Targeted drugs by olefin metathesis: piperidine-based iminosugars
Ileana
Dragutan
*a,
Valerian
Dragutan
a and
Albert
Demonceau
b
aInstitute of Organic Chemistry, Romanian Academy, 202B Spl. Independentei, P.O. Box 35-108, Bucharest 060023, Romania. E-mail: idragutan@yahoo.com
bMacromolecular Chemistry and Organic Catalysis, Institute of Chemistry (B6a), University of Liège, Sart Tilman, Liège 4000, Belgium
First published on 30th November 2011
Abstract
Examining advances in the class of natural and non-natural piperidine azasugars, important therapeutic agents and potent glycosidase inhibitors, this review looks at syntheses applying olefin metathesis as a highly efficient key step and gateway strategy for discovery of better iminosugar leads for treatment of widespread affections like viral and metabolic diseases. Amply illustrated is how ring-closing metathesis (RCM and RCEYM), promoted by commercial ruthenium alkylidene catalysts, manage to construct the common tetrahydropyridine core while cross-metathesis (CM), starting from this generic scaffold, provides general access to families of novel azasugars. Special consideration is given to high-profile iminosugar drugs of this class (1-deoxynojirimycin and congeners, adenophorine, fagomine, isofagomine and some of their N- and C-substituted analogues) stressing upon newest trends for enhancing biological activity and modulating previously unexploited targets in multispecific therapies.
1. Introduction
Polyhydroxylated piperidines constitute a successful group within the vast class of azasugars (also named iminosugars or iminocyclitols), the most attractive carbohydrate mimetics reported to date. Structurally diverse azasugars are frequently encountered in nature, in various wild plants or microorganisms. As is the case for all iminocyclitols, in polyhydroxylated piperidines an imino group is replacing the endocyclic oxygen of the parent sugar. Natural or synthetically produced 6-membered iminocyclitols effectively inhibit oligosaccharide processing enzymes, especially glycosidases and glycosyltransferases involved in a whole range of essential biological transformations.1 They also exert a profound effect on cellular processes such as N-linked glycoprotein processing and maturation, and cell-cell and cell-virus recognition.2 Specifically, polyhydroxylated piperidines inhibit exoglycosidases active in glycoprotein formation and catalysis, and not endoglycosidases.3 Due to their biological attributes, azasugars have gained clinical importance in the treatment of cancer,4 type II diabetes,5 viral diseases such as HIV, hepatitis B and C,6–9 Gaucher's disease,10,11 and other glycosphingolipid storage disorders12etc. These compounds are as well of interest in studying enzyme mechanisms in relation to metabolic diseases.In particular Hepatitis C received a lot of attention this year in the context of introduction of new drugs (Boceprevir, Telaprevir) that, by directly targeting hepatitis C virus (HCV) as protease inhibitors, are expected to improve the cure rates.13 However, the new drugs must be used in combination with the existing standard of care (dual: general antivirals plus immune-boosting proteins) because the virus can easily overcome a single direct-acting antiviral. A cure for all, well over a hundred million people infected with HCV, will take generations of therapies and combinations of antivirals with diverse modes of action so that the virus does not become resistant. Iminocyclitols may play an important role in such drug cocktails, like already proved for animals where several polyhydroxylated piperidine azasugars, added to the dual current therapy, managed to clear the virus.14
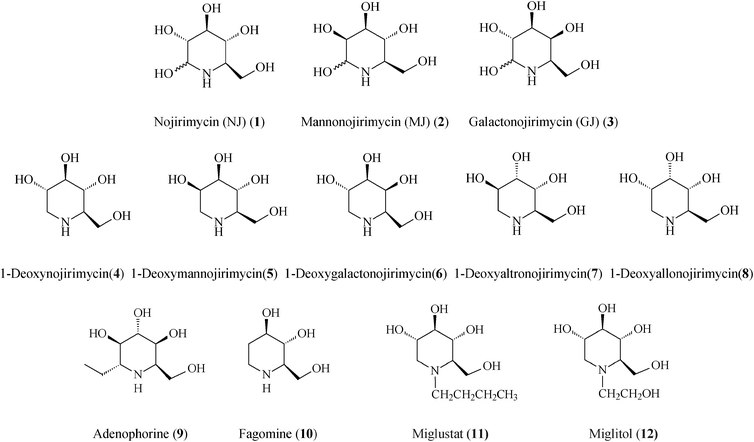
Of the piperidine-based azasugars class (Scheme 1), two members, N-butyl-1-deoxynojirimycin (Miglustat, 11) and N-hydroxyethyl-1-deoxynojirimycin (Miglitol, 12; FDA approved), have presently reached the market as drugs against, respectively, Gaucher's disease (the most common glycosphingolipid lysosomal storage disorder) and type II diabetes (diabetes mellitus).7 The primary pharmacological activity of Miglustat is inhibition of the enzyme glucosylceramide synthase, catalyzing the first step in the biosynthesis of glycosphingolipids, i.e. the formation of glucosylceramide. Therefore, the treatment of Gaucher's disease with Miglustat is based on the concept of substrate reduction therapy. In the body Miglustat exhibits a large volume of distribution having the capacity to access organs located deep within, however, it may only be used in the treatment of type 1 Gaucher patients for whom enzyme replacement therapy is unsuitable.15 Early studies have shown Miglustat to be also active against the HIV infection. The second commercial drug, Miglitol, is an oral antidiabetic drug which acts by inhibiting alpha-glucosidases (glycoside hydrolysis enzymes), and consequently the ability of the body to breakdown complex carbohydrates into glucose.
 | ||
| Scheme 1 Representative piperidine-based iminosugars. | ||
Generally, azasugars possess several relevant points of diversity: stereogenic diversity (configuration of hydroxyl groups), substituent diversity, and size diversity of the N-containing ring. In piperidine iminocyclitols, which have as signature core a 6-membered N-heterocycle, structural features such as the number, position and steric configuration of the hydroxyl groups determine the type of glycosidases that are inhibited. While the spatial arrangement of OH groups in polyhydroxylated piperidines serves for recognition by specific glycosidases, the endocyclic N-atom also plays an important role in the inhibition by affecting the conformation and electrostatic properties. All of these considerations pose considerable challenges for bottom-up design of synthetic strategies. Pathways amenable to produce libraries of iminocyclitols are eagerly sought after, since often even slight structural or stereochemical modifications can alter the degree of inhibition. Presently, many stereoselective syntheses of polyhydroxylated piperidines start from carbohydrates that are not only abundant sources but also provide the required chirality. The high density of the functional groups demands additional protection/deprotection steps, masterfully conducted so as to ensure the preordained chemoselectivity and stereochemistry.
Following the tremendous recent success of olefin metathesis in the rapidly growing field of natural product synthesis,16 this type of C–C forming reaction, especially the ruthenium-catalyzed ring-closing metathesis (RCM) and cross-metathesis (CM), has been increasingly used to refine and render more productive synthetic protocols for iminosugars.2,17 Mimicking nature which, from simple starting materials creates generic precursors of families of complex molecules, metathesis-based strategies often use a common molecular scaffold to access a whole range of iminosugars, just by playing on reaction conditions in post-metathesis transformations. Irrespective of the starting material, tactical approaches to polyhydroxylated piperidines use RCM to form the C3–C4 bond, from a prerequisite diene precursor, thus creating the essential 1,2,5,6-tetrahydropyridine building block. The other consequential stage in the overall synthesis is the diastereoselective dihydroxylation (or epoxidation) of the endocyclic double bond to yield the target product(s). From the vast library of piperidine azasugars existing to date, this review highlights the most promising representatives endowed with therapeutic activity that have been lately prepared by metathetical routes (Scheme 1).
2. 1-Deoxynojirimycin and analogues
Nojirimycin (NJ, 5-amino-5-deoxy-d-glucopyranose) (1), the first discovered natural D-glucose mimic is an antibiotic produced by Streptomyces strains, and also found in other bacterial cultures and plant sources. Since the configuration of the hydroxyl substituents in nojirimycin corresponds to that of glucose in the pyranose form, this azasugar is a powerful glucosidase inhibitor, preventing normal glycosylation processes. The α- and β-forms of nojirimycin existing in equilibrium in aqueous solution (60%:40%) are responsible for inhibition of the α- and β-glucosidase, respectively. However, intervention of hemiacetal (hemiaminal) structures (i.e. a labile N,O-acetal function, see Scheme 2) renders nojirimycin and the related mannonojirimycin (2, MJ or nojirimycin B) and galactonojirimycin (3, GJ or galactostatin) unstable, making them unsuitable for use as drugs or in other biological media.7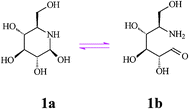 | ||
| Scheme 2 Hemiacetal structures of nojirimycin. | ||
To circumvent the lability problem, researchers thought of replacing the azasugar's pseudo-anomeric centre, CHOH, for a methylene group and thus create the stable 1-deoxy family, i.e.1-deoxynojirymicin (4, DNJ, 2R,3R,4R,5S)-2-hydroxymethyl-3,4,5-piperidinetriol) and analogs that, by better mimicking the target glycosides have enhanced inhibitory action. While nojirimycin (1) and 1-deoxynojirimycin (4) inhibit glucosidases, galactonojirimycin (3) and 1-deoxygalactonojirimycin (6) inhibit galactosidases, and 1-deoxygulonojirimycin inhibits fucosidases.18L-Enantiomers of DNJ, manno-DNJ, allo-DNJ, altro-DNJ, and gulo-DNJ occur naturally. One of DNJ's isomers, 1-deoxymannonojirimycin (5, DMJ), was found in Streptomyces lavandulaeSF-425 and Adenophora triphylla but 1-deoxygalactonojirimycin (6, DGJ) has not yet been obtained from natural sources.
Introduction of structural modifications on DNJ, in terms of epimerization or C- or N-substitution opens up possibilities for identification of novel azasugars with added therapeutic activities. Indeed, N-alkylated DNJ-s have found applications as antidiabetic drugs (Miglitol, Glyset; non-insulin dependent diabetes) or anti-HIV agents (Glycovir, SC 49483).19N-butyl DNJ (Miglustat, Zavesca) and N-nonyl DNJ) have been shown to also inhibit the hepatitis B virus, with the N-nonyl compound being considerably more potent.20
Many 1-deoxy iminosugars (e.g.5–8) having 1-deoxynojirimycin as a privileged motif have been prepared during the last few years by a variety of methodologies, either chemical21,22 or enzymatic. However, many of these, employing sugars or amino acids as chiral synthons, suffer from a lack of selectivity and a more general applicability.23
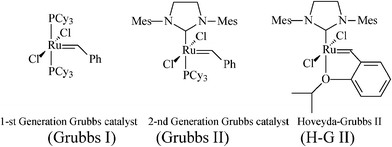
In this context, aiming at a more flexible and atom-economical access to piperidine iminosugars, synthetic chemists resorted to olefin metathesis—in time-correlation with the rapid developments in metathesis catalytic systems with improved efficiency and stability.24,25Metathesis-based approaches to polyhydroxylated piperidines imply as main steps the stereoselective synthesis of an N-containing diene precursor, its ring-closure to the tetrahydropyridine generic building block, and the introduction of additional OH substituents to finally attain, by diastereoselective dihydroxylation,26 the required number of hydroxy groups. Success in achieving the asymmetric total synthesis of piperidine azasugars featuring enantiopurity and defined stereochemistry greatly depends on the careful manipulation of all stereoselective pathways, as well as on the optimization of the RCM step by selecting an appropriate catalyst (ruthenium alkylidene catalysts shown in Scheme 3 have been mostly used). Moreover, before carrying out functionality-sensitive reactions, proper protection must be ensured. While the protection of OH groups is encountered in all protocols for iminocyclitols syntheses, RCM requires further attention to the basic amino group, incompatible with many metathesis catalysts.27 The NH group must be either temporarily protected (as N-Boc, N-Cbzetc.), masked by incorporation into a cleavable heteroatom-containing cycle (oxazolidine, cyclic ketalsetc.) or deactivated via conversion into stable ammonium salts,28amide or carbamate.
 | ||
| Scheme 3 Ru catalysts applied in RCM-based syntheses of piperidine iminosugars. | ||
Progress in the RCM strategy to polyhydroxylated piperidine greatly benefited from abundant work on the synthesis, involving metathesis, of N-heterocycles27,29 and of five-membered azasugars.17,30 Furthermore, in obtaining hydroxylated azacycles, a successful merger of the two crucial steps, metathesis and cis-dihydroxylation, into a one-pot single operation could be accomplished.31
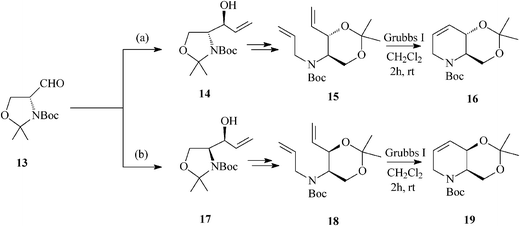
For constructing the piperidine ring of 1-deoxynojirimycin (4) and its congeners (5–8) by RCM, starting from D-serine-derived Garner's aldehyde 13, Takahata32 devised a scheme amenable to obtaining either enantiomeric protected terahydropyridine core (16 or 19) (Scheme 4) from which 1,2-trans- (4, 5, 7 and 8) (Schemes 5 and 6)32a and 1,2-cis-substituted (6, 25 and 27) azasugars (Scheme 7)32b have been prepared.
 | ||
| Scheme 4 Enantiomeric tetrahydropyridine cores 16 and 19 for the synthesis of 1-deoxynojirimycin and congeners. | ||
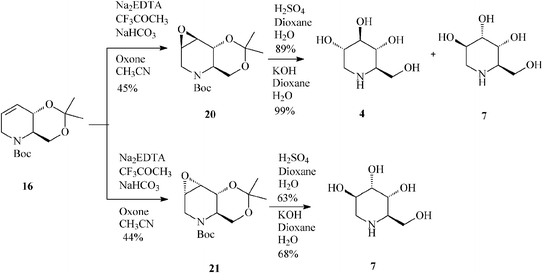 | ||
| Scheme 5 Synthesis of 1-deoxynojirimycin (4) and 1-deoxyaltronojirimycin (7) from the tetrahydropyridine core 16. | ||
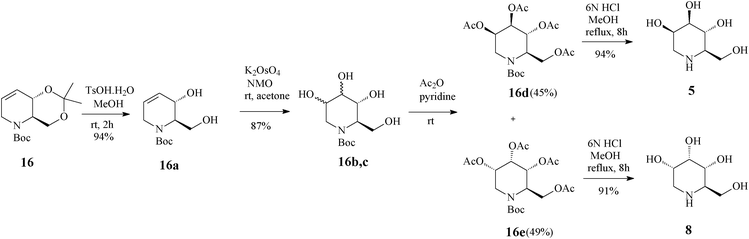 | ||
| Scheme 6 Access to 1-deoxymannonojirimycin (5) and 1-deoxyallonojirimycin (8) starting from the tetrahydropyridine core 16. | ||
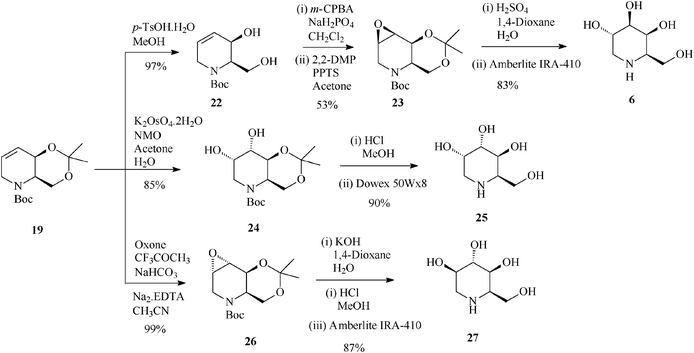 | ||
| Scheme 7 Stereoselective synthesis of 1-deoxygalactonojirimycin (6) and its congeners (25 and 27) from the protected tetrahydropyridine 19. | ||
Unfortunately, epoxidation of 16 occurred with low stereoselectivity,32a either when using oxone (45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :44 anti:syn-epoxide, 20:21)(Scheme 5) or m-CPBA (47:27 anti:syn-epoxide, 20:21) imposing separation of isomers by column chromatography.
:44 anti:syn-epoxide, 20:21)(Scheme 5) or m-CPBA (47:27 anti:syn-epoxide, 20:21) imposing separation of isomers by column chromatography.
It is worth mentioning that 19 (2,3-cis) undergoes epoxidation with much higher stereoselectivity (99% antiepoxide 26; Scheme 7) than its trans isomer 16, and so does the 19-derived diol 22, this time owing to a hydroxy-directed formation of the epoxide (synepoxide 23, Scheme 7).32b
Entirely differing from the procedure based on Garner's aldehyde illustrated in Schemes 4 and 6, preparation of 1-deoxymannojirimycin (5) alternatively succeeded from the bicyclic carbamate (−)-31 (Schemes 8, 9).33 This key intermediate was accessible starting with the 2,3-epoxy-4-penten-1-ol, (+)-28, in its turn obtained with high enantiomeric purity through Sharpless epoxidation of (E)-2,4-pentadien-1-ol. Later on in the synthesis came the RCM leitmotif that, applied on the diene (−)-30 (5% mol Grubbs I catalyst; CH2Cl2, rt), proceeded almost quantitatively (98% yield) to (−)-31.
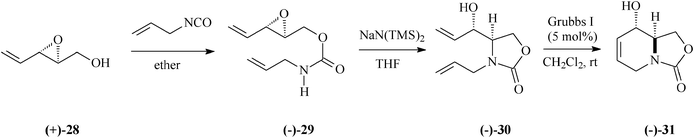 | ||
| Scheme 8 Preparation of the bicyclic carbamate (−)-31. | ||
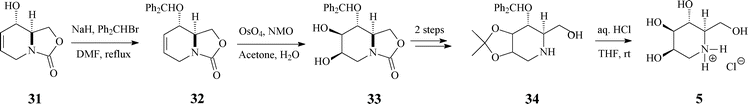 | ||
| Scheme 9 Synthesis of 1-deoxymannojirimycin (5.HCl) from the bicyclic carbamate 31. | ||
The next reaction sequence, involving standard dihydroxylation and OH protection/deprotection (Scheme 9), conducted to the targeted 1-deoxymannojirimycin (5) in 61% yield (based on (−)-31).
The strength of the concept in Scheme 8 is demonstrated through the expedient access (also from 31) to another valuable azasugar, the bicyclic swainsonine. However, as the stereogenic centers in the piperidine ring of swainsonine have the opposite configuration of those in mannojirimycin, the synthesis required starting from the enantiomer, (+)-31, prepared from (−)-28 by the same reaction sequence as in Scheme 8. Quite conveniently, (−)-28 is also readily available by Sharpless epoxidation as the source of chirality, simply by changing the configuration of the tartrate in the epoxidation step.
Considering the focus of this review, it is of particular interest that Riera et al. also followed recent trends for “greener” metathesis protocols by carrying out the ring-closing step (Scheme 8) in supercritical CO2 (scCO2), a medium with intrinsic eco-compatibility and unique physicochemical properties. Supercritical CO2 was first introduced by Fürstner and Leitner34 to metathesis reactions promoted by Grubbs catalysts and was later embraced by other researchers active in this field.35 As the Grubbs I and II catalysts behave like heterogeneous catalysts in scCO2, this medium provides convenient workup procedures while also acting as a protective environment for basic amine functions. In the hands of Riera and coworkers, RCM of (−)30 to (−)-31 (200 bar; 40 °C) rewardingly provided an 88% yield with only 2 mol% of the Grubbs I catalyst.
A fine recent addition to the previously reported synthesis of (+)-1-deoxynojirimycin (4)(Schemes 4, 5) is the elegant total synthesis disclosed by Poisson36 as proceeding through a one-pot protocol (24% overall yield) that implies tandem enol-ether RCM/hydroboration/oxidation (Scheme 10). The synthesis started from a non-chiral source, the diallyl alcohol 35, and proceeded with full stereocontrol over 11 steps. The RCM precursor, the diene 38, resulted from the asymmetric epoxidation of penta-1,4-dien-3-ol (35), Payne rearrangement to the enantiopure alcohol 36 (ee >99%), base-induced epoxide opening and benzoyl migration to give the oxazolidinone 37, its conversion to tert-butyl carbamate, concomitant cleavage of the oxazolidinone and benzoate moieties, protection of the resulted diol, and N-alkylation with 3-iodo-2-(O-methoxymethyl)prop-1-ene. In the presence of the Grubbs II catalyst (10 mol%) and benzoquinone (10 mol%) in refluxing toluene, 38 was converted into the cyclized enol ether 39 in 70% yield while the Hoveyda–Grubbs II catalyst afforded 39 in still better yield (85%). The enol ether 39 undergoes facially selective hydroboration/oxidation to produce the desired trans-diol derivative 40. Remarkably, the three chemical transformations RCM/hydroboration/oxidation (38 to 40) could be performed in a one-pot procedure to give a single isomeric product in 70% overall yield. Finally, simultaneous removal of the three protecting groups in 40 quantitatively afforded (+)-1-deoxynojirimycin (4).
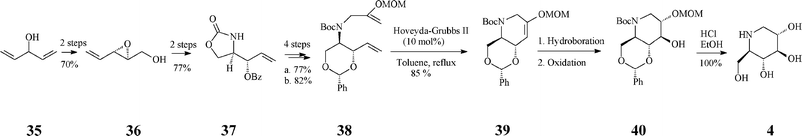 | ||
| Scheme 10 One-pot synthesis of 1-deoxynojirimycin 4. | ||
As opposed to the behaviour of 38, another potentially useful diene precursor resulting from a N-alkylation with 3-iodo-2-(OMOM)prop-1-ene directly on 37, escaped all attempts to achieve metathesis leading instead to either its recovery or diene isomerization.
Of particular interest is the asymmetric synthesis of an array of deoxyiminosugars (1-deoxymannonojirimycin 5, 1-deoxyaltronojirimycin 7, 1-deoxygulonojirimycin 41, 1-deoxyidonojirimycin 42). The divergent strategy assembled by Singh and Han37 takes advantage of the special features displayed by the achiral olefin 43 employed as starting material. Indeed, olefin 43 is prone to interact, electronically and by aryl–aryl stacking, with the regioselective osmium catalyst during asymmetric aminohydroxylation (regioselectivity >20:1; enantioselectivity >99% ee) eventually leading to the protected diene 44, the RCM precursor. Ring-closing metathesis promoted by the Grubbs I catalyst (10 mol%, toluene, 90 °C, 2 h) is showcased to efficiently produce the chiral tetrahydropyridine (45) (80% yield). The common framework 45 is then exploited to afford, via diastereoselective dihydroxylation and protection/deprotection, two of the looked for azasugars, 1-deoxymannonojirimycin (5) and 1-deoxygulonojirimycin (41). For the other two iminosugars, 1-deoxyaltronojirimycin (7) and 1-deoxyidonojirimycin (42), Singh and Han conceived an alternative pathway involving the cyclic sulphate 47, in turn obtained from 46 (diol mixture), the product of non-selective dihydroxylation and protection of 45 (Scheme 11, for 5 and 7).
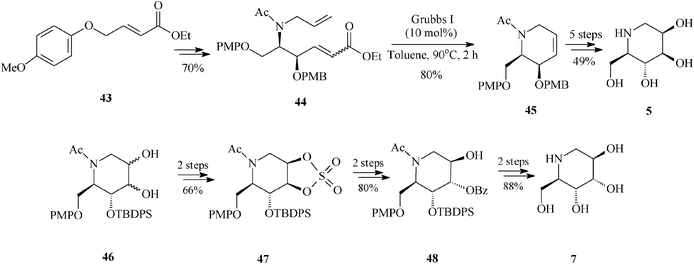 | ||
| Scheme 11 1-Deoxyiminosugars 5 and 7 as obtained by an RCM route. | ||
At about the same time with the robust entry to L-1-deoxyaltronojirimycin (7) published by Singh and Han, a quite different and shorter access to this iminocyclitol, starting now from L-serine, has been advanced by Park.38 In stark contrast with the Singh and Han synthesis, in this case the RCM-derived terahydropyridine 50 has a different configuration at positions 2,3 (Scheme 12) (vs. the parent 45, Scheme 11). The chiral diene 49 (four steps away from L-serine and obtained with high stereoselectivity) was subjected to RCM promoted by Grubbs II to produce an isomeric mixture (85:15) from which the desired chiral tetrahydropyridine 50 was separated chromatographically. Selective deprotection (Bu4NF) of 50 to 51 was followed by a H-bond directed epoxidation of this allylic alcohol proceeding exclusively syn to give 52. Regioselective oxirane ring-opening in 52, setting up the 4,5-antidiol motif, finally led to the targeted L-1-deoxyaltronojirimycin (7). Thus, this synthetic strategy manages to install four contiguous stereocentres in four overall steps, by three totally stereoselective transformations.
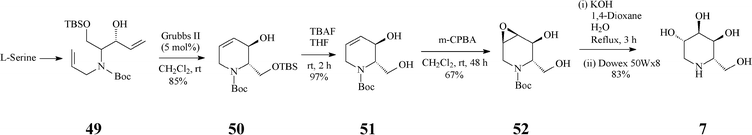 | ||
| Scheme 12 Synthesis of 1-deoxyaltronojirimycin (7) starting from L-serine. | ||
The much higher stereocontrol observed in epoxidation of 51 (Scheme 12), vs. that of the corresponding protected form 16 (Scheme 4),32a was rationalized assuming that H-bonding in 51 has a much more powerful effect than steric factors intervening in 16.38
In fact, the intermediate 51 (or its protected form 16), a crucial synthon in the synthesis of many polyhydroxylated piperidine iminosugars (L-manno-DNJ, L-allo-DNJ, L -altro-DNJ etc.), has been synthesized also by other routes, e.g. by a less stereoselective one starting from Garner's aldehyde (Scheme 4) or by another, highly diastereo- and enantioselective one where it resulted from the unprecedented RCM of a tert-butylsulfinyl allylamine (53) (Scheme 13).39
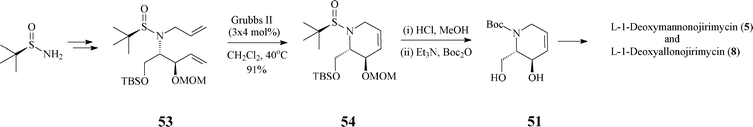 | ||
| Scheme 13 Alternative synthesis of 51 . | ||
A monosaccharide-based, stereochemically worked out pathway aiming at both the synthesis of D-1-deoxygulonojirimycin (41) and of L-1-deoxyallonojirimycin (L-8) emerged from the group of Ghosh40 (Scheme 14). Though they start from the same protected diacetone glucose derivative 55, the epimeric RCM precursors 57 and 60 are obtained via different kinds and number of reaction steps, first in the row being an inversion of the free hydroxyl group of 55 (to 56) or its transformation into an azide (59), respectively. The key ring-closing reaction of dienes 57 and 60 (Grubbs I catalyst, 10 mol%, CH2Cl2, under argon, 24 h at 50 °C; 90% yield) led to 58 and 61, with preserved configurations at the stereogenic centre that ensure the desired stereochemistry in the final products 41 and L-8.
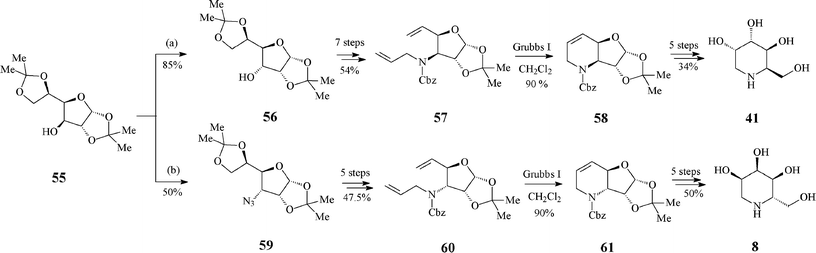 | ||
| Scheme 14 Synthesis of 1-deoxyiminosugars 41 and L-8 from protected diacetone glucose. | ||
It should be noted that an earlier stereoselective synthesis 1-deoxygulonojirimycin (41),41 employing D-serine as a chiral starting material, managed to build the RCM precursor 63via the trans-oxazolidine 62 (Scheme 15). RCM of 63 with the Grubbs I catalyst (5 mol%, CH2Cl2, 9 h at room temperature; 96% yield) afforded the tetrahydropyridine synthon 64 which, upon stereoselective dihydroxylation (OsO4/NMO), gave 65 as a single isomer. Full deprotection (75% yield) led to (2R,3S,4S,5S)-1-deoxygulonojirimycin, found to be identical with the natural compound apart from possessing the opposite sign of optical rotation. Remarkably, this finding enabled the unambiguous demonstration of the absolute configuration of the naturally occurring 1-deoxygulonojirimycin as being 2S,3R,4R,5R (Scheme 16).
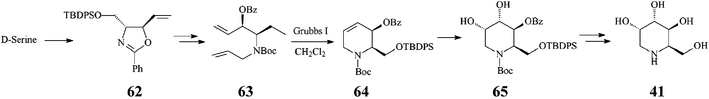 | ||
| Scheme 15 Synthesis 1-deoxygulonojirimycin (41) starting from D-serine. | ||
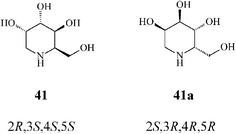 | ||
| Scheme 16 Synthetic (41) and natural 1-deoxygulonojirimycin (41a). | ||
The 2S,3R,4R,5R configuration was confirmed quite recently, through NMR techniques, in a refined synthesis protocol starting from D-mannitol.42 The RCM-derived synthon (70), stepwise constructed from the azide 67, acts as a pivotal intermediate prone to provide access to a variety of mono- and bicyclic azasugars (e.g. trihydroxylated indolizidinone), through suitable functionalisation of the double bond and manipulation of the protecting groups (Scheme 17).
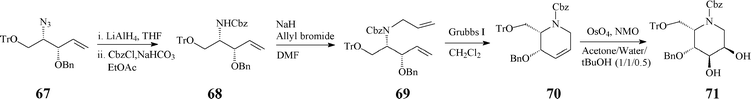 | ||
| Scheme 17 Synthesis involving RCM of the versatile synthon 70. | ||
Endeavours for development of new piperidine azasugars were also directed toward congeners containing only three hydroxyl groups (Schemes 18, 19), as in 5-des(hydroxymethyl)-1-deoxynojirimycin (79) and the corresponding mannose analog 77. Syntheses of these two compounds share the common intermediate 76 and have as starting point another easily available olefin, 72 (Scheme 18).43 The recurring RCM theme is found here in the transformation of 74 to 75. Interestingly, although the Grubbs II catalyst was quite productive (89% yield, in CH2Cl2), all attempts to employ the Grubbs I catalyst unexpectedly failed, supposedly because of an unfavourable steric environment during generation of the Ru carbene species from 74, as compared to 44 (Scheme 11; distinct N-protective groups). While the classical cis-dihydroxylation of the RCM product 75 opened a facile access to 77 (2,3-cis), for the case of its stereoisomer 79 (2,3-trans) the cyclic sulphate motif illustrated in Scheme 11 was again invoked to provide a useful synthetic handle. Trihydroxylated piperidines 77 and 79 are not only potent glycosidase inhibitors but also suitable intermediates for further, differently substituted iminocyclitols.
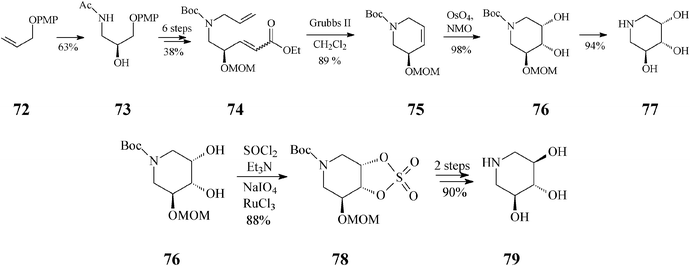 | ||
| Scheme 18 Synthesis of 5-des(hydroxymethyl)-1-deoxyiminosugars by the RCM route induced by the Grubbs II catalyst. | ||
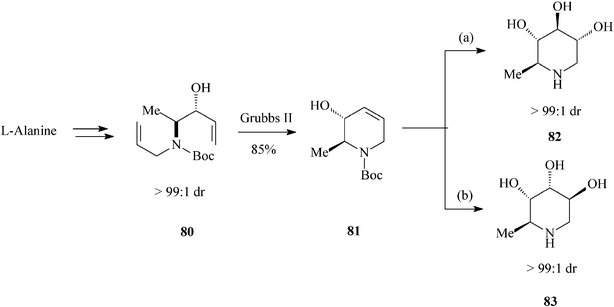 | ||
| Scheme 19 Stereoselective synthesis of dideoxyiminosugars 82 and 83. | ||
Looking further afield, Park et al.,44 in valuable investigations on the synthesis of ent-1,6-dideoxynojirimycin (82) and 5-amino-1,5,6-trideoxyaltrose (83) (Scheme 19), appealed to another chiral natural compound, the enantiopure amino acidL-alanine. Then, to prepare their starting material 80 (single diastereomer; (3R, 4S)-3-hydroxy-4-N-allyl-N-Boc-amino-1-pentene) they resorted to a vinyl Grignard addition to the N-Boc-N-allyl aminoaldehyde derived from the methyl ester of L-alanine. Diene 80 submitted to RCM (Grubbs II catalyst; 85% yield) produced the tetrahydropyridine core 81. Stereodivergent dihydroxylation of the latter resulted in the targeted hydroxylated piperidines, via a common epoxide intermediate and with excellent diastereoselectivity (> 99:1 dr), i.e.82 (ent-1,6-dDNJ) (28% overall yield from N-Boc-L-alanine methyl ester) and 83 (29% overall yield from N-Boc-L-alanine methyl ester). It is worth pointing out that this total synthesis of ent-1,6-dDNJ (82) is the most advantageous reported so far.
Recent findings by Vankar45 expand the scope of piperidine azasugars by elaborating on other points of diversity of this class, a larger number of OH groups and the substitution pattern, jointly expected to affect the biological activity. Thus, the high yielding syntheses of 1,4-dideoxy-1,4-iminoheptitols and 1,5-dideoxy-1,5-iminooctitols were devised to start from the inexpensive D-xylose. Exemplified here is the case of 1,5-dideoxy-1,5-iminooctitols (Scheme 20) where the RCM substrate 84 was obtained starting from the above mentioned chiral material and proceeded through a skilfully conducted sequence of seven isolated steps. As expected, ring-closing of diene 84 to the tetrahydropyridine building block 85 occurred smoothly and in good yield (88%) when promoted by the Grubbs II catalyst (3 mol%; in refluxing dichloromethane). Next in the row came the dihydroxylation step (OsO4, NMO) of 85, followed by OH-protection as acetonides using 2,2-dimethoxypropane (DMP) and p-toluenesulfonic acid in acetone. Noteworthy, dihydroxylation of this framework was not stereoselective leading to a mixture of diastereomers, isolated as a 1:1 mixture of the acetonides 86 and 87 that both serve quite well the purpose of the synthesis. Indeed, hydrogenolysis of 86 (10% Pd(OH)2/C) followed by acetylation and global deprotection with aq HCl afforded 1,5-dideoxy-1,5-imino-D-threo-L-galacto-octitol (88) while 1,5-dideoxy-1,5-imino-D-threo-L-allo-octitol (89) could be similarly obtained from 87. Iminoalditols 88 and 89 showed disappointingly low glycosidase inhibition activity at millimolar concentrations, although 88 did specifically inhibit α-galactosidase.
