
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIsotype heterojunction graphitic carbon nitride photocathode for photo-accelerated zinc-ion capacitors†
Xiaopeng
Liu
a,
Yijia
Zhu
a,
Firoz
Alam
b,
Tianlei
Wang
c,
Ivan P.
Parkin
 c,
Mingqing
Wang
a and
Buddha
Deka Boruah
*a
c,
Mingqing
Wang
a and
Buddha
Deka Boruah
*a
aInstitute for Materials Discovery, University College London, London WC1E 7JE, UK. E-mail: b.boruah@ucl.ac.uk
bDepartment of Electronic and Electrical Engineering, University College London, London, WC1E 6BT, UK
cDepartment of Chemistry, University College London, London WC1H 0AJ, UK
First published on 5th September 2024
Abstract
Photoelectrodes that combine light harvesting and energy storage within a single material represent an effective approach for developing standalone devices for solar energy capture and storage. However, achieving high-performance photoelectrodes requires precise tuning of optical, electrochemical and interface properties. In this study, we introduce an optimization approach designed to improve the charge storage, optical properties, and interface adjustments of graphitic carbon nitride (g-C3N4) when used as the photoelectrode material in photo-accelerated zinc-ion capacitors (Photo-ZICs). Through the adjustment of precursor ratios, we successfully introduced a g-C3N4 isotype heterojunction electrode via a thermal polycondensation method. This optimized electrode exhibited a performance level four times higher than that of pristine g-C3N4 synthesized from urea, even at a high specific current of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mA g−1. Additionally, it demonstrated an impressive capacity of 37.62 mA h g−1 with a coulombic efficiency of 99.9% after 10000 cycles. Furthermore, we explored the potential of a dual heterojunction structure by combining the optimized g-C3N4 isotype heterojunction with titanium dioxide to create highly efficient Photo-ZICs. These assembled devices exhibited remarkable capacity enhancements and cycling stability even under light exposure. As a proof-of-concept experiment, our findings underscore the significant benefits of isotype heterojunctions in enhancing both energy storage capabilities and solar harvesting efficiency, ultimately leading to the development of highly efficient Photo-ZICs.
000 mA g−1. Additionally, it demonstrated an impressive capacity of 37.62 mA h g−1 with a coulombic efficiency of 99.9% after 10000 cycles. Furthermore, we explored the potential of a dual heterojunction structure by combining the optimized g-C3N4 isotype heterojunction with titanium dioxide to create highly efficient Photo-ZICs. These assembled devices exhibited remarkable capacity enhancements and cycling stability even under light exposure. As a proof-of-concept experiment, our findings underscore the significant benefits of isotype heterojunctions in enhancing both energy storage capabilities and solar harvesting efficiency, ultimately leading to the development of highly efficient Photo-ZICs.
Introduction
As the global population continues to grow, so does the demand for energy consumption, leading to increased environmental pollution due to the extensive use of fossil fuels.1 This situation underscores the need for the intensive adoption of green energy sources such as solar and wind power. Solar energy, in particular, stands out for its abundance and environmental friendliness, making it a promising solution to address both environmental concerns and energy shortages.2 However, the intermittent nature of sunlight necessitates the integration of solar harvesting devices with electrochemical storage systems to store excess solar energy for use during periods of darkness or when needed.3 While this approach is effective for most applications, it comes with certain drawbacks. Integrating solar energy harvesters and storage systems can result in larger packaging volumes, increased costs, and the requirement for additional electronic components to ensure energy compatibility between the two separate components.4 In response to these challenges, the concept of solar-accelerated electrochemical energy storage has been extended to the field of electrode design, specifically in the development of photoelectrodes. This innovation aims to achieve highly efficient solar energy capture and storage within the same electrode components, offering a more integrated and streamlined solution.5Indeed, the utilization of photoelectrodes has garnered significant research attention lately, primarily because they offer the dual functionality of both capturing light and storing energy. Furthermore, this configuration has been observed to enhance the gravimetric energy density and electronic transfer within the device when exposed to light.6 The concept of photoelectrodes has already been successfully demonstrated in various energy storage systems, including sodium-ion batteries,7 Li–O2 batteries,8 zinc-ion batteries,3 and capacitors.9 Instead of relying solely on photo-active energy storage materials that can directly convert solar light into electricity through the photoelectric effect,10 some researchers have explored the use of photo-accelerated batteries with single-material electrodes. For instance, Wang et al. introduced the concept of a photo-accelerated Li-ion battery with a LiV2O5 photocathode, showcasing direct photo energy storage capabilities.11 However, these single-material electrode-based photo-accelerated batteries face limitations due to inefficient solar utilization and photo-charge separation. Therefore, there is a pressing need to develop efficient bi-functional photoelectrodes for photo-accelerated energy storage systems. In this context, the design of heterojunctions emerges as a promising approach to effectively suppress photo-charge recombination and enhance light absorption by tuning the band gap of two different semiconductor materials.12 Moreover, the presence of an internal electric field resulting from heterojunction structures at the interface can improve ion diffusion kinetics and material utilization, ultimately benefiting high-rate electrochemical performance.13 Recent advancements have led to the achievement of isotype heterojunctions, which involve the combination of different phases within a single material, such as zinc blende/wurtzite,14 TiO2 anatase/rutile,15 and WO3−x/WO3.16 For example, Dickon et al. reported that isotype homojunctions in MoO3 can regulate ion storage and electronic structure, resulting in higher capacity and improved rate performance.17 Therefore, the design of heterojunction-based photoelectrodes represents an efficient strategy to enhance the performance of photo-accelerated energy storage systems.
Although lithium-ion based energy storage systems have played an important role in electrical energy storage fields among the various energy storage systems available due to the lightweight design and high energy densities, they still have shortcomings that impede their application as emerging technologies, including safety risks, low power density, and high cost, which hinder their further application in mini-grid level and off-grid cases.18,19 Recognizing these issues underscores the necessity for further exploration of next-generation energy storage systems. In this context, zinc-ion capacitors (ZICs) have emerged as a promising alternative for energy storage systems. The combination of intrinsic safety, low cost, environmental compatibility, and competitive gravimetric energy density makes ZICs particularly appealing for mini-grid level and off-grid energy storage applications.20
In our previous research, our team introduced the concept of photo-accelerated zinc-ion capacitors (Photo-ZICs) employing g-C3N4 as the photocathode material, which was derived solely from urea as the precursor.21 However, the uncontrolled integration of active materials with charge transfer materials in this configuration limited the photo-accelerated electrochemical performance of the device. In this study, we took a dedicated approach to design g-C3N4 isotype heterojunctions based on triazine and heptazine moieties within the same photocathode composition. This was achieved by carefully adjusting the mass ratio of the different precursors, melamine and urea. As anticipated, the creation of g-C3N4 isotype heterojunctions resulted in significantly improved electrochemical performance compared to their pristine counterparts in the context of ZICs. Furthermore, we combined these optimized g-C3N4 isotype heterojunctions with TiO2 to enhance charge transfer and explored the feasibility of a dual heterojunction structure for achieving highly efficient Photo-ZICs. As a proof-of-concept experiment, this research not only underscores the beneficial impact of heterojunctions on energy storage but also lays the groundwork for straightforward band alignment strategies aimed at realizing highly efficient Photo-ZICs.
