
A highly conductive, non-flammable polymer–nanoparticle hybrid electrolyte†
Akanksha Agrawal‡
,
Snehashis Choudhury‡ and
Lynden A. Archer*
School of Chemical and Biomolecular Engineering, Cornell University, Ithaca, NY 14853, USA. E-mail: laa25@cornell.edu
First published on 12th February 2015
Abstract
We report on the physical properties of lithium-ion conducting nanoparticle–polymer hybrid electrolytes created by dispersing bidisperse mixtures of polyethylene glycol (PEG)-functionalized silica nanoparticles in an aprotic liquid host. At high particle contents, we find that the ionic conductivity is a non-monotonic function of the fraction of larger particles xL in the mixtures, and that for the nearly symmetric case xL ≈ 0.5 (i.e. equal volume fraction of small and large particles), the room temperature ionic conductivity is nearly ten-times larger than in similar nanoparticle hybrid electrolytes comprised of the pure small (xL ≈ 0) or large (xL ≈ 1) particle components. Complementary trends are seen in the activation energy for ion migration and effective tortuosity of the electrolytes, which both exhibit minima near xL ≈ 0.5. Characterization of the electrolytes by dynamic rheology reveals that the maximum conductivity coincides with a distinct transition in soft glassy properties from a jammed to partially jammed and back to jammed state, as the fraction of large particles is increased from 0 to 1. This finding implies that the conductivity enhancement arises from purely entropic loss of correlation between nanoparticle centers arising from particle size dispersity. As a consequence of these physics, it is now possible to create hybrid electrolytes with MPa elastic moduli and mS cm−1 ionic conductivity levels at room temperature using common aprotic liquid media as the electrolyte solvent. Remarkably, we also find that even in highly flammable liquid media, the bidisperse nanoparticle hybrid electrolytes can be formulated to exhibit low or no flammability without compromising their favorable room temperature ionic conductivity and mechanical properties.
Introduction
Significant research efforts have been devoted towards improving the portability, power, lifetime and safety of secondary batteries with extensive focus on electrolytes and ion conducting membranes.1–6 Polymer–nanoparticle composites have received particular attention because of their promise to create new electrolyte platforms that leverage the attractive transport properties, low flammability, and processability of polymers and the ability of nanostructures to disrupt polymer crystallization and facilitate ion transport in order to mitigate safety issues associated with conventional flammable liquid electrolytes.4–8 These nanocomposite electrolytes have been shown to also improve portability of batteries by preventing electrolyte leakage and by eliminating the need for a physical separator.9–15 The inorganic particles in such electrolytes also play an important role in ion transport mainly as passive fillers and sometimes as active fillers.9 As a passive filler, they act as plasticizers for the polymers preventing crystallization, thus speeding up segmental dynamics and enhancing ion transport.9,12,14–16 However, specific particle surface chemistries and an optimum nanoparticle loading are required to ensure a well-dispersed state of the particles, such that the polymer viscosity remains reasonable and ion transport pathways are not disrupted at high particle concentration. Active fillers directly participate in the ion transport process either by providing additional cations/anions or by surface reaction with mobile ions. They have been reported to improve the cation transference number, which ultimately results in significant increment in the cell-scale coulombic efficiency.9,11,12,15,17,18Perhaps the greatest benefit of nanocomposite electrolytes stems from the higher mechanical stability they provide to the entire battery design, which for example opens the way to batteries in a much wider range of form factors than available today. It has also been previously reported that a high modulus electrolyte can be effective in preventing dendrite-induced short circuit in a lithium metal battery, which provide opportunities for novel high-energy battery designs that take advantage of the factor of 10 or more improvement in anode capacity made possible by metallic lithium.19–25 Nanocomposite electrolyte-based batteries have shown encouraging results in this regard.16,26–28
Despite this promise, a persistent and important challenge to application of nanocomposite electrolytes in practical rechargeable batteries has been their low ambient ionic conductivity and high interfacial resistance.9,26,29,30 Weston and Steele31 were the first to make improvements towards developing a decently ion conducting and highly mechanical stable electrolyte by adding ceramic fillers in low volume fractions to polyethylene oxide polymer. However, achieving practical conductivity at high particle loadings, where the mechanical strength is maximized, was not possible, and remains an unsolved problem despite almost two decades of focused research. More recent studies on hybrid electrolytes based on polymer–tethered nanoparticles have shown a significant promise towards this step.32–34 These hybrid electrolytes based on hairy nanoparticles have good mechanical and electrochemical properties, and at the same time they provide enough room for nano-engineering to improve the current state of art even further. The polymer–grafted nanoparticles have been shown to exhibit interesting physical properties like viscoelasticity,35,36 thermal jamming,37 and star polymer like relaxation.38 Previously, studies on binary mixture of star polymers have gained significant attention by showing that addition of smaller star polymers to bigger ones leads to a transition from glassy state to liquid state.39,40 Theoretical studies have shown that at the critical size ratio of 0.4, a transition from glassy to liquid state is observed in binary hard spheres.41 Similar studies on the self-suspended binary mixtures of these hairy nanoparticles have demonstrated that addition of either small or bigger particles leads to un-jamming of the system.42 Currently, we focus on this jamming transition observed in binary hairy nanoparticles in the context of an electrolyte and try to utilize it to build a better battery.
In this article we report on ionic conductivity, mechanical properties, and structure of hybrid electrolytes comprised of a bidisperse blend of SiO2–PEG hairy nanoparticles dispersed in propylene carbonate (PC). The study focuses on silica particles with diameters of 10 nm and 25 nm (size ratio of 0.4) covalently functionalized with PEG oligomers. Also, on assuming a fcc packing arrangement for these particles we find that for large spheres of 25 nm diameter, the interstitial space is estimated to be 10 nm which is just sufficient to accommodate smaller particles. We thus propose that at this critical size ratio we can observe a transition from the jammed state of the material. Our specific interest is in understanding the effect of both the total particle volume fraction, Φ, and relative volume fraction of the bigger particles xL – at a fixed Φ – on suspension structure and physical properties. We find that the additional degree of freedom provided by xL allows the structure and transport properties of polymer–nanoparticle hybrid electrolytes to be tuned in novel ways to achieve both high ionic conductivity and good mechanical performance. To our knowledge, this is the first study to systematically investigate the effect of particle size dispersity on ionic conductivity of nanoparticle–polymer hybrid electrolytes.
