarizations, cannot be justified only by the increase of cathodic area if one assumes that hydrogen evolution occurs via the commonly accepted Volmer–Tafel mechanism.Generally, for high cathodic polarizations of pure samples of the metals found as noble impurities in Mg, which e.g. correspond to low anodic polarization of the Mg-matrix, the rate determining step for HER differs. The rate determining reaction at low cathodic polarization vs. OCP of e.g. pure Fe is typically diffusion limited O2 reduction. Cathodic polarization of e.g. Fe, Ni and Cu above the reverse hydrogen potential Erev,H induces the HER. The rate determining step for the HER is a Volmer-type reaction of electrochemical adsorption. The rate of this process increases with continuing polarization. Then the chemical recombination of the intermediate state adsorbed hydrogen atoms followed by the desorption of hydrogen molecules controls the kinetics according to the Tafel step. However, continuing to even higher cathodic polarization activates the electrochemical desorption of hydrogen according to the Heyrovsky mechanism,76 especially on 3d transition metals. Vilekar et al.77 performed a very fundamental study on the kinetics of hydrogen reactions on Pt under alkaline conditions, showing the Heyrovsky step to be rate-determining for cathodic polarization more than −0.2 V vs. Erev,H (RHE). Chialvo et al.78 studied the HER on iron in 2 M NaOH and explained the kinetics by a three step Volmer–Heyrovsky–Tafel mechanism. They showed that the apparent activation energy they measured corresponds to the Volmer or Heyrovsky step. Another investigation by Qian et al.79 on iron in 0.2 M NaOH showed the occurrence of control via the Heyrovsky pathway as well. Therefore it is reasonable to suggest that at high cathodic polarizations in alkaline electrolytes the HER on iron-containing zones on Mg occurs via the Heyrovsky mechanism. As a result a much higher HER rate can be observed than in the case of the Tafel step.
The potential (marked as ηel in Fig. 9) at which the Heyrovsky path takes over the rate (current) control is specific for each metal under certain conditions and depends on the metal–hydrogen (M–H) bond energy. The domination of the Volmer–Heyrovsky pathway was shown and clearly validated for iron even at moderate overpotentials.79 For nickel the rate control depends on the position of the cathodic branch. It was shown by Krstajić et al.80 that at certain cathodic overpotentials the Heyrovsky reaction is rate-determining as well. Further investigations on copper81 have also shown the same mechanism.
Thus, it is reasonable to assume that, at the corrosion (mixed) potential of Mg-based substrates, the noble (Fe, Cu, and Ni) inclusions can be highly cathodically polarized and might be exposed to conditions when the Heyrovsky step becomes rate-determining as shown in Fig. 9. Crossing the Heyrovsky branch of the impurity elements (here Fe) with the corrected anodic branch of α-Mg delivers the corrosion rate as marked in Fig. 9.
The upward shift of the cathodic branch is related to the amount of cathodically active surface area Aimp. In the case of dark area formation Aimp increases, and as a result the final corrosion current becomes higher. The explanation of Fig. 9 contributes to the interpretation of measured polarization curves of Mg with different amounts of impurities (here Fe). The enhanced cathodic activity (incl. HER and IR-drop), the shift of measured OCP and the corrosion rate are the physicochemical consequences of previously explained behaviour.
The HER kinetics itself strongly depends on the kind of impurity, alloying element and/or compound involved in the corrosion process. In order to validate the Heyrovsky contribution aspect, the cathodic polarization of bulk specimens of Fe as the most common impurity element was performed under corrosion conditions comparable to those occurring on Mg alloys (alkaline pH) as discussed in Section 3.1 and shown in Fig. 10. Very important information is received at approx. −1.0 VAg/AgCl during iron polarization. A peak occurs which is related to the reduction of iron oxides to metallic iron.43,44,52,68 The current density gets contribution due to newly formed metallic Fe. Heyrovsky reaction at this point arises on the impurities and on the re-deposited iron film (not on iron oxides).