Results and discussion
Fig. 1a illustrates a schematic representation of the synthesis process for the g-C3N4 isotype heterojunction, which includes photos of the precursor and product within the circular diagram. To provide a concise overview, melamine (M) and urea (U) were thoroughly mixed and subjected to a thermal condensation method in a crucible with a lid, resulting in the formation of the MUxy series. Here, ‘x’ and ‘y’ represent the mass ratio of melamine and urea, respectively. In contrast, pristine g-C3N4 samples were prepared through thermal polymerization of either melamine (PM) or urea (PU) alone. Additional details can be found in the Experimental section. Surface morphologies of the prepared samples were observed via scanning electron microscope (SEM) (Fig. 1b–e). Clearly, PM showcases a thick sheet-like morphology with a smooth surface (Fig. 1b). Conversely, PU reveals a porous sheet-like structure that results from the gas released during the decomposition of urea (Fig. 1c and d). When melamine is introduced alongside urea, it causes the formation of agglomerated nanoparticles on the surface of the thick sheet, likely due to the creation of an isotype heterojunction (Fig. 1e). The similar rough surface could also be observed in other MU samples, revealing the uniformity of our strategy (Fig. S1†). To further clarify the morphological characteristics of the g-C3N4 homojunction, TEM images of MU21 are depicted in Fig. 1f, g and S2.† In Fig. 1f, the colour variations aid in distinguishing different components within MU21. The dark regions may correspond to the stacked structure of melamine-derived g-C3N4 layers, while the lighter regions may represent urea-derived g-C3N4 porous nanosheets.22,23 The surface of these observations closely align with the SEM results, confirming the cohesive integration of melamine- and urea-derived components within the structure. Moreover, the interface between different components could be seen in the high-magnification TEM image in Fig. 1g. Fig. S2† also showed the uniformity of intimately contact between the boundaries of the two components, further could be confirmed the successful establishment of the homojunction.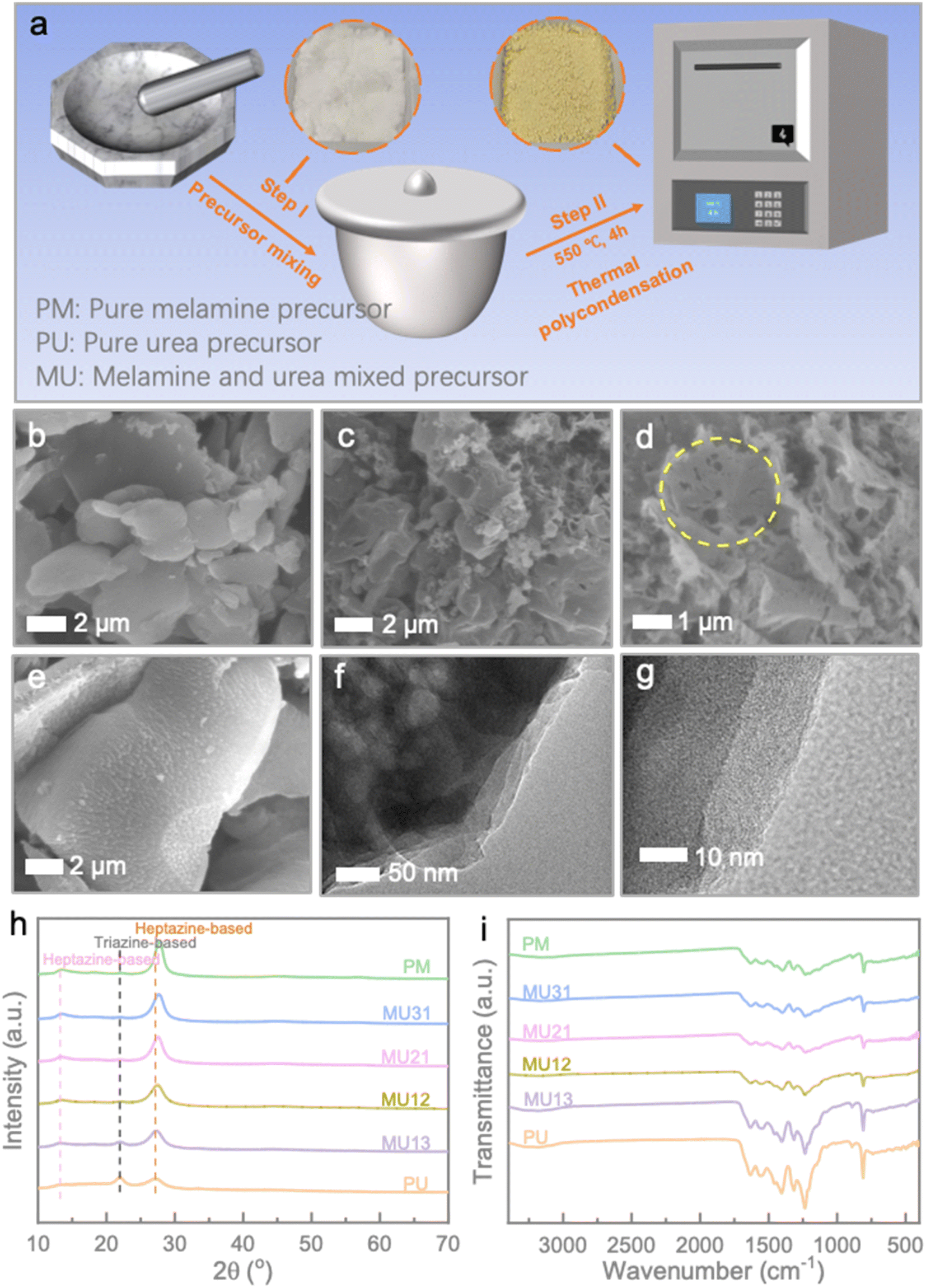 | ||
| Fig. 1 (a) Schematic illustration of the synthesis of the different g-C3N4-based samples. SEM images of (b) PM, (c and d) PU, and (e) MU21. (f) and (g) TEM images of MU21 at low and high magnifications. (h) XRD patterns and (i) FT-IR curves of synthesized samples. | ||
The crystalline phases of the various g-C3N4 samples were confirmed through X-ray diffraction analysis (XRD) patterns (Fig. 1h). The diffraction peaks at around 13.2° and 27.1° correspond to the (100) and (002) crystal planes of the heptazine-based phase, resulting from the in-plane packing motifs of heptazine units and the interlayer stacking of conjugated aromatic systems (JCPDS 87-1526), respectively.24,25 In the case of PU, the diffraction peak at 21.8° can be attributed to the triazine-based g-C3N4 phase.26 As the melamine content increases, a slight rightward shift is observed in the (002) plane peak, suggesting a reduction in interlayer distance.27 Also, the higher triazine content is accompanied by the lack of parallel stacking of g-C3N4 layer. The lone pair electrons of sp2 N within the triazine ring would repel each other and lead to the random rotation of the closed structural unit.28 This can be also illustrated by the thinner SEM morphology of PU than that of PM and MU21. Moreover, the MU series exhibits a less pronounced intensity of the triazine-based phase and a sharper intensity of the heptazine-based phase as the melamine/urea ratio increases, indicating the successful construction of g-C3N4 isotype heterojunction. The differences in local structures can be further analysed by Raman spectra. The characteristic peak around 975 cm−1 and 1160 cm−1 represented the symmetric N-breathing mode of heptazine units and the hydrogen bonding between heptazine-based layers, respectively. The samples with a high melamine/urea ratio exhibited stronger intensities at the two peaks, confirming the phase change of g-C3N4 demonstrated above (Fig. S3†).29,30 The FT-IR spectra of the obtained g-C3N4 samples exhibit similar characteristics (Fig. 1i). The absorption peak at approximately 808 cm−1 corresponds to the breathing mode of the triazine ring. Characteristic peaks in the range of 1200–1700 cm−1 arise from the stretching vibrations of aromatic C–N heterocycles. Additionally, the peaks at 891 cm−1 and in the range of 3100–3300 cm−1 can be attributed to the deformation mode of tris-triazine units and the stretching vibration of N–H in C–NH–C units.21
