Photo-responsive systems and biomaterials: photochromic polymers, light-triggered self-assembly, surface modification, fluorescence modulation and beyond
Francesca
Ercole
abc,
Thomas P.
Davis
a and
Richard A.
Evans
*abc
aCentre for Advanced Macromolecular Design, School of Chemical Sciences and Engineering, The University of New South Wales, Sydney, NSW 2052, Australia. E-mail: Richard.Evans@csiro.au
bCSIRO Molecular & Health Technologies, Bag 10, Clayton, VIC 3169, Australia
cThe Cooperative Research Centre for Polymers, 8 Redwood Drive, Notting Hill, VIC 3168, Australia
First published on 25th November 2009
Abstract
There has been considerable interest in the application of photochromism to photo-responsive systems which has led to the development of new tailored smart materials for photonics and biomedical fields. Within a polymeric matrix photochromic isomerizations can be stimulated by light to reversibly alter the physical and chemical properties of a material such as LC phase, shape, surface wettability, permeability, solubility, self-assembly, size and fluorescence. The underlying principles behind photo-responsive behavior, subsequent applications and relevant examples are discussed in this review.
1. Introduction
The ability to reversibly manipulate the physical and chemical properties of a material with an external stimulus forms the basis of stimuli-responsive systems. The use of light as a trigger is particularly attractive since its characteristics can be remotely and accurately controlled, quickly switched and easily focused into specific areas. New tailorable and ‘smart’ polymeric materials have emerged in areas such as nanotechnology, electronics, diagnostics and biomedical fields with properties, such as conformation, shape, phase, wettability, permeability and solubility, ability to be reversibly transformed with light stimulation. Many of the ideas for such photo-responsive systems have been inspired by nature which has evolved many complex biological systems able to exploit light as an external source of energy and information. For example, the light-induced cis–trans isomerization of the retinal molecule, bound to the membrane of proteinopsin, triggers a number of events including a change in the conformation of the protein and membrane permeability, leading to a neural signal and ultimately to the perception of light.2. Photochromism
Photochromism involves the reversible transformation of a chemical species between two isomeric forms induced by the absorption of light which results in a change in absorption spectra. The mechanisms include pericyclic reactions, cis–trans isomerizations, dissociation processes, intramolecular hydrogen transfers or group transfers, and electron transfers (oxidation–reduction).1 In addition to a color change, these transformations are accompanied by changes in the physical and chemical properties of the species involved, such as alterations to in the dipole moment, refractive index and geometrical structure. Importantly, these dynamic transformations can generate coincident changes in the optical, chemical, electrical and bulk properties of the system that incorporates them. Photochromic molecules therefore play a pivotal role within photo-responsive systems, being able to capture an optical signal and then convert this, via their isomerization, to a useful property change.This review is focused on the behavior of photo-responsive systems involving transformations shown in Fig. 1 which are notably unimolecular processes and reversible. The majority of the systems described are based on polymers. Other useful photo-active transformations of molecules such as triphenyl methane leuco derivatives, cinnamates, stillbenes and coumarins have also attracted considerable interest but are beyond the scope of this review.
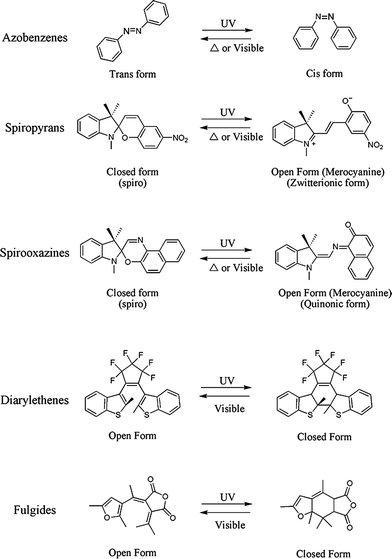 | ||
| Fig. 1 Families of photochromic compounds commonly used in polymeric systems. | ||
The transformations depicted in Fig. 1 can be described briefly as follows: ultraviolet (UV) irradiation of an azobenzene stimulates the conversion of the planar trans isomer to the bent cis isomeric form via the isomerization of a –N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– bond; UV irradiation of a spiropyran or spirooxazine initiates an electrocyclic ring opening reaction of a spiro form which results in the formation of an open, planar merocyanine form with an extended conjugated system able to absorb strongly in the visible region; for diarylethenes and fulgides , UV irradiation results in the closing of the six-membered ring within its core which results in the formation of thermally irreversible colored isomers. The photochromic inter-conversion between isomeric forms is often referred to as switching.
N– bond; UV irradiation of a spiropyran or spirooxazine initiates an electrocyclic ring opening reaction of a spiro form which results in the formation of an open, planar merocyanine form with an extended conjugated system able to absorb strongly in the visible region; for diarylethenes and fulgides , UV irradiation results in the closing of the six-membered ring within its core which results in the formation of thermally irreversible colored isomers. The photochromic inter-conversion between isomeric forms is often referred to as switching.
3. Photochromic polymers
3.1 Matrix effect
From a practical standpoint, polymers are important for applications that require phorochromic switching to occur in a robust matrix such as films, beads and moldable materials. The behavior of the photochromic and the polymer matrix are however connected in a relationship that can hold further significance. Both the characteristics of the polymer and the mode of incorporation, whether it be covalent attachment or dissolution into a polymer host, can affect photochromic behavior.2 The micro and nano-environmental properties of matrices, such as local polarity and free volume, as well as intermolecular interactions can also affect efficiencies. As a general concept, photochromic transitions are generally slower in a polymer matrix, as compared to solution. This effect is attributed to the limited free volume available in a polymeric medium, the reduced segmental motion of the macromolecules and steric restrictions imposed on the isomerizations. Furthermore, aggregation of photochromic moieties can also influence kinetic and spectral properties. Krogauz described these aspects as the ‘matrix effect’ with an investigation into the behavior of spiropyrans and azobenzenes in a review featuring much of the early research using polymer matrices.3The photochromic reactions of spirooxazines4 and spiropyrans5 both proceed by electrocyclic ring opening followed by a molecular rotation. The latter requires considerable mechanical movement considering the size of fragments that are shifted relative to one another. Overall, for coloration and discoloration the molecule has to convert between an orthogonal and planar conformation. The matrix effect for these photochromics is therefore particularly striking. Decoloration kinetics can deviate from the expected first order kinetics at temperatures below the glass transition temperature (Tg) of the polymer medium, attributed to the non-homogenous distribution of free volumes existing in a glassy, rigid medium.3 Furthermore, complexities can also arise from the merocyanine form interconverting between different geometrical conformations. Substitution patterns also have a marked effect on kinetics through steric and electronic contributions.
The reversible trans–cis isomerization of azobenzenes can be described as a geometrical isomerisation. The difference between the isomers is revealed by their separate λmax values, however their inter-conversions are not visualized as a distinct color change, as is the case for most other photochromic transformations. Instead, at a molecular level, the isomerization leads to a substantial change in geometric conformation and size. In the dark or under typical ambient illumination the azobenzenes are found predominantly in their more stable trans form. Once formed, cis isomers will thermally reconvert back to the more stable trans state with a timescale that depends greatly on the azobenzene substitution pattern as well as their environment. The thermal reversion is generally first-order, although a distribution of highly constrained configurations can arise in a glassy polymer matrix which can lead to fast decay components.6,7 Historically the kinetic behavior of azobenzenes has attracted considerable attention.8,9
The matrix effect also has a secondary meaning which is of prime importance to the area of photo-responsive polymers: the influence of photochromism on the matrix. This directly concerns the reversible property changes that are induced in the polymeric materials as a result of photochromic conversions. An interesting dimensional classification of matrix effects was proposed by Ichimura based on the level of orderedness of the molecular structures in the polymeric matrices.10 Each dimension is able to display a measurable optical effect, however matrices of higher dimensions exhibit a higher level of order such as phase separated states and self assembled systems. Zero dimensional orderedness deals specifically with photochromic transformations occurring in solution and amorphous polymers. Here the primary optical changes that take place are the absorption profile (color), emission, reflection and refraction characteristics which are valuable to areas such as the ophthalmic lens market and optical storage media. Birefringence, dichroism, circular dichroism and optical rotary power are optical manifestations arising from higher dimensional phases which have notable application in photonic and biomedical fields.
4. Reversible modulation of optical properities
Commercial demand for photochromic dyes is dominated by the ophthalmic lens industry which exploits their ability to induce a marked change in color and transmission in lens media on exposure to UV light. The dyes of choice are naphthopyran (chromenes)11 and spirooxanine4dyes whose photochromic characteristics include the ability to display a broad range of intense colors with excellent fatigue resistance. Photochromic switching of these dyes involves coloration which is stimulated by UV light and discoloration with visible light or thermal pathways. These inter-conversions involve substantial molecular mechanical movement therefore placement of the dye in a rigid cross-linked lens matrix represents the most severe test of a dye's photochromic performance. Given that there is still a commercial need for more rapid fading in lenses, this area continues to motivate research efforts. Our solution to overcoming the lens matrix effect has been to apply concepts developed in drug and gene delivery, where polymer conjugates are used to protect the drug from a harsh biological environment. We have found that when dyes have oligomers covalently attached to them they are essentially insulated from the rigid bulk environment, whereby entanglement and partitioning of polymer tails is able to provide a local environment of controlled viscosity (as depicted in Fig. 2).12–20 Controlled radical polymerization techniques can be exploited to synthesize photochromic macromolecules with both defined architectures and compositions. This methodology could be applied to other photo-responsive systems which demand efficient and controlled switching environments in otherwise rigid media.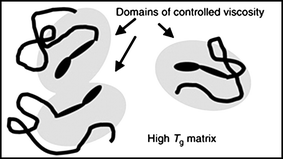 | ||
| Fig. 2 An illustration of how the tethering of oligomers with known properties (such as Tg or viscosity) to dyes controls their switching behavior by modifying its local environment.17 | ||
Photochromic dyes have also become promising candidates for new high density optical recording media in which data is reversibly stored in photon mode. The multiplexing of light characteristics such as wavelength, polarization, and phase provides an opportunity for high capacity photon-mode recording systems. Photochromic materials have been applied to three-dimensional (3D) optical recording systems based on two-photon absorption where data is written not on the material surface, but within the entire volume, resulting in increased storage density. These systems have been demonstrated using spiropyran, azobenzene and diarylethene dyes. Initially these were conceived using fluorescence emission as a read out method, however this often resulted in coincident loss of stored data by photo-stimulating the photochromics to revert back to their uncolored forms. A more promising method has been to detect the refractive-index changes that accompany the photoisomerizations induced by long-wavelength light.
So far the best candidates for 3D optical data storage are diarylethenes which are thermally irreversible therefore capable of archival storage (Fig. 3). The various optical systems for reading and writing 3D memories using photochromic materials have been reviewed by Kawata.22
 | ||
| Fig. 3 Readouts of bit patterns written into photochromic memory using a diarylethene derivative. A two-photon process was used to record the data and a reflection confocal microscope was used to read the data. Reprinted with permission from ref. 21. Copyright (1998) Optical Society of America. | ||
4.1 Photo-orientation
A change in color is not always the most spectacular effect arising from a photochromic transformation. For azobenzenes the absorption of light induces molecular motion which can lead to the process of photo-orienation.10,23 This gives rise to the optical properties of birefringence (anisotropy in refractive index) and dichroism (anisotropy in absorption spectrum), applicable to areas such as holography and reversible data storage.24–26 When irradiated by linearly polarized light, an azobenzene molecule will preferentially absorb light polarized along its transition dipole axis (long axis of the molecule). Repeated trans–cis–trans isomerization cycles result in a statistical depletion of the population of transchromophores that lie along the polarization direction, with an eventual enrichment of those lying perpendicular (as in Fig. 4). Irradiation with unpolarized or circularly polarized light can reverse the process by re-establishing isotropic orienation. The reversibility of the process enables subsequent photo-reorientations.Exposure to an interference pattern caused by intersecting two coherent laser beams, as a result of large-scale polymer chain migration can produce surface relief gratings which are coincident with the light interference pattern.27,28 Birefringence and surface relief gratings has been optically induced in various forms of materials, such as polymer matrices doped with azobenzenedyes29–31 as well as amorphous azobenzenepolymers.32–34Polymers doped with spiropyran, fulgide and diarylethenes have also been investigated, although azobenzene systems are by far the most studied.35–38
The extent of photoinduced anisotropic orientation can depend on many aspects, such as the chemical nature of the photochromic compound as well as the composition and viscosity of the host polymer.10 Having photochromic units covalently bound within the polymer backbone or attached as side groups has been found to enhance the stability of molecular orientation, especially in rigid polymer networks . Other aspects such as the composition and nature of the attached polymer have also been shown to have an influence on the extent of anisotropic orientation, longevity and also the kinetics of orientation. Some examples include acrylate amorphous copolymers, polyesters, polyimides and polyurethanes.39 A review by Natansohn and Rochon deals specifically with the dynamics of photoinduced motions occurring in azobenzene-containing polymers.40 The linear and nonlinear optical (NLO) properties of photochromic materials has also been reviewed by Delaire and Nakatani.41
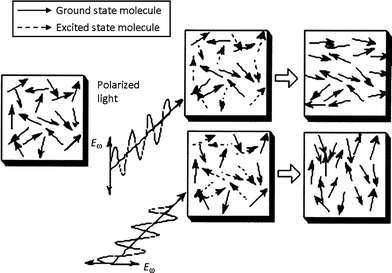 | ||
| Fig. 4 Generation of anisotropy with light: molecules excited with polarized light tend to align in a direction perpendicular to the polarization direction. Reprinted with permission from ref. 39. Copyright (2000) American Chemical Society. | ||
5. Photo-responsive liquid crystal polymer systems
Photochromic liquid crystal (LC) systems have a remarkable ability to change their long range ordering and optical properties in response to a light stimulus. Two types of photo-modulations of photochromic LC processes are possible; order–disorder phase transitions and order–order alignment changes of LC directors. Overall the response of these materials to light is supported by the ability of LCs to display cooperative motion, whereby, if a small portion of LC molecules change their orientation or conformation with light, this promotes other LC molecules to do the same. Overall the response of the LC polymer to light can be amplified into conversions of considerable length-scales.When a small amount of photochromic molecules such as azobenzenes, spiropyrans and fulgides are incorporated with LC molecules and the resulting ordered mixtures are irradiated to stimulate photochromic isomerizations, a LC to isotropic phase transition can be induced isothermally. The trans–cis photoisomerization of azobenzenes incorporated in nematic LC systems are the most classically studied where the distinct transformation of molecular shape from the rod-like shape of the trans isomer, which stabilizes the phase structure of the LC, to the bent cis isomer, which acts as an impurity, results in the destruction of ordered mesophase structures (Fig. 5).
