
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn efficient NiCoSe4/NiCo-LDH/CF catalyst for the co-production of value-added formate and hydrogen via selective methanol electro-oxidation†
Jiaxin
Li
,
Hongmei
Yu
 *,
Jun
Chi
,
Xu
Luo
,
Tongzhou
Li
and
Zhigang
Shao
*,
Jun
Chi
,
Xu
Luo
,
Tongzhou
Li
and
Zhigang
Shao
Fuel Cell System and Engineering Laboratory, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, Liaoning, China. E-mail: hmyu@dicp.ac.cn
First published on 6th September 2024
Abstract
The sluggish kinetics of the anodic oxygen evolution reaction (OER) and the valueless product O2 are tricky issues for water splitting. Replacing the OER with a more thermodynamically favorable selective methanol oxidation reaction (SMOR) to combine with the hydrogen evolution reaction can not only lower the applied voltage but also simultaneously produce value-added formate in the anode and hydrogen energy in the cathode. Herein, a mirror-like heterostructure of NiCoSe4@NiCo-LDH supported on cobalt foam (NCS/CF) was synthesized by partial selenidation of NiCo-LDH to facilitate the SMOR. Experimental tests and theoretical calculations revealed the “shuttle-like” role of Se in NCS/CF, by which electrons of Ni were first transferred to Se and then to Co. Moreover, the superiority of the bimetallic compound was also proved by comparing the activity of NCS/CF to that of the counterparts NiSe2@Ni(OH)2/CF (NS/CF) and CoSe2@Ni(OH)2/CF (CS/CF). Owing to the unique electronic effect of the strongly coupled hetero-interface and the bimetallic synergism, more accessible active sites, fast charge transfer ability, facilitated catalytic kinetics, and modulated electron redistribution were achieved. Therefore, improved SMOR performance was obtained with ∼100% faradaic efficiency of methanol-to-formate. The potential at 10 and 100 mA cm−2 was only 1.274 and 1.432 V vs. RHE, respectively, outperforming most existing catalysts. In addition, the two-electrode electrolyzer of methanol-assisted water electrolysis with NCS/CF as both the anode and cathode only required an applied cell voltage of 1.380 V to reach 10 mA cm−2, 253 mV less than that of pure water splitting. Meanwhile, outstanding catalytic stability was achieved by continuously delivering a current density of 100 mA cm−2 for 100 hours without obvious attenuation. The inference of the “shuttle-like” role of Se will provide new insight into the electronic redistribution in the NiCo-LDH derivative that is incorporated with foreign atoms. The integration of methanol upgradation and hydrogen generation exploits an energy-saving way to simultaneously obtain value-added organic substances and green hydrogen.
1. Introduction
Green hydrogen generated from water electrolysis by using renewable energy is significant for tackling the energy crisis and mitigating environmental pollution.1 However, the inevitable bottleneck of water electrolysis is the multistep proton-coupled electron transfer process of the anodic oxygen evolution reaction (OER), which leads to intrinsic slow kinetics and large overpotential.2,3 Though the equilibrium potential of water electrolysis is 1.23 V, the applied voltage even increases to 1.8–2.0 V in practical use to conquer the extra resistance and provide sufficient thermodynamic driving force.4 In addition, the anodic product O2 is less valuable and has the risk of explosion when mixed with H2 produced in the cathode.5 An effective way to address these drawbacks is to replace the OER with a more thermodynamically favorable organic upgrading reaction, which can simultaneously lower the overpotential and yield value-added products.6,7 In recent years, the nucleophile oxidation reaction (NOR) has been widely studied as a substitute for the OER,8,9 where nucleophilic groups (hydroxyl, aldehyde, amino groups, etc.) containing active hydrogen in organic substrates (alcohols, aldehydes, amines, etc.) act as the reaction site.10–13 Therein, the electro-reforming of methanol (CH3OH) to formate (HCOOH) attracts much attention due to the simple structure, high solubility, and good oxidization reactivity of methanol molecules.14–16 Moreover, the equilibrium potential of partial methanol oxidation to formate involving a 4-electron transfer process is 0.103 V vs. standard hydrogen electrode (SHE), largely reducing the energy input.17 Most attractively, the industrial value of formate (1300 USD per t) is much higher than that of methanol (350 USD per t),18,19 meanwhile, the electro-synthesis of formate under mild conditions is superior to the traditional multi-step and high energy-consuming industrial procedures.20 Therefore, the selective methanol oxidation reaction (SMOR) is an ideal alternative to the OER for the energy-saving co-generation of value-added chemicals and green hydrogen.Since noble metal-based catalysts are more inclined towards completely oxidizing methanol to the environmentally unfriendly CO2,21 non-noble metal-based catalysts are preferred to be used for the SMOR because their moderate activity is beneficial to control the methanol oxidation process.22–24 Among them, NiCo layered double hydroxide (LDH) and its derivatives were proved efficient for the SMOR with good activity and selectivity.25–28 NiCo-LDH nanosheets were reported to have 95% faradaic efficiency of methanol electrooxidation to formate (FEformate).27 The in situ formed Ni3+ species in Ni-containing hydroxides were believed to be the active sites. The 2D properties of LDH provide restrained space for the directed growth of functional materials via the additional ultra-edge strategy.29 However, LDH faces the restrictions of relatively low electronic conductivity.30 Considering these properties, strategies such as phase transition and heterostructure construction were employed to modify NiCo-LDH catalysts for further improving the activity. For example, a novel Ni(OH)2/MnCO3 heterostructure catalyst was fabricated for methanol oxidation; the synergism derived from the Jahn–Teller distortion effect overcomes the disadvantage of inactive MnCO3 and low intrinsic conductivity of Ni(OH)2, thereby yielding enhanced activity for methanol oxidation.31 In another research study, hetero-nanosheet arrays of CoxP@NiCo-LDH were synthesized by a combined electrodeposition and phosphorization method.32 Benefiting from the fast charge transfer induced by CoxP and the abundant active sites in the hetero-interface, a current density of 10 mA cm−2 could be yielded at a small potential of 1.24 V vs. RHE, the FEformate reached nearly 100%. Similarly, NiCoxP@NiCo-LDH nanosheets were also found to be excellent for the SMOR due to the synergistic effect between NiCoxP and NiCo-LDH.33 Nevertheless, these studies of NiCo-LDH derivatives mainly focused on the synergism of hetero-components, and the role of incorporated foreign elements (like P) in metallic compounds was not fully discussed. In particular, in bimetallic NiCo compounds, the similar electronic structures of Ni and Co make valence electron substitutions and charge transitions more likely to occur, which yield a synergistic effect to induce better electrochemical properties than monometallic compounds.34 Thus, the influence of the incorporated foreign element on the electronic interaction of bimetallic NiCo in NiCo-LDH derivatives is attractive to explore.
In recent years, metal chalcogenides have gained increasing attention as electrocatalysts due to their unique physical and chemical properties.35,36 Compared to O and S elements in the same main group, the mild electronegativity of Se permits a facile artificial modulation of electronic configuration and bonding state between Se and metal atoms, allowing for an appealing electrochemical reaction.37 In addition, the electronic properties of transition metal selenides (TMSe) are reported to be influenced by the filling of the d-bands of the transition metal.38,39 Meanwhile, TMSe displays more metallic properties since the 3d orbital of Se is involved in bonding with metal atoms, enhancing the ability of electron transport and charge transfer in the electrocatalytic process.40,41 Therefore, in this work, we synthesized a heterostructure of NiCoSe4@NiCo-LDH on cobalt foam (NCS/CF) via a hydrothermal method and the subsequent selenidation treatment for the SMOR and methanol–water hybrid electrolysis. Benefiting from the partial selenidation, largely exposed active sites, accelerated electrical conductivity, tuned electronic structure, and strong synergism of the coupled hetero-interface were achieved. Therefore, a high selectivity (∼100%) of methanol electrooxidation to value-added formate was achieved, and current densities of 10 and 100 mA cm−2 were reached at pretty low potentials of 1.274 and 1.432 V vs. RHE, respectively. In addition, the two-electrode system equipped with NCS/CF as both the anode and cathode delivered 10 mA cm−2 at a cell voltage of 1.380 V, which was 253 mV less than that of overall water splitting. A long-term stability of continuous 100 h operation at the current density of 100 mA cm−2 was also achieved.