To investigate the optical absorption characteristics of the prepared samples, UV-vis absorption spectroscopy was conducted (Fig. 2a). In comparison to PM, the MUxy series exhibits a noticeable redshift and enhanced light absorption capacity, with the degree of enhancement corresponding to the increase in urea content. The calculated band gap energies of the samples are presented in Fig. S4.† PM displays a band gap of 2.74 eV, while PU and MU21 exhibit band gaps of 2.43 eV and 2.63 eV, respectively. These band gaps offer promising opportunities for the construction of isotype heterojunction within the MUxy samples. Fig. 2b displays the photoluminescence (PL) spectra of the PM, PU, and MU21 samples. As anticipated from the UV-vis results, the PL peak of PU undergoes a red shift, attributed to the reduction in the optical band gap compared to that of PM. In contrast, MU21 exhibits an intermediate PL peak due to the formation of the isotype heterojunction. Furthermore, Fig. 2c illustrates the time-resolved PL (TRPL) of the samples. The time decay plots of the samples follow tri-exponential decay functions, and the calculated average time constants (τave = (A1τ12 + A2τ22 + A3τ32)/A1τ1 + A2τ2 + A3τ3) are found to be 0.61 ns, 0.58 ns, and 0.77 ns for PM, PU, and MU21 samples, respectively. Although the urea-based component is expected to make MU21 less crystalline than that of PM, the longer charge lifetime of MU21 should be attributed to the different band potentials of the two components in MU21. The homojunction structure of MU21 drive the conduction band (CB) electrons and valence band (VB) holes to the opposite direction rather than recombine back, leading to the longer charge lifetime. As an analogy, PM and PU with no such homojunction structure showed shorter carrier lifetime. Fig. 2d shows typical adsorption–desorption isotherms of nitrogen at 77 K for the prepared samples. In contrast to the surface area of 9.72 m2 g−1 observed for PM, PU and MU21 display significantly larger surface areas of 20.97 m2 g−1 and 18.78 m2 g−1, respectively. Notably, the larger surface area implies the presence of more exposed active sites, facilitating ion access and active participation in electrochemical redox reactions.31 It's worth highlighting the potential for charge separation in the MU21 electrode. When the two components of MU21 make contact, there is a bending of bands, resulting in the creation of an interfacial electric field at the interface (for more details, see below). This electric field acts as a driving force, facilitating the migration of charges and expediting ion transfer. As a result, it is anticipated that MU21 will demonstrate improved electrochemical performance (see further).32
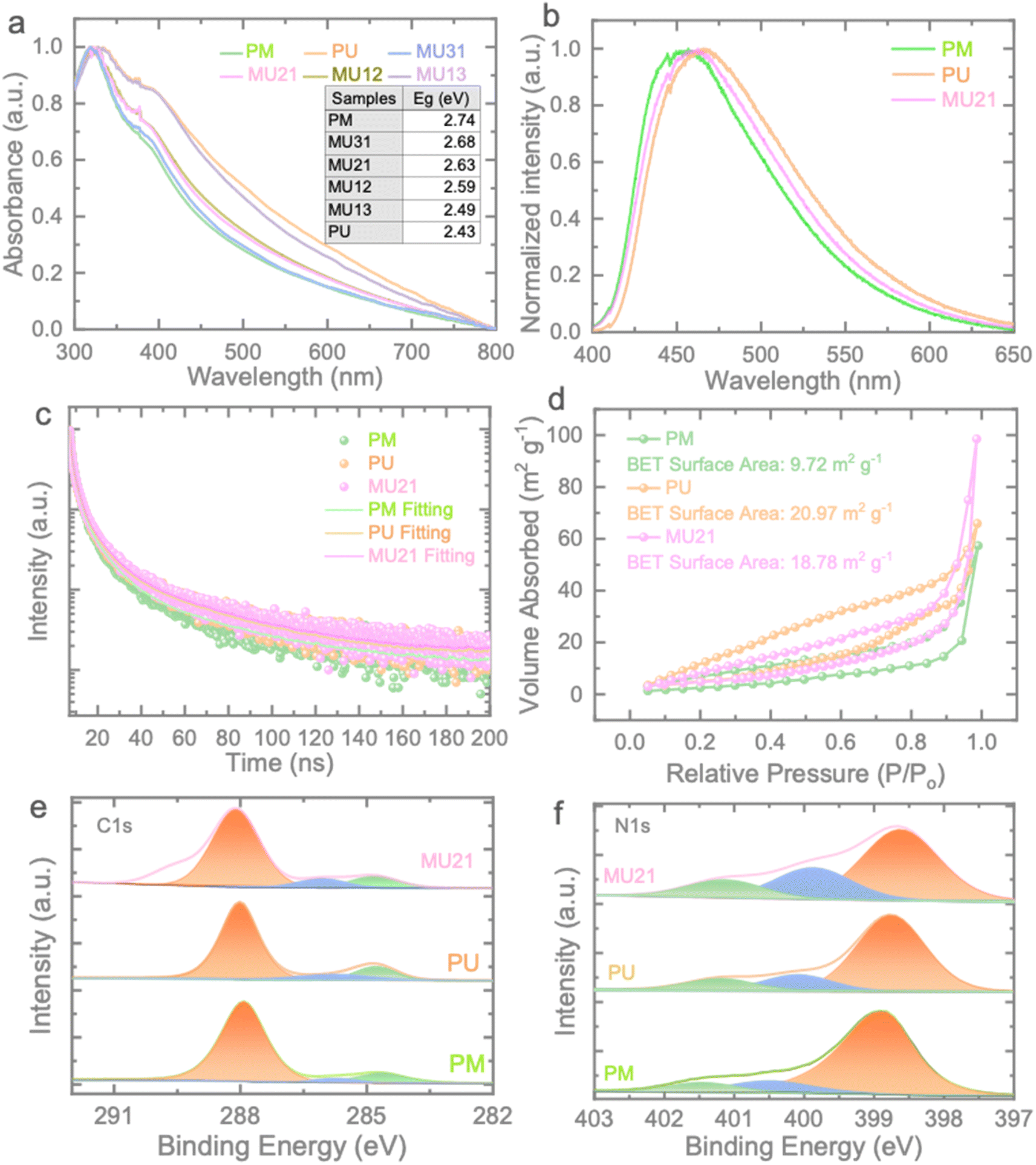 | ||
| Fig. 2 (a) UV-vis spectrum of the prepared samples. (b and c) PL and TRPL of PM, PU and MU21 samples. (d) N2 adsorption–desorption isotherms of PM, PU, and MU21. XPS spectra of MU21: (e) C 1s and (f) N 1s. | ||
The surface compositions of PM, PU and MU21 are examined by XPS (Fig. 2e and f). The C 1s spectrum of PM can be divided into three peaks within the range of 280–292 eV (Fig. 2e). The main peak centered at 288.11 eV could be attributed to N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N sp2 C, while the other two located at 284.8 eV and 286.0 eV could be assigned to the surface adventitious carbon and C–NHx group, respectively.33,34 These peaks can be also observed in the C 1s spectra of PU and MU21, indicating the typical structure features of the synthesized g-C3N4 samples. The corresponding N 1s spectra of the three samples are composed of three peaks (Fig. 2f). The peaks around 399.0 eV originate from sp2 N bonded within both triazine and heptazine rings and the peaks around 400.3 eV represent N–3C the in the heptazine unit.35 The binding energy around 401.4 eV corresponds to C–N–H group in the framework.27 Notably, the peak area of N–3C increases with the higher melamine content, which confirms the feasibility of our homojunction construction strategy.