Materials and methods
Synthesis
Silica nanoparticles (Ludox, SM-30 and TM-50; Sigma Aldrich) with diameters of 10 nm and 25 nm, respectively, were grafted by covalent attachment of a trimethoxysilane functionalized polyethylene glycol methyl ether (PEG, MW ∼ 500 g mol−1, Gelest chemicals) in aqueous solution using a previously reported silane chemistry43,44 (see schematic in Fig. 1(a)). The grafting densities were computed from analysis of the residual inorganic content using thermal gravimetric analysis (TGA) to be Σ ∼ 1.3 chains per nm2 and Σ ∼ 1.5 chains per nm2 for 10 nm and 25 nm particles, respectively. Following synthesis, the hairy particles were purified using a two step process, first by dialysis using a snake skin membrane to remove any dissolved salts along with unattached free PEG chains; and second by repeated centrifugation at 8500 rpm for 10 minutes using a chloroform(solvent)–hexane(non-solvent) mixture to completely remove any remaining unattached PEG oligomers. The particles were then subsequently dried in convection oven at 55 °C overnight and at least for 12 h under high vacuum. The dried sample was quickly transferred to Argon filled glove box for storage and subsequent modification. Electrolytes were prepared inside the glove box by suspending the hairy nanoparticles in the electrolyte solvent propylene carbonate (PC, Sigma Aldrich) at various core volume fractions, Φ ranging from 0.1 to 0.5. For each Φ, the relative fraction of bigger particles with respect to the overall particle volume fraction, i.e. ΦL/Φ = xL, in the SiO2–PEG/PC suspensions was varied. The resulting solution of hybrid nanoparticles in PC was doped with bis(trifluoromethanesulfone imide) (LiTFSi, Sigma Aldrich) salt to create SiO2–PEG/PC hybrid electrolytes containing 1 M LiTFSI based on the total organic content (i.e. PEG corona and PC electrolyte solvent).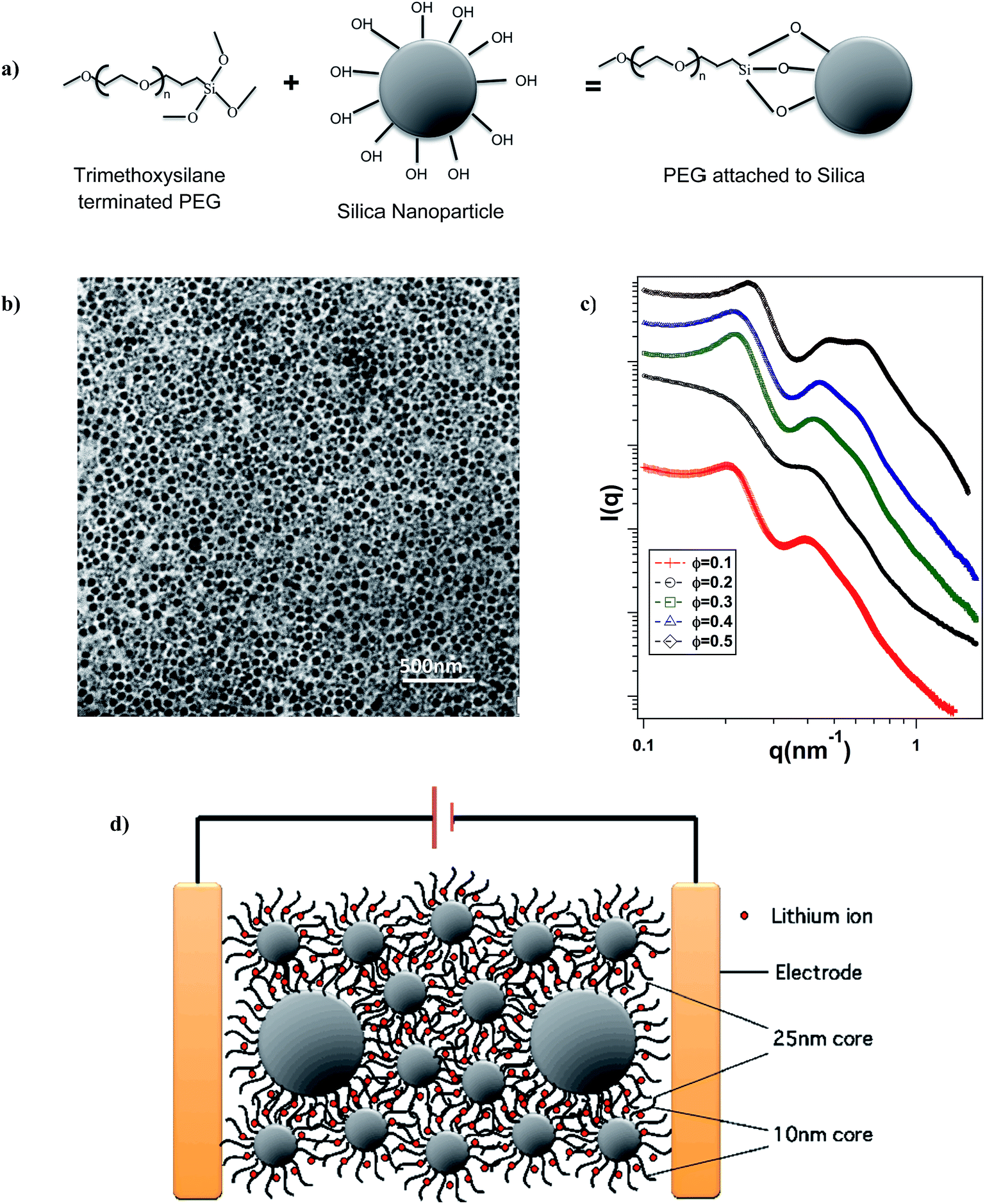 | ||
| Fig. 1 Physical Characterization – (a) schematic of hairy nanoparticle synthesis. (b) Transmission Electron Micrograph (TEM) image of binary hairy nanoparticle composite with xL = 0.5. (c) Intensity (I(q)) as a function of wave vector (q) for xL = 0.5 at different Φ, obtained from SAXS measurements. (d) Pictorial representation of a battery with binary hairy nanoparticle composite electrolyte. | ||
Characterization
The particle weight fraction in the SiO2–PEG/PC–LiTFSi hybrid electrolytes was determined from thermal gravimetric analysis (TGA) by heating the sample at 10 °C min−1 to 600 °C. The structure and dispersion state of the nanoparticles was characterized by angle-resolved Small Angle X-ray Scattering (SAXS) measurements at Station D1 of Cornell High Energy Synchrotron Source (CHESS) using a point collimated X-ray beam. All studied materials were smeared on a sample cell and the measurements performed at 30 °C.Dynamic rheological properties were studied using frequency- and strain-dependent oscillatory shear measurements at 30 °C on a MCR 301 rheometer outfitted with 10 mm diameter, 2° cone and plate fixtures. The frequency sweep measurements were performed at a low strain, γ = 0.05%, which is within the linear viscoelastic regime for the materials. The strain-dependent oscillatory shear measurements were performed at a fixed angular frequency of ω = 10 rad s−1.
Electrochemical measurements
The ionic conductivity of the SiO2–PEG/PC–LiTFSI hybrid electrolytes was measured as a function of temperature, ranging from 0 °C to 105 °C, using a Novocontrol Broadband Dielectric spectrometer. For each temperature, the frequency was varied from 0.1–3 × 106 Hz. The DC conductivity at each temperature was obtained from the plot of real part of the conductivity with frequency using the procedure described by Jonscher.45 The Re[conductivity] can be expressed as σ′(ω) = σDC + Aωs; where A is a constant. The DC conductivity can thus be estimated from the plateau value of the plot between Re[conductivity] and ω.Characterizing flammability
The flammability of electrolytes containing the hairy SiO2–PEG nanoparticles was studied by suspending the particles in a more flammable electrolyte mixture, ethylene carbonate/diethyl carbonate (EC:DEC, Sigma Aldrich). This approach was necessary because the PC solvent used for the other studies is flammable only at very high temperatures. The samples used for this component of the study were doped with 1 M of Lithium hexafluorophosphate (LiPF6, Sigma Aldrich). 0.2 g of SiO2–PEG/EC:DEC/LiPF6 hybrid electrolytes at xL = 0.5 and various Φ were transferred to aluminum pans. Material flammability was studied by igniting each electrolyte specimen with butane torch lighter and recording images at the time of ignition, 4 s after ignition, and at the time the flame self-extinguished.Results and discussion
Fig. 1(b) is a Transmission electron micrograph (TEM) of a SiO2–PEG/PC hybrid electrolyte with Φ = 0.5 and xL = 0.5. The particles are observed to be quite well dispersed in the PC host. Fig. 1(c) reports the wavenumber (q)-dependent scattering intensity (I(q)) for SiO2–PEG/PC hybrid electrolyte obtained using SAXS measurements. The results are reported for a fixed value of xL = 0.5 at different Φ. It is evident that at any Φ, in the low q region, I(q) is at best a very weak function of q, whereas in the high q region, I(q) varies as q−4. Both observations are characteristic of un-aggregated well-dispersed spherical nanoparticles confirming the good dispersion state of the materials inferred from the small-area TEM measurements.46,47Fig. 2 shows the temperature dependent DC conductivity of SiO2–PEG/PC–LiTFSi hybrid electrolytes at various particle volume fractions (Φ) and for a range of xL values (0 ≤ xL ≤ 1). The data is well described by the Vogel–Fulcher–Thamann (VFT) temperature dependent conductivity, S(T) = A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−Ea/k(T − T0)), over much of the temperature range and for all the electrolytes studied. Here the prefactor A, with units S cm−1, is proportional to the number of mobile ions present in the electrolyte, Ea is the activation energy for ion mobility in kJ mole−1, k is the gas constant in kJ mol −1 K−1, and To is the empirical reference Temperature in K (ref. 48) (Table ST1, ESI†).The fact that conductivity for all electrolyte compositions studied are well described by the VFT relation over the entire range of temperature implies that there is no melting or crystallization transition, and that there is no phase separation.