2. Experimental section
2.1 Synthesis of materials
2.2 Structural characterization
The crystal structures of the electrodes were characterized by X-ray diffraction (XRD, Empyrean-100) using Cu Kα radiation (λ = 0.1541 nm) with 2θ = 10o–90o. The Raman spectra were collected from a RENISHAW NanoWizard with 532 nm of laser beam. The morphology of the obtained electrodes was examined by field-emission scanning electron microscopy, energy dispersive X-ray spectroscopy (SEM & EDS, JEOL JSM-IT800), and transmission electron microscopy (TEM, JEOL JEM2200FS). X-ray photoelectron spectroscopy (XPS) was performed in a Shimadzu AXIS SUPRA+ spectrometer with a monochromatic Al Kα (hν = 1486.6 eV) X-ray resource.2.3 Electrochemical measurements and product analysis
The catalytic activity of methanol oxidation and electrolysis were tested in three-electrode and two-electrode systems by cyclic voltammetry (CV) and linear scan voltammetry (LSV) curves. The catalytic kinetics were explored from the Tafel slope and electrochemical impedance. The catalytic stability was examined by the chronopotentiometry test. The electrochemical surface area (ECSA) was determined by calculating the double-layer capacitance (Cdl). The detailed testing device and methods are shown in the ESI.†To analyze the products, chronoamperometry (CA) curves were recorded for 6 h in 1 M KOH solution containing 1 M methanol at various potentials of 1.38, 1.43, 1.48, 1.53, 1.58, 1.63, and 1.68 V vs. RHE. The electrolyte after the CA tests was collected and analyzed by 1H nuclear magnetic resonance (NMR) (Avance III 400 MHz, Bruker BioSpin) and Ion Chromatography (IC) (Eco IC 1, Metrohm) equipped with a Metrosep A Supp 5-250/4.0 anion chromatographic column. The Faraday efficiency of methanol to formate was calculated using the following equation.42
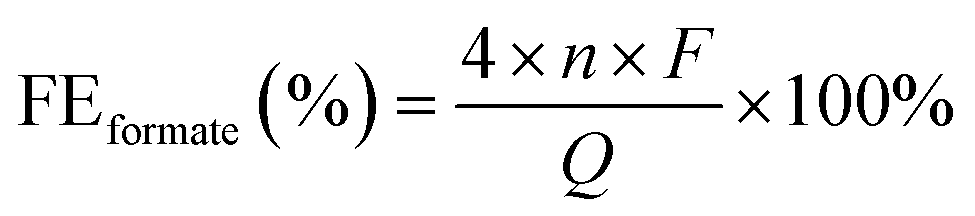
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485C mol−1), and Q is the total charge passed (C).
485C mol−1), and Q is the total charge passed (C).
2.4 Theoretical calculation method
Density functional theory (DFT) calculation was performed to give a deep insight into the electronic interaction of catalysts and the role of Se atoms. The detailed calculation method is provided in the ESI.†3. Results and discussion
3.1 Composition and microstructures
The target electrode NiCoSe4@NiCo-LDH loaded on Co foam (NCS/CF) was prepared by combined hydrothermal and selenidation methods. In the former process, NiCo-LDH was synthesized on cleaned CF via a hydrothermal method involving NH4F as a chelating agent and CO(NH2)2 as an alkaline environment creator, which has been commonly used in many other NiCo-LDH-relevant literature studies.43–45 In the latter process, a selenidation treatment was performed to realize the partial phase transition of NiCo-LDH. The other two counterparts, NiSe2@Ni(OH)2 on Co foam (NS/CF) and CoSe2@Co(OH)2 on Co foam (CS/CF), were fabricated in the same way, except for the difference in the metal salt. The crystal structures of all the as-prepared electrodes were probed by X-ray diffraction (XRD) (Fig. 1a). The well-identified typical diffraction peaks of cubic CoSe2 (PDF#09-0234),46 NiSe2 (PDF#41-1495),47 and NiCoSe4 (PDF#97-062-4483)48 were obviously observed in CS/CF, NS/CF, and NCS/CF, respectively. However, no characteristic peaks of hydroxide were detected in all three samples. Thus, we adopted the Raman scattering technique to collect information on molecular vibration or rotation to further probe the molecular structure of electrodes (Fig. 1b). In CS/CF, the Se–Se stretching mode of cubic CoSe2 at 190 cm−1 was detected.49 The peaks at 469, 514, and 670 cm−1 also confirmed the constitution of cubic CoSe2 phases.50,51 In NS/CF, the characteristic peaks located at around 151, 170, 214, and 242 cm−1 corresponded to NiSe2.52 They represent the Tg (libration), Eg (libration), Ag (stretching), and Tg (stretching) modes of NiSe2, respectively.53 Similarly, Guo et al. also used the Raman technique to study the structural transformation of Ni3Se4/NiSe2.54 They found that the peaks at low wavenumbers (149 and 169 cm−1) could be attributed to the libration mode and the peaks at high wavenumbers (212 and 240 cm−1) corresponded to the stretching modes of selenide. In another research study, Wang et al. obtained the characteristic peak at 210 cm−1 in the Raman spectrum, the authors attributed it to the stretching modes of cubic NiSe2,55 whereas in NCS/CF, both the Raman characteristic peaks of NiSe2 and CoSe2 were observed. The peaks at 152 and 175 cm−1 were assigned to the Tg and Eg modes of NiSe2, and the peaks centered at 192 and 671 were attributed to the crystalline structure of CoSe2.56 It was noteworthy that weak Raman peaks of Co–OH (610 cm−1),57 Ni–OH (527 cm−1),58 and NiCo-LDH involving the stretching modes of Co–O (513 cm−1) and Ni–O (468 cm−1)59,60 can be observed in CS/CF, NS/CF, and NCS/CF, respectively. This result confirmed that a small portion of the metal hydroxide remained after the partial selenidation treatment. Therefore, no obvious diffraction peaks of metal hydroxide were detected in XRD patterns. Moreover, the XRD signal of the cobalt foam (CF) support was almost negligible compared to that of the strong crystalline selenide due to its weak diffraction peaks (Fig. S1†).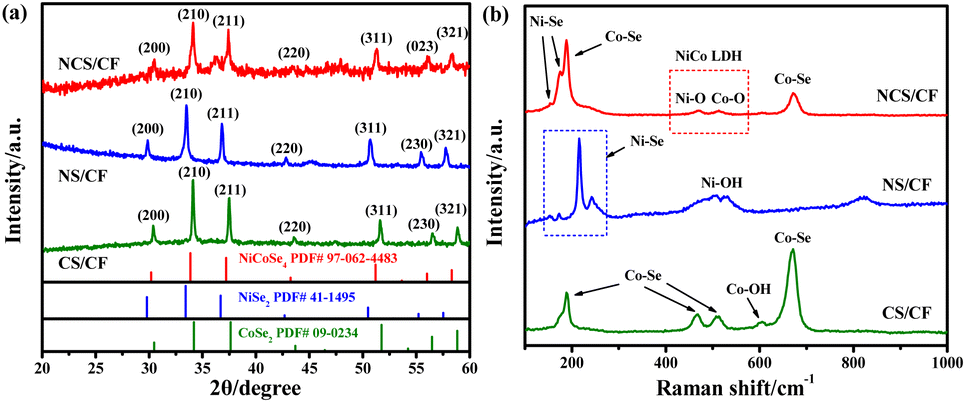 | ||
| Fig. 1 (a) XRD patterns and (b) Raman spectra of NCS/CF, NS/CF, and CS/NF. | ||
The fine morphology features of all the samples were studied via the electron microscopy technique. The cleaned CF support displayed a smooth skeleton structure with open pores (Fig. S2†). When loaded with hybrid metal hydroxide and selenide, unique morphologies of urchin-like CS/CF (Fig. 2a), hydrangea-like NS/CF (Fig. 2b), and mirror-like NCS/CF (Fig. 2c) were obtained. The magnified scanning electron microscopy (SEM) images showed that the urchin-like CS/CF was composed of individual nanoneedles with lengths of several microns and diameters ranging from ∼50 nm (needle tip) to ∼300 nm (needle root) (Fig. 2d and S3†). All the elements present, Co, Se, and O, were uniformly distributed throughout the CS/CF (Fig. S4†).