N sp2 C, while the other two located at 284.8 eV and 286.0 eV could be assigned to the surface adventitious carbon and C–NHx group, respectively.33,34 These peaks can be also observed in the C 1s spectra of PU and MU21, indicating the typical structure features of the synthesized g-C3N4 samples. The corresponding N 1s spectra of the three samples are composed of three peaks (Fig. 2f). The peaks around 399.0 eV originate from sp2 N bonded within both triazine and heptazine rings and the peaks around 400.3 eV represent N–3C the in the heptazine unit.35 The binding energy around 401.4 eV corresponds to C–N–H group in the framework.27 Notably, the peak area of N–3C increases with the higher melamine content, which confirms the feasibility of our homojunction construction strategy.
The electrochemical performance of the prepared g-C3N4 samples was investigated using 2032 type coin cells (Fig. S5†), and detailed information can be found in the Experimental section. Fig. 3a, b and S6† present the results of cyclic voltammetry (CV) measurements conducted at various scan rates ranging from 50 to 1000 mV s−1. At a low scan rate of 50 mV s−1, the CV curves of the samples exhibit a similar quasi-rectangular shape, spanning a voltage window between 0.2 and 1.7 V. As the scan rate increases, ion diffusion becomes limited, resulting in shifts of the anodic and cathodic peaks toward more negative and positive potentials, respectively. However, even at high scan rates (as shown in Fig. 3b), no significant distortion in the CV curve of MU21 is observed, indicating its potential for high-rate charge storage. Comparatively, MU21 displays the largest enclosed CV area (Fig. S7†) among all the tested scan rates, signifying the highest specific capacitance when compared to the other samples.
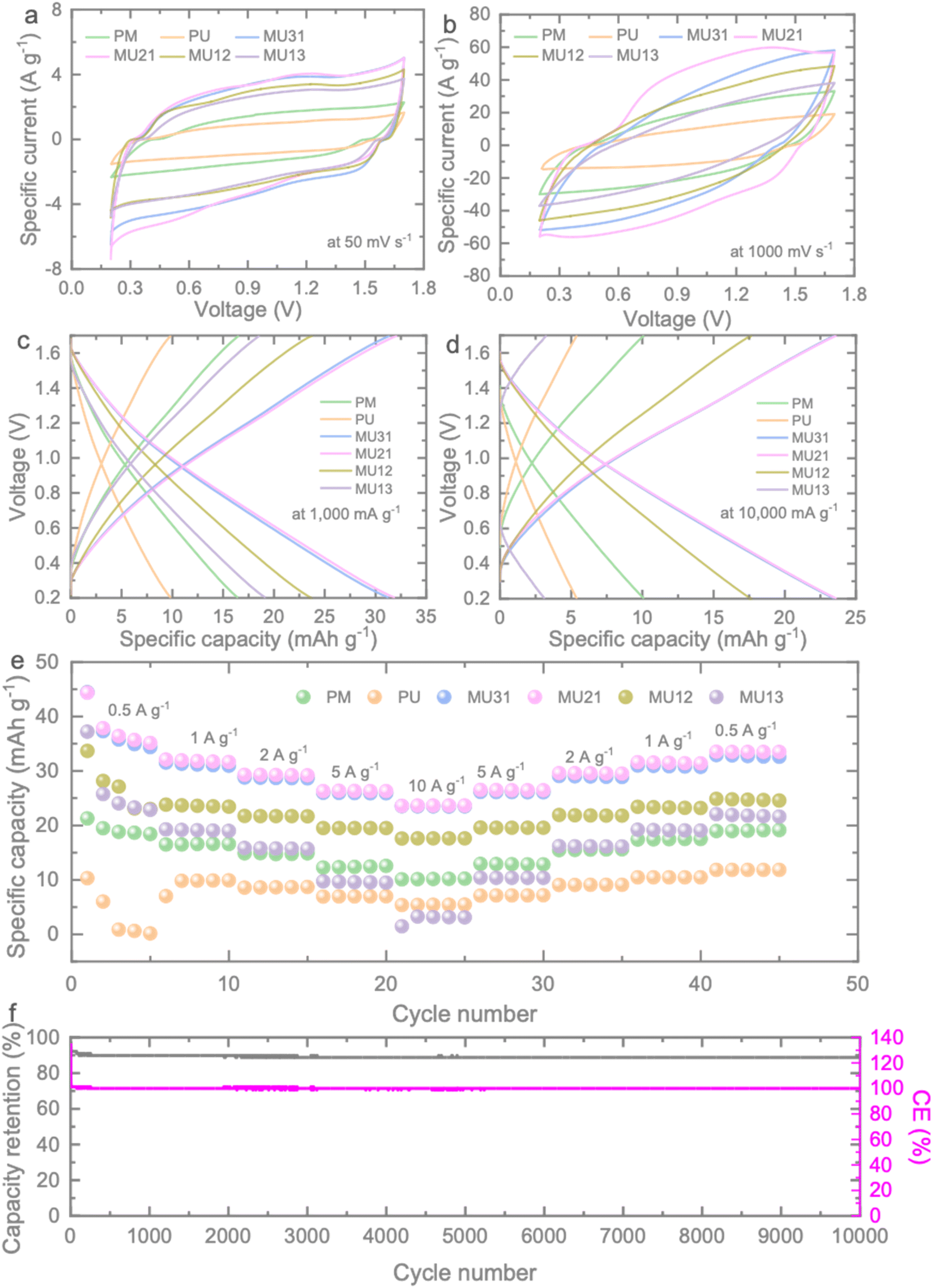 | ||
| Fig. 3 Comparative CV curves of the prepared ZICs at different scan rates of (a) 50 mV s−1, (b) 1000 mV s−1. Comparative GCDs of the ZICs at specific currents of (c) 1000 mA g−1 and (d) 10000 mA g−1. (e) Rate tests of the ZICs. (f) Cycling test at 5000 mA g−1 of the ZIC using MU21 as cathode. | ||
The electrochemical performance of the ZICs was further substantiated through galvanostatic charge–discharge (GCD) measurements conducted over a range of current densities from 500 to 10000 mA g−1. The near-triangular GCD curves align with the findings from the CV results (Fig. 3b and c). When examining the specific capacity at 1000 mA g−1, it's evident that MU21 outperforms PM (16.47 mA h g−1) and PU (9.81 mA h g−1), boasting the highest gravimetric capacity of 31.63 mA h g−1. Moreover, MU21-based ZIC maintains an impressive high-rate capacity of 23.64 mA h g−1 even at the challenging rate of 10000 mA g−1. This rate is four times higher than that of PU (5.42 mA h g−1) and twice that of PM (10.17 mA h g−1), as illustrated in Fig. 3d. Fig. 3e illustrates rate tests results that summarize the specific capacities of the ZICs with the different prepared g-C3N4 samples across all tested specific currents. MU21-based ZIC consistently outperforms the others under these conditions, corroborating the findings from the CV results. Furthermore, MU21 demonstrates a remarkable recovery rate of 99.3%, reaching a gravimetric capacity of 31.41 mA h g−1 when the specific current is reverted to 1000 mA g−1. This underscores the highly electrochemical reversibility of MU21. The enhanced specific capacity exhibited by MU21 can be attributed to the surface area and porous structure of the electrode, which provide a greater number of active sites for electrochemical reactions. Additionally, the optimized triazine-to-heptazine ratio in MU21 emphasizes the significance of isotype heterojunctions in augmenting the charge storage capabilities of the electrode. Consequently, the cycling stability of MU21 was assessed at 5000 mA g−1 (Fig. 3f). The material initiates with a capacity of 42.38 mA h g−1 and maintains a capacity of 37.62 mA h g−1 with a remarkable coulombic efficiency of 99.9% after 10000 cycles. This observation underscores the robust stability of the electrode during cycling.
