Thiol-ene “click” reactions and recent applications in polymer and materials synthesis
Andrew B.
Lowe
*
Centre for Advanced Macromolecular Design (CAMD), School of Chemical Sciences & Engineering, The University of New South Wales, Kensington, Sydney, NSW 2052, Australia. E-mail: a.lowe@unsw.edu.au
First published on 25th November 2009
Abstract
This review highlights examples of recent applications of both the radical-mediated and base/nucleophile-initiated thiol-ene reactions in polymer and materials synthesis. Initial discussion focuses on mechanistic aspects of these reactions and also notes some of the structural considerations, with respect to reactants, that should be considered when practising such chemistries. The review is not intended to be exhaustive but rather to serve as an illustration of the impressive versatility and clear potential of these thiol-based “click” reactions and to highlight examples demonstrating its broad utility.
1. Introduction
The introduction and development of the “click” approach to the design and preparation of complex, highly functional molecules, as first highlighted by Kolb et al.,1 has had a transformational effect on synthesis in areas as diverse as polymers and materials, small molecule organic chemistry and drug discovery. A “click” reaction, as defined by Kolb et al.,1 is one that is modular, wide in scope, gives (near) quantitative yields, generates inoffensive (if any) byproducts that are easily removed by non-chromatographic methods and is stereospecific. From a practical standpoint, such reactions should be simple to perform and preferably be insensitive to water and oxygen, be accomplished with readily available starting materials and reagents, be performed under solventless conditions or in environmentally benign media such as water and facilitate simple product isolation. While a series of general reactions were originally highlighted as “click” reactions, until relatively recently research and applications have focused almost exclusively on the Cu(I)-mediated Huisgen reaction between an alkyne and an azide.2–7 This has been due to its general ease of execution, facile reaction conditions and impressive orthogonality. However, the broad utility of the Cu(I)-mediated alkyne-azide reaction has encouraged researchers to more closely evaluate the potential of other reactions that possess “click” characteristics. In particular, attention has recently been paid to Diels–Alder reactions,8–12 metal-free dipolar cycloadditions,13,14 and a series of thiol-based reactions including the thiol-ene,8,13,15–19 thiol-yne,15,16,20–22 thiol-isocyanate,23,24 thiol-para-fluorostyrene25 and thiol-bromo26,27 processes.Within this review article the term “thiol-ene” will be used to denote the addition of a thiol to an ene bond regardless of reaction mechanism. While the phrase is typically associated with the radical version of the reaction this is for historical reasons and the term, clearly, does not indicate/specify a particular mechanistic pathway and as such is also applicable to base/nucleophile-mediated thiol additions with activated substrates. However, while the term is used herein in a general, all-encompassing manner it is noted that hydrothiolations of the latter type (base/nucleophile-mediated additions with activated enes) can also be more specifically described as conjugate additions or thiol-Michael reactions.28
The thiol-ene reaction, known for over 100 years,29 is, simply, the hydrothiolation of a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, Scheme 1. Historically, in the polymer/materials fields the reaction has been most widely employed as a means of preparing near-perfect networks and films as exemplified by the work of Hoyle and co-workers23,30–39 and Bowman et al.40–46
C bond, Scheme 1. Historically, in the polymer/materials fields the reaction has been most widely employed as a means of preparing near-perfect networks and films as exemplified by the work of Hoyle and co-workers23,30–39 and Bowman et al.40–46
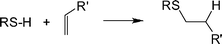 | ||
| Scheme 1 The hydrothiolation of a CC bond with anti-Markovnikov orientation. | ||
However, the thiol-ene reaction has recently attracted researchers in other areas of synthesis due to the recognition of its “click” characteristics, vide infra. There are several features associated with the thiol-ene reaction that make it a particularly attractive, facile and versatile process. Firstly, such hydrothiolation reactions can proceed under a variety of conditions including by a radical pathway,30via catalytic processes mediated by nucleophiles, acids, bases,16,47 in the apparent absence of an added catalyst in highly polar solvents such as water or DMF,48 or via supramolecular catalysis using β-cyclodextrin49 for example. Secondly, a wide range of enes serve as suitable substrates, including activated and non-activated species as well as multiply-subsituted olefinic bonds. However, reactivity can vary considerably depending on reaction mechanism and substitution pattern at the CC bond. Thirdly, virtually any thiol can be employed, including highly functional species, although reactivity can cover several orders of magnitude depending on the S–H bond strength and the cleavage mechanism, i.e. homolytic vs. heterolytic lysis. Finally, such reactions are generally extremely rapid and can be complete in a matter of seconds (even at ambient temperature and pressure), are tolerant to the presence of air/oxygen and moisture (provided the concentration of oxygen does not approach that of the thiol), and proceed with (near) quantitative formation of the corresponding thioether in a regioselective fashion.
Generally, the thiol-ene reaction has been conducted under radical conditions, often photochemically induced.30,50,51 Under such conditions it proceeds via a typical chain process with initiation, propagation and termination steps, Scheme 2. Initiation involves the treatment of a thiol with photoinitiator, under irradiation, resulting in the formation of a thiyl radical , RS˙, plus other byproducts. Simple thermal lysis of the S–H bond can also employed as a means of generating thiyl radicals .52 Propagation is a two step process involving first the direct addition of the thiyl radical across the CC bond yielding an intermediate carbon-centred radical followed by chain transfer to a second molecule of thiol to give the thiol-ene addition product, with anti-Markovnikov orientation, with the concomitant generation of a new thiyl radical . Possible termination reactions involve typical radical–radical coupling processes.
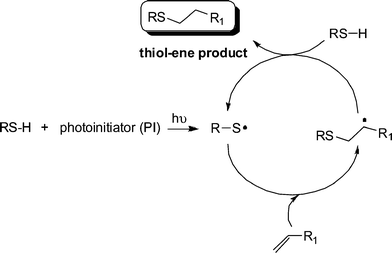 | ||
| Scheme 2 The mechanism for the hydrothiolation of a CC bond in the presence of a photoinitiator and hν. | ||
It is worth noting at this point that radical thiol-ene (photo)polymerization processes are actually radical step-growthpolymerizations—the only instance in which these two, typically considered exclusive processes, combine. As noted above, reactivity in the radical thiol-ene reaction can vary considerably depending on the chemical structure of the ene and thiol components. General trends are highlighted in several excellent papers.30,50 Briefly, as described by Hoyle et al., general ene reactivity with three typical thiol types (alkyl 3-mercaptopropionates, alkyl thioglycolates, and alkylthiols) under radical conditions, follows the order: norbornene > vinylether > propenyl > alkene ≈ vinylester > N-vinylamide > allylether ≈ allyltriazine ≈ allylisocyanurate > acrylate > N-substituted maleimide > acrylonitrile ≈ methacrylate > styrene > conjugated diene. With the exception of the first and last three species the ene reactivity falls with decreasing electron density of the double bond. The anomalously high reactivity of norbornenes can be attributed to bond angle distortion and associated loss of ring strain upon addition of a thiyl radical , whereas the low reactivity of methacrylates, styrenes and conjugated dienes is due to the relatively high stability of the intermediate carbon-centered radicals leading to low hydrogen abstraction rates from RS–H in the second stage of the propagation step, Scheme 2. As noted above, one salient feature of the radical thiol-ene reaction is that addition can proceed with virtually any olefinic bond. However, and not surprisingly, the degree of substitution has a significant effect on the overall rate of hydrothiolation. Generally, terminal enes are significantly more reactive towards hydrothiolation compared to internal enes. For example, Hoyle et al. reported that 1-hexene is 8× more reactive than trans-2-hexene and 18× more reactive than trans-3-hexene, clearly highlighting that steric effects are important when considering reactivity.30 However, these differences in reactivity are not due entirely to steric effects. As noted previously,53–55thiyl radical addition to cis CC bonds is reversible and is accompained with an isomerization process, i.e.thiyl radicals can be employed as a means of converting cis CC bonds to trans CC bonds with high efficiency, Scheme 3.
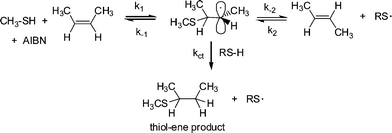 | ||
| Scheme 3 Thiyl radical induced cis–trans isomerization in 2-butene with concurrent thiol-ene product formation. | ||
In the reaction of excess CH3SH with cis-2-butene at 60 °C in the presence of 2,2′-azobis(2-methylpropionitrile) (AIBN), Walling and Helmreich53 reported that significant amounts of the corresponding trans diastereomer was detected in the unreacted olefin. Additionally, they reported that the rate constant, k−1, for the decomposition of the intermediate carbon-centred radical thioether into cis-2-butene, i.e. the starting materials, was 20× larger than the rate of chain transfer, ktc, yielding the desired hydrothiolation products, while k−2, the rate constant of decomposition yielding the less reactive trans-2-butene, was 80× larger than ktc. As such, while the desired thiol-ene products are obtained, the competing cis–trans equilibria significantly affects the rate at which this occurs.
Just as different enes exhibit varying propensities towards hydrothiolation not all thiols are equally reactive. Unfortunately, very little has been done with respect to examining thiol structure and its effect in radical thiol-ene reactions. The most common, general types, of thiols employed in such reactions are the alkyl 3-mercaptopropionates, the alkyl thioglycolates and simple alkylthiols. Of these three general families, the propionates and glycolates are significantly more reactive than the alkylthiols. While this has been attributed to a weakening of the sulfur–hydrogen bond due to intramolecular H-bonding to the estercarbonyl, little, if any, evidence supports this supposition and it seems more likely that the enhanced reactivity is due to polar effects.56
Aside from the radical mediated thiol-ene reactions, hydrothiolations can be readily accomplished under mild base or nucleophilic catalysis. Such reactions are slightly less versatile than the radical-mediated thiol-ene reaction since to be effective the CC must be activated, i.e. electron deficient, Scheme 4. However, given the large number of commercially available activated enes, including multifunctional species, there is clearly still considerable scope for the synthesis of novel and interesting materials.
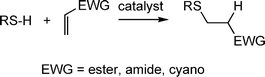 | ||
| Scheme 4 Hydrothiolation of a CC bearing an electron-withdrawing substituent. | ||
Fig. 1 shows examples of suitable activated ene substrates, and includes (meth)acrylates, fumarate esters and maleimide derivatives. As noted above, the base/nucleophile-mediated addition of a thiol to an activated CC bond can also be described as a thiol-Michael or conjugate addition reaction and as such can proceed under conditions typical of such reactions. For example, there are numerous examples in the literature of base catalyzed thiol-ene reactions.28 However, compared to more traditional Michael reactions, the use of weak base catalysts, such as NEt3, is usually sufficient to catalyze the process due to the readily accesible pKa of most thiols, Scheme 5. Reaction of a thiol with NEt3 results in deprotonation of the thiol to the corresponding thiolate anion and formation of the triethylammonium cation. The thiolate, a powerful nucleophile, adds into the activated CC bond at the electrophilic β-carbon forming an intermediate carbon-centered anion (or enolate ) which is, of course, a very strong base. This anion picks up a proton (either from a thiol or from the ammonium cation) yielding the thiol-ene product, again with regioselective formation of the anti-Markovnikov product. So, in this process, a relatively weak base (NEt3) is used to generate a much stronger base (the carbanion) in the catalytic cycle.