 | ||
| Fig. 2 SEM images of CS/CF (a and d), NS/CF (b and e), and NCS/CF (c, f and g). TEM image of NCS/CF (h). HRTEM image of NCS/CF (i). Elemental mapping and the overlapping images of NCS/CF (j–n). | ||
In addition to surface oxidation, the O element was also found to come from the remaining Co(OH)2 after partial selenidation. The hydrangea-like NS/CF was found to consist of interconnected nanosheets of Ni(OH)2 and the surface-embedded NiSe2 nanoparticles (NPs) (Fig. 2e and S5†). The distribution of the elements Ni, Se, and O evidenced the composition and heterostructure (Fig. S6†). As for the mirror-like NCS/CF, it seemed to combine the morphological characteristics of both CS/CF and NS/CF. The central NiCo-LDH nanosheets and the numerous surrounding NiCoSe4 nanoneedles formed the unique mirror-like structure (Fig. 2f and S7†). The diameter of NiCo-LDH nanosheets was similar to that of Ni(OH)2. Nevertheless, the length of NiCoSe4 nanoneedles (hundreds of nanometers) was shorter than that of CS/CF nanoneedles (Fig. 2g). By further observation using a transmission electron microscope (TEM), the NiCoSe4 nanoneedles were revealed to be composed of connected NPs with an average particle size of ∼58 nm (Fig. 2h). The spacing of the lattice fringe on the whole NPs was measured as 0.26 nm in high-resolution TEM (HRTEM) images, corresponding to the (2, 1, 0) plane of NiCoSe4 (Fig. S8†).61,62 Meanwhile, the selected area electron diffraction (SAED) pattern showed a regular single-crystal diffraction lattice with a relatively consistent lattice direction (Fig. S9†). Clear diffraction spots on crystal planes including (2, 1, 0), (−2, 5, 0), and (−4, 4, 0) can be observed. Combining the analysis of HRTEM and SAED, it can be proven that the NPs were constituted of a single crystal of NiCoSe4, mainly exposing the crystal plane of (2, 1, 0). The corresponding EDS mapping and line scan images confirmed the main constituent elements to be Ni, Co, and Se with negligible O (Fig. S10 and 11†). Moreover, an interplanar spacing of 0.20 nm was also measured in another HRTEM image (Fig. 2i), which was attributed to the (1, 0, 7) crystal plane of NiCo-LDH (PDF#33-0429).63,64 Note that the intensity of the (1, 0, 7) crystal plane was pretty low, this was the reason why the NiCo-LDH diffraction peaks cannot be detected in the XRD pattern. The elemental distribution images of the entire NCS/CF can clearly show the unique mirror-like heterostructure (Fig. 2j–n and S12†). Ni and Co elements were distributed on the whole surface of NCS/CF, while the O element was concentrated in the nanosheets at the centre, representing the NiCo-LDH. In sharp comparison, the Se element was only distributed in the surrounding area, corresponding to NiCoSe4. The EDS line scan images further demonstrated that the NCS/CF heterostructure was composed of surrounding selenides and central hydroxides (Fig. S13†). By quantitatively identifying all the concerned elements by EDS patterns (Fig. S14†), the atomic percentage of Ni, Co, Se, and O was 17.05, 16.34, 35.71, and 26.83%, respectively (Table S1†). The atomic ratio of Ni/Co/Se was close to 1/1/2, which was nearly consistent with the nominal composition. The O element came from both LDH and surface oxidation, while the C element was derived from the conductive adhesive used in the test.
The chemical environment and chemical states of all the samples were explored by the X-ray photoelectron spectroscopy (XPS) technique to study the role of the incorporated foreign element Se in the electron interaction of NCS/CF. In high-resolution Ni 2p and Co 2p spectra (Fig. 3a and b), two spin–orbit doublets of 2p1/2 and 2p3/2 components were observed, and oxidative state Ni and Co accompanied by the corresponding satellite peaks were fitted, indicating that Ni and Co mainly existed in the form of divalent and trivalent ions. It can be seen that some chemical shifts occurred because of the distinct chemical environment derived from the different compositions and structures. For NS/CF, the peaks of Ni2+ were located at 853.04 and 870.34 eV, and the Ni3+ peaks were situated at 855.48 and 873.91 eV (Table S2†),65 while the binding energies (BEs) of Ni2+ and Ni3+ for NCS/CF both shifted to higher values. The shift of BE can be used to evaluate the changes in electron density.66 It is known that the atomic nucleus and the surrounding electrons have a certain BE through the Coulomb effect, in the meantime, the BE is also influenced by the outer electrons.67 Thus, the increased BE of Ni indicated the decreasing electron density of Ni atom.68 That is to say Ni atoms in NCS/CF lost more electrons than the Ni atoms in NS/CF after the incorporation of Co. Then we found that all the peaks of Co2+ and Co3+ for NCS/CF are located at a slightly lower BE compared with CS/CF (Table S3†), revealing that Co gained electrons in the heterostructure NCS/CF. A similar phenomenon was also discovered in another study of NiCoSe4@CNFs.69 As for Se 3d spectra, the signal peaks of Se 3d5/2, Se 3d3/2, and oxidative state Se can be fitted in all three samples (Fig. 3c). The BEs of Se 3d5/2 and Se 3d3/2 in NS/CF were 54.51 and 55.35 eV (Table S4†),70 while the corresponding values in CS/CF were 54.86 and 55.62 eV due to the different chemical environments of NS/CF and CS/CF.71 It was an interesting finding that each sub-deconvolution peak of Se 3d in NCS/CF was located between the corresponding peaks of NS/CF and CS/CF.72 In particular, the BE of Se 3d peaks for NCS/CF was higher than those of NS/CF but lower than those of CS/CF. This chemical shift indicated that Se atoms in NCS/CF received more electrons in comparison with Se atoms in CS/CF after the introduction of Ni. But when compared with NS/CF, the Se atoms in NCS/CF showed a trend of losing electrons due to the existence of Co. Therefore, it was inferred that the Se atom in NCS/CF played a shuttle-like role where electrons of Ni were first transferred to Se and then flowed to Co. This conclusion revealed the effect of Se on the electron structure modulation in bimetallic NiCo in NiCo-LDH derivatives. Last but not least, metal–O (M–O), metal–OH (M–OH), and surface-adsorbed water (H2Oads) were also fitted in O 1s spectra (Fig. 3d), verifying the existence of metal hydroxide.73 The larger intensity of the M–OH peak in NCS/CF indicated that more metal hydroxides were exposed in the middle nanosheets.
 | ||
| Fig. 3 (a) XPS spectra of the Ni 2p region for NCS/CF and NS/CF. (b) XPS spectra of the Co 2p region for NCS/CF and CS/CF. XPS spectra of the Se 3d region (c) and O 1s region (d) for NCS/CF, NS/CF, and CS/CF. | ||
3.2 Evaluation of the electrocatalytic performance
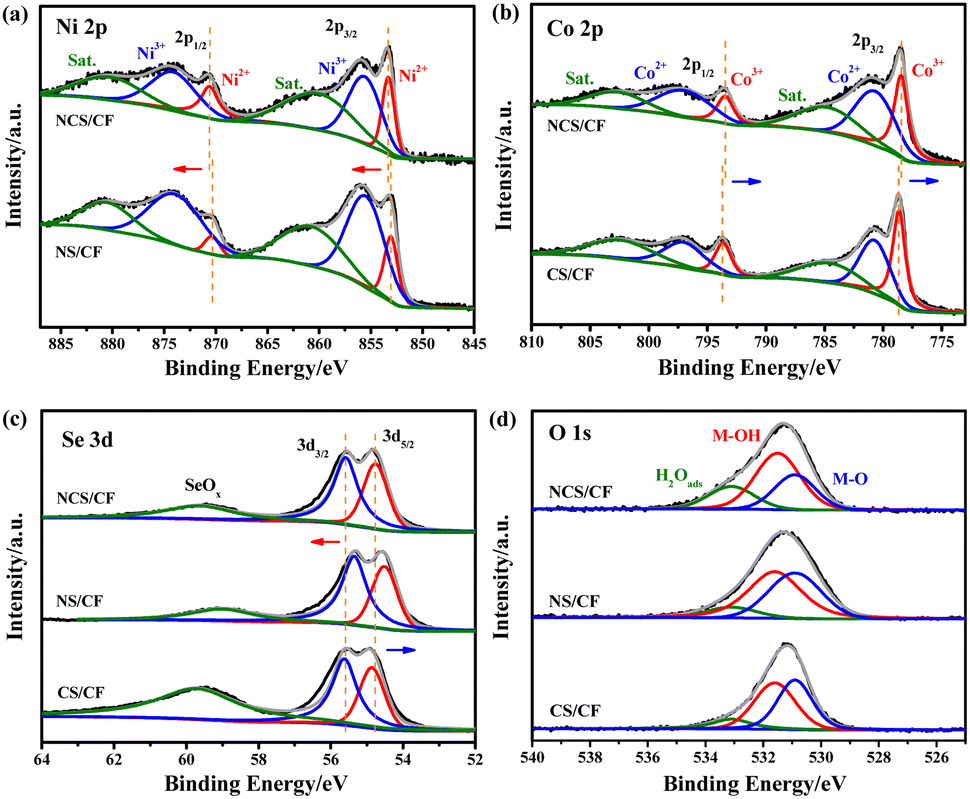