To investigate the enhanced photocharging capabilities of the optimized MU21 electrodes, we constructed Photo-ZICs, which featured MU21 photocathodes paired with Zn anodes in an optical coin cell design (Fig. S5b†). However, to ensure efficient photocharge generation, separation, and transportation kinetics, we applied a pre-coating of TiO2 electron transfer onto carbon felt before casting MU21 to form MU21/TiO2. This design is expected to facilitate efficient charge transfer kinetics, a requirement for Photo-ZICs (for more details, see below). To assess the performance of the Photo-ZICs, we conducted CV experiments, as depicted in Fig. 4a and b. The consistent shape of the CV curves confirms that the electrode structure remains unaltered under light conditions. Notably, the substantial increase in the CV curve area when exposed to light underscores the heightened photoactivity of the electrode materials.36 This heightened activity is evident in a 26.50% capacity increase at a scan rate of 200 mV s−1, which is higher than that of its PM (14.95%)/PU (16.88%) counterpart (Table S2 and Fig. S8†). Furthermore, the similar shape of the CV curves at a high scan rate of 1000 mV s−1 under different light conditions (Fig. 4b) emphasizes the robust stability of the MU21 photoelectrode. Fig. S9† illustrates capacity enhancements under light compared to dark conditions, and significant capacity improvements were recorded at each scan rate. Table S2† illustrates capacity enhancements under light compared to dark conditions, and significant capacity improvements were recorded at each scan rate. Similarly, the areal specific capacitance (Fig. 4c and d) and rate-based areal specific capabilities (Fig. 4e) of the photoelectrode under various current densities in both dark and illuminated conditions are revealed through galvanostatic CD tests. The capacity enhancements observed in the charge–discharge curves of the Photo-ZICs (Fig. 4c and d) align with the expectations set by the CV curves (Fig. 4a and b). The rate test measurements of Photo-ZICs clearly demonstrate significant capacity enhancements across different specific currents and cycles. For instance, the calculated capacity enhancement is approximately 26.23% under light compared to dark, even at very high specific currents of 10000 mA g−1. However, the initial increase in the specific capacity of our Photo-ZICs under light conditions during the first few cycles may be attributed to the further activation of materials when exposed to light. To further investigate the impact of light during CD, we conducted CD tests under various conditions: dark charge & dark discharge, light charge & light discharge, and light charge & dark discharge, all at the same specific current of 2000 mA g−1, as illustrated in Fig. 4f. As expected, under light, both charge and discharge times increase compared to dark conditions, indicating an increase in capacity. Notably, when the cell is charged in light but discharged in the dark, there is only a negligible decrease in discharge time compared to the light discharge measurement. This implies the ability to retain enhanced capacities under light even when drawing energy in dark conditions. The influence of light on Photo-ZIC, in terms of charge carrier migration and reaction kinetics, is evaluated through electrochemical impedance spectroscopy (EIS) measurements under both dark and light conditions (Fig. 4g). In the impedance spectra, the semicircle at the high-frequency section and the straight line at the low-frequency region correspond to charge transfer resistance (Rct) and Warburg impedance, respectively. It is well known that TiO2 and g-C3N4 have low conductivity. In this case, the generation of electron–hole pairs provided extra charge for the photo-cathode and thus increase its conductivity under illumination. Evidently, the MU21-based Photo-ZIC exhibits a smaller Rct and a more vertically aligned straight line in the low-frequency region under light illumination. These observations indicate faster reaction kinetics and more efficient ion migration, elucidating the higher capacity enhancement observed in high-rate performance scenarios.37
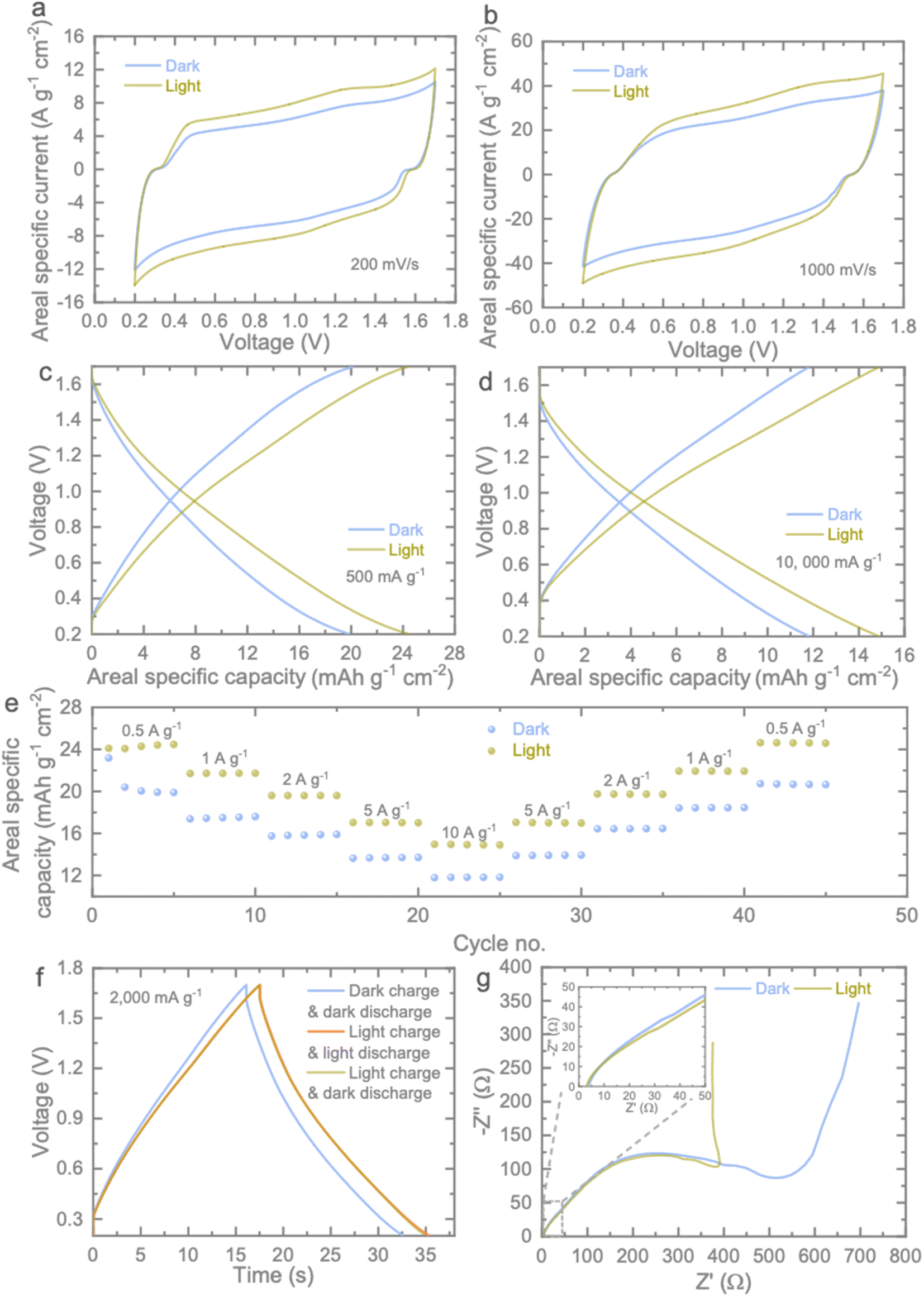 | ||