exp(−Ea/k(T − T0)), over much of the temperature range and for all the electrolytes studied. Here the prefactor A, with units S cm−1, is proportional to the number of mobile ions present in the electrolyte, Ea is the activation energy for ion mobility in kJ mole−1, k is the gas constant in kJ mol −1 K−1, and To is the empirical reference Temperature in K (ref. 48) (Table ST1, ESI†).The fact that conductivity for all electrolyte compositions studied are well described by the VFT relation over the entire range of temperature implies that there is no melting or crystallization transition, and that there is no phase separation.
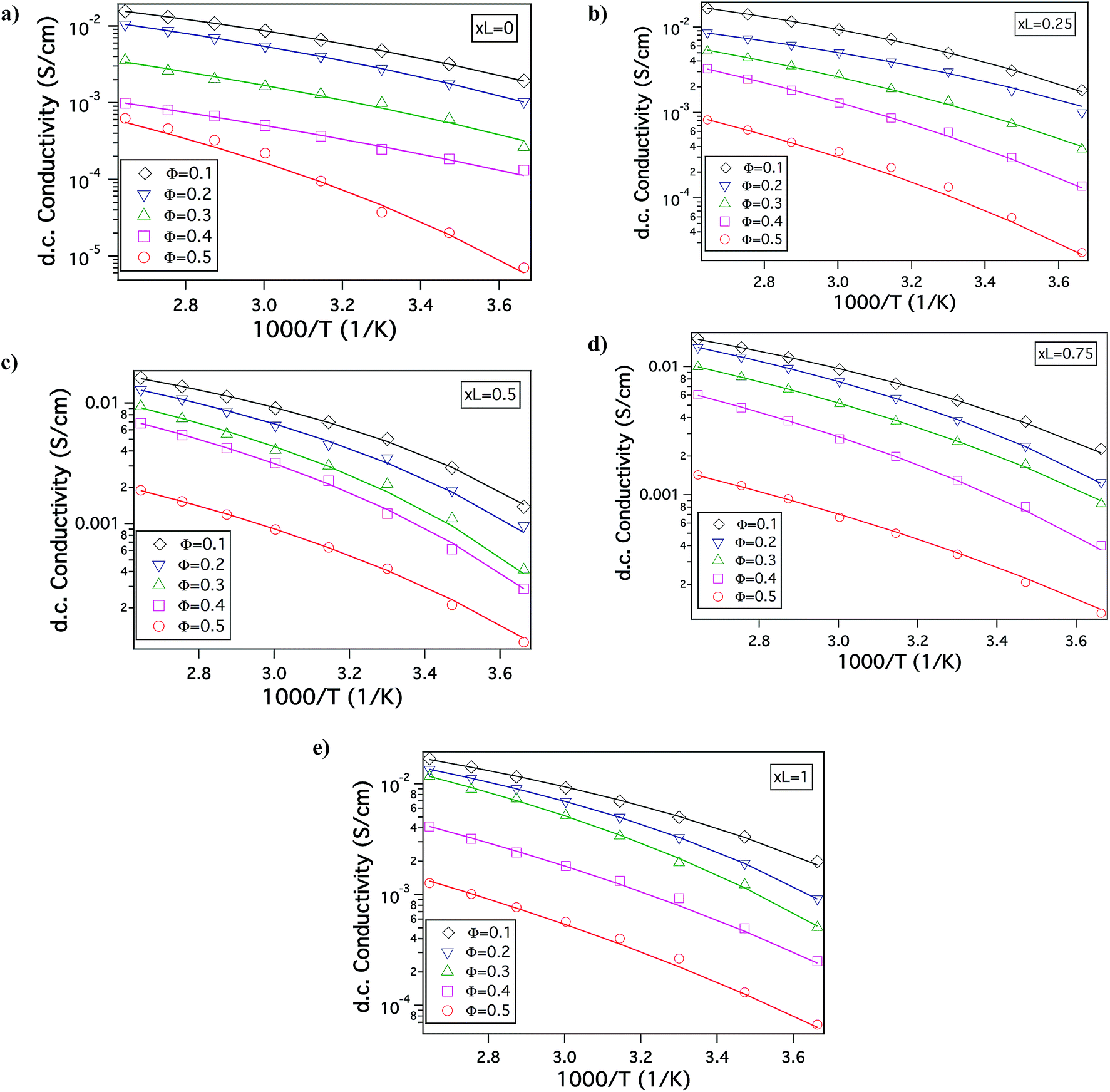 | ||
| Fig. 2 Electrochemical properties – conductivity as a function of inverse of temperature for different core volume fraction (Φ) at various fractions of bigger nanoparticles in the binary composite electrolyte indicated by xL, as, (a) xL = 0; (b) xL = 0.25; (c) xL = 0.5; (d) xL = 0.75; (e) xL = 1. | ||
In order to understand the effect of particle size dispersity on ionic conductivity, the DC conductivity measured at a fixed temperature T = 30 °C is plotted as a function of xL at different Φ, in Fig. 3(a). It can be observed that at Φ = 0.1 and 0.2, the conductivity does not vary much with changes in xL. However, for Φ ≥ 0.3 conductivity for the bidisperse SiO2–PEG/PC–LiTFSi hybrid electrolytes is noticeably higher than the conductivity of hybrid electrolytes comprised of the pure small (xL = 0) or big (xL = 1) SiO2–PEG hairy nanoparticles. This effect is most pronounced at Φ = 0.5, where a clear maximum is seen near xL = 0.5. The conductivity is also plotted as a function of overall particle volume fraction for different xL values in Fig. S1 of ESI.† There is a greater drop in the conductivity on increasing the particle volume fraction for pure small (xL = 0) and big (xL = 1) SiO2–PEG/PC–LiTFSi hybrid electrolytes than for the bidisperse hybrid electrolytes; the effect being particularly visible in electrolytes with xL = 0.5 and 0.75.
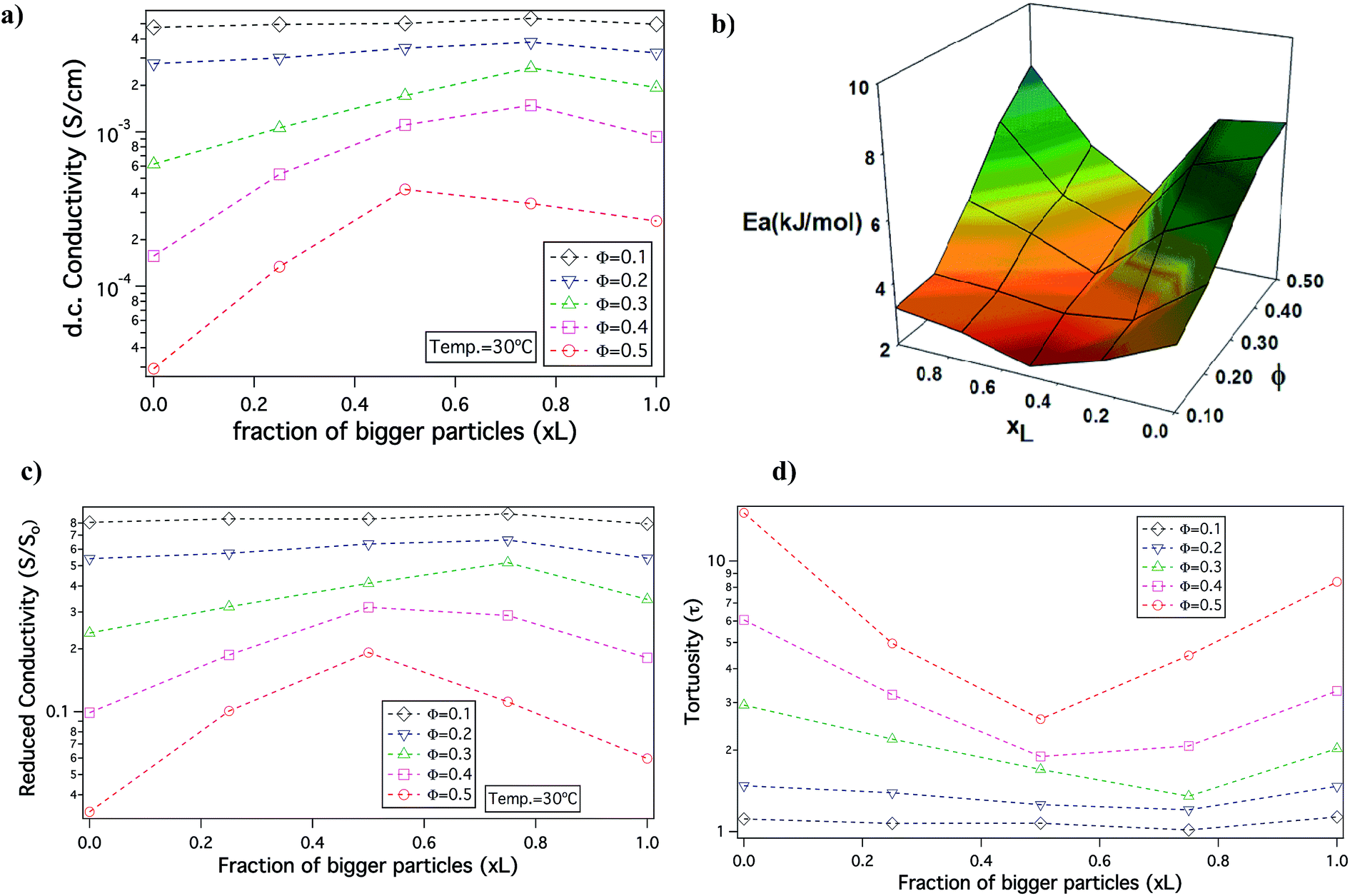 | ||
| Fig. 3 Analysis of ion transport – (a) conductivity as a function of xL at 30 °C at different Φ; (b) activation energy landscape at different xL values and corresponding Φ; (c) reduced conductivity given by the ratio of actual conductivity to that of the organic content, shown as a function of xL at 30 °C (d) tortuosity given as, τ = [So(1 − Φ)]/S, versus xL. | ||
Activation energies computed from the VFT fits of temperature-dependent conductivity provides additional insights about the nature of ion transfer processes in electrolytes. It is known that for electrolytes with higher activation energy there is a significant change in conductivity associated with temperature, which is correlated with more practical risks, such as thermal runaway and fire in cells in which there are exothermic side reactions.49 Fig. 3(b) reports the activation energy landscape obtained from VFT fits at different Φ and xL. It is seen that for high Φ the activation energy is greatest for the pure small (xL = 0) or pure big (xL = 1) hybrid electrolyte and that it is minimum near xL ≈ 0.5 (see also, Table ST1 in ESI†). Consistent with the conductivity data, the effect becomes weaker as the overall particle volume fraction, Φ, in the electrolytes is reduced. Thus, the bidisperse system of nanocomposite electrolytes can find wide applications as electrolytes that can be used at ambient temperatures, while sustaining sudden thermal shocks.
We hypothesize that the observed enhancement in conductivity in bidisperse SiO2–PEG/PC–LiTFSi hybrid electrolytes at high Φ and xL ≈ 0.5 may arise due to reduced correlation between particle-centers in the bidisperse materials. In particular at the high particle volume fractions, where bidispersity has the most noticeable effect on conductivity and activation energy, the positions of SiO2–PEG nanospheres in a monodisperse suspension are more correlated than in the bidisperse case. The tethered PEG corona chains are hence more crowed and confined in electrolytes containing either pure small or pure large SiO2–PEG particles. Such crowding of surface grafted chains has already been reported to lead to dramatically slower chain reorientation dynamics for cis-1,4-polyisoprene tethered to SiO2.35 The motion of ions mediated by the confined PEG chains would therefore be expected to be correspondingly impaired and sluggish, which is consistent with the higher activation energy. In contrast, for a bidisperse suspension of SiO2–PEG nanospheres, neighboring particles exert weaker constraints on each other because of the size dispersity. In other words, at high particle loadings, the system is less jammed due to the heterogeneities introduced by addition of a different sized species,42,50,51 thus loosening the packing of particles and reducing the confinement on tethered PEG chains, increasing their mobility and lowering the activation energy. This freedom would in turn increase the rate at which ions migrate in the electrolyte, reflected in a higher ionic conductivity. This hypothesis can also account for the weaker dependence of particle size dispersity at lower Φ where the tethered PEG chains are not confined and ion motion is controlled more in coordination with the mobile PC phase.
Since, the organic phase in the SiO2–PEG/PC–LiTFSi hybrid electrolytes is comprised of both PC and tethered PEG chains, it is useful to resolve the relative content of PEG and PC in different electrolytes to better understand the effect of bidispersity of the nanocores on electrolyte properties. The conductivity of PC/PEG–LITFSi liquid electrolyte mixtures was measured as a function of composition and the results are reported in Fig. S2.† A straight-line fit of the data allows us to determine how the content of PC/PEG influences ionic conductivity in the absence of particles. Using this knowledge, the conductivity contribution (So) attributable to the organic content for each electrolyte shown in Fig. 3(a) can be recovered. The relative PC content in each sample is tabulated in Table ST2 of the ESI.† In order to isolate the contribution of the nanocores, we plot the reduced conductivity as S/So versus the fraction of bigger particles (xL) in Fig. 3(c). The effect of particle size dispersity is now more profound, but again the maximum is most readily apparent for hybrids with the highest Φ and the maximum is clear. Hence, we conclude that the idea of unjamming a high volume fraction nanoparticle hybrid electrolyte by introducing bidispersity is not a function of the chemistry of the electrolyte solvent, making the concept a potentially powerful tool in other fields, such as ion exchange chromatography, desalination, and electroplating, where ion transport through complex media is relevant.
To understand the nature of the inter-particle spaces in the SiO2–PEG/PC hybrid electrolytes, we look at the trends in ‘tortuosity’ of these materials, as previously proposed by Carman.52 The conductivity of an electrolyte is a direct reflection of the tortuous nature of the conducting phase, where, tortuosity (τ) is defined as the ratio between the actual distance covered by the ions to travel across the bulk and the shortest distance between the two points. In the context of electrolytes, the conductivity of a neat electrolyte is suppressed by the addition of nanoparticles or other non-conducting entities depending upon the resulting porosity, which is equivalent to the volume fraction of the conductive phase in a suspension electrolyte. The experimental conductivity, S, is lower than that obtained empirically, So(1 − Φ), this discrepancy is quantified using the term effective tortuosity,53 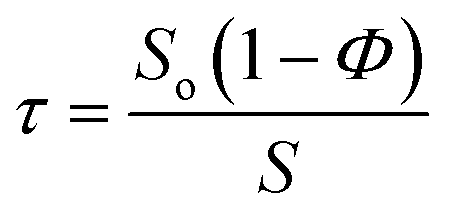 . Fig. 3(d) reports tortuosity as a function of xL for different volume fractions for the binary hairy nanoparticle electrolytes. It is seen that for low volume fractions, there is little change across the range of xL, and the tortuosity values remain close to unity. This is consistent with intuition that, at low particle volume fraction, the nature of the conducting pathway across the electrolyte is almost same as for the particle-free electrolyte and size dispersity of the particulate phase has no obvious effect on conductivity. On increasing the particle volume fraction, the tortuosity of the pure small or pure big SiO2–PEG/PC–LiTFSi electrolyte becomes greater than 10, however the bidisperse hybrid electrolytes are seen to have substantially lower effective tortuosity. This behavior can be attributed to the unjamming of densely packed SiO2–PEG nanoparticles, facilitating nearly unimpeded transport of ions in the electrolyte. Although empirical models for tortuosity for different shaped randomly spaced particles exist,53–57 such models do not yet exist for the surface-functionalized particles used in this study, making it difficult at the present time to understand the significance of the experimentally determined values for τ.