To evaluate the electrocatalytic performance for the SMOR, the as-prepared samples were directly used as self-supported electrodes in a typical three-electrode system. Cyclic voltammetry (CV) curves were tested in 1 M KOH containing 1 M CH3OH, and an ultra-slow scan rate of 0.1 mV s−1 was employed during the test to minimize the contribution of the electric double-layer of CF.74 A very strong current derived from the CF electric double-layer would interfere with the evaluation of the overpotential and overestimate the catalytic ability of the active phases, especially for the catalyst with a porous substrate. In addition, to clearly show the difference of activity, only the positive scanning curves are displayed (Fig. 4a). It can be seen that NCS/CF delivered the highest current density of 335.6 mA cm−2 at 1.73 V vs. RHE, about 1.60 times and 2.07 times higher than that of NS/CF (210.1 mA cm−2) and CS/CF (162.4 mA cm−2), respectively, showing its excellent catalytic activity for the SMOR. Note that CF as a support also had certain catalytic ability with a current density of 81.32 mA cm−2 at the same potential. However, it was achieved at the expense of a large overpotential. We further compared the potential needed for all the samples at the current density of 10 mA cm−2, which was the predicted current density for a 10% competent solar-to-fuel conversion device under 1 sun illumination.75,76 CF required a large potential of 1.491 V vs. RHE to obtain 10 mA cm−2 (inset of Fig. 4a), while NS/CF and CS/CF had similar values of 1.338 and 1.362 V vs. RHE, respectively. Surprisingly, after the heterostructure catalyst NCS/CF was assembled, the potential greatly decreased to 1.274 V vs. RHE, proving its immensely enhanced catalytic activity for the SMOR. In addition, to deliver 100 mA cm−2, NCS/CF only demanded a potential of 1.432 V vs. RHE, showing superior catalytic activity to many other catalysts (Table S5†). To probe the advantage of NCS/CF for the SMOR over the OER, CV curves were also conducted in 1 M KOH solution at a scan rate of 0.1 mV s−1 (Fig. 4b). The potential at 10 mA cm−2 for the OER was 1.457 V vs. RHE corresponding to the overpotential of 227 mV, which was comparable to other catalysts with similar elemental composition (Table S6†). By contrast, the potential used for the SMOR at the same current density was 183 mV lower than that of the OER, implying the energy-saving merit of the SMOR. Then, the catalytic kinetics of the as-prepared electrodes for the SMOR was evaluated from the Tafel slope and charge transfer resistance (Rct). NCS/CF showed the smallest Tafel slope of 115.28 mV dec−1 (Fig. 4c), lower than that of NS/CF (132.92 mV dec−1), CS/CF (174.71 mV dec−1), and CF (247.49 mV dec−1). A small Tafel slope indicates that a large current density increase can be achieved via a small potential change,77 therefore the NCS/CF electrode exhibited the most favorable reaction kinetics. The Rct was probed by electrochemical impedance spectroscopy (Fig. 4d). The recorded Nyquist plots were first fitted with an equivalent circuit model consisting of resistance, constant phase element, and capacitance (Fig. S15†). Results indicated that the modification of CF greatly reduced the resistance. The pristine CF had a large Rct of 32.52 Ω (Table S7†). The Rct values of NS/CF and CS/CF descended to 0.74 and 1.30 Ω, respectively. When it comes to the heterostructure NCS/CF, the Rct further decreased to 0.49 Ω, confirming its improved charge transfer ability. In addition, the solution resistance (Rs) of NCS/CF, NS/CF, CS/CF, and CF was quantified to be 0.99, 1.06, 1.00, and 0.98 Ω, respectively. The similar Rs values indicated the constant and consistent electrolyte conditions during the test. To explore the exposure of active sites derived from the unique morphology, the double-layer capacitance (Cdl) was calculated by recording CV curves in the non-faradaic regions with different scan rates to evaluate the electrochemical active surface area (ECSA) (Fig. 4e and S16†). The Cdl was determined by plotting the average of the positive and negative charging current densities at the middle potential against the scan rate, the linear slope represents Cdl and was proportional to the ECSA.78,79 CF only had a small Cdl of 0.05 mF cm−2, indicating its negligible contribution to the active area. However, NCS/CF increased the ECSA to 2.36 mF cm−2 due to the large exposure of active sites in the hetero-interface. In order to probe the influence of the different morphologies of NCS/CF, NS/CF, and CS/CF, we normalized the Cdl by the specific capacitance on metal foam in alkaline solution.80 The resultant ECSA values of NCS/CF, NS/CF, and CS/CF were 59.00, 50.00, and 29.25 cm2, respectively. It was noted that the ECSA of NCS/CF was 1.18 times and 2.02 times higher than that of NS/CF and CS/CF, respectively, which was lower than the corresponding multiplication of activity improvement revealed by the CV test (1.60 and 2.07 times). Therefore, though the morphologies of the samples differed significantly and may affect the kinetics of the SMOR process, the intrinsic activity of NCS/CF was still the highest. This demonstrated that the optimized shuttle effect and bimetallic synergism of NCS/CF can yield higher catalytic activity.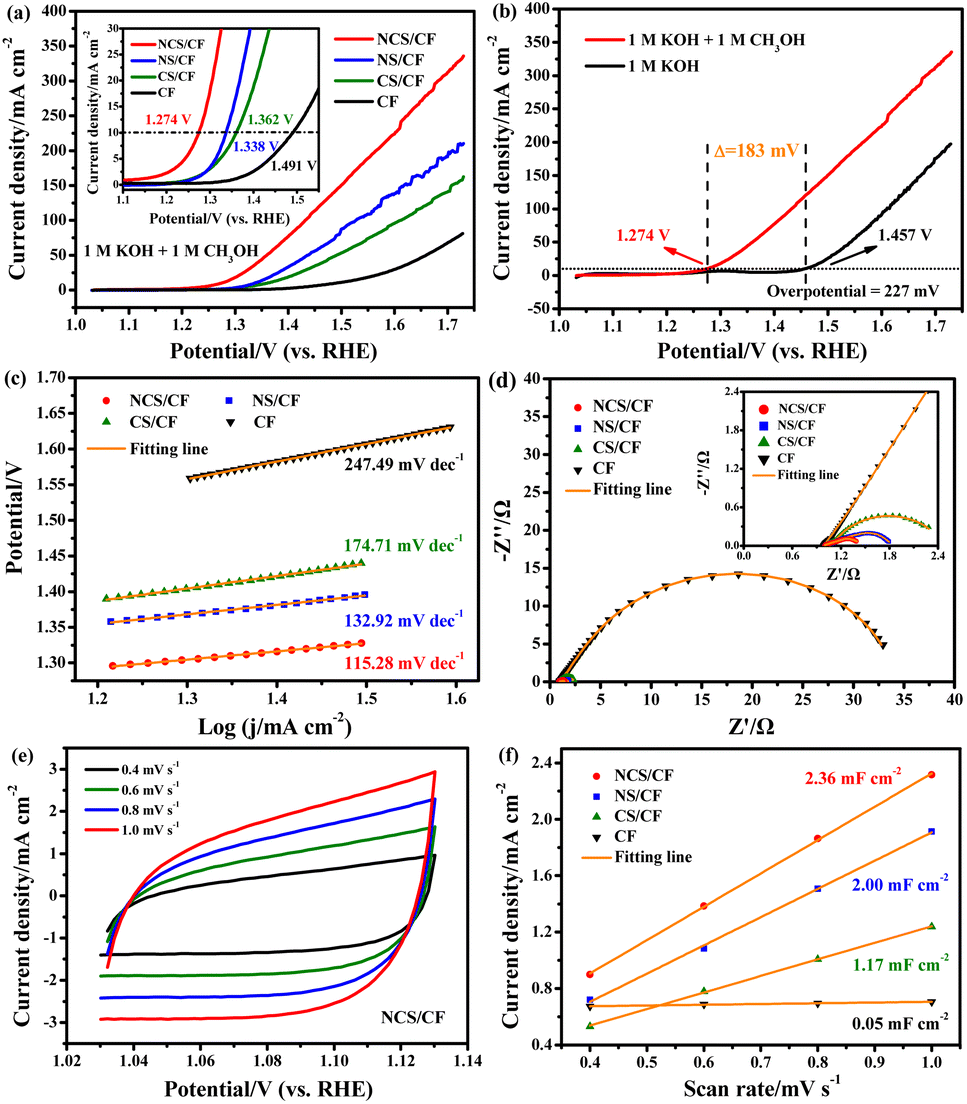 | ||
| Fig. 4 (a) Positive scanning curves of CV test measured at 0.1 mV s−1 without IR corrections (inset: potential at 10 mA cm−2). (b) Negative CV curves of NCS/CF in 1 M KOH with and without 1 M CH3OH without IR corrections. (c) Tafel slope and (d) EIS for all the samples. (e) Non-faradaic CV curves at different scan rates for NCS/CF. (f) Cdl values of NCS/CF, NS/CF, CS/CF, and CF. | ||
To further probe the influence of Se content on catalytic performance, different selenidation treatments of 1 and 5 h were carried out on NiCo-LDH/CF, and the corresponding NCS/CF-1 and NCS/CF-5 were obtained. Meanwhile, the catalytic activity of the non-selenized sample NiCo-LDH/CF was also probed and compared (Fig. S17†). It can be seen that the selenidation treatment optimized the onset potential and peak current density of NiCo-LDH/CF. The catalytic ability and activity of selenized samples were all better than those of NiCo-LDH/CF, proving the superiority of selenization. As for the influence of Se content, NCS/CF-1 exhibited worse catalytic performance with larger onset potential and smaller peak current density when compared to NCS/CF. This may be because the insufficient selenidation led to less generation of NiCoSe4 (Fig. S18†), thereby reducing the hetero-interface and weakening the synergistic effect, whereas NCS/CF-5 derived from long-time selenization showed a smaller onset potential but an even lower peak current density than that of NCS/CF-1. Hence one can see that excessive selenization can enhance the shuttle-like role of Se and increase the catalytic ability, however, the redundant NiCoSe4 may cover the hetero-interface and hinder its exposure, thus yielding a relatively lower current density (Fig. S19†). Furthermore, the Tafel slope proved the inferiority of NCS/CF-1 and NCS/CF-5 (Fig. S20†), their Tafel slope values were both smaller than that of NiCo-LDH/CF but higher than that of NCS/CF. This result implied that only appropriate selenization can balance the shuttle-like role and the hetero-interface effect, and then yielded the optimized catalytic performance.