| Fig. 4 CVs of the Photo-ZICs at scan rates of (a) 200 mV s−1 and (b) 1000 mV s−1 under both dark and light conditions. Comparative CD curves of Photo-ZICs at specific currents of (c) 500 mA g−1 and (d) 10000 mA g−1 in both dark and light conditions. (e) Rate tests of the Photo-ZIC in dark and light conditions reveal significant capacity enhancements under light compared to dark at each specific current. (f) CD curves at 2000 mA g−1 for dark charge & dark discharge, light charge & light discharge, and light charge & dark discharge conditions. (g) Nyquist plots of the Photo-ZICs in both dark and light conditions. | ||
To gain further insight into the photo-charging effect of the Photo-ZIC, we conducted an extended cycling experiment. After the initial activation, we cycled the device in the dark for 4000 cycles, followed by an additional 4000 cycles under light conditions, as illustrated in Fig. 5a. During the dark cycling phase, the observed areal specific capacity remained stable at approximately 15 mA h g−1 cm−2 over the initial 4000 cycles. Subsequently, the cell continued cycling for an additional 4000 cycles under light conditions. As anticipated, the cell exhibits a superior capacity compared to cycling in the dark, with an initial areal specific capacity of 18 mA h g−1 cm−2 and ultimately maintaining a areal specific capacity of 17.12 mA h g−1 cm−2 under light. These results underscore the exceptional cycling stability of our MU21 photocathode. Moreover, the slower self-discharge rates of the Photo-ZICs in light compared to those in the dark validate the concurrent photo-charging effect (Fig. 5b). It's important to note that our Photo-ZICs exhibit negligible self-discharge in comparison to supercapacitor devices.46,47 In contrast, the capacity enhancement steadily rises when the electrode is exposed to light. Furthermore, we conducted electrochemical performance assessments of our Photo-ZIC at various temperatures to investigate potential capacity improvements induced by photothermal effects. Fig. 5c presents CVs obtained at 100 mV s−1 with different temperatures, including 25 °C, 30 °C, and 40 °C. We maintained a maximum temperature of 40 °C, as our LED solar simulator does not generate significant heat (remaining below 40 °C) even during continuous illumination. Interestingly, we observed no significant increase in the enclosed CV area with rising temperatures, unlike the enhancement (∼28% at 100 mV s−1) seen under light illumination (Table S2†). Specifically, a mere 5.74% increase in capacity at 40 °C compared to 25 °C suggests that the capacity improvements are primarily attributed to the photocharging effect rather than photothermal effects. Fig. 5d presents a comparative Ragone plot, illustrating specific energy and specific power. In addition to the photo-enhanced characteristics of our devices, the Photo-ZICs deliver an energy density of 6.55 W h kg−1 at a power density of 748 W kg−1 and maintain 5.97 W h kg−1 at 1506 W kg−1 in dark which are increased to 8.3 W h kg−1 at 766 W kg−1 and 7.53 W h kg−1 at 1538 W kg−1 under light. These performance metrics surpass the majority of reported asymmetric supercapacitors. Fig. 5e provides insight into the photocharging mechanism of a Photo-ZIC, elucidated through the energy band structure of MU21. It is expected that when the photocathode is exposed to light under charging, the band positions of MU21, which are categorized into PU and PM, come into play. Photogenerated electrons in the conduction band (CB) of PU transfer to the CB of TiO2 and ultimately reach the Zn anode through the external circuit for deposition of Zn2+ ions from electrolyte on Zn anode. Conversely, photo-induced holes transfer in the opposite direction in the valence band (VB) of the composite for adsorption of cations from electrolyte. This differential band structure between components facilitates the movement of photo-induced charge carriers across various elements, resulting in the creation of built-in electric fields. These fields drive electrons to migrate through the isotype heterojunctions at the interface, effectively reducing the recombination of photogenerated electrons and holes when exposed to sunlight and offer photo-accelerated performance. Moreover, to demonstrate the practical feasibility of our capacitor, we successfully power a moisture sensor using our capacitors (Fig. 5f).