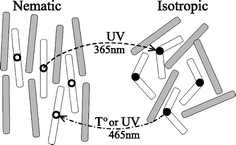 | ||
| Fig. 5 A schematic depiction of nematic–isotropic phase transformation in a LC containing photoisomerizable mesogenic molecules, which turn from a rod-like trans to a bent cis conformation under UV irradiation. Reprinted with permission from ref. 42. Copyright (2002) American Institute of Physics. | ||
The photochemical phase transitions between nematic and isotropic phases (order–disorder) are thermodynamically driven and also reversible. The most notable applications for this light driven process are optical switching devices and information storage systems.42–44
Extensive investigations have been made into the photo-induced mesophase alterations of nematic LC polymers; single component copolymers containing azobenzene molecules and mesogens in the same macromolecule; polymeric liquid crystals (PLCs) doped with low molecular weight (LMW) azobenzene guests and mixtures of PLCs, LMW LCs and LMW azobenzene guests. Photochemical phase transitions in PLCs have also been induced by other photochromic molecules, in addition to azobenzenes, such as, spiropyrans, spirooxazines and fulgides .45–50 Various polymeric types have also been trialed, such as azobenzene side-chain acrylates and polyesters.51,52 New photo-responsive synthetic materials have also emerged, like systems formed via hydrogen bonding53 and with novel architectures such as dendrimers54 and block copolymers.55–57 The relaxation time and response time for the photochemical phase transition of PLCs, which is of prime importance for application of the materials, has also been heavily evaluated.58–60 So far many copolymer LCs have been found to be ideal materials for optical image storage, able to be processed easily, to form robust films with high stability of the stored images and also able to display quick responses.61
As discussed for amorphous polymer systems, the molecular orientation of photochromic molecules can be built up under illumination with polarized light. This process can be applied to photochromic PLCs to reorient the LC director (an order–order change).62,63 Studies of azobenzene LC polymer systems have analyzed the factors which are important for this process. These include the structure of the polymer, the content and chemical nature of the photochromic moieties and the spacer lengths of the side chains. Linearly polarized light-induced reorientation of molecules in LC polymer layers can in fact be classified into three types64 (Fig. 6).
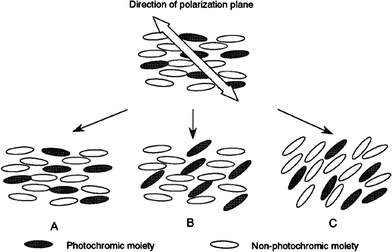 | ||
| Fig. 6 Three types of molecular reorientation of photochromic liquid-crystal polymers irradiated with linearly polarized light: (A) no photoreorientation; (B) selective photoreorientation of photochromic mesogens ; (C) cooperative photoreorientation. Reprinted with permission from ref. 65. Copyright (2000) American Chemical Society. | ||
The reorientation can be almost completely suppressed because of the very stable orientational order of a mesophasic matrix; the reorientation can also take place solely at azobenzene residues without the reorientation of non-photochromic side groups; or the reorientation of the photochromic moieties causes a continuous reorientation of non-photochromic mesogenic groups, even below the Tg of the polymer, to exhibit a large optical anisotropy.
The photoreorientation of azobenzenechromophores can therefore induce the cooperative reorientation of non-photoactive mesogens in copolymer systems and overall this photoalignment behavior can be optimized effectively by an appropriate choice of azobenzene units and polymer backbones. Single component copolymer systems have been developed in which the liquid crystallinity of the polymers along with the best azobenzenes in the side chains can provide larger optical anisotropy and thermal stability of photoalignment.65,66
Ichimura,65 Shibaev et al.,54 Yu66 and Ikeda66,67 have published comprehensive reviews covering much of the recent as well as historical advances in photo-responsive LC systems.
5.1 Photochromic surfaces for photo-regulated LC alignment
The alignment of LC molecules is governed by the nature of the substrate surface.68 The modification of the surface with amphiphilic compounds or long-chain alkyl substituted reagents can be used to generate homeotropic alignment of a LC cell (unidirectionally perpendicular to the substrate) and a homogeneous alignment (unidirectionally parallel) can be yielded by rubbing thin films of PLCs covering the surface of the substrate. Such surface induced phenomena led to the idea that photo-switching of photochromic molecules immobilized on a surface can also control the alignment of a LC cell.69 These photoactive LC surfaces are referred to as ‘command surfaces’ in which each photochromic unit is estimated to bring about the reversible alignment transformation of about 104 LC molecules situated on top. The four modes of the LC alignment controlled by such command surfaces, are illustrated in Fig. 7.65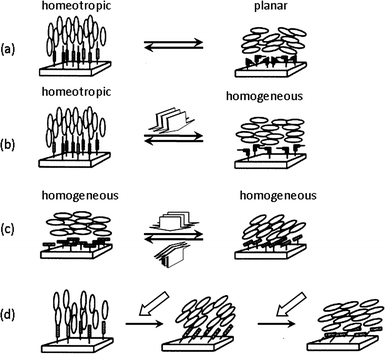 | ||
| Fig. 7 An illustrative representation of surface-assisted photoalignment control of LC molecules triggered by photochromic molecules tethered to a substrate surface. (a) Out-of-plane LC photoalignment between homeotropic and planar modes triggered by trans–cis photoisomerization of photochromic surface molecules upon alternate irradiation with nonpolarized light UV and visible light. (b) Out-of-plane LC photoalignment between homeotropic and homogeneous modes triggered by alternate irradiation with linearly polarized UV light and nonpolarized light. (c) In-plane photoalignment by irradiation with linearly polarized light. (d) Tilt-angle generation with slantwise photoirradiation. Reprinted with permission from ref. 65. Copyright (2000) American Chemical Society. | ||
One type of out-of-plane alignment that is triggered by photoisomerization involves the reversible change between homeotropic and planar alignment (Fig. 7a). For example, when filled with a nematic LC incorporating azobenzenes immobilized on the surface of the substrate, the LC cell shows a homeotropic alignment when azobenzenes are in the trans form. Trans–cis isomerization via photoirradiation enables repeatable changes in alignment from the homeotropic state to a planar state. Ichimura et al. employed azobenzene monolayers formed on glass substrates by silane coupling agents to demonstrate the possibility of such photoalignment control using photoactive surface layers.69 Out-of-plane LC photoalignment of azobenzene on surfaces, between homeotropic and homogeneous alignment modes can be triggered by alternate irradiation with linearly polarized UV light and nonpolarized light (Fig. 7b).70 The photocontrol of out-of-plane alignment has been demonstrated on spin coated films of poly(vinyl alcohol), either surface modified71 or partly modified on side groups72 with azobenzenes, as well as azobenzene-functionalized poly(acrylics) and poly(acrylamides).73,74 Photoalignment control of azobenzene LC command surfaces prepared by the Langmuir–Blodgett technique has also provided insights into the working mechanisms of surface assisted LC phenomena.75–77
In-plane alignment control of photochromic command surfaces (Fig. 7c–7d) can be achieved by means of linearly polarized light irradiation, giving rise to homogeneous, tilted or even biaxial alignment control of nematic LCs.78 This alignment control has a practical application in erasable optical data storage devices and laser display technologies and has been demonstrated by exploiting the surface photochromism of both azobenzenes79–83 and spiropyrans.84,85
5.2 Photo-deformable LC elastomers
Photochromic liquid crystal elastomers (LCE) are a relatively new and exciting class of photomechanical materials in which large deformations can be generated in response to light stimulation, such as reversible contractions and expansions, with changes in shape and volume. Using mono-domain nematic LCEs containing a polysiloxane main chain and azobenzenechromophores in the crosslinks, Finkelmann et al.86 reported a large and reversible photoinduced (UV) contraction (∼20%) of azobenzene-containing LCEs, caused by a decrease in order due to the photochemical LC phase transition. A variety of UV-responsive nematic LCEs containing different compositions and crosslinking topologies have been investigated with contractions found to be dependent on the proportion and the position of the azobenzene units in the crosslinked polymer network .42 In addition, this photomechanical effect has also been observed in side-on nematic LCEs.87LCE films prepared by copolymerization of an LC monomer and a diacrylate, both of which contain an azobenzene moiety, were expected to yield stronger photoresponses. Such films were found to undergo bending and unbending behavior on alternate exposure to unpolarized UV and visible light. This occurred only in the direction parallel to the rubbing direction of the alignment layers. A specialized LCE was also prepared consisting of many micro-sized domains of azobenzene liquid-crystal moieties aligned in one direction, although macroscopically the direction of alignment in the material was random overall. When exposed to linearly polarized UV light it was able to bend toward the light with bending occurring parallel to the direction of light polarization (Fig. 8).88–92
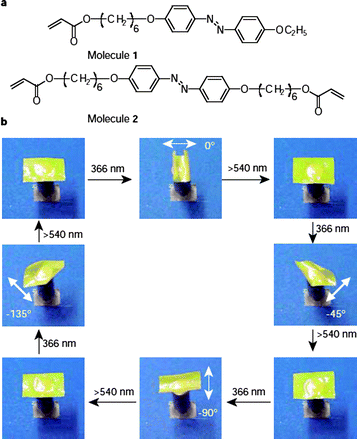 | ||
| Fig. 8 (a) Chemical structures of the LC monomer (molecule 1) and crosslinker (molecule 2) used for preparation of the LCE film; (b) photographic frames showing the precise control of the bending direction of a LCE film by linearly polarized light. Reprinted with permission from ref. 91. Copyright (2003) Nature Publishing Group. | ||
Palffy-Muhoray and co-workers found that by dissolving, rather than covalently bonding azobenzenedyes into a LCE sample, a mechanical deformation response was displayed to non-uniform illumination with visible light which was even larger than previously reported (>60° bending). The rapid light-induced deformations allowed the material to interact with its environment in a new way: when light was irradiated from above onto a dye-doped LCE sample floating on water, the elastomer was found to ‘swim’ away from the light, displaying an impressive propulsion mechanism.93
A functional light-driven plastic motor device was reported by Ikeda and co-workers,94 in which the rotation of a LCE belt on a homemade pulley system was induced by light. By irradiating the belt simultaneously with UV light from the right and visible light from left, the pulley could be driven in a counter-clockwise direction at room temperature. The plastic belt was a LCE laminated film connected at both ends and composed of a LCE film layer and a flexible polyethylene plastic sheet. The LCE film was prepared by photopolymerization of a mixture of azobenzene-functionalized monomers (a LC acrylate and a LC diacrylate) in a glass cell coated with rubbed polyimide alignment layers. Being able to directly convert light energy into mechanical work, without the aid of batteries or electrical connections, these systems have the potential to drive miniaturized systems such as micro and nanorobots.
5.3 Photo-responsive cholesteric LC systems
A cholesteric (chiral nematic) LC phase is typically composed of nematic mesogenic molecules which contain a chiral center. These form in layers with consecutive chiral molecules aligned at a slight angle and rotated relative to the one before. The cholesteric director rotates helically throughout the sample forming a helical supramolecular structure. Nematic LCs can also be converted into chiral nematic LC materials using chiral dopants (i.e. induced cholesteric). Cholesteric LCs are interesting because of their unique optical properties. For example, their ability to reflect visible light of a wavelength is dependant on the helical pitch of their structure.Photo-responsive chiral photochromic PLC systems have been reviewed by Shibaev et al.54,95 The main system described is based on photochromic cholesteric LC copolymers which contain mesogenic groups and combined chiral-photochromic groups, as separate side-chain segments. The mesogenic fragments form the nematic phase and the chiral groups provide the twisting of the phase, to form a complex helical structure. Light irradiation and subsequent isomerization of photochromic groups bound to chiral fragments influences both the configuration and shape of these side chain groups. With a variation of the helical twisting power, this results in dramatic changes in the optical properties of the LC polymer. Other systems described include combinations of different LC copolymers, different photochromic moieties in the same macromolecule and mixtures of LC polymers with chiral-photochromic dopants. The use of light to control the pitch of helix, the rate of helix twisting and untwisting, the spectral range and the width of the selective light reflection have all been investigated. The majority of these have involved the photoisomerization of menthone derivatives as well as azobenzenes.