After screening the catalysts, NCS/CF with the highest activity was employed to trace the anodic product of the SMOR and calculate the faradaic efficiency (FE). 6 h chronoamperometry (CA) tests were conducted at the potentials of 1.38, 1.43, 1.48, 1.53, 1.58, 1.63, and 1.68 V vs. RHE to collect the liquid product after the reaction (Fig. S21†). Then the final product was qualitatively identified by 1H nuclear magnetic resonance (NMR) spectroscopy and quantitatively analyzed by ion chromatography (IC). In the 1H NMR spectrum (Fig. 5a), the value-added formate was detected at around 8.27 ppm,81 it was evidenced as the only product of the SMOR. Afterward, the practical production of the formate was quantitatively calibrated with chromatographic grade formate of known concentrations via the IC technique (Fig. S22†). Until the potential of 1.63 V vs. RHE, the signal intensity of HCOO− increased gradually, but when the potential reached 1.68 V vs. RHE, the peak intensity began to decrease (Fig. 5b). Within the testing potential window, all the FEformate was greater than 80% (Fig. 5c). In particular, at the potential of 1.53 and 1.58 V vs. RHE, the FEformate was up to 103.06 and 99.65%, respectively, indicating the near 100% rate of conversion of methanol to formate. It can be explained that formate was not fully produced at a lower potential and the competitive OER will be involved at a higher potential, thereby leading to insufficient conversion of methanol to formate. Considering the high activity and good selectivity of NCS/CF for the SMOR, a two-electrode system was constructed by using NCS/CF as both the anode and cathode to conduct energy-saving SMOR-assisted water splitting (co-electrolysis) for simultaneous methanol upgradation and hydrogen harvest. The co-electrolysis showed an apparently small onset potential and high current density (Fig. 5d). NCS/CF‖NCS/CF only required a low cell voltage of 1.380 V to deliver 10 mA cm−2 for co-electrolysis, 253 mV lower than that of pure water electrolysis. Furthermore, only a low current density of 46.4 mA cm−2 was achieved at 1.8 V in 1 M KOH solution, while the current density was boosted to 149.2 mA cm−2 at the same voltage with the assistance of methanol, which was 3.2 times higher than that of traditional water splitting. This result confirmed the superiority of SMOR-assisted water splitting with low energy consumption and high current density. The reaction phenomenon can be seen in the digital photograph of the electrolytic cell (Fig. S23†). In the co-electrolysis process, H2 evolution on the cathode is significant with large amounts of bubbles. In contrast, the SMOR at the anode did not produce any bubbles, even with the current density at 100 mA cm−2. This further indicated that formate was the only product without the generation of O2 or CO2, demonstrating the high selectivity of SMOR and suppression of the competitive OER. Moreover, an exceptional robustness of NCS/CF‖NCS/CF was achieved. After a continuous operation of 100 h, the large current density of 100 mA cm−2 could still be achieved at a near-constant voltage, demonstrating its brilliant catalytic stability (Fig. 5e and Tabel S5†).
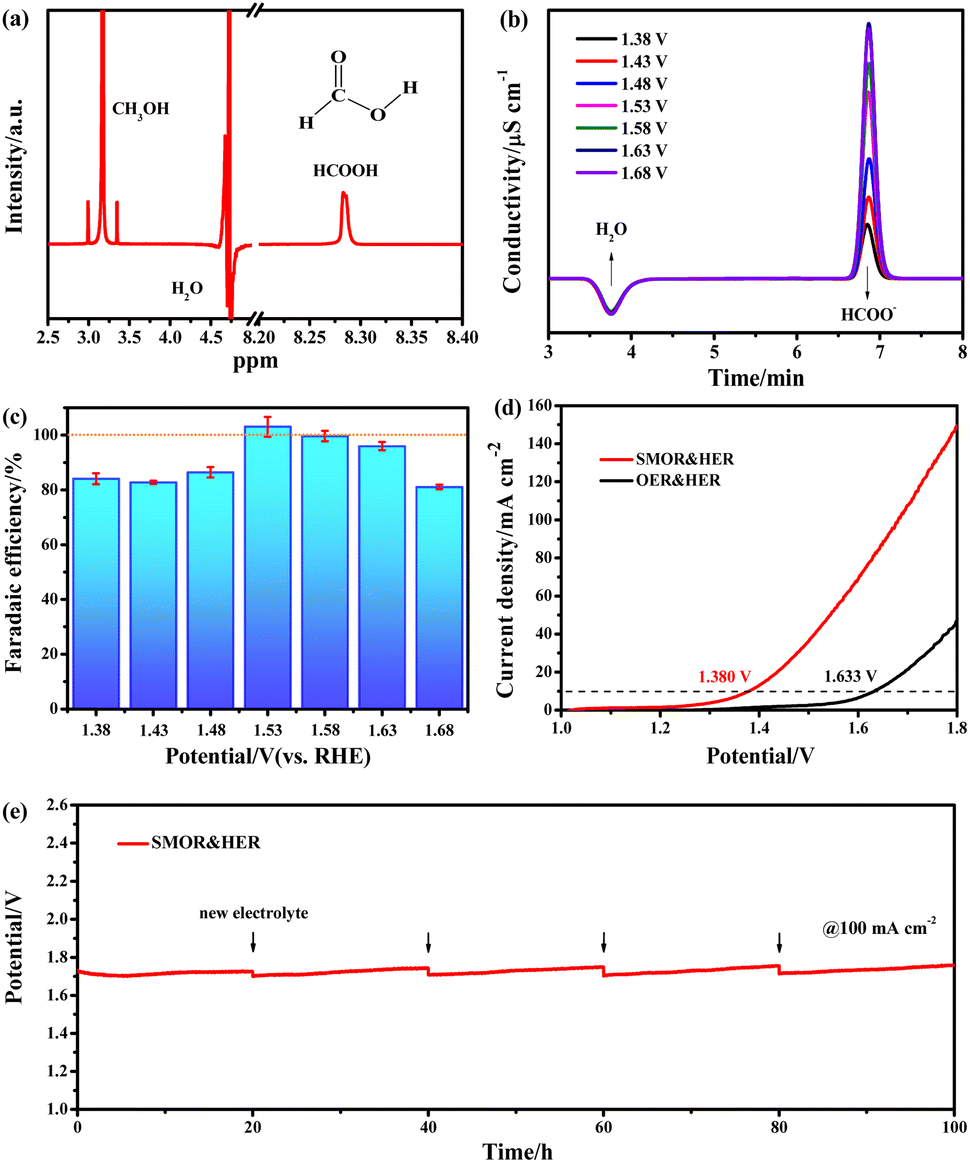 | ||
| Fig. 5 (a) The 1H NMR spectrum of the electrolyte after the electrochemical test. (b) IC traces of the electrolyte and (c) corresponding FEformate at different potentials. (d) LSV plots of methanol electrolysis and overall water splitting on the NCS/CF‖NCS/CF electrode without IR corrections. (e) 100 h durability test of the NCS/CF‖NCS/CF electrode for methanol electrolysis at 100 mA cm−2. | ||
3.3 Mechanism study
To explore the structural evolution and catalytic mechanism of the catalyst, the phase composition, morphology, and surface chemical state of NCS/CF were thus probed after the SMOR electrocatalytic reaction. The XRD pattern showed a very weak diffraction peak intensity of NiCoSe4 due to the surface reconstruction, but the signal of the (2, 1, 0) crystal plane can still be observed, confirming that it is the most exposed and primarily functional phase (Fig. S24†). The morphology of NCS/CF after the SMOR reaction underwent significant changes due to the harsh alkaline electrolyte and the strong oxidative environment (Fig. S25†).82 The long NiCoSe4 nanoneedles disappeared and only irregular particles were retained. The flat NiCo-LDH nanosheets also underwent deformation. This phenomenon indicated the existence of elemental dissolution or leaching during the reaction process, but the hetero-interface between NiCoSe4 and NiCo-LDH still existed. The variation of the elemental composition of NCS/CF before and after the reaction was investigated by EDS analysis (Fig. S26 and Table S8†). The increased O element meant the partial surface oxidation and the formation of metal oxyhydroxide. In contrast, the content of Se element decreased a lot, which was in consistence with the observation of SEM images, indicating the leaching out of Se. The atomic ratio of Ni and Co was nearly kept constant. The K element came from the KOH electrolyte. The surface chemical state change after the SMOR was also probed by XPS. In Ni 2p spectra (Fig. S27a†), Ni2+ peaks disappeared and Ni3+ became the dominant species, indicating the sufficient surface reconstruction of Ni element. As for Co 2p spectra (Fig. S27b†), though the content ratio of Co3+/Co2+ became larger, the Co2+ species still existed, suggesting that Co element also underwent surface reconstruction to some degree but not as strong as Ni. When it comes to Se 3d spectra (Fig. S27c†), the Se element at the surface of NCS/CF was oxidized, covering the internal metal–selenium bonds. The enhanced intensity of the M–OH peak in O 1s spectra also evidenced the surface oxidation and reconstruction (Fig. S27d†).83 These results revealed that the high valent metal oxyhydroxide was generated during the SMOR, including NiOOH and CoOOH. They were both involved in the catalytic process, and NiOOH was the species that played a more important role due to its complete reconstruction.To further explore the role of Se in bimetallic NiCo in the NiCo-LDH derivative, we performed density functional theory (DFT) calculations by building a heterostructure model composed of NiCoSe4 (2, 1, 0) and NiCo-LDH (1, 0, 7) faces (Fig. 6a and S28†). The differential charge density and Bader analysis demonstrated the electron redistribution. An intense disturbance of electrons was found at the interface of NiCoSe4 and NiCo-LDH, where the blue and yellow regions represented electron depletion and accumulation, respectively (Fig. S29†). It can be seen that the depletion domain was mainly concentrated on the NiCo-LDH side, meanwhile, the NiCoSe4 side exhibited both electron gain and electron loss phenomena. On the whole, there was a trend of electron flowing from NiCo-LDH to NiCoSe4. This electronic effect was beneficial for the generation of high valent active metal species, which was consistent with the results of post-catalysis XPS characterization. Bader analysis quantitatively determined the charge of all the atoms in the NCS model (Tables S9 and S10†). The total Bader charge indicated that both Ni and Co lost electrons and Se gained electrons as a whole. It was surprisingly found that the Se19 atom at the hetero-interface lost 0.06e. As shown in the local magnification of differential charge density around the Se19 atom (Fig. 6b), all the metal atoms connected to Se19 lost electrons, whereas, the Bader charge number of Co2 (0.31) was much lower than that of Co10 (1.22). Thus, we inferred that the electrons lost from Se19 flowed towards Co2, resulting in its smaller Bader charge. Furthermore, by probing the density of states (DOS) (Fig. 6c), the metallic properties of NCS/CF were revealed because its Fermi levels passed through the conduction band.84 This suggested that the modification of NiCo-LDH by partial selenidation was beneficial to improving the electrical conductivity and electron transfer, compensating for the disadvantage of LDH.
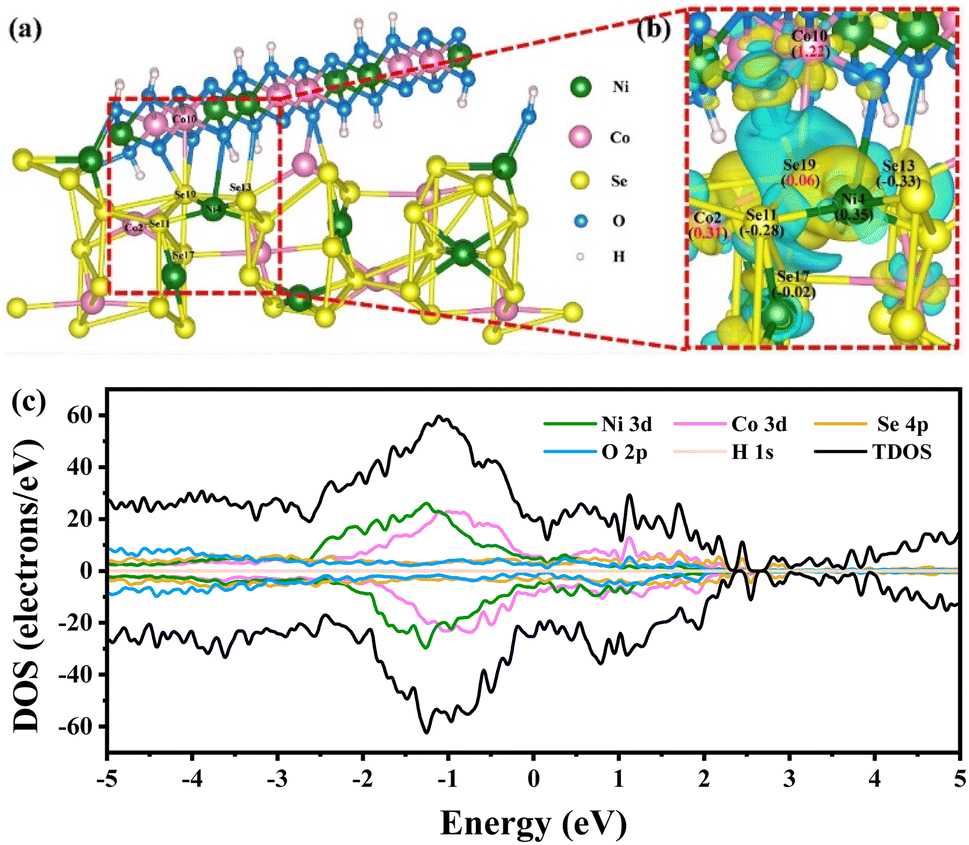 | ||
| Fig. 6 (a) The structural model of NCS. (b) The local magnification of differential charge density around the Se atom at the interface of NCS. (c) Density of states of NCS. | ||
4. Conclusion
In this study, a mirror-like heterostructure of NiCoSe4@NiCo-LDH loaded on cobalt foam (NCS/CF) was synthesized through a partial selenidation strategy to modify NiCo-LDH for the selective methanol oxidation reaction (SMOR). A shuttle-like role of Se in NCS/CF was found by observing electron transfer from Ni to Co via Se based on the experimental test and theoretical calculation. Except for this electronic effect, the interface formed between NiCoSe4 and NiCo-LDH was also proved to create more active sites, minimize the charge transfer resistance, and yield favorable synergism. In the end, a ∼100% faradaic efficiency of methanol upgradation to formate (FEformate) was achieved. When used as an anode in a three-electrode system, the potential needed to achieve a current density of 10 and 100 mA cm−2 for the SMOR was only 1.274 and 1.432 V vs. RHE, surpassing most of the catalysts reported in the literature. The potential of 1.274 V vs. RHE was much smaller than the 1.457 V vs. RHE of the OER, showing its strong possibility of replacing water oxidation from an energy saving point of view. Moreover, the SMOR activity of NCS/CF was 1.60 times and 2.07 times larger than that of NS/CF and CS/CF, respectively, highlighting the advantage of bimetallic synergy. When used as a bifunctional electrode in the two-electrode system, NCS/CF‖NCS/CF only required a cell voltage of 1.380 V at the current density of 10 mA cm−2 for the co-electrolysis of methanol and water, 253 mV lower than that of pure water splitting. In addition, its robust catalytic ability was also proved by continuously carrying out the operation at 100 mA cm−2 for 100 h at a near constant cell voltage, promising the satisfying stability of NCS/CF for the simultaneous harvest of value-added formate and green hydrogen.Data availability
Data supporting the findings of this study are available within the article ESI.†Author contributions
Jiaxin Li: conceptualization, methodology, investigation, validation, writing – original draft; Hongmei Yu: conceptualization, writing – review & editing, resources, funding acquisition. Jun Chi: writing – review & editing; Xu Luo: methodology and investigation; Tongzhou Li: investigation and formal analysis; Zhigang Shao: writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (22090030). Additionally, we sincerely appreciate the Liaoning Provincial Engineering Research Center of Electrolysis for Hydrogen Production and the Dalian Key Laboratory of Electrolysis for Hydrogen Production's support of our work.References
- J. Na, H. Yu, S. Jia, J. Chi, K. Lv, T. Li, Y. Zhao, Y. Zhao, H. Zhang and Z. Shao, J. Energy Chem., 2024, 91, 370–382 CrossRef CAS.
- Y. Kuang, R. He, X. Gu, F. Yang, X. Tian and L. Feng, Chem. Eng. J., 2023, 456, 141055 CrossRef CAS.
- J. Li, S. Wang, S. Sun, X. Wu, B. Zhang and L. Feng, J. Mater. Chem. A, 2022, 10, 9308–9326 RSC.
- J. Li, S. Wang, J. Chang and L. Feng, Adv. Powder Mater., 2022, 1, 100030 CrossRef.
- E. A. Toledo-Carrillo, M. García-Rodríguez, L. M. Sánchez-Moren and J. Dutta, Sci. Adv., 2024, 10, eadi3180 CrossRef CAS PubMed.
- T. Wang, X. Cao and L. Jiao, Angew. Chem., Int. Ed., 2022, 61, e202213328 CrossRef CAS.
- Q. Qian, Y. Zhu, N. Ahmad, Y. Feng, H. Zhang, M. Cheng, H. Liu, C. Xiao, G. Zhang and Y. Xie, Adv. Mater., 2024, 36, 2306108 CrossRef CAS PubMed.
- W. Chen, C. Xie, Y. Wang, Y. Zou, C. Dong, Y. Huang, Z. Xiao, Z. Wei, S. Du, C. Chen, B. Zhou, J. Ma and S. Wang, Chem, 2020, 6, 2974–2993 CAS.
- Y. Zhu, Q. Qian, Y. Chen, X. He, X. Shi, W. Wang, Z. Li, Y. Feng, G. Zhang and F. Cheng, Adv. Funct. Mater., 2023, 33, 2300547 CrossRef CAS.
- L. Xu, Z. Huang, M. Yang, J. Wu, W. Chen, Y. Wu, Y. Pan, Y. Lu, Y. Zou and S. Wang, Angew. Chem., Int. Ed., 2022, 61, e202210123 CrossRef CAS PubMed.
- G. Li, G. Han, L. Wang, X. Cui, N. K. Moehring, P. R. Kidambi, D.-e. Jiang and Y. Sun, Nat. Commun., 2023, 14, 525 CrossRef CAS PubMed.
- Y. Huang, X. Chong, C. Liu, Y. Liang and B. Zhang, Angew. Chem., Int. Ed., 2018, 57, 13163–13166 CrossRef CAS PubMed.
- Y. Chen, Y. Wen, Q. Zhou, L. Shen, F. Du, P. Peng, Y. Chen and J. Zheng, J. Energy Chem., 2023, 86, 609–619 CrossRef CAS.
- K. Xiang, Z. Song, D. Wu, X. Deng, X. Wang, W. You, Z. Peng, L. Wang, J. Luo and X. Fu, J. Mater. Chem. A, 2021, 9, 6316–6324 RSC.
- A. Sathyaseelan, K. Krishnamoorthy, P. Pazhamalai, N. U. H. Liyakath Ali and S. J. Kim, J. Mater. Chem. A, 2023, 11, 20712–20723 RSC.
- Q. Ling, Z. Zhao, Z. Li, K. Yan, C. Ding, P. Chen, Z. Sun, G. He, J. Lv and M. Zhang, J. Mater. Chem. A, 2023, 11, 2876–2888 RSC.
- X. Wei, Y. Li, L. Chen and J. Shi, Angew. Chem., Int. Ed., 2021, 60, 3148–3155 CrossRef CAS PubMed.
- Q. Mao, X. Mu, W. Wang, K. Deng, H. Yu, Z. Wang, Y. Xu, L. Wang and H. Wang, Nat. Commun., 2023, 14, 5679 CrossRef CAS PubMed.
- B. Zhu, B. Dong, F. Wang, Q. Yang, Y. He, C. Zhang, P. Jin and L. Feng, Nat. Commun., 2023, 14, 1686 CrossRef CAS.
- D. A. Bulushev and J. R. H. Ross, ChemSusChem, 2018, 11, 821–836 CrossRef CAS.
- F. Meng, C. Dai, Z. Liu, S. Luo, J. Ge, Y. Duan, G. Chen, C. Wei, R. R. Chen, J. Wang, D. Mandler and Z. J. Xu, eScience, 2022, 2, 87–94 CrossRef.
- S. Ding, J. Duan and S. Chen, EcoEnergy, 2024, 2, 45–82 CrossRef.
- J. Shi, J. Ma, E. Ma, J. Li, Y. Hu, L. Fan and W. Cai, Carbon Neutralization, 2024, 3, 285–312 CrossRef CAS.
- Y. Wang, X. Yang, Y. Zhang, C. Zhang, L. Yang, Q. Jiang, H. He, G. Ying and H. Huang, Mater. Today Energy, 2024, 40, 101495 CrossRef CAS.
- B. Liu, T. Xiao, X. Sun, H. Peng, X. Wang, Y. Zhao, W. Zhang and Y. Song, J. Mater. Chem. A, 2022, 10, 19649–19661 RSC.
- C. Xiao, L. Cheng, Y. Wang, J. Liu, R. Chen, H. Jiang, Y. Li and C. Li, J. Mater. Chem. A, 2022, 10, 1329–1335 RSC.
- H. Chi, J. Lin, S. Kuang, M. Li, H. Liu, Q. Fan, T. Yan, S. Zhang and X. Ma, J. Energy Chem., 2023, 85, 267–275 CrossRef CAS.
- B. Liu, X. Wang, S. Wang, H. Peng, T. Xiao, G. Liu, S. Bai, Y. Zhao, W. Zhang and Y. Song, Mater. Today Energy, 2022, 28, 101082 CrossRef CAS.
- D. P. Sahoo, K. K. Das, S. Mansingh, S. Sultana and K. Parida, Coord. Chem. Rev., 2022, 469, 214666 CrossRef CAS.
- L. Hu, M. Li, X. Wei, H. Wang, Y. Wu, J. Wen, W. Gu and C. Zhu, Chem. Eng. J., 2020, 398, 125605 CrossRef CAS.
- C. Liu, F. Yang, Y. Yang, S. Wang and L. Feng, Chem. Commun., 2024, 60, 1591–1594 RSC.
- M. Li, X. Deng, Y. Liang, K. Xiang, D. Wu, B. Zhao, H. Yang, J. Luo and X. Fu, J. Energy Chem., 2020, 50, 314–323 CrossRef.
- Y. Zhang, X. Wu, G. Fu, X. Fu and J. Luo, J. Alloys Compd., 2022, 906, 164305 CrossRef CAS.
- L. Du, N. Lv, J. Li, J. Zhang, Y. Chen, Y. Zhang, Z. Li, X. Huang and J. Luo, J. Ind. Eng. Chem., 2023, 120, 467–476 CrossRef CAS.
- A. Giri, G. Park and U. Jeong, Chem. Rev., 2023, 123, 3329–3442 CrossRef CAS PubMed.
- J. Zhou, C. Zhu, Y. Zhou, J. Dong, P. Li, Z. Zhang, Z. Wang, Y. Lin, J. Shi, R. Zhang, Y. Zheng, H. Yu, B. Tang, F. Liu, L. Wang, L. Liu, G. Liu, W. Hu, Y. Gao, H. Yang, W. Gao, L. Lu, Y. Wang, K. Suenaga, G. Liu, F. Ding, Y. Yao and Z. Liu, Nat. Mater., 2023, 22, 450–458 CrossRef CAS PubMed.
- P. Santhoshkumar, G. Nagaraju, N. Shaji, G. S. Sim, M. Nanthagopal, S. C. Sekhar, J. S. Yu and C. W. Lee, Electrochim. Acta, 2020, 356, 136833 CrossRef CAS.
- J. Sun, Z. Zhao, J. Li, Z. Li and X. Meng, J. Alloys Compd., 2022, 918, 165719 CrossRef CAS.
- H. Xiang, Q. Dong, M. Yang and S. Liu, Mater. Chem. Front., 2024, 8, 1888–1926 RSC.
- D. Chen, Z. Zhao, G. Chen, T. Li, J. Chen, Z. Ye and J. Lu, Coord. Chem. Rev., 2023, 479, 214984 CrossRef CAS.
- X. Xia, L. Wang, N. Sui, V. L. Colvin and W. W. Yu, Nanoscale, 2020, 12, 12249–12262 RSC.
- D. Wu, J. Hao, W. Wang, Y. Yu, X. Fu and J. Luo, ChemSusChem, 2021, 14, 5450–5459 CrossRef CAS PubMed.
- G. Chen, H. Xiang, Y. Guo, J. Huang, W. Chen, Z. Chen, T. Li and K. Ostrikov, Carbon Energy, 2024, e522, DOI:10.1002/cey2.522.
- W. Tan, M. Qin, G. Ma, Z. Fan, X. Li and X. Xin, ACS Sustain. Chem. Eng., 2024, 12, 5139–5149 CrossRef CAS.
- X. Han, Y. Niu, C. Yu, Z. Liu, H. Huang, H. Huang, S. Li, W. Guo, X. Tan and J. Qiu, Nano Energy, 2020, 69, 104367 CrossRef CAS.
- Z. Yuan, Q. Lin, Y. Li, W. Han and L. Wang, Adv. Mater., 2023, 35, 2211527 CrossRef CAS PubMed.
- X. Xu, J. Li, C. Zhang, S. Zhang, G. Su, Z. Shi, H. Wang and M. Huang, Appl. Catal., B, 2022, 319, 121949 CrossRef CAS.
- Z. Xie, D. Qiu, J. Xia, J. Wei, M. Li, F. Wang and R. Yang, ACS Appl. Mater. Interfaces, 2021, 13, 12006–12015 CrossRef CAS PubMed.
- K. Zhou, J. He, X. Wang, J. Lin, Y. Jing, W. Zhang and Y. Chen, Electrochim. Acta, 2017, 231, 626–631 CrossRef CAS.
- K. Ding, J. Hu, J. Luo, W. Jin, L. Zhao, L. Zheng, W. Yan, B. Weng, H. Hou and X. Ji, Nano Energy, 2022, 91, 106675 CrossRef CAS.
- Y. Guo, C. Shang and E. Wang, J. Mater. Chem. A, 2017, 5, 2504–2507 RSC.
- L. Zhai, T. W. Benedict Lo, Z.-L. Xu, J. Potter, J. Mo, X. Guo, C. C. Tang, S. C. Edman Tsang and S. P. Lau, ACS Energy Lett., 2020, 5, 2483–2491 CrossRef CAS.
- H. Zhou, Y. Wang, R. He, F. Yu, J. Sun, F. Wang, Y. Lan, Z. Ren and S. Chen, Nano Energy, 2016, 20, 29–36 CrossRef CAS.
- K. Guo, Y. Wang, J. Huang, H. Li, Y. Peng and C. Xu, Chem. Eng. J., 2023, 454, 140488 CrossRef CAS.
- B. Wang, X. Wang, B. Zheng, B. Yu, F. Qi, W. Zhang, Y. Li and Y. Chen, Electrochem. Commun., 2017, 83, 51–55 CrossRef CAS.
- S. Aftab, M. B. Tahir, M. S. Tahir and M. Sagir, J. Energy Storage, 2022, 56, 106111 CrossRef.
- H. V. S. R. M. Koppisetti, H. R. Inta, K. Mukhuti, A. Roy, S. Ghosh, V. G. Pol and V. Mahalingam, J. Energy Storage, 2024, 86, 111178 CrossRef.
- W. Chen, Y. Wang, B. Wu, J. Shi, Y. Li, L. Xu, C. Xie, W. Zhou, Y.-C. Huang, T. Wang, S. Du, M. Song, D. Wang, C. Chen, J. Zheng, J. Liu, C. Dong, Y. Zou, J. Chen and S. Wang, Adv. Mater., 2022, 34, 2105320 CrossRef CAS PubMed.
- H. Kuang, H. Zhang, X. Liu, Y. Chen, W. Zhang, H. Chen and Q. Ling, Carbon, 2022, 190, 57–67 CrossRef CAS.
- B. Fan, H. Wang, X. Han, Y. Deng and W. Hu, Chem. Commun., 2022, 58, 8254–8257 RSC.
- G. Yang, C. Pei, F. Xu, H. S. Park, X. Yu and H. Pang, Chin. Chem. Lett., 2023, 34, 108152 CrossRef CAS.
- X. Huang, S. Men, H. Zheng, D.-D. Qin and X. Kang, Chem. Asian J., 2020, 15, 1456–1463 CrossRef CAS PubMed.
- Q. Wang, C. Wang, X. Du and X. Zhang, Int. J. Hydrogen Energy, 2024, 51, 1154–1166 CrossRef CAS.
- D. D. Khumujam, T. Kshetri, T. I. Singh, N. H. Kim and J. H. Lee, Adv. Funct. Mater., 2023, 33, 2302388 CrossRef CAS.
- Z. Liu, C. Zhang, H. Liu and L. Feng, Appl. Catal., B, 2020, 276, 119165 CrossRef CAS.
- J. Li, S. Wang, B. Zhang, W. Wang and L. Feng, Int. J. Hydrogen Energy, 2017, 42, 12236–12245 CrossRef CAS.
- P. Zhang, Y. Li, Y. Zhang, R. Hou, X. Zhang, C. Xue, S. Wang, B. Zhu, N. Li and G. Shao, Small Methods, 2020, 4, 2000214 CrossRef CAS.
- J. Li, S. Sun, Y. Yang, Y. Dai, B. Zhang and L. Feng, Chem. Commun., 2022, 58, 9552–9555 RSC.
- D. He, C. Yue, L. Tang, B. Wang, H. Tang, X. Li, J. Chen, M. Gao and N. Liu, J. Alloys Compd., 2023, 957, 170449 CrossRef CAS.
- C. Xu, G. Li, W. Huang, X. Luo, M. Wei, H. Wen, Y. Li, X. Li, Y. Zhang and W. Chen, Electrochim. Acta, 2024, 475, 143683 CrossRef CAS.
- L. Liao, J. Sun, D. Li, F. Yu, Y. Zhu, Y. Yang, J. Wang, W. Zhou, D. Tang, S. Chen and H. Zhou, Small, 2020, 16, 1906629 CrossRef CAS PubMed.
- H. Liang, X. Li, Z. Liu, W. Yang, X. Liu, Y. Zhang and H. Fan, Mater. Chem. Front., 2022, 6, 194–202 RSC.
- H. Jiang, H. Qin, P. Zhou, L. Kong, C. Wang, Z. Ji, X. Shen, G. Zhu and A. Yuan, Int. J. Hydrogen Energy, 2024, 58, 892–901 CrossRef CAS.
- Y. Wu, G.-D. Li, Y. Liu, L. Yang, X. Lian, T. Asefa and X. Zou, Adv. Funct. Mater., 2016, 26, 4839–4847 CrossRef CAS.
- J. Luo, D. A. Vermaas, D. Bi, A. Hagfeldt, W. A. Smith and M. Grätzel, Adv. Energy Mater., 2016, 6, 1600100 CrossRef.
- J. Zhu, L. Hu, P. Zhao, L. Y. S. Lee and K. Y. Wong, Chem. Rev., 2020, 120, 851–918 CrossRef CAS PubMed.
- N. T. Suen, S. F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- Y. Kuang, W. Qiao, F. Yang and L. Feng, J. Energy Chem., 2023, 85, 447–454 CrossRef CAS.
- R. He, J. Li and L. Feng, Catal. Commun., 2022, 163, 106407 CrossRef CAS.
- D. Liu, H. Ai, J. Li, M. Fang, M. Chen, D. Liu, X. Du, P. Zhou, F. Li, K. H. Lo, Y. Tang, S. Chen, L. Wang, G. Xing and H. Pan, Adv. Energy Mater., 2020, 10, 2002464 CrossRef CAS.
- K. Deng, P. Liu, X. Liu, H. Li, W. Tian and J. Ji, Green Chem., 2023, 25, 9837–9846 RSC.
- J. Li, C. Yin, S. Wang, B. Zhang and L. Feng, Chem. Sci., 2024, 15, 13659–13667 RSC.
- C. Yin, J. Li, S. Wang, H. Wen, F. Yang and L. Feng, Carbon Energy, 2024, e553 CrossRef.
- C. Hou, J. Wang, W. Du, J. Wang, Y. Du, C. Liu, J. Zhang, H. Hou, F. Dang, L. Zhao and Z. Guo, J. Mater. Chem. A, 2019, 7, 13460–13472 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta04581e |
| This journal is © The Royal Society of Chemistry 2024 |