. Fig. 3(d) reports tortuosity as a function of xL for different volume fractions for the binary hairy nanoparticle electrolytes. It is seen that for low volume fractions, there is little change across the range of xL, and the tortuosity values remain close to unity. This is consistent with intuition that, at low particle volume fraction, the nature of the conducting pathway across the electrolyte is almost same as for the particle-free electrolyte and size dispersity of the particulate phase has no obvious effect on conductivity. On increasing the particle volume fraction, the tortuosity of the pure small or pure big SiO2–PEG/PC–LiTFSi electrolyte becomes greater than 10, however the bidisperse hybrid electrolytes are seen to have substantially lower effective tortuosity. This behavior can be attributed to the unjamming of densely packed SiO2–PEG nanoparticles, facilitating nearly unimpeded transport of ions in the electrolyte. Although empirical models for tortuosity for different shaped randomly spaced particles exist,53–57 such models do not yet exist for the surface-functionalized particles used in this study, making it difficult at the present time to understand the significance of the experimentally determined values for τ.
The changes in ionic conductivity are associated with important changes in mechanical properties of the electrolytes. It can be seen from Fig. 4(a) that for a given xL value, on changing Φ from 0.2 to 0.5, the system undergoes a well-studied transition from a viscoelastic liquid in which the loss modulus, G′′, is larger than the storage modulus G′, to a soft glass, characterized by G′ ≫ G′′ at low shear strains, and the tell tale maximum in G′′ at higher strains associated with yielding and flow (G′′ > G′) of the material.58,59 In particular, at low Φ and for xL = 0 or 1, G′′ > G′, indicating the electrolytes exhibit liquid-like behavior; remarkably, even under these conditions the bidisperse mixture with xL = 0.5 shows soft glassy behavior. In concentrated particulate suspensions, soft glassy rheology is understood to result from crowding and trapping of particles in cages formed due to the presence of neighboring particles. Upon increasing the shear strain, these cages break producing the burst of dissipated energy as particles move relative to each other, which manifests as a maximum in G′′.60,61 This implies that in such suspensions high shear strains can transform a jammed material to a less solid-like, processable form; removal of the strain causes the cage structure to reform and the solid-like jammed state is restored.
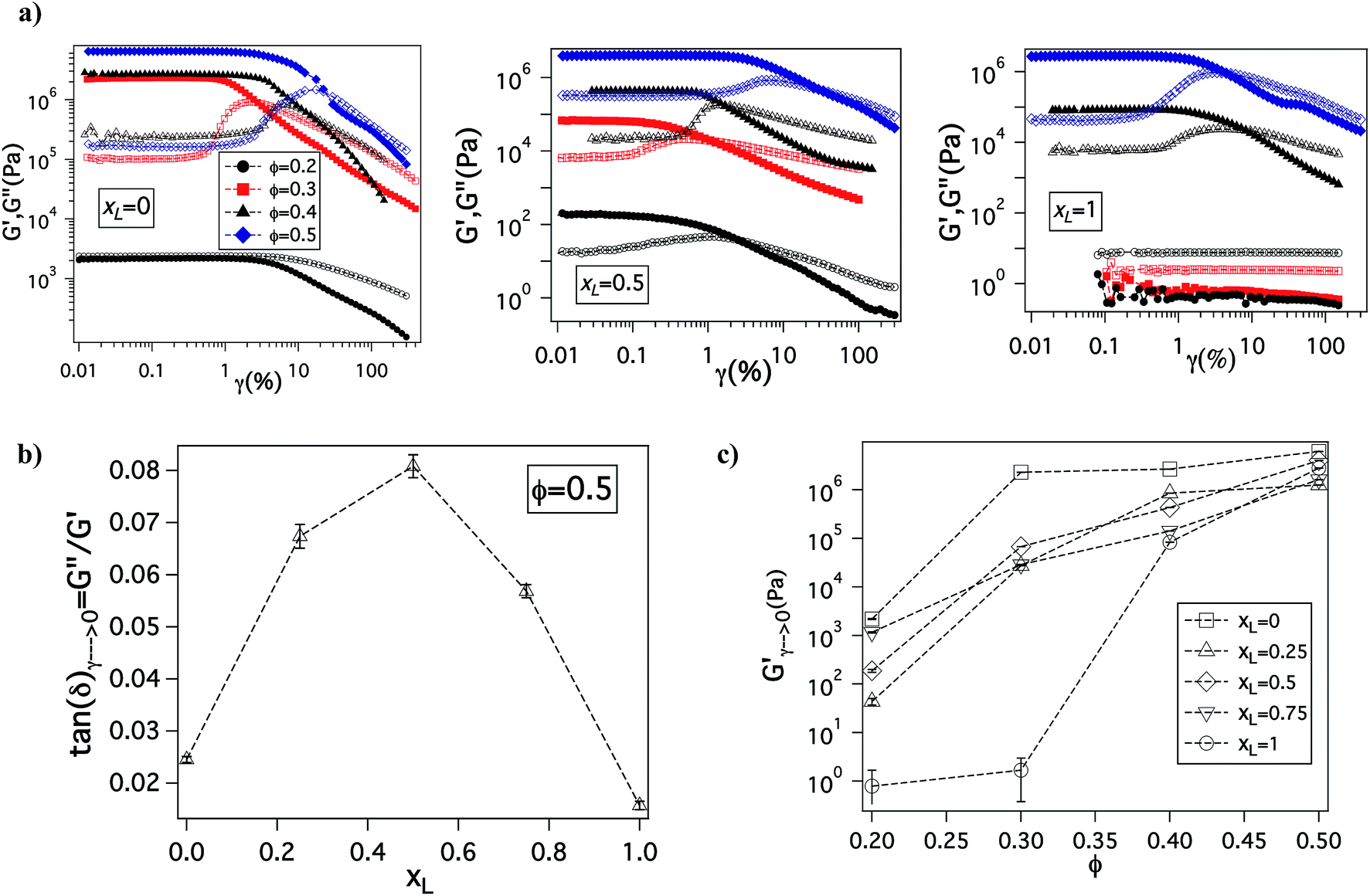 | ||
| Fig. 4 Rheological properties – (a) evolution of storage modulus, G′ (closed symbol) and loss modulus, G′′ (open symbol) as a function of amplitude strain, γ with particle volume fraction, Φ at xL = 0, 0.5 and 1 on going from left to right. (b) Variation of loss tangent, tan(δ) = G′′/G′ obtained at γ → 0 as a function of xL at Φ = 0.5. Since tan(δ) is the ratio of loss modulus to storage modulus, the higher the loss tangent the more fluid-like or un-jammed the system is. It can be observed that addition of either small or big particles leads to increase in un-jamming of the particles as compared to the pure species. (c) Storage modulus at γ → 0 varying as a function of Φ at different xL values. It can be seen that at high Φ, the modulus values are extremely high, more than 106. | ||
The degree of jamming in a soft glassy material can be quantified using the value of its loss tangent (tanδ = G′′/G′), measured at low stains in the linear viscoelastic regime of oscillatory shear measurements. Fig. 4(b) shows tanδ value as a function of xL, it is seen that addition of either the bigger or smaller particles to their pure cohorts leads to an increase in tanδ; implying that introduction of heterogeneity leads to less jamming of the system. Remarkably, at Φ = 0.5, where the electrolyte exhibits soft glassy rheology over the entire range of xL, tanδ exhibits a clear maximum near xL = 0.5, i.e. exactly where the maximum in ionic conductivity and minimum in activation energy for the electrolytes is observed. This finding succinctly shows that the enhanced conductivity for the bidisperse SiO2–PEG/PC hybrid electrolytes is a result of reduced jamming. Fig. 4(c) shows the viscoelastic storage modulus (G′) of these electrolytes as a function of volume fraction for various xL. At high particle loading, these electrolytes display G′ values around 5 MPa (Table ST2 in ESI†), which is only modestly lower than the modulus of hybrid electrolytes created using the pure small or pure large particles at Φ = 0.5.
The favorable ionic conductivity and mechanical properties of these bidisperse SiO2–PEG/PC–LiTFSi hybrid electrolytes at relatively high nanoparticle concentrations is already attractive for the application related investigations outlined in the introduction. Since silica and PEG are both non- or poorly-flammable materials, it is possible that at the high particle loadings where size dispersity has the greatest effect on conductivity and rheology of the electrolytes, it may also have a positive effect on their safety. To explore this aspect of the materials, we briefly studied their flammability using a previously disclosed protocol.13 The experiments are complicated by the fact that propylene carbonate, the electrolyte solvent used in the study so far, is only flammable at very high temperatures. In order to correctly assess the role of particles on flammability, we have created similar nanoparticle hybrid electrolytes in a more flammable electrolyte solvent (EC:DEC with 1 M LiPF6). Fig. 5 reports the results from these experiments in the form of snapshots of an electrolyte with xL = 0.5, with varying nanoparticle contents at different times following ignition. Fig. 5(a) shows the physical appearance of the electrolyte specimen at each volume fraction before ignition. Fig. 5(b) shows the same materials at the time of ignition, i.e. at t = 0. It can be seen that the electrolyte specimen with Φ = 0.4 initially catches fire, it is extinguished within one second of ignition; whereas the specimen with Φ = 0.5 is highly fire resistant. At the same time, the neat electrolyte without any particle or the samples with lower core volume fractions burn when ignited, as also seen in Fig. 5(c). Fig. 5(d) marks the time required for the fire to completely extinguish. Among the flammable samples, the particle-free electrolyte burns out the quickest, while the electrolytes with Φ = 0.3 take the longest to completely extinguish; implying that the degree of flammability decreases for higher volume fractions. The bidisperse hybrid electrolytes with Φ = 0.4 and 0.5 therefore appear to be good candidates for future applications-oriented studies.
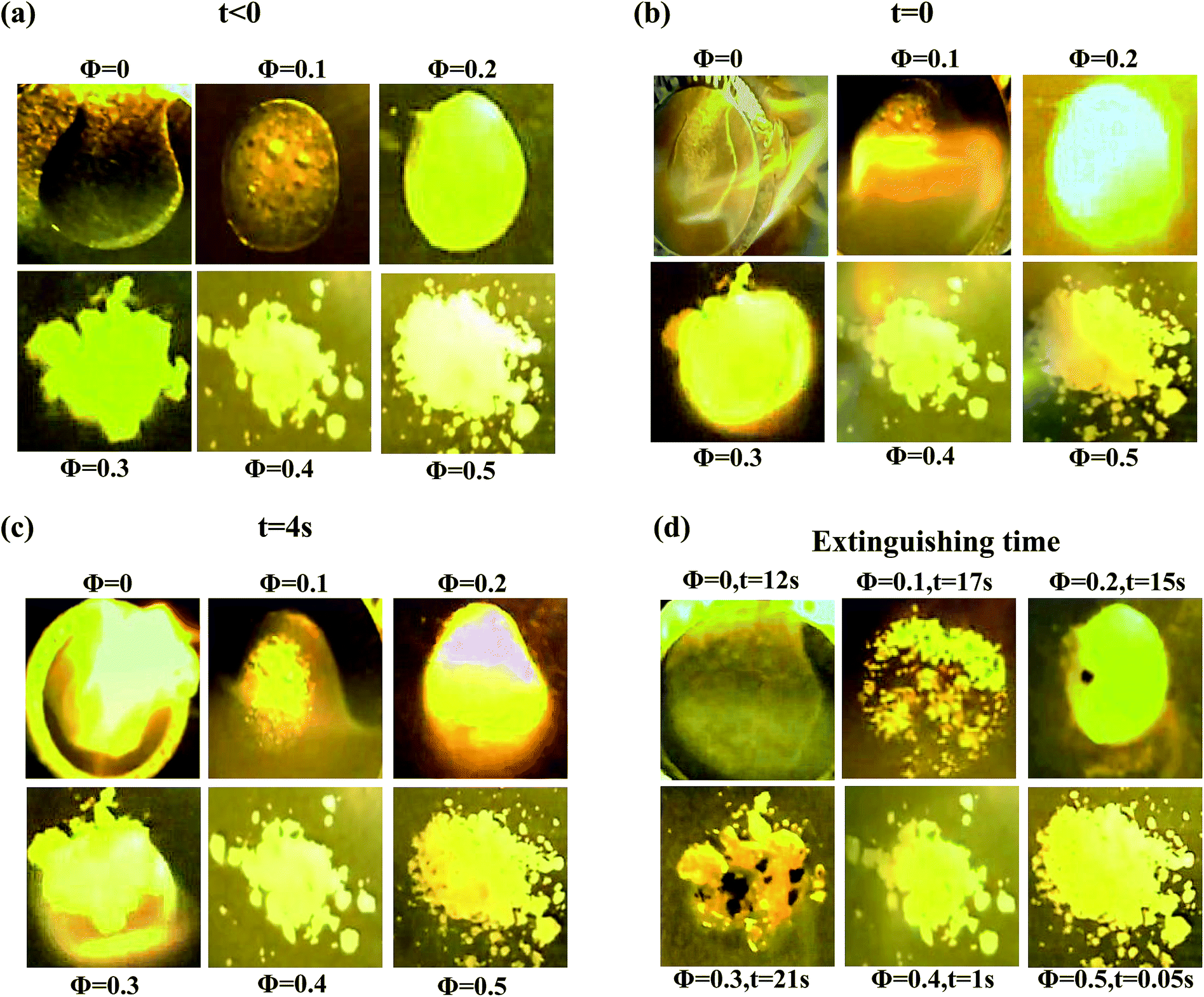 | ||
| Fig. 5 Flammability test – electrolyte samples with different particle volume fraction (a) before ignition i.e. t < 0. (b) At ignition time i.e. t = 0 (c) after ignition i.e. at t = 4 s (d) At the extinguishing time. (The binary electrolyte sample used here corresponds to xL = 0.5.). | ||
Conclusion
In summary, we have synthesized nanoparticle hybrid electrolytes based on bidisperse mixtures of PEG-functionalized silica nanoparticles in aprotic electrolyte solvents. The nanoparticles are found to be well dispersed and un-aggregated in their liquid hosts, even at high volume fractions. The conductivity of electrolytes containing nanoparticles with a bimodal size distribution is found to exhibit a pronounced maximum when the volume fraction of small and large particles are the same. This observation is attributed to the ability of large/small particles in a concentrated suspension of the opposite counterpart to produce disordering of the suspension by lowering correlation among the polydisperse particles. The tortuosity as well as activation energy values obtained from the measured conductivity show pronounced minima that correlate with the maximum conductivity, lending support to the idea that loose packing of the cores in a binary suspension is responsible for the observed enhancement in conductivity. Oscillatory shear rheology measurements show that the hybrid electrolytes transits from a jammed state to relatively unjammed and back to a jammed state when the fraction of bigger particles in the mixture is increased from 0 to 1. At high nanoparticle volume contents, where the effect of particle size dispersity on conductivity is greatest, little changes are seen in the elastic modulus of the materials. In particular, it is possible to create nanoparticle hybrid electrolytes with MPa mechanical modulus and mS cm−1 level ionic conductivity at room temperature. Preliminary studies of flammability show that even if the electrolyte solvent used in such bidisperse nanoparticle hybrid electrolytes is a high-flammability aprotic liquid, the electrolytes can be formulated to exhibit low flammability. Thus we conclude that nanoparticle hybrid electrolytes created using bidisperse mixtures of PEG-functionalized particles in conventional aprotic liquids provide an attractive platform for tuning multiple properties of contemporary interest for applications.Acknowledgements
This work was supported by the National Science Foundation, Award no DMR-1006323 and by Award no. KUS-C1-018-02, made by King Abdullah University of Science and Technology (KAUST).References
- B. Dunn, H. Kamath and J.-M. Tarascon, Electrical Energy Storage for the Grid: A Battery of Choices, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- P. Yang and J. M. Tarascon, Towards Systems Materials Engineering, Nat. Mater., 2012, 11, 560–563 CrossRef CAS PubMed.
- R. Bouchet, S. Maria, R. Meziane, A. Aboulaich, L. Lienafa, J. Bonnet, T. N. T. Phan, D. Bertin, D. Gigmes and D. Devaux, et al. Efficient Electrolytes for Lithium-Metal Batteries, Nat. Mater., 2013, 12, 452–457 CrossRef CAS PubMed.
- J. M. Tarascon and M. Armand, Issues and Challenges Facing Rechargeable Lithium Batteries, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- M. S. Dresselhaus and I. L. Thomas, Alternative Energy Technologies, Nature, 2001, 414, 332–337 CrossRef CAS PubMed.
- M. Armand and J.-M. Tarascon, Building Better Batteries, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- M. Zhou, T. Cai, F. Pu, H. Chen, Z. Wang, H. Zhang and S. Guan, Graphene/carbon-Coated Si Nanoparticle Hybrids as High-Performance Anode Materials for Li-Ion Batteries, ACS Appl. Mater. Interfaces, 2013, 5, 3449–3455 CAS.
- J. B. Goodenough and Y. Kim, Challenges for Rechargeable Li Batteries, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- S. Srivastava, J. L. Schaefer, Z. Yang, Z. Tu and L. A. Archer, 25th Anniversary Article: Polymer–Particle Composites: Phase Stability and Applications in Electrochemical Energy Storage, Adv. Mater., 2014, 26, 201–234 CrossRef CAS PubMed.
- F. Croce, S. Sacchetti and B. Scrosati, Advanced, Lithium Batteries Based on High-Performance Composite Polymer Electrolytes, J. Power Sources, 2006, 162, 685–689 CrossRef CAS PubMed.
- P. G. Bruce, B. Scrosati and J.-M. Tarascon, Nanomaterials for Rechargeable Lithium Batteries, Angew. Chem., Int. Ed., 2008, 47, 2930–2946 CrossRef CAS PubMed.
- F. Croce, G. B. Appetecchi, L. Persi and B. Scrosati, Nanocomposite Polymer Electrolytes for Lithium Batteries, 1998, 394, 456–458 CAS.
- T. Kashiwagi, F. Du, J. F. Douglas, K. I. Winey, R. H. Harris and J. R. Shields, Nanoparticle Networks Reduce the Flammability of Polymer Nanocomposites, Nat. Mater., 2005, 4, 928–933 CrossRef CAS PubMed.
- C. Tang, K. Hackenberg, Q. Fu, P. M. Ajayan and H. Ardebili, High Ion Conducting Polymer Nanocomposite Electrolytes Using Hybrid Nanofillers, Nano Lett., 2012, 12, 1152–1156 CrossRef CAS PubMed.
- F. Croce and B. Scrosati, Nanocomposite Lithium Ion Conducting Membranes, Ann. N. Y. Acad. Sci., 2003, 984, 194–207 CrossRef CAS PubMed.
- S. Liu, N. Imanishi, T. Zhang, A. Hirano, Y. Takeda, O. Yamamoto and J. Yang, Effect of Nano-Silica Filler in Polymer Electrolyte on Li Dendrite Formation in Li/poly(ethylene oxide)–Li(CF3SO2)2N/Li, J. Power Sources, 2010, 195, 6847–6853 CrossRef CAS PubMed.
- J. L. Schaefer, D. A. Yanga and L. A. Archer, High Lithium Transference Number Electrolytes via Creation of 3-Dimensional, Charged, Nanoporous Networks from Dense Functionalized Nanoparticle Composites, Chem. Mater., 2013, 25, 834–839 CrossRef CAS.
- S. Jung, D. W. Kim, S. D. Lee, M. Cheong and D. Q. Nguyen, Fillers for Solid-State Polymer Electrolytes: Highlight, Bull. Korean Chem. Soc., 2009, 30, 2355–2361 CrossRef CAS.
- C. Monroe and J. Newman, The Effect of Interfacial Deformation on Electrodeposition Kinetics, J. Electrochem. Soc., 2004, 151, A880 CrossRef CAS PubMed.
- C. Monroe and J. Newman, The Impact of Elastic Deformation on Deposition Kinetics at Lithium/Polymer Interfaces, J. Electrochem. Soc., 2005, 152, A396 CrossRef CAS PubMed.
- D. T. Hallinan, S. A. Mullin, G. M. Stone and N. P. Balsara, Lithium Metal Stability in Batteries with Block Copolymer Electrolytes, J. Electrochem. Soc., 2013, 160, A464–A470 CrossRef CAS PubMed.
- M. D. Tikekar, L. A. Archer and D. L. Koch, Stability Analysis of Electrodeposition across a Structured Electrolyte with Immobilized Anions, J. Electrochem. Soc., 2014, 161, A847–A855 CrossRef CAS PubMed.
- Z. Tu, Y. Kambe, Y. Lu and L. A. Archer, Nanoporous Polymer-Ceramic Composite Electrolytes for Lithium Metal Batteries, Adv. Energy Mater., 2014, 4, 1300654 Search PubMed.
- R. Khurana, J. L. Schaefer, L. A. Archer and G. W. Coates, Suppression of Lithium Dendrite Growth Using Cross-Linked Polyethylene/poly(ethylene Oxide) Electrolytes: A New Approach for Practical Lithium–Metal Polymer Batteries, J. Am. Chem. Soc., 2014, 136, 7395–7402 CrossRef CAS PubMed.
- Y. Lu, Z. Tu and L. A. Archer, Stable Lithium Electrodeposition in Liquid and Nanoporous Solid Electrolytes, Nat. Mater., 2014, 13, 961–969 CrossRef CAS PubMed.
- G. M. Stone, S. A. Mullin, A. A. Teran, D. T. Hallinan, A. M. Minor, A. Hexemer and N. P. Balsara, Resolution of the Modulus versus Adhesion Dilemma in Solid Polymer Electrolytes for Rechargeable Lithium Metal Batteries, J. Electrochem. Soc., 2012, 159, A222–A227 CrossRef CAS PubMed.
- I. Gurevitch, R. Buonsanti, A. A. Teran, B. Gludovatz, R. O. Ritchie, J. Cabana and N. P. Balsara, Nanocomposites of Titanium Dioxide and Polystyrene-Poly(ethylene Oxide) Block Copolymer as Solid-State Electrolytes for Lithium Metal Batteries, J. Electrochem. Soc., 2013, 160, A1611–A1617 CrossRef CAS PubMed.
- S. Liu, H. Wang, N. Imanishi, T. Zhang, A. Hirano, Y. Takeda, O. Yamamoto and J. Yang, Effect of Co-Doping Nano-Silica Filler and N-Methyl-N-Propylpiperidinium Bis(trifluoromethanesulfonyl)imide into Polymer Electrolyte on Li Dendrite Formation in Li/poly(ethylene Oxide)-Li(CF3SO2)2N/Li, J. Power Sources, 2011, 196, 7681–7686 CrossRef CAS PubMed.
- G. L. Baker and S. Colsons, Composite Polymer Electrolytes Using Fumed Silica Fillers, Rheology and Ionic Conductivity, 1994, 2359–2363 Search PubMed.
- M. Singh, O. Odusanya, G. M. Wilmes, H. B. Eitouni, E. D. Gomez, A. J. Patel, V. L. Chen, M. J. Park, P. Fragouli and H. Iatrou, et al. Effect of Molecular Weight on the Mechanical and Electrical Properties of Block Copolymer Electrolytes, Macromolecules, 2007, 40, 4578–4585 CrossRef CAS.
- J. E. Weston and B. C. H. Steele, Effects of Inert Fillers on the Mechanical and Electrochemical Properties of Lithium Salt-Poly(Ethylene Oxide) Polymer Electrolytes_I B., Solid State Ionics, 1982, 7, 75–79 CrossRef CAS.
- J. L. Nugent, S. S. Moganty and L. A. Archer, Nanoscale Organic Hybrid Electrolytes, Adv. Mater., 2010, 22, 3677–3680 CrossRef CAS PubMed.
- J. L. Schaefer, S. S. Moganty, D. A. Yanga and L. A. Archer, Nanoporous Hybrid Electrolytes, J. Mater. Chem., 2011, 21, 10094 RSC.
- S. Gowneni, K. Ramanjaneyulu, P. Basak, P. C. Division, C. Technology, N. Institutes, S. Energy and A. Pradesh, Polymer–Nanocomposite Brush-like Architectures as an All-Solid Electrolyte Matrix, ACS Nano, 2014, 8, 11409–11424 CrossRef CAS PubMed.
- P. Agarwal, H. Qi and L. A. Archer, The Ages in a Self-Suspended Nanoparticle Liquid, Nano Lett., 2010, 10, 111–115 CrossRef CAS PubMed.
- S. Choudhury, A. Agrawal, S. A. Kim and L. A. Archer, Self-Suspended Suspensions of Covalently Grafted Hairy Nanoparticles, submitted.
- P. Agarwal, S. Srivastava and L. A. Archer, Thermal Jamming of a Colloidal Glass, Phys. Rev. Lett., 2011, 107, 1–5 Search PubMed.
- P. Agarwal, S. A. Kim and L. A. Archer, Crowded, Confined, and Frustrated: Dynamics of Molecules Tethered to Nanoparticles, Phys. Rev. Lett., 2012, 109, 258301 CrossRef.
- C. Mayer, E. Zaccarelli, E. Stiakakis, C. N. Likos, F. Sciortino, A. Munam, M. Gauthier, N. Hadjichristidis, H. Iatrou and P. Tartaglia, et al. Asymmetric Caging in Soft Colloidal Mixtures, Nat. Mater., 2008, 7, 780–784 CrossRef CAS PubMed.
- E. Zaccarelli, C. Mayer, A. Asteriadi, C. Likos, F. Sciortino, J. Roovers, H. Iatrou, N. Hadjichristidis, P. Tartaglia and H. Löwen, et al. Tailoring the Flow of Soft Glasses by Soft Additives, Phys. Rev. Lett., 2005, 95, 268301 CrossRef CAS.
- T. Voigtmann, Multiple Glasses in Asymmetric Binary Hard Spheres, Europhys. Lett., 2011, 96, 36006 CrossRef.
- Agrawal, A.; Yu, H.; Srivastava, S.; Narayanan, S.; Archer, L. A. Jamming and Un-Jamming in Binary Soft Colloids, submitted.
- Q. Zhang and L. A. Archer, Poly(ethylene oxide)/Silica Nanocomposites: Structure and Rheology, Langmuir, 2002, 18, 10435–10442 CrossRef CAS.
- S. S. Moganty, N. Jayaprakash, J. L. Nugent, J. Shen and L. A. Archer, Ionic-Liquid-Tethered Nanoparticles: Hybrid Electrolytes, Angew. Chem., 2010, 122, 9344–9347 CrossRef.
- A. K. Jonscher, The “Universal” Dielectric Response, Nature, 1977, 267, 673–679 CrossRef CAS.
- O. Glatter and O. Kratky, Small angle X-ray scattering, Academic Press, New York, USA, 1982 Search PubMed.
- S. Srivastava, P. Agarwal and L. A. Archer, Tethered Nanoparticle-Polymer Composites: Phase Stability and Curvature, Langmuir, 2012, 28, 6276–6281 CrossRef CAS PubMed.
- M. Duclot and M. Levy, Salt-Polymer Complexes: Strong or Weak Electrolytes?, 1996, vol. 85, pp. 149–157 Search PubMed.
- D. H. C. Wong, J. L. Thelen, Y. Fu, D. Devaux, A. A. Pandya, V. S. Battaglia, N. P. Balsara and J. M. DeSimone, Nonflammable Perfluoropolyether-Based Electrolytes for Lithium Batteries, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 3327–3331 CrossRef CAS PubMed.
- T. Sentjabrskaja, E. Babaliari, J. Hendricks, M. Laurati, G. Petekidis and S. U. Egelhaaf, Yielding of Binary Colloidal Glasses, Soft Matter, 2013, 9, 4524 RSC.
- T. Sentjabrskaja, M. Hermes, W. C. K. Poon, C. D. Estrada, R. Castaneda-Priego, S. U. Egelhaaf and M. Laurati, Transient Dynamics during Stress Overshoots in Binary Colloidal Glasses, Soft Matter, 2014, 10, 6546–6555 RSC.
- P. C. Carman, Flow of Gases through Porous Media, New York Acad. Press, 1956 Search PubMed.
- R. Bouchet, T. N. T. Phan, E. Beaudoin, D. Devaux, P. Davidson, D. Bertin and R. Denoyel, Charge transport in nanostructured PS–PEO–PS triblock copolymer electrolytes Macromolecules, 2014, 47, 2659–2665 Search PubMed.
- E. Then, The Tortuosity Concept in Fixed and Fluidized Bed, Chem. Eng. Sci., 1993, 48, 2173–2175 CrossRef.
- S. Khirevich, A. Höltzel, A. Daneyko, A. Seidel-Morgenstern and U. Tallarek, Structure-Transport Correlation for the Diffusive Tortuosity of Bulk, Monodisperse, Random Sphere Packings, J. Chromatogr. A, 2011, 1218, 6489–6497 CrossRef CAS PubMed.
- R. Bouchet and R. Denoyel, Influence of Molecule Size on Its Transport, Anal. Chem., 2010, 82, 2668–2679 CrossRef PubMed.
- I. V. Thorat, D. E. Stephenson, N. A. Zacharias, K. Zaghib, J. N. Harb and D. R. Wheeler, Quantifying Tortuosity in Porous Li-Ion Battery Materials, J. Power Sources, 2009, 188, 592–600 CrossRef CAS PubMed.
- W. J. Frith, T. A. Strivens and J. Mewis, Dynamic Mechanical Properties of Polymerically Stabilized Dispersions, J. Colloid Interface Sci., 1990, 139, 55–62 CrossRef CAS.
- R. G. Larson, The Structure and Rheology of Complex Fluids, New York- Oxford University Press, 1999 Search PubMed.
- P. Sollich, F. Lequeux, P. Hébraud and M. Cates, Rheology of Soft Glassy Materials, Phys. Rev. Lett., 1997, 78, 2020–2023 CrossRef CAS.
- P. Sollich, Rheological Constitutive Equation for a Model of Soft Glassy Materials, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1998, 58, 738–759 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra01031d |
| ‡ These authors contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2015 |