Other reversible, light-induced changes in the optical properties of cholesteric LC polymers have been reported. Feringa et al. demonstrated the reversible, light-induced conversion of cholesteric to nematic LC phase and the reversible alteration of macroscopic helical pitch in a cholesteric LC system. Both processes could be controlled by modulation of the diastereomeric ratio of an overcrowded alkene dopant that was dependant on the wavelength of light used for irradiation.96
The reversible change in optical rotation of the cholesteric liquid-crystalline polymer, poly(benzyl)-L-glutamate, by the photochromism of 1–4% of spiropyran dissolved in the polymer was demonstrated by Ichimura et al.10,97
Reversible light-induced changes in the optical properties of a cholesteric LC system containing a chiral binaphthyl ether has also been achieved by doping with an achiral fulgide . Alternating irradiation resulted in a pitch change of the induced cholesteric LC of 30% due to ring opening and ring closure of the fulgide .98
A chiral diarylethene was also found to induce stable cholesteric phases when doped in nematic LC. In this system the switching between the two photoisomeric forms (open and closed) resulted in the disappearance and reappearance of the cholesteric phase.99
Feringa et al. report many developments made in chiroptical molecular switching systems based on photochromism.100
6. Photo-responsive biomaterials
6.1 Photo-regulation of biological properties
A large number of efficient light-switchable systems involving photo-responsive biomacromolecules such as polynucleic acids, proteins, cellular signaling molecules and lipid systems, which find application in a broad range of biomedical fields, have all been reported.The ability to control the structure and functions of biomaterials and biomolecular processes with light is of substantial interest in the development of photo-therapies and optobioelectronic systems.101,102 One approach involves the modification of biomaterials with photoisomerizable components which provides a general means to control with light the binding affinities and activities of complementary components. One photoisomeric state can, for instance, block binding by distorting the biomaterial's recognition site. Such photo-regulated ‘ON–OFF’ biomaterials provide a means to design targeted therapeutic agents which can be activated and deactivated by external light signals (Fig. 9a). The binding of saccharides to concanavalin A (con A), a globular lectin protein, modified with either thiophenefulgide103 or spiropyran104 components, was able to be regulated reversibly by light. Azobenzene-modified papain enzyme was also able to undergo photo-regulated substrate hydrolysis.105 In these systems, regulation of binding properties originated from structural perturbations of the proteins upon photoisomerization.
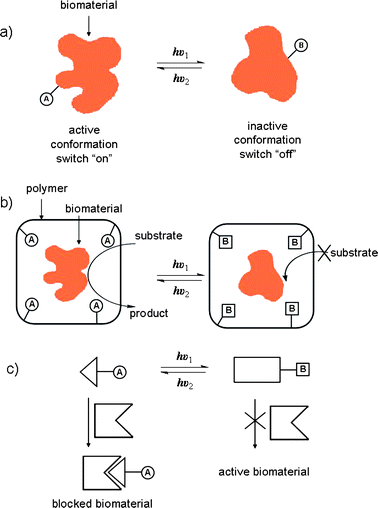 | ||
| Fig. 9 Methods for tailoring reversible photobiological switches. (a) By covalent attachment of photoisomerizable units to the biomaterial. (b) By embedding the biomaterial in a phtoisomerizable environment. (c) By using a low molecular weight photoisomerizable inhibitor. A and B are interchangeable photoisomers.101 | ||
Low molecular weight photochromic-functionalized enzyme inhibitors have also been designed where the geometry of one photo-isomeric state enhances affinity of the inhibitory moiety for the enzyme active site (Fig. 9c).
A dithienylethene-functionalized inhibitor scaffold, decorated with sulfonamide and copper(II) iminodiacetate moieties was used to reversibly control the activity of the enzyme, carbonic anhydrase, in solution using light.106 Similarly, azobenzene-based inhibitors have been used to reversibly control the activity of a bovine heart mitochondrial enzyme complex,107 with partial success, and also the activity of carbonic anhydrase.108 Reversible photo-regulation on a gold (Au) surface was demonstrated successfully for α-chymotrypsin using an azobenzene-based inhibitor. A terminal alkyne allowed attachment to surface-bound azides using click chemistry109 and an ethylene glycol tether was used for extension of the inhibitor into solution. Significantly more enzyme was found to bind to the surface cis forms after UV irradiation.110
Photo-reversible antibody–antigen reactions have also been investigated. Using fluorescence quenching and HPLC monitoring, the binding of a monoclonal antibody to an oligopeptide, hapten , carrying an azobenzene group, was found to occur effectively with trans forms. The antibody was found to release the azobenzene-hapten with UV light, corresponding to the formation of cis forms. Uptake and release occurred reversibly under alternating irradiation with UV and visible light.111 Similarly, an enzyme coupled reaction was found to be completely blocked when an azobenzene-functionalized-NAD+coenzyme was in the trans form (the OFF state). The ON state was provided by UV light which provided the cis form that freed the coenzyme from the antibody.112
Most of these approaches have involved the attachment of the photochromic directly to a protein/peptide. Another approach is to immobilize it into a photochromic-functionalized polymer. Photoisomerization cycles can then be used to regulate its biological function (Fig. 9b). This was exemplified with the enzyme, α-chymotrypsin, immobilized in a membrane made from a crosslinked acrylamide copolymer incorporating spiropyran units and bis-acrylamide crosslinks. Activation and deactivation of the enzyme activity arose from light controlled permeability of the enzyme substrate across the polymer membrane.105 A template-directed polymerization technique, known as molecular imprinting allows receptor (binding) sites that are capable of recognizing specific molecular species to be conveniently imprinted into rigid polymer matrices. Photo-responsive molecularly imprinted polymers have been fabricated using azobenzene-based monomers incorporated into 3D crosslinked networks. Photoisomerization then regulates the release and uptake of a substrate by altering the geometry and spatial arrangement of receptor binding sites imprinted in the polymer network .113,114
Shimoboji et al. developed an approach for controlling protein activity that utilizes both photo and thermo-responsive properties of copolymers incorporating azobenzene and N,N-dimethylacrylamide (DMA) components.115 These copolymer compositions displayed phase transitions within the desired temperature range of 40–45 °C where the enzyme is active and thermally stable. Within this temperature range, one polymer system existed as a soluble, extended coil under UV irradiation (with azobenzenecis forms) and collapsed into a compact, hydrophobic conformation under visible light (with trans forms). A corresponding azobenzenepolymer that incorporated only acrylamide-type monomers was found to switch between the two states in the opposite way. The investigators showed that the photoinduced size and hydration of the polymers, when conjugated just outside the active site of the enzyme endoglucanase, was determined by the isomeric state of the azobenzene form and could be used to regulate substrate access and subsequent hydrolysis. These photo-responsive polymers thus served to switch the polymer–enzyme conjugates ON and OFF and worked both when the conjugate was free in solution and when immobilized on beads.
The application of photo-stimulated biomaterials in bioelectronic devices requires the transduction of a recorded optical signal into a measurable response. The photo-stimulation of redox enzymes can be used to transduce recorded optical signals into amperometric responses by their electrical interaction with electrode surfaces. Photoisomerizable biomaterials assembled as monolayers on electrodes have provided the basis for such reversible, real-time biosensor devices which transduce a bio-recognition event into measurable electronic signal.101,102,116
This was accomplished using a spiropyran-modified flavoenzyme, glucose oxidase (SP-GOD), assembled as monolayer onto a Au electrode that revealed reversible photo-stimulated properties. In the presence of an electron-transfer mediator electrical communication between the SP-GOD monolayer and the electrode interface was attained, leading to the bioelectrocatalyzed oxidation of glucose and the observation of an electrocatalytic anodic current. Upon irradiation with UV light, merocyanine protonated forms (MRH+) were produced, so that the enzyme monolayer (MRH+-GOD) became deactivated and the bioelectrocatalyzed oxidation of glucose was substantially inhibited. This event could be carried out cyclically as an amperometric response upon optical switching of the spiropyran form.117
The modification of proteins with photoisomerizable components in positions which are remote from the active site can lead to only mild perturbations and incomplete photochemical deactivation of enzyme activity. The site-specific modification of the flavoenzyme GOD by a spiropyran component in the microenvironment of the redox active center yielded a fully ‘ON–OFF’ photo-switchable SP-GOD enzyme system. Its assembly on a Au electrode was used as an active biointerface for reversible electrochemical transduction of recorded optical signals.118
Amperometric immunosensors have also been developed by immobilization of monolayers of chemically-modified photoisomerizable antigens onto electrode surfaces. The association of an antibody onto the antigen monolayer electrode results in the insulation of the electrode surface which enables amperometric detection by a redox probe solublized in the electrolyte solution. The monolayer exhibits antigenic properties for the respective antibody in one photoisomer state and photoisomerization to the complementary state distorts the monolayer into a configuration lacking antigen properties, allowing the antibody to be released and washed off. The active antigen electrode can be regenerated by photoisomerization to the original state, facilitating the cyclic, reusable operation (Fig. 10).102,116,119,120
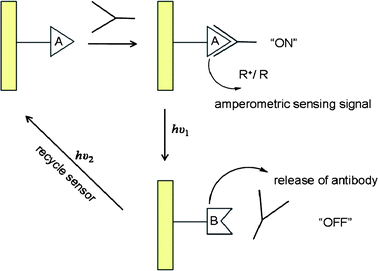 | ||
| Fig. 10 Cyclic operation of an amperometric immunosensor.116,119,120 | ||
In one example a reversible monolayer electrode was realized using a dinitro-spiropyran antigen monolayer on a Au electrode which underwent reversible photoisomerization between a dinitro-spiropyran closed spiro state and the protonated merocyanine form, MRH+, which lacked affinity for the antibody.119,121
Photoisomerizable spiropyran monolayers assembled on electrodes have been used to develop other sensor systems. A mixed monolayer of mercapto-pyridine and spiropyran units assembled on Au electrodes provided one workable system. The pyridine components of the electrode associated with the protein, cytochrome c (cyt c) to allow electrical communication to be established between the heme site of the protein and the electrode. Photoisomerization of the spiro form to the positively charged MRH+ form meant that cyt c was electrostatically repelled from the monolayer and electrical interaction at the electrode was blocked. The amplification of the amperometrically transduced optical signal was provided by the inter-protein reduction of cyt c oxidase and the bioelectrocatalysed reduction of oxygen.122 Other electrode systems involving photo-switchable electrical communication have also been investigated.117,123,124 Recent studies have demonstrated that the use of photoisomerizable monolayers immobilized onto conductive surfaces as active interfaces can also be used for optical recording and surface patterning.125
As stated in previous examples, the open merocyanine state of a spiropyran can exist either as a zwitterionic form with a phenolate group or a positively charged protonated form (MRH+) with a phenol group. This largely depends on the pH of the medium. The ability of the phenolate group to bind to metal ions has been investigated for sensor technologies.126 The zwitterionic merocyanine form has also been found to bind to amino acids such as L-tryptophan, L-tyrosine and L-dopamine through complementary two-point electrostatic interactions.127
6.2 Photo-responsive peptides and polynucleic acids
From the perspective of molecular structure, polypeptides are specialized macromolecular polymers able to exist in disordered or regularly folded arrangements, such as the α-helix and β-structures found in proteins. When photochromic molecules, like azobenzene or spiropyran units, are attached to polypeptides these systems can be made to respond to light by undergoing large photoinduced structural changes.128 A number of photochromic-functionalized polypeptides have been investigated as chiroptical switches in which structural changes can be induced reversibly with light by manipulation of α-helix and β-structures. Photochromism can result in helix reversals, random coil to α-helix transitions, modulation of redox processes or a change in aggregation and disaggregation of the system, as reviewed by Feringa et al.129In this context polypeptides containing azobenzene130,131 and spiropyran132,133 units in their side chains, cyclic peptides and polyamide-oligomers with backbone azobenzene moieties134,135 have also been examined. The use of azobenzenecross-linking reagents to form intramolecular bridges in an engineered peptide system has been found to reversibly induce conformational changes and control helix stability upon photoisomerization.136,137 Photochromics can be anchored to polypeptides with diverse structures as a way to probe conformational dynamics of structural motifs such as protein folding mechanisms and functional aspects of specific amino-acid sequences.138 The use of light-sensitive polypeptides as intelligent molecular materials has been proposed for various applications such as micro- and nanoelectronics, biomedicine, ecology, and other related areas of science.139
Photo-responsive polypeptide monolayers prepared at air–water interfaces have also been of interest for probing photo-mechanical responses such as surface-pressure changes and surface-area changes upon exposure to light. These can be related to the molecular shape and orientation of the photochromics in the assembled structures. For example, a monolayer consisting of poly(L-lysine) containing azobenzene as side chains, displayed a decrease in surface pressure at constant area upon UV irradiation, with an increase in surface pressure with visible light which restored the trans-azobenzene monolayer.140 These changes were ascribed to the different monolayer structures—the trans isomers exhibiting an extended structure of higher surface area and the cis isomer existing in an α-helix configuration. Monolayers of hairy-rod type polymers containing poly(glutamate) with azobenzene units on their side chains have been shown to display reversible expansions upon irradiation with UV light.141,142
The photochemical control of cell adhesion has been achieved using RGD-functionalized azobenzene poly(methyl methacrylate), poly(MMA), surfaces and rationalized in terms of changes occurring in the distance of the RGD ligand from the surface via light-induced isomerizations. All peptides tested led to enhanced cell adhesion on poly(MMA) disks when azobenzenes were their trans form, whereas the plating efficiency was found to decrease with cis forms on UV irradiation by shortening the distance of the RGD-containing peptides from the surface.143