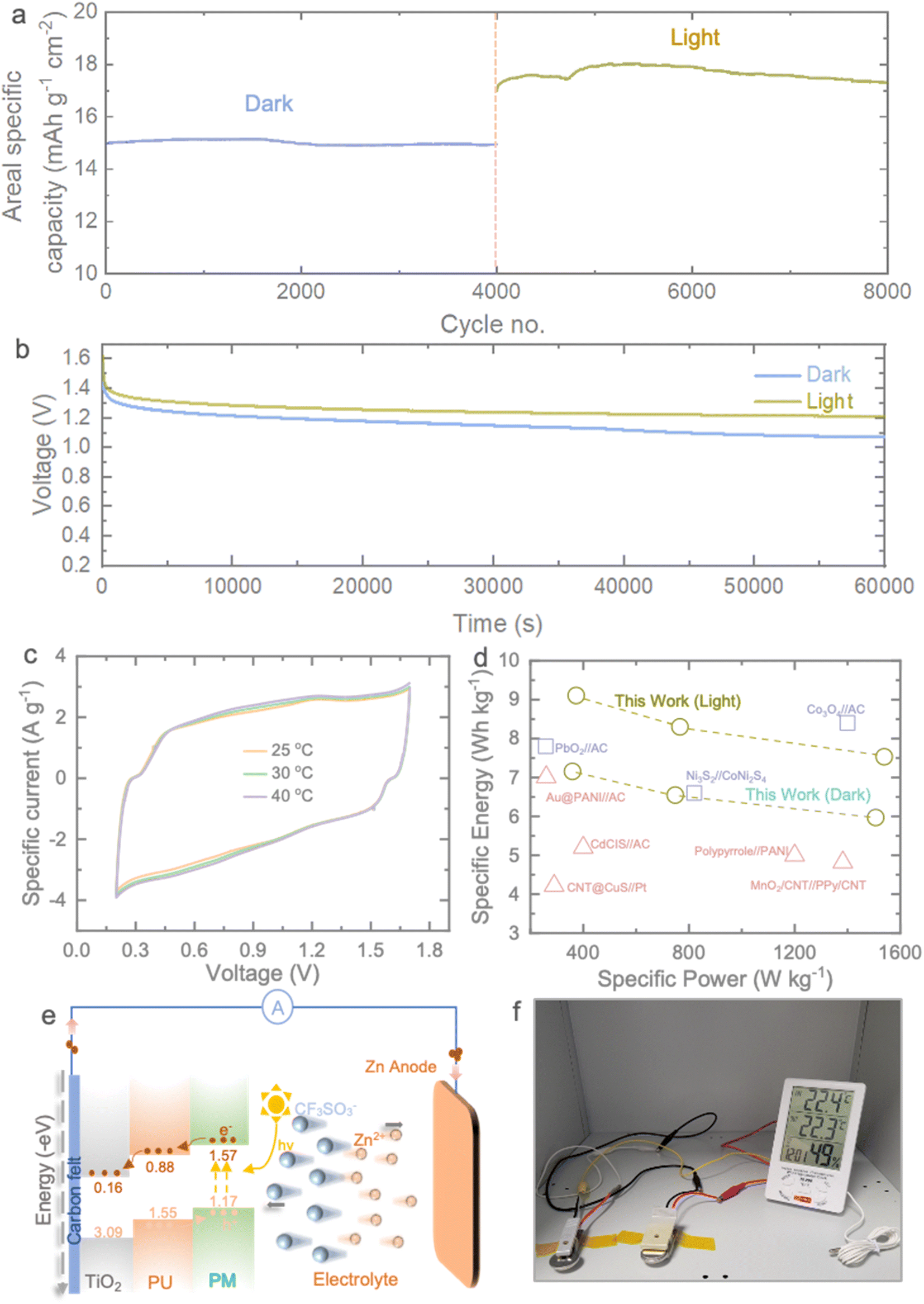 | ||
| Fig. 5 (a) Long-term cycling test at 5000 mA g−1 of the Photo-ZIC in the dark for 4000 cycles and light for another 4000 cycles conditions. (b) Self-discharge behaviour of the Photo-ZICs in dark and light conditions. (c) Comparative CVs of the Photo-ZIC at different temperatures of 25 °C, 30 °C and 40 °C, respectively. (d) Comparative Ragone plot of our Photo-ZIC with other asymmetric supercapacitors reported in the literature: PbO2//AC,38 Ni3S2//CoNi2S4,39 Co3O4//AC,40 Au@PANI//AC,41 CdCSI//AC,42 CNT@CuS//Pt,43 PPy@PANI,44 MnO2/CNT//PPy/CNT.45 (e) Schematic illustration Photo-charge enhanced capacity of the Photo-ZIC under light. (f) Photograph of powering a moisture sensor with the prepared Photo-ZICs. | ||
Conclusion
In summary, this report outlines a strategy for developing high-performance semiconductor isotype heterojunctions based on g-C3N4. This strategy serves two main purposes: enhancing capacity and regulating interfacial electronic structures to facilitate ion diffusion for rapid reaction kinetics. The presented Photo-ZIC system, utilizing the isotype g-C3N4 heterojunction electrode coupled with a TiO2-coated carbon felt current electrode, demonstrates effective photo-accelerated charge storage performance. This results in a significant increase in capacity under illumination compared to dark conditions. Furthermore, the Photo-ZIC system exhibits a slow self-discharge rate, both in the absence of light and when exposed to it, which is considerably lower than that of conventional supercapacitors along with improved energy density and power density. Therefore, as a proof-of-concept experiment, the introduction of the isotype heterojunction strategy represents a promising approach for optimizing light absorption and charge storage activities in highly efficient metal-ion capacitors.Data availability
All scientific data has been reported in the main manuscript and ESI.† The raw data files are available upon request from the corresponding authors.Conflicts of interest
The authors declare no competing financial interest.References
- A. Sharma, A. Hosseini-Bandegharaei, N. Kumar, S. Kumar and K. Kumari, J. CO2 Util., 2022, 65, 102205 CrossRef CAS.
- N. H. Alotaibi, J. Phys. Chem. Solids, 2022, 171, 110984 CrossRef CAS.
- B. Deka Boruah, A. Mathieson, S. K. Park, X. Zhang, B. Wen, L. Tan, A. Boies and M. De Volder, Adv. Energy Mater., 2021, 11, 2100115 CrossRef CAS.
- Z. Tian, Z. Sun, Y. Shao, L. Gao, R. Huang, Y. Shao, R. B. Kaner and J. Sun, Energy Environ. Sci., 2021, 14, 1602–1611 RSC.
- J. Lv, J. Xie, A. G. Abdelkader Mohamed, X. Zhang and Y. Wang, Chem. Soc. Rev., 2022, 51, 1511–1528 RSC.
- X. Liu, H. Andersen, Y. Lu, B. Wen, I. P. Parkin, M. De Volder and B. D. Boruah, ACS Appl. Mater. Interfaces, 2023, 15, 6963–6969 CrossRef CAS.
- J. Li, Y. Zhang, Y. Mao, Y. Zhao, D. Kan, K. Zhu, S. Chou, X. Zhang, C. Zhu, J. Ren and Y. Chen, Angew. Chem., Int. Ed., 2023, 62, e202303056 CrossRef CAS.
- L. Li, F. Ma, C. Jia, Q. Li, X. He, J. Sun, R. Jiang, Z. Lei and Z.-H. Liu, Adv. Sci., 2023, 10, 2301682 CrossRef CAS.
- P. Chen, C. Cao, C. Ding, Z. Yin, S. Qi, J. Guo, M. Zhang and Z. Sun, J. Power Sources, 2022, 521, 230920 CrossRef CAS.
- S. Mozaffari and M. R. Nateghi, Top. Curr. Chem., 2022, 380, 28 CrossRef CAS PubMed.
- J. Wang, Y. Wang, C. Zhu and B. Liu, ACS Appl. Mater. Interfaces, 2022, 14, 4071–4078 CrossRef CAS PubMed.
- Y. Zhao, X. Linghu, Y. Shu, J. Zhang, Z. Chen, Y. Wu, D. Shan and B. Wang, J. Environ. Chem. Eng., 2022, 10, 108077 CrossRef CAS.
- H. Fu, Q. Wen, P.-Y. Li, Z. Wang, Z. He, C. Yan, J. Mao, K. Dai, X. Zhang and J. Zheng, Small Methods, 2022, 6, 2201025 CrossRef CAS PubMed.
- Y.-H. Chew, B.-J. Ng, X. Y. Kong, L. K. Putri, J.-Y. Tang, L.-L. Tan and S.-P. Chai, Sustain. Energy Fuels, 2020, 4, 1822–1827 RSC.
- L. Ma, Y. Zhang, S. Zhang, L. Wang, C. Zhang, Y. Chen, Q. Wu, L. Chen, L. Zhou and W. Wei, Adv. Funct. Mater., 2023, 33, 2305788 CrossRef CAS.
- F. Zhan, Y. Liu, K. Wang, X. Yang, M. Liu, X. Qiu, J. Li and W. Li, ACS Appl. Mater. Interfaces, 2019, 11, 39951–39960 CrossRef CAS PubMed.
- D. H. L. Ng, S. Li, J. Li, J. Huang, Y. Cui, J. Lian and C. Wang, Nanomaterials, 2022, 12, 3762 CrossRef CAS PubMed.
- E. I. Come Zebra, H. J. van der Windt, G. Nhumaio and A. P. C. Faaij, Renew. Sustain. Energy Rev., 2021, 144, 111036 CrossRef CAS.
- S. Paul Ayeng'o, T. Schirmer, K.-P. Kairies, H. Axelsen and D. Uwe Sauer, Sol. Energy, 2018, 162, 140–152 CrossRef.
- X. Lu, L. Tao, K. Qu, A. Amardeep and J. Liu, Adv. Funct. Mater., 2023, 33, 2211736 CrossRef CAS.
- B. D. Boruah, A. Mathieson, B. Wen, C. Jo, F. Deschler and M. De Volder, Nano Lett., 2020, 20, 5967–5974 CrossRef CAS.
- L. X. Nong, V. H. Nguyen, T. Lee and T. D. Nguyen, Top. Catal., 2023, 66, 275–288 CrossRef CAS.
- G. Liao and W. Yao, Diamond Relat. Mater., 2022, 128, 109227 CrossRef CAS.
- C. Liu, H. Huang, W. Cui, F. Dong and Y. Zhang, Appl. Catal., B, 2018, 230, 115–124 CrossRef CAS.
- J. Wang, Y. Yu, J. Cui, X. Li, Y. Zhang, C. Wang, X. Yu and J. Ye, Appl. Catal., B, 2022, 301, 120814 CrossRef CAS.
- H. Jing, M. You, S. Yi, T. Li, H. Ji, Y. Wang, Z. Zhang, R. Zhang, D. Chen and H. Yang, ChemSusChem, 2020, 13, 827–837 CrossRef CAS PubMed.
- X. Zhang, P. Ma, C. Wang, L. Gan, X. Chen, P. Zhang, Y. Wang, H. Li, L. Wang, X. Zhou and K. Zheng, Energy Environ. Sci., 2022, 15, 830–842 RSC.
- M. Deifallah, P. F. McMillan and F. Corà, J. Phys. Chem. C, 2008, 112, 5447–5453 CrossRef CAS.
- L. Yue, C. Fang, Z. Yan, Z. Xu, G. Wang and Q. Liu, J. Environ. Chem. Eng., 2022, 10, 107881 CrossRef CAS.
- W.-J. Ong, L.-L. Tan, S.-P. Chai and S.-T. Yong, Dalton Trans., 2014, 44, 1249–1257 RSC.
- X. Liu, Y. Hou, M. Tang and L. Wang, Chin. Chem. Lett., 2023, 34, 107489 CrossRef CAS.
- S. Yang, C. Liu, H. Li, S. Wang, J. Choi and L. Li, Energy Storage Mater., 2021, 39, 278–286 CrossRef.
- W. Wang, L. Du, R. Xia, R. Liang, T. Zhou, H. Kay Lee, Z. Yan, H. Luo, C. Shang, D. Lee Phillips and Z. Guo, Energy Environ. Sci., 2023, 16, 460–472 RSC.
- Y. Chai, Q. Liu, L. Zhang, J. Ren and W.-L. Dai, Chin. J. Chem., 2017, 35, 173–182 CrossRef CAS.
- C. Yang, Z. Xue, J. Qin, M. Sawangphruk, X. Zhang and R. Liu, Appl. Catal., B, 2019, 259, 118094 CrossRef CAS.
- B. D. Boruah, B. Wen and M. De Volder, ACS Nano, 2021, 15, 16616–16624 CrossRef CAS PubMed.
- P. Hu, X. Luo, T. Hu, S. Chen, D. Li, Y. Chen and F. Li, ACS Appl. Mater. Interfaces, 2023, 15, 5180–5190 CrossRef CAS.
- N. Yu, L. Gao, S. Zhao and Z. Wang, Electrochim. Acta, 2009, 54, 3835–3841 CrossRef CAS.
- S. J. Patil, J. H. Kim and D. W. Lee, Chem. Eng. J., 2017, 322, 498–509 CrossRef CAS.
- B. Vidyadharan, R. A. Aziz, I. I. Misnon, G. M. Anil Kumar, J. Ismail, M. M. Yusoff and R. Jose, J. Power Sources, 2014, 270, 526–535 CrossRef CAS.
- Y. Tan, Y. Zhang, L. Kong, L. Kang and F. Ran, J. Alloys Compd., 2017, 722, 1–7 CrossRef CAS.
- M. Farzi, M. Moradi, S. Hajati, J. Toth and A. Kazemzadeh, Synth. Met., 2021, 278, 116815 CrossRef CAS.
- T. Zhu, B. Xia, L. Zhou and X. W. David Lou, J. Mater. Chem., 2012, 22, 7851–7855 RSC.
- F. Fusalba, P. Gouérec, D. Villers and D. Bélanger, J. Electrochem. Soc., 2001, 148, A1 CrossRef CAS.
- C. Ren, Y. Yan, B. Sun, B. Gu and T.-W. Chou, J. Colloid Interface Sci., 2020, 569, 298–306 CrossRef CAS PubMed.
- N. R. Chodankar, D. P. Dubal, Y. Kwon and D.-H. Kim, NPG Asia Mater., 2017, 9, e419 CrossRef CAS.
- C. Zhou, Y. Zhang, Y. Li and J. Liu, Nano Lett., 2013, 13, 2078–2085 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section; SEM images (Fig. S1); TEM images (Fig. S2); Raman spectra (Fig. S3); Tauc plots (Fig. S4); digital images of coin cell and optical coin cell (Fig. S5); CVs (Fig. S6); CV area with respect scan rate plots (Fig. S7); CVs (Fig. S8), capacity enhancement under light (Fig. S9). See DOI: https://doi.org/10.1039/d4ta03938f |
| This journal is © The Royal Society of Chemistry 2024 |