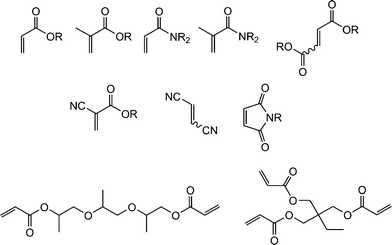 | ||
| Fig. 1 Examples of activated substrates susceptible to hydrothiolation via a base/nucleophile-mediated process. | ||
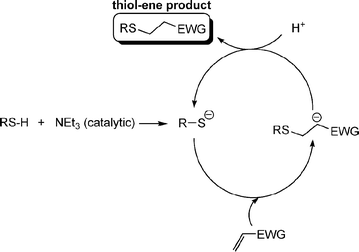 | ||
| Scheme 5 The proposed base-catalyzed mechanism for the hydrothiolation of an activated CC bond. | ||
Additionally, such thiol-ene reactions can be readily conducted under nucleophilic catalysis using simple primary/secondary amines or certain phosphines, although the use of such reagents is not always recognized as a nucleophilic process. Evidence for a nucleophile-based mechanism is obtained, for example, from the kinetics for the reaction between hexyl acrylate (3.3 M) and hexanethiol (3.3 M) catalyzed by 0.4 M hexylamine (C6H13NH2), di-n-propylamine (NH(C3H7)2), and NEt3 at room temperature under bulk conditions for a period of 500 s, Fig. 2, with reactions being monitored by following the decrease of the intensity of the band associated with the thiol by real time FTIR spectroscopy.57
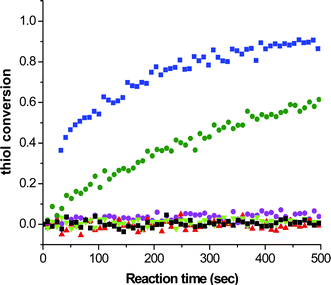 | ||
| Fig. 2 Real time kinetics, monitored by FTIR spectroscopy, for the reaction between hexylacrylate (3.3 M) and hexanethiol (3.3 M) and a range of amine-based catalysts (0.4 M): blue squares: hexylamine, green circles: di-n-propylamine, purple circles: triethylamine, black squares: pyridine, red triangles: aniline, and green triangles: proton sponge. | ||
It is clear that the fastest reaction is that mediated by hexylamine with ∼95% conversion being obtained after the 500 s reaction time. In contrast, the reaction catalysed by NEt3 reaches < 1% conversion after the same time period. The reaction mediated by di-n-propylamine exhibits the second fastest kinetics and reaches ∼60% conversion after 500 s. In addition, reactions performed in the presence of pyridine (pKa = 5.14), aniline (pKa = 9.34) and proton sponge (1,8-bis(dimethylamino)naphthalene, pKa = 12.1), all weak nucleophiles but of broad basicity, yield less than 1% conversion after 500 s. If the reactions were proceeding by purely base catalysis such a dramatic difference in the kinetics would not be expected. Consider the three alkylamines employed as catalysts: the pKa's of these three species are very similar and range from 10.56 (C6H13NH2) to 11.00 (NH(C3H7)2) with NEt3 having a pKa of 10.75, i.e. the secondary amine is actually the strongest base. As such, the order of reactivity is not consistent with the basic characteristics of the aminecatalysts. However, this observed pattern of reactivity is readily rationalized if the amines are considered to be reacting not as bases but as nucleophiles. Indeed, there is literature precendent for such reactivity. Stewart et al.58 described the phosphine-catalyzed hydration and hydroxylation of activated enes and proposed a nucleophile-based mechanism. Only recently, has a similar mechanism been proposed for the thiol-ene reaction with activated substrates.15,16Scheme 6 shows the proposed nucleophile-based mechanism for the phosphine-mediated thiol-ene reaction with an acrylate—the mechanism is assumed to be the same for 1°/2° amine-mediated reactions with other activated CC bonds.
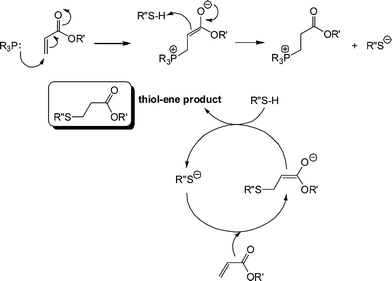 | ||
| Scheme 6 Proposed anionic chain mechanism for the hydrothiolation of an acrylic CC bond under phosphine catalysis. | ||
In contrast to the base-catalyzed system, where a weak base is used to generate a strong base, here a strong nucleophile is used to generate the strong base. Nucleophilic attack at the activated ene generates the intermediate, strong (zwitterionic)59,60enolate base that is responsible for deprotonating thiol, generating the thiolate anion. Once the thiolate is formed the anionic chain process begins with extremely rapid formation of the thiol-ene product. It should be noted that while 1° and 2° amines are very potent catalysts for such reactions, it was recently highlighted that weakly basic dimethylphenylphosphine (Me2PPh) is a far superior nucleophilic catalyst and its use will be noted below.47 One other salient feature of this proposed anionic chain process for hydrothiolation, mediated by amines or phosphines, is that no special precautions need to be taken to exclude moisture. The combination of the typically low pKa's of thiols coupled with the high reactivity of the thiolate towards conjugate addition enables such reactions to be performed with 100% efficiency in the presence of other protic species, including water.
Maleimides are common activated substrates for such thiol-ene reactions and deserve a special comment. Due to the presence of two activating carbonyl groups in a cis-conformation coupled with ring-strain/bond angle distortion, the CC bond in maleimides is especially reactive and as such thiol-ene reactions occur extremely rapidly. Indeed, the high efficiency of these reactions is evident from their wide-spread use as a bioconjugation tool.61–69 Two special points must be made regarding this chemistry – one practical, one mechanistic. While arguably the most efficient of such thiol-ene reactions maleimides are highly potent neurotoxins and should be treated/handled with extreme care. From a mechanistic viewpoint, a cursory inspection of the literature indicates that it is common for such thiol-maleimide reactions to be conducted in solvents such as DMF or buffered aqueous solutions at ∼pH 7.5 in the apparent absence of an added (organo)catalyst. While such reactions are, technically, “catalyst free”48 there is no apparent evidence for a concerted addition of RSH across the activated maleimide CC bond. However, there is evidence that in solvents of high dielectric constant, such as DMSO and DMF, that some degree of spontaneous dissociation of thiol into thiolate occurs. In other words, in highlypolar solvents, in the absence of an apparent added catalyst, it is the solvent that is promoting the formation of the thiolate anion which is the active species, Schemes 5 and 6, initiating the anionic chain reaction. Therefore, thiol-maleimide coupling reactions that proceed under such conditions (high dielectric constant solvent, no added catalyst) should, perhaps, be more accurately described as solvent-promoted processes.
As noted for the radical-mediated thiol-ene reaction, thiol structure has an effect on the kinetics of the hydrothiolation reaction, i.e. the rate of hydrogen abstraction from thiol, or homolytic cleavage of the RS–H bond, is a rate determining factor. Under either base or nucleophile catalysis the ease with which the thiol is deprotonated to the thiolate, i.e. the pKa of the thiol, is also a factor to be considered. Thiols are, generally, significantly more acidic than the corresponding alcohols, however, pKa values can span an impressive range from, for example, 4.13 for 2,4,6-trinitrothiophenol to 11.2 for tert-pentylmercaptan.70–72
2. Applications of the radical thiol-ene reaction
Beyond the synthesis and evaluation of essentially perfect films/networks, several research groups have recently been evaluating the radical-mediated thiol-ene reaction as a facile and convenient tool for the post-polymerization modification of well-defined reactive precursor (co)polymers and for the construction of complex (macro)molecules. For example, Schlaad and co-workers have described the post-polymerization modification of well-defined homopolymers of 1,2-polybutadiene (1,2-PB) and AB diblock copolymers of 1,2-PB and poly(ethylene oxide) (PEO) with a range of functional thiols, Fig. 3.73–75 The selection of this range of thiols, T1–T10, also serves to reinforce the excellent functional group tolerance of such radical-mediated thiol-ene reactions.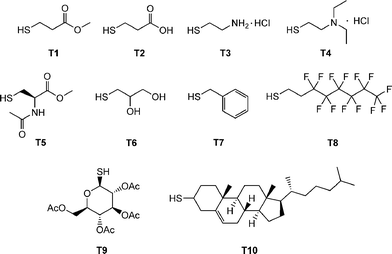 | ||
| Fig. 3 Chemical structures of thiols employed in the thiol-ene modification of 1,2-polybutadienes and polyoxazolines. | ||
1,2-PB (co)polymers were typically treated with a 10 fold excess of thiol, relative to ene bonds, in the presence of AIBN at 70 °C for 24 h under an inert atmosphere. Generally, the products were free of CC bonds, as evidenced by a combination of 1H NMR and FTIR spectroscopies, however, the degree of modification was always <100% and more commonly in the range 70–80%. The less than quantitative functionalisation was shown to be due to the occurrence of competing intramolecular cyclisation reactions, Scheme 7. As is evident, the addition of a single thiyl radical to a pendent CC bond, followed by cyclisation, actually consumes two CC bonds thus accounting for the complete loss of unsaturation but less than quantitative modification. The degree of cyclisation was also shown to be dependent on the size of the thiol-functional molecule with larger, bulkier thiols resulting in a higher degree of cyclisation. This phenomena was attributed to a steric effect whereby addition of the first, bulky, thiyl radical significantly hinders the subsequent chain transfer step thus increasing the probability of intramolecular cyclisation. However, such less than quantitative functionalisation of 1,2-PB should not be dismissed and can lead to the formation of interesting assembled structures in aqueous environments such as uni- and multilamellar vesicles.76
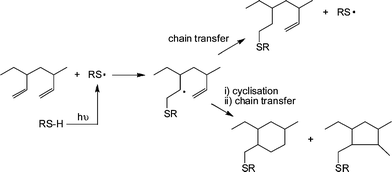 | ||
| Scheme 7 The thiol-ene modification of 1,2-polybutadiene under photochemical conditions highlighting possible competing intramolecular cyclisation reactions. | ||
While these reports highlighted a potential problem with modifying 1,2-PB Schlaad et al.75 did subsequently note that the use of such 1,2-PBhomo- and copolymers as reactive scaffolds for thiol-ene modification and the synthesis of novel functional (co)polymers should not be excluded, in part due to their commercial availability, as exemplified by the recent work of Lotti et al. who modified low molecular weight 1,2-PB with either N-acetyl-L-cysteine or its methyl ester in the presence of AIBN at 70 °C.77 Schlaad and co-workers also highlighted the fact that similar degrees of modification can be achieved under considerably milder conditions than those originally reported when initiated by either UV light or even sunlight employing significantly lower amounts of thiol.75
While the thiol-ene modification of 1,2-PB is not, strictly, a “click” process it did inspire the same group to seek a route in which such post-polymerization modifications on an ene-containing (co)polymer could be accomplished without competing, and undesirable, cyclisation reactions thus establishing a formal thiol-ene “click” modification approach to highly functional materials. To this end, Gress et al. described the synthesis and controlled cationic isomerization (co)polymerization of a novel oxazoline, 2-(3-butenyl)-2-oxazoline, followed by its thiol-ene functionalisation, Scheme 8.18
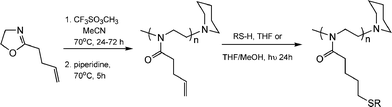 | ||
| Scheme 8 The controlled cationic isomerization polymerization of a novel ene-functional oxazoline and its subsequent post-polymerizationthiol-ene functionalisation. | ||
A series of homopolymers were prepared of varying molecular weight and modified with a range of thiols including T1, T6, T8 and T9, Fig. 3. Thiol-ene reactions were performed with 1.2–1.5 equivalents of thiol, significantly less than employed in the modification of 1,2-PB, at ambient temperature under UV irradiation. In almost all instances the post-polymerizationthiol-ene reactions were demonstrated to be quantitative with no evidence of any cyclisation products, or to contain products derived from other undesirable side-reactions, thus verifying the “click” nature of these facile functionalisations. Expanding on these initial findings, Diehl and Schlaad examined the ability to prepare a range of functional copolyoxazolines employing the radical thiol-ene reaction as a means of synthesizing materials with widely tunable lower critical solution temperatures (LCSTs).78Statistical copolymers of varying molar composition derived from 2-isopropyl-2-oxazoline and 2-(3-butenyl)-2-oxazoline were prepared by cationic isomerization copolymerization. The copolymers were subsequently quantitatively modified, under UV irradiation, with a series of thiols, including, T2, T6 and T9, yielding a range of materials with LCSTs spanning the range 5–90 °C.
Hawker and co-workers have also been actively investigating the application of the radical thiol-ene “click” reaction in polymer and materials synthesis. Killops et al. described the highly efficient synthesis of 4th-generation dendrimers in which radical thiol-ene chemistry was employed to both construct the backbone and to modify chain ends, Scheme 9.19 Treatment of 2,4,6-triallyloxy-1,3,5-triazine and thioglycerol, T6Fig. 3, with a photoinitiator (2,2-dimethoxy-2-phenylacetophenone, DMPA) under bulk conditions for 30 min gave the 6-functional alcohol ([G1]-OH6) in quantitative yield. Esterification of the 1° and 2° alcohol functionalities with pentenoic anhydride in the presence of DMAP yielded the corresponding 6-functional ene, [G1]-ene6. Subsequent thiol-ene reaction of [G1]-ene6 with thioglycerol under UV conditions yields the corresponding 2nd generation 12-functional alcohol, [G2]-OH12. Repeating this sequence of esterification/thiol-ene reactions the authors prepared, with high efficiency, both the [G4]-OH48 and [G4]-ene48dendrimers. In the case of the [G4]-ene48dendrimer, the authors also modified the peripheral CC bonds via hydrothiolation reactions with the thiolsT11–T13, Fig. 4.
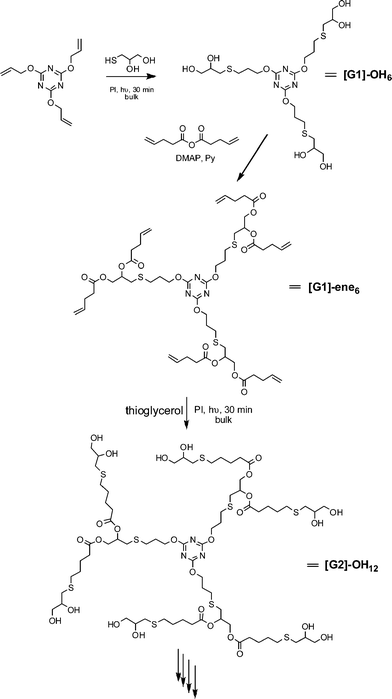 | ||
| Scheme 9 Dendrimer synthesis via sequential esterification/radical thiol-ene reactions. | ||
![Examples of thiols (T11–T13) employed to modify the outer ene bonds in a [G4]-ene48dendrimer and for the side chain modification of ene-containing (co)polymers.](/image/article/2010/PY/b9py00216b/b9py00216b-f4.gif) | ||
| Fig. 4 Examples of thiols (T11–T13) employed to modify the outer ene bonds in a [G4]-ene48dendrimer and for the side chain modification of ene-containing (co)polymers. | ||
Campos et al.79 demonstrated the broad utility of both the thermal and photochemical radical thiol-ene reactions for the post-polymerization modification of a series of ene-functional (co)polymers with the reactive olefinic functionality being statistically incorporated as pendent groups along the copolymer backbone or as single chain end species. Specifically, reversible addition-fragmentation chain transfer (RAFT) radical polymerization was employed to prepare a statistical copolymer of styrene (Sty) with 1-[(3-butenyloxy)methyl]-4-vinylbenzene (BOMVB), atom transfer radical polymerization (ATRP) for a statistical copolymer of methyl methacrylate (MMA) with but-3-enyl methacrylate (BYMA) and Sn-mediated ring-opening polymerization for a copolymer of ε-caprolactone (CL) with 6-allyl-ε-caprolactone (ACL). In all instances copolymerization yielded well-defined materials with 10–17 mol% incorporation of the ene functional monomers. Subsequent thiol-ene reactions mediated thermally with AIBN or photochemically with DMPA with T6, T11, T13–T15 (Fig. 3 and 4) (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of thiol:ene) yielded the corresponding thioether derivatives in generally excellent yields although some thermally-mediated reactions were less than quantitative, Scheme 10.
:1 molar ratio of thiol:ene) yielded the corresponding thioether derivatives in generally excellent yields although some thermally-mediated reactions were less than quantitative, Scheme 10.
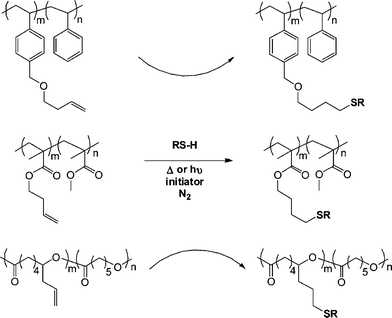 | ||
| Scheme 10 Photochemical and thermal radical thiol-ene modification of pendent side-chains in styrenic, methacrylic and polyester-based copolymers. | ||
Employing the same approach the authors also demonstrated the ability to prepare telechelic materials as well as the ability to conduct sequential thiol-ene/alkyne-azide reactions in an orthogonal manner, see Scheme 11 for example.79
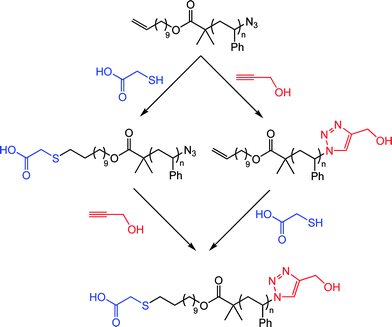 | ||
| Scheme 11 α,ω-Functional polystyrenevia sequential thiol-ene/alkyne-azide couplings, or vice versa. | ||
Campos et al. have also described the application of radical thiol-ene chemistry for the preparation of high quality soft imprint lithographic stamps based on polysiloxane and poly(ethylene glycol) (PEG) with features of ∼200 nm and < 100 nm readily obtained within a matter of minutes. The general approach to stamp preparation is shown in Fig. 5.80
![Outline for the preparation of poly[(mercaptopropyl)methylsiloxane] (PMMS) stamps: A) drop casting of thiol-ene mixture onto a patterned hard surface; B) photopolymerization at 365 nm, and C) peeling of patterned soft stamp. Reproduced by permission of WILEY-VCH from ref. 80: Campos et al., J. Adv. Mater., 2008, 20, 3728–3733. DOI: 10.1002/adma.200800330.](/image/article/2010/PY/b9py00216b/b9py00216b-f5.gif) | ||
| Fig. 5 Outline for the preparation of poly[(mercaptopropyl)methylsiloxane] (PMMS) stamps: A) drop casting of thiol-ene mixture onto a patterned hard surface; B) photopolymerization at 365 nm, and C) peeling of patterned soft stamp. Reproduced by permission of WILEY-VCH from ref. 80: Campos et al., J. Adv. Mater., 2008, 20, 3728–3733. DOI: 10.1002/adma.200800330. | ||
Two different hard masters were examined—silicon wafers with features of ca. 200 nm and highly ordered porous aluminium oxide with features on the order of 50 nm. A variety of thiol-ene mixtures were drop cast onto these masters followed by curing at 365 nm for 2 min in the presence of <0.1 wt% DMPA. For example, the polymeric siloxane, poly[(mercaptopropyl)methylsiloxane] (PMMS), was copolymerized with a range of multifunctional enes and ene mixtures including copolymers with triallyl cyanurate (TAC), TAC and the diacrylate of ethoxylated bisphenol A (BPDMA) and a TAC/BPDMA/ethyleneglycol diacrylate 4-component mixture. These multicomponent mixtures were prepared to examine the effect of comonomer(s) on the Young's modulus and water-contact angle of the resulting materials. As an example of the efficiency, i.e. high pattern replication, of this process Fig. 6 shows the scanning electron microscope (SEM) images of the silicon master (6A) along with the stamp (6B) obtained from a PMMS/TAC/BPDMA mixture, at a 6:4:1 wt ratio with a corresponding Young's modulus of ∼24 MPa. Importantly, it is clear that the nanometer scale features are perfectly reproduced without any need for applied pressure and that in the case of this material, with an intermediate Young's modulus, the stamp is easily removed by peeling without any damage to the master or any evidence of adhesion of the stamp to the master making this an extremely facile process.
 | ||
| Fig. 6 SEM images of (A) the hard silicon master with depth and width noted inset, and (B) the soft stamp cast obtained from photocuring the PMMS/TAC/BPDMA mixture. Reproduced by permission of WILEY-VCH from ref. 80: Campos et al., J. Adv. Mater., 2008, 20, 3728–3733. DOI: 10.1002/adma.200800330. | ||
Rissing and Son have exploited radical thiol-ene chemistry in the preparation of branched organosilanes as well as in the synthesis of carbosilane-thioetherdendrimers.81,82 In the case of the branched organosilanes, tetravinylsilane (TVS) and thiol were dissolved in a solvent, typically MeOH, at a molar ratio of 1:4, TVS:thiol. In some instances a photoinitiator was added, although it was not necessarily required. Samples were irradiated for between 2 and 4 h yielding the corresponding 4-armed silanes in essentially quantitative yield, Scheme 12. In all syntheses no precautions were taken to exclude atmospheric oxygen nor were any rigorous purification steps required in the isolation of the products. The authors reported the successful preparation of nine such species including products derived from T2, T3, T6, T11 and T14. In addition, products were obtained from T16–T20, Fig. 7. The carbosilane-thioetherdendrimers were constructed by alternating photochemical thiol-ene and nucleophilic substitution reactions starting from a TVS core, Scheme 13.
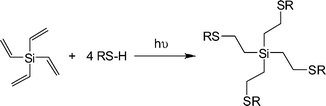 | ||
| Scheme 12 Synthetic route to 4-armed silanesvia photochemical induced thiol-ene coupling with tetravinylsilane. | ||
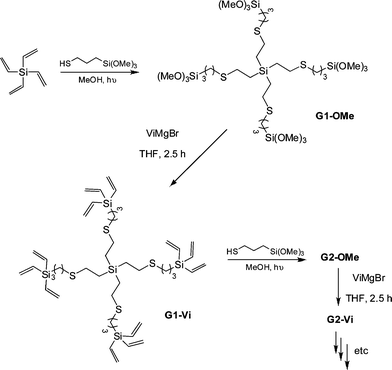 | ||
| Scheme 13 Sequential photochemical thiol-ene/nucleophilic substitution reactions as a facile route to carbosilane-thioetherdendrimers. | ||
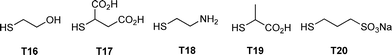 | ||
| Fig. 7 Chemical structures of examples of thiols employed in the photochemically induced thiol-ene synthesis of 4-armed silanes. | ||
The authors were able to readily prepare the 5th generation G5-OMe and G5-Videndrimers however the preparation of G6-OMe was not successful. While the thiol-ene-based steps proceeded smoothly the vinylation steps required slightly more care and reactions left for prolonged periods of time or employing >50% excess of ViMgBr resulted in the formation of high molecular weight impurities. Importantly however, the dendrimers were easily purified by simple precipitation protocols.
The synthesis of biomimetic sugarpolymers, so-called glycopolymers, can be achieved by two general routes: direct polymerization of a sugar-containing monomer (in either a protected or non-protected form), or via the post-polymerization modification of an appropriate reactive (co)polymer with a sugar molecule.83 Given the facile and orthogonal nature of the thiol-ene reaction it is not, therefore, surprising that such chemistry has been employed as a means of preparing sugar-containing materials. Chen et al. recently described the preparation of neoglycopolymers via the thiol-ene coupling reaction between ene-functional precursor (co)polymers and glucothiose under photochemical conditions with DMPA as the photoinitiator.84
Initially, a commercially available poly(2-hydroxyethyl methacrylate) was treated with 4-pentenoic anhydride to yield the corresponding ene-side chain species poly(2-(methacryloyloxy)ethyl pent-4-enoate), which when reacted with glucothiose under photochemcial conditions yielded the corresponding glycopolymer in quantitative yield as judged by a combination of NMR and FTIR spectroscopies. Following this proof-of-concept demonstration Chen et al.84 prepared a well-defined AB diblock of di(ethylene glycol)methylether methacrylate (DEGMA) and 2-hydroxyethyl methacrylate (HEMA) viaRAFT polymerization using cumyl dithiobenzoate as the chain transfer agent. Subsequent cleavage of the thiocarbonylthio end-groups by treatment with AIBN was followed by treatment of the HEMA residues as described above to introduce ene functionality. Finally, photochemical reaction of glucothiose with the ene groups yielded the corresponding AB diblock glycopolymer, Scheme 14. By virtue of the readily accessible LCST of ∼29 °C associated with polyDEGMA such DEGMA-glucothiose-based diblock copolymers were shown to undergo temperature-induced micellisation, Fig. 8. Heating a solution of the AB diblock copolymer to 40 °C resulted in self-assembly in which the now-hydrophobic DEGMA residues were located in the micelle interior stabilised by the hydrophilic sugar residues. Such self-assembly was verified by a combination of dynamic light scattering (DLS) and transmission electron microscopy (TEM). The glycopolymers were also demonstrated to efficiently bind with Concanavalin A, a glucose-specific lectin .
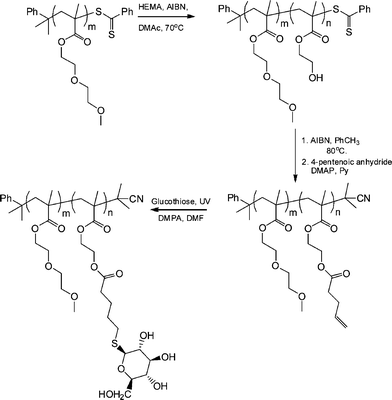 | ||
| Scheme 14 Synthetic approach to AB diblock copolymers of DEGMA-HEMA followed by post-polymerization modification to yield the target neoglycopolymers. | ||
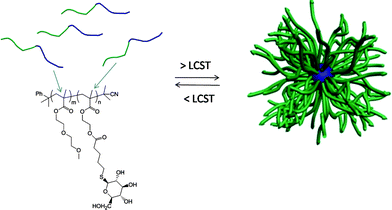 | ||
| Fig. 8 Temperature-induced self-assembly of AB diblock copolymers comprised of DEGMA and glucothiose-modified HEMA residues. | ||
Jonkheijm et al.85 highlighted the bioorthogonality of the radical thiol-ene reaction in the preparation of patterned surfaces via the immobilization of biomolecules. The covalent attachment of polyamidoaminedendrimers to a silicon oxide surface followed by treatment with aminocaproic acid (to introduce a spacer group) and subsequent coupling with cystamine yielded, after disulfide reduction, the corresponding thiol-functional surface. The surface was coated with a solution of the ene functional biomolecules, such as an ene-bearing biotin, followed by immediate coverage with a photomask and irradiation. After removal of the mask and washing the biotin patterns were visualized by treatment with a Cy5-labeled streptavidin (SAv), yielding the corresponding SAv-patterned surface, Fig. 9.
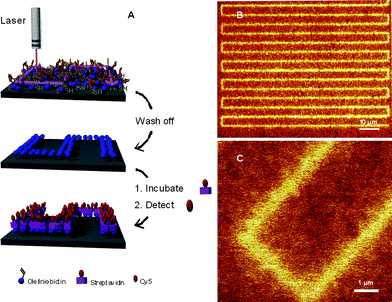 | ||
| Fig. 9 (A) General method of photochemical nanopatterning in which ene-functional biotin is deposited onto a thiol-modified surface and laser-irradiated at 411 nm. The pattern is visualized by incubating the surface with SAv-Cy5, and (B), (C) scanning confocal fluorescence microscopy images of the nanopatterns. Reproduced by permission of WILEY-VCH from ref. 85: Jonkheijm et al., Angew. Chem., Int. Ed., 2008, 47, 4421–4424. DOI: 10.1002/anie.200800101. | ||
Connal and co-workers recently described the layer-by-layer assembly of hydrogen-bonded multilayers comprised of polyvinylpyrrolidone (PVP) and poly(methacrylic acid) (PMAA, functionalized with either ene or thiol functional groups) onto silica nanoparticles, Scheme 15. After the assembly of nine layers the coated particles were irradiated with UV at 256 nm for 2 h, inducing crosslinking between the PMAA layers viathiol-ene coupling.86 Efficient crosslinking was achieved without added photoinitiator or exclusion of atmospheric oxygen. Successful crosslinking was verified by fluorescence microscopy in a pH 7 solution—conditions that in the absence of crosslinking would result in the disintegration of the multi-layer assembly due to ionization of the PMAA residues and a disruption of H-bonding. After HF etching (to remove the silica core) and incubation at pH 7 (to remove the PVP), crosslinked PMAA capsules were obtained as verified by a combination of SEM and TEM, Fig. 10. Finally, the authors demonstrated the ability to functionalise the outer layer of the polymer multilayer-coated particles by reacting residual thiol-functional groups with acrylate or maleimide-functionalised PEG.
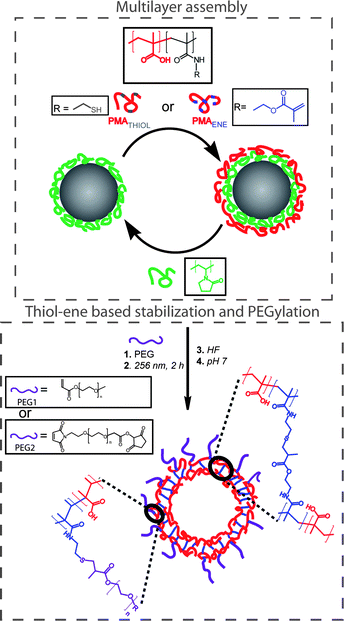 | ||
| Scheme 15 Preparation of (PVP/PMAAthiol/PVP/PMAAene)-coated particles, PEGylation, and stabilization viathiol-ene chemistry and generation of PMAA capsules upon removal of silica and PVP. Reproduced by permission of The American Chemical Society from ref. 86: Connal et al., Chem. Mater., 2009, 21, 576–578. DOI: 10.1021/cm803011w. Copyright 2009 American Chemical Society. | ||
 | ||
| Fig. 10 (A) Transmission electron micrographs and (B) scanning electron micrographs of poly(methacrylic acid) capsules at pH 7. Reproduced by permission of The American Chemical Society from ref. 85: Connal et al., Chem. Mater., 2009, 21, 576–578. DOI: 10.1021/cm803011w. Copyright 2009 American Chemical Society. | ||
Goldmann et al. described the surface modification of micron-sized polydivinylbenzene (PDVB) particles employing both thiol-ene and alkyne-azide “click” chemistries.87 In the case of thiol-ene coupling, a precursor homopolymer of N-isopropylacrylamide was prepared by RAFT polymerization with a degree of polymerization of 45 (PNIPAm45). After end-group cleavage the thiol-terminated PNIPAm45polymers were reacted with residual vinyl groups on the surface of the PDVB microspheres in the presence of AIBN at 70 °C for 48 h, ultimately yielding the grafted-to PNIPAm45-PDVBnanoparticles, Scheme 16 and Fig. 11. The PNIPAm45-modified PDVBnanoparticles were characterized, and successful grafting-to verified, using a variety of techniques such as elemental microanalysis, SEM, and X-ray photoemission spectroscopy (XPS).
 | ||
| Scheme 16 General grafting-to approach for the surface modification of PDVBnanoparticles. | ||
 | ||
| Fig. 11 Scanning electron micrographs of (a) PDVB microspheres and (b) PDVB microspheres grafted with PNIPAM45. The surface of the NIPAm-grafted PDVB microspheres is clearly coarser than the precursor particles. Reproduced by permission of The American Chemical Society from ref. 87: Goldmann et al., Macromolecules, 2009, 42, 3707–3714. DOI: 10.1021/ma900332d. Copyright 2009 American Chemical Society. | ||
Harth and co-workers recently detailed the synthesis and post-assembly modification of polyesternanoparticles containing reactive functional groups including allylic species suitable for thiol-ene reactions.88Linear polymers were prepared viaring-opening copolymerization of δ-valerolactone, α-allyl-δ-valerolactone and 2-oxepane-1,5-dione, Fig. 12.
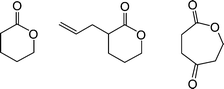 | ||
| Fig. 12 Chemical structures of lactones employed in polyesternanoparticle synthesis. | ||
Subsequently, the allylic side groups were partially oxidized, using meta-chloroperbenzoic acid, introducing epoxy functionality in the side-chains that was used as a cross-linking handle in reactions with ethylenediamine yielding allyl-functional nanoparticles, with an average size of ∼300 nm as determined by TEM, Fig. 13. Initially, benzylmercaptan was conjugated to the free allylic groups at between 25 and 35 °C in the absence of an added radical (photo)initiator yielding, under optimized conditions, approximately 75% hydrothiolation product. Such “non-forcing” conditions were intentionally employed since the primary motivating factor for preparing reactive nanoparticles was to establish a facile conjugation protocol for biologically relevant moieties for cancer targeting/delivery applications. Having optimized conjugation conditions, a variety of thiol-containing peptidic targeting species were conjugated as well as thiolated dendritic transporter molecules containing guanidine functional groups.
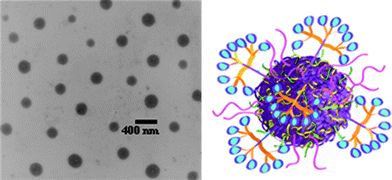 | ||
| Fig. 13 TEM image (left) of polyesternanoparticles and idealized image (right) of a polyesternanoparticle modified with dendron carrier units and peptide targeting groups. Reproduced by permission of The Royal Society of Chemistry from ref. 88: van der Ende et al.,Soft Matter, 2009, 5, 1417–1425. 10.1039/b820379b. | ||
Hawker and co-workers89 recently highlighted an interesting route to functional multimodal, hybrid organic–inorganic nanoparticles, Scheme 17. MnFe2O4 and Au nanoparticles, grafted with short polystyrene ligands, were initially dispersed in divinylbenzene (DVB) monomer followed by emulsification in aqueous media with cetyltrimethylammonium chloride as surfactant. Subsequent free radical polymerization with 2,2′-azobis(2-amidinopropane) dihydrochloride (V-50) yielded the crosslinked PDVBlatex particles embedded with the inorganic Fe and Au particles, Fig. 14. While these particles were stable in an aqueous environment they exhibited poor dispersability in THF. To address this issue, short thiol-terminated poly(ethylene glycol) chains (Mn ≈ 2000) were grafted to the particle surfaces viathiol-ene coupling, taking advantage of the presence of residual CC bonds from the miniemulsion process.
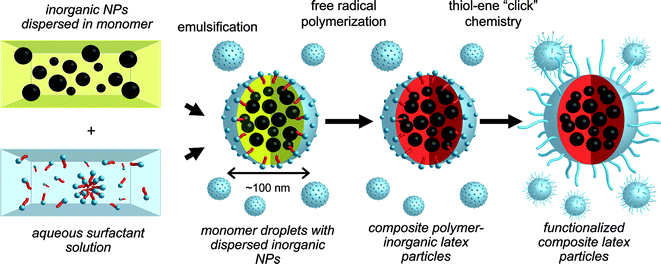 | ||
| Scheme 17 Synthesis of multimodal latex nanoparticlesviaminiemulsion polymerization followed by surface functionalisation with poly(ethylene glycol)viathiol-ene ‘click’ chemistry. | ||
 | ||
| Fig. 14 TEM images of Fe–Au hybrid organic–inorganic nanoparticles prepared by coencapsulation of MnFe2O4 (10 wt% loading) with an average diameter of 13.0 nm and Au nanoparticles (19 wt% loading) of average diameter 13.0 nm (a), 18.0 nm (b), 24.0 nm (c), and 46.0 nm (d). Reproduced by permission of The American Chemical Society from ref. 89: van Berkel et al., Macromolecules, 2009, 42, 1425–1427. DOI: 10.1021/ma802849f. Copyright 2009 American Chemical Society. | ||
3. Applications of the thiol-ene reaction with activated substrates
Though not as widely examined as the radical-mediated thiol-ene reaction in polymer/materials synthesis there are recent examples of the base- and nucleophile-mediated thiol-ene reaction with activated substrates that clearly proceed with “click” characteristics. It should be noted, however, that the majority of these reports have thus far focused on end-group functionalisation of precursor (co)polymers with fewer examples describing the synthesis of complex (macro)molecules. Perhaps not surprisingly much of the work in this area has focused on the modification of (co)polymers prepared by RAFT polymerization90–98 by virtue of the fact that materials prepared by this method serve as convenient masked macromolecular thiols. Indeed, the thiocarbonylthio end-groups present in RAFT-prepared (co)polymers can be readily cleaved to the corresponding thiolvia a number of different routes.99–105Qiu and Winnik106 described the RAFT synthesis of an α,ω-functional thiocarbonylthio PNIPAmhomopolymer prepared from a difunctional trithiocarbonate RAFT agent with a measured Mn of 13000. Subsequently, one-pot end-group transformations were achieved viaaminolysis with butylamine (a 5-fold excess based on thiocarbonylthio groups) in the presence of a small amount of tris(2-carboxyethyl)phosphine hydrochloride (TCEP·HCl) under a nitrogen atmosphere for a period of 1 h followed by the direct addition of a 10-fold excess (based on thiocarbonylthio groups) of either butyl acrylate (ENE1, Fig. 15) or 2-hydroxyethyl acrylate (ENE2, Fig. 15). Successful end-group cleavage and subsequent thiol-ene reaction was verified using a combination of 1H NMR spectroscopy, size exclusion chromatography and UV-vis spectrophotometry.
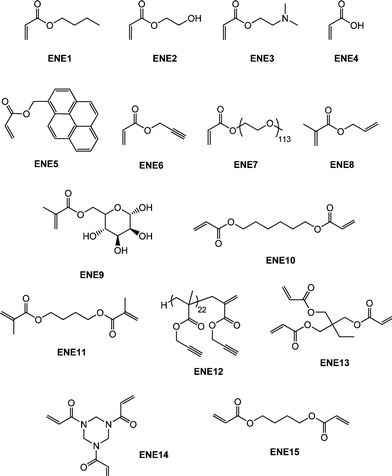 | ||
| Fig. 15 Chemical structures of activated enes employed in base/nucleophilic thiol-ene reactions. | ||
Scales et al.107 reported the RAFT synthesis of an intermediate molecular weight NIPAmhomopolymer (Mn = 38500, Mw/Mn = 1.07) using a trithiocarbonate RAFT agent. End-group cleavage with NaBH4 followed by purification by dialysis and subsequent treatment with TCEP·HCl yielded the thiol-terminated NIPAmhomopolymer. Reaction with pyrenemaleimide (MAL1, Fig. 16) for 24 h at 50 °C in the presence of a catalytic amount of ethylenediamine in DMF yielded the fluorescent, ω-modified PNIPAm in an 84% yield.
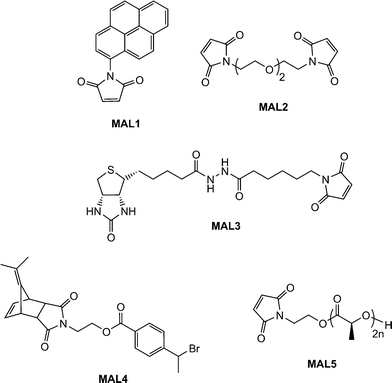 | ||
| Fig. 16 Chemical structures of maleimide, and protected maleimides, used as substrates in base/nucleophilic thiol-ene conjugation reactions. | ||
Li and co-workers8 described the first example of sequential thiol-ene reactions, both involving NEt3-catalyzed thiol-maleimide coupling, as a route to end-functional polymers as well as a viable modular approach to AB diblock copolymers and also as a means of combining thiol-ene reactions with highly efficient Diels–Alder reactions. Treatment of a precursor NIPAmhomopolymer, prepared by RAFT with the trithiocarbonate chain transfer agent 2-dodecylsulfanylthiocarbonylsulfanyl-2-methylpropionic acid, with a 10-fold excess of 2-ethanolamine (based on thiocarbonylthio end-groups) in the presence of tributylphosphine for 2 h under a nitrogen atmosphere in 1,4-dioxane yielded the thiol-terminated homopolymer (PNIPAm-SH) quantitatively as determined by a combination of UV-vis spectrophotometry and FT-IR spectroscopy. Reaction of PNIPAm-SH with a 10-fold molar excess of bismaleimidodiethyleneglycol (MAL2, Fig. 16) in the presence of NEt3, Scheme 18, resulted in hydrothiolation of one of the maleimide groups yielding the maleimide end-functional PNIPAm, PNIPAm-M. Clearly the presence of this reactive end-group facilitates further modification. Indeed, the authors demonstrated that reaction of PNIPAm-M with either 1-dodecanethiol or 4-methoxybenzyl mercaptan under base catalysis yielded the corresponding thioether products in essentially quantitative yield as determined by end-group analysis and 1H NMR spectroscopy. Extending this approach, the use of a low molecular weight thiol-terminated polystyrene (PS-SH), also prepared by RAFT followed by aminolysis of the end-groups, in place of a low-molecular-weight thiol facilitated the modular synthesis of PNIPAm-b-PScopolymers. A large excess of PS-SH was employed to help drive block copolymer formation with unreacted PS-SH being easily removed by immobilization onto an iodoacetate resin. Successful block copolymer formation was verified by gel permeation chromatography, Fig. 17.
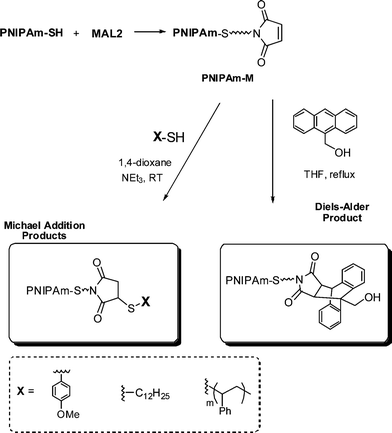 | ||
| Scheme 18 Synthesis outline for the preparation of maleimide end-functionalized PNIPAm and subsequent thiol-ene and Diels–Alder modification. | ||
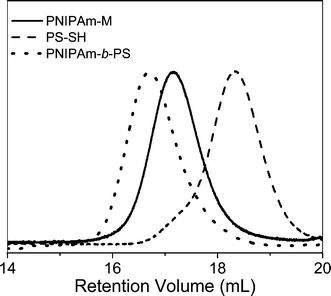 | ||
| Fig. 17 Gel permeation chromatographic traces (RI signals) for precursor PNIPAm-M and PS-SHhomopolymers and the resulting AB diblock copolymer after thiol-ene coupling. | ||
Spruell et al.108 described the RAFT synthesis of PShomopolymers and poly(methyl methacrylate)-block-PS (PMMA-b-PS) copolymers and their subsequent end-group cleavage and thiol-Michael addition to a range of functional acrylic monomers (ENE1–7, Fig. 15). End-group cleavage/conjugation was performed in a one-pot process using a variety of approaches including NaBH4 reduction (in the presence and absence of PBu3) and also viaaminolysis, with propylamine, again with and without PBu3. The importance of adding PBu3 to prevent disulfide formation was demonstrated. Generally, very high degrees of end-group functionalization were achieved as determined by NMR spectroscopy. This particular study clearly highlighted the functional group tolerance of this thiol-ene chemistry with the employed enes, ENE1–7, bearing, for example, tertiary amino, acid, propargyl and hydroxyl functional groups.
Boyer et al.109,110 detailed the end-group modification of three different RAFT-synthesised homopolymers, namely PMMA, PNIPAm and poly(N-2-(hydroxypropyl) methacrylamide) (PHPMA). The dithioester or trithiocarbonate end-groups were cleaved viaaminolysis in DMF with hexylamine while in the presence of a range of functional enes and NEt3 including the sugar methacrylate, ENE9, the di-(meth)acrylates ENE10 and ENE11, Fig. 15, as well as the biotin-maleimide, MAL3Fig. 16. Typically end-group modification efficiencies of >90% were obtained and clearly the use of ENE10 and ENE11 as activated ene substrates offers an attractive route to (meth)acrylic macromonomers.
As described above, both Li8 and co-workers and Boyer et al.109 have used difunctional activated enes (a bis maleimide and bis (meth)acrylates respectively) in end-group modifications with RAFT-prepared precursors. While efficient these approaches necessitate the use of a large excess of di-ene since in all instances both CC bonds are identical and thus equally reactive towards hydrothiolation. Recently, Yu et al.15 described an approach enabling sequential thiol-ene/thiol-ene (or thiol-ene/thiol-yne) reactions using an initial substrate in which the two CC bonds had inherently different reactivity, i.e. they exhibited orthogonality. Specifically, the approach took advantage of the use of allyl methacrylate (ENE8, Fig. 15) in which the pendent allylic group readily undergoes radicalthiol-ene reactions but not base/nucleophilic thiol-ene reactions. Homopolymerization of NIPAm in the presence of CPDB and AIBN in DMF at 70 °C, Scheme 19, yielded the well-defined PNIPAmhomopolymer with an average degree of polymerization of 50. The one-pot treatment of the NIPAmhomopolymer with octylamine in CH2Cl2 with dimethylphenylphosphine (Me2PPh) as catalyst in the presence of ENE8 under a nitrogen atmosphere at RT results in the quantitative formation of the allyl-end-functionalized PNIPAm (PNIPAm50-ALMA). Similarly, replacing ENE8 with propargyl acrylate, ENE6, results in the formation of the corresponding yne-end-functional PNIPAm (PNIPAm50-PROPA). Successful modification, without any apparent detrimental side-reactions, was confirmed using a combination of 1H NMR spectroscopy and gel permeation chromatography. Subsequently, the ene, and yne, end groups were reacted under photochemical radical conditions (RT, air atmosphere) in the presence of benzil dimethyl ketal (Irgacure 651) with 6-mercaptohexan-1-ol, hexane-1-thiol, and iso-butyl 3-mercaptopropyl polyhedral oligomeric silsesquioxane (POSS-SH) yielding the corresponding mono and bis addition products in essentially quantitative yields.
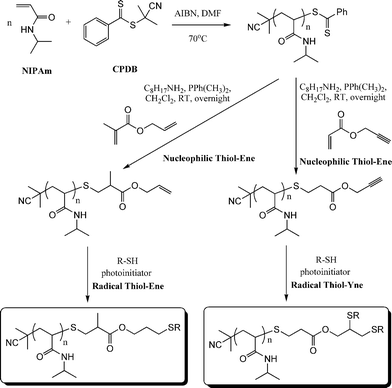 | ||
| Scheme 19 Synthetic approach to mono and bis end-functionalised PNIPAmvia sequential nucleophilic thiol-ene/radical thiol-ene or nucleophilic thiol-ene/radical thiol-yne reactions. | ||
This is still the only example in which sequential thiol-ene/thiol-ene reactions operating via inherently different mechanisms have been employed in polymer/materials synthesis/modification. Importantly, aside from offering a facile route to mono and bis-end-functional polymers, in the case of polymeric substrates such as PNIPAm it also allows for an easy tuning of the LCST. For example, in the study detailed above, the measured LCST of the end-modified PNIPAm50homopolymers ranged between 2 and ∼10 °C lower than that typical of NIPAmhomopolymers, and in the case of the homopolymer modified with two POSS-SH molecules, the resulting material was not soluble as a 1 wt% solution in water even at temperatures approaching 0 °C.
In addition to the preparation of telechelic (co)polymers and the modular synthesis of block copolymers, the nucleophilic thiol-ene reaction has also been employed in the synthesis of advanced architecture materials such as star polymers , Scheme 20. In keeping with the inherent ability of RAFT-prepared (co)polymers to act as macromolecular thiols after end-group cleavage, Chan et al.47,111 detailed the convergent synthesis of 3-arm star polymers based on N,N-diethylacrylamide (DEAm) and n-butyl acrylate (BA). Low molecular weight polyDEAm (PDEAm, Mn ∼ 4000 by end-group analysis) and polyBA (PBA, Mn ∼ 3300 by end-group analysis) were treated with hexylamine (end-group cleavage agent) in the presence of Me2PPh (hydrothiolation catalyst) and the multifunctional ene trimethylolpropane triacrylate, TMPTA (ENE13, Fig. 15). Using a combination of FT-IR and 1H/13C NMR spectroscopies and MALDI-TOF MS the authors demonstrated that such convergent syntheses were rapid, and quantitative. For example, Fig. 18 shows 1H NMR spectra, between the range 5.5–6.5 ppm, for a mixture of PBA and ENE13 (top spectra) highlighting the presence of the ene bonds and the same sample after the addition of hexylamine and Me2PPh. In the short period of time required for the addition of these two reagents and the subsequent recording of a spectrum (∼5 min), the signals associated with the CC bonds had completely disappeared highlighting the unusually fast nature of such convergent syntheses.
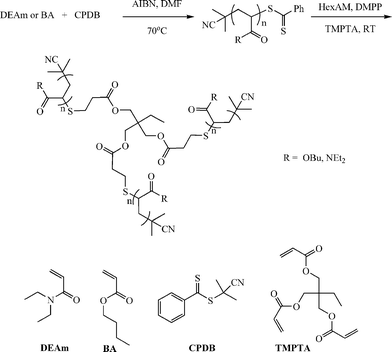 | ||
| Scheme 20 Synthetic outline for the convergent preparation of 3-arm star polymers under phosphine catalysis with RAFT-prepared precursor homopolymers. Reproduced by permission of Elsevier from ref. 111: Chan et al., The nucleophilic, phosphine-catalyzed thiol-ene click reaction and convergent star synthesis with RAFT-prepared homopolymers, Polymer, 2009, 50, 3158–3168. DOI: 10.1016/j.polymer.2009.04.030. Copyright 2009 Elsevier. | ||
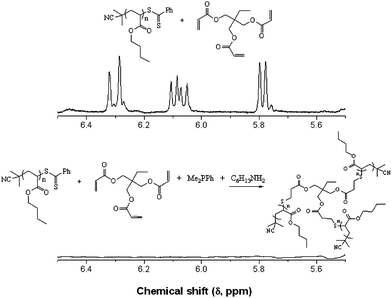 | ||
| Fig. 18 1H NMR spectra, recorded in CDCl3, of PBA + ENE13 (top) and the sample after the addition of hexylamine and dimethylphenylphosphine (bottom) highlighting the disappearance of the ene bonds. Reproduced by permission of Elsevier from ref. 111: Chan et al., The nucleophilic, phosphine-catalyzed thiol-ene click reaction and convergent star synthesis with RAFT-prepared homopolymers, Polymer, 2009, 50, 3158–3168. DOI: 10.1016/j.polymer.2009.04.030. Copyright 2009 Elsevier. | ||
The thiol-ene-based synthesis of star shaped oligomers has been described by Rim and Son.112 Reaction of the trifunctional amide, ENE14Fig. 15, with 2-mercaptoethanol in the presence of n-propylamine resulted in hydrothiolation of the activated CC bonds yielding the corresponding triol, Scheme 21. Subsequent acylation of the OH groups with acryloyl chloride regenerated a triene which was subsequently reacted with additional 2-mercaptoethanol.
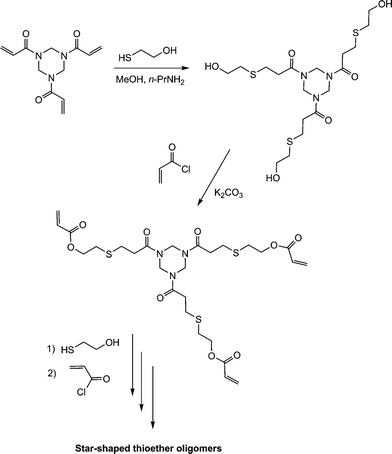 | ||
| Scheme 21 Synthesis route to 3-arm star thioether oligomers via sequential thiol-ene/acylation reactions. | ||
Repeating this sequence of reactions resulted in the formation of the star-shaped thioether oligomers.
An alternative approach to telechelic polymers was described by Tolstyka et al.113 who prepared PS end functionalised with benzyl and cysteine residuesvia the protected maleimide ATRP initiator MAL4, Fig. 16. Styrene was homopolymerized under standard ATRP conditions (CuBr, CuBr2 (5%), PMDETA, 80 °C) yielding a precursor PS with an Mn of 3,840 and polydispersity index of 1.17. The PShomopolymer was then dimerized via an atom transfer radical coupling reaction yielding an α,ω-dimethylfulvene-protected maleimide PS, Scheme 22. Subsequent treatment with either benzyl mercaptan or N-acetyl-L-cysteine methyl ester in refluxing toluene for 24 h gave the hydrothiolation products in quantitative yield via a tandem [4 + 2] cycloreversion, liberating the free maleimide, followed by thiol-ene coupling.
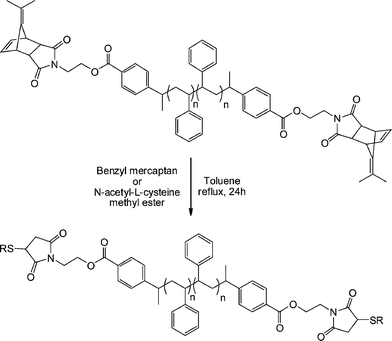 | ||
| Scheme 22 One-pot cycloreversion/thiol-ene coupling as a route to α,ω-functional polystyrenevia a precursor dimethylfulvene-protected maleimide. | ||
Employing a similar strategy, Dove and co-workers114,115 reported the synthesis of maleimide-end functionalized polylactide, MAL5, Fig. 16, suitable for thiol-ene coupling using a furan-protected initiator, Scheme 23.
![The synthesis of α-functional thioetherpolylactidesviaring-opening polymerization of lactide with a furan-protected maleimide alcohol initiator followed by a [4 + 2] cycloreversion and base-mediated thiol-ene coupling.](/image/article/2010/PY/b9py00216b/b9py00216b-s23.gif) | ||
| Scheme 23 The synthesis of α-functional thioetherpolylactidesviaring-opening polymerization of lactide with a furan-protected maleimide alcohol initiator followed by a [4 + 2] cycloreversion and base-mediated thiol-ene coupling. | ||
The authors examined the coupling of a range of functional thiols including T2, T3, T6 and T7 (Fig. 3) as well as thiophenol (T21), dodecylmercaptan (T22), iso-propylmercaptan (T23), tert-butylmercaptan (T24), cysteine ethyl ester (T25) and glutathione (T26), Fig. 19. In all instances coupling reactions were left until >99.5% conversion had been reached as determined by 1H NMR spectroscopy. This highly versatile approach was also extended to the preparation of telechelic and star shaped polylactides.
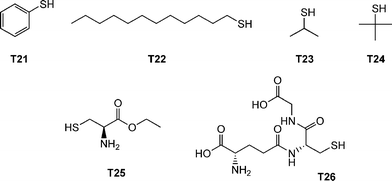 | ||
| Fig. 19 Chemical structures of thiols used in the α-functionalisation of polylactide. | ||
In a complimentary approach to the radical thiol-ene modification of ene side-chain functional polyoxazolines as described by Gress et al.,18 Cesana et al.116 reported the synthesis and controlled ring-opening copolymerization of a thiol-protected oxazoline monomer, namely 2-[2-(4-methoxybenzylsulfanyl)ethyl]-2-oxazoline, with 2-ethyl-2-oxazoline yielding well-defined statistical copolymers with narrow molecular weight distributions. After quantitative deprotection, in a mixture of anisole and trifluoroacetic acid, the liberated free thiol groups were reacted with, N-phenylacrylamide, benzylmaleimide to yield the side-chain-modified copolymers quantitatively, as well as with acrylic and maleimide α-functional poly(2-methyl-2-oxazoline)s to yield novel graft copolymers, Scheme 24. Interestingly, the thiol-Michael reactions were performed in the absence of an added catalyst although were left for 14 h to facilitate quantitative reaction.
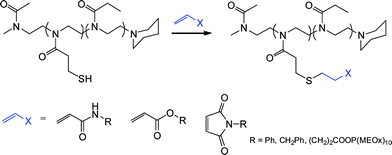 | ||
| Scheme 24 Synthesis of side-chain-modified poly(2-oxazoline)svia a thiol-Michael polymer analogous reaction. | ||
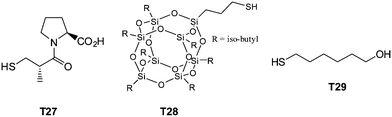 | ||
| Fig. 20 Chemical structures of examples of thiols employed in the synthesis of multifunctional thioethers. | ||
Haddleton and co-workers117 described the synthesis of glycopolymers employing catalytic chain transfer polymerization (CCTP) to prepare alkyne-functional macromonomers (ENE12, Fig. 15) which were subsequently modified via a combination of Me2PPh-catalyzed thiol-ene coupling with either T7 (Fig. 3) or T16 (Fig. 7) followed by Cu-catalyzed alkyne-azide coupling with a series of azide functional sugars, Scheme 25. These synthetic glycopolymers were examined with respect to their ability to be recognised by certain lectins . For example, the mannose-functional glycopolymer was demonstrated to bind to the mannose-specific lectin Concanavalin A, while the galactose-bearing polymer was shown to bind to the galactose-specific lectin Ricinus communis agglutinin I (RCA I).
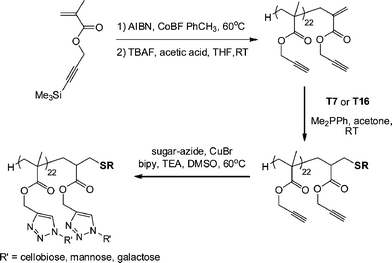 | ||
| Scheme 25 Synthetic route to novel glycopolymers employing a combination of thiol-ene and alkyne-azide “click” chemistries. | ||
Highly functional branched thioethers were reported by Chan et al.via a sequential route involving initial phosphine-catalyzed nucleophilic thiol-ene reaction followed by a radical thiol-yne coupling process, Scheme 26.16 Reaction of the commercially available tetrathiol, pentaerithryol tetra(3-mercaptopropionate) with propargyl acrylate (ENE6, Fig. 15) in the presence of Me2PPh under ambient conditions gave the corresponding tetra-yne in quantitative yield. The tetra-yne was subsequently employed, without purification, in the reaction with a range of functional thiols including T2, T6, T14, T15 and T17 as well as captopril (an angiotensin converting enzyme (ACE) inhibitor), iso-butyl mercaptopropyl polyhedral oligomeric silsesquioxane, and 6-mercaptohexanol (T27–T29, Fig. 20) under photochemical conditions with α,α-dimethoxy-α-phenylacetophenone at 365 nm under a normal air atmosphere.
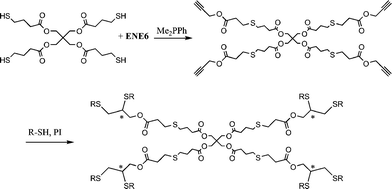 | ||
| Scheme 26 Synthesis of multifunctional thioethersvia sequential thiol-ene/thiol-yne reactions (generated stereocentres denoted by *). | ||
The high efficiency of these sequential thiol-based reactions was confirmed by MALDI-TOF MS. For example, Fig. 21 shows the recorded mass spectrum for the product derived from the reaction of the tetra-yne with thioglycerol (T6, Fig. 3). Assuming quantitative reaction for the nucleophilic thiol-ene and radical thiol-yne processes the resulting 16-functional polyol would have an expected molecular mass of 1794.4 Da. The mass spectrum indicates a primary peak at m/z = 1816.4 that is due to the Na+ cationized polyol, i.e. [16-functional polyol + Na]+. Inset is shown the measured (black) and calculated (grey) isotopic distributions associated with [16-functional polyol + Na]+. These agree perfectly with each other and further serve to confirm the quantitative nature of both of these thiol-based reactions. Similar results were obtained for the products derived from other functional thiols.
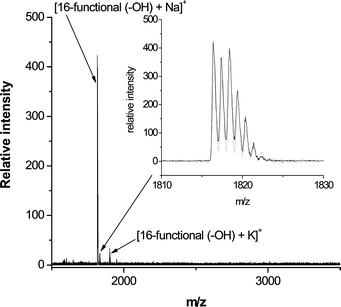 | ||
| Fig. 21 MALDI-TOF mass spectrum of the 16-functional polyol derived from the thiol-yne reaction of the tetra-yne with T6. Reproduced by permission of The American Chemical Society from ref. 16: Chan et al., J. Am. Chem. Soc., 2009, 131, 5751–5753. DOI: 10.1021/ja8099135. Copyright 2009 American Chemical Society. | ||
In another example of sequential thiol-based “click” reactions, Shin et al.23 described the synthesis and thermal/mechanical properties of novel segmented polythiourethane elastomers employing a combination of phosphine-catalyzed thiol-ene chemistry along with NEt3-catalyzed thiol-isocyanate coupling, Scheme 27. Reaction of a slight excess of 1,6-hexanedithiol (HDT) with butanediol diacrylate (ENE15, Fig. 15) under Me2PPh catalysis in DMAc resulted in the rapid and quantitative formation of a thiol-terminated oligomeric species that served as the soft, flexible segment in the final polythiourethanes. The molecular weight of such oligomers is readily controlled by varying the ratio of HDT:ENE15. Subsequent reaction of the prepolymer and HDT with 0.1 wt% NEt3 in DMAc with a range of commercially available diisocyanates yielded, quantitatively, the target segmented polythiourethanes. Thermal and dynamic mechanical property measurements indicated that the respective soft and hard phases could be separated with the degree of phase mixing easily controlled by varying the length of each segment, the ratio of the two phases, and the chemical structure of the hard segment, i.e. that derived from the diisocyanate. The tensile properties were measured and were shown to be dependent on the degree of microphase separation/mixing of the two segments.
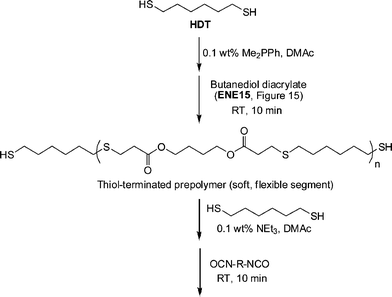 | ||
| Scheme 27 Outline for the preparation of segmented polythiourethanesvia sequential thiol-ene/thiol-isocyanate processes. | ||
Recently, Jones and co-workers118 described the preparation of polymer–protein conjugates via a one-pot phosphine-mediated thiol-ene reaction between poly(monomethoxy ethyleneglycol) (meth)acrylates and thiol groups in salmon calcitonin (sCT), Scheme 28. In this particular instance the added phosphine, TCEP·HCl, served two equally important roles. In the first step it served as a reducing agent, converting the disulfide bonds in sCT to the corresponding free thiols, while after the addition of the poly(monomethoxy ethyleneglycol) (meth)acrylates the TCEP·HCl subsequently acted as the nucleophilic catalyst mediating the thiol-Michael reaction. While not the first example highlighting the dual capabilities of phosphines as both reducing agents and nucleophilic catalysts it serves as another important demonstration of the potency of this synthetic approach.
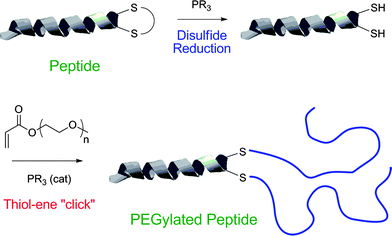 | ||
| Scheme 28 The synthesis of polymer–proteinbioconjugates based on salmon calcitonin and poly(monomethoxy ethylene glycol) (meth)acrylates constructed viaphosphine-mediated thiol-Michael addition. Reproduced by permission of The Royal Society of Chemistry from ref. 118: Jones et al., Chem. Commun., 2009, 5272–5274. 10.1039/b906865a. | ||
In another example of polymer–protein bioconjugation, Boyer and Davis119 reported a one-pot synthetic protocol for the attachment of sugar moieties to a polymer side-chain with simultaneous end-group modification via thiol-Michael reaction with a maleimide-functional biotin, Scheme 29. Pentafluorophenyl acrylate was polymerized by RAFT in benzene in the presence of 3-(benzylsulfanylthio-carbonylsulfanyl) propionic acid and AIBN at 70 °C yielding homopolymers with molecular weights in the range ∼3500–15000 and polydispersity indices of 1.14–1.20. Subsequent treatment of the homopolymers with an aminosugar in the presence of maleimide-functional biotin and NEt3 led to the formation of the target biotin-modified glycopolymers. The use of an excess of the aminosugar is important since beside serving as the reagent that effects the acyl substitution on the polymer sidechain, as a primary amine it also effectively and rapidly cleaves the thiocarbonylthio end-group liberating the free thiol. In the absence of a suitable activated substrate this can result in disulfide bond formation and other undesirable side reactions. However, when conducted in the presence of the maleimide-biotin the liberated free thiol at the ω-chain terminus is immediately “captured” by the maleimide-biotinvia a thiol-Michael reaction yielding the doubly-modified homopolymers.
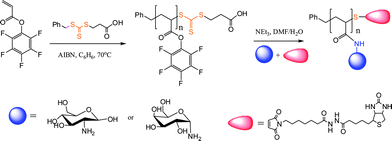 | ||
| Scheme 29 One-pot synthesis of ω-biotin functional glycopolymersvia simultaneous substitution/thiol-ene reactions. | ||
In an approach similar to that described by Goldmann et al.,87 Gu and co-workers120 recently described a route to sugar-modified microspheres based on ethyleneglycol dimethacrylate (EGDMA). EGDMA-based microspheres were prepared by suspension polymerization using PVP as stabilizer yielding methacrylate functional particles susceptible to thiol-Michael addition. The treatment of the ene-functional microspheres with glucothiose in DMF with TCEP·HCl yielded the corresponding sugar-modified spheres. Importantly, such surface-modified species still retained their biological-relevant activity as evidenced by their ability to strongly bind to Concanavalin A. This straightforward approach was extended to commercially available Wang resin which, after acylation of the surface OH groups with acryloyl chloride and thiol-Michael coupling with glucothiose was also shown to strongly bind Concanavalin A.
4. Recent applications of the thiol-ene reaction in (bio)organic chemistry
While the primary aim of this review is to highlight/emphasize recent reports of the thiol-ene reaction in polymer/materials science it should be noted that it has also been employed in more traditional (bio)organic synthesis.Wittrock et al.121 described an approach to immune-compatible antitumor vaccines in which model studies were performed highlighting the efficacy of the radical thiol-ene reaction in the coupling between amino acids and peptides. Such reactions were shown to proceed readily in water and water/alcohol mixtures with quantitative conversion if an excess of thiol-containing species was used. The approach was also shown to be useful for the introduction of spacer molecules, fluorescent labels and biotin markers.
Triola et al. reported a racemisation-free route to S-alkylated cysteinesviaAIBN-mediated radical thiol-ene reactions.122 For example, starting from the disulfide of the N-Fmoc, O-tert-butyl-protected cysteine, 1Scheme 30, the authors reported that a 3-step process involving initial reduction of the disulfide bond with dithiothreitol (DTT), followed by AIBN-mediated thiol-ene coupling with hexadecene in dichloroethane (DCE), with a final acid hydrolysis step (with trifluoroacetic acid (TFA) and triethylsilane (TES)) yielded the target N-Fmoc-protected hexadecylated thioether product in an overall 42% yield with a measured 99% ee. The general applicability of this approach was demonstrated in reactions on N-Fmoc-cysteine (Trt)-OH in which the trityl (Trt) group was first removed by acid hydrolysis followed by reaction of the free thiol with 1-octene, 2-methyl-1-hexene, trans-2-octene, an ene-bearing fluorescent dansyl derivative as well as an ene-biotin in the presence of AIBN at 90 °C, giving the target thioethers in yields ranging from 28–91%.
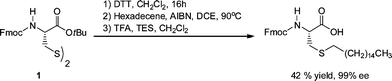 | ||
| Scheme 30 Synthesis approach to S-hexadecylated N-Fmoc cysteinevia a three step reduction, radical thiol-ene coupling, hydrolysis sequence. | ||
Xu et al.66 described the synthesis of a maleimido-functional CHX-A″ DTPA chelator, 2Scheme 31, designed specifically to take advantage of facile thiol-maleimide coupling with biomolecules. Such chelators are important, and have been studied, as molecules that are both suitable for binding to proteins as well as being able to chelate radioactive nuclei for radioimmunotherapy and radioimmunoimaging applications. The chelator2 was prepared and conjugated viathiol-ene coupling to thiolated Herceptin (a monoclonal antibody approved by the FDA to treat breast cancer that is HER2 positive)123 yielding, on average, two chelate molecules per Herceptin. The Herceptin-chelate conjuagte was successfully radiolabelled (95%) with 111In at RT within 30 min.
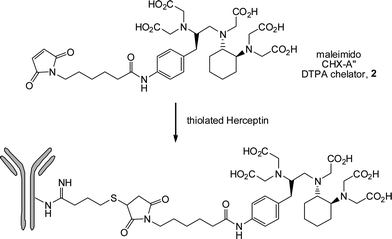 | ||
| Scheme 31 Conjugation of a maleimide-functional chelator to the monocolonal antibody Herceptin as a facile route to bioconjugates for radioimmunotherapy and radioimmunoimaging applications. | ||
Fiore et al.124 reported the synthesis of a range of unnatural thioglycosidesvia UV-initiated radical thiol-ene coupling between thiol and ene-functional sugars, see Scheme 32 for a general example. After optimization of the reaction conditions such facile couplings proceeded rapidly with >97% conversion as determined by 1H NMR spectroscopic analysis. The scope and versatility of this approach to novel disaccharides was demonstrated with ten examples given using a variety of thiol and ene sugar substrates.
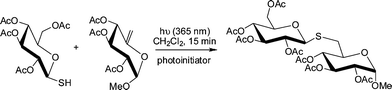 | ||
| Scheme 32 Radical-induced thiol-ene coupling between a peracetylated glucosylthiol and a hex-5-enepyranoside under photochemical conditions. | ||
Bantchev and co-workers125 described the photochemical reaction of butanethiol with the internal cis CC bonds in vegetable and canola oils, Scheme 33, with the motivation being to produce novel lubricating oils from natural renewable resources. Low ratios (1.5–3) of butanethiol to ene bonds resulted in degrees of hydrothiolation in the range 18–55% as determined by 1H NMR spectroscopy after 8 h of reaction. This is perhaps not surprising given, as described above, the reversibility of the thiyl radical addition reaction with internal CC bonds coupled with steric considerations. Indeed, the occurrence of such reversible reactions was noted by the presence of trans double bonds in the product mixture as determined by FTIR spectroscopy (the natural precursor oils contain 100% cis CC bonds). The yields were readily increased to ≥89% (for both oils) by increasing the butanethiol:ene ratio to 6 (with or without added photoinitiator). Interestingly, lowering the temperature from ambient to −78 °C resulted in an essentially quantitative modification (97%) after only 2 h of reaction and was attributed to the low-temperature suppression on the reversibility of thiyl radical addition.
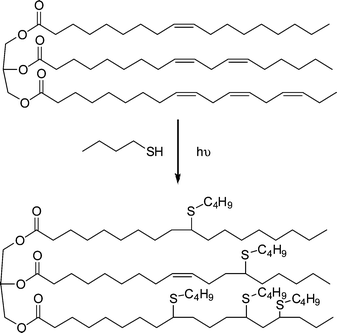 | ||
| Scheme 33 Reaction of butanethiol with the cis-ene bonds in vegetable oil under photochemical conditions. | ||
Hong et al.126 reported the preparation of a new class of reactive, selective and fluorogenic probes, based on 7-oxanorbornadiene, suitable for thiol coupling in aqueous media, examples of which are given in Fig. 22.
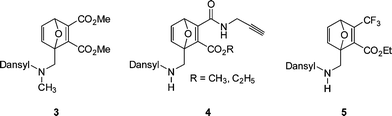 | ||
| Fig. 22 Examples of dansyl-functional 7-oxanorbornadienes susceptible to thiol-Michael hydrothiolation. | ||
Similar to propargyl acrylate (ENE6Fig. 15), the 7-oxanorbornadienes bear CC bonds with potential orthogonal activity. Whereas the disubstituted bond is susceptible to radical thiol-ene chemistry (and metathesis ring opening reactions), the tetra-substituted bond containing ester/amide or fluoro functionality is electron deficient and therefore susceptible to base/nucleophile mediated thiol-ene reactions. For example, the dimethylester derivative, 3Fig. 22, reacts readily at the activated CC bond with 2-mercaptoethanol in the presence of di-isopropylethylamine as catalyst at RT for 5 min giving the thiol-ene product in a >80% isolated yield. Given the tetra-substituted nature of this CC bond such rapid reaction and high yields is clearly impressive. Indeed, the approach was extended beyond simple low molecular weight thiols to peptides and proteins, such as thiolated BSA, Fig. 23. Interestingly, the precursor dansyl-labeled 7-oxanorbornadienes exhibit no fluorescence due to quenching associated with the activated CC bond. However, after thiol-ene coupling, and consumption of this bond, the fluorescence properties are restored.
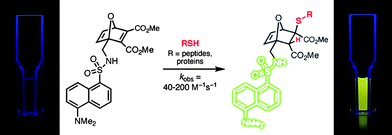 | ||
| Fig. 23 Thiol-Michael coupling to a dansyl-labeled 7-oxanorbornadiene as a means of promoting fluorescence. | ||
Conclusions
While only recently recognized and exploited as a “click” process, the thiol-ene reaction, in both its radical and base/nucleophilic forms, has already been demonstrated to be a powerful and versatile method for site specific functionalisation, the construction of complex (macro)molecules and as a convenient conjugation tool. Given that research efforts are still in their infancy with respect to exploiting this reaction in advanced polymer/materials synthesis, it is likely that its true potential has yet to be realised.Acknowledgements
This review is dedicated to the memory of Professor Charles E. Hoyle who sadly passed away in early September 2009.Professors Craig J. Hawker, Christof Niemeyer, Frank Caruso, Axel H. E. Müller, M. G. Finn, Eva Harth, Brent S. Sumerlin, and David M. Haddleton, and their respective group members, are kindly thanked for providing graphics and/or images used in Fig. 5, 6, 9, 10, 11, 13, 14, 17 and 23 and Schemes 15, 17, 18 and 28.
Notes and references
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2007, 28, 15–54 CrossRef CAS.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2008, 29, 952–981 CrossRef CAS.
- B. L. Droumaguet and K. Velonia, Macromol. Rapid Commun., 2008, 29, 1073–1089 CrossRef.
- R. A. Evans, Aust. J. Chem., 2007, 60, 384–395 CrossRef CAS.
- D. Fournier, R. Hoogenboom and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 1369–1380 RSC.
- J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249–1262 RSC.
- M. Li, P. De, S. R. Gondi and B. S. Sumerlin, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5093–5100 CrossRef CAS.
- S. Sinnwell, C. V. Synatschke, T. Junkers, M. H. Stenzel and C. Barner-Kowollik, Macromolecules, 2008, 41, 7904–7912 CrossRef CAS.
- A. J. Inglis, S. Sinnwell, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Macromolecules, 2008, 41, 4120–4126 CrossRef CAS.
- S. Sinnwell, M. Lammens, M. H. Stenzel, F. E. Du Prez and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2207–2213 CrossRef CAS.
- G. Franc and A. K. Kakkar, Chem.–Eur. J., 2009, 15, 5630–5639 CrossRef CAS.
- C. R. Becer, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 4900–4908 CrossRef CAS.
- I. Singh, Z. Zarafshani, J.-F. Lutz and F. Heaney, Macromolecules, 2009, 42, 5411–5413 CrossRef CAS.
- B. Yu, J. W. Chan, C. E. Hoyle and A. B. Lowe, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3544–3557 CrossRef CAS.
- J. W. Chan, C. E. Hoyle and A. B. Lowe, J. Am. Chem. Soc., 2009, 131, 5751–5753 CrossRef CAS.
- A. Dondoni, Angew. Chem., Int. Ed., 2008, 47, 8995–8997 CrossRef CAS.
- A. Gress, A. Volkel and H. Schlaad, Macromolecules, 2007, 40, 7928–7933 CrossRef CAS.
- K. L. Killops, L. M. Campos and C. J. Hawker, J. Am. Chem. Soc., 2008, 130, 5062–5064 CrossRef CAS.
- B. D. Fairbanks, T. F. Scott, C. J. Kloxin, K. S. Anseth and C. N. Bowman, Macromolecules, 2009, 42, 211–217 CrossRef CAS.
- J. W. Chan, H. Zhou, C. E. Hoyle and A. B. Lowe, Chem. Mater., 2009, 21, 1579–1585 CrossRef CAS.
- A. B. Lowe, C. E. Hoyle and C. N. Bowman, J. Mater. Chem., 2010 10.1039/b917102a.
- J. Shin, H. Matsushima, J. W. Chan and C. E. Hoyle, Macromolecules, 2009, 42, 3294–3301 CrossRef CAS.
- H. Li, B. Yu, H. Matsushima, C. E. Hoyle and A. B. Lowe, Macromolecules, 2009, 42, 6537–3542 CrossRef CAS.
- C. R. Becer, K. Babiuch, D. Pilz, S. Hornig, T. Heinze, M. Gottschaldt and U. S. Schubert, Macromolecules, 2009, 42, 2387–2394 CrossRef CAS.
- B. M. Rosen, G. Lligadas, C. Hahn and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3931–3939 CrossRef CAS.
- B. M. Rosen, G. Lligadas, C. Hahn and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3940–3948 CrossRef CAS.
- B. D. Mather, K. Viswanathan, K. M. Miller and T. E. Long, Prog. Polym. Sci., 2006, 31, 487–531 CrossRef CAS.
- T. Posner, Ber. Dtsch. Chem. Ges., 1905, 38, 646–657 CrossRef.
- C. E. Hoyle, T. Y. Lee and T. Roper, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5301–5338 CrossRef CAS.
- Q. Li, H. Zhou, D. A. Wicks and C. E. Hoyle, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5103–5111 CrossRef CAS.
- A. F. Senyurt, H. Wei, C. E. Hoyle, S. G. Piland and T. E. Gould, Macromolecules, 2007, 40, 4901–4909 CrossRef CAS.
- A. F. Senyurt, H. Wei, B. Phillips, M. Cole, S. Nazarenko, C. E. Hoyle, S. G. Piland and T. E. Gould, Macromolecules, 2006, 39, 6315–6317 CrossRef CAS.
- J. Shin, S. Nazarenko and C. E. Hoyle, Macromolecules, 2008, 41, 6741–6746 CrossRef CAS.
- H. Wei, A. F. Senyurt, S. Jonsson and C. E. Hoyle, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 822–829 CrossRef CAS.
- Z. Yang, D. A. Wicks, C. E. Hoyle, H. Pu, J. Yuan, D. Wan and Y. Liu, Polymer, 2009, 50, 1717–1722 CrossRef CAS.
- H. Zhou, Q. Li, J. Shin and C. E. Hoyle, Macromolecules, 2009, 42, 2994–2999 CrossRef CAS.
- T. Clark, L. Kwisnek, C. E. Hoyle and S. Nazarenko, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 14–24 CrossRef CAS.
- Q. Li, H. Zhou and C. E. Hoyle, Polymer, 2009, 50, 2237–2245 CrossRef CAS.
- J. A. Carioscia, J. W. Stansbury and C. N. Bowman, Polymer, 2007, 48, 1526–1532 CrossRef CAS.
- N. B. Cramer, S. K. Reddy, A. K. O'Brien and C. N. Bowman, Macromolecules, 2003, 36, 7964–7969 CrossRef CAS.
- V. S. Khire, D. S. W. Benoit, K. S. Anseth and C. N. Bowman, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 7027–7039 CrossRef CAS.
- V. S. Khire, A. W. Harant, A. W. Watkins, K. S. Anseth and C. N. Bowman, Macromolecules, 2006, 39, 5081–5086 CrossRef CAS.
- T. Y. Lee, Z. Smith, S. K. Reddy, N. B. Cramer and C. N. Bowman, Macromolecules, 2007, 40, 1466–1472 CrossRef CAS.
- A. E. Rydholm, N. L. Held, C. N. Bowman and K. S. Anseth, Macromolecules, 2006, 39, 7882–7888 CrossRef CAS.
- A. E. Rydholm, S. K. Reddy, K. S. Anseth and C. N. Bowman, Polymer, 2007, 48, 4589–4600 CrossRef CAS.
- J. W. Chan, B. Yu, C. E. Hoyle and A. B. Lowe, Chem. Commun., 2008, 4959–4961 RSC.
- H. Kakwere and S. Perrier, J. Am. Chem. Soc., 2009, 131, 1889–1895 CrossRef CAS.
- N. S. Krishnaveni, K. Surendra and K. R. Rao, Chem. Commun., 2005, 669–671 RSC.
- C. R. Morgan, F. Magnotta and A. D. Ketley, J. Polym. Sci., Polym. Chem. Ed., 1977, 15, 627–645 CrossRef CAS.
- K. Griesbaum, Angew. Chem., Int. Ed. Engl., 1970, 9, 273–287 CAS.
- M. G. Voronkov and E. N. Deryagina, Russ. Chem. Rev., 1990, 59, 778–791 Search PubMed.
- C. Walling and W. Helmreich, J. Am. Chem. Soc., 1959, 81, 1144–1148 CrossRef CAS.
- C. Chatgilialoglu, A. Altieri and H. Fischer, J. Am. Chem. Soc., 2002, 124, 12816–12823 CrossRef CAS.
- C. Ferreri, A. Samadi, F. Sassatelli, L. Landi and C. Chatgilialoglu, J. Am. Chem. Soc., 2004, 126, 1063–1072 CrossRef CAS.
- L. M. Stock, J. Chem. Educ., 1972, 49, 400–404 CAS.
- J. W. Chan, A. B. Lowe, C. E. Hoyle, manuscript in preparation.
- I. C. Stewart, R. G. Bergman and F. D. Toste, J. Am. Chem. Soc., 2003, 125, 8696–8697 CrossRef CAS.
- P. Klemarczyk, Polymer, 2001, 42, 2837–2848 CrossRef CAS.
- S. M. Heilmann, J. K. Rasmussen and L. R. Krepski, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 3655–3677 CrossRef CAS.
- T. Miyadera and E. M. Kosower, J. Med. Chem., 1972, 15, 534–537 CrossRef CAS.
- L. C. Radu, J. Yang and J. Kopecek, Macromol. Biosci., 2009, 9, 36–44 CrossRef CAS.
- M. de Kort, B. Gianotten, J. A. J. Wisse, E. S. Bos, M. H. M. Eppink, E. Mattaar, G. M. T. Vogel, W. H. A. Dokter, M. Honing, S. Vonsovic, M.-J. Smit, J. C. H. M. Wijkmans and C. A. A. van Boeckel, ChemMedChem, 2008, 3, 1189–1193 CrossRef CAS.
- M. E. Gindy, S. Ji, T. R. Hoye, A. Z. Panagiotopoulos and R. K. Prud'homme, Biomacromolecules, 2008, 9, 2705–2711 CrossRef CAS.
- H.-Y. Yeh, M. V. Yates, A. Mulchandani and W. Chen, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17522–17525 CrossRef CAS.
- H. Xu, K. E. Baidoo, K. J. Wong and M. W. Brechbiel, Bioorg. Med. Chem. Lett., 2008, 18, 2679–2683 CrossRef CAS.
- R. Singh, Bioconjugate Chem., 1994, 5, 348–351 CrossRef CAS.
- G. Mantovani, F. Lecolley, L. Tao, D. M. Haddleton, J. Clerx, J. J. L. M. Cornelissen and K. Velonia, J. Am. Chem. Soc., 2005, 127, 2966–2973 CrossRef CAS.
- I. C. Reynhout, J. J. L. M. Cornelissen and R. J. M. Nolte, Acc. Chem. Res., 2009, 42, 681–692 CrossRef CAS.
- M. M. Kreevoy, E. T. Harper, R. E. Duvall, H. S. Wilgus and L. T. Ditsch, J. Am. Chem. Soc., 1960, 82, 4899–4902 CrossRef CAS.
- I. V. Koval, Russ. J. Org. Chem., 2005, 41, 631–648 CrossRef CAS.
- M. M. Kreevoy, B. E. Eichinger, F. E. Stary, E. A. Katz and J. H. Sellstedt, J. Org. Chem., 1964, 29, 1641–1642 CrossRef CAS.
- J. Justynska and H. Schlaad, Macromol. Rapid Commun., 2004, 25, 1478–1481 CrossRef CAS.
- J. Justynska, Z. Hordyjewicz and H. Schlaad, Polymer, 2005, 46, 12057–12064 CrossRef CAS.
- N. t. Brummelhuis, C. Diehl and H. Schlaad, Macromolecules, 2008, 41, 9946–9947 CrossRef.
- Z. Hordyjewicz-Baran, L. You, B. Smarsly, R. Sigel and H. Schlaad, Macromolecules, 2007, 40, 3901–3903 CrossRef CAS.
- L. Lotti, S. Coiai, F. Ciardelli, M. Galimberti and E. Passaglia, Macromol. Chem. Phys., 2009, 210, 1471–1483 CrossRef CAS.
- C. Diehl and H. Schlaad, Macromol. Biosci., 2009, 9, 157–161 CrossRef CAS.
- L. M. Campos, K. L. Killops, R. Sakai, J. M. J. Paulusse, D. Damiron, E. Drockenmuller, B. W. Messmore and C. J. Hawker, Macromolecules, 2008, 41, 7063–7070 CrossRef CAS.
- L. M. Campos, I. Meinel, R. G. Guino, M. Schierhorn, N. Gupta, G. D. Stucky and C. J. Hawker, Adv. Mater., 2008, 20, 3728–3733 CrossRef CAS.
- C. Rissing and D. Y. Son, Organometallics, 2008, 27, 5394–5397 CrossRef CAS.
- C. Rissing and D. Y. Son, Organometallics, 2009, 28, 3167–3172 CrossRef CAS.
- V. Ladmiral, E. Melia and D. M. Haddleton, Eur. Polym. J., 2004, 40, 431–449 CrossRef CAS.
- G. Chen, S. Amajjahe and M. H. Stenzel, Chem. Commun., 2009, 1198–1120 RSC.
- P. Jonkheijm, D. Weinrich, M. Kohn, H. Engelkamp, P. C. M. Christianen, J. Kuhlmann, J. C. Mann, D. Nusse, H. Schroeder, R. Wacker, R. Breinbauer, C. M. Niemeyer and H. Waldmann, Angew. Chem., Int. Ed., 2008, 47, 4421–4424 CrossRef CAS.
- L. A. Connal, C. R. Kinnane, A. N. Zelikin and F. Caruso, Chem. Mater., 2009, 21, 576–578 CrossRef CAS.
- A. S. Goldmann, A. Walther, L. Nebhani, R. Joso, D. Ernst, K. Loos, C. Barner-Kowollik, L. Barner and A. H. E. Muller, Macromolecules, 2009, 42, 3707–3714 CrossRef CAS.
- A. van der Ende, T. Croce, S. Hamilton, V. Sathiyakumar and E. Harth, Soft Matter, 2009, 5, 1417–1425 RSC.
- K. Y. van Berkel, A. M. Piekarski, P. H. Kierstead, E. D. Pressly, P. C. Ray and C. J. Hawker, Macromolecules, 2009, 42, 1425–1427 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- C. Barner-Kowollik, M. Buback, B. Charleux, M. L. Coote, M. Drache, T. Fukuda, A. Goto, B. Klumperman, A. B. Lowe, J. B. McLeary, G. Moad, M. J. Monteiro, R. D. Sanderson, M. P. Tonge and P. Vana, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5809–5831 CrossRef CAS.
- A. B. Lowe and C. L. McCormick, Prog. Polym. Sci., 2007, 32, 283–351 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2006, 59, 669–692 CrossRef CAS.
- Handbook of RAFT Polymerization, ed. C. Barner-Kowollik, WILEY-VCH, Weinheim, 2008 Search PubMed.
- C. W. Scales, Y. A. Vasilieva, A. J. Convertine, A. B. Lowe and C. L. McCormick, Biomacromolecules, 2005, 6, 1846–1850 CrossRef CAS.
- A. B. Lowe, M. Torres and R. Wang, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5864–5871 CrossRef CAS.
- C. Barner-Kowollik, T. P. Davis, J. P. A. Heuts, M. H. Stenzel, P. Vana and M. Whittaker, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 365–375 CrossRef CAS.
- A. B. Lowe, B. S. Sumerlin, M. S. Donovan and C. L. McCormick, J. Am. Chem. Soc., 2002, 124, 11562–11563 CrossRef CAS.
- B. S. Sumerlin, A. B. Lowe, P. A. Stroud, M. W. Urban and C. L. McCormick, Langmuir, 2003, 19, 5559–5562 CrossRef CAS.
- J. W. Hotchkiss, A. B. Lowe and S. G. Boyes, Chem. Mater., 2007, 19, 6–13 CrossRef CAS.
- J. Xu, J. He, D. Fan, X. Wang and Y. Yang, Macromolecules, 2006, 39, 8616–8624 CrossRef CAS.
- Y. K. Chong, G. Moad, E. Rizzardo, M. Skidmore and S. H. Thang, Aust. J. Chem., 2006, 59, 755–762 CrossRef CAS.
- A. Postma, T. P. Davis, G. Moad and M. S. O'Shea, Macromolecules, 2005, 38, 5371–5374 CrossRef CAS.
- G. Moad, Y. K. Chong, A. Postma, E. Rizzardo and S. H. Thang, Polymer, 2005, 46, 8458–8468 CrossRef CAS.
- X.-P. Qiu and F. M. Winnik, Macromol. Rapid Commun., 2006, 27, 1648–1653 CrossRef CAS.
- C. W. Scales, A. J. Convertine and C. L. McCormick, Biomacromolecules, 2006, 7, 1389–1392 CrossRef CAS.
- J. M. Spruell, B. A. Levy, A. Sutherland, W. R. Dichtel, J. Y. Cheng, J. F. Stoddart and A. Nelson, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 346–356 CrossRef CAS.
- C. Boyer, A. Granville, T. P. Davis and V. Bulmus, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3773–3794 CrossRef CAS.
- C. Boyer, V. Bulmus and T. P. Davis, Macromol. Rapid Commun., 2009, 30, 493–497 CrossRef.
- J. W. Chan, B. Yu, C. E. Hoyle and A. B. Lowe, Polymer, 2009, 50, 3158–3168 CrossRef CAS.
- C. Rim and D. Y. Son, Tetrahedron Lett., 2009, 50, 4161–4163 CrossRef CAS.
- Z. P. Tolstyka, J. T. Kopping and H. D. Maynard, Macromolecules, 2008, 41, 599–606 CrossRef CAS.
- R. J. Pounder, M. J. Stanford, P. Brooks, S. P. Richards and A. P. Dove, Chem. Commun., 2008, 5158–5160 RSC.
- M. J. Stanford and A. P. Dove, Macromolecules, 2009, 42, 141–147 CrossRef CAS.
- S. Cesana, A. Kurek, M. A. Baur, J. Auernheimer and O. Nuyken, Macromol. Rapid Commun., 2007, 28, 608–615 CrossRef CAS.
- L. Nurmi, J. Lindqvist, R. Randev, J. Syrett and D. M. Haddleton, Chem. Commun., 2009, 2727–2729 RSC.
- M. W. Jones, G. Mantovani, S. M. Ryan, X. Wang, D. J. Brayden and D. M. Haddleton, Chem. Commun., 2009, 5272–5274 RSC.
- C. Boyer and T. P. Davis, Chem. Commun., 2009, 6029–6031 RSC.
- W. Gu, G. Chen and M. H. Stenzel, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5550–5556 CrossRef CAS.
- S. Wittrock, T. Becker and H. Kunz, Angew. Chem., Int. Ed., 2007, 46, 5226–5230 CrossRef CAS.
- G. Triola, L. Brunsveld and H. Waldmann, J. Org. Chem., 2008, 73, 3646–3649 CrossRef CAS.
- http://www.herceptin.com/ .
- M. Fiore, A. Marra and A. Dondoni, J. Org. Chem., 2009, 74, 4422–4425 CrossRef CAS.
- G. B. Bantchev, J. A. Kenar, G. Biresaw and M. G. Han, J. Agric. Food Chem., 2009, 57, 1282–1290 CrossRef CAS.
- V. Hong, A. A. Kislukhin and M. G. Finn, J. Am. Chem. Soc., 2009, 131, 9986–9994 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |