Aromatic hydroxyl group—a hydrogen bonding activator in bifunctional asymmetric organocatalysis
Pankaj
Chauhan
and
Swapandeep Singh
Chimni
*
Department of Chemistry, U.G.C. Centre of Advance Studies in Chemistry, Guru Nanak Dev University, Amritsar, 143005, India. E-mail: sschimni@yahoo.com; Fax: (+)91-183-2258820
First published on 2nd December 2011
Abstract
Recent progress in asymmetric catalysis has contributed to the development of a diverse range of chiral organocatalysts that differ in their origin, structure and mode of activation. The bifunctional organocatalysts bearing aromatic hydroxyl (or phenolic) groups have emerged as a privileged class of organocatalyst. In these bifunctional organocatalysts, the aromatic hydroxyl group functions as a weak Brønsted acid or hydrogen bonding site and a nucleophilic or basic moiety such as amine and phosphine serve as a base or hydrogen bond acceptor. The bifunctional organocatalysts having these moieties have not been reviewed. In this review we are presenting the asymmetric transformations catalyzed by the bifunctional organocatalyst bearing an aromatic hydroxyl group.
 Pankaj Chauhan | Pankaj Chauhan was born in 1984 at Bindal, a small village in Shimla District of Himachal Pradesh in India. He obtained his B.Sc. from Himachal Pradesh University in 2004. After completing his M.Sc. from Guru Nanak Dev University, Amritsar in 2007, he commenced his Ph.D. under the supervision of Dr Swapandeep Singh Chimni in the same University. His research interests include synthesis and application of chiral bifunctional organocatalysts for the development of new asymmetric transformation and green chemistry. |
 Swapandeep Singh Chimni | Swapandeep Singh Chimni was born in 1962 at Amritsar, India. He received his M.Sc.(Hons. Sch.) in Chemistry in 1985 and Ph.D. in 1991 from the Guru Nanak Dev University, Amritsar. After two years as Lecturer in Regional Engineering College (now NIT) Jalandhar, he joined the Department of Chemistry, Guru Nanak Dev University as Lecturer in 1992. He is presently working as Professor in the same department. He has 21 years of research experience and published over 80 publications. He works in the area of synthetic organic chemistry with emphasis on asymmetric organocatalysis, biocatalysis and phase transfer catalysis as well as green chemistry. |
1. Introduction
Organocatalysis has emerged as a dynamic area of research in the domain of catalytic asymmetric synthesis.1,2 Over the years, many chiral organocatalysts have been developed and explored for a wide array of organic transformations. Among these diverse organocatalysts, bifunctional organocatalysts have been found to be highly efficient for bringing out a variety of useful asymmetric transformations. The bifunctional organocatalysts provide synergic activation to nucleophile and electrophile with two activating sites, either through hydrogen bonding or Brønsted acid-Lewis/Brønsted base activation.3 Several natural catalytic processes also proceed through hydrogen bonding of the active site of the enzyme with the substrate, but it is only recently that chemists have shown keen interest in exploiting the hydrogen bonding tendency of small organic molecules for catalyzing a variety of asymmetric transformations. Asymmetric organocatalysis with hydrogen bonding activation have been carried out using chiral urea/thiourea derivatives, chiral phosphoric acids, amides including phosphoramides and sulphonamides and phenol based chiral catalysts. Recent reviews on this mode of activation have illustrated the importance of proline and its analogues,4 urea/thiourea5 and phosphoric acid based organocatalysts.6 Beside these useful chiral organocatalysts there exist another class of valuable bifunctional catalysts bearing aromatic hydroxyl group in combination with a basic nitrogen or phosphorous moiety. The hydrogen bonding capability or Brønsted acid character of phenolic OH is more than that of aliphatic hydroxyl group. Enzymes such as class II aldolase, epoxide hydrolase (Fig. 1) and Δ5-3-ketosteroid isomerase (KSI) also activate the substrate through hydrogen bonding with an aromatic hydroxyl group.7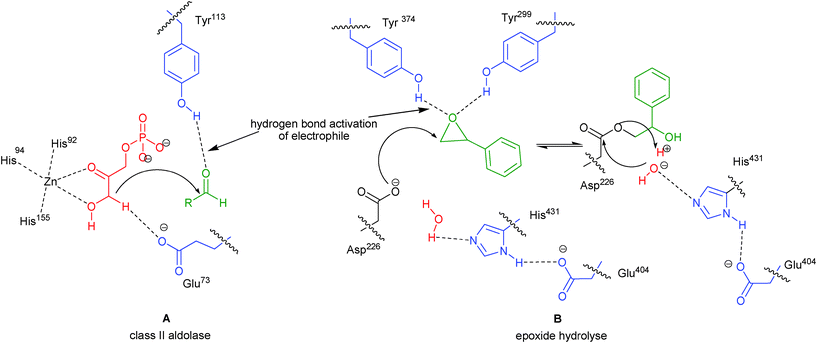 | ||
| Fig. 1 Mode of activation in enzymes (A) class II aldolase and (B) epoxide hydrolyse. | ||
Most hydrogen bonding chiral catalysts have a basic or nucleophilic moiety along with hydrogen bonding group. Tertiary ‘Group V elements’ mainly nitrogen and phosphorous are basic in nature viz tertiary amines and phosphines. These tertiary amines and phosphines serve as a Lewis base for hydrogen bonding or a Brønsted base for proton abstraction from nucleophile (Fig. 2). These moieties have been successfully exploited in bifunctional asymmetric catalysis in cooperation with hydrogen bonding moiety such as urea/thiourea, hydroxyl and aromatic hydroxyl for the synergic activation of substrates.
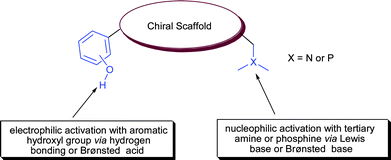 | ||
| Fig. 2 Synergic mode of activation. | ||
In the last decade, bifunctional organocatalysts bearing aromatic hydroxyl groups have emerged as a powerful tool for bringing out a wide range of organic transformations. The aromatic hydroxyl group as a hydrogen bonding unit has not been reviewed, there is only one review on 6′–OH Cinchona alkaloids and their modified thiourea derivatives published in 2006.8c Since then, significant progress has been made in the application of 6′–OH Cinchona alkaloids as bifunctional organocatalysts. Other than 6′–OH Cinchona alkaloids, the BINOL derived organocatalysts bearing aromatic hydroxyl groups and amine/phosphine moieties, have emerged as efficient chiral catalysts in asymmetric synthesis. In this review we are presenting the catalytic potential of chiral bifunctional organocatalysts bearing aromatic hydroxyl groups as hydrogen bond donors and nitrogen or phosphorous moiety as basic units or hydrogen bond acceptors. For simplicity in presentation as well as understanding, this review has been classified into three major classes, based on the chiral scaffold of the catalysts: 1) Cinchona alkaloid derivatives 2) BINOL derivatives and 3) miscellaneous organocatalysts.
2. Cinchona alkaloid derivatives (Cinchona derived organocatalysts bearing aromatic hydroxyl and tertiary amino groups)
The multifunctional Cinchona alkaloids have unique molecular recognition abilities that render them and their derivatives as invaluable molecules in nearly every field of chemistry dealing with chirality: they are used as asymmetric catalysts, surface modifiers and selectors for chromatographic separations, bases for the resolution of racemates, ligands for transition-metal complexes and building blocks for supramolecular architectures. These alkaloids also find applications in asymmetric metal catalysis as well as organocatalysis. The Cinchona alkaloids are valuable motif in asymmetric organocatalysis both in their native form and modified form. They find applications in enamine catalysis, iminium catalysis, Lewis base catalysis, phase transfer catalysis as well as in bifunctional Lewis base-Brønsted acid catalysis.8 The latter property of these molecules arises from their structure as they possess a secondary alcohol and tertiary nitrogen. These features render them to be used as bifunctional organocatalysts.Selective demethylation of quinine and quinidine generate C6′–OH species, that are known as cupreines and cupreidines, respectively. The demethylated form of the Cinchona derivatives or 6′–OH Cinchona alkaloids such as cupreines, cupreidines and β-isocupreidine (β-ICPD) and other simpler congeners containing a free hydroxyl group at C6 positions were recently shown to be powerful chiral organocatalysts for a wide array of asymmetric reactions (Fig. 3). Like parent alkaloids these catalysts feature two different sites for simultaneous activation of both nucleophiles and electrophiles. The cupreines and cupreidines also have increased acidity of the Brønsted acid site due to the presence of phenolic OH and a free site for further functionalization (C9–OH) that can be exploited to fine-tune properties such as basicity and conformation, therefore affecting the catalytic performance. In the following sections, the catalytic applications of 6′–OH Cinchona alkaloids for asymmetric transformations are being discussed.
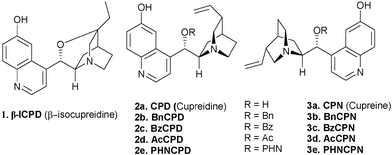 | ||
| Fig. 3 Structures and abbreviations of some 6′–OH Cinchona alkaloids. | ||
2.1 Carbon–carbon bond formations
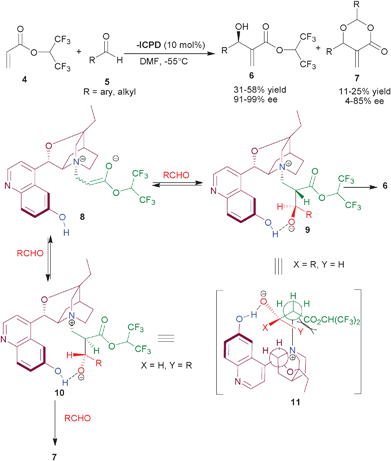 | ||
| Scheme 1 β-ICPD catalyzed MBH reaction of hexatrifluoroisopropyl acrylate (4) and aldehydes (5). | ||
In the mechanistic studies, they highlighted the prerequisite of the aromatic hydroxyl group for the observed stereochemistry. The reaction proceeds through initial nucleophilic addition of tertiary amine of the catalyst to acrylate, resulting in an enolate (8), which undergoes aldol type addition on the aldehyde to provide an equilibirium mixture of several diastereomers. The intramolecular hydrogen bonding between oxy-anion and aromatic OH, stabilizes the betaine intermediates, (9) and (10). The intermediate (9) can easily adopt the anti-periplanar arrangement (11) of the quaternary nitrogen and the α-hydrogen atom, required for the elimination to take place, leading to the preferential formation of one single enantiomer of the MBH adduct. The other structure (10), experiences severe steric repulsions in doing this and therefore reacts with another molecule of aldehyde, yielding dioxanone (7).
Later in 2006, they established, that water bound β-ICPD leads to the partial hydrolysis of HFiPA and found that azotropically dried β-ICPD showed remarkable catalytic activity, in particular, for aromatic aldehydes resulting in MBH adduct in excellent enantioselectivity (94–100% ee) and improved yield (27–82%).10b
The application of this methodology was extended for the total synthesis of (−)-mycestericin E (14),11 a potent immunosuppressant and the formal synthesis of epopromycin B (17) (Scheme 2).12 In the latter case chiral aldehyde (15) was used and the role of the catalyst was envisioned to achieve high levels of diastereoselectivity.
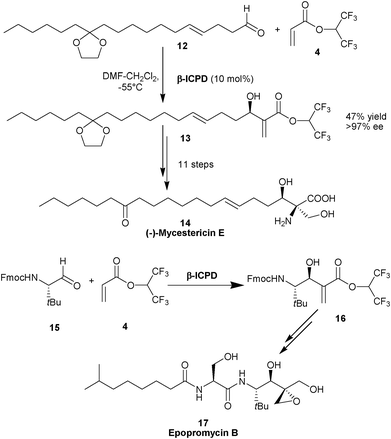 | ||
| Scheme 2 Synthesis of (–)-mycestericin E (14) and epopromycin B (17). | ||
Shi and co-workers have extended the substrate scope for β-ICPD catalyzed MBH reaction for methyl vinyl ketone (MVK) and α-naphthyl acrylate (18) with aldehydes (5) (Scheme 3).13 The β-ICPD provides a poor to moderate level of enantioselectivity (7–49% ee) when MVK was used. Although, α-naphthyl acrylate reacts with aromatic and aliphatic aldehydes to provide MBH product (19) with moderate to good level of enantioselectivity (33–92% ee) but, the reaction rate was very slow in the reaction conditions affording high enantioselectivity.
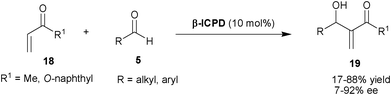 | ||
| Scheme 3 β-ICPD catalyzed MBH reaction of (18) with aldehydes (5). | ||
Very recently, three reports have been published that describe the catalytic application of β-ICPD for the synthesis of enantiomerically enriched 3-substituted-3-hydroxyoxindole derivatives via Morita–Baylis–Hillman reaction of unsaturated nucleophile (20) with isatin (21) (Scheme 4).14 Zhou and co-workers have developed a β-ICPD catalyzed asymmetric MBH reaction of acrolein with isatin derivatives to procure chiral quaternary hydroxylated carbon at C3-position of oxindole (22) in good to excellent yield (65–97%) and high enantioselectivity (90–98% ee) at −20 °C in dichloromethane as solvent.14a The product of this transformation has been utilized for the synthesis of chiral lactone and (+)-madindoline analogue precursor. Shi and co-workers have reported the β-ICPD catalyzed asymmetric MBH reaction of acrylate with isatin derivatives to generate quaternary hydroxylated stereocenter on oxindole with moderate to excellent yield (17–99%) and good enantioselectivity (46–94% ee) in dichloromethane at room temperature.14b Lu et al. have also reported a similar transformation in good yield (61–96% yield) and high enantioselectivity (86–96% ee) catalyzed by 10 mol% of β-ICPD in chloroform with 4 Å molecular sieves (MS) as additives at room temperature.14c All these reactions seem to proceed with a mechanism similar to that of the β-ICPD catalyzed MBH reaction of aldehydes with HFiPA. The synergic activation of unsaturated compound with tertiary amine and the activation of isatin carbonyl group with aromatic hydroxyl group of the catalyst via hydrogen bonding leads to the formation of MBH adduct with quaternary stereocenters with high ee.
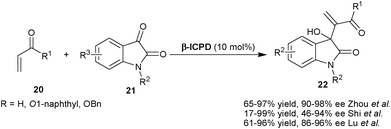 | ||
| Scheme 4 β-ICPD catalyzed asymmetric MBH reaction of isatin (21). | ||
Balana and Adolfsson later modified this protocol with in situ generation of N-tosyl imine with titanium isopropoxide and molecular sieves from aldehydes and p-toluenesulfonamide. β-ICPD catalyzes the three component aza-MBH reaction with low to good yield (12–95%) and moderate level of enantioselectivity (49–74% ee).17
Zhu and co-workers have demonstrated the reversal of enantioselectivity of aza-MBH product of MVK and N-sulfonyl imines on using β-naphthol as an additive in the β-ICPD catalyzed asymmetric aza-MBH reaction.18
Hatakeyama and co-worker have developed β-ICPD catalyzed asymmetric aza-MBH reaction of HFiPA (4) and aromatic phosphonyl imines (26) (Scheme 5).19 The S isomer of N-protected-α-methylene-β-amino acid esters (27) were obtained in moderate to high yield (42–90%) and moderate enantioselectivity (54–73% ee). The enantioselectivity was enriched to 99% after single crystallization with quite a satisfactory yield. On the basis of observed stereochemistry of the product, the proposed transition state has been illustrated to involve two hydrogen bonded stable betaine intermediates (28) and (29). The ammonium cation and α-hydrogen of ester group, adopt anti-periplanar geometry in both the intermediates, since (28) is free from the steric interaction, so it affords the major (S) enantiomer. The chiral aza-MBH product of this transformation was used as a precursor for the synthesis of β-lactam.
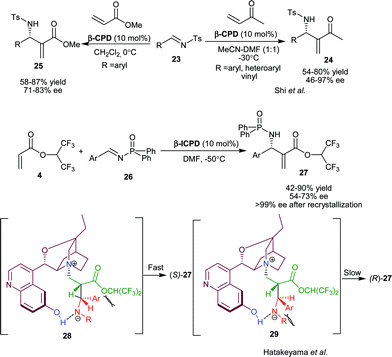 | ||
| Scheme 5 β-ICPD catalyzed aza-MBH reaction. | ||
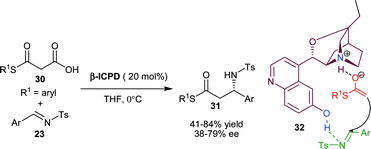 | ||
| Scheme 6 β-ICPD catalyzed decarboxylative Mannich reaction of malonic half thioestsers (30) with N-tosyl imines (23). | ||
On achieving remarkable success employing α-isothiocyano imides in catalytic asymmetric aldol reaction with thiourea tertiary amine catalyst, Seidel and co-workers employed the α-isothiocyanato imides (33) as a nucleophile for Mannich reactions with sulfonyl imines (23) (Scheme 7).22 The qunidine derived catalyst AcCPD catalyzes the formation of Mannich product (34) in good to high yield (53–99%), with good to excellent syn-diastereoselectivity (72![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :28–95:5 dr) and high enantioselectivity (89–99% ee) using a diverse range of imines (Ts, Bs and Ns) with substituted aromatic, heteroaromatic and α,β-unsaturated groups. The reactions with Bs- and Ns-imines were much faster than Ts-imine and the reaction of Bs- and Ns-imines proceeds at a remarkably low catalyst loading (0.25–1.0 mol%) with a reasonable reaction rate.
:28–95:5 dr) and high enantioselectivity (89–99% ee) using a diverse range of imines (Ts, Bs and Ns) with substituted aromatic, heteroaromatic and α,β-unsaturated groups. The reactions with Bs- and Ns-imines were much faster than Ts-imine and the reaction of Bs- and Ns-imines proceeds at a remarkably low catalyst loading (0.25–1.0 mol%) with a reasonable reaction rate.
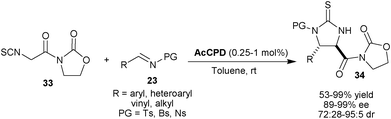 | ||
| Scheme 7 AcCPD catalyzed asymmetric Mannich reaction α-isothiocyanato imides (33) with sulfonyl imines (23). | ||
The similiar transformation catalyzed by BzCPN, has been developed by Zhong and co-workers, where 2.5 mol% BzCPN catalyzes the Mannich reaction of α-isothiocyanato imides (33) with N-tosyl imines (23) to provide (34) in 80–99% yield, 86–99% ee and 67:33–97:3 dr.23
Chen and co-workers have reported the 6′–OH Cinchona alkaloid catalyzed direct anti-selective Mannich reaction of 2-oxindoles (35) with N-tosyl imines (23) (Scheme 8).24 The 10 mol% of quinine derived catalyst PHNCPN efficiently catalyzes the addition of (35) to (23) leading to the formation of anti-3,3-disubstituted 2-oxindoles (36) with vicinal quaternary and tertiary stereogenic centers in good to excellent diastereoselectivity (79:21–95:5) and good enantioselectivity (70–89% ee). The transition state (37) involves the initial formation of enolate that is stabilized by the tertiary amine, which directs the re-face attack of the enolate to the re-face of the imine, which is oriented by the hydrogen bonding with 6′–OH of the catalyst.
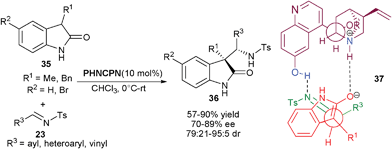 | ||
| Scheme 8 BnCPN catalyzed anti-Mannich reaction of 2-oxindoles (35) with N–tosyl imines (23). | ||
:1 ratio, 91% yield and 91% ee.
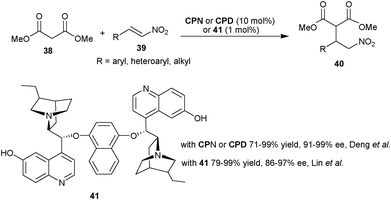 | ||
| Scheme 9 6′–OH Cinchona alkaloids catalyzed Michael addition of malonic ester (38) to nitro-olefins (39). | ||
Very recently, Lin and co-workers have developed a new bifunctional bis 6′–OH Cinchona alkaloid catalyst (41) for asymmetric conjugate addition of malonate (38) to nitroalkenes (39) (Scheme 9).27 With 1 mol% of catalyst (41) and its pseudoenantiomeric form, the Michael adducts (40) with opposite configuration were obtained in high yield (79–99%) and very good enantioselectivity (86–97% ee).
The extension of application of 6′–OH Cinchona alkaloids for the asymmetric conjugate addition reaction to generate vicinal stereocenters was reported by the Deng et al. in 2005. The C9 protected 6′–OH Cinchona alkaloids catalyze the enantioselective Michael addition of racemic tri-substituted carbon nucleophiles (42) to nitro-olefins (39) to generate adjacent quaternary and tertiary chiral carbon in the addition products (43) (Scheme 10).28 Various cyclic and acyclic β-ketoester, diketones, nitroester and cyanoester react with substituted aromatic, heteroaromatic and aliphatic nitroolefins in the presence of BnCPN or BnCPD to provide Michael adduct (43) in excellent diastereoselectivity (82:18–98:2 dr) and enantioselectivity (89–99% ee). The catalyst β-ICPD provides Michael adduct with yield and stereoselectivity comparable to BnCPD. This result led them to propose that in the transition state BnCPD being a flexible molecule, adopts a gauche-open conformation, like more rigid β-ICPD and simultaneously activate and orient the Michael donor with tertiary amine moiety and the acceptor via a network of hydrogen-bonding interactions with aromatic hydroxyl group (44). In the transition state the substituents of the two bond-forming carbons remained in a staggered rather than eclipsed arrangement.
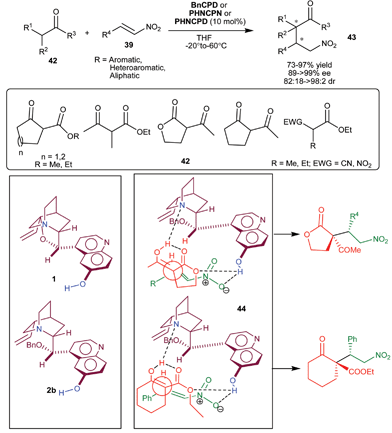 | ||
| Scheme 10 6′–OH Cinchona alkaloids catalyzed Michael addition of racemic tri-substituted carbon nucleophiles (42) to nitro-olefins (39). | ||
Young and White have synthesized many racemic trisubstituted nitroacetates with Pd(II) catalyzed allylic C–H alkylation reaction.29 The synthetic utility of nitroacetate (45) was demonstrated by BnCPN catalyzed Michael addition of (45) to the nitrostyrene (39) to obtain 1,4-adduct (46) in 88% yield, and 95% ee and 12:1 dr (Scheme 11).
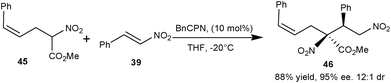 | ||
| Scheme 11 BnCPN catalyzed asymmetric Michael addition of nitroacetate (45) to the nitrostyrene (39). | ||
Wang and co-workers developed the organocatalytic asymmetric 1,4-conjugate addition of α-fluoroketoester to nitro-olefins catalyzed by 9–OH protected cupreine (Scheme 12).30 1 mol% of PHNCPN catalyzes the formation of non-enolizable ketoesters (48) with vicinal fluorinated quaternary and tertiary stereocenters from α-fluoroethyl acetoacetate (47) and nitro-olefins (39) in high yield (75–98%) and excellent enantioselectivity (94–99% ee) with acceptable diastereoselectivity (1.7:1–4:1 dr).
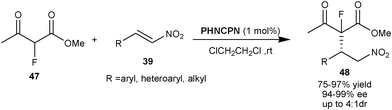 | ||
| Scheme 12 PHNCPN catalyzed asymmetric synthesis of non-enolizable ketoesters (48) with fluorinated quaternary stereocenter. | ||
They have also developed PHNCPN catalyzed asymmetric Michael addition of 2-fluoromalonate with nitro-olefin derivatives to provide corresponding 1,4-adduct in good to high yield (65–98%) and high enantioselectivity (86–99% ee).31
Yan and co-workers utilized the catalytic potential of 6′–OH Cinchona alkaloids for the synthesis of enantioenriched nitrocyclopropane (50) viaCPN catalyzed initial asymmetric conjugate addition of bromomalonate (49) to aromatic and heteroaromatic nitro-olefins (39) followed by DABCO mediated intramolecular cyclopropanation (Scheme 13).32 The substituted nitrocyclopropane products (50) were obtained in moderate to good yield (47–78%) and excellent stereoselectivity (75–99% ee and >99:1 dr).
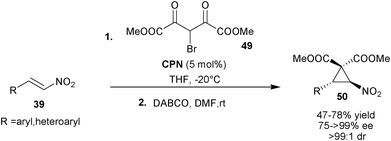 | ||
| Scheme 13 Asymmetric cyclopropanation reaction catalyzed with CPN. | ||
The application of asymmetric 1,4-addition to nitroalkenes for the synthesis of functionalized cyclopentanes (52), with oxime unit have been reported by Rodriguez et al. in a one-pot three step procedure.33 The one pot sequence involves Takemoto's catalyst (53) or CPN catalyzed initial asymmetric Michael reaction of substituted malonates (51) with nitro-olefins (39) followed by carbohydroxylation via an intramolecular [3 + 2]-silylnitronate olefin cyclization (ISOC)-fragmentation sequence with Me3SiCl and triethyl amine, followed by fragmentation with tetrabutylammonium fluoride (TBAF) (Scheme 14). Many aryl and heteroaryl substituted cyclopentanes (52) have been synthesized in good to high yield (60–99%) and high enantioselectivity (88–97% ee).
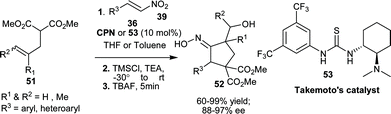 | ||
| Scheme 14 Asymmetric synthesis of functionalized cyclopentanes (52). | ||
Recently, Feng and co-workers have developed the asymmetric synthesis of multifunctionalized tetrahydroindan (55) with four contiguous stereocenters via two sequential conjugate addition reaction, involving initial BnCPN catalyzed stereoselective addition of cyclic γ,δ-unsaturated-β-ketoester (54) to substituted nitroalkenes (39), followed by 1,1,3,3-tetramethylguanidine (TMG) catalyzed intramolecular Michael reaction (Scheme 15).34 The aromatic, heteroaromatic, cinnamic as well as aliphatic nitro-olefins provide chiral indan (55) in good to high yield (50–99%) with excellent enantioselectivity (95–99% ee) and diastereoselectivity (>99:1) with low catalyst loading of BnCPN (0.5–2 mol%).
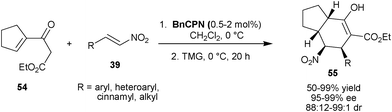 | ||
| Scheme 15 Asymmetric synthesis of multifunctionalized tetrahydroindans (55). | ||
Wang et al. reported the asymmetric conjugate addition of nitroalkanes (56) to nitro-olefins (39) using cupreine as catalysts (Scheme 16).35 The 10 mol% of CPN catalyzes the Michael addition of nitroalkanes such as nitropropane, nitroethane and cyclic nitroalkanes to aromatic and heteroaromatic nitro-olefins to yield 1,3-dinitro compounds (57) in good yield (73–82%) and enantioselectivity (64–88% ee) under neat condition.
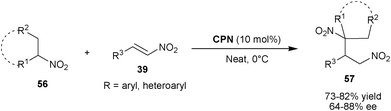 | ||
| Scheme 16 CPN catalyzed Michael addition of nitroalkanes (56) to nitroalkenes (39). | ||
Shi and co-workers reported the Michael addition of anthrone (58) to nitro-olefins (39) catalyzed by BzCPN to provide (59) in excellent yield (91–99%) and high enantioselectivity (80–99% ee) (Scheme 17).36 It is noteworthy that the anthrone acts as an active nucleophile rather than as an active diene.
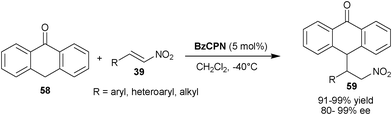 | ||
| Scheme 17 BzCPN catalyzed asymmetric Michael reaction of anthrone (58) with nitroalkenes (39). | ||
Very recently, Chauhan and Chimni have developed a highly regio- and stereoselective route for the synthesis of vicinal quaternary and tertiary stereocenters via 6′–OH Cinchona alkaloid catalyzed Michael addition of tri-substituted racemic nucleophiles (42) to nitrodienes (60) (Scheme 18).37 At low catalyst loading (0.5–5 mol%) of BnCPN or BnCPD, the nucleophiles such as cyclic and acyclic β-ketoesters as well as cyclic diketone were successfully added to aromatic and aliphatic nitrodienes to provide adduct (61) with adjacent quaternary and tertiary stereocenters in good to excellent yield (57–99%), high enantioselectivity (88–99% ee) and good to high diastereoselectivity (56:44–99:1 dr).
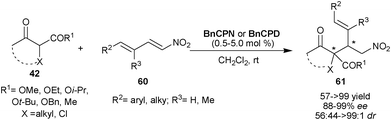 | ||
| Scheme 18 6′–OH Cinchona alkaloids catalyzed Michael addition of tri-substituted carbon nucleophiles (42) to nitrodienes (60). | ||
Deng and co-workers demonstrated the practical as well as preparative utility of PHNCPD and PHNCPN catalysts for asymmetric 1,4-conjgate addition of a diverse range of cyclic and acyclic α-substitued β-ketoesters to various α,β-unsaturated ketones and various α,β-unsaturated aldehydes to yield Michael adducts (62) and (63) bearing quaternary stereocenters in excellent yield (82–99%) and enantioselectivity (85–99% ee) (Scheme 19).38 In case of β-substituted α,β-unsaturated aldehydes a very good diastereoselectivity ranging from 18:1 to 25:1 has been observed. This approach was utilized for the concise total synthesis of (+)-tanikolide. The new cupreidine derived catalyst (66) catalyzes the Michael addition of α-aryl/heteroaryl α-cyanoacetate (64) to acrolein to provide (65) in quantitative yield and up to 95% ee (Scheme 19).
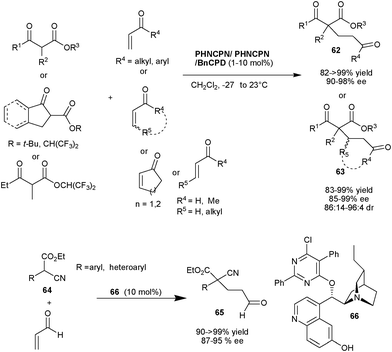 | ||
| Scheme 19 6′–OH Cinchona alkaloids catalyzed Michael addition of tri-substituted carbon nucleophiles to enones and enals. | ||
Rigby and Dixon reported the PHNCPD catalyzed 1,4-addition of various β-keto ester pro-nucleophiles to acrylic esters, thioesters and N-acryloyl pyrrole (Scheme 20).39 The 10 mol% of catalyst provides an access to all-carbon quaternary chiral carbon containing 1,4-adducts (68) from a diverse range of tri-substituted racemic nucleophiles (42) and unsaturated acceptor (67) in moderate to high yield (52–96%) and good to excellent enantioselectivity (67–98% ee).
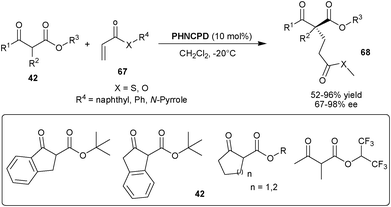 | ||
| Scheme 20 6′–OH Cinchona alkaloids catalyzed Michael addition to acrylic esters, thioesters and N-acryloyl pyrrole (67). | ||
Deng and co-workers developed the first enantioselective 1,4-addition of α-substituted α-cyanoacetates (64) to arylvinyl sulfones (69) (Scheme 21). The catalysts PHNCPN or PHNCPN provide conjugate adduct (70) with all-carbon quaternary stereogenic centre from α-aryl or heteroaryl α-cyanoacetate and phenyl vinyl sulfone in high yield (80–96%) and excellent enantioselectivity (88–97% ee).40 The reaction of α-alkyl α-cyanoacetate with phenylvinyl sulfone proceeded with slow rate but in case of 3,5-bis-(triflouromethyl)phenyl vinyl sulfone, a higher yield was obtained, which is attributed to the enhanced electrophilicity of sulfone. The corresponding 1,4-adduct was obtained in up to 94% ee. Later in 2009, they extended the scope of this reaction to α-cyanoketone and β-ketoesters. These nucleophiles add to the unsubstituted and β-substituted sulfones to yield 1,4-adduct in high stereoselectivity.41
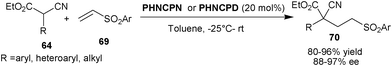 | ||
| Scheme 21 6′–OH Cinchona alkaloids catalyzed Michael addition of α-cyanoacetates (64) and vinyl sulfones (69). | ||
Very recently, Marini and co-workers have described an elegant organocatalytic protocol for the synthesis of chiral spirolactones (73) from cyclic β-ketoesters (71) and vinyl selenone (72) by a one-pot Michael addition-cyclization reaction (Scheme 22).42PHNCPN catalyzes the initial Michael reaction of β-ketoesters (71) with (72) in a highly enantioselective manner. The corresponding Michael adduct undergoes lactonization in the presence of silica gel to provide (73) in good to excellent yield (71–99%).
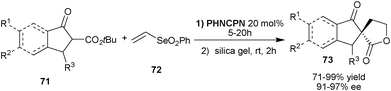 | ||
| Scheme 22 One-pot Michael addition-cyclization reaction catalyzed by PHNCPN. | ||
Loh et al. reported the highly efficient vinylogous Michael addition of α,α-dicyanoolefins (74) to the maleimides (75) using 6′–OH Cinchona alkaloids (Scheme 23).43 The BnCPD or 2-naphthyl cupreidine (NpCPD) catalysts have shown to be equally efficient for the addition from the γ-carbon of the various nucleophiles (74) to the N-protected maleimides (75) to provide the desired product (76) with anti-conformation in moderate to good yield (35–90%) and high enantioselectivity (80–99% ee).
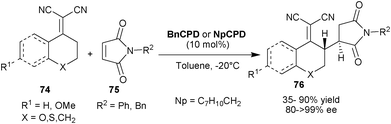 | ||
| Scheme 23 Asymmetric vinylogous Michael addition of α,α-dicyanoolefins (74) to maleimides (75). | ||
Deng and co-workers developed 6′–OH Cinchona alkaloid catalyzed organocatalytic asymmetric tandem Michael addition-protonation for the direct synthesis of two non adjacent quaternary and tertiary stereogenic centers (Scheme 24).44 The PHNCPN or PHNCPD (for cyclic nucleophile) or AcCPD (for acyclic nucleophile) catalyzes the asymmetric tandem reaction between carbon nucleophiles (77) and (78) with α-chloroacrylonitrile (79) in very good yield (60–95%) and good to excellent stereoselectivity (85–99% ee and 2:1–25:1 dr). This strategy was further extended for the asymmetric synthesis of (−)-manzacidin.
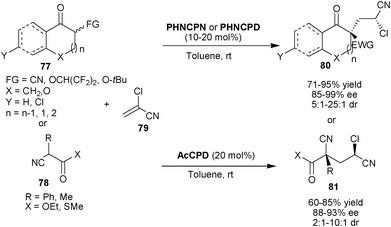 | ||
| Scheme 24 6′–OH Cinchona alkaloids catalyzed organocatalytic asymmetric tandem Michael addition-protonation reaction. | ||
Yuan and co-workers have developed cupreine catalyzed enantioselective construction of spiro[4H-pyran-3,3′-oxindoles] (84) via two or three component domino Knoevenagel–Michael-cyclization sequence (Scheme 25).45 A wide range of optically active (84) were obtained in excellent yield (up to 99%) with good to excellent enantioselectivity (up to 97%) from simple and readily available starting materials under mild reaction conditions. The two component reaction involves α,α-dicyano olefin derived from isatin (21) and malononitrile (83), and active methylene compounds (82) as reactant that tune into a CPN catalyzed domino sequence providing high yield and enantioselectivity. The three component reaction involves the in situ formation of α,α-dicyano olefin followed by Knoevenagel–Michael-cyclization reaction providing an access to spiro compound in good to high enantioselectivity.
![Cupreine catalyzed enantioselective synthesis of spiro[4H-pyran-3,3′-oxindoles] (84).](/image/article/2012/RA/c1ra00872b/c1ra00872b-s25.gif) | ||
| Scheme 25 Cupreine catalyzed enantioselective synthesis of spiro[4H-pyran-3,3′-oxindoles] (84). | ||
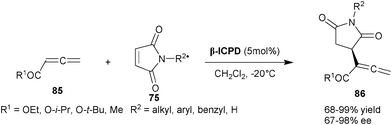 | ||
| Scheme 26 Enantioselective Rauhut–Currier reaction catalyzed by β-ICPD. | ||
 | ||
| Scheme 27 β-ICPD catalyzed asymmetric allylic alkylation of Morita–Baylis–Hilmann carbonates (87). | ||
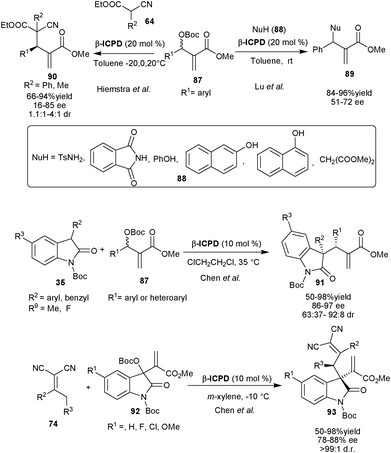
Later in 2007, Hiemstra et al. extended this approach to generate adjacent quaternary and tertiary stereocenters. The β-ICPD catalyzed the allylic alkylation of (87) with α-phenyl or methyl cyano esters (64) to generate vicinal quaternary and tertiary stereocenters in high yield (up to 94%) and moderate to good stereoselectivity (16–85% ee and 1.1:1–4:1 dr).49 The kinetic resolution of carbonate (87) has also been observed with 90% ee, when used in excess.
Chen and co-workers have reported allylic alkylation of 87 with N-protected oxindoles (35) catalyzed by β-ICPD.50 The various aromatic and heteroaromatic carbonates react with N-protected oxindole derivatives providing access to vicinal quaternary and tertiary stereocenters (91) with good to high yield (50–98% yield) and stereoselectivity (88–97% ee and 63:37–92:8 dr).
Very recently, the same research group has utilized the MBH carbonate of the isatin (92) for β-ICPD catalyzed asymmetric allylic alkylation with α,α-dicyanoolefins (74) to obtain 3-substituted oxindole derivatives (93) bearing vinical quaternary and tertiary stereocenter in good to high yield (50–98%) with good to excellent enantioselectivity (86–97% ee) and excellent diastereoselectivity (>99:1 dr).51
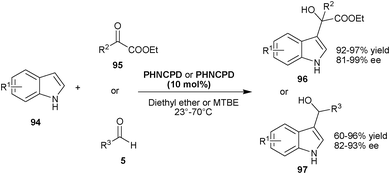 | ||
| Scheme 28 6′–OH Cinchona catalyzed asymmetric Friedel–Crafts reaction of indole (94) with carbonyl compounds (95) and (5). | ||
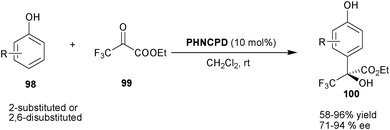
Chen and co-workers have reported an elegant protocol for the synthesis of chiral trifluoromethyl-substituted tertiary alcohols via asymmetric organocatalytic Friedel–Crafts reaction of various 2-substituted and 2,6-disubstituted phenols (98) with trifluoropyruvates (99) (Scheme 29).54 With 10 mol% PHNCPD, alkylation of substituted phenols have been performed regioselectively at the 4-position of phenol to afford the Friedel–Crafts adducts (100) in moderate to good yield (58–96%) and good enantioselectivity (71–94% ee).
 | ||
| Scheme 29 Asymmetric synthesis trifluoromethyl-substituted tertiary alcohols via Friedel–Crafts reaction (100). | ||
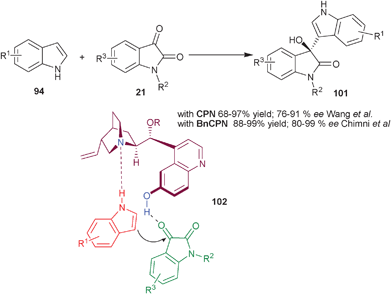
Wang and co-workers have reported a highly efficient CPN catalyzed Friedel–Crafts reaction of indole and isatin derivatives to provide 3-indolyl-3-hydroxyoxindoles (101) (Scheme 30).55 20 mol% of CPN and benzoic acid catalyzes the Friedel–Crafts reaction of indole derivatives with different isatins in dioxane yielding chiral (101) in high yield (68–97%) and good enantioselectivity (76–91% ee).
 | ||
| Scheme 30 6′–OH Cinchona alkaloids catalyzed asymmetric synthesis of 3-indolyl-3-hydroxyoxindole derivatives (101). | ||
Concurrently, Chauhan and Chimni have developed a highly enantioselective Friedel–Crafts reaction of indole and isatin derivatives catalyzed by BnCPN (Scheme 30).56 The 3-indolyl-3-hydroxyoxindole derivatives (100) were synthesized in high yield (88–99%) and good to excellent enantioselectivity (80–99% ee) under mild reaction conditions. The methodology was free from competing double addition of indole to isatin. On the basis of several experiments the bifunctional mode of activation of the catalyst was demonstrated. The protection of either 6′–OH or quinuclidine N results in the failure of the reaction. This was further proven by the lack of product formation when N-methyl indole was used. The proposed transition state (102) involves a ternary complex between the catalyst, isatin and indole, in which tertiary amine activate and orient the indole by forming hydrogen bond with NH of indole and aromatic OH group activate the isatin with hydrogen bonding.
Very recently, Wang et al. have reported the organocatalytic asymmetric aza-Friedel–Crafts reaction of 1-naphthols (103) with N-sulfonyl aldimines (23) catalyzed by PHNCPD to provide chiral aminonaphthol derivatives (104) (Scheme 31).57PHNCPD catalyzes the Friedel–Crafts reaction of 1-naphthol derivatives with aryl, heteroaryl and alkyl sulfonyl imines to provide (104) with good to quantitative yield (62–100%) and high enantioselectivity (80–96% ee). Only a moderate level of enantioselectivity was achieved in the case of 2-naphthol (105).
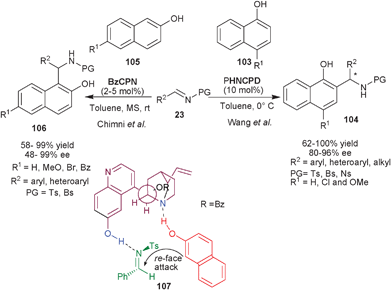 | ||
| Scheme 31 6′–OH Cinchona alkaloids catalyzed asymmetric aza-Friedel–Crafts reaction of naphthols. | ||
Chauhan and Chimni have applied the catalytic potential of 6′–OH Cinchona alkaloids for the aza-Friedel–Crafts reaction of 2-naphthols (105) with N-sulfonyl imines (23) (Scheme 31).58BzCPN catalyzes the formation of aza-Friedel–Crafts products (106) from 2-naphthol derivatives and N-tosyl imines in good to high yield (58–99%) and moderate to excellent enantioselectivity (48–99%). This methodology was successfully extended for aza-Friedel–Crafts reaction of 1-naphthols (103) with N-tosyl imines as well, the aminonaphthol adducts (104) were obtained in very good yield (68–76%) and enantioselectivity (80–98% ee). The bifunctional mode of catalysis has been established on the basis of designed experiments and the transition state (107) was proposed to involve the ternary complex of catalyst, naphthol and imine in which the phenolic group of the catalyst activates the imine through hydrogen bonding and the tertiary amine moiety activate and orient the naphthol for the re-face attack to provide S-enantiomer of the product.
 | ||
| Scheme 32 BnCPD catalyzed asymmetric Henry reaction of aldehydes. | ||
Deng and co-workers have developed the first organocatalytic asymmetric Henry reaction of nitromethane with α-ketoesters (95) (Scheme 33).61 With BzCPN or BzCPD, the chiral β-nitro alcohols (109) were obtained in high yield (84–99%) and excellent enantioselectivity (93–97% ee). The remarkable finding of this study was that, the β,γ-unsaturated α-ketoesters undergo 1,2-addition of nitromethane, instead of 1,4-addition with high chemoselectivity. The high enantioselectivity was not only obtained for alkenyl α-ketoesters, but a wide variety of aryl and alkyl α-ketoesters also provide access to chiral tertiary alcohols. The chiral nitroaldol product so formed was utilized for the new and concise asymmetric synthesis of aziridines (110), β-lactams (111) and α-methylcysteine derivatives (112).
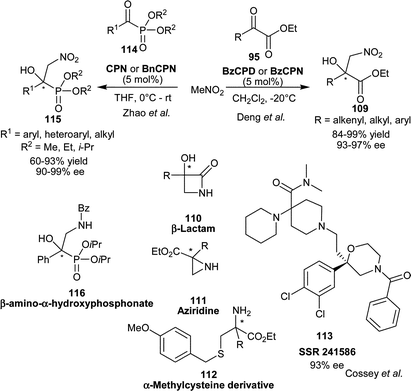 | ||
| Scheme 33 6′–OH Cinchona alkaloids catalyzed asymmetric Henry reaction of α-ketoesters (95) and α-ketophosphonates (114). | ||
Cossy and co-worker have extended the application of BzCPN catalyzed Henry reaction of α-ketoester with nitromethane for enantioselective synthesis of SSR 241586 (113) a 2,2-disubstituted morpholine active in the treatment of schizophrenia and irritable bowel syndrome (IBS) (Scheme 33).62
Zhao and co-workers have reported CPN or BnCPN catalyzed organocatalytic asymmetric nitroaldol reaction of α-ketophosphonates (114) and nitromethane.63 Both catalysts proved to be equally efficient, providing α-hydroxy-β-nitrophosphonates (115) in good yield (60–93%) and excellent enantioselectivity (90–99% ee) using aromatic as well as aliphatic α-ketophosphonates. The chiral nitro-alcohol (115) was transformed into enantioenriched β-amino-α-hydroxyphosphonate (116) by reduction and subsequent benzylation (Scheme 33).
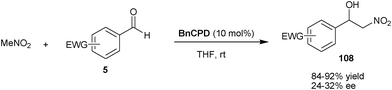
Bandini and co-workers developed the asymmetric organocatalytic nitroaldol reaction of fluoromethyl ketones (117) utilizing the catalytic applications of 6′–OH Cinchona alkaloids (Scheme 34).64 They found remarkable enantioenrichment by introduction of an electron withdrawing group on the benzoyl group at C9 of cupreine. A newly developed catalyst (119), catalyzes the Henry reaction of nitromethane with tri- and di-fluoromethyl ketone (117), resulting in the formation nitroaldol product (118) in moderate to high yield (67–99%) and excellent enantioselectivity (76–99% ee).
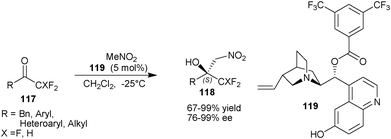 | ||
| Scheme 34 Asymmetric organocatalytic nitroaldol reaction of fluoromethyl ketones (117). | ||
Very recently, Wei Wang and co-workers of the University of New Mexico have reported the first asymmetric Henry reaction of isatin derivatives catalyzed by cupreine (Scheme 35).65CPN catalyzes the nitro-aldol reaction of nitroalkanes such as nitromethane, nitroethane and nitropropane (56) with various isatin derivatives (21) to provide an easy access to chiral 3-substituted 3-hydroxyoxindole derivatives (120) in excellent yield and good to high stereoselectivity.65a The chiral adduct of this reaction was successfully transformed to oxindole alkaloids such as (+)-dioxibrassinin and (S)-(−)-spirobrassinin.
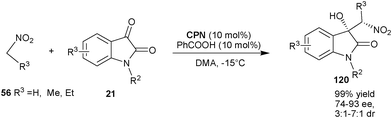 | ||
| Scheme 35 CPN catalyzed Henry reaction of isatin derivatives (21). | ||
Simultaneously with this, two independent reports have been published on similar transformations catalyzed by 6′–OH Cinchona alkaloids. Xing-Wang Wang and co-workers have reported the synthesis of hydroxylated quaternary chiral carbon on oxindole viaBnCPN catalyzed asymmetric Henry reaction of nitromethane with isatin derivatives.65b The corresponding nitro-aldol product was isolated in good to high yield (90–98%) and moderate to good enantioselectivity (72–95% ee). Wei Wang et al. at Lanzhou University have reported the 6′–OH Cinchona alkaloid (119) catalyzed asymmetric Henry reaction of nitromethane with isatin derivatives in very high yield (95–97%) and moderate to good enantioselectivity (71–92% ee).65c
 | ||
| Scheme 36 Hydroxymethylation of α-substituted nitroacetates (121). | ||
2.2 Carbon-hetero-atom bond formation
2.2.1.1 Aminaton reaction. Jørgensen and co-workers have reported highly enantioselective organocatalytic amination of α-substituted α-cyanoacetate (64) with di-tert-butyl azodicarboxylate (124) catalyzed by β-ICPD to yield aminated product (125) with quaternary stereocenter (Scheme 37).67 The diverse range of variation in the nucleophile provided (125) in excellent yield (84–99%) and enantioselectivity (91–98% ee) with remarkably low catalyst loading (0.1–5 mol%). β-ICPD also catalyzes the amination of open chain as well as cyclic β-ketoesters and β-diketones with di-tert-butyl azodicarboxylate (DTAD) to provide the aminated adducts (126) and (127) with high yield (90–99%) and enantioselectivity (83–90% ee).
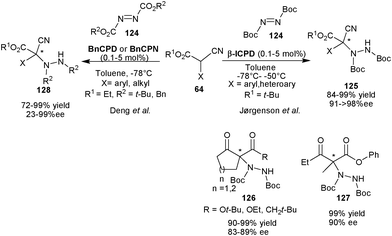 | ||
| Scheme 37 6′–OH Cinchona alkaloids catalyzed asymmetric amination reaction. | ||
In 2005, Deng et al. have used BnCPD or BnCPN to obtain both enantiomers of the aminated product (128) in high yield (72–99%) and moderate to high enantioselectivity (23–99% ee) by asymmetric amination of α-aromatic substituted α-cyanoacetate (64) with tert-butyl/benzyl azodicarboxylate (124) (Scheme 37).68a The aliphatic α-cyanoacetate resulted in low enantiomeric excess. Later in 2009, Deng's group established a highly enantioselective amination of acyclic α-alkyl β-keto thioesters and trifluoroethyl α-methyl α-cyanoacetate with as low as 0.05 mol% of BnCPN or BnCPD.68b The corresponding aminated products were isolated in 91–99% yield and 81–90% ee.
Jørgensen and co-workers have reported an elegant protocol for the asymmetric catalytic Friedel–Crafts amination of 8-amino 2-naphthol (129) with azodicarboxylate (124) using 20 mol% demethylated quinine (131) catalyst in excellent yield (87–98%) and enantioselectivity (94–98% ee) (Scheme 38).69a The authors have envisioned that cupreidine and cupreine might themselves be substrates for this reaction.69b Although cupreidines do not undergo reaction with DTAD (124) under the optimized conditions, but they could be efficiently aminated at the C′–5 position simply by changing solvent and temperature. Remarkably, the resulting compounds (132) and (133) were shown to be even better catalysts for the amination of aminonaphthols. C5–Aminated cupreidines were also shown to promote enantioselective Michael addition of cyclic β-ketoesters to acrolein and methyl vinyl ketones with good enantiomeric excess, although with no real advantage over the parent catalysts.
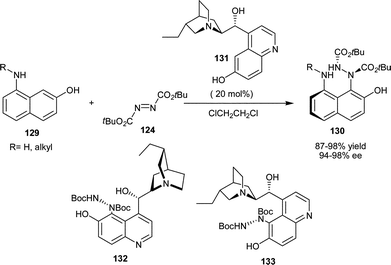 | ||
| Scheme 38 Asymmetric amination of amino-naphthol catalyzed by 6′–OH Cinchona alkaloids. | ||
Very recently, Shi and co-workers have disclosed 6′–OH Cinchona alkaloid catalyzed biomimetic transamination of α-ketoesters (95) using benzylamines derivatives (Scheme 39).70 With 10 mol% of catalyst (135) a wide variety of α-amino esters (134) containing various functional groups have been synthesized in high enantioselectivity (88–92% ee) and reasonable yield (47–71%).
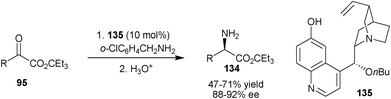 | ||
| Scheme 39 Enantioselective transamination of α-ketoesters (95). | ||
2.2.1.2 aza-Michael reaction. The asymmetric organocatalytic addition of amine to unsaturated acceptors i.e. the aza-Michael reaction is a convenient method for the synthesis of chiral amine derivatives.71 The application of cupreine for the asymmetric aza-Michael reaction of nitrogen heterocycles to nitro-olefins (39) was explored by Wang and co-workers (Scheme 40).72CPN catalyzes the conjugate addition of benzotriazole (136) from N1 to aromatic, heteroaromatic and aliphatic nitro-olefins (39) in moderate to good yield (64–90%) and moderate to high enantioselectivity (57–94% ee). The conjugate addition of other N–heterocycles such as 1H–[1,2,3]triazole and 5-phenyl-1H-tetrazole to the nitro-olefin took place with moderate yield (65% and 76%, respectively) and with good enantioselectivity (84% and 85%, respectively).
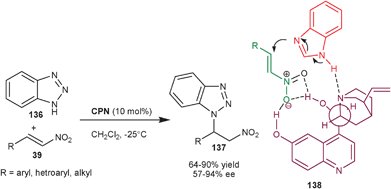 | ||
| Scheme 40 CPN catalyzed enantioselective aza-Michael reaction of benzotriazole with nitroalkenes. | ||
2.2.1.3 N-Nitroso-aldol reaction. Chen and co-workers have reported an organocatalytic asymmetric N-nitroso-aldol reaction of 2-oxindoles (35) with nitrosobenzene (139) catalyzed by CPN (Scheme 41).73 The corresponding products (140) having a quaternary stereocenter were obtained in good to excellent yield (71–100%) and moderate enantioselectivity (59–72% ee).
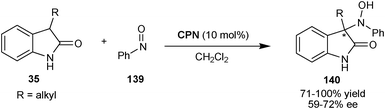 | ||
| Scheme 41 Asymmetric N-nitroso-aldol reaction catalyzed by CPN. | ||
Zhao et al. reported asymmetric synthesis of fluorinated flavanone (142) by an organocatalytic intramolecular oxa-Michael addition of phenol to chalcone followed by base mediated electrophilic fluorination with N-fluorobenzenesulfonimide (NFSI) (Scheme 41).76 The quinidine derived catalyst (143) catalyzes the intramolecular tandem reaction of substituted alkylidene β-ketoesters (141) in good to excellent yield (56–99%) and moderate to high enantioselectivity (17–96% ee) of single diastereomer. In the proposed transition state (144) bifunctional catalyst simultaneously activate the oxygen nucleophile with tertiary amine assisting its deprotonation, while 6′–OH activate the two carbonyl oxygen in unsaturated part of the substrate by hydrogen bonding, thus facilitating the re-face attack to generate the (R) enantiomer.
Recently, the same research group have reported an intramolecular oxa-Michael addition/decarboxylation reaction (Scheme 42).77 The alkylidene β-ketoesters (141) undergoes oxa-Michael addition in the presence of BnCPD, followed by an acid mediated decarboxylation reaction to provide an easy access to a series of chiral flavanone derivatives (145) with moderate to good enantioselectivity (60–90% ee) and good to high yield (85–97%). A simple extension of this work to synthesize flavanones (146) with varied functionalities, the base catalyzed electrophilic cascade reaction was successfully performed using electrophiles such as methyl vinyl ketone (MVK), N–bromosuccinimide (NBS) and N–chlorosuccinimide (NCS).
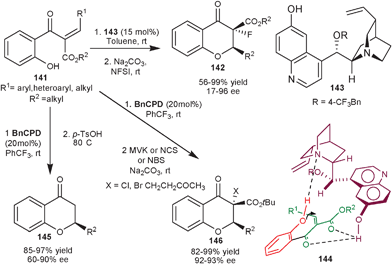 | ||
| Scheme 42 6′–OH Cinchona alkaloids catalyzed synthesis of chiral flavanone derivatives involving the oxa-Michael addition as key step. | ||
Xiao and co-workers have developed asymmetric organocatalytic conjugate addition of oximes (147) to trisubstituted β-nitroacrylate (148) (Scheme 43).78 The 6′–OH Cinchona alkaloids derived from quinine (151) and quinidine (150) provide the adduct (149) in good to high yield (91–98%) and moderate to high enantioselectivity (61–93% ee). The formation of (S) enantiomer of the product from quinidine derived catalyst was elucidated by the formation of ternary complex in the transition state (152) involving oxime, β-nitroacrylate and the catalyst. The 6′–OH group of the catalyst act as hydrogen bond donor that activate the nitroalkene and the tertiary amine activates and orients the nucleophilic attack of oxime on the si-face of the β-nitroacrylate rather than re-face attack. The later attack seems to be disfavoured due to steric reasons.
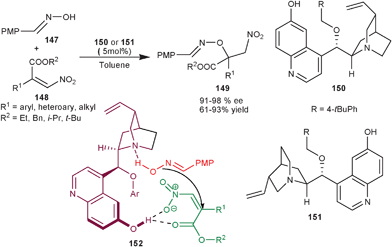 | ||
| Scheme 43 Asymmetric synthesis of protected tertiary alcohol (149) via 6′–OH Cinchona alkaloids catalyzed oxa-Michael reaction. | ||
:35–77:23 dr) and good enantioselectivity (72–86% ee). The thiochromone derivatives with high diastereoselectivity (87:13–9:1 dr) and enantioselectivity (85–99% ee) were obtained, after single crystalization. In the proposed transition state (155), CPN acts as a bifunctional catalyst which activates the nitro-group of nitroalkene with 9–OH and 6′–OH group and the tertiary amine of the catalyst activates the sulfur nucleophile for the re-face attack.
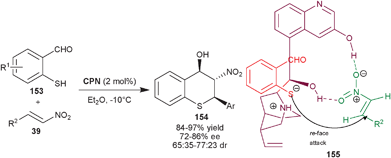 | ||
| Scheme 44 CPN catalyzed tamdem sulfa-Michael–Henry reaction. | ||
Very recently, Melchiorre and co-workers have developed diastereodivergent asymmetric sulfa-Michael additions to α-branched enones using a single organocatalyst, by applying an external chemical stimulus (Scheme 45).80a The quinidine derived organocatalyst (159) bearing primary-tertiary amine and phenolic moiety and 2-flourobenzoic acid as additive catalyzes the Michael addition of various aliphatic sulfur nucleophiles (157) to the various α-branched enones (156) to provide the major syn diastereomer of Michael adducts (158) in moderate to good yield (40–79%), good to high enantioselectivity (61–90% ee) and good diastereoselectivity (syn:anti 2.8:1–9.3:1dr). The anti Michael adducts (158′) were obtained in moderate to good yield (43–80%), high enantioselectivity (83–99% ee) and good diastereoselectivity (anti:syn 2.0:1–7.2:1 dr) using phosphoric acid (160) or diphenyl phosphoric acid (DPP) as additive. The cooperative catalytic system of (159) and (160) has also been used for direct asymmetric γ-alkylation of α-branched enals.80b
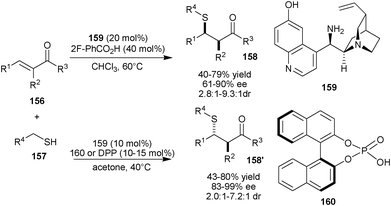 | ||
| Scheme 45 Diastereodivergent asymmetric sulfa-Michael additions to α-branched enones. | ||
2.3 Cycloaddition reaction
Deng and co-workers developed the asymmetric Diels–Alder reaction of pyrones (156) with various unsaturated dienophiles catalyzed by bifunctional 6′–OH Cinchona alkaloids, The catalysts simultaneously raised the energy of the HOMO of the diene and lowered the energy of the LUMO of the dienophile (Scheme 46).81 The 6′–OH Cinchona alkaloid catalysts (164) or (165) catalyzes synthesis of [4 + 2] adduct (163) with adjacent quaternary chiral carbon in good to high yield (63–100%) and stereoselectivity (82–94% ee and 85:15–95:5 dr), by the reaction of pyrone derivatives (161) with unsaturated ester/ketones (162). The catalyst (164) and (165) catalyzes the reaction of pyrone with α-chloroacrylonitrile (79) to provide the cycloaddition product in high yield (85% and 90%, respectively) and good stereoselectivity (85% and 75% ee; 87:13 and 78:22 exo:endo, respectively).
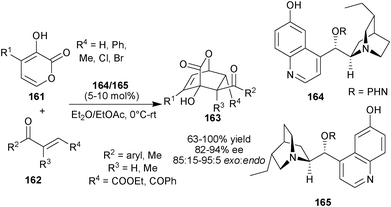 | ||
| Scheme 46 Asymmetric organocatalytic Diels–Alder reaction catalyzed by 6′–OH Cinchona alkaloids. | ||
Very recently, Deng and co-workers have developed a new series of bifunctional 6′–OH Cinchona alkaloids bearing bulky silylated groups at C9, for the first asymmetric [4 + 2] cycloaddition reaction between 2-pyrones (161) and aliphatic nitroalkenes (39) (Scheme 47).82 The corresponding endo-[2.2.2] bicyclic adducts (166) were obtained in good yield (56–86%) with excellent diastereo- and enantioselectivity (95–98% ee) using 167 as the catalyst. On the basis of a carbon kinetic isotope effect study, it was proved that the reaction did not occur through a concerted pathway, but instead involves a stepwise pathway.
![Asymmetric [4 + 2] cycloaddition reaction of 2-pyrones (156) with aliphatic nitroalkenes (39).](/image/article/2012/RA/c1ra00872b/c1ra00872b-s47.gif) | ||
| Scheme 47 Asymmetric [4 + 2] cycloaddition reaction of 2-pyrones (156) with aliphatic nitroalkenes (39). | ||
Chen and co-workers have reported the first organocatalytic enantioselective 1,3-dipolar cycloaddition of cyclic enones (168) and azomethine imines (169), by using multi-functional primary amine catalysts (159) or (171) derived from Cinchona alkaloids (Scheme 48).83 A wide range of cycloaddition products (170) have been synthesized in good to excellent yield and with excellent stereoselectivity by using 10 mol% of catalyst and 2,4,6-triisopropylbenzenesulfonic acid as acid additive in THF. The phenolic OH at the C′–6 position provide an additional site for hydrogen bonding interaction of catalyst and 1,3-dipole to introduce high stereocontrol (85–95% ee, >99:1 dr). In plausible transition state (172), the ketiminium cation adopt trans conformation between catalyst and enone. The aromatic OH activates ketiminium cation with hydrogen bonding and the steric hindrance exerted by the ion pair of the tertiary amine moiety enforce endo- and re-face selectivity to give the desired cycloaddition product.
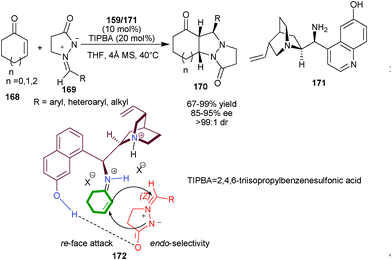 | ||
| Scheme 48 Organocatalytic enantioselective 1,3-dipolar cycloaddition reaction. | ||
Gong and co-workers have developed elegant asymmetric [3 + 2] cycloaddition reaction of isocyanoesters (173) to nitroolefins (39) and aldehydes (5) leading to the formation of highly substituted 2,3-dihydropyrrole (174) and 2-oxazoline (175) derivatives, respectively (Scheme 49).84–85 The C9 benzoyl ester of cupreine and cupreidine (BzCPN and BzCPD) efficiently catalyzes the cycloaddition of aryl or benzyl isocyanoacetate (169) with aryl, heteroaryl as well alkyl nitroalkenes (39) giving access to various dihydropyrrole derivatives (174) in moderate to high yield (52–99%) and excellent stereoselectivity (90–99% ee and 4:1–20:1 dr).84 The C9 α-naphthyl ester of cupreine (α-C7H10COCPN) catalyzes the asymmetric synthesis of oxazolines (175) via [3 + 2] cycloaddition of (173) with various aromatic, heteroaromatic and aliphatic aldehydes (5) in moderate to good yield (18–95%) and with moderate to high stereoselectivity (31–90% ee, 1:1–18:1 dr).85 The reaction mechanism seems to involve the initial tertiary amine catalyzed Michael addition of isocyanoester to nitro-olefin or aldol type addition to aldehyde, which is activated and positioned by the 6′–OH of the catalyst.
![[3 + 2] cycloaddition reaction of isocyanoesters catalyzed by 6′–OH Cinchona alkaloids.](/image/article/2012/RA/c1ra00872b/c1ra00872b-s49.gif) | ||
| Scheme 49 [3 + 2] cycloaddition reaction of isocyanoesters catalyzed by 6′–OH Cinchona alkaloids. | ||
Recently, Cai and co-workers reported a highly enantioselective [4 + 2] cycloaddition reaction of β,γ-unsaturated α-keto esters (162) with oxazolones (176) catalyzed by 6′–OH Cinchona alkaloid (Scheme 50).86 Using 20 mol% of α-C7H10COCPN, a series of highly functionalized δ-lactones (177) with vicinal quaternary and tertiary stereocenters have been synthesized in high yield (44–96%) and moderate to excellent enantioselectivity (20–97% ee).
![Enantioselective [4 + 2] cycloaddition reaction of β,γ-unsaturated α-keto esters (157) with oxazolones (174).](/image/article/2012/RA/c1ra00872b/c1ra00872b-s50.gif) | ||
| Scheme 50 Enantioselective [4 + 2] cycloaddition reaction of β,γ-unsaturated α-keto esters (157) with oxazolones (174). | ||
In 2001, Romo and co-workers have developed an elegant procedure for an intramolecular ketene-aldehyde formal [2 + 2] cycloaddition for the synthesis of bicyclic cis-lactones (178) catalyzed by O–acetylquinidine (Scheme 51).87 With β-ICPD complete reversal of asymmetric induction with identical level of stereoinduction was observed.
![Asymmetric intramolecular ketene-aldehyde formal [2 + 2] cycloaddition.](/image/article/2012/RA/c1ra00872b/c1ra00872b-s51.gif) | ||
| Scheme 51 Asymmetric intramolecular ketene-aldehyde formal [2 + 2] cycloaddition. | ||
2.4 Rearrangement
Toste and co-workers utilized the catalytic potential 6′–OH Cinchona alkaloids for the synthesis of γ-hydroxyenones (180) via Kornblum DeLaMare rearrangement (Scheme 52).88AcCPN and AcCPD catalyzes the enantioselective desymmetrization of various substituted bicyclic endoperoxides (179) to γ-hydroxyenones (180) in good to excellent yield (76–99%) and moderate to excellent enantioselectivity (50–99% ee). On the basis of their experimental observation they proposed a transition state (181) in which quinuclidine N and 6′–OH group of the catalyst interact with the endoperoxide via hydrogen bonding, in such a manner that the alkene gets positioned in the sterically demanding pocket covered by the quinoline ring.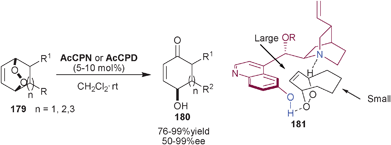 | ||
| Scheme 52 Asymmetric synthesis of γ-hydroxyenones (180) via Kornblum DeLaMare rearrangement. | ||
Tu and co-workers developed catalytic enantioselective vinylogous α-ketol rearrangement of vinylogous α-hydroxy enones (182) by the application of iminium ion catalysis affording spirocyclic ketones (183) with an all-carbon stereogenic centre (Scheme 53).89 The N-Boc-L-phenylglycine salt of 6′–OH Cinchona alkaloid derived 9-amino (171) catalyzes semipinacol-type 1,2-carbon sigmatropic migration that converts cyclobutanol moiety into a cyclopentanone with good to high yield (57–95%) and moderate to high enantioselectivity (48–97% ee) and high diastereoselectivity (2.3:1–29:1% dr).
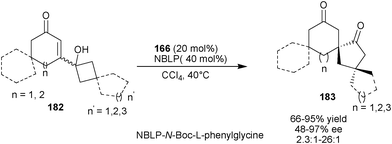 | ||
| Scheme 53 Asymmetric semipinacol-type 1,2-carbon sigmatropic migration catalyzed by 6′–OH Cinchona alkaloids. | ||
2.5 Oxaziridination
Jørgensen and co-workers have developed the first catalytic enantioselective oxaziridination reaction of N-tosyl imines (23) catalyzed by 6′–OH Cinchona alkaloid (Scheme 54).90 The oxidation of aryl and alkyl aldimines with m-CPBA in the presence of cupreidine derivative (164), to provide the optically active oxaziridine (184) in good yield (50–97%) and moderated to high enantioselectivity (46–94% ee).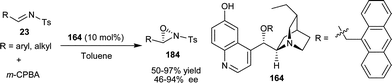 | ||
| Scheme 54 Asymmetric oxaziridination of N-tosyl imines. | ||
2.6 Isomerization
Recently, Deng and co-workers have developed an unprecedented and general method for enantioselective olefin isomerization via biomimetic proton transfer catalysis with a new 6′–OH Cinchona alkaloid organocatalyst (187) (Scheme 55).91 A broad range of mono- and disubstituted β,γ-unsaturated butenolides (185) were transformed into the corresponding chiral α,β-unsaturated butenolides (186) in good to high yield (56–95%) and good enantioselectivity (81–94% ee) using as low as 0.1 mol% of catalyst.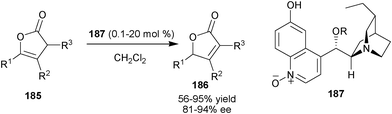 | ||
| Scheme 55 Enantioselective isomerisation of disubstituted β,γ-unsaturated butenolides (185). | ||
2.7 Dynamic kinetic resolution (DKR)
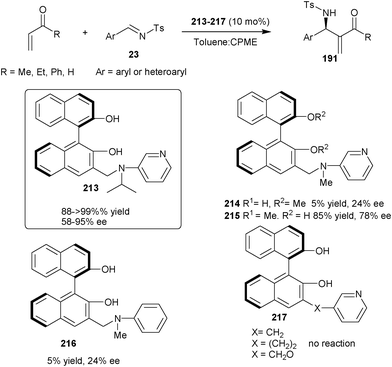
Very recently, Connon and co-workers have developed a new library of Cinchona alkaloids derived bifunctional organocatalyst bearing phenolic moiety at the C9 position (Scheme 56).92 The newly developed organocatalyst (189) catalyzes the DKR of azlactone (172) to provide (188) with good to excellent yield (91–96%) and high enantioselectivity (83–100% ee).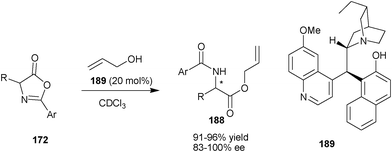 | ||
| Scheme 56 Asymmetric DKR of azlactone (172) catalyzed with (189). | ||
3. BINOL derivatives
The enantiomeric atropisomers of 1,1′-binaphthyl-2,2′-diol (BINOL) are the best-known representatives of axially chiral molecules, that are widely used for both stoichiometric and catalytic asymmetric reactions.93 In recent years, BINOL and its derivatives have been shown to be a powerful organocatalyst for a wide variety of organic transformations.3.1 BINOL derived organocatalysts bearing aromatic hydroxyl and tertiary phosphino groups
The catalytic potential of the bifunctional organocatalysts bearing tertiary phosphine in cooperation with aromatic OH groups derived BINOL are being discussed in this section.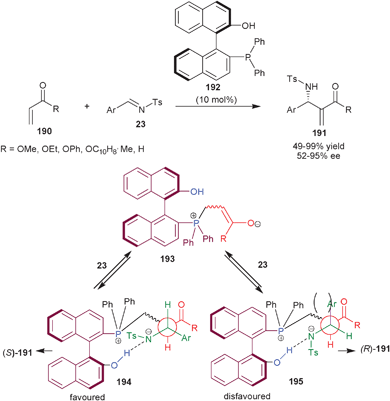 | ||
| Scheme 57 Asymmetric aza-MBH reaction catalyzed by (192). | ||
Shi and co-workers have demonstrated the application of dimethyl phosphine catalyst (198) with enhanced nucleophilicity at a phosphorous centre for catalyzing aza-MBH reaction of less reactive substrate such as 2-cyclopenten-1-one (196) and 2-cyclohexene-1-one with N-tosyl imines (23) (Scheme 58).94b The aza-MBH adducts (197) in good to high yield and moderate enantioselectivity were obtained with 10 mol% of the catalyst (198).
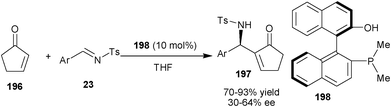 | ||
| Scheme 58 Asymmetric aza-MBH reaction of cyclic enones (196). | ||
After the successful establishment of this nucleophilicity effect Shi and co-workers synthesized a new series of bifunctional catalysts (199) having aromatic hydroxyl group and alkylated phosphine moiety in order to effect the nucleophilicity and the steric hindrance at Lewis base site of the catalyst.95 The catalyst bearing butyl chain efficiently catalyzes the aza-MBH reaction of MVK with N-tosyl imines in short duration with high yield (80–96%) and moderate to high enantioselectivity (44–88% ee) (Fig. 4).
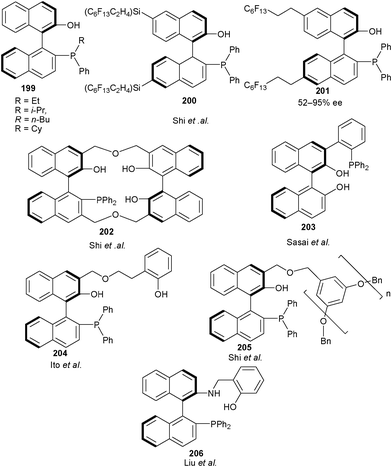 | ||
| Fig. 4 BINOL derived bifunctional organocatalysts for asymmetric aza-MBH reaction. | ||
The more efficient catalyst bearing long perfluoroalkane chains so called ‘pony tail’ at the naphthalene framework of the chiral phosphine catalysts (200) and (201) catalyzes aza-MBH reaction of N-tosyl imines with MVK giving aza-MBH adduct in good yield (53–98%) and moderate to high enantioselectivity (52–95% ee).96
Working on the hypothesis that the presence of multiple phenolic groups on the catalyst can stabilize the phosphonium enolate of aza-MBH reaction via hydrogen bonding, Shi and co-workers synthesized chiral phosphine catalyst (202) bearing three phenolic groups.97 This multiple phenolic catalyst, catalyzes aza-MBH reaction of MVK, ethyl vinyl ketone or acrolein with N-sulfonyl imines with good to high yield (67–97%) and enantio-induction (72–99% ee).
Sasai and co-workers have developed new organocatalysts by functionalizing the 3-position of BINOL with a series of aryl phosphines.98 10 mol% of catalyst (203) was found to catalyze the aza-MBH reaction of substituted aromatic and heteroaromatic N-tosyl imines with alkyl and phenyl vinyl ketone in good to quantitative yield (85–99%) and good to high enantioselectivity (82–95% ee).
In 2007, Ito et al. developed biphenol based bifunctional catalyst (204) for aza-MBH reaction of MVK with various N-tosyl imines.99 Remarkably, low catalyst loading of 1 mol% provides the aza-MBH adducts in good to excellent yield (84–100%) and high enantioselectivity (87–96% ee).
Shi and Liu developed a more efficient dendrimer immobilized bifunctional catalyst (205) for aza-MBH reaction of N-sulfonyl imine with vinyl ketone or acrolein.100 The catalyst could be easily separated by simple filtration and recycled without much loss in the catalytic activity.
The application of asymmetric counterion directed organocatalysis for aza-MBH reactions was explored by Liu and co-workers using new trifunctional organocatalyst (206).101 The protonation of Brønsted base part of catalyst with benzoic acid switches the activity of the catalyst, thus catalyzing the aza-MBH reactions between electron-deficient or electron rich aromatic N-tosyl imines and MVK at ambient temperature in short duration in moderate to high yield (26–99%) and good to high enantioselectivity (76–97% ee).
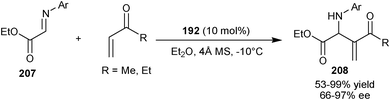
Shi and Zhao found that catalyst (192) could not give good enantiomeric excess in the reaction of N-arylmethylidenediphenylphosphinamides (26) with activated alkenes such as MVK, acrylonitrile, or phenyl acrylate.102 However, later in 2007, they developed bifunctional chiral phosphine Lewis base catalyzed asymmetric aza-MBH reaction of ethyl (arylimino)acetates (207) with α,β-unsaturated ketones (Scheme 59).103 10 mol% bifunctional catalyst (192) in the presence of 4 Å molecular sieves (MS) additive, catalyzes the aza-MBH reaction of (207) with MVK or EVK providing corresponding adducts (208) in moderate to good yield (53–99%) and enantioselectivity (66–97% ee).
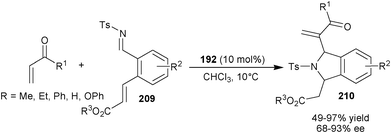
Recently, Sasai and co-workers have reported first domnio process involving aza-MBH/intramolecular aza-Michael reaction of electron deficient alkenes and N-tosyl imines (209) catalyzed by (192) (Scheme 60).104 The corresponding cis 1,3-disubstituted isoindolin (210) were obtained in good to excellent yield (49–97%) with high diastereo- and enantioselectivity (63–93% ee).
 | ||
| Scheme 60 Asymmetric organocatalytic aza-MBH/intramolecular aza-Michael reaction. | ||
3.2 BINOL derived organocatalysts bearing aromatic hydroxyl and tertiary amino group
 | ||
| Scheme 62 (S)-3-(N-isopropyl-N-3-pyridinylaminomethyl)BINOL (213) catalyzed asymmetric aza-MBH reaction. | ||
Later on, in 2010, they developed two new bifunctional catalysts (219) and (220) bearing phenolic hydroxyl groups and imidazole unit (Scheme 63).108 Both the catalyst catalyzes the aza-MBH reaction of nitroalkenes (39) with N-tosyl imines (23) to afford the MBH adduct (218) in low to good yield (14–98%) and low to moderate enantioselectivity (21–57% ee).
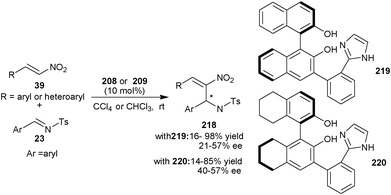 | ||
| Scheme 63 Asymmetric organocatalytic aza-MBH reaction of nitroalkenes (39) with N-tosyl imines (23). | ||
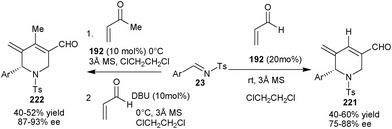 | ||
| Scheme 64 Asymmetric organocatalytic aza-MBH/aza-Michael/aldol/dehydration reaction. | ||
Very recently, Chen and co-workers have reported asymmetric variant of cross-Rauhut–Currier/Michael/aldol condensation triple domino reaction between acrylaldehyde and alkene (223) catalyzed by (192) (Scheme 65). The adduct (224) was obtained in 35% yield and 32% and 4% ee with cis-selectivity.110
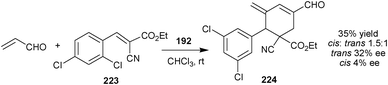 | ||
| Scheme 65 Cross-Rauhut–Currier/Michael/aldol condensation triple domino reaction. | ||
4. Miscellaneous organocatalysts
Sasai and coworkers have developed a unique spiro-type organocatalyst (225) bearing an aromatic hydroxyl group and phosphine as Brønsted acid and Lewis base unit (Scheme 66).111 This newly synthesized bifunctional organocatalyst catalyzes the aza-Morita–Baylis–Hillman reactions of unsaturated ketones with N-tosyl imines (23) in good to high yield (72–99%) and enantioselectivity (85–98% ee). Interestingly, spiro-type organocatalyst (225) provides aza-MBH adduct (191) with an enantioselectivity higher than those achieved using a BINOL derived catalyst (192). The spiro organocatalyst was assumed to provide a geometrically distinct and more rigid chiral pocket than its BINOL-derived counterpart. The rigid spiro catalyst backbone could reduce the conformational flexibility in the transition state for the catalyzed reaction resulting in the formation of the aza-MBH adducts with excellent enantioselectivity.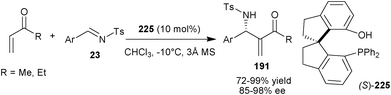 | ||
| Scheme 66 Asymmetric aza-MBH reaction catalyzed with (225). | ||
A highly enantio- and diastereoselective direct vinylogous Mannich reaction of α,α-dicyanoolefins (74) and alkyl N-sulfonyl alkylimines (226) has been reported by Chen and co-workers (Scheme 67).112 A new family of bifunctional organocatalysts (228) have been developed by merging chiral BINOLs and 9-amino-9-deoxy-epi-Cinchona alkaloid skeletons for the asymmetric synthesis of chiral amino compounds (227) in good to high yield (58–98%) and moderate to high enantioselectivity (52–97% ee) and good diastereoselectivity (70:30–91:9 dr).
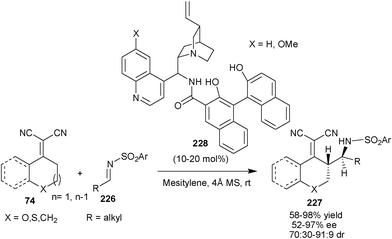 | ||
| Scheme 67 Vinylogous Mannich reaction catalyzed with (228). | ||
Recently, Zhu, Cheng and co-workers have developed a new phenolic OH–primary amine–imine organocatalyst (232), generated in situ from a chiral diamine (231) under acidic conditions (Scheme 68).113 This newly synthesized catalyst has been found to catalyze the direct asymmetric aldol reaction between ketones (229) and α-keto esters to afford aldol product (230) in high yield (65–88%) and with excellent enantioselectivity (87–97% ee).113a
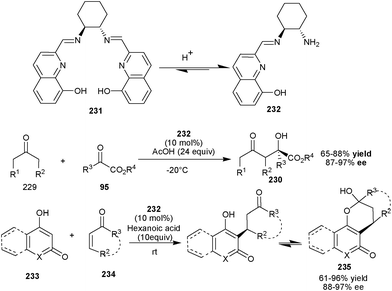 | ||
| Scheme 68 In situ generated aromatic OH–primary amine–imine catalyst (231) catalyzed asymmetric aldol and Michael reaction. | ||
Further, in situ generated aromatic OH–primary amine–imine catalyst (232) efficiently catalyze the enantioselective Michael addition reaction of substituted 4-hydroxycoumarin derivatives (233) with cyclic and acyclic enones (234) (Scheme 68).113b High yield (61–96%) and excellent enantioselectivity (88–97% ee) were achieved for a series of addition products (235) bearing a 4-hydroxycoumarin unit.
Irie and co-workers have developed an optically active tripodal amine (238), as a potent chiral catalyst for methanolytic asymmetric desymmetrization of cyclic meso-anhydrides (236) to hemiesters (237) (Scheme 69).114 Moderate to high yield (33–99%) and a good level of enantioselectivity (52–81% ee) were achieved for various cyclic anhydrides, some of which are considered as difficult substrates in the Cinchona alkaloid-mediated ring-opening.
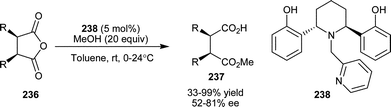 | ||
| Scheme 69 Asymmetric desymmetrization of cyclic meso-anhydrides (236) catalyzed with (238). | ||
Conclusions
Since the origin of asymmetric organocatalysis, tremendous progress has been achieved in the development of new synthetic methodologies and new chiral organocatalysts. This review summarized the application of bifunctional organocatalysts bearing aromatic hydroxyl groups and tertiary amines/phosphines in asymmetric synthesis. The unique properties of these organocatalysts not only enable them to be broadly used in a wide spectrum of simple carbon–carbon, carbon–hetero atom bond formations but also enable them as successful organocatalysts in more complex tandem, cascade and domino reactions as well as multicomponent asymmetric transformations. These chiral catalysts are mild and their pseudoenantiomeric catalysts can be easily obtained. The superior reactivity and stereoselectivity of these catalysts over other types of bifunctional organocatalysts have been observed in several cases. Considering the inherent structural advantages as well as increased interest in catalytic potential at the current stage, we believe that these bifunctional organocatalysts will give rise to more promising findings in the near future. Further, the mechanistic investigations could be beneficial for future development of new bifunctional organocatalysts having aromatic hydroxyl groups in cooperation with a basic unit.Acknowledgements
Our research work was supported by the research project sanctioned to S. S. C. by the University Grant Commission (UGC), India. Financial support from the Department of Science and Technology (DST), India under FIST program and UGC, India, under CAS-I is gratefully acknowledged.References
- (a) For pioneer reviews on asymmetric organocatalysis see: P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2001, 40, 3726 CrossRef CAS; (b) B. List, Tetrahedron, 2002, 58, 5573 CrossRef CAS; (c) B. List, Synlett, 2001, 1675 CrossRef CAS; (d) P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2004, 43, 5138 CrossRef CAS; (e) W. Notz, F. Tanaka and C. F. Barbas III, Acc. Chem. Res., 2004, 37, 580 CrossRef CAS; (f) J. Seayad and B. List, Org. Biomol. Chem., 2005, 3, 719 RSC; (g) M. Marigo and K. A. Jorgensen, Chem. Commun., 2006, 2001 RSC; (h) C. M. Kleiner and P. R. Schreiner, Chem. Commun., 2006, 4315 RSC; (i) A. Erkkila, I. Majander and P. M. Pihko, Chem. Rev., 2007, 107, 5416 CrossRef; (j) S. Mukherjee, J. W. Yang, S. Hoffmann and B. List, Chem. Rev., 2007, 107, 5471 CrossRef CAS; (k) M. J. Gaunt, C. C. C. Johansson, A. McNally and N. T. Vo, Drug Discovery Today, 2007, 12, 8 CrossRef CAS; (l) R. M. de Figueiredo and M. Christmann, Eur. J. Org. Chem., 2007, 2575 CrossRef CAS; (m) D. Enders, C. Grondal and M. R. M. Huttl, Angew. Chem., Int. Ed., 2007, 46, 1570 CrossRef CAS; (n) H. Pellisier, Tetrahedron, 2007, 63, 9267 CrossRef; (o) A. Dondoni and A. Massi, Angew. Chem., Int. Ed., 2008, 47, 4638 CrossRef CAS; (p) P. Melchiorre, M. Marigo, A. Carlone and G. Bartoli, Angew. Chem., Int. Ed., 2008, 47, 6138 CrossRef CAS.
- (a) For leading books on organocatalysis see: Books: A. Berkessel and H. Groger, Metal-Free Organic Catalysis in Asymmetric Synthesis, Wiley-VCH, Weinheim, 2004 Search PubMed; (b) A. Berkessel and H. Groger, Asymmetric Organocatalysis–From Biomimetic Concepts to Applications in Asymmetric Synthesis, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2005 Search PubMed; (c) P. I. Dalko, Enantioselective Organocatalysis, Wiley-VCH, Weinheim, 2007 Search PubMed; (d) B. List, Asymmetric Organocatalysis, Springer, 2009 Search PubMed.
- (a) For comprehensive reviews on bifunctional organocatalysis see: A. G. Doyle and E. N Jacobsen, Chem. Rev., 2007, 107, 5713 CrossRef CAS; (b) P. S. Bhadury, B.-A. Song, S. Yang, D.-Y. Hu and W. Xue, Curr. Org. Synth., 2009, 6, 380 CrossRef CAS.
- (a) For selected reviews on bifunctional catalysis with proline its analogues, see: T. Kano and K. Maruoka, Chem. Commun., 2008, 5465 RSC; (b) A. Lattanzi, Chem. Commun., 2009, 1452 RSC; (c) X. Liu, L. Lin and X. Feng, Chem. Commun., 2009, 6145 RSC.
- (a) For selected reviews on chiral thiourea derivatives as organocatalyst, see: S. J. Connon, Chem.–Eur. J., 2006, 12, 5418 CrossRef; (b) S. J. Connon, Chem. Commun., 2008, 2499 RSC; (c) H. Miyabe and Y. Takemoto, Bull. Chem. Soc. Jpn., 2008, 81, 785 CrossRef CAS; (d) M. S. Taylor and E. N. Jacobsen, Angew. Chem., Int. Ed., 2006, 45, 1520 CrossRef CAS.
- (a) For recent reviews on chiral phosphoric acid catalysis, see: T. Akiyama, Chem. Rev., 2007, 107, 5744 CrossRef CAS; (b) D. Kampen, C. M. Reisinger and B. List, Top. Curr. Chem., 2010, 291, 395 CrossRef CAS; (c) M. Terada, Synthesis, 2010, 1929 CrossRef CAS; (d) G. Adair, S. Mukhe and B. List, Aldrichim. Acta, 2008, 41, 31 CAS.
- (a) K. Drauz and H. Waldmann, Enzyme Catalysis in Organic Synthesis, Wiley-VCH, Weinheim, 2002 Search PubMed; (b) T. D. Machajewski and C.-H. Wang, Angew. Chem., Int. Ed., 2000, 39, 1352 CrossRef CAS; (c) A. Archelas and R. Furstoss, Curr. Opin. Chem. Biol., 2001, 5, 112 CrossRef CAS; (d) S. Steinreiber and K. Faber, Curr. Opin. Biotechnol., 2001, 12, 552 CrossRef; (e) E. J. D. Vries and D. B. Janssen, Curr. Opin. Biotechnol., 2003, 14, 414 CrossRef; (f) C. Morisseau and B. D. Hammock, Annu. Rev. Pharmacol., 2005, 45, 311 CrossRef CAS; (g) M. S. Smit and M. Labuschagné, Curr. Org. Chem., 2006, 10, 1145 CrossRef CAS; (h) E. Y. Lee, Biotechnol. Lett., 2008, 30, 1509 CrossRef CAS; (i) N. Bala and S. S. Chimni, Tetrahedron: Asymmetry, 2010, 21, 2879 CrossRef CAS; (j) L. Xue, P. Talalay and A. S. Mildvan, Biochemistry, 1990, 29, 7491 CrossRef CAS; (k) J. C. Austin, A. Kuliopulos, A. S. Mildvan and T. G. Spiro, Protein Sci., 1992, 1, 259 CrossRef CAS.
- (a) For recent reviews on Cinchona alkaloids as organocatalysts, see: S.-K. Tian, Y.-G. Chen, J.-F. Hang, L. Tang, P. McDaid and L. Deng, Acc. Chem. Res., 2004, 37, 621 CrossRef CAS; (b) H. Hoffmann, R. Martin and J. Frackenpohl, Eur. J. Org. Chem., 2004, 4293 CrossRef CAS; (c) T. Marcelli, J. H. van Maarseveen and H. Hiemstra, Angew. Chem., Int. Ed., 2006, 45, 7496 CrossRef CAS; (d) T. Marcelli and H. Hiemstra, Synthesis, 2010, 1229 CrossRef CAS; (e) T. E. M. O. Yeboah, S. O. Yeboah and G. S. Singh, Tetrahedron, 2011, 67, 1725 CrossRef; (f) T. Marcelli, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 142 CrossRef CAS.
- (a) For selected reviews on MBH reaction, see: D. Basavaiah, P. D. Rao and R. S. Hyma, Tetrahedron, 1996, 52, 8001 CrossRef CAS; (b) P. Langer, Angew. Chem., Int. Ed., 2000, 39, 3049 CrossRef CAS; (c) D. Basavaiah, A. J. Rao and T. Satyanarayana, Chem. Rev., 2003, 103, 811 CrossRef CAS; (d) D. Basavaiah, K. V. Rao and R. J. Reddy, Chem. Soc. Rev., 2007, 36, 1581 RSC; (e) G. Masson, C. Housseman and J. Zhu, Angew. Chem., Int. Ed., 2007, 46, 4614 CrossRef CAS; (f) V. Singh and S. Batra, Tetrahedron, 2008, 64, 4511 CrossRef CAS; (g) D. Basavaiah, B. S. Reddy and S. S. Badsara, Chem. Rev., 2010, 110, 5447 CrossRef CAS.
- (a) Y. Iwabuchi, M. Nakatani, N. Yokoyama and S. Hatakeyama, J. Am. Chem. Soc., 1999, 121, 10219 CrossRef CAS; (b) A. Nakano, S. Kawahara, S. Akamatsu, K. Morokuma, M. Nakatani, Y. Iwabuchi, K. Takahashi, J. Ishihara and S. Hatakeyama, Tetrahedron, 2006, 62, 381 CrossRef CAS.
- Y. Iwabuchi, M. Furukawa, T. Esumi and S. Hatakeyama, Chem. Commun., 2001, 2030 RSC.
- Y. Iwabuchi, T. Sugihara, T. Esumi and S. Hatakeyama, Tetrahedron Lett., 2001, 42, 7867 CrossRef CAS.
- M. Shi and J.-K. Jiang, Tetrahedron: Asymmetry, 2002, 13, 1941 CrossRef CAS.
- (a) Y.-L. Liu, B.-L. Wang, J.-J. Cao, L. Chen, Y.-X. Zhang, C. Wang and J. Zhou, J. Am. Chem. Soc., 2010, 132, 15176 CrossRef CAS; (b) X.-Y. Guan, Y. Wei and M. Shi, Chem.–Eur. J., 2010, 16, 13617 CrossRef CAS; (c) F. Zhong, G.-Y. Chen and Y. Lu, Org. Lett., 2011, 13, 82 CrossRef CAS.
- (a) For selected reviews on aza-MBH reactions, see: Y.-L. Shi and M. Shi, Eur. J. Org. Chem., 2007, 2150 Search PubMed; (b) V. Declerck, J. Martinez and F. Lamaty, Chem. Rev., 2009, 109, 1 CrossRef CAS.
- (a) M. Shi and Y.-M. Xu, Angew. Chem., Int. Ed., 2002, 41, 4507 CrossRef CAS; (b) M. Shi, Y.-M. Xu and Y. L. Shi, Chem.–Eur. J., 2005, 11, 1794 CrossRef CAS.
- D. Balan and H. Adolfsson, Tetrahedron Lett., 2003, 44, 2521 CrossRef CAS.
- N. Abermil, G. Masson and J. Zhu, Org. Lett., 2009, 11, 4648 CrossRef CAS.
- S. Kawahara, A. Nakano, T. Esumi, Y. Iwabuchi and S. Hatakeyama, Org. Lett., 2003, 5, 3103 CrossRef CAS.
- (a) For selected reviews on asymmetric Mannich reactions, see: A. Cordova, Acc. Chem. Res., 2004, 37, 102 CrossRef CAS; (b) J. M. M. Verkade, L. J. C. van Hemert, P. J. L. M. Quaedflieg and F. P. J. T. Rutjes, Chem. Soc. Rev., 2008, 37, 29 RSC; (c) R. G. Arrayás and J. C. Carretero, Chem. Soc. Rev., 2009, 38, 1940 RSC.
- A. Ricci, D. Pettersen, L. Bernardi, F. Fini, M. Fochi, R. P. Herrera and V. Sgarzani, Adv. Synth. Catal., 2007, 349, 1037 CrossRef CAS.
- (a) L. Li, E. G. Klauber and D. Seidel, J. Am. Chem. Soc., 2008, 130, 12248 CrossRef CAS; (b) L. Li, M. Ganesh and D. Seidel, J. Am. Chem. Soc., 2009, 131, 11648 CrossRef CAS.
- Z. Shi, P. Yu, P. J. Chua and G. Zhong, Adv. Synth. Catal., 2009, 351, 2797 CrossRef CAS.
- L. Cheng, L. Liu, H. Jia, D. Wang and Y.-J. Chen, J. Org. Chem., 2009, 74, 4650 CrossRef CAS.
- (a) For recent reviews of asymmetric Michael additions, see: O. M. Berner, L. Tedeschi and D. Enders, Eur. J. Org. Chem., 2002, 1877 CrossRef CAS; (b) R. Ballini, G. Bosica, D. Fiorini, A. Palmieri and M. Petrini, Chem. Rev., 2005, 105, 933 CrossRef CAS; (c) S. B. Tsogoeva, Eur. J. Org. Chem., 2007, 1701 CrossRef CAS; (d) J. Christoffers and A. Baro, Angew. Chem., Int. Ed., 2003, 42, 1688 CrossRef CAS; (e) D. Almasi, D. A. Alonso and C. Nájera, Tetrahedron: Asymmetry, 2007, 18, 299 CrossRef CAS; (f) S. Sulzer-Mosse and A. Alexakis, Chem. Commun., 2007, 3123 RSC.
- H. Li, Y. Wang, L. Tang and L. Deng, J. Am. Chem. Soc., 2004, 126, 9906 CrossRef CAS.
- F. Li, Y.-Z. Li, Z.-S. Jia, M.-H. Xu, P. Tian and G.-Q. Lin, Tetrahedron, 2011 DOI:10.1016/j.tet.2011.08.070.
- H. Li, Y. Wang, L. Tang, F. Wu, X. Liu, C. Guo, B. M. Foxman and L. Deng, Angew. Chem., Int. Ed., 2005, 44, 105 CrossRef CAS.
- A. J. Young and M. C. White, J. Am. Chem. Soc., 2008, 130, 14090 CrossRef CAS.
- H. Li, S. Zhang, C. Yu, X. Song and W. Wang, Chem. Commun., 2009, 2136 RSC.
- H. Li, L. Zu, H. Xie and W. Wang, Synlett, 2009, 1525 CAS.
- Y. Xuan, S. Nie, L. Dong, J. Zhang and M. Yan, Org. Lett., 2009, 11, 1583 CrossRef CAS.
- W. Raimondi, G. Lettieri, J.-P. Dulcere, D. Bonne and J. Rodriguez, Chem. Commun., 2010, 46, 7247 RSC.
- P. He, X. Liu, J. Shi, L. Lin and X. Feng, Org. Lett., 2011, 13, 936 CrossRef CAS.
- J. Wang, H. Li, L. Zu, W. Jiang and W. Wang, Adv. Synth. Catal., 2006, 348, 2047 CrossRef CAS.
- M. Shi, Z.-Y. Lei, M.-X. Zhao and J.-W. Shi, Tetrahedron Lett., 2007, 48, 5743 CrossRef CAS.
- P. Chauhan and S. S. Chimni, Adv. Synth. Catal., 2011, 353, 3203 CrossRef CAS.
- (a) F. Wu, H. Li, R. Hong and L. Deng, Angew. Chem., Int. Ed., 2006, 45, 947 CrossRef CAS; (b) F. Wu, R. Hong, J. Khan, X. Liu and L. Deng, Angew. Chem., Int. Ed., 2006, 45, 4301 CrossRef CAS.
- C. L. Rigby and D. J. Dixon, Chem. Commun., 2008, 3798 RSC.
- H. Li, J. Song, X. Liu and L. Deng, J. Am. Chem. Soc., 2005, 127, 8948 CrossRef CAS.
- H. Li, J. Song and L. Deng, Tetrahedron, 2009, 65, 3139 CrossRef CAS.
- S. Sternativo, A. Calandriello, F. Costantino, L. Testaferri, M. Tiecco and F. Marini, Angew. Chem., Int. Ed., 2011, 50, 9382 CrossRef CAS.
- J. Lu, W.-J. Zhou, F. Liu and T.-P. Loh, Adv. Synth. Catal., 2008, 350, 1796 CrossRef CAS.
- B. Wang, F. Wu, Y. Wang, X. Liu and L. Deng, J. Am. Chem. Soc., 2007, 129, 768 CrossRef CAS.
- W.-B. Chen, Z.-J. Wu, Q.-L. Pei, L.-F. Cun, X.-M. Zhang and W.-C. Yuan, Org. Lett., 2010, 12, 3132 CrossRef CAS.
- (a) For general review on RC reaction, see for example: C. E. Aroyan, A. Dermenci and S. J. Miller, Tetrahedron, 2009, 65, 4069 CrossRef CAS; (b) J. L. Methot and W. R. Roush, Adv. Synth. Catal., 2004, 346, 1035 CrossRef CAS.
- Q.-Y. Zhao, C.-K. Pei, X.-Y. Guan and M. Shi, Adv. Synth. Catal., 2011, 353, 1973 CrossRef CAS.
- Y. Du, X. Han and X. Lu, Tetrahedron Lett., 2004, 45, 4967 CrossRef CAS.
- D. J. V. C. van Steenis, T. Marcelli, M. Lutz, A. L. Spek, J. H. van Maarseveen and H. Hiemstra, Adv. Synth. Catal., 2007, 349, 281 CrossRef CAS.
- K. Jiang, J. Ping, H.-L. Cui and Y.-C. Chen, Chem. Commun., 2009, 3955 RSC.
- J. Peng, X. Huang, H.-L. Cui and Y.-C. Chen, Org. Lett., 2010, 12, 4260 CrossRef CAS.
- (a) For selected reviews on asymmetric Friedel–Crafts reaction, see: K. A. Jørgensen, Synthesis, 2003, 1117 CrossRef; (b) M. Bandini, A. Melloni and A. Umani-Ronchi, Angew. Chem., Int. Ed., 2004, 43, 550 CrossRef CAS; (c) M. Bandini, A. Melloni, S. Tommasi and A. Umani-Ronchi, Synlett, 2005, 1199 CrossRef CAS; (d) Y.-F. Sheng, A. J. Zhang, X.-J. Zheng and S.-L. You, Chin. J. Org. Chem., 2008, 28, 605 CAS; (e) T. Poulsen and K. A. Jørgensen, Chem. Rev., 2008, 108, 2903 CrossRef CAS; (f) S.-L. You, Q. Cai and M. Zeng, Chem. Soc. Rev., 2009, 38, 2190 RSC; (g) M. Bandini and A. Eichholzer, Angew. Chem., Int. Ed., 2009, 48, 9608 CrossRef CAS; (h) V. Terrasson, R. M. de Figueiredo and J. M. Campagne, Eur. J. Org. Chem., 2010, 2635 CAS; (i) M. Zen and S.-L. You, Synlett, 2010, 1289 Search PubMed.
- H. Li, Y. Wang and L. Deng, Org. Lett., 2006, 8, 4063 CrossRef CAS.
- J.-L. Zhao, L. Liu, C.-L. Gu, D. Wang and Y.-J. Chen, Tetrahedron Lett., 2008, 49, 1476 CrossRef CAS.
- J. Deng, S. Zhang, P. Ding, H. Jiang, W. Wang and J. Li, Adv. Synth. Catal., 2010, 352, 833 CAS.
- P. Chauhan and S. S. Chimni, Chem.–Eur. J., 2010, 16, 7709 CrossRef CAS.
- G. Liu, S. Zhang, H. Li, T. Zhang and W. Wang, Org. Lett., 2011, 13, 828 CrossRef CAS.
- P. Chauhan and S. S. Chimni, Eur. J. Org. Chem., 2011, 1636 CrossRef CAS.
- (a) For selected reviews on asymmetric Henry reaction, see: C. Palomo, M. Oiarbide and A. Mielgo, Angew. Chem., Int. Ed., 2004, 43, 5442 CrossRef CAS; (b) J. Boruwa, N. Gogoi, P. P. Saikia and N. C. Barua, Tetrahedron: Asymmetry, 2006, 17, 3315 CrossRef CAS; (c) C. Palomo, M. Oiarbide and A. Laso, Eur. J. Org. Chem., 2007, 2561 CrossRef CAS.
- T. Marcelli, R. N. S. van der Haas, J. H. van Maarseveen and H. Hiemstra, Synlett, 2005, 2817 CAS.
- H. Li, B. Wang and L. Deng, J. Am. Chem. Soc., 2006, 128, 732 CrossRef CAS.
- (a) A. Cochi, T.-X. Metro, D. G. Pardo and J. Cossy, Org. Lett., 2010, 12, 3693 CrossRef CAS; (b) T.-X. Metro, A. Cochi, D. G. Pardo and J. Cossy, J. Org. Chem., 2011, 76, 2594 CrossRef CAS.
- T. Mandal, S. Samanta and C.-G. Zhao, Org. Lett., 2007, 9, 943 CrossRef CAS.
- M. Bandini, R. Sinisi and A. Umani-Ronchi, Chem. Commun., 2008, 4360 RSC.
- (a) L. Liu, S. Zhang, F. Xue, G. Lou, H. Zhang, S. Ma, W. Duan and W. Wang, Chem.–Eur. J., 2011, 17, 7791 CrossRef CAS; (b) M. Q. Li, J.-X. Zhang, X.-F. Huang, B. Wu, Z.-M. Liu, J. Chen, X.-D. Li and X.-W. Wang, Eur. J. Org. Chem., 2011, 5237 CrossRef CAS; (c) Y. Zhang, Z. J. Li, H. S. Xu, Y. Zhang and W. Wang, RSC Adv., 2011, 1, 389 RSC.
- C.-B. Ji, Y.-L. Liu, Z.-Y. Cao, Y.-Y. Zhang and J. Zhou, Tetrahedron Lett., 2011, 52, 6118 CrossRef CAS.
- S. Saaby, M. Bella and K. A. Jørgensen, J. Am. Chem. Soc., 2004, 126, 8120 CrossRef CAS.
- (a) X. Liu, H. Li and L. Deng, Org. Lett., 2005, 7, 167 CrossRef CAS; (b) X. Liu, B. Sun and L. Deng, Synlett, 2009, 1685 CAS.
- (a) S. Brandes, M. Bella, A. Kjærsgaard and K. A. Jørgensen, Angew. Chem., Int. Ed., 2006, 45, 1147 CrossRef CAS; (b) S. Brandes, B. Niess, M. Bella, A. Prieto, J. Overgaard and K. A. Jørgensen, Chem.–Eur. J., 2006, 12, 6039 CrossRef CAS.
- X. Xiao, Y. Xie, C. Su, M. Liu and Y. Shi, J. Am. Chem. Soc., 2011, 133, 12914 CrossRef CAS.
- For review on asymmetric organocatalytic aza-Michael reaction, see: D. Enders, C. Wang and J. X. Liebich, Chem.–Eur. J., 2009, 15, 11058 CrossRef CAS.
- J. Wang, H. Li, L. Zu and W. Wang, Org. Lett., 2006, 8, 1391 CrossRef CAS.
- T. Zhang, L. Cheng, L. Liu, D. Wang and Y.-J. Chen, Tetrahedron: Asymmetry, 2010, 21, 2800 CrossRef CAS.
- (a) For recent reviews on asymmetric organocatalytic oxa-Michael reaction, see: Y.-L. Shi and M. Shi, Org. Biomol. Chem., 2007, 5, 1499 RSC; (b) C. F. Nising and S. Brase, Chem. Soc. Rev., 2008, 37, 1218 RSC.
- E. G. Taylor, DuPont Patent, WO2003040083, p. 2003 Search PubMed.
- H.-F. Wang, H.-F. Cui, Z. Chai, P. Li, C.-W. Zheng, Y.-Q. Yang and G. Zhao, Chem. Eur. J., 2009, 15, 13299 CAS.
- H.-F. Wang, H. Xiao, X.-W. Wang and G. Zhao, Tetrahedron, 2011, 67, 5389 CrossRef CAS.
- F.-G. Zhang, Q.-Q. Yang, J. Xuan, H.-H. Lu, S.-W. Duan, J.-R. Chen and W.-J. Xiao, Org. Lett., 2010, 12, 5636 CrossRef CAS.
- R. Dodda, J. J. Goldman, T. Mandal, C.-G. Zhao, G. A. Broker and E. R. T. Tiekink, Adv. Synth. Catal., 2008, 350, 537 CrossRef CAS.
- (a) X. Tian, C. Cassani, Y. Liu, A. Moran, A. Urakawa, P. Galzerano, E. Arceo and P. Melchiorre, J. Am. Chem. Soc., 2011, 133, 17934 CrossRef CAS; (b) G. Bergonzini, S. Vera and P. Melchiorre, Angew. Chem., Int. Ed., 2010, 49, 9685 CrossRef CAS.
- Y. Wang, H. Li, Y.-Q. Wang, Y. Liu, B. M. Foxman and L. Deng, J. Am. Chem. Soc., 2007, 129, 6364 CrossRef CAS.
- K. J. Bartelson, R. P. Singh, B. M. Foxman and L. Deng, Chem. Sci., 2011, 2, 1940 RSC.
- W. Chen, W. Du, Y.-Z. Duan, Y. Wu, S.-Y. Yang and Y.-C. Chen, Angew. Chem., Int. Ed., 2007, 46, 7667 CrossRef CAS.
- C. Guo, M.-X. Xue, M.-K. Zhu and L.-Z. Gong, Angew. Chem., Int. Ed., 2008, 47, 3414 CrossRef CAS.
- X.-M. Xue, C. Guo and L.-Z. Gong, Synlett, 2009, 2191 Search PubMed.
- Y. Ying, Z. Chai, H.-F. Wang, P. Li, C.-W. Zheng, G. Zhao and Y.-P. Cai, Tetrahedron, 2011, 67, 3337 CrossRef CAS.
- (a) G. S. Cortez, R. L. Tennyson and D. Romo, J. Am. Chem. Soc., 2001, 123, 7945 CrossRef CAS; (b) G. S. Cortez, S. H. Oh and D. Romo, Synthesis, 2001, 1731 CrossRef CAS.
- S. T. Staben, X. Linghu and F. D. Toste, J. Am. Chem. Soc., 2006, 128, 12658 CrossRef CAS.
- E. Zhang, C.-A. Fan, Y.-Q. Tu, F.-M. Zhang and Y.-L. Song, J. Am. Chem. Soc., 2009, 131, 14626 CrossRef CAS.
- L. Lykke, C. Rodríguez-Escrich and K. A. Jørgensen, J. Am. Chem. Soc., 2011, 133, 14932 CAS.
- Y. Wu, R. P. Singh and L. Deng, J. Am. Chem. Soc., 2011, 133, 12458 CrossRef CAS.
- C. Quigley, Z. Rodriguez-Docampo and S. J. Connon, Chem. Commun., 2012 10.1039/C1CC14684J.
- (a) For reviews on BINOL as a versatile chiral reagent, see J. M. Brunel, Chem. Rev., 2005, 105, 857 CrossRef CAS; (b) J. M. Brunel, Chem. Rev., 2007, 107, PR1–PR45 CrossRef CAS.
- (a) M. Shi and L.-H. Chen, Chem. Commun., 2003, 1310 RSC; (b) M. Shi, L.-H. Chen and C.-Q. Li, J. Am. Chem. Soc., 2005, 127, 3790 CrossRef CAS.
- Z.-Y. Lei, G.-N. Ma and M. Shi, Eur. J. Org. Chem., 2008, 3817 CrossRef CAS.
- M. Shi, L.-H. Chen and W.-D. Teng, Adv. Synth. Catal., 2005, 347, 1781 CrossRef CAS.
- Y.-H. Liu, L.-H. Chen and M. Shi, Adv. Synth. Catal., 2006, 348, 973 CrossRef CAS.
- K. Matsui, S. Takizawa and H. Sasai, Synlett, 2006, 761 CAS.
- K. Ito, K. Nishida and T. Gotanda, Tetrahedron Lett., 2007, 48, 6147 CrossRef CAS.
- Y.-H. Liu and M. Shi, Adv. Synth. Catal., 2008, 350, 122 CrossRef CAS.
- (a) J.-M. Garnier, C. Anstiss and F. Liu, Adv. Synth. Catal., 2009, 351, 331 CrossRef CAS; (b) J.-M. Garnier and F. Liu, Org. Biomol. Chem., 2009, 7, 1272 RSC.
- M. Shi and G.-L. Zhao, Adv. Synth. Catal., 2004, 346, 1205 CrossRef CAS.
- M. Shi, G.-N. Ma and J. Gao, J. Org. Chem., 2007, 72, 9779 CrossRef CAS.
- S. Takizawa, N. Inoue, S. Hirata and H. Sasai, Angew. Chem., Int. Ed., 2010, 49, 9725 CrossRef CAS.
- M. Shi, Y.-H. Liu and L.-H. Chen, Chirality, 2007, 19, 124 CrossRef CAS.
- Z.-Y. Lei, X.-G. Liu, M. Shi and M. Zhao, Tetrahedron: Asymmetry, 2008, 19, 2058 CrossRef CAS.
- (a) K. Matsui, S. Takizawa and H. Sasai, J. Am. Chem. Soc., 2005, 127, 3680 CrossRef CAS; (b) K. Matsui, K. Tanaka, A. Horii, S. Takizawa and H. Sasai, Tetrahedron: Asymmetry, 2006, 17, 578 CrossRef CAS.
- S. Takizawa, A. Horii and H. Sasai, Tetrahedron: Asymmetry, 2010, 21, 891 CrossRef CAS.
- S. Takizawa, N. Inoue and H. Sasai, Tetrahedron Lett., 2011, 52, 377 CrossRef CAS.
- P. Xie, Y. Huang, W. Lai, X. Meng and R. Chen, Org. Biomol. Chem., 2011, 9, 6707 CAS.
- S. Takizawa, K. Kiriyama, K. Ieki and H. Sasai, Chem. Commun., 2011, 47, 9227 RSC.
- X.-F. Xiong, Z.-J. Jia, W. Du, K. Jiang, T.-Y. Liu and Y.-C. Chen, Chem. Commun., 2009, 6994 RSC.
- (a) X. Zhu, A. Lin, L. Fang, W. Li, C. Zhu and Y. Cheng, Chem.–Eur. J., 2011, 17, 8281 CrossRef CAS; (b) X. Zhu, A. Lin, Y. Shi, J. Guo, C. Zhu and Y. Cheng, Org. Lett., 2011, 13, 4382 CrossRef CAS.
- T. Okamatsu, R. Irie and T. Katsuki, Synlett, 2007, 1569 CAS.
| This journal is © The Royal Society of Chemistry 2012 |