The presented experimental results in Fig. 10 agree well with the literature82 and also validate the “pseudo-Tafel” description of Heyrovsky reactions. In particular the measured slopes bHey clearly fit to the theory where bHey follows the relation 2 × 2.303RT/F ≤ bHey ≤ 3 × 2.303RT/F.83 More details on the scientific background, about the switch to the Heyrovsky pathway, can be found in the work of Fang et al. or Vilekar et al.77,84
In the case of alkaline conditions observed at a corroding Mg surface this pathway can be represented in the following way:
| M + H2O + e− → M–Had + OH− (Volmer – V – electrochemical adsorption of hydrogen) |
| 2Had → H2 (Tafel – T – chemical desorption of hydrogen) |
| Had + H2O + e− → H2 + OH− (Heyrovsky – H – electrochemical desorption of hydrogen) |
The three possible reaction steps are schematically illustrated in Fig. 11, which also illustrates the main idea of the mechanism behind the negative difference effect. Water molecules are discharged according to Volmer on the metal M followed by a subsequently Tafel or/and Heyrovsky reaction (V–T, V–H or V–TH pathway). The kinetics of the whole process depend on the cathodic overpotential, the Gibbs energy of adsorbed hydrogen Had at the top layer and the hydrogen surface coverage.79 If one compares previously shown experimental results, especially from TEM and XRD, on magnesium with the theoretical background of Heyrovsky reactions, the logical conclusion is that electrochemical desorption according to the Heyrovsky pathway drives hydrogen generation during Mg corrosion in the presence of Fe, Ni, and Cu impurities.
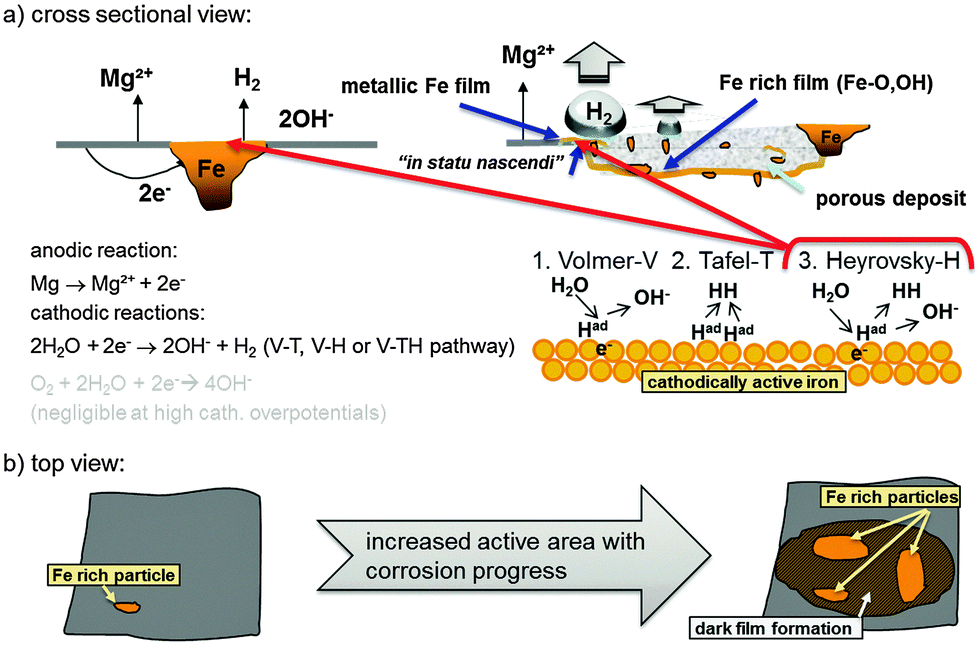 | ||
| Fig. 11 Schematic representation of a typical Mg system with Fe rich impurities showing the initial increase of cathodic active (dark) area based on findings of Curioni et al.32 (a) shows a cross-sectional view and involved anodic and cathodic reactions especially on the redeposited Fe film. (b) Anodic dissolution of Mg results in the increase of active area as shown by Taheri et al.85 and subsequent film formation based on the mechanism proposed by Flis-Kabulska et al.49 | ||
Thus, the additive combination of two effects, namely the fast Heyrovsky HER with continuously enhanced surface fraction of the cathodic area especially due to the iron re-deposition, ensures high hydrogen evolution at the Mg surface subjected to relatively low anodic polarization. In addition the very low polarization resistance of magnesium contributes to this mechanism, since it prevents the occurrence of low hydrogen overpotentials at cathodic areas where the rate control should change between the Heyrovsky reaction and the Tafel reaction (V–T → V–TH → V–H). As a result, according to the mixed potential theory, the global anodic net-polarization point can only slightly shift towards more positive values. Despite the strong dependency of the local mixed potential on the surface state and the amount of cathodically active area, on the amount and condition of alpha magnesium, and on the electrolyte composition, true cathodic overpotentials at the impurities, which induce Tafel reaction control of hydrogen generation or even control by oxygen reduction reaction, cannot occur.
At this point oxygen reduction gives main contribution to the oxidation of iron species. Thus, oxygen plays an important role in Mg corrosion, which is in contrast to common assumptions. Minor contribution can be considered for hydrogen evolution by the Tafel reaction at the α-Mg surface because of the very low hydrogen evolution exchange current at the Mg surface.
A theoretical contradiction to the approach occurs at higher anodic polarization where HER still increases. In that situation the contribution of Heyrovsky reactions should theoretically decrease since cathodic overpotential should decrease, but it does not. Explanation of this discrepancy requires the discussion of process kinetics. King86 and Frankel's7 work studied the IR-drop through the electrolyte. The strength of such a drop on active electrodes due to gas bubbles has been investigated in the literature87,88 and can locally exceed hundreds of mV. If one considers even higher anodic polarization an increased gas evolution is observed. Under these conditions a truly anodic polarization which hinders hydrogen evolution cannot be reached. The IR-drop increases fast in correlation with the current, since the anodic reaction is kinetically fast, based on the very low polarization resistance of magnesium. As a result, the developing mixed potential is always very close to the OCP of the less noble phase (α-Mg) and the Heyrovsky reaction remains active. Thus, the V–H pathway cannot be switched off by the increase of applied positive potentials simply because magnesium is not anodically polarizable.
The remaining question of the contribution to cathodic reactions by the α-Mg surface can be partially answered as well. As explained within the introduction, compared to transition metals such as Fe or Ni, which are known as efficient electrodes for hydrogen production, magnesium does not exhibit d-orbitals which can interact with the hydrogen anti-bonding orbital and thus facilitate the dissociation of molecular hydrogen.89 The preferred reaction pathway is V–T. Thus, pure α-Mg surfaces do not drive hydrogen dissociation on the V–H pathway30 and as a result, just slightly contribute to the strong hydrogen gas evolution, which is typically measured for impurities containing magnesium during corrosion. At low impurity levels the dopant effect31 might work. Hence, the water reduction during cathodic polarization is even more efficient on impurities and dark areas, which corresponds to the literature findings.16,22,26
5. Conclusions
The present work contributes to the better understanding of the corrosion mechanisms of Mg substrates containing iron impurities. For the first time the enhanced hydrogen evolution is correlated with the possible electrochemical re-deposition of a thin iron (patch) film on the magnesium surface. Re-deposition accelerates HER and becomes the determining process after an initial period. The assumed formation of re-deposited Fe was confirmed by the results of in situ GIXRD and application of ferrofluids according to the Bitter decoration method.Additionally, the Heyrovsky mechanism of HER on the redeposited iron surface is discussed as a potential reason for the enhanced rate of hydrogen evolution even at high pH values.
Based on the progress being achieved in related research and by adding the new aspect of this study, the following conclusions can be drawn:
(1) Dark film formation (Mg(OH)2 + Fe rich film) is related to the deposition of thin metallic iron layers especially at the moving corrosion front as a result of impurity self-corrosion and subsequent reduction of iron species at higher cathodic polarizations of approx. −1 VAg/AgCl (in combination with the generation of highly reactive atomic hydrogen) and subsequent corrosion product deposition (magnesium hydroxide precipitation).
(2) SEM-EDS analysis of the distribution of ferrofluid around the black areas formed on the Mg sample placed on the magnet shows that the dark areas influence the local magnetic field which can be explained by their enrichment with Fe and/or iron oxides/hydroxides, which was also shown by TEM studies. The results of GIXRD indicate the formation of Fe(OH)2 and γ-FeOOH in the dark areas.
(3) As corrosion progresses, re-deposited Fe films “in-statu nascendi” at the corrosion forefront drive the cathodic current and become the most active H2 generator.
(4) The initial HER and its kinetics are related to the amount, and the effective surface fraction of metallic impurities and/or intermetallics, and their respective electrochemical hydrogen desorption kinetics according to the Heyrovsky mechanism. The electrochemical desorption process has much faster kinetics than the chemical desorption described by the Tafel mechanism.
(5) The re-deposition mechanism combined with electrochemical desorption kinetics according to the Heyrovsky pathway is an adequate candidate for the explanation of observed phenomena generally known as the negative difference effect – NDE.
(6) Restricting the Fe re-deposition offers the ability to create new strategies for corrosion control of Fe-containing Mg substrates.
Appendix
The calculation of the number of deposited Fe monolayers “in statu nascendi”, formed during the growth of the dark area100 ml of 0.5 M FeCl2 (FeCl2·4H2O (198.83 g mol−1)) was used for the tests. A rough approximation can be carried out:
• A total of ∼2.8 g of Fe was dissolved which corresponds to ∼3 × 1022 atoms
• The size of the immersed Mg sample was approx. 3 cm2. Upon considering the increase due to surface roughness, we can simply assume that about 10 cm2 are covered.
• By taking the “area” of one iron atom in a bcc structure (a = 0.287 nm) to be 8.24 × 10−16 cm2 we get a number of atoms of 8.24 × 1017, which is theoretically required to cover the sample with a monolayer
• According to 3 × 1022/8.24 × 1017 = 3.6 × 104 Fe monolayers could be deposited on the Mg surface.
For the used test samples (in a worse scenario of low deposition rate) we simply assume that one of the thousand atoms is re-deposited. As a result, the surface still can be fully covered (theoretically) by an active film. And in reality the surface composition is a mixture of iron, Mg and Mg(OH)2.
In the case of impurities this approximation can give the contribution of Fe particles of e.g. 1 μm diameter. Such a particle has a weight of about 5 ng and is constituted of 5 × 1013 atoms. This leads to a surface coverage of ∼0.05 cm2 per particle. In other words, 20 of such iron particles are sufficient to cover 1 cm2 with active iron. An experimental showcase can be found in the study of Samaniego et al.21
Acknowledgements
Dr Höche would like to thank Dr W. Dietzel for input and discussions, Dr A. Schwarz from University of Hamburg for assisting the ferrofluid application, Dr Marc Willinger and Michael Scherzer from the Department of Inorganic Chemistry at Fritz Haber Institute of the Max Planck Society for their contribution to the diffraction experiments and Eng. U. Burmester for setting up the experimental equipment. Dr Lamaka acknowledges the financial support of Alexander von Humboldt Foundation via the Experienced Researcher Grant.References
- G. Williams, N. Birbilis and H. N. McMurray, Electrochem. Commun., 2013, 36, 1–5 CrossRef CAS.
- A. Atrens and W. Dietzel, Adv. Eng. Mater., 2007, 9, 292–297 CrossRef CAS.
- J. A. Boyer, The Corrosion of Magnesium and of the Magnesium Aluminum Alloys Containing Manganese, Report 248, United States National Advisory Committee for Aeronautics, Washington, 1926.
- R. Tunold, H. Holtan, M.-B. H. Berge, A. Lasson and R. Steen-Hansen, Corros. Sci., 1977, 17, 353–365 CrossRef CAS.
- W. J. James, M. E. Straumanis, B. K. Bhatia and J. W. Johnson, J. Electrochem. Soc., 1963, 110, 1117–1120 CrossRef CAS.
- J. L. Robinson and P. F. King, J. Electrochem. Soc., 1961, 108, 36–41 CrossRef CAS.
- G. S. Frankel, A. Samaniego and N. Birbilis, Corros. Sci., 2013, 70, 104–111 CrossRef CAS.
- N. T. Kirkland, G. Williams and N. Birbilis, Corros. Sci., 2013, 77, 407–409 CrossRef CAS.
- Z. Shi and A. Atrens, Corros. Sci., 2013, 77, 403–406 CrossRef CAS.
- M. E. Straumanis and Y. N. Wang, J. Electrochem. Soc., 1955, 102, 304–310 CrossRef CAS.
- M. Garreau and P. L. Bonora, J. Appl. Electrochem., 1977, 7, 197–209 CrossRef CAS.
- D. M. Dražić and J. P. Popić, J. Appl. Electrochem., 1999, 29, 43–50 CrossRef.
- L. Rossrucker, K. J. J. Mayrhofer, G. S. Frankel and N. Birbilis, J. Electrochem. Soc., 2014, 161, C115–C119 CrossRef CAS.
- N. Birbilis, G. Williams, K. Gusieva, A. Samaniego, M. A. Gibson and H. N. McMurray, Electrochem. Commun., 2013, 34, 295–298 CrossRef CAS.
- N. T. Kirkland, G. Williams and N. Birbilis, Corros. Sci., 2012, 65, 5–9 CrossRef CAS.
- M. Taheri, J. R. Kish, N. Birbilis, M. Danaie, E. A. McNally and J. R. McDermid, Electrochim. Acta, 2014, 116, 396–403 CrossRef CAS.
- M. Curioni, Electrochim. Acta, 2014, 120, 284–292 CrossRef CAS.
- S. Bender, J. Göllner, A. Heyn and S. Schmigalla, Mater. Corros., 2012, 63, 707–712 CAS.
- A. Atrens, G.-L. Song, F. Cao, Z. Shi and P. K. Bowen, J. Magnesium Alloys, 2013, 1, 177–200 CrossRef CAS.
- G. S. Frankel, S. Fajardo and B. M. Lynch, Faraday Discuss., 2015, 180, 11–33 RSC.
- A. Samaniego, N. Birbilis, X. Xia and G. S. Frankel, Corrosion, 2015, 71, 224–233 CrossRef CAS.
- N. Birbilis, A. D. King, S. Thomas, G. S. Frankel and J. R. Scully, Electrochim. Acta, 2014, 132, 277–283 CrossRef CAS.
- S. Lebouil, O. Gharbi, P. Volovitch and K. Ogle, Corrosion, 2015, 71, 234–241 CrossRef CAS.
- S. Thomas, N. V. Medhekar, G. S. Frankel and N. Birbilis, Curr. Opin. Solid State Mater. Sci., 2015, 19, 85–94 CrossRef CAS.
- S. M. Nelson, A. C. D. Newman, T. E. Tomlinson and L. E. Sutton, Trans. Faraday Soc., 1959, 55, 2186–2202 RSC.
- S. Fajardo and G. S. Frankel, Electrochim. Acta, 2015, 165, 255–267 CrossRef.
- D. M. Bird, L. J. Clarke, M. C. Payne and I. Stich, Chem. Phys. Lett., 1993, 212, 518–524 CrossRef CAS.
- S. Trasatti, J. Electroanal. Chem., 1972, 39, 163–184 CrossRef.
- J. K. Nørskov, A. Houmøller, P. K. Johansson and B. I. Lundqvist, Phys. Rev. Lett., 1981, 46, 257–260 CrossRef.
- T. Vegge, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 035412 CrossRef.
- M. Pozzo and D. Alfè, Int. J. Hydrogen Energy, 2009, 34, 1922–1930 CrossRef CAS.
- M. Curioni, F. Scenini, T. Monetta and F. Bellucci, Electrochim. Acta, 2015, 166, 372–384 CrossRef CAS.
- G. Williams, N. Birbilis and H. N. McMurray, Faraday Discuss., 2015, 180, 313–330 RSC.
- M. P. Brady, G. Rother, L. M. Anovitz, K. C. Littrell, K. A. Unocic, H. H. Elsentriecy, G. L. Song, J. K. Thomson, N. C. Gallego and B. Davis, J. Electrochem. Soc., 2015, 162, C140–C149 CrossRef CAS.
- D. Höche, J. Electrochem. Soc., 2015, 162, C1–C11 CrossRef.
- C. Deslouis, D. Festy, O. Gil, V. Maillot, S. Touzain and B. Tribollet, Electrochim. Acta, 2000, 45, 1837–1845 CrossRef CAS.
- L. Rossrucker, A. Samaniego, J. P. Grote, A. M. Mingers, C. A. Laska, N. Birbilis, G. S. Frankel and K. J. J. Mayrhofer, J. Electrochem. Soc., 2015, 162, C333–C339 CrossRef CAS.
- T. Cain, S. B. Madden, N. Birbilis and J. R. Scully, J. Electrochem. Soc., 2015, 162, C228–C237 CrossRef CAS.
- L. Yang, X. Zhou, M. Curioni, S. Pawar, H. Liu, Z. Fan, G. Scamans and G. Thompson, J. Electrochem. Soc., 2015, 162, C362–C368 CrossRef CAS.
- D. Lysne, S. Thomas, M. F. Hurley and N. Birbilis, J. Electrochem. Soc., 2015, 162, C396–C402 CrossRef CAS.
- S. H. Salleh, S. Thomas, J. A. Yuwono, K. Venkatesan and N. Birbilis, Electrochim. Acta, 2015, 161, 144–152 CrossRef.
- K. S. Williams, J. P. Labukas, V. Rodriguez-Santiago and J. W. Andzelm, Corrosion, 2014, 71, 209–223 CrossRef.
- H. H. Le and E. Ghali, J. Appl. Electrochem., 1993, 23, 72–77 CrossRef CAS.
- R. D. Armstrong and I. Baurhoo, J. Electroanal. Chem., 1972, 40, 325–338 CrossRef.
- P. Dauphin-Ducharme and J. Mauzeroll, Anal. Chem., 2015, 87, 7499–7509 CrossRef CAS PubMed.
- F. Bitter, Phys. Rev., 1931, 38, 1903–1905 CrossRef.
- M. Scherzer, PhD thesis, Fritz Haber Institut der Max Planck Gesellschaft, 2015.
- M. Sipiczki, E. Kuzmann, Z. Homonnay, J. Megyeri, K. Kovács, I. Pálinkó and P. Sipos, Hyperfine Interact., 2013, 217, 145–149 CrossRef CAS.
- I. Flis-Kabulska, J. Flis, Y. Sun and T. Zakroczymski, Electrochim. Acta, 2015, 167, 61–68 CrossRef.
- X. Zou, S. Gu, H. Cheng, X. Lu, Z. Zhou, C. Li and W. Ding, J. Electrochem. Soc., 2015, 162, D49–D55 CrossRef CAS.
- A. Allanore, H. Lavelaine, G. Valentin, J. P. Birat, P. Delcroix and F. Lapicque, Electrochim. Acta, 2010, 55, 4007–4013 CrossRef CAS.
- A. Allanore, H. Lavelaine, G. Valentin, J. P. Birat and F. Lapicque, J. Electrochem. Soc., 2007, 154, E187–E193 CrossRef CAS.
- B. Beverskog and I. Puigdomenech, Corros. Sci., 1996, 38, 2121–2135 CrossRef CAS.
- S. Hofmann, Surf. Interface Anal., 1986, 9, 3–20 CrossRef CAS.
- M. Descostes, F. Mercier, N. Thromat, C. Beaucaire and M. Gautier-Soyer, Appl. Surf. Sci., 2000, 165, 288–302 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- A. P. Grosvenor, B. A. Kobe, M. C. Biesinger and N. S. McIntyre, Surf. Interface Anal., 2004, 36, 1564–1574 CrossRef CAS.
- M. S. Mel'gunov, V. B. Fenelonov, E. A. Mel'gunova, A. F. Bedilo and K. J. Klabunde, J. Phys. Chem. B, 2003, 107, 2427–2434 CrossRef.
- G. Baril and N. Pébère, Corros. Sci., 2001, 43, 471–484 CrossRef.
- J. H. Nordlien, S. Ono, N. Masuko and K. Nisancioglu, J. Electrochem. Soc., 1995, 142, 3320–3322 CrossRef CAS.
- R. Zboril, M. Mashlan and D. Petridis, Chem. Mater., 2002, 14, 969–982 CrossRef CAS.
- B. J. Lemaire, P. Davidson, J. Ferré, J. P. Jamet, P. Panine, I. Dozov and J. P. Jolivet, Phys. Rev. Lett., 2002, 88, 125507 CrossRef CAS PubMed.
- O. Perales Perez, Y. Umetsu and H. Sasaki, Hydrometallurgy, 1998, 50, 223–242 CrossRef CAS.
- J. D. Bernal, D. R. Dasgupta and A. L. Mackay, Clay Miner. Bull., 1959, 4, 15 CAS.
- B. Yuan and G. M. Haarberg, ECS Trans., 2009, 16, 31–37 CrossRef CAS.
- G. M. Haarberg and B. Yuan, ECS Trans., 2014, 58, 19–28 CrossRef.
- A. Allanore, L. Yin and D. R. Sadoway, Nature, 2013, 497, 353–356 CrossRef CAS PubMed.
- I. Flis-Kabulska and J. Flis, Int. J. Hydrogen Energy, 2014, 39, 3597–3605 CrossRef.
- D. Fray, Nature, 2013, 497, 324–325 CrossRef CAS PubMed.
- I. Flis-Kabulska, J. Flis and T. Zakroczymski, Electrochim. Acta, 2008, 53, 3094–3101 CrossRef CAS.
- T. Misawa, Corros. Sci., 1973, 13, 659–676 CrossRef CAS.
- M. Stratmann, K. Bohnenkamp and H. J. Engell, Corros. Sci., 1983, 23, 969–985 CrossRef CAS.
- R. G. Buchheit, R. P. Grant, P. F. Hlava, B. McKenzie and G. L. Zender, J. Electrochem. Soc., 1997, 144, 2621–2628 CrossRef CAS.
- K. A. Yasakau, M. L. Zheludkevich, S. V. Lamaka and M. G. S. Ferreira, J. Phys. Chem. B, 2006, 110, 5515–5528 CrossRef CAS PubMed.
- A. A. E. Warraky, A. M. El-Aziz and K. A. Soliman, Anti-Corros. Methods Mater., 2007, 54, 155–162 CrossRef.
- M. R. Gennero de Chialvo and A. C. Chialvo, Electrochim. Acta, 1998, 44, 841–851 CrossRef CAS.
- S. A. Vilekar, I. Fishtik and R. Datta, J. Electrochem. Soc., 2010, 157, B1040–B1050 CrossRef CAS.
- M. R. Gennero de Chialvo and A. C. Chialvo, Phys. Chem. Chem. Phys., 2001, 3, 3180–3184 RSC.
- S. Y. Qian, B. E. Conway and G. Jerkiewicz, J. Chem. Soc., Faraday Trans., 1998, 94, 2945–2954 RSC.
- N. Krstajić, M. Popović, B. Grgur, M. Vojnović and D. Šepa, J. Electroanal. Chem., 2001, 512, 16–26 CrossRef.
- S. Sharifi-Asl and D. D. Macdonald, J. Electrochem. Soc., 2013, 160, H382–H391 CrossRef CAS.
- M. R. Gennero de Chialvo and A. C. Chialvo, Electrochem. Commun., 1999, 1, 379–382 CrossRef.
- M. R. G. de Chialvo and A. C. Chialvo, J. Electroanal. Chem., 1994, 372, 209–223 CrossRef.
- Y.-H. Fang, G.-F. Wei and Z.-P. Liu, J. Phys. Chem. C, 2013, 117, 7669–7680 CAS.
- M. Taheri, R. C. Phillips, J. R. Kish and G. A. Botton, Corros. Sci., 2012, 59, 222–228 CrossRef CAS.
- A. D. King, N. Birbilis and J. R. Scully, Electrochim. Acta, 2014, 121, 394–406 CrossRef CAS.
- G. Kreysa and H. J. Külps, J. Electrochem. Soc., 1981, 128, 979–984 CrossRef CAS.
- D. Kiuchi, H. Matsushima, Y. Fukunaka and K. Kuribayashi, J. Electrochem. Soc., 2006, 153, E138–E143 CrossRef CAS.
- K.-F. Aguey-Zinsou and J.-R. Ares-Fernandez, Energy Environ. Sci., 2010, 3, 526–543 CAS.
| This journal is © the Owner Societies 2016 |