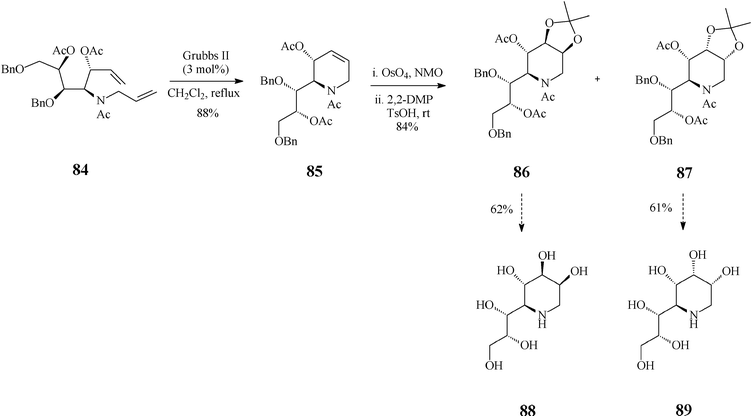 | ||
| Scheme 20 Synthesis of 1,5-dideoxy-1,5-iminooctitols 88 and 89. | ||
To further diversify the applications of the metathesis-based methodology in synthesis of azasugars, Overkleeft,46 exploring the large chiral pool of potentially useful starting materials, imaginatively selected a single chiral cyanohydrin, 90, available via a chemoenzymatic approach involving almond hydroxynitrile lyase (paHNL). This cyanohydrin is at the base of a quite attractive synthesis of three 1-deoxynojirimycin-related azasugars, L-1-deoxyaltronojirimycin (7), D-1-deoxyallonojirimycin (8), and D-1-deoxygalactonojirimycin (6), in which the key steps consist of the cascade Dibal reduction/transimination/NaBH4 reduction effected on the enantiomerically pure 90, followed by the crucial, productive RCM stage (CH2Cl2, 3.5 mol% Grubbs I, reflux under Ar for 48 h) and Upjohn dihydroxylation (Scheme 21 for 7).
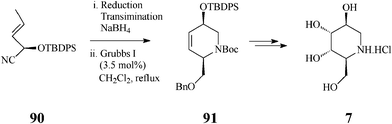 | ||
| Scheme 21 Synthesis of L-1-deoxyaltronojirimycin (7). | ||
3. Adenophorine
Adenophorine (α-1-deoxy-1-C-methyl-homonojirimycin, trans-6-ethyl-2-hydroxymethyl-1,2,5,6-tetrahydropyridine) is a further important, naturally occurring, member of the polyhydroxylated piperidine class. First isolated from Adenophora spp. roots, by Asano, adenophorine was shown to be a good α-galactosidase inhibitor, only a moderate to poor inhibitor of α-glucosidases (IC50: 34 μM) and not at all of β–galactosidase.47 As this homonojirimycin analog is endowed with a lipophilic substituent (ethyl) at the anomeric position, its biologic activity is proof that α-alkylation at C1 does not suppress the glycosidase inhibitory effect.Although the structure and relative configuration shown in 9 were determined by Asano,47 the assignment of the absolute configuration of the natural (+) isomer became possible only a few years later when Davis completed the first synthesis of (−)-adenophorine.48 However, it was Lebreton and coworkers49 who, by tackling a metathesis-related approach, successfully carried out the first asymmetric total synthesis of (+)-adenophorine. The synthesis proceeded in 14 steps and a 3.5% overall yield, relative to the (+) enantiomer 13 of Garner's aldehyde, in turn prepared from D-methyl serinate hydrochloride (Scheme 22). In the reaction sequence RCM (Grubbs II catalyst, 5 mol%) was again key for building the 1-C-alkylated N-heterocyclic core 93 from a mixture of protected dienes 92 (with the NH masked as oxazolidinone) corresponding to amino alcoholstrans-92 and cis-92. After RCM, the separation of the diastereomers by flash chromatography became possible providing the pure tetrahydropyridinetrans-93 in 74% yield. Epoxidations first on the enantiopure trans-93 and then on 94, followed by regioselective epoxide opening (with a selenium-boron complex and water, respectively), provided 9 with good stereoselectivity.
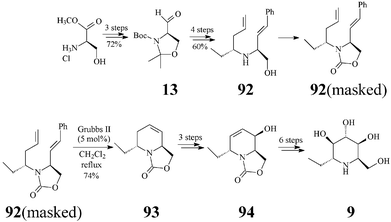 | ||
| Scheme 22 Total synthesis of (+)-adenophorine (9). | ||
The Lebreton group50–52 also investigated in detail the synthesis of a series of substituted piperidine iminosugars. Like adenophorine, these compounds were isolated from Adenophora spp.47 This range of natural products display an unusual structural trait accommodating a hydrophobic α-1-C-alkyl substituent such as a undecyl, heptyl, butyl and ethyl group. The strategy for (+)-5-deoxyadenophorine (98a) and its analogues (98b–e) starts again from D-Garner's aldehyde 13 and uses as well the versatile RCM (Grubbs II, 5 mol%; CH2Cl2, reflux 1 h) for creating the chiral trans-2,6-disubstituted-1,2,5,6-tetrahydropyridine system 96 (72–88% yield) (Scheme 23). Stereochemically controlled epoxidation and epoxide ring opening, followed by basic hydrolysis, finally afforded the targeted 5-deoxyadenophorine analogues 98b–e.
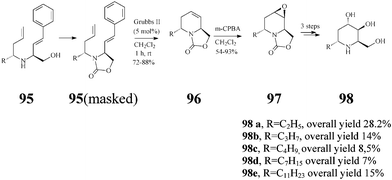 | ||
| Scheme 23 Total synthesis of (+)-5-deoxyadenophorine (98a) and analogues (98b–e). | ||
(+)-5-Deoxyadenophorine was found to have a potent effect on α-galactosidase (IC50: 6.4 lM) and β-galactosidase (IC50: 34 lM).47 Such activity was not observed with compounds (±)-98b–e in experiments conducted by the Lebreton group52 suggesting that in this series, under the tested conditions, the lengthening of the α-1-C hydrophobic tail has no beneficial influence on the inhibitory action.
4. Fagomine and its isomers
There are four possible isomers of the azasugar fagomine (Scheme 24). D-Fagomine (10;1,2,5-trideoxy-1,5-imino-D-arabinitol, 1,2,5-trideoxy-1,5-imino-D-arabino hexitol, 1,2-dideoxynojirimycin or 2-deoxy-DNJ) and its stereoisomers 100 (3-epi-fagomine) and 101 (3,4-di-epi-fagomine) are natural iminosugars present in seeds, roots and leaves of a variety of plants (Leguminosae, Solanaceae, Moraceae etc). Fagomine has been isolated inter alia from mulberry leaves. The fagomine-rich buckwheat (Fagopyrum esculentum) is widely used in traditional recipes as an efficient anti-hyperglycemic agent for preventing sharp blood glucose peaks after intake of refined carbohydrates,53 and for positively influencing intestinal microbiota by favouring adhesion of probiotics. It is supposed that fagomine enhances glucose-induced insulin secretion by accelerating processes following the glyceraldehyde 3-phosphate formation in the glycolytic pathway.54 Fagomine and 3-epi-fagomine have activity against α-glucosidase and β-galactosidase while 3,4-di-epi-fagomine has no inhibitory effect.55 The fourth fagomine isomer, 102 (4-epi-fagomine or 1,2-dideoxygalactonojirimycin), not occurring in nature, was first prepared by Asano from fagomine through chemical epimerization of the 4-OH group, and was reported to inhibit in vitro and intracellularly enhance the lysosomal α-galactosidase A activity in Fabry lymphoblasts (Fabry disease is a lysosomal storage disorder caused by a deficient lysosomal α-galactosidase A activity).56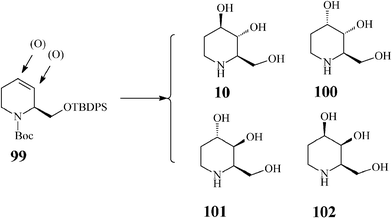 | ||
| Scheme 24 Fagomine (10) and isomers (100–102). | ||
Although fagomine had previously been synthesized by alternative routes, Takahata et al.54 managed to perform the asymmetric synthesis of fagomine (10) and its three isomers (100–102) starting from Garner's aldehyde 13 (derived from D-serine) and using a common structural motif, the chiral 1,2,5,6-tetrahydropyridine scaffold 99. The latter productive intermediate was constructed by an effective RCM step promoted (97% yield) by the Grubbs I catalyst (Scheme 25). Then, playing on the stereochemistry of different oxidation procedures applied on the double bond in 99, as illustrated in Scheme 26, opened access to 10 and 100–102. This divergent synthesis of all fagomine isomers is yet again an important achievement supported by the versatile metathesis reaction.
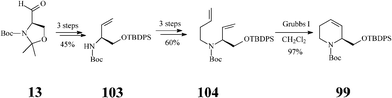 | ||
| Scheme 25 Synthesis of the common 1,2,5,6-tetrahydropyridine scaffold 99. | ||
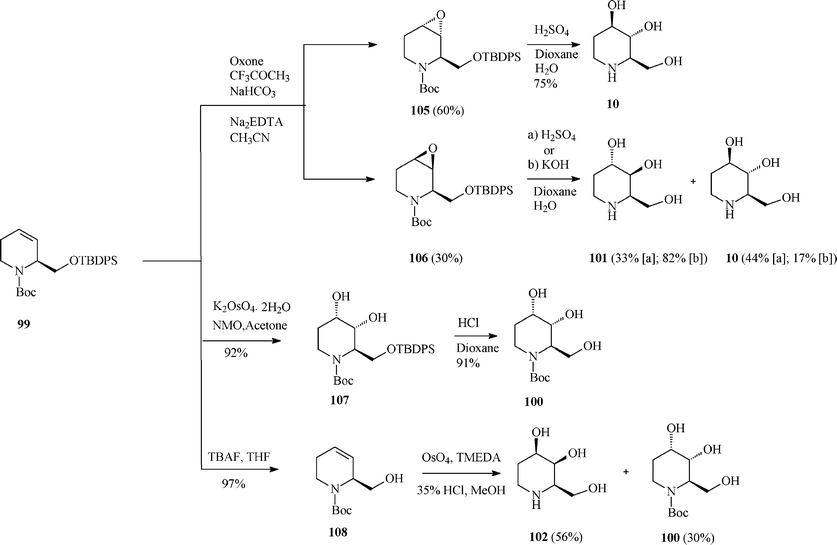 | ||
| Scheme 26 Total synthesis of fagomine (10) and isomers. | ||
For fagomine 10 and its isomer 101 (trans-diols in the 3 and 4 position) Takahata et al.54 (Scheme 26) invoked the epoxidation (Oxone; CF3COCH3) of the double bond in 99 to give with low diastereoselectivity a mixture of anti (105; 60%) and syn (106; 30%) epoxides, easily separated by medium-pressure chromatography. Acid hydrolysis of the antiepoxide led selectively to 10 (75% yield), while the syn stereoisomer gave a mixture of fagomine and 3,4-di-epi-fagomine (in 77% combined yield; ratio 10:101 = 1.3:1). Interestingly, under basic hydrolysis conditions the ratio is reversed in favour of 3,4-di-epi-fagomine (10:101 = 0.2:1). The mixtures of fagomine and 3,4-di-epi-fagomine were separated by ion-exchange resin chromatography.
The cleanest pathway of the protocol (Scheme 26) was the dihydroxylation of 99 under modified Upjohn conditions proceeding with very good yield (92%) to 107 as a single diastereoisomer. The rationale behind this high stereoselectivity was supposed to be an attack occurring exclusively from the less hindered side, anti to the siloxymethyl substituent that has to adopt an axial conformation because of the allylic strain in the tetrahydropyridine moiety.
Therefore, this methodology solely afforded 3-epi-fagomine 100 resulting after deprotection of 107 in acidic medium. As should have been expected the same high degree of stereoselectivity was observed on carrying out the modified Upjohn dihydroxylation on the alcohol 108 derived from 99.
Finally, benefiting from the earlier reported dihydroxylation of homoallylic alcohols using the complex OsO4 with tetramethylethylenediamine, the oxidation of 108 and deprotection conducted to the fourth fagomine isomer, the D-4-epi-fagomine (or (2R,3S,4R)-2-hydroxymethylpiperidine-3,4-diol; 102). However, only moderate selectivity could be attained and a mixture of 102 (56%) and 100 (30% yields) was formed from which 102 was separated by ion-exchange resin chromatography. It should be remarked that the above described routes represent the first total syntheses of 3-epi-fagomine (100) and 3,4-di-epi-fagomine (101).54a
5. Isofagomine and congeners
The pharmacological chaperone isofagomine (109) (Scheme 27) was designed for the treatment of Gaucher's disease, the most common of the lysosomal storage syndromes characterized by genetic mutations resulting in the production of a defective key enzyme, β-glucocerebrosidase. At sub-inhibitory concentrations, competitive inhibitors of the mutant enzyme like isofagomine help it fold correctly, by selectively binding to the misfolded β-glucocerebrosidase, and thus facilitating trafficking to lysosomes and restoring the activity of this enzyme.57 Consequently, the mechanism of action of isofagomine differs from that established for the other major azasugar involved in the treatment of Gaucher's disease, Miglustat (11), which acts by inhibiting biosynthesis of macromolecular substrates accumulating pathologically in glycosphingolipidoses.58 As isofagomine tartrate [(3R,4R,5R)-3,4-dihydroxy-5-(hydroxymethyl)piperidine L-(+)-tartrate] this potent iminosugar reached phase II clinical development.59 Also, it has been proved that iminosugars like isofagomine (IC50 = 0.7 μM) bind strongly to the active site of glycogen phosphorylase (the main regulatory enzyme in the liver responsible for the control of blood glucose levels), recently identified as a new target for treatment of type 2 diabetes.60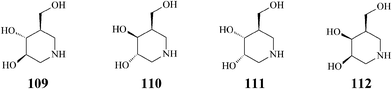 | ||
| Scheme 27 Isofagomine (109) and isomers (110–112). | ||
Isofagomine, a potent β-glycosidase inhibitor,61 is a non-natural azasugar first synthesized in 1994 via a strategy not implying olefin metathesis.62 However, only a few years later, olefin metathesis gained a well-deserved place in an array of synthetic protocols targeting isofagomine and congeners that employ different key building blocks accessed by ring-closing metathesis.
Along these lines, Guanti and Riva63 used the asymmetrized 113 (readily obtained in both enantiomeric forms through a chemoenzymatic procedure). Multistep sequences entailing the critical RCM on diene 114, opened the way to several enantiopure branched tetrahydropyridines (115), as precursors of isofagomine (Scheme 28).
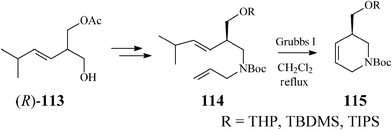 | ||
| Scheme 28 Synthesis of isofagomine precursors 115. | ||
As RCM of 114 proceeded with excellent yield (over 95%) in the presence of Grubbs catalyst, irrespective of the R substituent, the authors concluded that the steric hindrance in the branched substrates as well as and the release of isopropylethylene have no influence on the reaction outcome. Classical deprotection to the alcohol 116, followed by its epoxidation gave diastereomeric epoxides 117 and 118 whose hydrolysis led to an easily separable mixture of isofagomine (109) and its “gulo” stereoisomer 110 (Scheme 29). The ratio in which the two isomers (109/110) are found differs if resulting from acidic or basic hydrolysis (3:1 and 1:1, respectively).
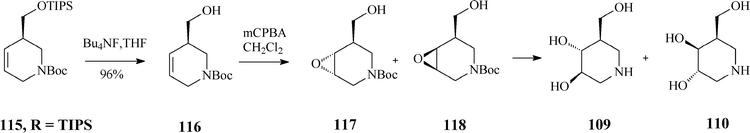 | ||
| Scheme 29 Synthesis of isofagomine and its “gulo” isomer 110. | ||
In a productive synthesis of isofagomine Takahata64 (Scheme 30) uses RCM only to prepare its starting material, the N-Boc-5-hydroxy-1,2,5,6-tetrahydropyridine (119), from 1,3-butadiene monoepoxide and allylamine. Noteworthy is, however, that along with isofagomine (obtained by epoxidation of 119 to 120 and regioselective epoxide ring opening of the latter to 121 by means of a “higher order” cuprate (CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH)2CuCNLi2)), the procedure also allowed synthesis of two isofagomine congeners, homoisofagomine (122) (from 121) and 5-deoxyisofagomine (123) (from 120), all of them in 44–50% overall yields.
CH)2CuCNLi2)), the procedure also allowed synthesis of two isofagomine congeners, homoisofagomine (122) (from 121) and 5-deoxyisofagomine (123) (from 120), all of them in 44–50% overall yields.
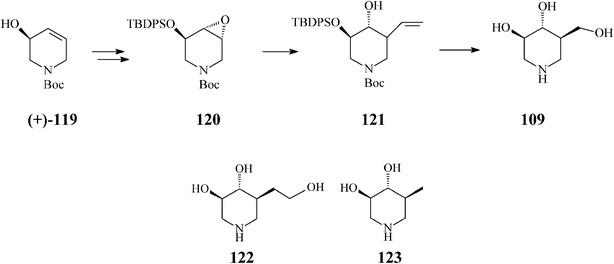 | ||
| Scheme 30 Synthesis of isofagomine, homoisofagomine (122) and 5-deoxyisofagomine (123). | ||
In contrast to synthetic strategies for piperidine azasugars discussed so far, that all employ ring-closing olefin metathesis (RCM) on a diene for constructing the general tetrahydropyridine framework, Imahori and Takahata65 provide a conceptual advance by suggesting to this end ring-closing enyne metathesis (RCEYM). Indeed, in the synthesis of (+)-isofagomine (Scheme 31), they demonstrated that RCEYM on alkyne substrates 124 proceeds smoothly, even under argon, provided they are endowed with an allylic hydroxy substituent (124, ROH). In this case, the ethylene atmosphere (usually applied for speeding up the rather slow RCEYM of terminal alkynes with the Grubbs I initiator) is no longer required. More detailed investigations66 proved that the acceleration effect in RCEYM is specific to an allylic free hydroxy group (because its protection as OBn or OTBDPS resulted in a considerable decrease of the reaction efficiency) and that the reaction rate, under argon, of 124, ROH (99% yield after 1.5 h) is comparable to that of 124, RH in an ethylene atmosphere (96% yield after 1.5 h) while in the absence of ethylene, 124, RH gave just 32% 125, RH (after a much longer reaction time, 41 h). From kinetic NMR studies effected on the RCEYM of 124 (ROH), the authors infer a “ene-then-yne” mechanistic pathway, based on comparison of the signals at 19.53 and 18.92 ppm with those of Ru-carbenes generated, in the presence of the same catalyst, from 125, ROH and 1-propen-3-ol, respectively, and additionally supported by an early detection of styrene in the NMR spectra.
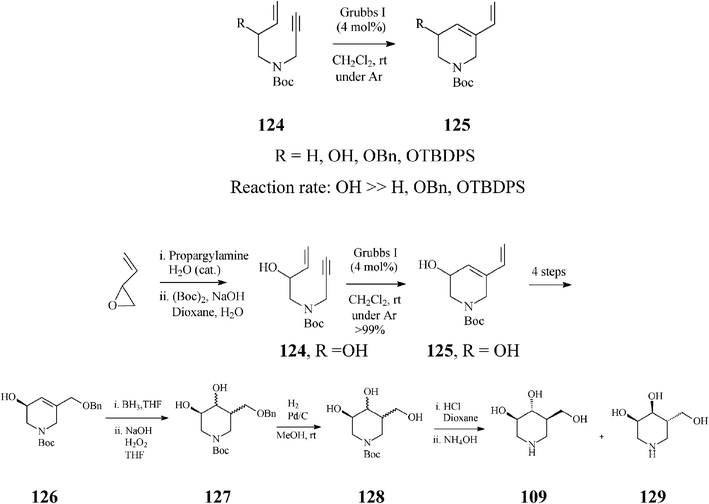 | ||
| Scheme 31 Synthesis of (+)-isofagomine (109) based on ring-closing enyne metathesis. | ||
Following RCEYM, four reaction steps (the last of which being enzymatic) provided almost enantiomerically pure (+)-126 (99% ee). Hydroboration of the double bond in 126 and subsequent oxidation (NaOH/H2O2) afforded diols 127. The mixture of (+)-isofagomine (109) and (−)-3-epi-isofagomine (129), obtained after removal of the benzyl and Boc protective groups, was purified by column chromatography (silica gel; MeOH/10% NH4OH, 10:1) to afford pure (+)-isofagomine (78% yield) and (−)-3-epi-isofagomine (20% yield). The overall yield in (+)-isofagomine was 11.5%, starting from the commercially available 1,3-butadiene-1,2-epoxide.
As a recent proof of principle, the Japanese group chose the allylic hydroxy group accelerated RCEYM for performing the asymmetric synthesis of 2-propylisofagomine with high diastereoselectivity, in 13% overall yield starting from the commercially available (E)-hex-2-ol.67
6. Structural modifications. Diversification of activity
6.1. N-alkylation
The fact that some of the earlier synthesized polyhydroxylated piperidine azasugars have either unacceptable low activity or poor selectivity has shifted the interest of researchers to inventing structural modifications that might result in improvement of potency and enzyme specificity. Adding structural elements that resemble the aglycon of the enzyme glycoside substrate should promote supplementary interactions with the aglycon binding site and therefore a more selective binding. One such synthetic approach is N-alkylation providing access to lipophilic piperidine iminocyclitols, readily available by reductive amination or alkylation reactions from commercial 1-deoxyiminosugars such as DNJ. Strong support for the N-alkylation concept came from investigations aimed at defining the structural features needed for antiviral activity of polyhydroxylated piperidines.68,69 Experiments indicated that N-substituted 1-deoxynojirimycin derivatives, especially N-butyl-DNJ, were more potent in vivo than the natural DNJ itself, an effect tentatively assigned to an increased uptake by the cells. Another rationalization was put forward by Asano70 who demonstrated that N-alkylation led to a conformational change at C6 determining a more specific inhibition of the endoplasmic reticulumα-glucosidase I which is implicated in several viral processes including human immunodeficiency virus (HIV) and hepatitis B virus. In addition to its glycoprotein processing α-glucosidase activity, N-butyl–DNJ was also proved to inhibit ceramide glycosyltransferase, an enzyme which catalyzes the first step in the glycosphingolipid biosynthetic pathway.71In attempts to determine the molecular basis for the above two activities and better study the contribution of N-alkyl and N-alkenyl side chains (C4, C9 or C18) to the inhibitory behaviour vs.ceramide glycosyltransferase, Butters et al. synthesized and tested a series of N-substituted DNJ analogs (130) (Scheme 32).72 They concluded that: (i) inhibition of ceramide glucosyltransferase was paralleling inhibition by ceramide itself thus indicating that the inhibitory activity is due to ceramide mimicry; (ii) α-glycosidase inhibition did not depend on the presence or length of an N-alkyl chain; (iii) an N-alkyl hydrophobic tail was mandatory for transferase inhibition but increases in alkyl chain length provided only a modest increase in inhibitory potency; (iv) the C3 hydroxyl group is of crucial importance for the activity of both enzymes.71b Today N-butyl-deoxynojirimycin (11), a drug approved for treatment of viral diseases and lysosomal storage disorders (such as Gaucher's disease), is produced and commercialized by Glaxo as Miglustat. Also, this research group discovered that the uptake of all compounds was extremely rapid (complete inhibition of glucosphingolipid biosynthetic pathway) and not dependent upon the length of the N-alk(en)yl moiety. Greatly increased cellular retention of N-cis-13-octadecenyl-DNJ was observed, relative to the shorter-chain compounds N-butyl-DNJ and N-nonyl-DNJ. The obvious conclusion was that lengthening the side chain leads to improving imino sugar retention and, therefore, the inhibitory efficacy for ceramide glycosyltransferase in cultured cells.
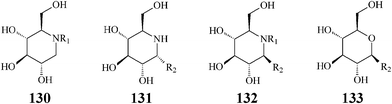 | ||
| Scheme 32 N- and C-alkylated iminosugars vs. C-alkylated sugars. | ||
Along similar considerations, Aerts et al. synthesized, by conventional methods, a series of lipophilic azasugars with large hydrophobic N-substituents promoting insertion in membranes. Included here is the N-[(5-adamantane-1-yl-methoxy)pentyl]-DNJ 134 (Scheme 33), a very powerful inhibitor (IC50: 1.7 nM) of non-lysosomal glucosylceramidase, also acting on α-glucosidases at micromolar concentrations (IC50: 0.87 μM).73 As will be seen below, iminosugar 134 is the lead compound in later studies by Aerts and Overkleeft74 aiming at more selective inhibitors of the enzymes involved in glucosylceramide metabolism.
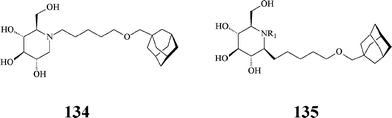 | ||
| Scheme 33 Hydrophobically modified azasugars. | ||
Not always, however, is N-substitution added value for potency. For instance, in the isofagomine series modification of the imino group by a hydrophobic moiety greatly decreased the inhibitory activity (IC50 > 44 μM and IC50 > 100 μM for N-butyl isofagomine and N-nonyl isofagomine, respectively). The potent inhibitory activity of isofagomine has been assigned to a salt bridge formation between the protonated isofagomine and the catalytic carboxylate of β-glucocerebrosidase. Or, an alkyl group attached to nitrogen might interfere with this salt formation resulting in less potent inhibitors.75
6.2. Aza-C-glycosides by metathetic routes
Azasugar C-glycosides,6 stable iminoanalogues of glycosides and glycoconjugates, have been synthesized since the 80s. Today they form a vast class with interesting biological and therapeutic properties to which, as one of its simplest members, α-homonojirimycin belongs. In comparison with the N-substituted 1-deoxyazasugars described above, azasugar C-glycosides are more difficult to prepare yet offer more beneficial features as antiviral agents and inhibitors of carbohydrate processing enzymes in regard to potency and selectivity. Hereinafter only some reports on azasugar C-glycosides synthesis implying olefin metathesis will be discussed.The same adamantyl-substituted motif as in 134 and 135 appears in the synthesis of an array of adamant-1-ylmethoxy-functionalized α-aza-C-glycosides valorizing cross-metathesis (CM) as a key strategy.74 The methodology is amenable to yielding different products, starting from one of three 6-propenyl-substituted iminocyclitols (D-gluco, L-ido or D-xylo α-allyl-aza-C-glycoside) and using as cross-metathesis partners adamant-1-ylmethoxy-functionalized terminal alkenes incorporating spacers with different lengths (C1–C3) (Scheme 34). Additional libraries of the targeted glycosides could be generated by Birch reduction of the cross-metathesis products (137, 139 and 141).
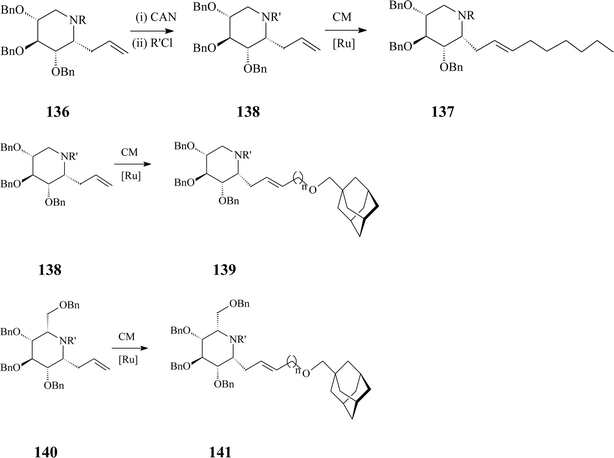 | ||
| Scheme 34 Cross-metathesis (CM) route to C-alkylated iminosugars (R = p-methoxybenzyl (PMB), R' = benzyloxy carbamate, n = 1–3; Grubbs I catalyst, 25 mol%, DCM, 45 °C, 24 h, 65–88% yields). | ||
In fact, cross-metathesis had been used before in diversity oriented iminosugar C-glycosides synthesis. Such compounds, with various aglycones, were obtained by cross-metathesis, promoted by the Grubbs II catalyst and occurring with excellent E/Z selectivity, between α-1-C-allyl-1-deoxynojirimycin derivatives and variously functionalized alkenes76 (Scheme 35). Noteworthy, the authors established that α-vinyl-aza-C-glycosides are not suitable substrates in cross-metathesis reactions.
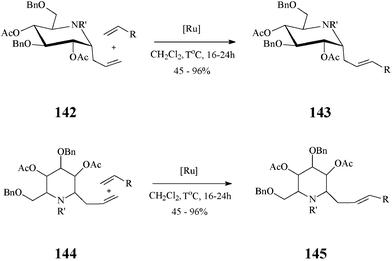 | ||
| Scheme 35 Cross-metathesis route to C-alkylated iminosugars (R = ester, phosphonate, sulphone, aryl or alkyl; 142: R’ = Troc; 144: R’ = CHO). | ||
Aza-C-linked disaccharides possessing a second sugar unit have been communicated as well (146).26 Their additional O-heterocycle potentially mimics the departing sugar in a glycosidase-catalysed reaction and, consequently, these iminosugars may be more potent inhibitors of glycosidases. Furthermore, as in their structure a methylene group replaces the exo-oxygen of the glycosidic linkage, thus circumventing the labile acetal functionality, aza-C-linked disaccharide mimetics are highly resistant to chemical and enzymatic hydrolysis.
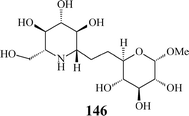
Though not related to metathesis, some remarkable recent research endeavours for modulating the affinity for glycosidases and enhancing inhibition effects are worth mentioning here, e.g. multivalent iminosugars based on oligoethylene scaffolds and N-substituted deoxynojirymicin epitopes synthesized by click chemistry,77 as well as a dodecavalent iminosugar derivative with a fullerene core showing, in glycosidase inhibition assays, a binding enhancement of up to three orders of magnitude over the corresponding monovalent ligand.78
It is now clear that the best option for identifying more potent inhibitors of different classes of enzymes involved in viral and bacterial infections is the design and synthesis of new iminosugars.79
By virtue of the diversity of these widespread diseases and the multiple biological targets of drugs, this area is still poorly served by existing therapies. New compounds (a second-generation leads from iminosugars)80 should be developed that, based on other mechanisms in vivo, may offer more tolerable alternatives to current treatment. The real boom presently experienced by chiral metathesis catalysts and asymmetric ring-closing metathesis might guide toward enantiospecific syntheses of piperidine iminosugars by simpler routes and ultimately to discovery of better drug leads.
7. Conclusions
This article critically examines olefin metathesis as a robust entry to synthesis of biologically most relevant piperidine azasugars, reputed as potent inhibitors of carbohydrate processing enzymes and useful therapeutic agents against viral and metabolic diseases. A recurring theme in all protocols reviewed is application of olefin metathesis (RCM, RCEYM) for building with high efficiency a versatile intermediate. The power of this concept has been demonstrated through the expedient creation from this intermediate of iminosugar families where each member has the same tetrahydropyridine core structure but with chemical modifications that endow it with different properties. Examples where cross-metathesis (CM) facilitates diversity oriented approaches, thus contributing to the discovery of novel drug leads, are also discussed. Light is shed on how the high-yielding metathesis reaction, promoted by the Grubbs I, Grubbs II or Hoveyda-Grubbs catalysts, allows remarkable molecular construction while preserving the stereogenic centres already carefully installed.Acting in conjunction with other stereoselective processes implied in the overall strategy, such as dihydroxylation or epoxidation, the key metathesis step supports a rigorous management of the stereocontrol, essential for access to piperidine iminosugars with specific biological activity.
Acknowledgements
I.D. and V.D. acknowledge this article as part of the research on olefin metathesis carried out at the Centre of Organic Chemistry “C. D. Nenitzescu” under the auspices of the Romanian Academy.References
- (a) Iminosugars: From Synthesis to Therapeutic Applications, ed. P. Compain and O. R. Martin, Wiley-VCH, Weinheim, Germany, 2007; (b) A.E. Stütz, Iminosugars as Glycosidase Inhibitors: Nojirimycin and Beyond, Wiley-VCH: Weinheim, Germany, 1999 Search PubMed; (c) E. Moreno-Clavijo, A. T. Carmona, A. J. Moreno-Vargas, L. Molina and I. Robina, Curr. Org. Synth., 2011, 8, 102–133 CrossRef CAS; (d) F. Oulaïdi, S. Front-Deschamps, E. Gallienne, E. Lesellier, K. Ikeda, N. Asano, P. Compain and O. R. Martin, ChemMedChem, 2011, 6, 353–361 CrossRef.
- D. J. Wardrop and S. L. Waidyarachchi, Nat. Prod. Rep., 2010, 27, 1431–1468 RSC.
- A. A. Watson, G. W. J. Fleet, N. Asano, R. J. Molyneux and R. J. Nash, Phytochemistry, 2001, 56, 265–295 CrossRef CAS.
- Iminosugars: Recent Insights into Their Bioactivity and Potential as Therapeutic Agents, ed. O. R. Martin and P. Compain, Curr. Top. Med. Chem., 2003, 3, 471–591 Search PubMed.
- B. G. Davis, Tetrahedron: Asymmetry, 2009, 20, 652–671 CrossRef CAS.
- P. Compain, V. Chagnault and O. R. Martin, Tetrahedron: Asymmetry, 2009, 20, 672–711 CrossRef CAS.
- M. S. M. Pearson, M. Mathé-Allainmat, V. Fargeas and J. Lebreton, Eur. J. Org. Chem., 2005, 2159–2191 CrossRef CAS.
- P. Compain and O. R. Martin, Curr. Top. Med. Chem., 2003, 3, 541–560 CrossRef CAS.
- V. H. Lillelund, H. H. Jensen, X. Liang and M. Bols, Chem. Rev., 2002, 102, 515–554 CrossRef CAS.
- R. A. Dwek, T. D. Butters, F. M. Platt and N. Zitzmann, Nat. Rev. Drug Discovery, 2002, 1, 65–75 CrossRef CAS.
- B. L. Stocker, E. M. Dangerfield, A. L. Win-Mason, G. W. Haslett and M. S. M. Timmer, Eur. J. Org. Chem., 2010, 1615–1637 CrossRef CAS.
- B. Winchester and G. W. J. Fleet, Glycobiology, 1992, 2, 199–210 CrossRef CAS.
- Nature Outlook: Hepatitis C, Nature, 474, (No. 7350_supp), S1–S48, http://www.nature.com/nature/outlook/hepatitis-c/index.html.
- S. D. Woodhouse, C. Smith, M. Michelet, N. Branza-Nichita, M. Hussey, R. A. Dwek and N. Zitzmann, Antimicrob. Agents Chemother., 2008, 52, 1820–1828 CrossRef CAS.
- T. M. Cox, J. M. F. G. Aerts, G. Andria, M. Beck, N. Belmatoug, B. Bembi, R. Chertkoff, S. Vom Dahl, D. Elstein, A. Erikson, M. Giralt, R. Heitner, C. Hollak, M. Hrebicek, S. Lewis, A. Mehta, G. M. Pastores, A. Rolfs, M. C. Sa Miranda and A. Zimran, J. Inherited Metab. Dis., 2003, 26, 513–526 CrossRef CAS.
- (a) Metathesis in Natural Product Synthesis, ed. J. Cossy, S. Arseniyadis and C. Meyer, Wiley-VCH: Weinheim, Germany, 2010; (b) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4490–4527 CrossRef CAS; (c) T. Gaich and J. Mulzer, Curr. Top. Med. Chem., 2005, 5, 1473–1494 CrossRef CAS.
- I. Dragutan, V. Dragutan, C. Mitan, H. C. M. Vosloo, L. Delaude and A. Demonceau, Beilstein J. Org. Chem., 2011, 7, 699–716 CrossRef CAS.
- (a) A. Kato, N. Kato, E. Kano, I. Adachi, K. Ikeda, L. Yu, T. Okamoto, Y. Banba, H. Ouchi, H. Takahata and N. Asano, J. Med. Chem., 2005, 48, 2036–2044 CrossRef CAS; (b) S.-J. Sung-Jae Pyun, K.-Y. Lee, C.-Y. Oh, J.-E. Joo, S.-H. Cheon and W.-H. Ham, Tetrahedron, 2005, 61, 1413–1416 CrossRef.
- N. Asano, Glycobiology, 2003, 13, 93R–104R CrossRef CAS.
- A. Mehta, N. Zitzmann, P. M. Rudd, T. M. Block and R. A. Dwek, FEBS Lett., 1998, 430, 17–32 CrossRef CAS.
- (a) See e.g. S. K. Bagal, S. G. Davies, J. A. Lee, P. M. Roberts, A. J. Russell, P. M. Scott and J. E. Thomson, Org. Lett., 2010, 12, 136–139 CrossRef CAS; (b) E. N. Tzanetou, K. M. Kasiotis, P. Magiatis and S. A. Haroutounian, Molecules, 2007, 12, 735–744 CrossRef CAS; (c) B.-G. Wei, J. Jie Chen and P.-Q. Huang, Tetrahedron, 2006, 62, 190–198 CrossRef CAS; (d) C. Boucheron, P. Compain and O. R. Martin, Tetrahedron Lett., 2006, 47, 3081–3084 CrossRef CAS; (e) C. Boglio, S. Stahlke, S. Thorimbert and M. Malacria, Org. Lett., 2005, 7, 4851–4854 CrossRef CAS.
- (a) A. Singh, B. Kim, W. K. Lee and H.-J. Ha, Org. Biomol. Chem., 2011, 9, 1372–1380 RSC; (b) D. Best, C. Wang, A. C. Weymouth-Wilson, R. A. Clarkson, F. X. Wilson, R. J. Nash, S. Miyauchi, A. Kato and G. W. J. Fleet, Tetrahedron: Asymmetry, 2010, 21, 311–319 CrossRef CAS; (c) S. K. Bagal, S. G. Davies, J. A. Lee, P. M. Roberts, A. J. Russell, P. M. Scott and J. E. Thomson, Org. Lett., 2010, 12, 136–139 CrossRef CAS; (d) M. Ganesan and N. G. Ramesh, Tetrahedron Lett., 2010, 51, 5574–5576 CrossRef CAS; (e) O. K. Karjalainen and A. M. P. Koskinen, Org. Biomol. Chem., 2011, 9, 1231–1236 RSC.
- E. N. Tzanetou, K. M. Kasiotis, P. Magiatis and S. A. Haroutounian, Molecules, 2007, 12, 735–744 CrossRef CAS.
- (a) Handbook of Metathesis, ed. R. H. Grubbs, Wiley-VCH, Weinheim, Germany, 2003, Vol. 1; (b) R. R. Schrock and A. H. Hoveyda, Angew. Chem., Int. Ed., 2003, 42, 4592–4633 CrossRef CAS; (c) H. Wakamatsu and S. Blechert, Angew. Chem., Int. Ed., 2002, 41, 2403–2405 CrossRef CAS; (d) D. Burtscher and K. Grela, Angew. Chem., Int. Ed., 2008, 48, 442–454 CrossRef; (e) M. Bieniek, A. Michrowska, D. L. Usanov and K. Grela, Chem.–Eur. J., 2008, 14, 806–818 CrossRef CAS; (f) S. P. Nolan and H. Clavier, Chem. Soc. Rev., 2010, 39, 3305–3316 RSC; (g) G. C. Vougioukalakis and R. H. Grubbs, Chem. Rev., 2010, 110, 1746–1787 CrossRef CAS; (h) S. Monfette and D. E. Fogg, Chem. Rev., 2009, 109, 3783–3816 CrossRef CAS; (i) J. M. Blacquiere, R. McDonald and D. E. Fogg, Angew. Chem., Int. Ed., 2010, 49, 3807–3810 CrossRef CAS; (j) F. Nicks, R. Aznar, D. Sainz, G. Muller and A. Demonceau, Eur. J. Org. Chem., 2009, 5020–5027 CrossRef CAS; (k) X. Sauvage, Y. Borguet, A. F. Noels, L. Delaude and A. Demonceau, Adv. Synth. Catal., 2007, 349, 255–265 CrossRef CAS; (l) K. Ferré-Filmon, L. Delaude, A. Demonceau and A. F. Noels, Eur. J. Org. Chem., 2005, 3319–3325 CrossRef.
- (a) V. Dragutan, I. Dragutan, L. Delaude and A. Demonceau, Coord. Chem. Rev., 2007, 251, 765–794 CrossRef CAS; (b) V. Dragutan and I. Dragutan, J. Organomet. Chem., 2006, 691, 5129–5147 CrossRef CAS; (c) V. Dragutan, I. Dragutan and H. Fischer, J. Inorg. Organomet. Polym. Mater., 2008, 18, 18–31 CrossRef CAS; (d) I. Dragutan, V. Dragutan and H. Fischer, J. Inorg. Organomet. Polym. Mater., 2008, 18, 311–324 CrossRef CAS; (e) F. Ding, Y. G. Sun, S. Monsaert, I. Dragutan, V. Dragutan and F. Verpoort, Curr. Org. Synth., 2008, 5, 291–304 CrossRef CAS; (f) I. Dragutan, V. Dragutan, L. Delaude, A. Demonceau and A. F. Noels, Rev. Roumaine Chim., 2007, 52, 1013–1025 CAS; (g) I. Dragutan and V. Dragutan, Platinum Met. Rev., 2006, 50, 81–94 CrossRef CAS; (h) I. Dragutan, V. Dragutan and P. Filip, ARKIVOC, 2005, x, 105–129 Search PubMed; (i) V. Dragutan, I. Dragutan and A. T. Balaban, Platinum Metals Rev., 2000, 44(3), 112–118 CAS; (j) V. Dragutan, I. Dragutan and A. Demonceau, Platinum Met. Rev., 2005, 49, 123–137 CrossRef CAS.
- A. Kennedy, A. Nelson and A. Perry, Beilstein J. Org. Chem., 2005, 1(2) DOI:10.1186/1860-5397-1-2.
- F.-X. Felpin and J. Lebreton, Eur. J. Org. Chem., 2003, 3693–3712 CrossRef CAS.
- C. P. Woodward, N. D. Spiccia, W. R. Jackson and A. J. Robinson, Chem. Commun., 2011, 47, 779–781 RSC.
- (a) C. M. Huwe, O. C. Kiehl and S. Blechert, Synlett, 1996, 65–66 CrossRef; (b) S. Randl and S. Blechert, J. Org. Chem., 2003, 68, 8879–8882 CrossRef CAS; (c) A. Deiters and S. F. Martin, Chem. Rev., 2004, 104, 2199–2238 CrossRef CAS; (d) I. Nakamura and Y. Yamamoto, Chem. Rev., 2004, 104, 2127–2198 CrossRef CAS; (e) M. Arisawa, A. Nishida and M. Nakagawa, J. Organomet. Chem., 2006, 691, 5109–5121 CrossRef CAS; (f) B. Schmidt and J. Hermanns, Curr. Org. Chem., 2006, 10, 1363–1396 CrossRef CAS; (g) S. K. Chattopadhyay, S. Karmakar, T. Biswas, K. C. Majumdar, H. Rahaman and B. Roy, Tetrahedron, 2007, 63, 3919–3952 CrossRef CAS; (h) P. Compain, Adv. Synth. Catal., 2007, 349, 1829–1846 CrossRef CAS; (i) V. Cadierno and P. Crochet, Curr. Org. Synth., 2008, 5, 343–364 CrossRef CAS; (j) H. Liu, D. Su, G. Cheng, J. Xu, X. Wang and Y. Hu, Org. Biomol. Chem., 2010, 8, 1899–1904 RSC; (k) K. C. Majumdar, B. Chattopadhyay and K. Ray, Curr. Org. Synth., 2010, 7, 153–176 CrossRef CAS; (l) K. M. Kuhn, T. M. Champagne, S. H. Hong, W.-H. Wei, A. Nickel, C. W. Lee, S. C. Virgil, R. H. Grubbs and R. L. Pederson, Org. Lett., 2010, 12, 984–987 CrossRef CAS; (m) V. Mahajan and H.-J. Gais, Chem.–Eur. J., 2011, 17, 6187–6195 CrossRef CAS.
- (a) C. M. Huwe and S. Blechert, Tetrahedron Lett., 1995, 36, 1621–1624 CrossRef CAS; (b) C. M. Huwe and S. Blechert, Synthesis, 1997, 61–67 CrossRef CAS.
- S. Beligny, S. Eibauer, S. Maechling and S. Blechert, Angew. Chem., Int. Ed., 2006, 45, 1900–1903 CrossRef CAS.
- (a) H. Takahata, Y. Banba, M. Sasatani, H. Nemoto, A. Kato and I. Adachi, Tetrahedron, 2004, 60, 8199–8205 CrossRef CAS; (b) H. Takahata, Y. Banba, H. Ouchi and H. Nemoto, Org. Lett., 2003, 5, 2527–2529 CrossRef CAS.
- R. Martín, C. Murruzzu, M. A. Pericàs and A. Riera, J. Org. Chem., 2005, 70, 2325–2328 CrossRef.
- (a) A. Fürstner, D. Koch, K. Langemann, W. Leitner and C. Six, Angew. Chem., Int. Ed. Engl., 1997, 36, 2466–2469 CrossRef; (b) A. Fürstner, L. Ackermann, K. Beck, H. Hori, D. Koch, K. Langemann, M. Liebl, C. Six and W. Leitner, J. Am. Chem. Soc., 2001, 123, 9000–9006 CrossRef; (c) W. Leitner, Acc. Chem. Res., 2002, 35, 746–756 CrossRef CAS.
- (a) see e.g. F. Gallou, S. Saim, K. J. Koenig, D. Bochniak, S. T. Horhota, N. K. Yee and C. H. Senanayake, Org. Process Res. Dev., 2006, 10, 937–940 CrossRef CAS; (b) F. Michalek, D. Mädge, J. Rühe and W. Bannwarth, Eur. J. Org. Chem., 2006, 577–581 CrossRef CAS; (c) H. Clavier, K. Grela, A. Kirschning, M. Mauduit and S.P. Nolan, Angew. Chem., Int. Ed., 2007, 46, 6786–6801 CrossRef CAS and ref. therein; (d) M. Selva, A. Perosa, M. Fabris and P. Canton, Green Chem., 2009, 11, 229–238 RSC; (e) S. Monfette and D. E. Fogg, “ Ring-Closing Metathesis Synthesis of Medium and Large Rings: Challenges and Implications for Sustainable Synthesis”, in: Green Metathesis Chemistry: Great Challenges in Synthesis, Catalysis and Nanotechnology), V. Dragutan, A. Demonceau, I. Dragutan and E. Sh. Finkelshtein ed., NATO Science for Peace and Security Series A: Chemistry and Biology Series, Springer-Verlag, New York, 2010, 129–156 Search PubMed.
- G. Danoun, J. Ceccon, A. E. Greene and J.-F. Poisson, Eur. J. Org. Chem., 2009, 4221–4224 CrossRef CAS.
- O. V. Singh and H. Han, Tetrahedron Lett., 2003, 44, 2387–2391 CrossRef CAS.
- R. Rengasamy, M. J. Curtis-Long, H. W. Ryu, K. Y. Oh and K. H. Park, Bull. Korean Chem. Soc., 2009, 30, 1531–1534 CrossRef CAS.
- F. Ferreira, C. Botuha, F. Chemla and A. Perez-Luna, J. Org. Chem., 2009, 74, 2238–2241 CrossRef CAS.
- S. Ghosh, J. Shashidhar and S. K. Dutta, Tetrahedron Lett., 2006, 47, 6041–6044 CrossRef CAS.
- S. J. Pyun, K. Y. Lee, C. Y. Oh, J. E. Joo, S. H. Cheon and W. H. Ham, Tetrahedron, 2005, 61, 1413–1416 CrossRef CAS.
- P. Gupta, A. P. J. Pal, Y. S. Reddy and Y. D. Vankar, Eur. J. Org. Chem., 2011, 1166–1175 CrossRef CAS.
- H. Han, Tetrahedron Lett., 2003, 44, 1567–1569 CrossRef CAS.
- R. Rengasamy, M. J. Curtis-Long, W. D. Seo, S. H. Jeong, I.-Y. Jeong and K. H. Park, J. Org. Chem., 2008, 73, 2898–2901 CrossRef CAS.
- A. Kumar, M. Abrar Alam, S. Rani and Y. D. Vankar, Carbohydr. Res., 2010, 345, 1142–1148 CrossRef CAS.
- A. M. C. H. van den Nieuwendijk, M. Ruben, S. E. Engelsma, M. D. P. Risseeuw, R. J. B. H. N. van den Berg, R. G. Boot, J. M. Aerts, J. Brussee, G. A. van der Marel and H. S. Overkleeft, Org. Lett., 2010, 12, 3957–3959 CrossRef CAS.
- K. Ikeda, M. Takahashi, M. Nishida, M. Miyauchi, H. Kizu, Y. Kameda, M. Arisawa, A. A. Watson, R. J. Nash, G. W. J. Fleet and N. Asano, Carbohydr. Res., 2000, 323, 73–80 CrossRef CAS.
- M. A. T. Maughan, I. G. Davies, T. D. W. Claridge, S. Courtney, P. Hay and B. G. Davis, Angew. Chem., Int. Ed., 2003, 42, 3788–3792 CrossRef CAS.
- M. S. M. Pearson, M. Evain, M. Mathé-Allainmat and J. Lebreton, Eur. J. Org. Chem., 2007, 4888–4894 CrossRef CAS.
- (a) F.-X. Felpin, K. Boubekeur and J. Lebreton, J. Org. Chem., 2004, 69, 1497–1503 CrossRef CAS; (b) F.-X. Felpin and J. Lebreton, Tetrahedron Lett., 2003, 44, 527–530 CrossRef CAS.
- F.-X. Felpin, K. Boubekeur and J. Lebreton, Eur. J. Org. Chem., 2003, 4518–4527 CrossRef CAS.
- M. S. M. Pearson, R. O. Saad, T. Dintinger, H. Amri, M. Mathé-Allainmat and J. Lebreton, Bioorg. Med. Chem. Lett., 2006, 16, 3262–3267 CrossRef CAS.
- S. Taniguchi, N. Asano, F. Tomino and I. Miwa, Horm. Metab. Res., 1998, 30, 679–683 CrossRef CAS.
- (a) Y. Banba, C. Abe, H. Nemoto, A. Kato, I. Adachi and H. Takahata, Tetrahedron: Asymmetry, 2001, 12, 817–819 CrossRef CAS; (b) H. Takahata, Y. Banba, H. Ouchi, H. Nemoto, A. Kato and I. Adachi, J. Org. Chem., 2003, 68, 3603–3607 CrossRef CAS.
- A. Kato, N. Asano, H. Kizu, K. Matsui, A. A. Watson and R. J. Nash, J. Nat. Prod., 1997, 60, 312–314 CrossRef CAS.
- N. Asano, S. Ishii, H. Kizu, K. Ikeda, K. Yasuda, A. Kato, O. R. Martin and J.-Q. Fan, Eur. J. Biochem., 2000, 267, 4179–4186 CrossRef CAS.
- R. Khanna, E. R. Benjamin, L. Pellegrino, A. Schilling, B. A. Rigat, R. Soska, H. Nafar, B. E. Ranes, J. Feng, Y. Lun, A. C. Powe, D. J. Palling, B. A. Wustman, R. Schiffmann, D. J. Mahuran, D. J. Lockhart and K. J. Valenzano, FEBS J., 2010, 277, 1618–1638 CrossRef CAS and references therein..
- T. M. Cox, J. M. F. G. Aerts, G. Andria, M. Beck, N. Belmatoug, B. Bembi, R. Chertkoff, S. Vom Dahl, D. Elstein, A. Erikson, M. Giralt, R. Heitner, C. Hollak, M. Hrebicek, S. Lewis, A. Mehta, G. M. Pastores, A. Rolfs, M. C. Sa Miranda and A. Zimran, J. Inherited Metab. Dis., 2003, 26, 513–526 CrossRef CAS.
- C. Dulsat and N. Mealy, Drugs Future, 2009, 34, 23 CrossRef CAS.
- L. Somsak, V. Nagy, Z. Hadady, T. Docsa and P. Gergely, Curr. Pharm. Des., 2003, 9, 1177–1189 CrossRef CAS.
- (a) O. Lopez and M. Bols, “Isofagomine, Noeuromycin and other 1-Azasugars, Iminosugar-Related Glycosidase Inhibitors”, Ch. 6 in Iminosugars: From Synthesis to Therapeutic Applications, ed. P. Compain and O. R. Martin, Wiley-VCH, Weinheim, Germany, 2007 Search PubMed; (b) A. Kato, S. Miyauchi, N. Kato, R. J. Nash, Y. Yoshimura, I. Nakagome, S. Hirono, H. Takahata and I. Adachi, Bioorg. Med. Chem., 2011, 19, 3558–3568 CrossRef CAS.
- (a) T. M. Jespersen, W. Dong, M. R. Sierks, T. Skrydstrup, I. Lundt and M. Bols, Angew. Chem., Int. Ed. Engl., 1994, 33, 1778–1779 CrossRef; (b) T. M. Jespersen, M. Bols, M. R. Sierks and T. Skrydstrup, Tetrahedron, 1994, 50, 13449–13460 CrossRef CAS.
- (a) G. Guanti and R. Riva, Tetrahedron Lett., 2003, 44, 357–360 CrossRef CAS; (b) L. Banfi, G. Guanti, M. Paravidino and R. Riva, Org. Biomol. Chem., 2005, 3, 1729–1737 RSC.
- H. Ouchi, Y. Mihara, H. Watanabe and H. Takahata, Tetrahedron Lett., 2004, 45, 7053–7056 CrossRef CAS.
- T. Imahori and H. Takahata, Tetrahedron Lett., 2008, 49, 265–268 CrossRef CAS.
- T. Imahori, H. Ojima, Y. Yoshimura and H. Takahata, Chem.–Eur. J., 2008, 14, 10762–10771 CrossRef CAS.
- H. Takahata, T. Taguchi, T. Imahori, Y. Yoshimura, A. Kato, I. Adachi, M. Kawahata and K. Yamaguchi, Heterocycles, 2012, 84 DOI:10.3987/COM-11-S(P)74.
- G. W. J. Fleet, A. Karpas, R. A. Dwek, L. E. Fellows, A. S. Tyms, S. Petursson, S. K. Namgoong, N. G. Ramsden, P. W. Smith, J. C. Son, F. Wilson, D. R. Witty, G. S. Jacob and T. W. Rademacher, FEBS Lett., 1988, 237, 128–132 CrossRef CAS.
- A. Karpas, G. W. J. Fleet, R. A. Dwek, S. Petursson, S. K. Namgoong, N. G. Ramsden, G. S. Jacob and T. W. Rademacher, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 9229–9233 CrossRef CAS.
- N. Asano, H. Kizu, K. Oseki, E. Tomioka, K. Matsui, M. Okamoto and M. Baha, J. Med. Chem., 1995, 38, 2349–2356 CrossRef CAS.
- (a) T. D. Butters, R. A. Dwek and F. M. Platt, Chem. Rev., 2000, 100, 4683–4696 CrossRef CAS; (b) T. D. Butters, L. A. G. M. van den Broek, G. W. J. Fleet, T. M. Krulle, M. R. Wormald, R. A. Dwek and F. M. Platt, Tetrahedron: Asymmetry, 2000, 11, 113–124 CrossRef CAS.
- H. R. Mellor, D. C. A. Neville, D. J. Harvey, F. M. Platt, R. A. Dwek and T. D. Butters, Biochem. J., 2004, 381, 861–866 CrossRef CAS.
- H. S. Overkleeft, G. H. Renjema, J. Neele, P. Vianello, I. O. Hung, A. Strijland, A. M. van der Burg, G.-J. Joomen, U. K. Pandit and J. M. F. G. Aerts, J. Biol. Chem., 1998, 273, 26522–26527 CrossRef CAS.
- T. Wennekes, R. J. B. H. N. van den Berg, T. J. Boltje, W. E. Donker-Koopman, B. Kuijper, G. A. van der Marel, A. Strijland, C. P. Verhagen, J. M. F. G. Aerts and H. S. Overkleeft, Eur. J. Org. Chem., 2010, 1258–1283 CrossRef.
- X. Zhu, K. A. Sheth, S. Li, H.-H. Chang and J.-Q. Fan, Angew. Chem., Int. Ed., 2005, 44, 7450–7453 CrossRef CAS.
- G. Godin, P. Compain and O.R. Martin, Org. Lett., 2003, 5, 3269–3272 CrossRef CAS.
- J. Diot, M. I. García-Moreno, S. G. Gouin, C. Ortiz Mellet, K. Haupt and J. Kovensky, Org. Biomol. Chem., 2009, 7, 357–363 CAS.
- P. Compain, C. Decroocq, J. Iehl, M. Holler, D. Hazelard, T. M. Barragán, C. Ortiz Mellet and J.-F. Nierengarten, Angew. Chem Int. Ed., 2010, 59, 5753–5756 CrossRef.
- P. Greimel, J. Spreitz, A. E. Stütz and T. M. Wrodnigg, Curr. Top. Med. Chem., 2003, 3, 513–523 CrossRef CAS.
- G. Home, F. X. Wilson, J. Tinsley, D. H. Williams and R. Storer, Drug Discovery Today, 2011, 16, 107–118 CrossRef.
| This journal is © The Royal Society of Chemistry 2012 |