
Optical, electrochemical, third order nonlinear optical, and excited state dynamics studies of bis(3,5-trifluoromethyl)phenyl-zinc phthalocyanine†
Abstract
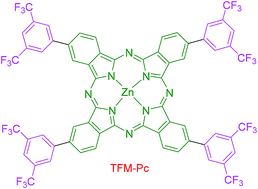
A zinc phthalocyanine with bis(3,5-trifluoromethyl)phenyl groups at peripheral positions has been synthesized and its optical, emission, electrochemical and third-order nonlinear optical properties were investigated. Both the Soret and Q bands were red-shifted and obeyed Beer–Lambert's law. Electrochemical properties indicated that both oxidation and reduction processes were ring centred. Emission spectra were recorded in different solvents and the fluorescence yields obtained were in the range of 0.29 to 0.10, while the time-resolved fluorescence data revealed lifetimes of typically a few ns. Nonlinear optical coefficients were retrieved from the Z-scan measurements using picosecond and femtosecond pulses. Excited state lifetime was deduced from the femtosecond degenerate pump–probe measurements. Our data suggests that this compound has potential for applications in the field of photonics.
 Please wait while we load your content...
Please wait while we load your content...