Photochromic responses have been coupled with deoxyribonucleic acid (DNA) functionality in novel ways. For example, the formation and dissociation of an azobenzene-tethered DNA duplex was able to be reversibly photo-regulated via isomerization. The transazobenzene could intercalate between base pairs stabilizing the binding of the two strands, whereas the cisazobenzene was found to disrupt the duplex.144 Furthermore, an azobenzene-tethered T7 promoter was used efficiently for photo-regulating transcription and gene expression.145
Hayashi et al. reported the combination of an azobenzene-containing peptide and its ribonucleic acid (RNA) aptamer pair as a molecular photo-switching system, where binding could be turned ON and OFF with light via the structural change of azobenzene units in the peptide backbone. When the peptide was covalently immobilized on a Au surface it also showed photo-responsive binding to the target RNA.146
A related phenomenon inspired by the photo-regulation capability of azobenzene-tethered DNA was a single-molecule DNA nanomotor. This hairpin structure integrated azobenzene moieties on DNA bases and was able to undergo reversible intramolecular extension-contraction behavior in response to trans–cis isomerization.147
6.3 Micelles and vesicles—photo-regulated delivery
Photochromic polymers can be logically used for the design of light sensitive self-assembled delivery vehicles for biomedical areas which include micelles, vesicles, gels , nanocomposite systems and various supramolecular architectures.148,149Surfactant and amphiphilic block copolymer micelles are commonly applied to controlled delivery systems. Light-controlled micelle disruption can be provided by incorporation of a photochromic chromophore into the hydrophobic block (Fig. 11). The photoreaction results in a conformational or structural change that shifts the hydrophilic/hydrophobic balance toward the destabilization of the micelles. Reversible dissociation upon illumination with UV/visible or near infrared (NIR) light has been achieved using various chromophores.57,150 As mentioned previously, the spiropyran photoconversion results in the largest change in polarity, since the merocyanine form is normally zwitterionic (depending on pH) compared to the relatively non polar spiro form. The azobenzenetrans form is planar and non-polar compared to the cis form, with an estimated dipole moment of ∼4.4.151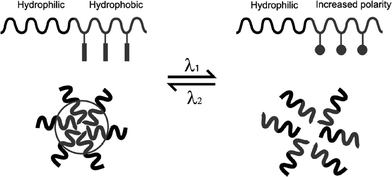 | ||
| Fig. 11 A schematic illustration of block copolymer micelles that can be reversibly dissociated and formed upon absorption of photons of two different wavelengths. The process is controlled by a reversible photoisomerization reaction of photochromic groups between two isomeric forms of different polarities. From ref. 150. Reproduced with permission of the Royal Society of Chemistry. | ||
A diblock copolymer containing, as its hydrophilic block, a random copolymer of tert-butyl acrylate and acrylic acid and an azobenzene-methacrylate as the hydrophobic block, was assembled into micelles. Upon irradiation with UV light the non-polar trans azobenzene groups were converted to the more polar cis forms, to significantly increase the hydrophilicity of the originally hydrophobic block. Therefore the micelles could be dissociated with UV light and reassembled using visible light.151,152 In a more recent example, micelles made from a block copolymer composed of poly(ethylene glycol) as the hydrophilic section and poly(spiropyran methacrylate) as the hydrophobic section displayed the same reversible dissociation using UV and visible light.153
Liu et al. reported reversible photoinduced micellization and a micelle-hollow sphere transition arising from hydrogen bonding associations in an azobenzenecopolymer system.154 On the basis of photoinduced polarity change, photocontrolled and reversible swelling-shrinking behavior of micron sized vesicles was observed, with a self-assembled poly(N-isopropylacrylamide), poly(NIPAAM), and azopyridine-containing diblock copolymer.155
Lin et al. also reported similar photoinduced behavior of copolymeric vesicles composed of an azopyridine-containing amphiphilic diblock copolymer. This included fusion, damage, defect formation, disruption, disintegration and rearrangements. All of these processes are expected to increase the permeability of the vesicular bilayer membranes.156
Specialized liposomes composed of dipalmitoyl phosphatidyl choline bearing two azobenzeneacyl chains were also found to release entrapped solutes due to the conformational changes brought on by photoisomerization cycles. The fine control of drug release from these systems was achieved by adjusting the liposome composition.157–159 Photo-induced ON–OFF release of internal material in a liposome was also demonstrated using a spiropyran-functionalized lipid molecule by perturbation of the membrane on UV irradiation.160
The conformation and polarity of photochromic groups can have a direct influence on inter and intramolecular interactions of their copolymers and therefore also on aggregative properties. This in turn can be used to regulate permeability and transport of certain solutes, such as proteins, through the polymer network . A copolymer made from an azobenzene methacrylate and DMA was found to display significant concentration dependant photoviscosity effects in aqueous solution without macrophase separation. Trans to cis isomerization with UV light led to partial dissociation of azobenzene aggregates that acted as connective junctions. This led to a loss of viscoelasticity, especially in concentrated solutions of the polymer.161
Other amphiphilc systems have been studied, such as hydrophobically modified polymers (HMPs). These macromolecules contain alkyl side-chains on a hydrophilic backbone and undergo intra or inter-chain associations in water. Noncovalent binding associations can occur between hydrophobic moieties of the polymer as well as with amphiphilic additives, such as surfactants and proteins, leading to aggregation and gelling behavior.162,163 Concentrated solutions of alkyl-modified poly(acrylic acid), poly(AA), can form highly viscous mixtures with proteins as a result of hydrophobic associations. Azobenzene-modified poly(AA)s (AMPs) have been studied as photo-responsive HMPs where light induced polarity changes that occur in hydrophobic side-groups can modulate aggregation and binding behavior of the polymeric systems. Khoukh et al. reported the photo-responsive association between AMPs and nonionic surfactants. On exposure to UV light, the formation of azobenzenecis forms weakened the association of the polymer with the surfactant.164 They also reported the reversible light-triggered control of emulsion type which could be switched from a direct emulsion (dispersion of oil droplets into the water phase) to the inverse (dispersion of water droplets into the oil phase), using poly(sodium acrylate) grafted with azobenzene units.165 The reversible photo-switching of viscosity and binding of bovine serum albumin (BSA) protein was also achieved using an aqueous solution of an AMP incorporating a few mol percent of hydrophobic moieties.166 In the dilute regime, BSA/polymer complexes were formed in equilibrium with unbound BSA. In the semi-dilute regime, a viscosity enhancement was obtained, ascribed to physical cross-linking of BSA with the polymer. Reversible release of BSA (by up to 80% of the protein) was obtained by exposure to UV light, due to the lower binding of cis form to the protein.
Low molecular weight photo-responsive surfactants have been developed to drive molecular changes in interfacial and colloidal systems. In these systems light can induce dramatic changes in hydrophilic head-groups and hydrophobic tails leading to changes in the surface activity, aggregation structure, viscosity, microemulsion separation and solubility.167
6.4 Nanocomposites—photo-regulated delivery
The combination of light-sensitive photochromic polymers and inorganic substrates within a single system has been investigated as potential photo-responsive biomedical drug delivery vehicle.168,169 Azobenzene chains tethered within the pores of mesoporous silica nanoparticles can act as impellers and nanovalves by reversibly changing their conformation with light. In one particulate system the continuous excitation at a wavelength where both the cis and trans isomers absorb resulted in the continual dynamic wagging of tethered azobenzenes, with the expulsion of luminescent probe molecules.170 Using the same light activated motion, azobenzene functionalized mesostructured silica nanoparticles have also been used to deliver and release anticancer drugs into living cells on demand (Fig. 12).171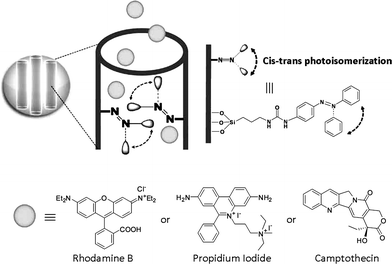 | ||
| Fig. 12 Light-activated mesostructured silica nanoparticles functionalized with azobenzene derivatives. Continuous illumination at 413 nm causes a constant trans–cis photoisomerization about the NN bond causing dynamic wagging motion of the azobenzene derivatives and results in the release of the molecules through and out of the mesopores. Reproduced with permission from ref. 171. Copyright Wiley-VCH Verlag GmbH & Co. KGaA. | ||
The same concept can be applied to control the behavior of nanocomposite membranes. A photo-responsive nanoporous membrane composed of monosized pores modified with azobenzene ligands, was prepared on an indium tin oxide working electrode which exhibited size-selective and photo-regulated mass transport of probing molecules to the electrode surface.172
Zeolite membranes containing adsorbed azobenzene have also shown photo-switchable gas permeation properties as a result of trans–cis isomerization cycles.173
6.5 Gels —photo-regulated swelling and shrinking
Hydrogels , including crosslinked versions, consist of elastic networks able to uptake substantial amounts of water in their interstitial spaces. By incorporating photo-responsive units in these gel systems, light can be used to control swelling and shrinkage. Such volume changes of gels can be applied to actuators, sensors, controllable membranes for separations, as well as modulators for drug delivery. The triggered release of therapeutic molecules from implantable hydrogel -based devices has also been considered.A common polymer used to develop such systems is poly(NIPAAM). Aqueous solutions exhibit a lower critical solution temperature (LCST) transition normally at 32 °C. Above this temperature it undergoes a volume phase transition as the solubility of the polymer is significantly reduced. This temperature depends on composition and, as a general guideline, the incorporation of more hydrophobic groups will lead to a lower LCST.174–177 For lower molecular weight homopolymer samples (normally <50![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 000 g mol−1), small variations in LCST have been correlated with changes in end-group structure and polarity.178 Other aspects that can influence LCST are poly(NIPAAM)'s tacticity,179 architecture175 as well as solution components, such as added salts.180
000 g mol−1), small variations in LCST have been correlated with changes in end-group structure and polarity.178 Other aspects that can influence LCST are poly(NIPAAM)'s tacticity,179 architecture175 as well as solution components, such as added salts.180
A functional copolymer composed of poly(NIPAAM) with a small proportion of pendant spiropyrans displayed photosensitive solubility characteristics by switching between neutral spiro and polar zwitterionic forms. Changes in the solubility of the material were also found to occur with temperature and pH.181 A corresponding crosslinked version of the copolymer displayed photo-responsive shrinking in an acidic solution and photocontrolled permeability as a porous membrane.182 Photo, thermally and pH-responsive microgel particles have also been investigated consisting of poly(NIPAAM), free amine groups and spiropyran moieties existing as polar merocyanine forms in the dark. Visible irradiation corresponded to a reduced particle size as a result of spiropyran merocyanines reverting to less polar, closed forms. Increasing temperature resulted in a volume contraction under all light conditions and, due to the presence of amine groups, the swelling capability of the microgel was found to diminish with increasing pH.183 Dual responsive (to temperature and light), crosslinked hydrogels incorporating poly(NIPAAM) and pendant spirooxazines have also been reported.184 Irradiation with UV light was found to enhance their water absorption and cause swelling, while an increase in temperature was found to do the opposite. In this system the spirooxazine open form is charge separated and of a higher dipole moment compared to its closed form, and therefore stabilized by an association with water. Rewritable microrelief formation on photo-responsive hydrogel layers composed of poly(NIPAAM) and spiropyrans has been demonstrated by means of micropatterned light irradiation.185 Spiropyrans have also been found to effect dextran, such as a two-phase gel system, which exhibited reversible photoinduced phase separation.186
In a related application, photo-responsive bioconjugates have been used as tools for the specific capture of biologicals using a process of photoaffinity precipitation. An affinity macroligand was synthesized comprising poly(NIPAAM) functionalized on its side-chains with azobenzene groups and at its end group with a biotin ligand. The resulting polymer showed a dependency of LCST in pure water on the isomeric state of the azobenzene groups. The precipitation of the polymer could therefore be stimulated isothermally with light, allowing the specific capture and recovery of avidin from a serum-containing cell-culture.187
Photo-responsive supramolecular gels have also been developed, with sol –gel transitions able to be induced reversibly via photoisomerization.188 These have been mostly based on azobenzene photochromism however other systems involving organogels of chromene and dithienylethene derivatives have also been reported. The photo-responsive self-assembly involving host–guest complexation has also been investigated using cyclodextrin/polymer systems with gel -to-sol and sol -to-gel transitions found to be dependant on the binding affinities of azobenzene guests with the α-cyclodextrin host.189
6.6 Photo-regulated wettability
The ability to control the properties of a surface has become a goal of biomimetic research, finding insight in many phenomena that occur in nature. Wettability is a fundamental property of a solid surface, which plays an active role in many facets of life. New functional surfaces with wettability properties are arising much interest from industry.190 The modification of surfaces with photochromic moieties whose switching can be used to alter the affinity of a liquid to the surface is a way to produce surfaces with switchable wettability.191Photoisomerizable monolayers applied to surfaces can provide two-dimensional membrane mimetic microenvironments on which interactions with liquids can be effectively controlled with light. The following examples demonstrate how the switching of polarity between photochromic forms on a surface is the main criterion. In these systems an increase in polarity results in an increase in surface energy which is displayed by the spreading of a liquid droplet on the surface.
A layer of photo-switchable spiropyrans covalently bound to a glass surface along with a mixture of organo silanes exhibited reversible wettability changes when irradiated with UV and visible light.192 The water contact angle was found to be lower for dry spiropyran surfaces after irradiation with UV light, as the water droplet spread out, compared to visible irradiation. Water in capillary tubes coated with the photosensitive layer was also observed to rise when the incident light was switched from visible to UV. This was ascribed to changes in the surface energy of the surfaces, correlated to the switching of the surface-bound spiropyran molecules between polar, zwitterionic and nonpolar spiro forms.
Water droplets have also been found to move, especially on very rough surfaces, using gradients of UV and visible light. This was studied using fractally rough silicon nanowire surfaces grown on a silicon substrate and coated with a hydrophobic monolayer of spiropyrans.193 Ichimura et al. studied surface-immobilized azobenzene systems with striking results. In one example a liquid droplet was placed on a silica substrate modified with a calix[4]resorcinarene derivative with pendent azobenzene units and was irradiated with UV and blue light (436 nm) to trigger the isomerization of the azobenzenes assembled on the surface. Initially the spreading/retraction motion of the liquid droplet was studied (Fig. 13). Gradients in surface free energy were then applied by asymmetrical illumination of the surface. The resulting spatially controlled changes in the photoisomerization of azobenzenes allowed the directional motion of the droplet. The direction of motion could be tailored by manipulating the direction of the light intensity gradient.194,195
![Lateral (a) and top (b) views of in situ spreading/retraction motion of droplets on plates modified with a monolayer of azobenzene terminated calix[4]resorcinarenes upon homogeneous irradiation with UV and blue light from ref. 194. Reproduced with permission of the Royal Society of Chemistry.](/image/article/2010/PY/b9py00300b/b9py00300b-f13.gif) | ||
| Fig. 13 Lateral (a) and top (b) views of in situ spreading/retraction motion of droplets on plates modified with a monolayer of azobenzene terminated calix[4]resorcinarenes upon homogeneous irradiation with UV and blue light from ref. 194. Reproduced with permission of the Royal Society of Chemistry. | ||
A monolayer of a polymeric material containing azobenzene side chains was used to form light-induced micropatterns. This was illustrated by the water microdroplet formation of a cis/trans pattern generated by light through a mask.196
Photomechanical effects observed in photochromic monolayers at the air–water interface have also been explained in terms of changes to polarity on photoisomerization.197,198 Monolayers of poly(vinyl alcohol) with azobenzene side chains were found to expand with UV light and shrink with visible light as a result of the different affinities of trans and cis forms for the water surface.199,200 Monolayers composed of poly(MMA) containing spiropyran units in the side chains have also been investigated showing a pH dependant increase in surface pressure on UV irradiation and a recovery in the dark. In these systems the photogenerated merocyanine species are strongly attracted by the water and can penetrate the monolayer more deeply than neutral SP forms, resulting in a marked surface pressure change.201
6.7 Membranes—photo-regulated permeability
The reversible changes of polarity and conformation imparted to a photochromic polymer system by photoirradiation can be applied to controlling mass transfer through porous and non-porous barriers such as membranes. Systems based on azobenzene, spiropyran and diarylethenes polymers are the most highly investigated.202Those developed early on were focused on crown ethers funtionalized with azobenzene, where selective ion complexation could be controlled by their butterfly-like motion, induced by light irradiation.203 This was extended to a photo-switchable ion carrier based on a spiropyran.204Metal ion complexation to a crown ether–spiropyran copolymer was found to enhance the aggregation of merocyanine forms, which resulted in the reversible precipitation of polymer205 Analogues containing both crown ether and spiropyran moieties in the same pendant side chains showed photo-responsive ion conducting behavior.206 Neutral-carrier-type ion-selective electrodes based on LC ion-sensing membranes were also prepared containing azobenzenes. These showed remarkable changes in their ion selectivity on irradiation, induced by the LC phase transition of the membranes.207
Later examples focused on the control of hydrophobic/hydrophilic balance brought about by photoisomerization. The swelling degree of a membrane in water made from an amphiphilic copolymer, poly(hydroxyethyl methacrylate) functionalized with azobenzene side groups, was found to decrease with UV irradiation but could be recovered to its original level by irradiating with visible light. The decrease in the swelling degree could not be interpreted simply as a result of an increase in polarity and instead was explained by the interaction of polymerhydroxyl groups with the dipole of cisazobenzene forms. This was thought to strip away the solvating water molecules from the hydroxyl groups and consequently from the membrane.208 These functional hydrogels were extended further to show photoinduced permeation control of proteins where the polymer membrane was permeable in the dark and semi-permeable for low molar mass compounds under UV irradiation due to the decreased swelling of the polymer membrane.209
As exemplified previously, the photo-regulation of a material’s function can be brought about by its immobilization in a photoisomerizable polymer. This approach, applied extensively to photo-responsive hydrogels , has also found relevance to membranes. For example, ion permeable membranes have been developed using azobenzene groups incorporated into chloromethylstyrene/divinylbenzene crosslinked polymers. Properties, with respect to ion exchange capacity, water content and electrical resistance were found to change with UV irradiation. Notably, transport rates in electrodialysis of various anions were found to increase with UV irradiation. This was ascribed to the conversion of the nonpolar trans forms into the more polar cis forms, which increased the water content and effective pore size of the membranes.210
A porous membrane grafted with spiropyran methacrylate and acrylamide showed an increase in permeability to a mixture of water and methanol with UV irradiation and a decrease with visible light. This was related to the extension of grafted copolymer chains, where polar zwitterionic groups caused a decrease in solubility with chain contraction resulting in more open pores.211
A macroporous polyethylene membrane coated with a copolymer carrying crown ether and spiropyran side chains also worked as a functional membrane to control solvent permeation rate photochemically. UV light increased the polarity of membrane pores as a result of polar merocyanine forms and decreased the permeation rate of hexane. Visible light restored the permeation rate by isomerization back to the electrically neutral spiropyran form. By contrast, the permeation of ethanol was enhanced by UV light with an increase in apparent pore size as a result of polymer chain contraction and the opposite was found to occur with visible light.212 The UV-grafting of a spiropyran monomer onto a poly(ether sulfone) commercial ultrafiltration membrane also resulted in a photo-switchable membrane with reversible polarity.213
In a more recent example, the modification of the surface of nanoporous alumina membranes with mixtures of a spiropyran and hydrophobic molecules provided photocontrol over the admission of water into the membrane. When the spiropyrans existed as the less polar spiro forms the membrane was not wet by an aqueous solution. Upon exposure to UV light, the more polar merocyanine forms allowed water to enter the pores and cross the membrane. If the aqueous solution contained ions, then the membrane acted as an electrical switch to allow a current to flow, with photoisomerization leading to a two-order-of-magnitude increase in ionic conductance.214
Surface control of hydrophilic/hydrophobic properties with light stimuli has also been used to control surface environments for attachment and proliferation of cells. The light-induced detachment of platelets and mesenchymal stem cells on glass plates coated with a copolymer containing methyl methacrylate and spiropyran units, was demonstrated upon simple and patterned UV irradiation. This also correlated with decreased water contact angles and increased water drop diameter relative to the unexposed surfaces.215 A photo-responsive culture surface which allowed photocontrol of cell adhesion was prepared with a copolymer of poly(NIPAAM) with spiropyran moieties in its side chains. Cell adhesion to the surface was drastically enhanced with polar zwitterionic forms via irradiation with UV light.216
7. Photo-switchable supramolecular assemblies
Supramolecular self-assembly is considered a powerful tool for the construction of well-defined nanoarchitectures. The photoisomerization of photochromic molecules in these self-assemblies is a popular method to non-invasively manipulate the degree of aggregation and supramolecular architecture using light. A variety of photo-responsive self-assemblies have been established using azobenzene, for instance, where the cis isomer is morphologically less aggregative because of its bent conformation and unfavorable for dense molecular packing. Furthermore, its increased polarity compared to the trans-isomer makes it less aggregative in polar media.158,217 In a classical example of photocontrolled self-assembled order, two cyclic peptides joined by an azobenzene linker acted as a photo-responsive material giving different hydrogen bonded networks depending on the isomeric state. In the trans state, the subunits organized into extended linear chains via hydrogen-bonding and with UV irradiation the extended network was disrupted.218Another unique photo-responsive system involved hydrogen-bonded macrocycles called rosettes, formed from azobenzene-appended melamines and barbiturates/cyanurates that could be hierarchically organized by irradiation. The rosettes stacked into columns which then intertwined to form fibers. UV irradiation was used to destroy the ordering, progressively disrupting the higher-order structures. The system displayed photo-reversible solid to liquid phase-transitions.219
8. Fluorescence switching
The photoinduced inter-conversion of the two states of a photochromic compound can be exploited to modulate the emission of a fluorescent partner. This photo-response can be facilitated through the Förster resonance energy transfer (FRET) process where both fluorescent and photochromic components are normally integrated within the same macromolecular or nanostructured construct.The mechanism initially involves UV irradiation to stimulate the transformation of the photochromic from a spiro form to a merocyanine form. This activates the energy transfer process from the excited state of the fluorophore (donor) to the merocyanine form (acceptor) thereby switching off its emission. The spectral overlap between the emission band of the fluorophore and the absorption band of the merocyanine form is therefore essential. The regeneration of the original spiro form of the photochromic allows suppression of the quenching process and switches the emission of the fluorophore back on. Thus, the emission of these systems can be repeatedly turned ON and OFF simply by switching the photochromic back and forth between its two states. Several recent reviews address the mechanisms involved in modulating the fluorescence of nanostructured constructs using photochromic switches.220,221
Several nanoparticle systems have been investigated which demonstrated fluorescence modulation using photochromic switching. A hybrid organic–inorganic nanosystem was prepared by attaching spiropyrans via a thiol linkage to core–shell semiconductive CdSe/ZnS nanocrystals (Fig. 14). Fluorescence from the hybrid nanocrystals was observed when the dye was in its spiro form but was strongly quenched by the dye in its merocyanine form.222
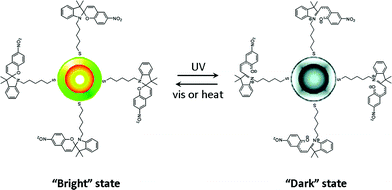 | ||
| Fig. 14 Nanocrystals functionalized with spiropyrans with demonstrate ON–OFF fluorescence modulation. Reprinted with permission from ref. 222. Copyright (2005) American Chemical Society. | ||
The switching of a spiropyran bound to a protein has also been used successfully to reversibly modulate quantum dot fluorescence, demonstrating the potential of this system to be applied to bioassays and biosensors .223
Using emulsion polymerization, a fluorescent dye and a spiropyran dye were able to be embedded within the hydrophobic cavities of polymeric nanoparticles composed of lightly crosslinked poly(NIPAAM) and polystyrene. Using this system, photochromic switching allowed the fluorescence of the nanoparticles to be switched reversibly ON and OFF.224
In another example dual-color (red–green) fluorescence was demonstrated. When the spiro-containing nanoparticles were stimulated with UV light, the FRET process was able to convert the green emission from the fluorescent dyes to a red emission from the merocyanine forms. These nanoparticles were applied successfully to image live-cells.225
Other optically switchable fluorescent nanoparticle systems have also been developed to provide smaller particle sizes, with higher dye loading and with more controlled quantities of the two dyes.226
Other examples have appeared in literature, using other photochromic dyes and constructs,227,228 demonstrating that this photo-responsive system is valuable to many areas such as biomedical imaging, sensing as well as information processing and storage.
9. Conductivity switching
Photochromic molecules are attractive candidates for use in molecular electronics. In these systems molecules are attached to metallic leads and an electronic current passes through them. Dithienylethene and diarylethene derivatives have been the most heavily investigated due to the thermal irreversibility of their photoinduced forms. The two molecular isomeric states (open and closed) are different in the nature and extent of conjugation path and therefore have distinct electronic transport properties when the molecule serves as a molecular wire in an electroactive system.229,230The fluorescence intensity and electrical conductivity of a photochromic conducting polymer, based on a diarylethene was able to be modulated by alternative irradiation with UV and visible light.232 Similarly photocurrent was measured in a diarylethene film which displayed ON–OFF switching according to the isomerization state of the photochromic.233 The reversible switching of conductivity of monolayers of dithienylethene assembled on gold has also been demonstrated (Fig. 15).231 Analogously, dithienylethene single molecule resistance changes have been reported on gold electrodes which varied depending on the photoisomerization state of the molecule.234
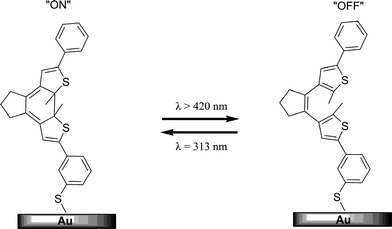 | ||
| Fig. 15 A schematic representation of the reversible switching of 1,2-dithienylethene on a Au surface where the open form is less conductive than the closed form.191,231 | ||
10. Conclusions and outlook
Photo-responsive systems based on photochromism have generated substantial research interest, proposing a wide range of potential applications. Remarkable advances have been made over the past 30 years largely as a result of interdisciplinary efforts. In the area of LCs, for example, many structural and external parameters strongly influence the ability of polymeric systems to achieve the highest and most stable birefringence and dichroism and indeed this area has prospered from contributions and insights of both polymer and photonics fields.A variety of novel photo-responsive biomaterials have been developed by modifying biopolymers or by synthesizing bio-inspired macromolecules incorporating photoisomerizable units. The design of such intelligent biomaterials and surfaces continues to nourish ideas inspired from biological processes occurring in nature. This area has greatly exploited spiropyran photochromism because of their remarkable ability to undergo a significant change in polarity on isomerizing. Whilst these dyes are particularly notable for this, their sensitivity to fatigue should also be recognized and may limit their long-term applicability. Therefore the development of other photoisomerizable dyes that undergo large polarity changes without such large fatigue problems are required. At the very least, a greater understanding of the fatigue mechanisms that spiropyrans undergo and methodologies aimed at minimizing such effects would be advantageous.
Another obstacle in the application of photo-responsive systems in biomedical therapeutic areas, such as implantable devices, is the requirement of UV stimulation. One questions how such irradiation could be administered within the body. This may only be practical for peripheral tissues where penetration depth and lack of transparency is not as significant. In these instances the incorporation of thermally irreversible photochromic dyes that absorb in the NIR and IR region would make in vivo activity more realistic.
In this review many photo-responsive systems have been described which do not necessarily exploit polymers. Noteworthy are those systems based on a defined arrangement and organization of building blocks comprising photochromic units. This includes self-assembled monolayers, supramolecular assemblies, phase separated structures, such as bilayer membranes, and ordered mesophases of LCs. In these systems the geometrical conformational change that accompanies the photosiomerization of each photochromic molecule has such a profound effect on the material properties. The molecular motions can give rise to changes of a much larger-scale than the molecular level, such as the coordinated bending motion of an elastomeric film. It would be interesting to see these systems applied with other photochromic classes which also undergo substantial geometrical alterations on isomerizing such as chromenes and spirooxazines.
What is also apparent from an overview of existing literature is that the main photochromic dyes that have been considered for the development and design of reversible photo-responsive systems are azobenzenes, spiropyrans and diarylethenes. This is a rather limited domain and therefore efforts should be aimed at making further improvements to these available photoresponsive units.
On a very positive concluding note, photochromic materials are finding new opportunities in applications that in the past seemed only idealistic. This has arisen along with recent developments in nanosciences and nanotechnologies and with new avenues to make engineered polymers as novel macromolecular constructs. This has led to ongoing improvements in the design of photo-responsive systems in which photochromics can be intuitively located within specialized nanoenvironments.
Overall we expect that collaborative efforts amongst different scientific disciplines will be a major factor that determines the full potential of any photo-responsive system.
Acknowledgements
The authors thank Dr Ian Dagley and the Cooperative Research Centre for Polymers for funding; the Centre for Advanced Macromolecular Design for research support and CSIRO, Molecular and Health Technologies for laboratory facilities. T.P.D acknowledges the receipt of a Federation Fellowship from the Australian Research Council.Notes and references
- H. Dürr, General Introduction, in Photochromism: Molecules and Systems, ed. H. Dürr and H. Bouas-Laurent, Elsevier Science Publishing House, Amsterdam, 1990, p. 1 Search PubMed.
- G. Such, R. A. Evans, L. H. Yee and T. P. Davis, J. Macromol. Sci., Polym. Rev., 2003, 43, 547.
- V. Krongauz, Environmental Effects on Organic Photochromic Systems in Photochromism: Molecules and Systems, ed. H. Dürr and H. Bouas-Laurent, Elsevier Science Publishing House, Amsterdam, 1990, p. 793 Search PubMed.
- S. Maeda, Spirooxazines, in Organic Photochromic and Thermochromic Compounds Volume 1: Main Photochromic Families, ed. J. Crano and R. Guglielmetti, Plenum Publishing, New York, 1999, pp. 85 Search PubMed.
- R. C. Bertelson, Spiropyrans, in Organic Photochromic and Thermochromic Compounds Volume 1: Main Photochromic Families, ed. J. Crano and R. Guglielmetti, Plenum Publishing, New York, 1999, pp. 11 Search PubMed.
- C. Barrett, A. Natansohn and P. Rochon, Chem. Mater., 1995, 7, 899 CrossRef CAS.
- C. Barrett, A. Natansohn and P. Rochon, Macromolecules, 1994, 27, 4781 CrossRef CAS.
- K. Horie and I. Mita, Adv. Polym. Sci., 1989, 88, 77 CAS.
- S. Xie, A. Natansohn and P. Rochon, Chem. Mater., 1993, 5, 403 CrossRef CAS.
- K. Ichimura, Photochromic Polymers, in Organic Photochromic and Thermochromic Compounds Volume 2: Physicochemical Studies, Biological Applications, and Thermochromism, ed. J. Crano and R. Guglielmetti, Kluwer Academic/Plenum Publishers, New York, 1999, p. 9 Search PubMed.
- B. Van Gembert, Benzo and Napthopyrans (Chromenes), in Organic Photochromic and Thermochromic Compounds Volume 1: Main Photochromic Families, ed. J. Crano and R. Guglielmetti, Plenum Publishing, New York, 1999, p. 111 Search PubMed.
- G. K. Such, R. A. Evans and T. P. Davis, Macromolecules, 2004, 37, 9664 CrossRef CAS.
- G. K. Such, R. A. Evans and T. P. Davis, Mol. Cryst. Liq. Cryst., 2005, 430, 273 CrossRef CAS.
- G. K. Such, R. A. Evans and T. P. Davis, Macromolecules, 2006, 39, 1391 CrossRef CAS.
- G. K. Such, R. A. Evans and T. P. Davis, Macromolecules, 2006, 39, 9562 CrossRef CAS.
- R. A. Evans, T. L. Hanley, M. A. Skidmore, T. P. Davis, G. K. Such, L. H. Yee, G. E. Ball and D. A. Lewis, Nat. Mater., 2005, 4, 249 CrossRef CAS.
- R. A. Evans and G. K. Such, Aust. J. Chem., 2005, 58, 825 CrossRef CAS.
- F. Ercole, T. P. Davis and R. A. Evans, Macromolecules, 2009, 42, 1500 CrossRef CAS.
- F. Ercole, N. Malic, T. P. Davis and R. A. Evans, J. Mater. Chem., 2009, 19, 5612 RSC.
- F. Ercole, N. Malic, S. Harrisson, R. A. Evans and T. P. Davis, Macromolecules, 2009 DOI:10.1021/ma901830b.
- A. Toriumi, S. Kawata and M. Gu, Opt. Lett., 1998, 23, 1924 CrossRef CAS.
- S. Kawata and Y. Kawata, Chem. Rev., 2000, 100, 1777 CrossRef CAS.
- K. G. Yager and C. J. Barrett, J. Photochem. Photobiol., A, 2006, 182, 250 CrossRef CAS.
- I. G. Naydenova, T. J. Petrova, N. K. Tomova, V. D. Dragostinova, L. P. Nikolova and T. A. Todorov, Abstr. Paper Am. Chem. Soc., 1998, 216, 434.
- I. Naydenova, T. Petrova, N. Tomova, V. Dragostinova, L. Nikolova and T. Todorov, Pure Appl. Opt., 1998, 7, 723 CrossRef CAS.
- T. Todorov, L. Nikolova, K. Stoyanova and N. Tomova, Appl. Opt., 1985, 24, 785 Search PubMed.
- P. Rochon, E. Batalla and A. Natansohn, Appl. Phys. Lett., 1995, 66, 136 CrossRef CAS.
- T. M. Geue, A. G. Saphiannikova, O. Henneberg, U. Pietsch, P. L. Rochon and A. L. Natansohn, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2002, 65, 052801 CrossRef CAS.
- T. Todorov, L. Nikolova and N. Tomova, Appl. Opt., 1984, 23, 4309 CrossRef CAS.
- M. Ivanov, L. Nikolova, T. Todorov, N. Tomova and V. Dragostinova, Opt. Quantum Electron., 1994, 26, 1013 CrossRef CAS.
- T. Buffeteau, F. L. Labarthet, M. Pezolet and C. Sourisseau, Macromolecules, 1998, 31, 7312 CrossRef CAS.
- A. Natansohn, P. Rochon, J. Gosselin and S. Xie, Macromolecules, 1992, 25, 2268 CrossRef CAS.
- A. Natansohn, P. Rochon, M. S. Ho and C. Barrett, Macromolecules, 1995, 28, 4179 CrossRef CAS.
- L. Lasker, T. Fischer, J. Stumpe, S. Kostromin, S. Ivanov, V. Shibaev and R. Ruhmann, Mol. Cryst. Liq. Cryst., 1994, 246, 347 CrossRef CAS.
- M. Dumont and A. El Osman, Chem. Phys., 1999, 245, 437 CrossRef CAS.
- H. Ishitobi, Z. Sekkat and S. Kawata, Chem. Phys. Lett., 1999, 300, 421 CrossRef CAS.
- P. Jones, W. J. Jones and G. Williams, J. Chem. Soc., Faraday Trans., 1990, 86, 1013 RSC.
- G. Berkovic, V. Krongauz and V. Weiss, Chem. Rev., 2000, 100, 1741 CrossRef CAS.
- J. A. Delaire and K. Nakatani, Chem. Rev., 2000, 100, 1817 CrossRef CAS.
- A. Natansohn and P. Rochon, Chem. Rev., 2002, 102, 4139 CrossRef CAS.
- J. Delaire and K. Nakatani, Chem. Rev., 2000, 100, 1817 CrossRef CAS.
- P. M. Hogan, A. R. Tajbakhsh and E. M. Terentjev, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2002, 65, 041720 CrossRef CAS.
- M. Eich and J. H. Wendorff, Makromol. Chem. Rapid Commun., 1987, 8, 467 CrossRef CAS.
- T. Ikeda, S. Horiuchi, D. B. Karanjit, S. Kurihara and S. Tazuke, Chem. Lett., 1988, 1679 CAS.
- S. Kurihara, T. Ikeda, S. Tazuke and J. E. Seto, J. Chem. Soc., Faraday Trans., 1991, 87, 3251 RSC.
- I. Cabrera and V. Krongauz, Macromolecules, 1987, 20, 2713 CrossRef CAS.
- I. Cabrera and V. Krongauz, Nature, 1987, 326, 582 CrossRef.
- I. Cabrera, V. Krongauz and H. Ringsdorf, Angew. Chem., Int. Ed. Engl., 1987, 26, 1178 CrossRef.
- V. Krongauz, Mol. Cryst. Liq. Cryst., 1994, 246, 339 CrossRef CAS.
- I. Cabrera, A. Dittrich and H. Ringsdorf, Angew. Chem., Int. Ed. Engl., 1991, 30, 76 CrossRef.
- A. S. Angeloni, D. Caretti, C. Carlini, E. Chiellini, G. Galli, A. Altomare, R. Solaro and M. Laus, Liq. Cryst., 1989, 4, 513 CrossRef CAS.
- Y. K. Han, D. Y. Kim and Y. H. Kim, Mol. Cryst. Liq. Cryst., 1994, 254, 445 CrossRef CAS.
- V. A. Mallia, M. George and S. Das, Chem. Mater., 1999, 11, 207 CrossRef.
- V. Shibaev, A. Bobrovsky and N. Boiko, Prog. Polym. Sci., 2003, 28, 729 CrossRef CAS.
- X. H. He, H. L. Zhang and X. Y. Wang, Polym. J., 2002, 34, 523 CrossRef CAS.
- J. del Barrio, L. Oriol, R. Alcala and C. Sanchez, Macromolecules, 2009, 42, 5752 CrossRef CAS.
- Y. Zhao and J. He, Soft Matter, 2009, 5, 2686 RSC.
- O. Tsutsumi, Y. Demachi, A. Kanazawa, T. Shiono, T. Ikeda and Y. Nagase, J. Phys. Chem. B, 1998, 102, 2869 CrossRef CAS.
- S. Kurihara, T. Ikeda, T. Sasaki, H. B. Kim and S. Tazuke, J. Chem. Soc., Chem. Commun., 1990, 1751 RSC.
- T. Ikeda, T. Sasaki and H. B. Kim, J. Phys. Chem., 1991, 95, 509 CrossRef CAS.
- T. Sasaki, T. Ikeda and K. Ichimura, Macromolecules, 1992, 25, 3807 CrossRef CAS.
- S. T. Sun, W. M. Gibbons and P. J. Shannon, Liq. Cryst., 1992, 12, 869 CAS.
- W. M. Gibbons, P. J. Shannon, S. T. Sun and B. J. Swetlin, Nature, 1991, 351, 49 CrossRef CAS.
- T. Fischer, L. Lasker, J. Stumpe and S. G. Kostromin, J. Photochem. Photobiol., A, 1994, 80, 453 CrossRef CAS.
- K. Ichimura, Chem. Rev., 2000, 100, 1847 CrossRef CAS.
- Y. L. Yu and T. Ikeda, J. Photochem. Photobiol., C, 2004, 5, 247 CrossRef CAS.
- T. Ikeda, J. Mater. Chem., 2003, 13, 2037 RSC.
- J. Cognard, Mol. Cryst. Liq. Cryst., 1982, S1, 1.
- K. Ichimura, Y. Suzuki, T. Seki, A. Hosoki and K. Aoki, Langmuir, 1988, 4, 1214 CrossRef CAS.
- Y. Kawanishi, T. Tamaki, M. Sakuragi, T. Seki, Y. Suzuki and K. Ichimura, Langmuir, 1992, 8, 2601 CrossRef CAS.
- K. Aoki, T. Seki, M. Sakuragi and K. Ichimura, Makromol. Chem., 1992, 193, 2163 CAS.
- K. Ichimura, Y. Suzuki, T. Seki, Y. Kawanishi and K. Aoki, Makromol. Chem. Rapid Commun., 1989, 10, 5 CrossRef CAS.
- Y. Kawanishi, T. Seki, T. Tamaki, K. lchimura, M. lkeda and K. Aoki, Polym. Adv. Technol., 1990, 1, 311 CrossRef.
- Y. Kawanishi, T. Tamaki, T. Seki, M. Sakuragi, Y. Suzuki, K. Ichimura and K. Aoki, Langmuir, 1991, 7, 1314 CrossRef CAS.
- T. Seki, M. Sakuragi, Y. Kawanishi, Y. Suzuki, T. Tamaki, R. Fukuda and K. Ichimura, Langmuir, 1993, 9, 211 CrossRef CAS.
- T. Seki, M. Sakuragi, Y. Kawanishi, Y. Suzuki, T. Tamaki, K. Ichimura, R. Fukuda and H. Hiramatsu, Thin Solid Films, 1992, 210–211, 836 CrossRef.
- T. Ubukata, T. Seki and K. Ichimura, Macromol. Symp., 1999, 137, 25 CAS.
- O. Yaroschuk, T. Sergan, J. Lindau, S. N. Lee, J. Kelly and L. C. Chien, J. Chem. Phys., 2001, 114, 5330 CrossRef CAS.
- K. Ichimura, H. Akiyama, K. Kudo, N. Ishizuki and S. Yamamura, Liq. Cryst., 1996, 20, 423 CAS.
- K. Ichimura, Y. Hayashi, H. Akiyama and N. Ishizuki, Langmuir, 1993, 9, 3298 CrossRef CAS.
- K. Ichimura, Y. Hayashi, Y. Kawanishi, T. Seki, T. Tamaki and N. Ishizuki, Langmuir, 1993, 9, 857 CrossRef CAS.
- K. Ichimura, Y. Hayashi, H. Akiyama, T. Ikeda and N. Ishizuki, Appl. Phys. Lett., 1993, 63, 449 CrossRef CAS.
- H. Akiyama, M. Momose, K. Ichimura and S. Yamamura, Macromolecules, 1995, 28, 288 CrossRef CAS.
- K. Ichimura, Y. Hayashi, K. Goto and N. Ishizuki, Thin Solid Films, 1993, 235, 101 CrossRef CAS.
- K. Ichimura, Y. Hayashi and N. Ishizuki, Chem. Lett., 1992, 1063 CAS.
- H. Finkelmann, E. Nishikawa, G. G. Pereira and M. Warner, Phys. Rev. Lett., 2001, 87, 015501 CrossRef CAS.
- M. H. Li, P. Keller, B. Li, X. G. Wang and M. Brunet, Adv. Mater., 2003, 15, 569 CrossRef CAS.
- T. Ikeda, M. Nakano, Y. L. Yu, O. Tsutsumi and A. Kanazawa, Adv. Mater., 2003, 15, 201 CrossRef CAS.
- M. Nakano, Y. L. Yu, A. Shishido, O. Tsutsumi, A. Kanazawa, T. Shiono and T. Ikeda, Mol. Cryst. Liq. Cryst., 2003, 398, 1 CrossRef CAS.
- Y. L. Yu, M. Nakano and T. Ikeda, Pure Appl. Chem., 2004, 76, 1467 CrossRef CAS.
- Y. L. Yu, M. Nakano and T. Ikeda, Nature, 2003, 425, 145 CrossRef CAS.
- Y. L. Yu, M. Nakano, A. Shishido, T. Shiono and T. Ikeda, Chem. Mater., 2004, 16, 1637 CrossRef CAS.
- M. Camacho-Lopez, H. Finkelmann, P. Palffy-Muhoray and M. Shelley, Nat. Mater., 2004, 3, 307 CrossRef CAS.
- M. Yamada, M. Kondo, J. I. Mamiya, Y. L. Yu, M. Kinoshita, C. J. Barrett and T. Ikeda, Angew. Chem., Int. Ed., 2008, 47, 4986 CrossRef CAS.
- V. Shibaev, A. Bobrovsky and N. Boiko, J. Photochem. Photobiol., A, 2003, 155, 3 CrossRef CAS.
- B. L. Feringa, N. P. M. Huck and H. A. Vandoren, J. Am. Chem. Soc., 1995, 117, 9929 CrossRef CAS.
- Y. Suzuki, K. Ozawa, A. Hosoki and K. Ichimura, Polym. Bull., 1987, 17, 285 CAS.
- S. Z. Janicki and G. B. Schuster, J. Am. Chem. Soc., 1995, 117, 8524 CrossRef CAS.
- C. Denekamp and B. L. Feringa, Adv. Mater., 1998, 10, 1080 CrossRef CAS.
- B. L. Feringa, R. A. van Delden, N. Koumura and E. Geertsema, Chem. Rev., 2000, 100, 1789 CrossRef CAS.
- I. Willner and S. Rubin, Angew. Chem., Int. Ed. Engl., 1996, 35, 367 CrossRef CAS.
- I. Willner, Acc. Chem. Res., 1997, 30, 347 CrossRef CAS.
- I. Willner, S. Rubin, J. Wonner, F. Effenberger and P. Bauerle, J. Am. Chem. Soc., 1992, 114, 3150 CrossRef CAS.
- S. Rubin and I. Willner, Mol. Cryst. Liq. Cryst., 1994, 246, 201 CrossRef CAS.
- I. Willner and S. Rubin, React. Polym., 1993, 21, 177 CrossRef CAS.
- D. Vomasta, C. Hogner, N. R. Branda and B. Konig, Angew. Chem., Int. Ed., 2008, 47, 7644 CrossRef CAS.
- D. Fujita, M. Murai, T. Nishioka and H. Miyoshi, Biochemistry, 2006, 45, 6581 CrossRef CAS.
- J. H. Harvey and D. Trauner, ChemBioChem, 2008, 9, 191 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS.
- D. Pearson, A. J. Downard, A. Muscroft-Taylor and A. D. Abell, J. Am. Chem. Soc., 2007, 129, 14862 CrossRef CAS.
- M. Harada, M. Sisido, J. Hirose and M. Nakanishi, FEBS Lett., 1991, 286, 6 CrossRef CAS.
- T. Hohsaka, K. Kawashima and M. Sisido, J. Am. Chem. Soc., 1994, 116, 413 CrossRef CAS.
- C. B. Gong, M. H. W. Lam and H. X. Yu, Adv. Funct. Mater., 2006, 16, 1759 CrossRef CAS.
- C. Gomy and A. R. Schmitzer, Org. Lett., 2007, 9, 3865 CrossRef CAS.
- T. Shimoboji, E. Larenas, T. Fowler, S. Kulkarni, A. S. Hoffman and P. S. Stayton, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16592 CrossRef CAS.
- I. Willner and B. Willner, Adv. Mater., 1995, 7, 587 CrossRef CAS.
- I. Willner, M. Liondagan, S. Marxtibbon and E. Katz, J. Am. Chem. Soc., 1995, 117, 6581 CrossRef CAS.
- I. Willner, R. Blonder, E. Katz, A. Stocker and A. F. Buckmann, J. Am. Chem. Soc., 1996, 118, 5310 CrossRef CAS.
- I. Willner, R. Blonder and A. Dagan, J. Am. Chem. Soc., 1994, 116, 9365 CrossRef CAS.
- E. Kaganer, R. Pogreb, D. Davidov and I. Willner, Langmuir, 1999, 15, 3920 CrossRef CAS.
- I. Willner, M. LionDagan and E. Katz, Chem. Commun., 1996, 623 RSC.
- M. Lion-Dagan, E. Katz and I. Willner, J. Chem. Soc., Chem. Commun., 1994, 2741 RSC.
- A. N. Shipway and I. Willner, Acc. Chem. Res., 2001, 34, 421 CrossRef CAS.
- I. Willner, A. Doron, E. Katz, S. Levi and A. J. Frank, Langmuir, 1996, 12, 946 CrossRef CAS.
- I. Willner and R. Blonder, Thin Solid Films, 1995, 266, 254 CrossRef CAS.
- R. J. Byrne, S. E. Stitzel and D. Diamond, J. Mater. Chem., 2006, 16, 1332 RSC.
- B. I. Ipe, S. Mahima and K. G. Thomas, J. Am. Chem. Soc., 2003, 125, 7174 CrossRef CAS.
- O. Pieroni, A. Fissi, N. Angelini and F. Lenci, Acc. Chem. Res., 2001, 34, 9 CrossRef CAS.
- B. L. Feringa, R. A. van Delden, N. Koumura and E. M. Geertsema, Chem. Rev., 2000, 100, 1789 CrossRef CAS.
- H. Yamamoto and A. Nishida, Polym. Int., 1991, 24, 145 CrossRef CAS.
- H. Yamamoto, A. Nishida and T. Kawaura, Int. J. Biol. Macromol., 1990, 12, 257 CrossRef CAS.
- O. Pieroni, A. Fissi, A. Viegi, D. Fabbri and F. Ciardelli, J. Am. Chem. Soc., 1992, 114, 2734 CrossRef CAS.
- A. Fissi, O. Pieroni, F. Ciardelli, D. Fabbri, G. Ruggeri and K. Umezawa, Biopolymers, 1993, 33, 1505 CrossRef CAS.
- R. Behrendt, C. Renner, M. Schenk, F. Q. Wang, J. Wachtveitl, D. Oesterhelt and L. Moroder, Angew. Chem., Int. Ed., 1999, 38, 2771 CrossRef CAS.
- C. Tie, J. C. Gallucci and J. R. Parquette, J. Am. Chem. Soc., 2006, 128, 1162 CrossRef CAS.
- D. G. Flint, J. R. Kumita, O. S. Smart and G. A. Woolley, Chem. Biol., 2002, 9, 391 CrossRef CAS.
- Z. H. Zhang, D. C. Burns, J. R. Kumita, O. S. Smart and G. A. Woolley, Bioconjugate Chem., 2003, 14, 824 CrossRef CAS.
- C. Renner and L. Moroder, ChemBioChem, 2006, 7, 869.
- G. Popova, A. Bobrov and M. Vantsyan, J. Photochem. Photobiol., A, 2008, 196, 246 CrossRef CAS.
- B. R. Malcolm and O. Pieroni, Biopolymers, 1990, 29, 1121 CrossRef CAS.
- H. Menzel, Macromol. Chem. Phys., 1994, 195, 3747 CrossRef CAS.
- H. Menzel, B. Weichart and M. L. Hallensleben, Polym. Bull., 1992, 27, 637 CrossRef CAS.
- J. Auernheimer, C. Dahmen, U. Hersel, A. Bausch and H. Kessler, J. Am. Chem. Soc., 2005, 127, 16107 CrossRef CAS.
- H. Asanuma, X. Liang, H. Nishioka, D. Matsunaga, M. Liu and M. Komiyama, Nat. Protoc., 2007, 2, 203 Search PubMed.
- M. Z. Liu, H. Asanuma and M. Komiyama, J. Am. Chem. Soc., 2006, 128, 1009 CrossRef CAS.
- G. Hayashi, M. Hagihara, C. Dohno and K. Nakatani, J. Am. Chem. Soc., 2007, 129, 8678 CrossRef CAS.
- H. Z. Kang, H. P. Liu, J. A. Phillips, Z. H. Cao, Y. Kim, Y. Chen, Z. Y. Yang, J. W. Li and W. H. Tan, Nano Lett., 2009, 9, 2690 CrossRef CAS.
- C. Alvarez-Lorenzo, L. Bromberg and A. Concheiro, Photochem. Photobiol., 2009, 85, 848 CrossRef CAS.
- J. F. Mano, Adv. Eng. Mater., 2008, 10, 515 CrossRef CAS.
- Y. Zhao, J. Mater. Chem., 2009, 19, 4887 RSC.
- X. Tong, G. Wang, A. Soldera and Y. Zhao, J. Phys. Chem. B, 2005, 109, 20281 CrossRef CAS.
- G. Wang, X. Tong and Y. Zhao, Macromolecules, 2004, 37, 8911 CrossRef CAS.
- H. i. Lee, W. Wu, J. K. Oh, L. Mueller, G. Sherwood, L. Peteanu, T. Kowalewski and K. Matyjaszewski, Angew. Chem., Int. Ed., 2007, 46, 2453 CrossRef CAS.
- X. K. Liu and M. Jiang, Angew. Chem., Int. Ed., 2006, 45, 3846 CrossRef CAS.
- K. Han, W. Su, M. C. Zhong, Q. Yan, Y. H. Luo, Q. J. Zhang and Y. M. Li, Macromol. Rapid Commun., 2008, 29, 1866 CrossRef CAS.
- L. Lin, Z. Yan, J. S. Gu, Y. Y. Zhang, Z. Feng and Y. L. Yu, Macromol. Rapid Commun., 2009, 30, 1089 CrossRef CAS.
- R. H. Bisby, C. Mead and C. C. Morgan, Biochem. Biophys. Res. Commun., 2000, 276, 169 CrossRef CAS.
- S. Yagai, T. Karatsu and A. Kitamura, Chem.–Eur. J., 2005, 11, 4054 CrossRef CAS.
- X. M. Liu, B. Yang, Y. L. Wang and J. Y. Wang, Biochim. Biophys. Acta, Biomembr., 2005, 1720, 28 CrossRef CAS.
- Y. Ohya, Y. Okuyama, A. Fukunaga and T. Ouchi, Supramol. Sci., 1998, 5, 21 CrossRef CAS.
- S. Deshmukh, L. Bromberg, K. A. Smith and T. A. Hatton, Langmuir, 2009, 25, 3459 CrossRef CAS.
- F. M. Winnik and S. T. A. Regismond, Colloids Surf., A, 1996, 118, 1 CrossRef CAS.
- R. Borrega, C. Tribet and R. Audebert, Macromolecules, 1999, 32, 7798 CrossRef CAS.
- S. Khoukh, R. Oda, T. Labrot, P. Perrin and C. Tribet, Langmuir, 2007, 23, 94 CrossRef CAS.
- S. Khoukh, C. Tribet and P. Perrin, Colloids Surf., A, 2006, 288, 121 CrossRef CAS.
- G. Pouliquen and C. Tribet, Macromolecules, 2006, 39, 373 CrossRef CAS.
- J. Eastoe and A. Vesperinas, Soft Matter, 2005, 1, 338 RSC.
- S. Sortino, Photochem. Photobiol. Sci., 2008, 7, 911 RSC.
- Y. Klichko, M. Liong, E. Choi, S. Angelos, A. E. Nel, J. F. Stoddart, F. Tamanoi and J. I. Zink, J. Am. Ceram. Soc., 2009, 92, S2 CrossRef CAS.
- S. Angelos, E. Choi, F. Vogtle, L. De Cola and J. I. Zink, J. Phys. Chem. C, 2007, 111, 6589 CrossRef CAS.
- J. Lu, E. Choi, F. Tamanoi and J. I. Zink, Small, 2008, 4, 421 CrossRef CAS.
- N. G. Liu, D. R. Dunphy, P. Atanassov, S. D. Bunge, Z. Chen, G. P. Lopez, T. J. Boyle and C. J. Brinker, Nano Lett., 2004, 4, 551 CrossRef CAS.
- K. Weh, M. Noack, K. Hoffmann, K. P. Schroder and J. Caro, Microporous Mesoporous Mater., 2002, 54, 15 CrossRef CAS.
- M. Shibayama and T. Tanaka, Adv. Polym. Sci., 1993, 109, 1 CAS.
- R. Plummer, D. J. T. Hill and A. K. Whittaker, Macromolecules, 2006, 39, 8379 CrossRef CAS.
- L. D. Taylor and L. D. Cerankowski, J. Polym. Sci., Part A: Polym. Chem., 1975, 13, 2551 Search PubMed.
- C. K. Chee, S. Rimmer, D. A. Shaw, I. Soutar and L. Swanson, Macromolecules, 2001, 34, 7544 CrossRef CAS.
- S. Furyk, Y. J. Zhang, D. Ortiz-Acosta, P. S. Cremer and D. E. Bergbreiter, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 1492 CrossRef CAS.
- B. Ray, Y. Isobe, K. Matsumoto, S. Habaue, Y. Okamoto, M. Kamigaito and M. Sawamoto, Macromolecules, 2004, 37, 1702 CrossRef CAS.
- Y. Xia, X. C. Yin, N. A. D. Burke and H. D. H. Stover, Macromolecules, 2005, 38, 5937 CrossRef CAS.
- K. Sumaru, M. Kameda, T. Kanamori and T. Shinbo, Macromolecules, 2004, 37, 4949 CrossRef CAS.
- K. Sumaru, K. Ohi, T. Takagi, T. Kanamori and T. Shinbo, Langmuir, 2006, 22, 4353 CrossRef CAS.
- A. Garcia, M. Marquez, T. Cai, R. Rosario, Z. B. Hu, D. Gust, M. Hayes, S. A. Vail and C. D. Park, Langmuir, 2007, 23, 224 CrossRef CAS.
- E. U. Kulawardana, T. Kuruwita-Mudiyanselage and D. C. Neckers, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3318 CrossRef CAS.
- A. Szilagyi, K. Sumaru, S. Sugiura, T. Takagi, T. Shinbo, M. Zrinyi and T. Kanamori, Chem. Mater., 2007, 19, 2730 CrossRef CAS.
- J. Edahiro, K. Sumaru, T. Takagi, T. Shinbo and T. Kanamori, Langmuir, 2006, 22, 5224 CrossRef CAS.
- A. Desponds and R. Freitag, Biotechnol. Bioeng., 2005, 91, 583 CrossRef CAS.
- N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821 RSC.
- I. Tomatsu, A. Hashidzume and A. Harada, Macromolecules, 2005, 38, 5223 CrossRef CAS.
- T. L. Sun, L. Feng, X. F. Gao and L. Jiang, Acc. Chem. Res., 2005, 38, 644 CrossRef CAS.
- N. Katsonis, M. Lubomska, M. M. Pollard, B. L. Feringa and P. Rudolf, Prog. Surf. Sci., 2007, 82, 407 CrossRef CAS.
- R. Rosario, D. Gust, M. Hayes, F. Jahnke, J. Springer and A. A. Garcia, Langmuir, 2002, 18, 8062 CrossRef CAS.
- R. Rosario, D. Gust, A. A. Garcia, M. Hayes, J. L. Taraci, T. Clement, J. W. Dailey and S. T. Picraux, J. Phys. Chem. B, 2004, 108, 12640 CrossRef CAS.
- S. K. Oh, M. Nakagawa and K. Ichimura, J. Mater. Chem., 2002, 12, 2262 RSC.
- K. Ichimura, S. K. Oh and M. Nakagawa, Science, 2000, 288, 1624 CrossRef CAS.
- G. Moller, M. Harke, H. Motschmann and D. Prescher, Langmuir, 1998, 14, 4955 CrossRef.
- T. Kinoshita, J. Photochem. Photobiol., B, 1998, 42, 12 CrossRef CAS.
- T. Seki, Supramol. Sci., 1996, 3, 25 CrossRef CAS.
- T. Seki, H. Sekizawa, R. Fukuda, T. Tamaki, M. Yokoi and K. Ichimura, Polym. J., 1996, 28, 613 CrossRef CAS.
- T. Seki and T. Tamaki, Chem. Lett., 1993, 1739 CrossRef CAS.
- R. Vilanove, H. Hervet, H. Gruler and F. Rondelez, Macromolecules, 1983, 16, 825 CrossRef CAS.
- D. M. He, H. Susanto and M. Ulbricht, Prog. Polym. Sci., 2009, 34, 62 CrossRef CAS.
- S. Shinkai, T. Nakaji, T. Ogawa, K. Shigematsu and O. Manabe, J. Am. Chem. Soc., 1981, 103, 111 CrossRef.
- T. Shimidzu and M. Yoshikawa, J. Membr. Sci., 1983, 13, 1 CrossRef CAS.
- K. Kimura, M. Sumida and M. Yokoyama, Chem. Commun., 1997, 1417 RSC.
- K. Kimura, H. Sakamoto and R. M. Uda, Macromolecules, 2004, 37, 1871 CrossRef CAS.
- S. Oosaki, H. Hayasaki, Y. Sakurai, S. Yajima and K. Kimura, Chem. Commun., 2005, 5226 RSC.
- K. Ishihara, N. Hamada, S. Kato and I. Shinohara, J. Polym. Sci., Part A: Polym. Chem., 1984, 22, 121 Search PubMed.
- K. Ishihara and I. Shinohara, J. Polym. Sci., Part C: Polym. Lett., 1984, 22, 515 Search PubMed.
- T. Sata, Y. Shimokawa and K. Matsusaki, J. Membr. Sci., 2000, 171, 31 CrossRef CAS.
- D. J. Chung, Y. Ito and Y. Imanishi, J. Appl. Polym. Sci., 1994, 51, 2027 CrossRef.
- K. Kimura, H. Sakamoto and T. Nakamura, J. Nanosci. Nanotechnol., 2006, 6, 1741 CrossRef CAS.
- A. Nayak, H. W. Liu and G. Belfort, Angew. Chem., Int. Ed., 2006, 45, 4094 CrossRef CAS.
- I. Vlassiouk, C. D. Park, S. A. Vail, D. Gust and S. Smirnov, Nano Lett., 2006, 6, 1013 CrossRef CAS.
- A. Higuchi, A. Hamamura, Y. Shindo, H. Kitamura, B. O. Yoon, T. Mori, T. Uyama and A. Umezawa, Biomacromolecules, 2004, 5, 1770 CrossRef CAS.
- J. Edahiro, K. Sumaru, Y. Tada, K. Ohi, T. Takagi, M. Kameda, T. Shinbo, T. Kanamori and Y. Yoshimi, Biomacromolecules, 2005, 6, 970 CrossRef CAS.
- S. Yagai and A. Kitamura, Chem. Soc. Rev., 2008, 37, 1520 RSC.
- M. S. Vollmer, T. D. Clark, C. Steinem and M. R. Ghadiri, Angew. Chem., Int. Ed., 1999, 38, 1598 CrossRef CAS.
- S. Yagai, T. Nakajima, K. Kishikawa, S. Kohmoto, T. Karatsu and A. Kitamura, J. Am. Chem. Soc., 2005, 127, 11134 CrossRef CAS.
- J. Cusido, E. Deniz and F. M. Raymo, Eur. J. Org. Chem., 2009, 2031 CrossRef.
- I. Yildiz, E. Deniz and F. M. Raymo, Chem. Soc. Rev., 2009, 38, 1859 RSC.
- L. Y. Zhu, M. Q. Zhu, J. K. Hurst and A. D. Q. Li, J. Am. Chem. Soc., 2005, 127, 8968 CrossRef CAS.
- I. L. Medintz, S. A. Trammell, H. Mattoussi and J. M. Mauro, J. Am. Chem. Soc., 2004, 126, 30 CrossRef CAS.
- M. Q. Zhu, L. Y. Zhu, J. J. Han, W. W. Wu, J. K. Hurst and A. D. Q. Li, J. Am. Chem. Soc., 2006, 128, 4303 CrossRef CAS.
- L. Y. Zhu, W. W. Wu, M. Q. Zhu, J. J. Han, J. K. Hurst and A. D. Q. Li, J. Am. Chem. Soc., 2007, 129, 3524 CrossRef CAS.
- J. Chen, F. Zeng, S. Z. Wu, J. Su and Z. Tong, Small, 2009, 5, 970 CrossRef CAS.
- E. Jares-Erijman, L. Giordano, C. Spagnuolo, K. Lidke and T. M. Jovin, Mol. Cryst. Liq. Cryst., 2005, 430, 257 CrossRef CAS.
- J. Folling, S. Polyakova, V. Belov, A. van Blaaderen, M. L. Bossi and S. W. Hell, Small, 2008, 4, 134 CrossRef.
- J. Li, G. Speyer and O. F. Sankey, Phys. Rev. Lett., 2004, 93, 248302 CrossRef.
- G. Speyer, J. Li and O. F. Sankey, Phys. Status Solidi B, 2004, 241, 2326 CrossRef CAS.
- N. Katsonis, T. Kudernac, M. Walko, S. J. van der Molen, B. J. van Wees and B. L. Feringa, Adv. Mater., 2006, 18, 1397 CrossRef CAS.
- T. Kawai, Y. Nakashima, T. Kunitake and M. Irie, Curr. Appl. Phys., 2005, 5, 139 CrossRef.
- T. Tsujioka, K. Masui and F. Otoshi, Appl. Phys. Lett., 2004, 85, 3128 CrossRef CAS.
- J. He, F. Chen, P. A. Liddell, J. Andreasson, S. D. Straight, D. Gust, T. A. Moore, A. L. Moore, J. Li, O. F. Sankey and S. M. Lindsay, Nanotechnology, 2005, 16, 695 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |