
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal–organic frameworks and their derivatives for the electrochemical CO2 reduction reaction: insights from molecular engineering
Xiaoming
Liu
 a,
Xuan-He
Liu
*a,
Xiangrui
Zhang
a,
Huan
Wang
b and
Qinglan
Zhao
*c
a,
Xuan-He
Liu
*a,
Xiangrui
Zhang
a,
Huan
Wang
b and
Qinglan
Zhao
*c
aSchool of Science, China University of Geosciences (Beijing), Beijing 100083, China. E-mail: liuxh@cugb.edu.cn
bKey Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), Nankai University, Tianjin 300071, China
cDepartment of Chemical and Biological Engineering, The Hong Kong University of Science and Technology, Clear Water Bay, Kowloon, Hong Kong, China. E-mail: keqzhao@ust.hk
First published on 29th June 2024
Abstract
Excessive fossil fuel consumption has led to a rapid increase in CO2 concentration, posing a threat to the global environment. The electrochemical conversion of CO2 back into valuable carbon-containing products offers a promising solution; however the lack of efficient electrocatalysts remains a challenge for high-efficiency CO2 reduction reaction (CO2RR). Metal–organic frameworks (MOFs) have emerged as promising electrocatalysts owing to their superior activity and well-defined active sites and are recognized as model electrocatalysts for the fundamental study of electrocatalytic reaction mechanisms. In this review, focusing on the roles of metals and the coordination environment, we have discussed the molecular engineering of MOF electrocatalysts for the CO2RR, including regulation of metal nodes, modulation of surrounding organic ligands, and post-modification of MOFs. In addition, stability of MOFs and transformation strategies through the wet chemistry method and pyrolysis treatment to obtain MOF derivatives are summarized. Metal centers and the coordination environment of N atoms and other heteroatoms of MOF derivatives are discussed for their performance in the CO2RR. Furthermore, computational simulations and advanced characterization studies are also summarized to understand the correlation between the structure and performance of MOFs and their derivatives. By overviewing the progress and current challenges of MOF electrocatalysts and their derivatives for the CO2RR, we are aiming to provide insights into design principles and propose future directions for high-efficiency electrocatalysts, paving the way toward carbon neutrality.
1. Introduction
Since the industrial revolution, there has been a dramatic increase in human demand for energy,1 leading to a great energy crisis. Furthermore, the excessive consumption of fossil fuels such as coal, oil and natural gas has resulted in a rapid increase in the global concentration of CO2 and related environmental issues.2 For example, the high concentration of greenhouse gases reduces the infrared radiation emitted into space, leading to the warming of the earth, melting glaciers, and ultimately an increase in sea levels.3 As a result, the concept of carbon neutrality was proposed by the Paris Agreement,4 to convert the released CO2 back to carbon products to achieve net zero emission by 2050.In recent decades, tremendous efforts have been dedicated to CO2 conversion with various strategies including photochemical,5 thermochemical,6 biochemical,7,8 chemical reforming,9–11 mineralization,12 electrochemical13–15 methods, and so on. Among these, the electrochemical CO2 reduction reaction (CO2RR) stands out as a promising approach to achieving carbon neutrality, as it can be conducted under mild aqueous solution conditions with the input of renewable electricity.16,17 In this regard, numerous constructive research studies have been reported to achieve high-performance electrocatalytic CO2RR toward formation of valuable reduced carbon products, by enhancing selectivity,18,19 energy efficiency,20,21 and stability.22,23 In the past few years, significant efforts have been devoted to developing efficient electrocatalysts for the CO2RR, such as metal nanoparticles (e.g., Ag, Au, Cu, Zn and Sn),24–29 metal complexes (e.g., oxides, sulfides and nitrides),30–32 carbonaceous materials,33 and metal–organic frameworks.34,35
Metal–organic frameworks (MOFs), a type of crystalline porous material with a periodic network structure formed by the self-assembly of transition metal ions and organic ligands, have been intensively studied as electrocatalysts for the CO2RR36,37 due to their large specific surface area, tunable structure and ordered structure.38 Furthermore, their well-defined structures and uniform active centers make it easy to construct reliable structural models to understand the mechanism of the CO2RR.39,40 It has been reported that both metal species41,42 and organic linkers43,44 of MOFs can serve as active sites or modifiers to drive electrocatalytic reactions. Besides MOFs, MOF-derived electrocatalysts, especially those with precisely active centers, also play a crucial role in influencing CO2RR activity and selectivity through the manipulation of the electronic structure.45–48
Based on the understanding of the roles of active metal nodes and coordinating organic ligands of MOF electrocatalysts, we discuss the molecular engineering strategy including regulation of metal nodes, modulation of surrounding organic ligands, and post-modification of MOFs for the CO2RR (Fig. 1). Regarding MOF derivatives, we discuss transformation strategies including the wet chemistry method at mild temperature and pyrolysis treatment at high temperature. In particular, the significant impacts of metal centers and the coordination environment surrounded by N atoms and other heteroatoms on performance of the CO2RR are discussed. Advanced structural characterization studies combined with the first-principle density functional theory (DFT) calculations to unravel the active sites for MOFs at atomic and molecular levels are summarized to help understand the relationship between their structure and performance.55–58 Finally, the current challenges and future directions are proposed for advancing MOF-based electrocatalysts for the CO2RR.
 | ||
| Fig. 1 Scheme of the molecular engineering strategy including regulation of metal nodes, modulation of surrounding organic ligands and post-modification of MOFs; in situ characterization and reconstruction; the wet chemistry method and pyrolysis treatment for MOF derivatives for the CO2RR (molecular structures shown in the scheme have been cited from ref. 49–54 with copyright approval from Wiley-VCH and American Chemical Society). | ||
2. Fundamentals of the CO2 reduction reaction
The σ and π34 bonds of linear CO2 molecules endow the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond with a certain degree of triple-bond character, which is thermodynamically stable and chemically inert.3 Therefore, the CO2RR is typically kinetically sluggish,59 especially toward high-energy and value-added carbon products formation with multi-proton and multi-electron transfer steps, such as CH4 (8e−), CH3OH (6e−), C2H4 (12e−), and C2H5OH (12e−).60 Up to now, at least 16 products have been identified in the CO2RR, with their corresponding half reactions listed in Table 1,61–63 leading to the occurrence of complex reactions yielding various carbon products. Moreover, the hydrogen evolution reaction (HER) with an equilibrium potential of 0 V vs. the reversible hydrogen electrode (RHE) hinders the CO2RR as a competition reaction. Therefore, it is a significant challenge to achieve certain desired reduction products with high selectivity and activity.
O bond with a certain degree of triple-bond character, which is thermodynamically stable and chemically inert.3 Therefore, the CO2RR is typically kinetically sluggish,59 especially toward high-energy and value-added carbon products formation with multi-proton and multi-electron transfer steps, such as CH4 (8e−), CH3OH (6e−), C2H4 (12e−), and C2H5OH (12e−).60 Up to now, at least 16 products have been identified in the CO2RR, with their corresponding half reactions listed in Table 1,61–63 leading to the occurrence of complex reactions yielding various carbon products. Moreover, the hydrogen evolution reaction (HER) with an equilibrium potential of 0 V vs. the reversible hydrogen electrode (RHE) hinders the CO2RR as a competition reaction. Therefore, it is a significant challenge to achieve certain desired reduction products with high selectivity and activity.
| Half reactions | Equilibrium potentials (V vs. RHE) | Number |
|---|---|---|
| CO2 + 2H+ + 2e− → HCOOH (aq) | −0.02 | (1) |
| CO2 + 2H+ + 2e− → CO (g) +H2O (l) | −0.10 | (2) |
| CO2 + 8H+ + 8e− → CH4 (g) + 2H2O (l) | 0.17 | (3) |
| CO2 + 4H+ + 4e− → HCHO (aq) + H2O (l) | −1.31 | (4) |
| CO2 + 4H+ + 4e− → C (s) + H2O (l) | 0.21 | (5) |
| CO2 + 6H+ + 6e− → CH3OH (aq) +H2O (l) | 0.03 | (6) |
| 2CO2 + 12H+ + 12e− → C2H4 (g) + 4H2O (l) | 0.08 | (7) |
| 2CO2 + 12H+ + 12e− → C2H5OH (aq) + 3H2O (l) | 0.09 | (8) |
| 2CO2 + 8H+ + 8e− → CH3COOH (aq) + 2H2O (l) | −0.26 | (9) |
| 2CO2 + 10H+ + 10e− → CH3CHO (aq) + 3H2O (l) | 0.05 | (10) |
| 2CO2 + 14H+ + 14e− → C2H6 (aq) + 4H2O (l) | 0.14 | (11) |
| 2CO2 + 2H+ + 2e− → H2C2O4 (aq) | −0.91 | (12) |
| 2CO2 + 16H+ + 16e− → C2H5CHO (aq) + 5H2O (l) | 0.14 | (13) |
| 3CO2 + 18H+ + 18e− → C2H5CH2OH (aq) + 5H2O (l) | 0.21 | (14) |
| 3CO2 + 20H+ + 20e− → C3H8 (g) + 6H2O (l) | 0.09 | (15) |
| 3CO2 + 16H+ + 16e− → (CH3)2CO (aq) + 5H2O (l) | −0.26 | (16) |
| 2H+ + 2e− → H2 (g) | 0 | (17) |
As summarized in Table 2, CO and HCOOH in 2-electron transfer reactions are common reduction products, whose faradaic efficiencies (FEs) reach up to 100% in MOF-based catalysts.50,81,90,110 However, FEs usually drop when multiple steps with more electrons are involved in the reaction system. For example, CH4, an 8-electron transfer product, needs a complex multiple proton-coupled electron transfer (PCET) process, which has lower selectivity for CO2-to-CH4 electroreduction.112,148 At this stage, the reported FEs for CH4 can reach 92% on [Cu4ZnCl4(btdd)3] (Cu4-MFU-4l, H2btdd = bis(1H-1,2,3-triazolo-[4,5-b],[4′,5′-i])dibenzo-[1,4]-dioxin) with trigonal pyramidal Cu(I)N3 active sites.121 C2H4 and C2H5OH, 12-electron transfer products, exhibit even more descending FEs (mostly below 60%) on MOF-based catalysts.125–127,149
| Sample | Main product | FE (%) | Potential (V vs. RHE) | Stability (h) | Ref. |
|---|---|---|---|---|---|
| Ag@UiO-66-SH | CO | 74.0 | −1.10 | 10 | 64 |
| Fe2–N6–C-o | CO | >80.0 | −0.5 to −0.9 | 21 | 65 |
| ZIF-8 | CO | 81.0 | −1.10 | — | 66 |
| CoPc–Cu–O | CO | 85.0 | −0.63 | 10 | 67 |
| PcCu–O8–Zn | CO | 88.0 | −0.70 | >10 | 68 |
| Cu0.5Zn0.5/ZIF-8 | CO | 88.5 | −1.00 | >6 | 69 |
| Fe@BIF-73-NS | CO | 88.6 | −1.8 V vs. Ag/AgCl | 14 | 70 |
| ZIF-A-LD | CO | 90.57 | −1.10 | 10 | 71 |
| Zr-BTB@Hemin-TMA | CO | 92.0 | −1.20 | 3.0 | 72 |
| Ga–N3S–PC | CO | 92.0 | −0.30 | 24 | 73 |
| NCMSH | CO | 92.68 | −0.70 | >24 | 74 |
| Fe–N2+2–C8 | CO | 93.0 | −0.47 | 20 | 75 |
| Fe–N5/DPCF | CO | 93.1 | −0.50 | 25 | 76 |
| Ag/Zr-fcu-MOF-NDC | CO | 94.0 | −1.00 | 1 | 77 |
| CALF20 | CO | 94.5 | −0.97 | ∼2.2 | 78 |
| Co–N2 | CO | 95.0 | −0.68 | 60 | 79 |
| O–Fe–N–C | CO | 95.0 | −0.50 | 30 | 54 |
| Ni-SNC | CO | 95.0 | −0.80 | 24 | 80 |
| Bi-BTC-D | CO | 95.5 | −0.86 | 12 | 81 |
| Ni–N3–C | CO | 95.6 | −0.65 | 10 | 48 |
| 1-NH2 | CO | 96.0 | −2.30 | 300 | 82 |
| CuN3O/C | CO | 96.0 | −0.80 | 15 | 83 |
| Fe1–Ni1–N–C | CO | 96.2 | −0.50 | 10 | 46 |
| CoCp2@MOF-545-Co | CO | 97.0 | −0.70 | 8 | 84 |
| Ni/HNC | CO | 97.2 | −0.70 | 10 | 85 |
| ZIF–NC–Ni–Fe | CO | 97.8 | −0.60 | 50 | 86 |
| NiSA–N2–C | CO | 98.0 | −0.80 | 10 | 34 |
| Cu–Fe–N6–C | CO | 98.0 | −0.70 | 10 | 87 |
| S/Fe–poN4–C | CO | 98.2 | −0.58 | >12 | 88 |
| MOF Ni–Fe | CO | 98.2 | −0.50 | 30 | 50 |
| Ni SAC-1000 | CO | 98.24 | −0.80 | 24 | 89 |
| NiPc–NiO4 | CO | 98.4 | −0.85 | 10 | 90 |
| K-defect-C-1100 | CO | 99.0 | −0.45 | 10 | 91 |
| Fe/Cu–N–C | CO | ∼99.0 | −0.80 | 60 | 92 |
| Fe–S1N3 | CO | 99.02 | −0.50 | 40 | 47 |
| Ni/Cu–N–C | CO | 99.2 | −0.79 | 60 | 93 |
| FeTCPP⊂UiO-66 | CO | ∼100 | −0.56 | 2.5 | 94 |
| H-3DOM-ZnN4/P–C | CO | ∼100 | −0.60 | >30 | 95 |
| ZnN4S1/P-HC | CO | ∼100 | −0.60 | >30 | 96 |
| Bi/UiO-66 | HCOOH | 70–85 | −0.4 to −0.7 | 1.1 | 97 |
| In-BDC | HCOO− | 88.0 | −0.669 | 21 | 98 |
| (Me2NH2+){InIII-[Ni(C2S2(C6H4COO)2)2]}·3DMF·1.5H2O | HCOO− | 89.6 | −1.30 | 12 | 99 |
| Bi-HHTP | HCOOH | 90.0% | 2.60 (cell voltage) | >30 | 100 |
| FJU-127-CH3 | HCOO− | 90.2 | −1.57 | 5 | 101 |
| Bi-ZMOF | HCOOH | 91.0 | −1.10 | 12 | 102 |
| BiZn-MOF | HCOO− | 92.0 | −0.90 | 13 | 103 |
| Bi NS | HCOO− | 92.0 | −1.10 | 10 | 104 |
| Sb2.5/Bi@C | HCOOH | 94.8 | −1.40 | 30 | 105 |
| Bi(btb) | HCOO− | 95.0 | −0.97 | 32 | 52 |
| Bi-HHTP | HCOO− | >95.0 | −0.70 | >30 | 106 |
| CAU-17 MOFs | HCOOH | 95.5 | −1.10 | 10 | 107 |
| Bi-BTC | HCOO− | 96.0 | −0.90 | 24 | 108 |
| Ce2–Bi@C | HCOO− | 97.2 | −1.10 | 48 | 109 |
| In2O3−x@C | HCOOH | 98.0 | −1.20 | 120 | 110 |
| Bi-ene | HCOO− | ∼100 | −0.8 to −1.2 | 12 | 111 |
| Cu-PorOH | CH4 | 51.3 | −1.50 | 6 | 112 |
| Cu/a-C | CH4 | 55.0 | −1.40 | 12.5 | 113 |
| Cu-DBC | CH4 | 56.0 | −1.40 | 4 | 114 |
| Cu–I | CH4 | 57.2 | −1.08 | 10 | 58 |
| Cu2O@Cu-MOF | CH4 | 63.2 | −1.71 | 1 | 115 |
| 2D-vc-MOF(Cu) | CH4 | 65.0 | −1.40 | 4 | 116 |
| MCH-3 | CH4 | 76.7 | −1.00 | 100 min | 117 |
| HATNA-Cu-MOF | CH4 | 78.0 | −1.50 | 12 | 118 |
| Cu/CeO2@C | CH4 | 80.3 | −1.50 | 9 | 119 |
| 2Bn-Cu@ UiO-67 | CH4 | 81.0 | −1.50 | 6 min | 120 |
| Cu4-MFU-4l | CH4 | 92.0 | −1.20 | 24 | 121 |
| BIF-102NSs | C2H4 | 11.3 | −1.00 | 5 | 122 |
| Fe-TCPP@Cu | C2H4 | 33.42 | −1.17 | 5 | 123 |
| CuN2/Cu(111) | C2H4 | 41.5 | — | 6 | 124 |
| Cu–MOF–CF | C2H4 | 48.6 | −1.10 | 12 | 125 |
| C/HKUST-1/Cu/PTFE | C2H4 | 54.0 | −4.00 | 65 | 126 |
| PcCu–Cu–O | C2H4 | 50.0 | −1.20 | 4 | 127 |
| Cutrz | C2H4 | 50.0 | −1.20 | 80 | 128 |
| Cu-HITP@PDA | C2H4 | 51.0 | −1.20 | 10 | 129 |
| MAF-2E, MAF-2E MAF-2P | C2H4 | 51.2 ± 2.3 | −1.10–1.50 | 10 | 49 |
| CuTrz-109 | C2H4 | 55.4 | −1.15 | 10 | 130 |
| Ag/Cu/Cu2OAg0.1/HKUST-1 | C2H4 | 57.2 | −1.30 | 10 | 131 |
| S-HKUST-1 | C2H4 | 60.0 ± 2.0 | −1.32 | 8 | 132 |
| KB@Cu3(HITP)2 | C2H4 | 70.0 | −1.37 | 10 | 133 |
| Cu–SAs@Ir–PCN-222-PA | C2H4 | 70.9 | −1.00 | 16 | 53 |
| CuPz2-Act-30 | C2H4 | 70.2 ± 1.7 | −1.03 | 12 | 134 |
| Cu–Cu2O@CC | CH3OH | 37.4 | −0.70 | 6 | 135 |
| OD-Cu/C | CH3OH | ∼43.2 | −0.30 | 15 | 136 |
| Cu@Cu2O-400 °C | CH3OH | 45.0 | −0.70 | 10 | 137 |
| Cu3(HHTQ)2 | CH3OH | 53.6 | −0.40 | 10 | 138 |
| CuNi SAs/UiO-66(Hf) | CH3OH | ∼98.0 | — | — | 139 |
| Cutrz | CH3COOH | 2.20 | −1.20 | 80 | 128 |
| CuN2/Cu(111) | C2H5OH | 18.2 | — | 6 | 124 |
| Cutrz | C2H5OH | 19.7 | −1.20 | 80 | 128 |
| Cu-MMT–H2O | C2H5OH | 20.8 | −1.15 | — | 140 |
| Cu2−xSe-450 | C2H5OH | 42.0 | −0.74 | — | 141 |
| CuAg5@NC | C2H5OH | 51.8 | −1.00 | 20 | 142 |
| CuSn–HAB | C2H5OH | 56.0 | −0.57 | 65 | 143 |
| OM–Cu–NiSNC | C2H5OH | 63.0 | −0.76 | 50 | 144 |
| Cu GNC-VL | C2H5OH | 70.52 | −0.87 | 12 | 145 |
| Cu2N4/Cu(111) | n-C3H7OH | 3.40 | — | 6 | 124 |
| CuN2/Cu(111) | n-C3H7OH | 5.10 | — | 6 | 124 |
| Cu–SA/NPC | CH3COCH3 | 36.70 | −0.36 | 10 | 146 |
| Cu0.85Zn0.15/C | CH3COCH3 | 38.10 | −0.40 | — | 147 |
Fig. 2 shows the possible intricate pathways of the CO2RR classified using various reduction products. Variations in the final products – such as CO, CH4, CH3OH, C2H4, C2H5OH and so on can happen on electrocatalysts with the same or different metal surfaces. For example, CO is a common product obtained from metals including Cu, Au, Ni, and Co during the electrocatalytic process.150–153 Furthermore, differences in facets, sizes, and oxidation states of Cu can lead to various products 154–156 such as CO, CH3OH and C2H4. Another example is formate, which is mostly produced on Bi and sometimes on In and Sb catalysts.98,110,157 Here, the possible reaction pathways are categorized using various reduction products, as shown in Fig. 2. In the CO2RR, *H, *CO, and *COOH (* denotes the surface-bond site) are typically key intermediates158 that can further convert into various reaction intermediates. *H is involved in the PCET process in reduction pathways. *CO is a common intermediate for forming deeply reduced products. The electrophilicity of C and O in *CO dictates the subsequent reduction pathways. When the O atom in *CO first accepts a proton to generate *COH, it undergoes further reduction by accepting multiple protons and losing one water molecule to generate CH4. On the other hand, when the C atom first accepts a proton to generate *CHO, the double bond between C and O opens to generate *CHOH, followed by further hydrogenation on C to produce methanol. Similar cases are also applied for *COOH and *HCOO. In terms of the *COOH intermediate, it is formed by transferring protons to O atoms of CO2, followed by removing one water molecule to generate *CO. As for *HCOO, it is formed by transferring protons to the C atom of CO2, followed by further hydrogenation to produce formate.
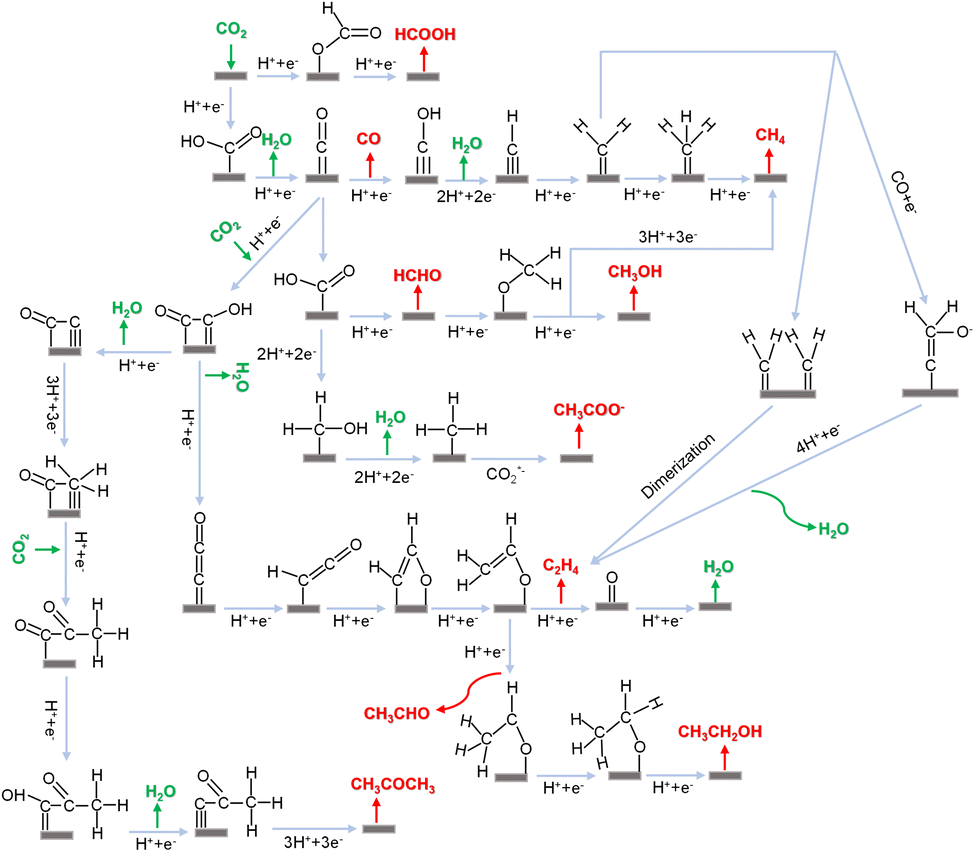 | ||
| Fig. 2 Possible pathways of the CO2RR toward various reduction products. | ||
3. Electrochemical measurements and performance assessments
Evaluating the performance of electrocatalysts is crucial and is typically conducted using an electrochemical three-electrode system. Commonly employed testing techniques, including chronoamperometry/chronovoltammetry, linear sweep voltammetry (LSV), and electrochemical impedance spectroscopy (EIS) are utilized to determine parameters such as Faraday efficiency (FE), current density (J), overpotential (η), Tafel slope (b), turnover frequency (TOF), electrochemical surface area (ECSA) and stability. The key electrochemical characterization methods and evaluation parameters for electrocatalysts are summarized below.3.1 Electrochemical measurements
3.2 Performance assessments
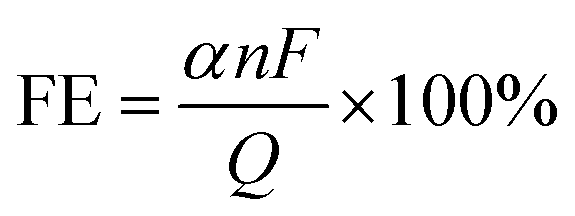 | (18) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485C mol−1); Q is the total charge consumed in the entire reaction.
485C mol−1); Q is the total charge consumed in the entire reaction.
| Jt = J/S | (19) |
| Jp = FE × Jt | (20) |
| η = E0 − Ecat | (21) |
| η = b × logjp + a | (22) |
| TOF = (Jt × FE)/(nFna) | (23) |
485C mol−1) and the number of active sites on the electrode.
| ECSA = Cdl/Cs | (24) |
4. Insights into catalyst design: why MOFs?
As discussed, there are so many possible pathways toward various products that occur during the CO2RR. Developing high-selectivity electrocatalysts is still a great challenge in this field. A deep insight into the structure-reactivity correlations is thus highly demanded to provide principles for designing high-performance electrocatalysts. MOFs as a type of material with well-defined structures are suitable to be studied as model catalysts for the fundamental study to unravel the true active sites for the CO2RR. Furthermore, given the nearly infinite number of potential metal nodes and coordinating organic linker combinations, MOFs display a wide range of chemical and structural variability, which makes them attractive catalyst candidates with tailorable properties for the CO2RR. However, low conductivity and stability remain the bottlenecks of MOFs, which restricts their application in the electrochemical CO2RR.84 In the past few decades, it has been demonstrated that electrocatalytic performance can be improved by regulating atomically dispersed metal catalytic sites and the coordination environment around them in MOFs.163–166 In this section, we focus on the molecular engineering of MOFs for enhanced CO2RR activity and selectivity toward desired products mainly based on the roles of metal nodes and coordinating organic ligands.4.1 Regulation of metal nodes
MOFs with a periodic network structure possess uniformity and nearly 100% atomically dispersed sites, whose porous structure facilitates the adsorption and transport of CO2.167 The metal nodes of MOFs coordinated with organic ligands serve as active sites to speed up reaction rates, selectively transforming CO2 into the desired products in a certain pathway. A family of typical two-dimensional (2D) flat MOFs [TM3(HAB)2, TM = Fe, Co, Ni and Cu; HAB = hexaaminobenzene] was reported by Tang et al.168 in which the TM bonded with four N atoms. According to the calculations using DFT methods combined with the computational hydrogen electrode (CHE) model, Fe3(HAB)2 exhibited the strongest adsorption of *COOH and *H among these compounds, leading to the highest activity toward CO2 production. The reaction pathway was proven to be a proton-assisted mechanism through a pathway of “CO2 → *COOH → *CO → *CHO → *CHOH → *CH2OH → CH3OH”. Thus, Fe3(HAB)2 was identified as a promising material catalyzing CO2 to CH3OH via the “RWGS (reverse water gas shift) + CO-hydro (CO hydrogenation)” process. Similarly, Cui et al.169 also performed the calculation of conductive [TM3(HAB)2, TM = Fe, Co, Ni, Cu, and Mo]. Fe-, Co- and Ni-based MOFs presented weak adsorption of CO2, while Cu-based MOFs exhibited strong adsorption energies of CO2 and H2O, with a higher interaction with H2O than with CO2. In terms of Mo-based MOFs, they were identified as favorable candidates for adsorbing more CO2 molecules than H2O. The different charge density of CO2 adsorbed on Mo-based MOFs displayed a distorted CO2 molecule, indicating the good activation and selectivity of CO2.M3(hexaiminotriphenylene)2, one type of conductive MOF, known as M3(HITP)2 (M = Ni2+, Cu2+, HITP = hexaiminotriphenylene), has also been applied to the CO2RR. The selectivity of M3(HITP)2 for the CO2RR can be regulated by varying the metal nodes. A series of M3(HITP)2 (M = Fe, Co, Ni, Ru, Rh, and Pd) samples were investigated for the CO2RR using DFT calculations. However, only Co3(HITP)2 and Rh3(HITP)2 exhibited superior selectivity toward CH3OH formation.170 This was attributed to the strong chemical activity toward *COOH species and high *CO adsorption strength, which indicated that the adsorbed CO2 molecule can be activated and further reduced without desorption. Co3(HITP)2 and Rh3(HITP)2 with respective overpotentials of 0.67 V and 0.46 V exhibited superior catalytic activity. Similarly, the CO2RR performance of bis(iminothiolato)metals (marked as TMIT, TM = Mn, Fe, Co, Ni, Cu, Ru, Rh, and Pd, IT = C6S3N3H3) can also be affected by the metal nodes as simulated by spin-polarized calculations.171 All these catalysts catalyzed CO2 to produce HCOOH except that RuIT and RhIT only catalyzed CO production. This was because the strong adsorption of CO on the RuIT and RhIT catalyst surfaces poisoned the catalysts. π-conjugated metal bis(dithiolene) complex nanosheets (M3C12S12, M = Fe, Co, Ni, Ru, Rh, and Pd)172 within spin-polarized frameworks were another typical type of MOF. However, Fe3C12S12, Co3C12S12, Ni3C12S12, and Pd3C12S12 sheets exhibited weak chemical activity toward *COOH and *HCOO intermediates due to their high free energies (ΔG > 0.74 eV), and thus these four catalysts were not considered to suit CO2RR applications. CO desorption and further hydrogenation on Ru3C12S12 were indicated to be thermodynamically disadvantageous, while Rh3C12S12 was suggested to be a potential catalyst for the CO2RR due to its low free energy barrier and thus thermodynamic advantages. Additionally, a porous 2D graphene-like sheet M3(HHTQ)2 (M = Cu, Ni) was also constructed from 2,3,7,8,12,13-hexahydroxytricycloquinazoline with Ni2+ and Cu2+ (ref. 138) (Fig. 3a). Cu3(HHTQ)2 showed stronger CO2 adsorption than Ni3(HHTQ)2 and higher FECH3OH up to 53.6% at an overpotential of −0.4 V.
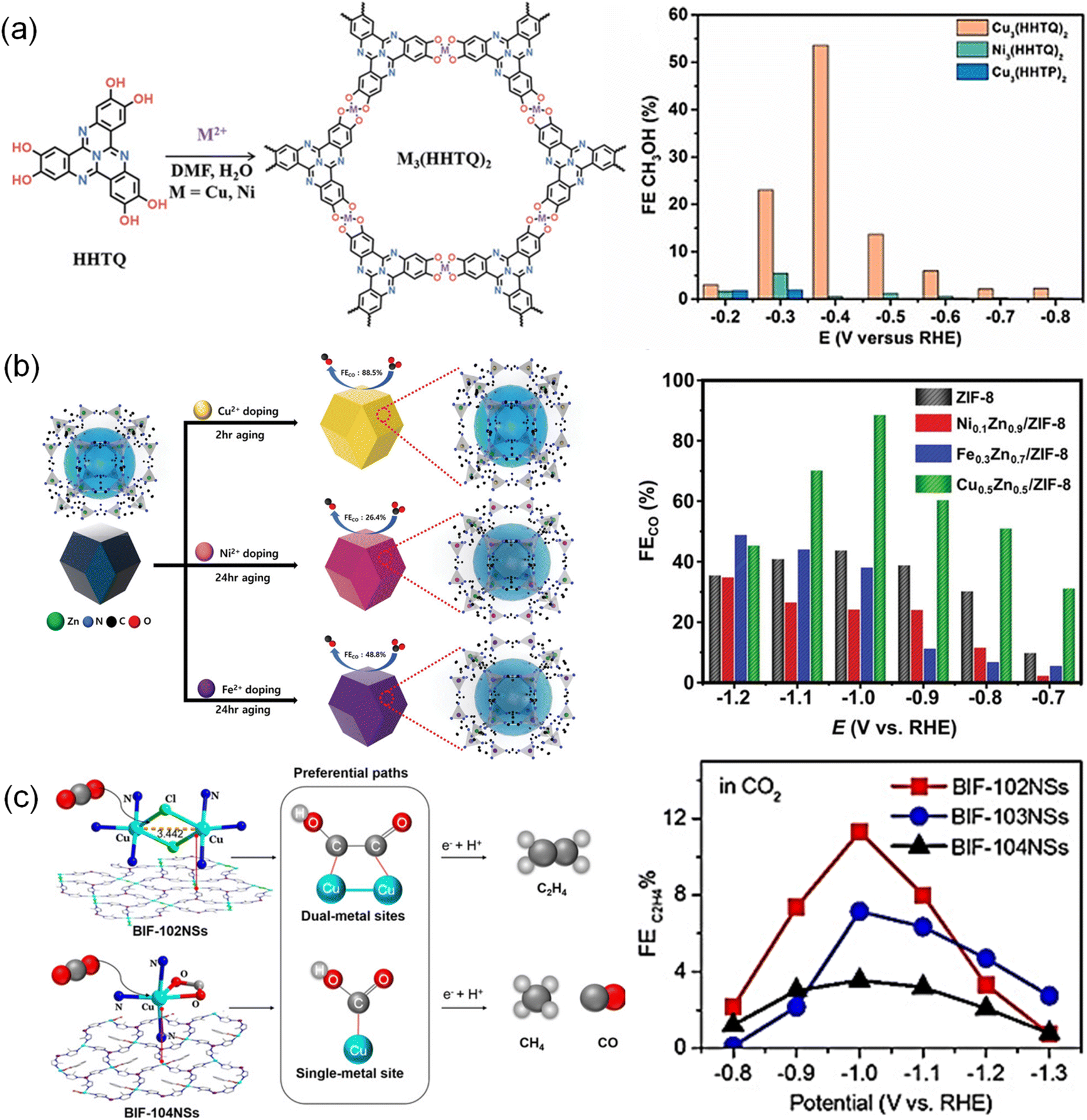 | ||
| Fig. 3 (a) Schematic synthesis of M3(HHTQ)2 (M = Cu, Ni) and FE of CH3OH.138 Copyright 2021, Wiley-VCH. (b) Schematic synthetic strategy of the MxZny/ZIF-8 catalyst and FE of CO.69 Copyright 2023, Wiley-VCH. (c) Coordination environment and reaction pathways for BIF-102NSs and BIF-104NSs as well as FE of C2H4.122 Copyright 2021, Wiley-VCH. | ||
Doping diverse metal ions into MOFs is an attractive synthetic strategy to enhance the performance of the CO2RR due to the unique characteristics introduced by additional metal nodes, which may have synergistic effects.67,68,173–175 For instance, Cho et al.69 reported a series of zeolite-imidazolate frameworks (ZIF-8) containing Ni2+, Fe2+, and Cu2+ (Fig. 3b). Cu0.5Zn0.5/ZIF-8 achieved a higher FE of 88.5% for CO than pristine ZIF-8 (43.7%), Ni0.1Zn0.9/ZIF-8 (34.7%) and Fe0.3Zn0.7/ZIF-8 (48.8%). This was because the doped Cu affected the electronic structures of electron-rich sp2 C sites via a local effect between the zinc-nitrogen (Zn–N4) and copper-nitrogen (Cu–N4) sites, which facilitated *COOH adsorption and promoted the reduction reaction. Iqbal et al.50 reported bimetallic Ni and Fe in MOFs (MOF Ni–Fe). The incorporated combination of Ni and Fe metal centers increased the active surface area and active sites for the adsorption of intermediates, promoting the CO2RR process. As a result, the Ni and Fe bimetallic centers lowered the kinetic energy barrier, which achieved a FECO of 98.2%, superior to single metal MOF-Ni (74.3%) and MOF-Fe (62.6%).
The construction of heteronuclear metal pair sites in MOFs can be used to regulate the kinetic energy barrier of intermediate formation for enhanced CO2RR performance. For instance, Zhong et al.68 developed a layer-stacked 2D conjugated MOF (PcCu-O8-Zn) with CuN4 (copper-phthalocyanine) and ZnO4 (zinc-bis(dihydroxy)) to promote CO2 into syngas (CO and H2). The molar ratio of H2/CO (1:7 to 4:1) was tuned by varying metal nodes (Cu and Zn) and applied potentials. ZnO4 showed high catalytic activity in converting CO2 to CO, while the CuN4 unit promoted the HER process. Synergistic catalytic effects were achieved using ZnO4 and CuN4 metal nodes. Another example was the integration of 2,3,9,10,16,17,23,24-octa-substituted metallophthalocyanine (MPc-XH, M = Co and Ni, X = N and O) into conductive MOFs as electrocatalysts for the CO2RR.67 The activity and selectivity were tailored by the selection of metal nodes of MPcs and further adjustment of heteroatomic linkages. CoPc–Cu–O achieved an FECO of 79% and a current density of −9.5 mA cm−2. This is because CoPc-based and O-linked MOFs lowered the activation energies toward *COOH formation, resulting in higher activity and selectivity. CuSn-HAB,143 a conductive 2D π-conjugated MOF with hexaiminobenzene (HAB) ligands and planar Cu–N4 nodes, had a pair of SnN2O2 and CuN4 sites bridged by μ-N atoms and demonstrated an FE of 56% toward CH3CH2OH formation. Further investigations revealed that the SnN2O2 site exhibited a higher affinity for O atoms compared with the copper site, playing an important role in promoting the formation of the *OCH2 intermediate. Therefore, the dual sites were more thermodynamically favorable for the C–C coupling between *CO and *OCH2. In addition, Cu–Ni dual-metal sites also showed similar synergistic effects for high selectivity.139 The UiO-66(Hf) matrix helped stabilize the single atoms and facilitating ionizing radiation conversion, while atomic Cu–Ni dual-metal sites contributed to high CH3OH selectivity. Under radiation conditions, CuNi SAs/UiO-66(Hf) realized a selectivity of ∼98% for CH3OH and an energy efficiency of ∼1.5 × 10−7 mol J−1. The spectral data and calculations showed that three C1 intermediate adsorption states may occur at the Cu site and the final two at the Ni site, leading to selective CH3OH formation.
Similarly, the same nuclear metal pair also showed synergetic effects on the promoted performance of the CO2RR. (2,3,9,10,16,17,23,24-octahydroxyphthalo-cyaninato) copper(II) (PcCu–(OH)8) ligands and the CuO4 nodes were used to construct a MOF catalyst (PcCu–Cu–O).127 PcCu–Cu–O with dual active sites exhibited high performance to convert CO2 to C2H4 with a FE of 50% and a current density of 7.3 mA cm−2. It was also found that dimer-copper catalysts CuII2[BH(mim)3]2Cl2 (BIF-102, BIF = boron imidazolate frameworks, mim = 2-methylimidazole) with Cl− bridged dimer copper (Cu2) units delivered high activity and selectivity for C2H4 (ref. 122) (Fig. 3c). The enhanced performance was due to the enriched charge around the dual Cu centers in CuII2[BH(mim)3]2Cl2 which performed as regulators for varying the energy barriers and afforded distinct reaction pathways. Therefore, CuII2[BH(mim)3]2Cl2 BIF-102NSs (NS = nanosheet) exhibited improved FE of C2H4 (11.3%), superior to iso-reticular BIF-103 (CuII2[BH(mim)3]2(HCOO)2, 7.15%) and single-metal BIF-104 (CuII[BH(mim)3](BA), 3.55%).
MOFs containing multi-metallic redox-active secondary building units (SBUs) have also been reported to be good candidates for high-performing electrocatalysts. Huang et al. reported an alkali-resisting MOF (Cu1Ni-1,4-benzenedipyrazolate, Cu1Ni-BDP) featuring pyrazolate-stabilized asymmetric Ni/Cu clustersites for the CO2RR.176 The unique Ni–Cu hybrid sites, characterized by an asymmetric electronic structure, orbital interaction and close distance (2.8 Å), synergistically catalyzed the conversion of CO2 to C2H4. In an alkaline environment (1.0 M KOH), Cu1Ni-BDP exhibited a notable C2H4 selectivity, yielding an FE of 52.7% and a partial current density of 278 mA cm−2 at −1.3 V. Moreover, only a 4.5% decrease in FE was observed after 25 h of electrolysis. Operando analyses and theoretical calculations indicated that the asymmetric Ni/Cu sites effectively reduced the energy barrier associated with the *COH–COH intermediate, the rate-limiting step in CO2 reduction to C2H4. MOFs containing heterobimetallic cluster sites offer the potential for synergistic catalysis or tandem catalysis toward C2+ production. Similarly, two-dimensional Ru2(OAc)4Ni(CN)4, featuring Ru and Ni bimetallic metal sites was reported to exhibit high activity and selectivity for 1-C4H8 production.177 At room temperature, it achieved a production rate of 1.3 mol gcat−1 h−1 and conversion efficiency of 97%. Trinuclear clusters within MOFs have also been proven to be effective in the electrochemical CO2RR to produce value-added products. Huang et al.130 reported a low-cost metal-azolate framework [Cu3(μ3-OH)(μ3-trz)3(OH)2(H2O)4](Cutrz) constructed from 1H,1,2,4-triazole (Htrz) organic ligands and Cu2SO4·5H2O salts. The Cutrz MOF with trinuclear clusters as active sites displayed a high FE of 50% for selective electroreduction of CO2 to C2H4. The trinuclear cluster {Cu3(μ3-OH)(μ3-trz)3}2+ enabled the simultaneous adsorption of three *CO intermediates on its surface, positioning closely at only 3.3 Å apart. This close proximity led to higher *CO coverage and facilitated C–C dimerization coupling with a reduced energy barrier. By tailoring the size of the monodispersed CuTrz, the catalyst's performance was further enhanced to 55.4%.130 CuTrz particles with an average size of 109 nm showed superior activity compared to their larger counterparts. Extensive characterization studies revealed that the remarkable selectivity was attributed to the uniform small size of CuTrz and the presence of abundant grain boundaries.
4.2 Modulation of surrounding organic ligands
Though metal nodes of MOFs play a significant role in determining the activity and selectivity of the CO2RR, it is important to note that the same type of metal nodes in different MOFs may result in different pathways toward different carbon products. The variation in catalytic behaviors may be attributed to differences in the electronic properties of metal nodes in different types of MOFs, including their oxidation state178,179 and electron density,180 which are significantly influenced by the metal coordination geometry and ligand environment. In this section, the important contribution of the ligands surrounding the metal nodes of MOFs to the CO2RR is discussed. In general, organic ligands in MOFs play an essential role in regulating specific sizes, geometries, and symmetries.181 It is considered that MOFs can achieve outstanding catalytic activity and stability for the CO2RR through rational design of building blocks with specific functional groups, side structures and modification with small molecules.78,182 The regulation strategy generally includes the modulation of side groups and the body structure of organic ligands.By changing the size of ligand side groups, Cu(I) triazolate frameworks (MAF-2ME, MAF-2E, and MAF-2P) were tailored to realize high-efficiency conversion of CO2 to C2H4/CH453 (Fig. 4a). The C2H4/CH4 selectivity ratio can be tuned and inversed from 11.8:1 to 1:2.6 with selectivity up to 51% (C2H4), 56% (CH4), and 77% (hydrocarbon), respectively. Computational simulations showed that the geometry structure of Cu(I) changed from triangular to tetrahedral due to the change of ligand side groups, with two adjacent Cu(I) cooperating for C–C coupling to form C2H4. Recently, Mao et al.183 synthesized Cu-based MOFs with an optimal Cu–Cu distance, named MIL-53 (Cu) (MIL = Materials of Institute Lavoisier) (Fig. 4b). Ten different ligand side groups from electron-donating groups (NH2, CH3, NHOH, OH, and H) to electron-withdrawing groups (COOH, Cl, Br, CN, and NO2) were chosen to decorate the organic ligand in MIL-53 (Cu) to modulate the Cu–Cu distance and charge of Cu. COOH-ligand-decorated MIL-53 (Cu) was located at the top of the volcano plot and exhibited the highest catalytic activity and selectivity toward CH3CH2OH production. COOH-ligand-decorated MIL-53 (Cu) exhibited high catalytic performance with a FE of 55.5%, which was superior to that of the Cu electrode (35.7% at −1.05 V vs. RHE). Similarly, Nam et al.77 added side groups of –NH2 in a face-centered cubic (fcu) MOF to strengthen the adsorption capacity of CO2 (Fig. 4c). As a result, FECO increased and FEH2 decreased on Ag/Zr-fcu-MOF-NDC (H2BDC: 1,4-benzenedicarboxylic acid) compared to Ag/Zr-fcu-MOF-BDC and Ag/Zr-fcu-MOF-NH2BDC. They also explored the role of the body structure of ligands in modifying the binding modes of *CO intermediates, which played a pivotal role in product selectivity. The high CObridge population in Ag/Zr-fcu-MOF-NDC (H2NDC: 1,4-naphthalenedicarboxylic acid) was modulated for enhanced CO selectivity. Aromatic amine groups were also coupled into a silver chalcogenolate cluster-based MOF (Ag12(StBu)8(CF3COO)4(bpy-NH2)4, denoted as 1-NH2, aka-Ag12bpy-NH2, bpy-NH2 = 3-amino-4,4′-bipyridine) to capture CO2 molecules in a simulated flue gas and served as CO2 catalytic sites82 (Fig. 4d). The 1-NH2 MOF could catalyze the adsorbed CO2 into CO with an ultra-high CO2 conversion of 60% and a FFCO of 96% due to the introduction of the aromatic amine groups, better than [Ag12(StBu)8(CF3COO)4(bpy)4] (bpy: 4,4′-bipyridine, 1, aka Ag12bpy) and [Ag12(StBu)8(CF3COO)4(bpy-CH3)4] (bpy-CH3: 3-methyl-4,4-bipyridine, 1-CH3, aka Ag12bpy-CH3).
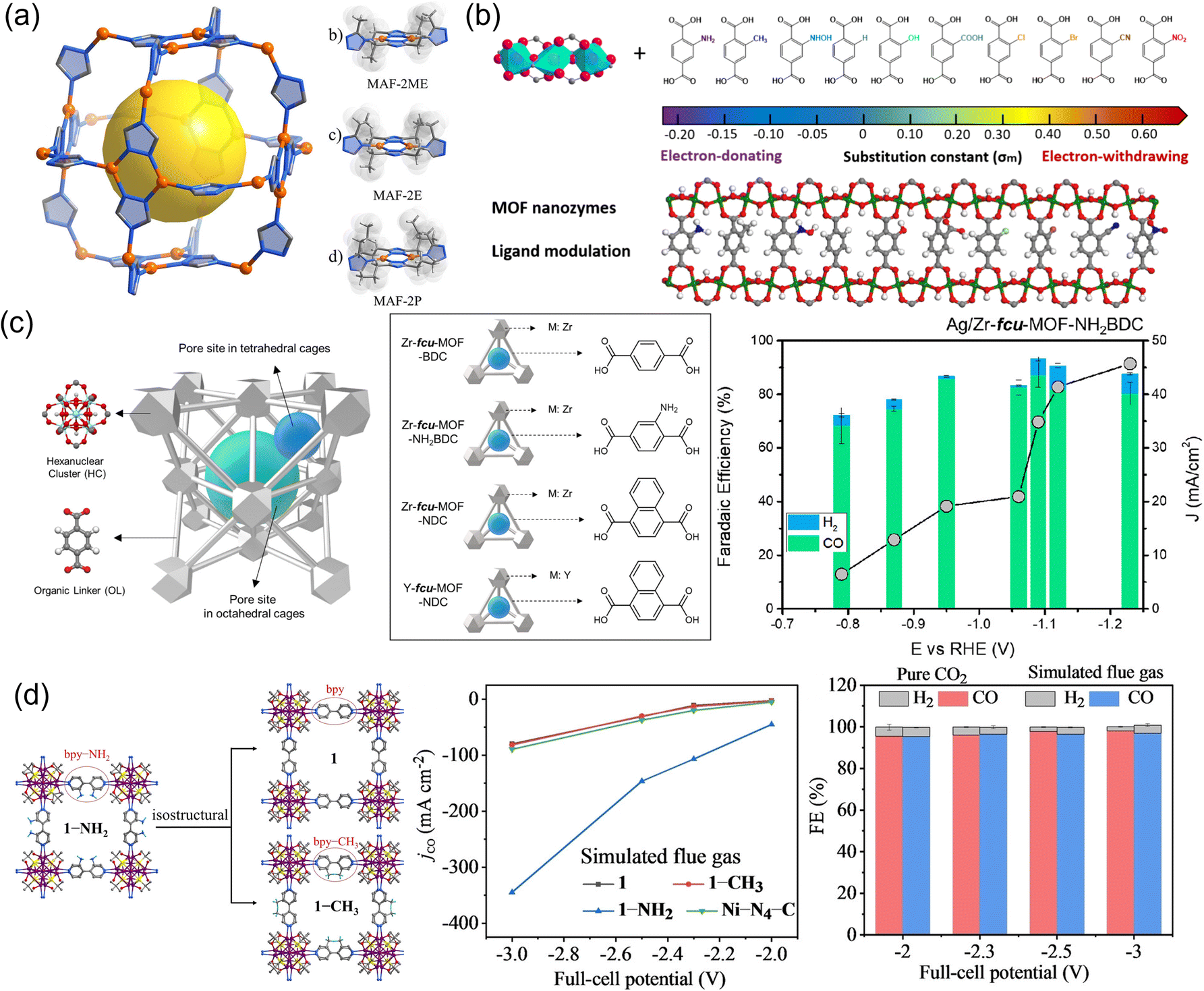 | ||
| Fig. 4 (a) Crystal structures and local coordination environments of MAF-2 analogs.49 Copyright 2022, Wiley-VCH. (b) Ten possible ligand-decorated 1,4-benzenedicarboxylic acid linkers with their substituent constant values.183 Copyright 2023, American Chemical Society. (c) MOF design strategies based on the Zr-fcu-MOF-BDC MOF and FE of CO for Ag/Zr-fcu-MOF-NH2BDC.77 Copyright 2020, American Chemical Society. (d) Schematic of 1, 1-CH3 and 1-NH2, jCO of 1, 1-CH3, 1-NH2 and Ni–N4–C catalysts under simulated flue gas and FECO for 1-NH2 in CO2 and simulated flue gas.82 Copyright 2023, Wiley-VCH. | ||
Modifying the body structure of organic ligands has also been reported as an effective strategy to enhance CO2RR performance. For instance, changing the ligand body structure of ligand linkers in Cu naphthalenedicarboxylate (Cu-UNDC) and Cu benzenedicarboxylate (Cu-UBDC) can regulate the MOF structure and electronic environment of Cu centers, thus changing the selectivity toward C2 products184 (Fig. 5a). The two catalysts showed different FEs for C2 products. FEs for Cu-UBDC were 13.2% in the dark and 26.2% in the light. FEs for Cu-UNDC were 24.3% in the dark and 21.8% in the light. Moreover, in-built heteroatoms within the molecular structure of organic ligands lead to significant variations in the electrocatalytic CO2RR. Liu et al.118 designed and synthesized a 2D conjugated Cu MOF (HATNA-Cu-MOF) by coordination of an electron-deficient N-containing conjugated molecule HATNA-6OH (hexaazatrinaphthylene) and Cu(NO3)2. The symmetric aromatic heterocycle of HATNA-6OH with 6 hydroxyl groups makes it selectively coordinate with Cu2+ in a 2D framework, with three phenanthroline units as additional sites for guest binding. The synergistic effects of HATNA and copper catecholate nodes enabled the conjugated HATNA-Cu-MOF to selectively convert CO2 to CH4 with an FE of 78% at −1.5 V. The tailoring of ligands can adjust both the coordination microenvironment of metal nodes and geometry structure of MOFs, thereby affecting their electronic structure and interaction with intermediates. Recently, a series of Cu(I)-based MOFs ([Cu4X(TIPE)3]·3X, [X = Cl, Br, I, TIPE = 1,1,2,2-tetrakis(4-(imidazole-1-yl)phenyl)ethene]) with different coordination microenvironments (Cu–Cl, Cu–Br, and Cu–I) were synthesized by Sun et al.58 (Fig. 5b). With the increasing radius of halogen atoms from Cl to I, the CO2 adsorption increased and the d-band center of Cu positively shifted to the Fermi level, resulting in enhanced selectivity of CO2 to CH4. The shifted d-band center reduced the formation energies of *CH2O and *CH3O species from Cl to I and thus enhanced the electrocatalytic activity. The MOF with a Cu4I cluster (X = Cl, Br and I) gave a partial current density of 60.7 mA cm−2 and FE of 57.2% for CH4 production at −1.08 V compared with Cu–Cl (32.9%) and Cu–Br (40.2%) at −1.28 V. Al-Attas et al.78 explored Zn-based MOFs with two different azolate functional ligands, 1,2,4-triazole (Calgary Framework 20, CALF20, [Zn2(Tz)2Ox]) and 2-methylimidazole (zeolitic imidazolate framework-8, ZIF-8, [Zn(MeIm)2]) for the CO2RR (Fig. 5c). CALF20 showed higher FE for CO (94.5%) compared to ZIF-8 (61.1%). The sp2 carbon atoms in 1,2,4-triazole ligands coordinated with the Zn(II) centers were claimed as the active sites for the CO2RR, as 3d orbitals of Zn(II) centers were fully occupied. In addition, ab initio investigation showed that the N-sp2 C adsorption site was the most favorable adsorption sites in CALF20 and ZIF-8. The 1,2,4-triazole ligand in CALF20 with more electrons induced in the adjacent active sites enhanced the charge transfer and facilitated *COOH formation, promoting CO production with high current density and FE. The researchers further assembled CALF20 into a gas diffusion electrode and realized a 95% FE for converting N2-diluted CO2 streams to CO.185
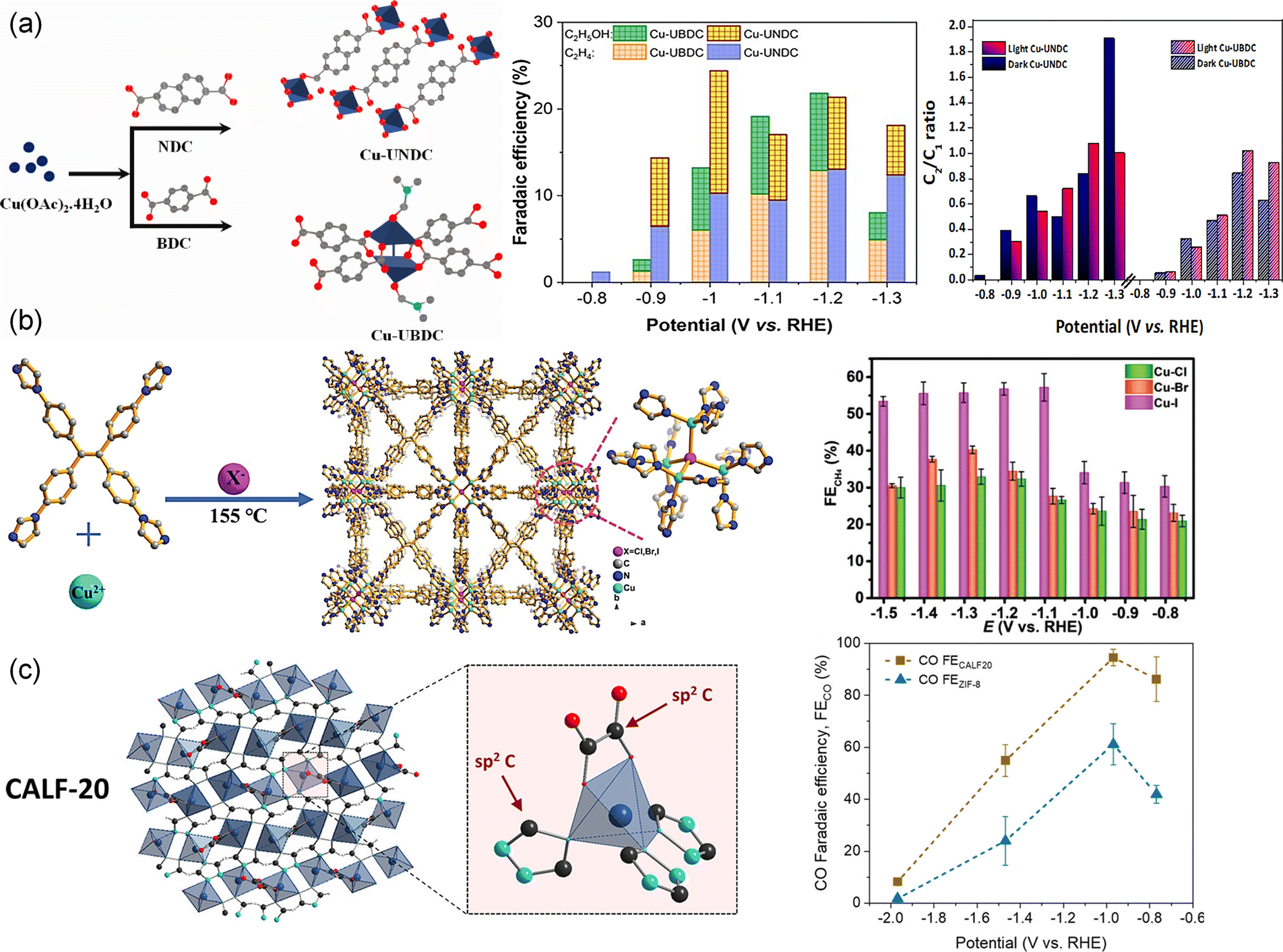 | ||
| Fig. 5 (a) Schematic, FE toward C2 products and the C2/C1 ratio of Cu-UNDC and Cu-UBDC in CO2-saturated 0.1 M KHCO3.184 Copyright 2023, American Chemical Society. (b) Schematic illustration of the structure and FECH4 of Cu-X (X = Cl, Br and I).58 Copyright 2022, Wiley-VCH. (c) Schematic illustration of the crystal structure of CALF20 and FECO of ZIF-8 and CALF20.78 Copyright 2021, American Chemical Society. | ||
4.3 Post-modification
As a matter of fact, incorporation of functional modules into MOFs for enhancing CO2RR performance can be realized not only through direct synthesis but also through post-modification. Therefore, doping extraneous ligands in MOFs is one of the most effective strategies. For example, ZIFs, a subclass of MOFs assembled using metal nodes and imidazole ligands, exhibited excellent thermal and chemical stability with various topologies.66 Active sites were considered to be the imidazolate ligands coordinated with the Zn(II) nodes in ZIFs with ZIF-8 showing the highest FECO of 81.0% and a current density of 12.8 mA cm−2.66 Later, Dou et al.71 developed a ligand doping strategy by introducing 1,10-phenanthroline into ZIF-8 using a post-treatment process. The conjugated structure of 1,10-phenanthroline might benefit from electron delocalization and increased the electron density around nitrogen, thereby facilitating π electron transfer and enhancing σ electron donation between nitrogen and the metal.186 In this study, 1,10-phenanthroline acted as an electron donor, induced the adjacent sp2 C atoms in imidazolate and thus strengthened the ability of *COOH formation, delivering a high FE of 90.57%. Also, metallocene84 had a similar impact on the improvement of electron conductivity and electron-donating ability. Using the chemical vapor deposition method, metallocene was implanted in MOF-545-Co. The as-prepared CoCp2@MOF-545-Co selectively converted CO2 to CO with an FE of 97% due to its strong binding interaction, which reduced the adsorption energy of CO2. This method can be applied universally to other materials. For instance, diphenyl sulfide immobilized onto graphene served as an axial ligand for tetraphenylporphyrin cobalt (PCo), while [PCo]*− acted as active sites promoting CO2 reduction.187 The benzene rings of diphenyl sulfide stacked with graphene in a face-to-face manner, serving as a mediator for the electron transfer communication between PCo and graphene.Doping extra metal elements in MOFs was an effective method to prepare high-efficiency MOF electrocatalysts. Recently, a new 2D Fe-based BIF-73-NS (BIF = boron imidazolate framework, NS = nanosheet) MOF was successfully synthesized through ultrasonication exfoliation of bulk BIF.70 Benefiting from the functional –OH groups, Fe ions were anchored, and molecular-level Fe2O3 was formed in 2D BIF-73-NS. 2D BIF-73-NS with Fe2O3 fragments exhibited a high FECO of 88.6% at −1.8 V vs. Ag/AgCl. In addition, Zhu et al.121 prepared CuII4-MFU-4l by exchanging partial Zn(II) ions in MFU-4l ([Zn5Cl4(btdd)3], H2btdd = bis(1H-1,2,3-triazolo-[4,5-b], [4′,5′-i])dibenzo-[1,4]-dioxin)) with Cu(II) irons (Fig. 6a). In situ X-ray absorption spectroscopy (XAS) and infrared (IR) spectroscopy spectra revealed that Cu(I)N3 was in situ formed from Cu(II) species, acting as an active site. Cu(I)N3 showed strong coordination ability and synergistic effects with adjacent aromatic hydrogen atoms, playing an important role in stabilizing the key intermediates and suppressing the competitive HER, thus showing high electrocatalytic activity and selectivity for CH4 formation (FE = 92%).
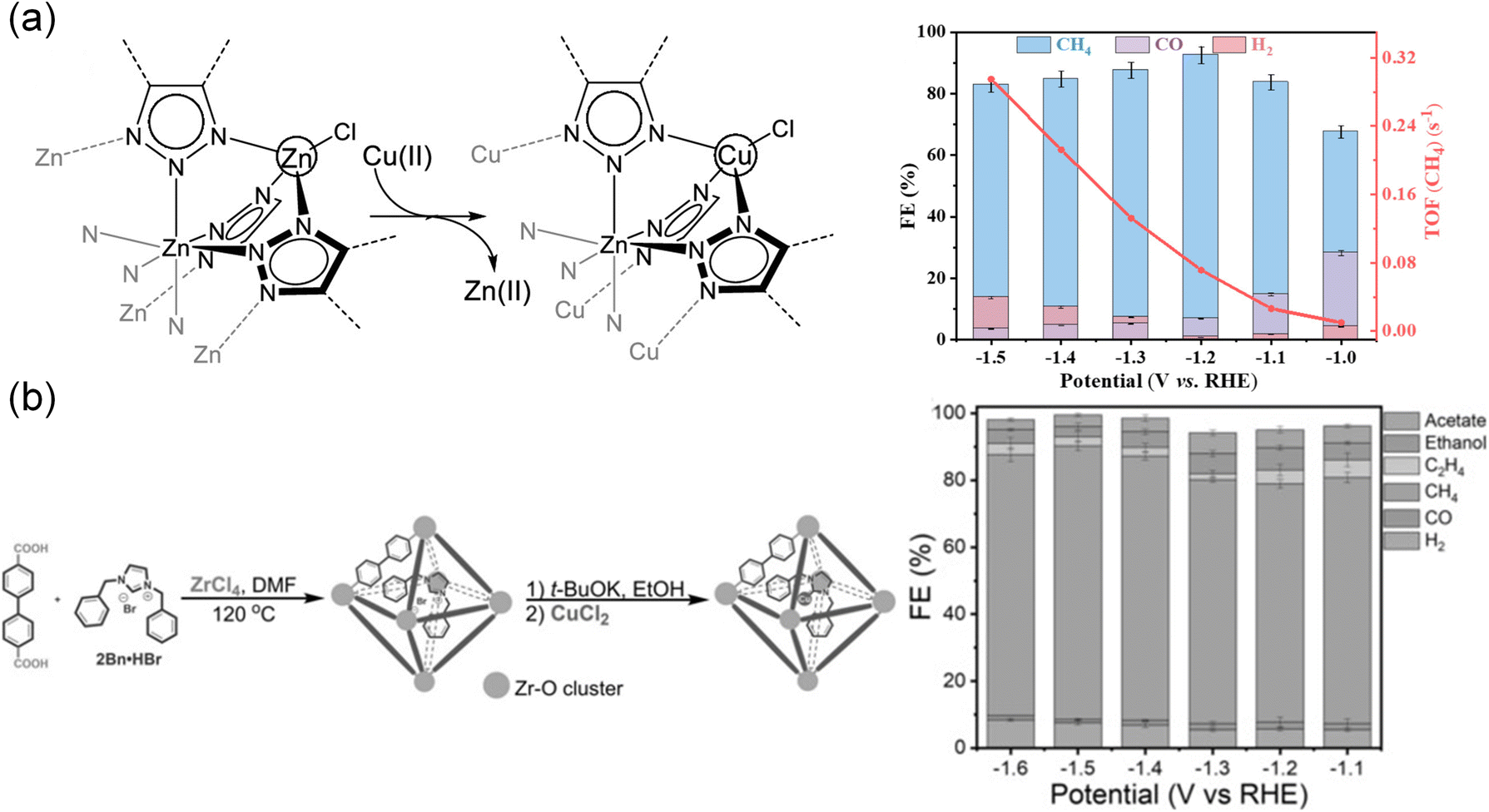 | ||
| Fig. 6 (a) Schematic of the metal transformation process, and FECH4 and TOF of CuII4-MFU-4l.121 Copyright 2021, American Chemical Society. (b) Schematic synthesis route and FE of 2Bn-Cu@UiO-67.120 Copyright 2021, Wiley-VCH. | ||
Additionally, an encapsulation strategy was also proposed to prepare MOF electrocatalysts with enhanced performance in the CO2RR. Chen et al.120 encapsulated N-heterocyclic carbene molecules (NHCs, 1,3-dibenzyl-1H-imidazole-3-ium bromide, 2Bn-HBr) into UiO-67 for their well-matched molecular size. Cu was added to the alkaline medium to synthesize 2Bn-Cu@UiO-67 (Fig. 6b). Due to the acidic C–H bond of the imidazole, NHCs with a lone pair of electrons tended to bond with the metal to generate a stable NHC-metal complex. 2Bn-Cu@UiO-67 achieved 81% FE of CH4 and a high TOF of 16.3 s−1. It was suggested that σ donation of NHC enhanced the surface electron density of Cu catalytic sites and optimized the adsorption of *CHO species.
A novel 3D framework 1′ {[Ni3Zr6(μ3-O)4(μ3-OH)4(IN)12(H2O)6]·Cl6·4DMF·18H2O}n with [Zr48Ni6] nano-cages was constructed to confine Au nanoparticles (Fig. 7a).51 The porous framework 1′ exhibited high surface area and good stability which allowed selectively extracting AuCl4− ions from electronic waste. The AuCl4− was further reduced into Au nanoparticles and thus formed Au nanoparticles@1′ hybrid materials (Au NPs@1′-x, x = 1, 2, 3, and 4) with different Au nanoparticle sizes. By controlling loading amounts, the sizes of Au NPs were tunable. Au NPs@1′-3 exhibited a FECO of 95.2% with a current density of 102.9 mA cm−2 at −1.1 V and remained stable for more than 15 h. The excellent stability was attributed to the confinement effect of 1′, which avoided the agglomeration of Au NPs. Inspired by the CO generator of Au and coupling of Cu–N4 active sites, Au nanoneedles were impregnated into PCN-222 MOF (PCN = porous coordination networks) to construct AuNN@PCN-222(Cu) with the assistance of reducibility of carboxylate ligands (Fig. 7b).188 AuNN@PCN-222(Cu) with metalloporphyrin Cu centers achieved an FE of 52.5% for C2H4 and exhibited better structural stability. Through operando X-ray spectroscopy, in situ infrared spectroscopy and DFT calculations, the enhanced selectivity was ascribed to the C–C coupling in a tandem catalysis mechanism. CO was generated at the Au nanoneedles and then abducted to another *CHO at the Au-activated N motifs. The catalyst thus exhibited excellent C2H4 selectivity and high FE. Furthermore, the Au-inserted catalyst also showed improved structural stability, due to the altered charge conduction pathway bypassing the reticular network. A similar case of an electron-conductive polypyrrole (PPy) molecule was also reported, which was inserted into the channel of MOFs through the in situ low-temperature polymerization of pyrrole in the pore of PPy@MOF-545-M (M = Fe, Co, and Ni) (Fig. 7c).189 MOF-545-Co with increased electron-transfer ability presented excellent electrocatalytic CO2RR performance. The FECO of PPy@MOF-545-Co reached 98% at −0.8 V, which was approximately two times higher than that of bare MOF-545-Co. Such high performance was attributed to the incorporation of PPy that serves as an electric cable in the channel of MOFs, facilitating electron transfer during the CO2RR process.
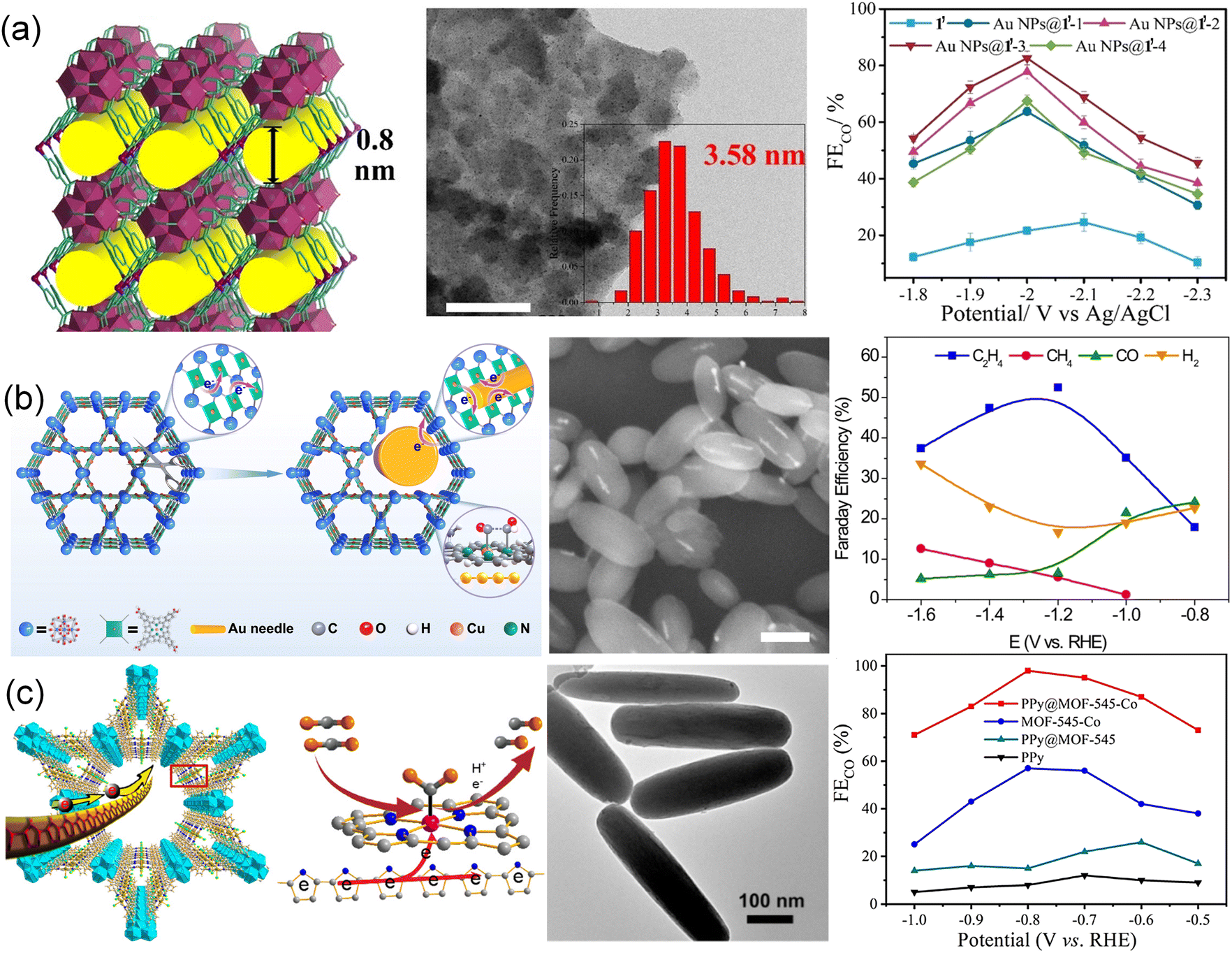 | ||
| Fig. 7 (a) Three-dimensional framework of 1′, TEM image of Au-NPs@1′-3 and FECO of Au NPs@1′-x (x = 1, 2, 3, and 4).51 Copyright 2022, Wiley-VCH. (b) Schematic illustration, the SEM image and FE for different products of the AuNN@PCN-222(Cu) catalyst.188 Copyright 2022, Springer Nature. (c) Schematic illustration for the advantages of PPy in MOF-545-Co, the TEM image of PPy@MOF-545-Co and FECO of PPy@MOF-545-Co at different voltages.189 Copyright 2021, American Chemical Society. | ||
5. MOF derivatives
MOFs usually face many challenges, such as limited active sites, unsatisfactory electrical conductivity, and stability issues. This results in the generation and development of MOF derivatives with robust structures and good conductivity as electrocatalysts for the CO2RR.5.1 MOF derivatives through the wet chemistry method
The structural transformation strategy by the wet chemistry method to obtain MOF derivatives at mild temperature is fascinating as it maximizes the preservation of the original coordination structure and geometric morphology. As an example, S-HKUST-1 was prepared by immersing HKUST-1 in an ethanol solution containing thioacetamide (Fig. 8a).132 Thioacetamide reacted with the water in HKUST-1 to produce H2S, facilitating the formation of Cu–S bonds. The element distribution, XRD and Fourier transform infrared spectra indicated that sulfur is distributed throughout the S-HKUST-1 particle, with the framework structure and organic bonding of HKUST-1 well maintained. Operando X-ray absorption and systematic characterization confirmed the stable Cu–S motif of S-HKUST-1 during the CO2 reduction reaction. The atomically dispersed Cu–S units in S-HKUST-1 exhibited high selectivity to C2H4 with a maximum FE of 60.0% in an H-type cell and up to 57.2% in a flow cell. The partially oxidized Cuδ+ at the Cu/CuxSy interfaces and the optimized geometric and electronic structures for *CO dimerization, played a crucial role in reducing the kinetic barriers for the CO2 reduction process. Utilizing a “reduction-cleavage-recrystallization” method (Fig. 8b),190 Cu(II)-BTC (HKUST-1) underwent a conversion to Cu(I)-BTC facilitated by methanol vapor, followed by reconstruction in a methanol solution. The liquid/solid–gas interface played an important role in the transformation into Cu(I)-BTC, altering the oxidation state of Cu and the coordination environment of the metal node. Cu(I)-BTC showed improved C2H4 selectivity with an FE up to 57% at −1.6 V and exceptional durability for over 38 h, superior to both the flower-like HKUST-1 (F-HKUST-1) and HKUST-1 catalysts. The superior performance was attributed to the Cu(I)–O coordinated structure and the presence of free carboxyl groups, which enhanced the dimerization of *CO intermediates and hindered the hydrogenation of *CO intermediates. In another report, Cu single atoms were incorporated into the Ir–PCN-222 MOF to create Cu–SAs@Ir–PCN-222–PA by stirring a mixture of metal precursor Cu–PA (PA = 4-picolinic acid) and Ir–PCN-222–PA at 140 °C for 12 h (Fig. 8c).53 The dual active sites of Ir-porphyrin and Cu–SAs enabled tandem catalysis for conversion of CO2 to C2H4. CO was initially produced on Ir-porphyrin and then transferred to the nearby adsorbed *CO intermediate on Cu–SAs to facilitate the C–C coupling process. The tandem catalysis led to the production of C2H4 from CO2 with an FE of 70.9% in a flow cell and 66.9% in an H-cell.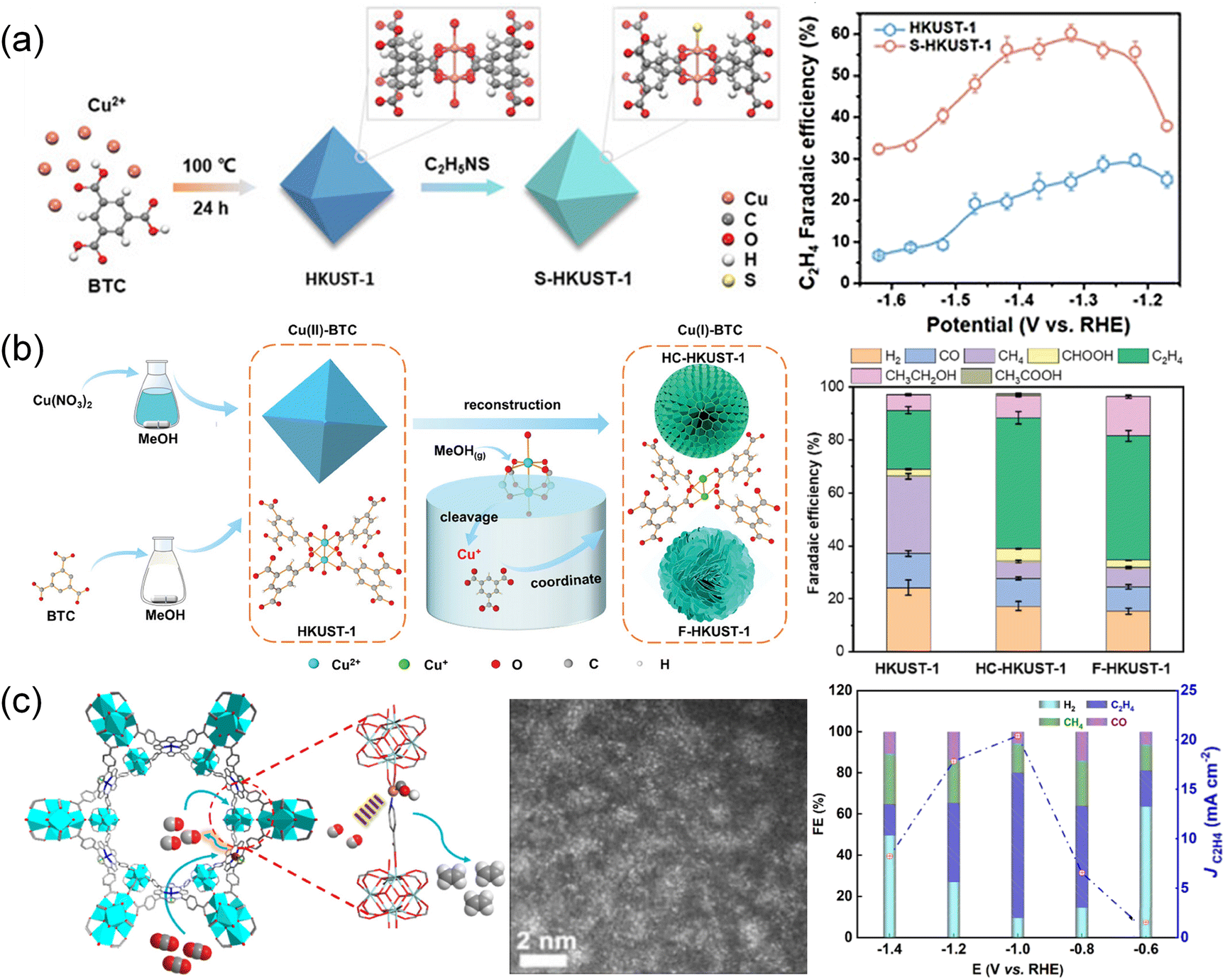 | ||
| Fig. 8 (a) Schematic illustration depicting the preparation of S-HKUST-1 and FE of C2H4 at different potentials.132 Copyright 2021, Wiley-VCH. (b) Schematic illustration of the synthetic procedures for HC-HKUST-1 and F-HKUST-1.190 Copyright 2024, Wiley-VCH. (c) Schematic illustration of the Cu–SAs@Ir–PCN-222-PA tandem electrocatalyst.53 Copyright 2024, American Chemical Society. | ||
5.2 MOF-derivatives through pyrolysis treatment
The process of structural transformation occurring at elevated temperatures is commonly referred to as pyrolysis or annealing. This process involves not only the reduction of ions to atoms but also the conversion of organic frameworks into carbon skeletons. High-temperature pyrolysis plays a crucial role in the creation of single-atom catalysts (SACs) and the introduction of heteroatoms through doping. This strategy allows for the regulation of the coordination environment of MOF-derived carbonaceous SACs for enhancement of CO2RR performance.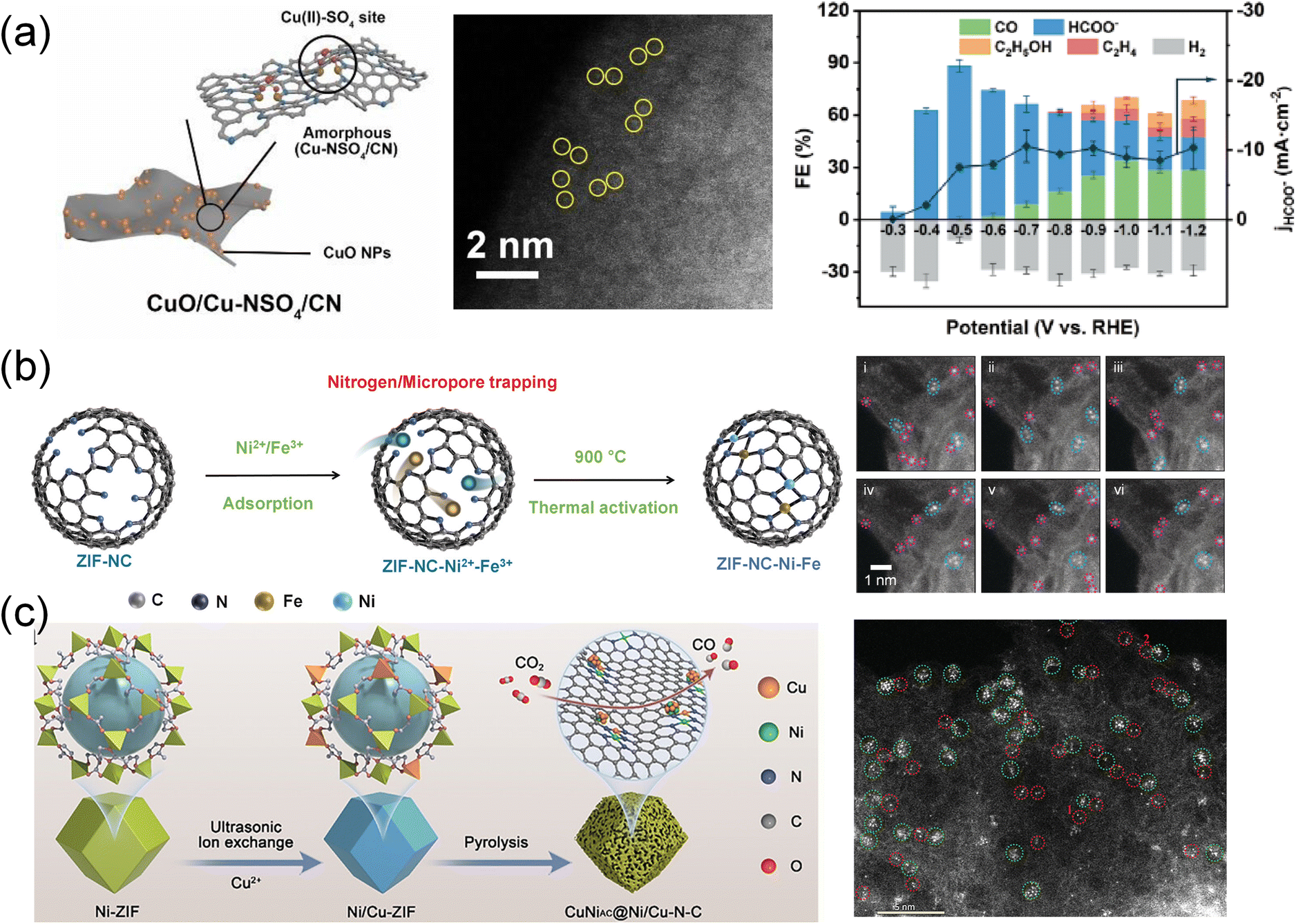 | ||
| Fig. 9 (a) Schematic illustration of the structure, aberration-corrected HAADF-STEM and FE of CuO/Cu–NSO4/CN.191 Copyright 2024, Wiley-VCH. (b) Schematic illustration and HAADF-STEM of dual metal Ni–Fe sites in ZIF–NC–Ni–Fe.86 Copyright 2022, Wiley-VCH. (c) Schematic diagram depicting the preparation of CuNiAC@Ni/Cu–N–C.192 Copyright 2024, Wiley-VCH. | ||
In addition to the active sites, recognized as independent units, the adjacent metal sites can largely affect the performance of the CO2 reduction reaction. Jiao et al.46 prepared Fe and Ni single-atom pairs on MOF-derived N-doped carbons (Fe1–Ni1–N–C) by the direct pyrolysis of MOFs. Due to the synergistic effect of Fe and Ni atom pairs, the Fe1–Ni1–N–C catalyst presented better selectivity for electrocatalytic reduction of CO2 to CO than Fe1–N–C and Ni1–N–C catalysts. Theoretical simulations revealed that single Fe atoms can be highly activated by adjacent Ni via non-bonding interactions in Fe1–Ni1–N–C, significantly facilitating the formation of COOH* intermediates and thereby accelerating the overall CO2 reduction. Similarly, a series of atomically dispersed and nitrogen coordinated dual-metal sites (ZIF–NC–Ni–Fe, ZIF–NC–Fe–Co, and ZIF–NC–Ni–Co) were designed (Fig. 9b).86 Among these, the ZIF–NC–Ni–Fe catalyst exhibited the most efficient CO2RR activity, achieving a maximum FECO of 97.8% at −0.6 V, surpassing ZIF–NC–Fe–Co (FECO = 76.3%) and ZIF–NC–Ni–Co (FECO = 68.9%). After characterization and theoretical prediction, it was confirmed that the most active N-coordinated dual-metal site was 2N-bridged (Fe–Ni)N6, where FeN4 and NiN4 moieties were shared with two N atoms. The redistributed electrons on the Fe and Ni atoms in ZIF–NC–Ni–Fe led to an optimal synergistic effect between the two metals, which facilitated *COOH adsorption and *CO desorption kinetics while suppressing the HER. In another report, a CuNi atomic cluster embedded Ni/Cu dual atomic site catalyst (CuNiAC@Ni/Cu–N–C) was designed (Fig. 9c).192 The introduction of Cu3Ni atomic clusters into N4Ni/CuN3 dual sites enhanced the electronic conductivity and decreased the energy barrier. Therefore, CuNiAC@Ni/Cu–N–C showed excellent performance with a maximum FECO of 98.2% at −0.7 V and maintained continuous electrocatalysis for 70 h.
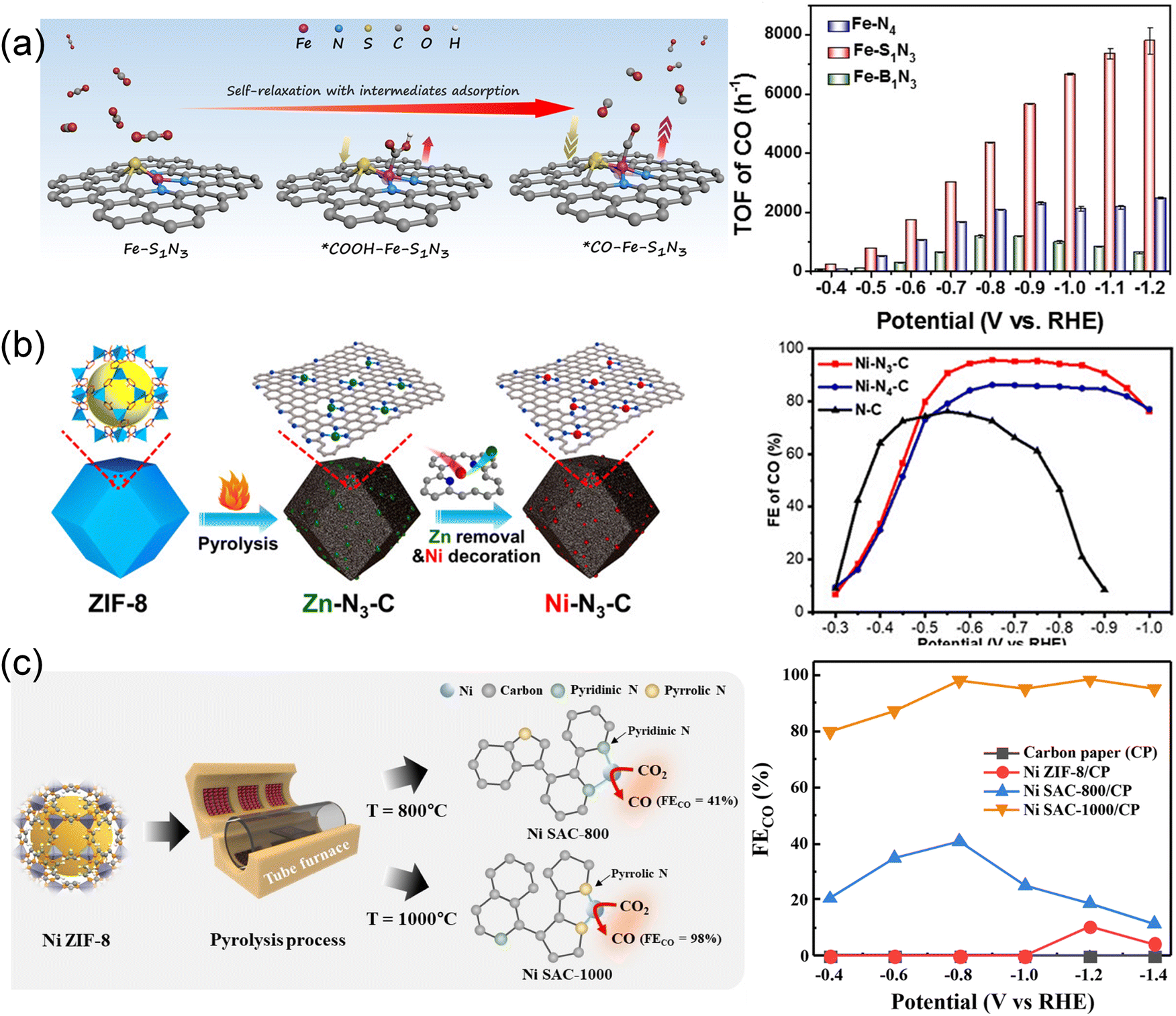 | ||
| Fig. 10 (a) Schematic illustration showcasing the structural distortion of the Fe–S1N3 site with various adsorbed intermediates.47 Copyright 2023, Wiley-VCH. (b) Schematic illustration for the fabrication of low-coordination single-atom Ni electrocatalysts.48 Copyright 2021, Wiley-VCH. (c) Schematic illustration for the fabrication of pyrrolic N-stabilized low-coordinated Ni SAC and FECO at different potentials.89 Copyright 2024, Royal Society of Chemistry. | ||
By manipulating pyrolysis temperatures, low-coordinated Ni–Nx sites were synthesized by replacing the atomically dispersed Zn sites with Ni atoms in a post-synthetic metal substitution strategy (Fig. 10b).48 The prepared Ni–N3–C catalyst showed excellent CO selectivity with an FECO of 95.6% and a TOF of 1425 h−1 at −0.65 V, superior to the Ni–N4–C catalyst. DFT calculations revealed that low-coordinated Ni–N3–C active sites significantly enhanced COOH* formation, thereby promoting CO2 conversion. By varying pyrolysis temperatures, significantly fewer low-coordinated Ni–N2–C sites were synthesized through a host-guest cooperative protection strategy involving the introduction of polypyrrole into a bimetallic MOF (MgNi–MOF-74).34 The resulting catalyst, NiSA–N2–C, displayed high FECO (98%) and TOF (1622 h−1), surpassing the performance of NiSA–N3–C and NiSA–N4–C. The coordination environment, which encompasses not only the amounts but also the types of N atoms, played a crucial role in these disparities. Through the pyrolysis of Ni ZIF-8 within the temperature range of 800 °C to 1000 °C (Fig. 10c), the ratio of pyrrolic N/pyridinic N increased from 0.37 to 1.01 as well as Ni–Nx sites with a decreased coordination number from 3.14 to 2.63.89 The pyrrolic N-stabilized Ni SAC-1000 showed heightened activity with an FECO of 98.24% at −0.8 V, surpassing Ni SAC-800 with an FECO of 40.76% at −0.8 V. DFT calculations revealed that the synergistic effect of the pyrrolic N and low-coordinated Ni could decreased the desorption energy barrier of *CO during the CO2RR, promoting the conversion of CO2 to CO.
Recent literature has brought attention to the often-overlooked O atoms. Zhang et al.54 reported an O-coordinated FeN4 active site (FeN4–O) through the pyrolysis of an O- and N-rich MOF (Fe-IRMOF-3) as shown in Fig. 11a. The atomically dispersed FeN4–O active sites broke the symmetrical structure of Fe–N4 sites and exhibited a superior catalytic performance compared to FeN4, reaching an FECO of 95% at −0.50 V and showing excellent stability over 30 h. The enhanced performance was attributed to the axial O coordination, which not only regulated the binding energy of intermediates and shifted the d-band center of Fe-3d orbitals, facilitating CO desorption, but also inhibited competitive hydrogen production. A similar modulation of the asymmetric atomic interface was also observed in Cu single-atom catalysts prepared by pyrolyzing Cu–Zn/MOF-74.83 The catalyst with CuN3O/C active sites delivered a higher FECO of 96% at −0.8 V compared to CuCO3/C (FECO of 20.0% at −0.5 V) and a higher TOF up to 2782.6 h−1. DFT calculations revealed that the incorporation of N and O atoms in CuN3O/C helped regulate the asymmetric atomic interface, leading to reduction in the Gibbs free energy of CO* desorption and ultimately improving catalytic performance.
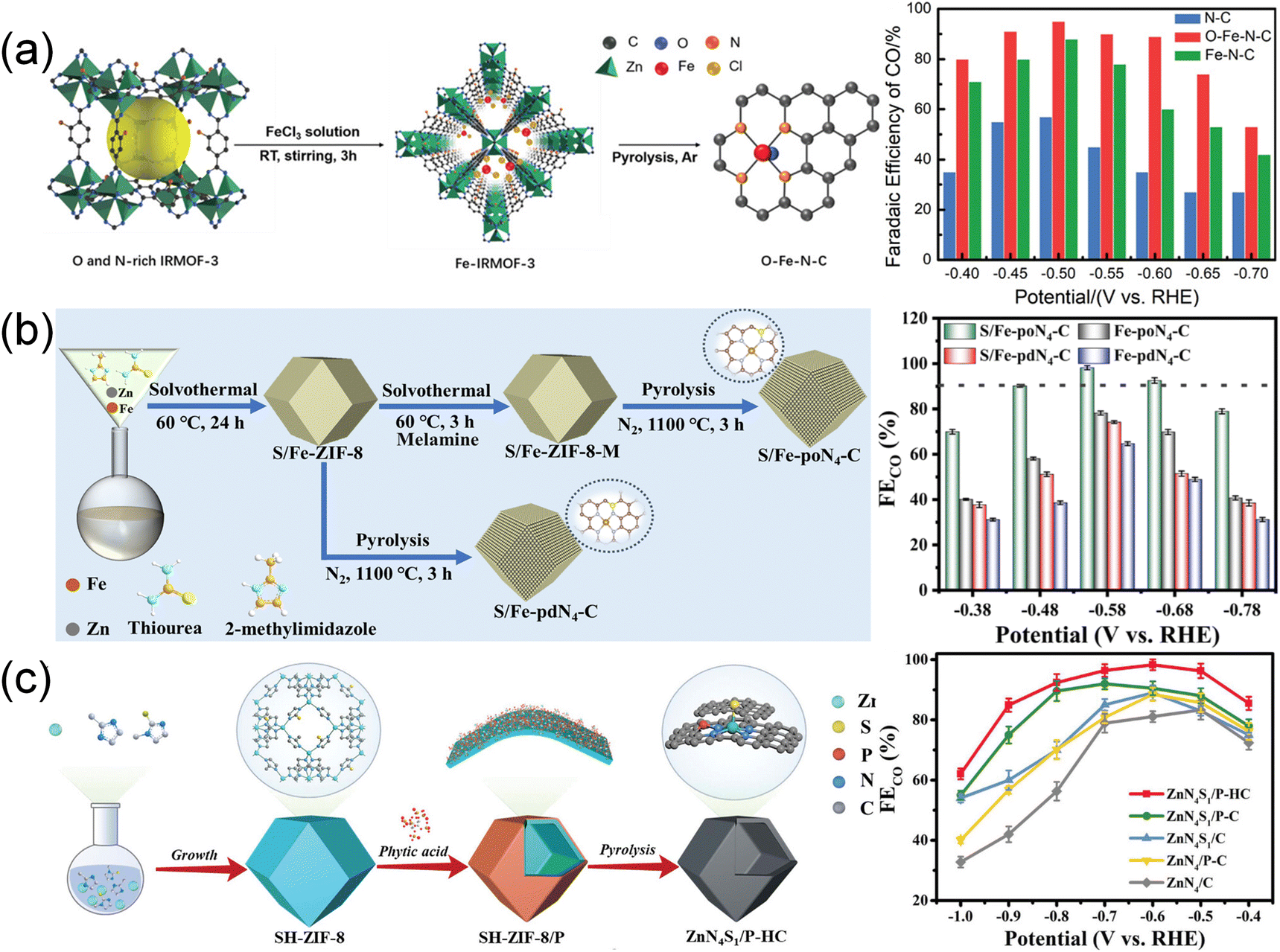 | ||
| Fig. 11 (a) Schematic synthesis of O–Fe–N–C and FECO in CO2-saturated 0.5 M KHCO3. Copyright 2022, Wiley-VCH.54 (b) Schematic illustration of preparing S/Fe-poN4-C. Copyright 2024, Royal Society of Chemistry.88 (c) Schematic illustration of the synthesis of ZnN4S1/P-HC. Copyright 2023, Wiley-VCH.96 | ||
S atoms, possessing lower electronegativity to N atoms, can also alter the electronic structure through the formation of M–S bonds. Yang et al.80 synthesized a S/N co-coordinated Ni single-atom catalyst (Ni-SNC) by pyrolysis of a SO42− doped Zn/Ni ZIF. The Ni atom interacted with 3 N atoms and a S atom to form an unsaturated Ni–N3–S active site. The unsaturated Ni–N3–S active site reduced the energy barrier of CO2 → COOH* conversion in the CO2RR process and S further improved the current density. A similar approach involved the incorporation of S and N atoms into the atomically dispersed pyrrole-type Fe–N4 through a series of “solvothermal–solvothermal–pyrolysis” treatments, featuring p–d orbital hybridization in the second coordination layer (Fig. 11b).88 The S-doped pyrrole-type Fe–N4 electrocatalyst (S/Fe-poN4-C) not only modulated the electron density of the Fe site, facilitating the desorption of *CO, but also compressed H2 evolution. This catalyst exhibited outstanding performance, achieving an FECO of 98% and a TOF of 4621.2 h−1. In situ characterization and calculations indicated that the p–d orbital hybridization balanced the adsorption of *COOH and *CO and accelerated the proton transfer.
P atoms also exhibit a lower electronegativity compared to N atoms, often enhancing the catalytic activity through alterations in the coordination environment and optimization of the adsorption process.196,197 Reports have highlighted similar catalytic sites of Ni–P1N3 and Fe–P nanocrystals anchored in N-doped carbon polyhedra.197,198 Recently, Hu et al.95 constructed a three-dimensional ordered microporous structure with ZnN4 sites embedded in P-functionalized carbon (H-3DOM-ZnN4/P–C). The rich hollow walls exposed more active sites while P atoms could adjust the electronic structure of ZnN4 sites, optimizing the adsorption of *COOH intermediates. H-3DOM-ZnN4/P–C showed approximately 100% FECO at −0.6 V and a high TOF up to 7.8 × 104 h−1 at −1.0 V. Subsequently, they reported a S and P co-doped CO2RR electrocatalyst, denoted as ZnN4S1/P-HC, employing a “synergistically near- and long-range regulation” strategy (Fig. 11c).96 Zn–N4 was decorated with an axial S ligand in the first coordination and adjacent P atom in the second coordination, leading to an enhancement of charge density in Zn sites. The optimization improved the adsorption of *COOH intermediates, resulting in exceptional performance with FECO close to 100% at −0.6 V.
6. Stability and reconstruction issues
The CO2RR is usually performed at various reduction potentials, which may lead to the reduction of metal ions of MOFs under certain circumstances. For example, bismuth (Bi) based electrocatalysts are considered environmentally friendly for producing formates due to low toxicity and inert HER activity.199–201 Lamagni et al.52 reported that Bi(1,3,5-tris(4-carboxyphenyl)benzene) MOF, denoted as Bi(btb), underwent an in situ structural rearrangement under CO2RR conditions, forming well-dispersed and highly active Bi nanoparticles for CO2 conversion (Fig. 12a). Interestingly, the inherent poor conductivity of Bi(btb) was overcome by the structural rearrangement. Following CO2RR testing, FE of formates reached 95% at an overpotential of 770 mV, with a mass activity of up to 261(13) A g−1 achieved. Cao et al.111 prepared atomically thin bismuthene (Bi-ene) by in situ electrochemical transformation of bismuth-based metal–organic layers (Fig. 12b). The as-obtained Bi-ene exhibited a high selectivity (∼100% FE) for formates, good stability in a broad potential range exceeding 0.35 V and ultrahigh current densities over 300 mA cm−2. Attenuated Total Reflection Fourier-Transform Infrared (ATR-FTIR) spectroscopy and DFT calculations indicated that formate was produced via the OCHO* intermediate, and the adsorbed HCO3− was found to have key roles in the CO2RR. Yao et al.104 investigated a Bi-MOF constructed from bismuth oxyiodide and H3BTC (benzene-1,3,5-tricarboxylic acid), demonstrating that electrolyte and potential mediated the restructuring processes (Fig. 12c). The Bi-MOF transformation into Bi2O2CO3 and subsequent reduction to Bi played a pivotal role in determining morphology, defects, composition, and valence states, leading to enhanced formate yields and partial current densities. The resulting Bi nanosheets showed a maximum FEHCOO− of 92% at −1.1 V and a partial current density of −15 mA cm−2 at −1.3 V. The presence of unsaturated Bi atoms facilitated *OCHO intermediate adsorption, contributing to improved reaction efficiency. Huang et al.202 further studied the transformation process of Bi-BTC (formed using Bi irons and H3BTC) and its CO2RR performance. The transformation of the column Bi-BTC structure into nanosheets was influenced by the breaking of the Bi–O bond. And the exposed (020) crystal plane of restructured Bi2O2CO3 favored *OCHO intermediate interactions. As a result, it achieved an FE of 96% and a current density of 25 mA cm−2. These studies underscored the significance of surface restructuring via electrochemical methods for developing high-performance and durable electrocatalysts.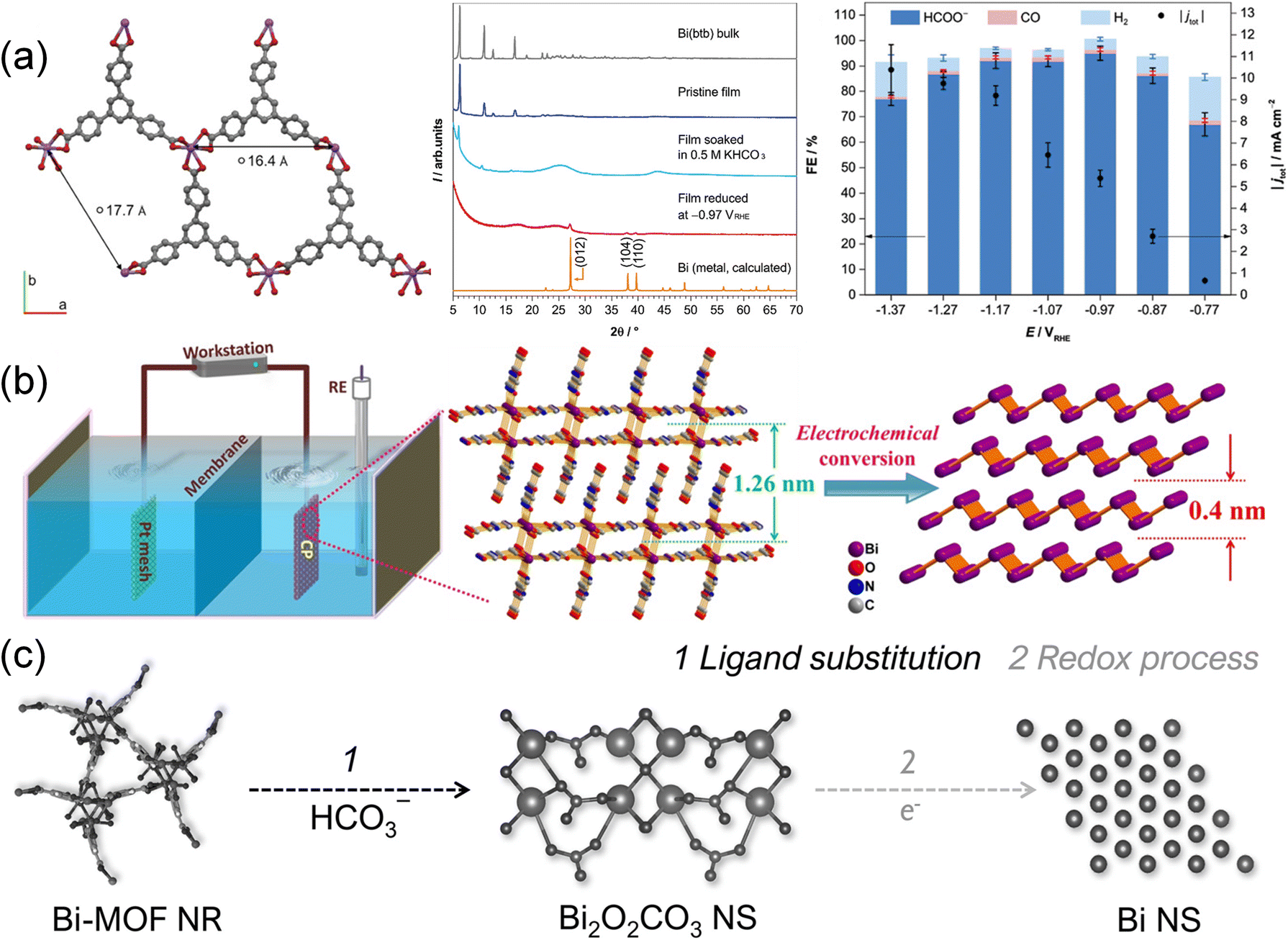 | ||
| Fig. 12 (a) Structural representation, XRD spectra and FE data of Bi(btb).52 Copyright 2020, Wiley-VCH. (b) Schematic illustration depicting the preparation of Bi-ene.111 Copyright 2020, Wiley-VCH. (c) Schematic illustration depicting the in situ reconstruction process of Bi-MOF NRs to Bi NSs.104 Copyright 2021, Wiley-VCH. | ||
Recently, the reconstruction of Cu-based MOFs was also investigated. Cu with low valence usually exhibits enhanced reactivity for C2 products. Initially, utilizing an electrochemical conversion method (Fig. 13a), CoS2 was obtained from a Co-ZIF-L precursor.203 Subsequently, the CoS2 template was converted into Cu2S nanocrystals through an electrochemical cation exchange process. The resulting Cu2S maintained its original three-dimensional morphology and a high density of grain boundaries. When applied in the CO2RR process, the catalyst demonstrated notable efficiency in converting CO2 to formates, achieving an impressive FE of 87.3%. Zhang et al.204 reported coordinatively unsaturated Cu paddle wheel (CU-CPW) clusters in defect-containing HKUST-1 (Cu(II)-BTC) prepared by an atomized trimesic acid and Cu(OH)2 strategy (Fig. 13b). Cu2(HCOO)3 in CU-CPW is proven to be maintained after the electrochemical structural transformation and serve as the active site. CU-CPW accelerated the PCET process compared with coordinatively saturated Cu2(HCOO)4, producing hydrocarbons from CO2. A highly active and selective C2H4 electrocatalyst (CuPz2-Act-30) derived from MOF CuPz2 (Pz = Pyrazole) with dispersive Cu/Cu2O nanoclusters was prepared by in situ electrochemical reduction (Fig. 13c).134 It achieved a high FE of 70.2 ± 1.7% at −1.03 V toward C2H4 generation. In situ ATR-FTIR spectroscopy and DFT calculations indicated that the nanoclusters transformed by in situ reconstruction had a preference to adsorb local CO and promote C–C coupling and hydrogenation to produce C2H4. Moreover, restructuring multi-phase interfaces of Ag/Cu/Cu2OAg0.1/HKUST-1 in Ag0.1/HKUST-1 was achieved by 1 h of electroreduction.131 By leveraging the higher standard redox potential of Ag and its less negative formation enthalpy of oxides, Ag was expected to stabilize neighboring Cu(I). Tandem electrolysis was enabled by the weak bonding of Ag *CO to Cu(0)–Cu(I) sites. The derived electrocatalysts showed a high FE of 57.2% at −1.3 V for CO2 conversion to C2H4. Characterization and calculations further highlighted the crucial role of Ag in stabilizing Cu(I) and increasing CO surface coverage, while the Cu/Cu2O interface reduced the energy barrier of C–C coupling. Tandem electrolysis and electrochemical reduction were also successfully carried out on a [CuI(im)] (1) MOF (Im = imidazole),124 resulting in the synthesis of CuN2 and Cu2N4. The CuN2/Cu(111) derivatives with Cu–N coordination exhibited superior performance compared to Cu2N4/Cu(111) with Cu–N and Cu–Cu coordination. The C2+ products including C2H4, C2H5OH and n-C3H7OH were detected and the total FEC2+ of CuN2 reached 64.8%, surpassing the 43.9% efficiency of Cu2N4. This study emphasized the crucial role of pristine MOFs as catalysts for facilitating the conversion of CO2 to CO.
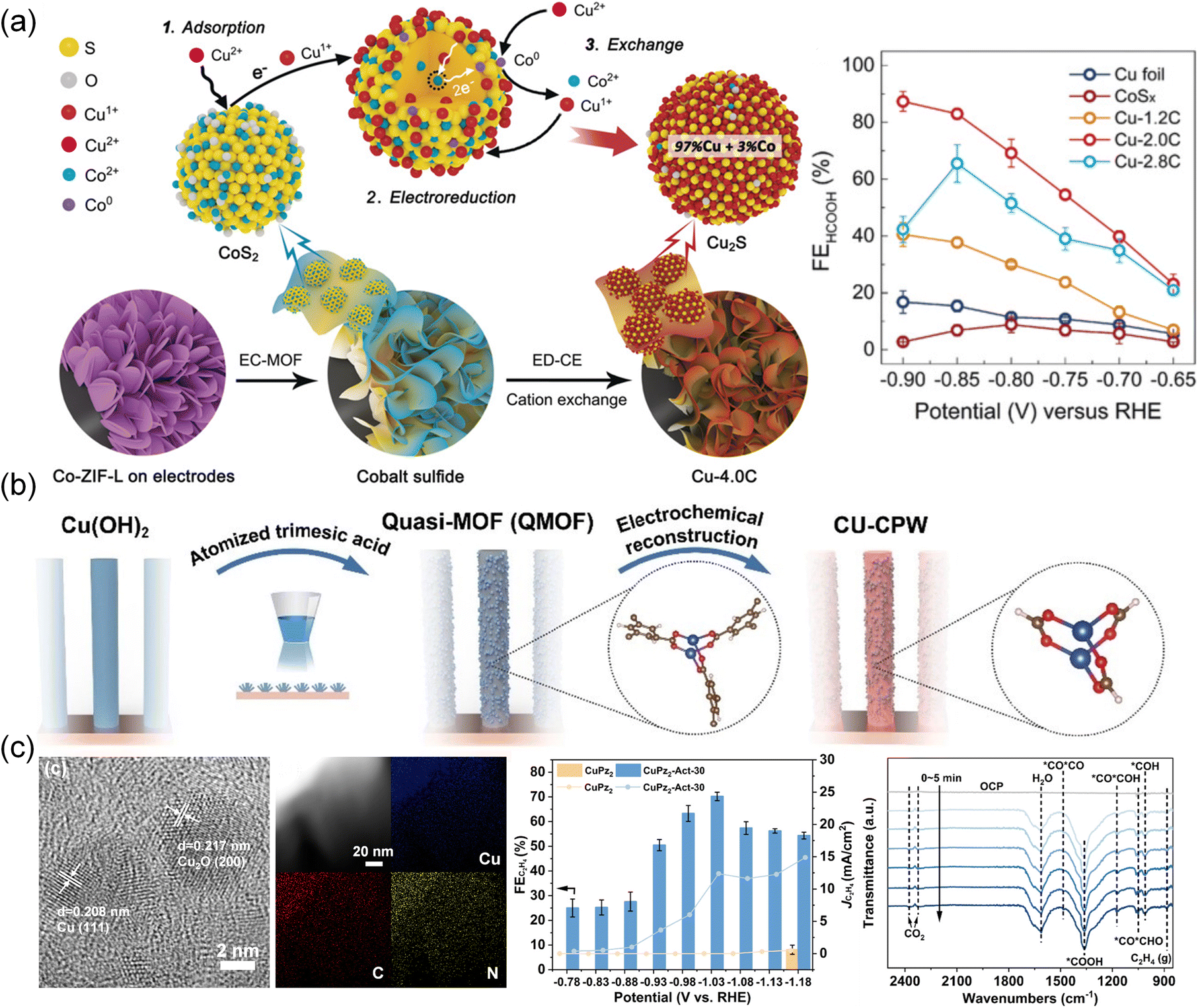 | ||
| Fig. 13 (a) Schematic illustration depicting the pathways/mechanisms for electrochemically driven cation exchange and FE of different catalysts.203 Copyright 2020, Wiley-VCH. (b) Schematic illustration of the synthetic procedures for CU-CPW.204 Copyright 2021, Wiley-VCH. (c) HRTEM images of CuPz2-Act-30 with the corresponding elemental mapping, FE of C2H4, and in situ ATR-FTIR spectra of CuPz2-Act-30.134 Copyright 2022, American Chemical Society. | ||
7. Advanced characterization techniques
Though the well-defined structure of MOFs can be tailored to help understand the structure–activity relationship, the electrochemical stability of MOFs would be a major concern for their application. Under the reduction conditions of the CO2RR, it is common for electrocatalysts undergoing reconstruction to build new active sites.104,134,205,206 For example, Cu centers in Cu3(HITP)2 would be irretrievably reduced to Cu0 at negative potential.133 Cu clusters were detected during the CO2RR from Cu dimers in HKUST-1, and Cu/CuxSy interfaces were reconstructed from S-HKUST-1 (with a Cu–S motif on HKUST-1) under CO2RR conditions.104,126,132 Therefore, it is important to monitor the structural changes of electrocatalysts during reactions to probe into the origin of the CO2RR on MOFs using in situ techniques. In addition, dynamic electrode/electrolyte interfaces can also be monitored to shed light on the reaction mechanisms.7.1 In situ XAS for probing active sites at the atomic level
XAS has emerged as a popular technique to detect the local structure around single metal atoms, which is not limited to the state of samples under testing. It can provide valuable information on the oxidation state, the local coordination environment, and the electronic structure of the material.207 The changes in metal nodes and the coordination environment surrounding metal active centers can be reflected in the in situ XAS spectra to monitor the CO2RR dynamics at the atomic level.208As shown in Fig. 14a, (5,10,15,20-tetrakis(4-carboxyphenyl)porphyrinato)-Fe(III) chloride (FeTCPPCl) was introduced into UiO-66 to form a robust active MOF (FeTCPP⊂UiO-66) with two carboxylic ligands.94 FeTCPP⊂UiO-66 realized nearly 100% selectivity for converting CO2 to CO. In situ near edge XAS displayed almost unchanged curves at different potentials compared to the initial state, which demonstrated the structural stability of FeTCPP⊂UiO-66 in an electrochemical environment. However, the wavenumber of the oscillation curve for UiO-66/FeTCPP (the physical mixture of FeTCPP and UiO-66) descended distinctly at −0.51 V, indicating its structural collapse. Due to the robust structure and protonation facilitator of FeTCPP⊂UiO-66, the PCET pathway on the iron porphyrin sites was facilitated, leading to a low overpotential. Similarly, no significant changes in the in situ XAS spectra of the PcCu-Cu-O MOF catalyst confirmed its stability during the CO2RR as shown in Fig. 14b.127 The stability of Ag/Zr-fcu-MOF-BDC with Ag-incorporated was also investigated by operando XAS (Fig. 14c).77 The oxidation states, coordination number and atomic distance showed no observable difference during and after the reaction as shown in the Zr K-edge X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) spectra, indicating a constant Zr oxidation state and robust structure of Ag/Zr-fcu-MOF-BDC. This was also found in the Y K-edge XANES and EXAFS spectra of Ag/Y-fcu-MOF-NDC, suggesting that the Zr- and Y-based MOFs with varying organic linkers and the metal nodes remained stable during the CO2RR.
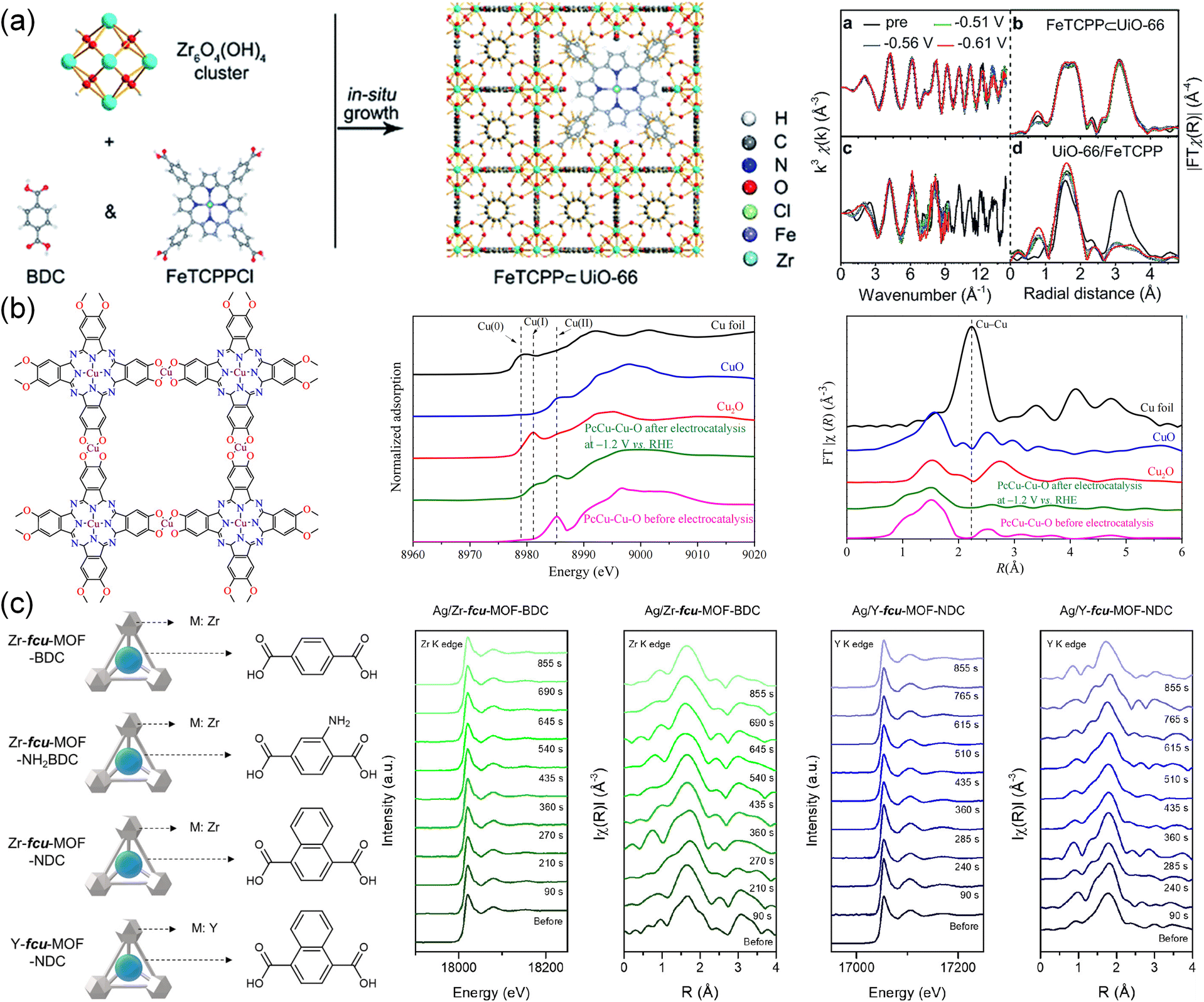 | ||
| Fig. 14 (a) Schematic of FeTCPP⊂UiO-66 MOF and the Zr K-edge in situ EXAFS for FeTCPP⊂UiO-66 and UiO-66/FeTCPP.94 Copyright 2019, Royal Society of Chemistry. (b) Illustration of the structure, normalized Cu K-edge XANES and Fourier transform EXAFS of PcCu-Cu-O MOF.127 Copyright 2021, American Chemical Society. (c) Schematic of Zr-fcu-MOF-BDC, Zr-fcu-MOF-NH2BDC, Zr-fcu-MOF-NDC and Y-fcu-MOF- NDC. Operando XAS analysis of the Zr K-edge XANES and EXAFS of Ag/Zr-fcu-MOF-BDC and the Y K-edge XANES and EXAFS of Ag/Y-fcu-MOF-NDC.77 Copyright 2020, American Chemical Society. | ||
7.2 In situ FTIR and Raman spectroscopy for monitoring dynamic interfaces
FTIR (Fourier-transform infrared spectroscopy) and Raman spectroscopy are useful spectroscopic techniques for capturing reaction intermediates on the surface of catalysts and providing information related to reaction products and pathways. By using FTIR and Raman spectroscopy, the vibration modes of chemical bonds and structural coordination of the intermediates during the CO2RR can be acquired. However, FTIR is usually interfered with by the water absorption peak in aqueous solution, which makes it a great challenge to monitor intermediates when applied in aqueous solution. Instead of measuring the absorption of light like FTIR, Raman spectroscopy based on the scattering of light can compensate for this drawback due to weak light scattering by water.210
In situ FTIR spectroscopy was reported to capture intermediates on the catalyst surface during the CO2RR. Fig. 15a shows the in situ FTIR spectra of Cu-HITP.129 The bands at 1387 and 1400 cm−1 were assigned to the *COOH and *COO, while the band at 2079 cm−1 was assigned to the chemisorbed *CO. The absorption bands of the *CH2 intermediate were also observed (3108 cm−1 and 995 cm−1). The band at 1150 cm−1 was attributed to the *COH, indicating the hydrogenation of *CO into *COH. Additionally, the band at 1575 cm−1 indicated the existence of *OCCOH, which meant that *OCCOH was a key intermediate for CO2 conversion to C2+ products. Similarly, a MOF-on-MOF (Cu@Bi1/2, CAU-17 and ZJU-199) copper-based catalytic electrode was prepared by in situ synthesis on a foamed copper substrate209 and studied using in situ FTIR to reveal the mechanism of HCOOH formation. As shown in Fig. 15b, the characteristic band located at 1363 cm−1 corresponded to the HCOO− which displayed a trend of increasing and then decreasing with the potential from −0.35 to 0.95 V. In addition, there was a HCOO− band that appeared at 1743 cm−1 different from that of Bi@Cu1/2, indicating different reaction ways to generate formic acid. The FE was further improved using a bismuth-based metal–organic framework (PZH-1), which was synthesized through a ligand-directing strategy using pyrazole-3,5-dicarboxylic acid (Fig. 15c).102 The bismuth-based ZMOF showed an ACO topological structure and strong coordination bonding within the Bi-based cages. In situ FTIR spectra of Bi-ZMOF showed a characteristic band of two-oxygen bridged *HCOO (1414 cm−1) at −0.4 V, which was intensified as the potential shifted from −0.4 V to −1.4 V. The band in the range of 1675–1685 cm−1 associated with the CO stretching mode became more prominent as the potential decreased. These findings indicated that the surface-bound Bi–*HCOO species played a crucial role as active intermediates for forming HCOOH.
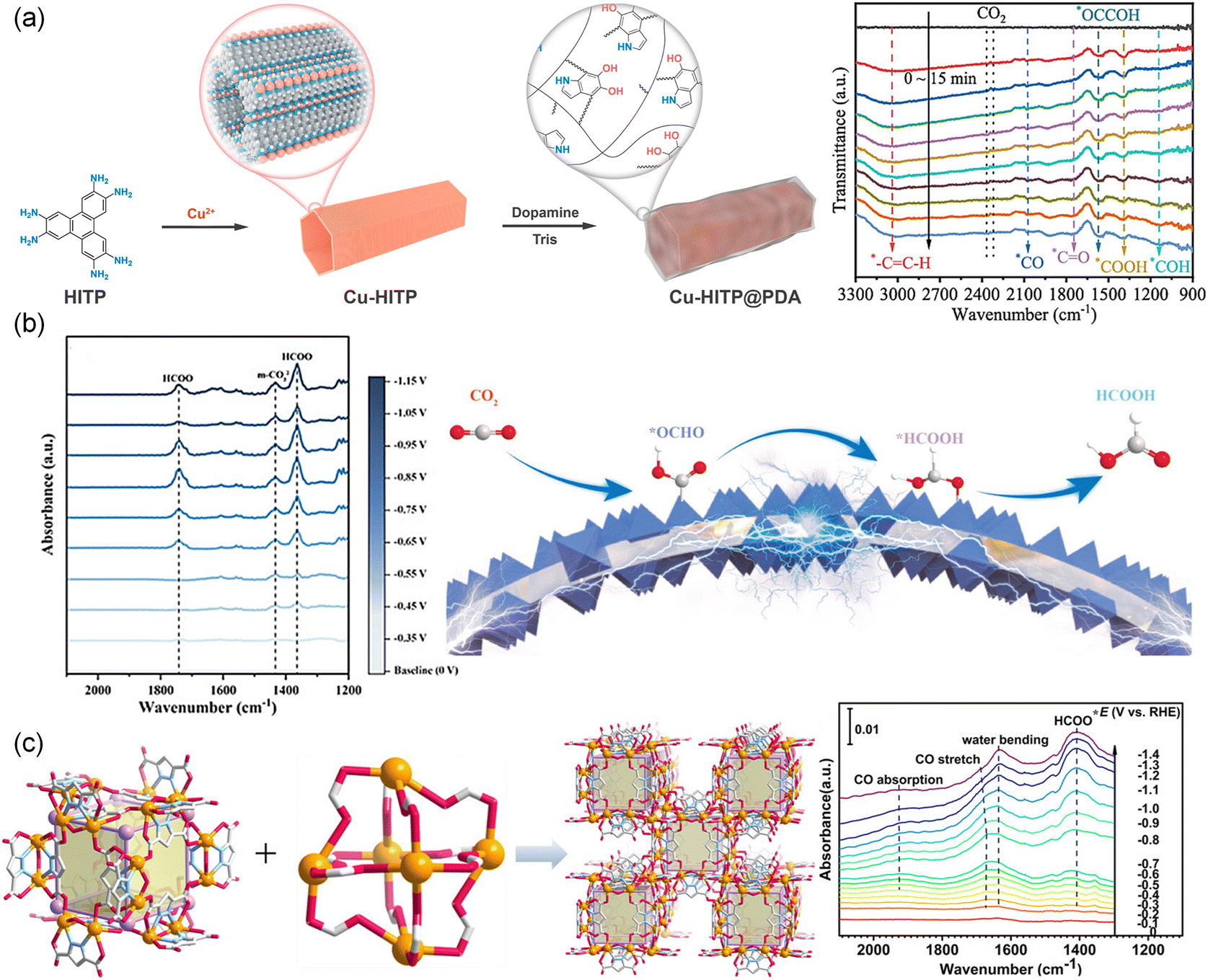 | ||
| Fig. 15 (a) Schematic of Cu-HITP@PDA and in situ FTIR spectra on Cu-HITP.129 Copyright 2022, American Chemical Society. (b) In situ FTIR spectra of Cu@Bi1/2 at various potentials and the schematic diagram of the CO2RR.209 Copyright 2023, Wiley-VCH. (c) Schematic of PZH-1 and in situ FTIR spectra of PZH-1.102 Copyright 2023, Wiley-VCH. | ||
The use of in situ Raman spectroscopy also provided valuable insights into the stability and reaction mechanisms of various electrocatalytic systems. Isomorphic MOFs (Me2NH2+){InIII-[Ni(C2S2(C6H4COO)2)2]}·3DMF·1.5H2O (1′′, DMF = N,N-dimethylformamide) and (Me2NH2+)[InIII-(TTFTB)]·0.7C2H5OH·DMF (2, TTFTB = tetrathiafulvalene-tetrabenzoate) were used as electrocatalysts for the CO2RR.99 The [NiS4] site in 1′′ was considered to be the catalytic center and CO2 binding site. Therefore, 1′′ showed enhanced CO2RR performance (FEHCOO− = 89.6%) compared with 2 (FEHCOO− = 54.7%). As shown in Fig. 16a, in situ Raman spectra exhibited no visible change in the CO2RR process, indicating the stability of the catalytic sites.
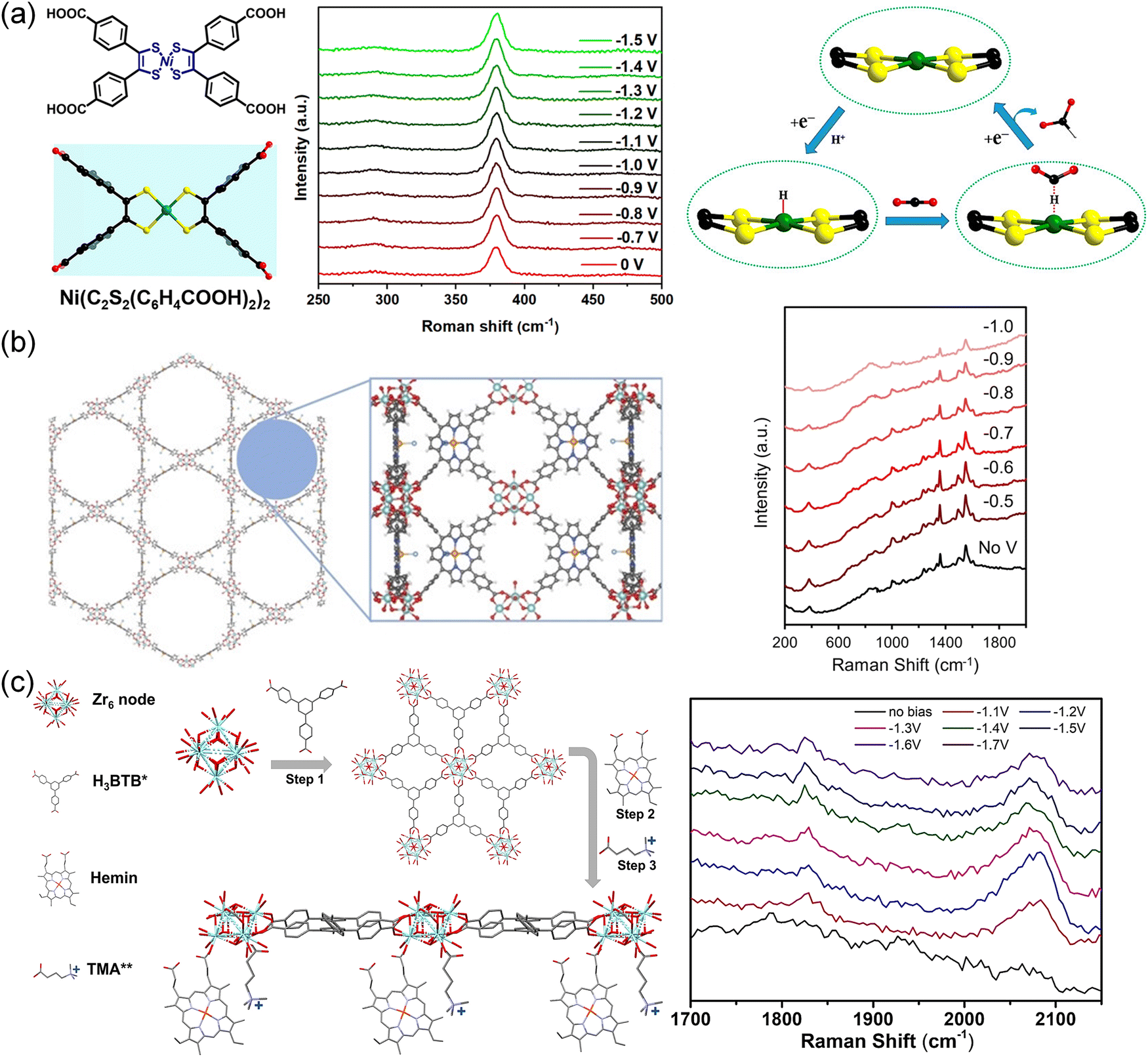 | ||
| Fig. 16 (a) Ligand structures of [Ni(C2S2(C6H4COOH)2)2], in situ Raman spectra of the Ni–S bond in 1′′ during the CO2RR process and proposed reaction paths for the formation of HCOO− on the [NiS4] sites.99 Copyright 2021, American Chemical Society. (b) Immobilized metalloporphyrin in MOF and in situ Raman spectra for each potential for PCN-222(Fe).211 Copyright 2022, Wiley-VCH. (c) Schematic of Zr-BTB@Hemin-TMA, and in situ Raman spectra recorded at different potentials for Zr-BTB@Hemin-TMA.72 Copyright 2022, Wiley-VCH. | ||
By immobilizing iron–porphyrin complexes into MOFs (Fig. 16b), PCN-222(Fe) exposed more active sites for the CO2RR toward CO production, which showed an increasing current trend as the potential increased from −0.5 to −1.0 V.211In situ Raman spectra suggested that the structure was maintained under electrochemical bias. The characteristic bands of PCN-222(Fe) did not attenuate under electrochemical bias, indicating that no structural reconstruction occurred during the CO2RR.
As shown in Fig. 16c, a cationic functional group (3-carboxypropyl trimethylammonium) was immobilized to Fe-porphyrin (Hemin)-modified Zr-BTB (termed Zr-BTB@Hemin-TMA) to enhance the activity and selectivity (∼100%).72In situ Raman spectroscopy conducted at different potentials suggested that two bands at 1840 cm−1 and 2060 cm−1 were attributed to catalyst-bounded CO stretching, indicating the presence of a weakly bounded CO intermediate. The enhanced selectivity resulted from the electrostatic stabilization provided by the tethered functional group, which facilitated the swift release of CO as a product.
8. Challenges and perspectives
8.1 Challenges
Metal–organic frameworks (MOFs), known for their orderly arrangement, extensive surface area, and adaptable structure, have been applied as electrocatalysts together with their derivatives for the CO2 reduction reaction (CO2RR). Different metal centers and surrounding atoms yield distinct reduction products. By selecting appropriate metals and meticulously adjusting the coordination environment, desired reduced carbon products can be obtained. However, their application still faces many challenges due to inherent limitations such as poor conductivity, low chemical stability, and limited density of active sites. Some issues need to be considered in designing highly active and selective electrocatalysts, which we list as follows:(1) The issue of poor conductivity arises from organic ligands in MOFs, hindering electron transfer during the electrocatalytic reduction process. Despite efforts to enhance conductivity through strategies such as morphological and structural regulation of MOFs, creation of unsaturated metal coordination sites, incorporation of functionalized ligands and/or multi-metal components in MOFs, and integration into conductive active materials, the conductivity of two-dimensional (2D) MOFs remains inferior to that of 2D graphene. Improving the conductivity of MOFs is still an important research topic for future advancements in this field.
(2) MOFs bonded with a coordination bond are prone to dissolution in the acidic or alkaline solution, underscoring the critical need to enhance the intrinsic stability of MOFs. Therefore, designing robust MOF catalysts adaptable to diverse environments is highly desirable. It may be considered to use that strategies involve creating acid-resistant MOFs with high-valence state metals (hard acids) and carboxylate ligands (hard bases); developing alkali-resistant MOFs with low-valence state metals (soft acids) and azolate ligands (soft bases); improving the connectivity of building units; tightening or stiffening the ligands; and fortifying MOFs by substituting unstable building units with resilient counterparts for improving stability.
(3) The density of active sites within MOFs is crucial for assessing CO2RR performance. Active sites within MOFs typically encompass metal centers and/or the surrounding ligands as illustrated in the above-mentioned sections, which have certain and thus limited quantities in a certain MOF structure. Therefore, enhancing the density of active sites in pristine MOFs is also effective in improving the CO2RR performance, which demands the exploration of newly designed MOFs. In addition, we also propose to incorporate functional guest molecules or elements into the pore channels of MOFs for enhancing the catalytic activity of MOFs toward the CO2RR, which may increase active sites in another way. Similarly, for MOF derivatives, their active densities can be increased through impregnation methods or other approaches with optimized loading followed by high-temperature pyrolysis.
8.2 Perspectives
Aligned with these challenges and issues to be addressed in the application of MOFs and their derivatives for the CO2RR, perspectives and future directions are proposed in this section. The metal nodes of MOFs or metal centers of MOF derivatives typically dictate the pathway toward various reduction products, while coordinated organic ligands or the surrounding heteroatoms usually influence the selectivity by adjusting the electron density around the metal nodes or metal centers. Regulation of the metal oxidation state and surrounding coordination environment has been demonstrated to effectively affect the electrochemical performance. Despite considerable efforts in applying MOFs and their derivatives for electrocatalytic CO2 reduction, designing novel structures with high performance still remains challenging and desirable. Furthermore, mechanisms of existing MOFs and their derivatives need to be clearly studied to guide future structure design and performance improvement. In summary, we believe that future directions for designing and developing novel MOF-based catalysts can be focused on the following:(1) Rationally designing metal centers: the selection of appropriate metal centers is essential for achieving desired products. The CO2RR can be extended from single-metal MOF-based catalysts to bimetallic, trimetallic or polymetallic MOFs, which often exhibit enhanced performance due to synergistic effects from the neighboring metal centers.
(2) Engineering the coordination environment: the microenvironment surrounding the metal centers significantly impacts product yield and selectivity. Modifying ligands with coordination functional groups or introducing new functional coordinating ligands by direct synthesis or post-modification within MOFs may be used to improve electrocatalytic CO2RR performance.
(3) Optimization of conductive substrates or inserted nanocomposites: integrating conductive substrates or nanocomposites with MOFs can significantly decrease interfacial resistance, thereby improving the electron transfer process. Consequently, suitable conductive substrates and/or nanocomposites may be developed for the future synthesis of conductive MOF-based catalysts.
(4) Advanced characterization techniques: utilizing advanced characterization techniques provides an opportunity to unravel the intrinsic active sites and capture reaction intermediates, offering insight into the reaction mechanism. It is desired to further develop advanced in situ characterization techniques for monitoring the dynamic changes of MOF-based electrocatalysts and electrode/electrolyte interfaces during the CO2RR, thereby facilitating the design and synthesis of highly active and selective MOF-based electrocatalysts.
(5) Computational calculations: to support mechanistic studies, computational simulations such as DFT calculations and molecular dynamics are also needed from the perspective of thermodynamics and kinetics of the CO2RR. Machine learning may also be involved as an effective method for screening MOF-based catalysts.212 The integration of computational tools will effectively pave the way for rational design of MOF-based catalysts in the CO2RR.
Data availability
All the data supporting this review have been cited from reported publications, which are indicated throughout this review with copyrights obtained.Author contributions
Xiaoming Liu: investigation, conceptualization, formal analysis, visualization, writing – original draft review and editing. Xuan-He Liu: supervision, funding acquisition, writing – review and editing. Xiangrui Zhang: writing – review. Huan Wang: review. Qinglan Zhao: supervision, funding acquisition, writing – review and editing.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21603194 and 21874120) and Fundamental Research Funds for the Central Universities (No. 2652018056 and 2652018004). QZ thanks the Hong Kong Postdoctoral Fellowship Scheme (HKUST PDFS2021-4S12).References
- X. Li, S. Wang, L. Li, Y. Sun and Y. Xie, J. Am. Chem. Soc., 2020, 142, 9567–9581 CAS.
- C. Hu, S. Bai, L. Gao, S. Liang, J. Yang, S.-D. Cheng, S.-B. Mi and J. Qiu, ACS Catal., 2019, 9, 11579–11588 CrossRef CAS.
- H. Liu, Y. Zhu, J. Ma, Z. Zhang and W. Hu, Adv. Funct. Mater., 2020, 30, 1910534 CrossRef CAS.
- Z. Liu, Z. Deng, G. He, H. Wang, X. Zhang, J. Lin, Y. Qi and X. Liang, Nat. Rev. Earth Environ., 2022, 3, 141–155 CrossRef.
- S. Bhattacharjee, M. Rahaman, V. Andrei, M. Miller, S. Rodríguez-Jiménez, E. Lam, C. Pornrungroj and E. Reisner, Nat., Synth., 2023, 2, 182–192 CrossRef.
- X. Chen, X. Su, H.-Y. Su, X. Liu, S. Miao, Y. Zhao, K. Sun, Y. Huang and T. Zhang, ACS Catal., 2017, 7, 4613–4620 CrossRef CAS.
- A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille and P. J. Kenis, Chem. Rev., 2013, 113, 6621–6658 CrossRef CAS PubMed.
- J. Shi, Y. Jiang, Z. Jiang, X. Wang, X. Wang, S. Zhang, P. Han and C. Yang, Chem. Soc. Rev., 2015, 44, 5981–6000 RSC.
- E. Le Saché and T. Reina, Prog. Energy Combust. Sci., 2022, 89, 100970 CrossRef.
- D. He, J. Yan, K. Chen, L. Zhang, J. Lu, J. Liu and Y. Luo, ACS Catal., 2023, 13, 12114–12124 CrossRef CAS.
- M. Li, Z. Sun and Y. H. Hu, Chem. Eng. J., 2022, 428, 131222 CrossRef CAS.
- J. W. Ko, S.-W. Kim, J. Hong, J. Ryu, K. Kang and C. B. Park, Green Chem., 2012, 14, 2391–2394 RSC.
- E. W. Lees, B. A. Mowbray, F. G. Parlane and C. P. Berlinguette, Nat. Rev. Mater., 2022, 7, 55–64 CrossRef CAS.
- W. Xia, Y. Xie, S. Jia, S. Han, R. Qi, T. Chen, X. Xing, T. Yao, D. Zhou and X. Dong, J. Am. Chem. Soc., 2023, 145, 17253–17264 CrossRef CAS.
- D. Song, Y. Lian, M. Wang, Y. Su, F. Lyu, Z. Deng and Y. Peng, eScience, 2023, 3, 100097 CrossRef.
- Z. Ma, T. Wan, D. Zhang, J. A. Yuwono, C. Tsounis, J. Jiang, Y.-H. Chou, X. Lu, P. V. Kumar and Y. H. Ng, ACS Nano, 2023, 17, 2387–2398 CrossRef CAS PubMed.
- S. Yang, M. Jiang, W. Zhang, Y. Hu, J. Liang, Y. Wang, Z. Tie and Z. Jin, Adv. Funct. Mater., 2023, 33, 2301984 CrossRef CAS.
- P. Saha, S. Amanullah and A. Dey, Acc. Chem. Res., 2022, 55, 134–144 CrossRef CAS PubMed.
- S. Banerjee, C. S. Gerke and V. S. Thoi, Acc. Chem. Res., 2022, 55, 504–515 CrossRef CAS PubMed.
- G. Wen, B. Ren, Y. Zheng, M. Li, C. Silva, S. Song, Z. Zhang, H. Dou, L. Zhao and D. Luo, Adv. Energy Mater., 2022, 12, 2103289 CrossRef CAS.
- S. Jin, Z. Hao, K. Zhang, Z. Yan and J. Chen, Angew. Chem., Int. Ed., 2021, 60, 20627–20648 CrossRef CAS PubMed.
- W. Lai, Y. Qiao, Y. Wang and H. Huang, Adv. Mater., 2023, 35, 2306288 CrossRef CAS PubMed.
- X. Wang, Y. Jiang, K. Mao, W. Gong, D. Duan, J. Ma, Y. Zhong, J. Li, H. Liu and R. Long, J. Am. Chem. Soc., 2022, 144, 22759–22766 CrossRef CAS PubMed.
- D. Sun, X. Xu, Y. Qin, S. P. Jiang and Z. Shao, ChemSusChem, 2020, 13, 39–58 CrossRef CAS PubMed.
- Y. Kim, B. Kim, H. Choi, S. Kim, Y. Yun and J. Oh, Chem. Eng. J., 2023, 461, 142126 CrossRef CAS.
- Z. Ma, C. Tsounis, C. Y. Toe, P. V. Kumar, B. Subhash, S. Xi, H. Y. Yang, S. Zhou, Z. Lin and K.-H. Wu, ACS Catal., 2022, 12, 4792–4805 CrossRef CAS.
- L. Gao, S. Bai, Y. Zhang and C. Hu, ChemCatChem, 2022, 14, e202200383 CrossRef CAS.
- Y. Yao, W. Zhuang, R. Li, K. Dong, Y. Luo, X. He, S. Sun, S. Al-Faifi, X. Sun and W. Hu, Chem. Commun., 2023, 59, 9017–9028 RSC.
- M. Zhang, M. Lu, M.-Y. Yang, J.-P. Liao, Y.-F. Liu, H.-J. Yan, J.-N. Chang, T.-Y. Yu, S.-L. Li and Y.-Q. Lan, eScience, 2023, 3, 100116 CrossRef.
- A. Xu, S.-F. Hung, A. Cao, Z. Wang, N. Karmodak, J. E. Huang, Y. Yan, A. Sedighian Rasouli, A. Ozden and F.-Y. Wu, Nat. Catal., 2022, 5, 1081–1088 CrossRef CAS.
- S. Liu, H. Tao, Q. Liu, Z. Xu, Q. Liu and J.-L. Luo, ACS Catal., 2018, 8, 1469–1475 CrossRef CAS.
- R. Zhao, Y. Wang, G. Ji, J. Zhong, F. Zhang, M. Chen, S. Tong, P. Wang, Z. Wu and B. Han, Adv. Mater., 2023, 35, 2205262 CrossRef CAS.
- T. Tang, Z. Wang and J. Guan, Adv. Funct. Mater., 2022, 32, 2111504 CrossRef CAS.
- Y. N. Gong, L. Jiao, Y. Qian, C. Y. Pan, L. Zheng, X. Cai, B. Liu, S. H. Yu and H. L. Jiang, Angew. Chem., Int. Ed., 2020, 59, 2705–2709 CrossRef CAS PubMed.
- Y. Zhou, R. Abazari, J. Chen, M. Tahir, A. Kumar, R. R. Ikreedeegh, E. Rani, H. Singh and A. M. Kirillov, Coord. Chem. Rev., 2022, 451, 214264 CrossRef CAS.
- X. Liu, M. Zhuo, W. Zhang, M. Gao, X.-H. Liu, B. Sun and J. Wu, Ultrason. Sonochem., 2020, 67, 105179 CrossRef CAS PubMed.
- X.-H. Liu, W.-L. Hu, W.-J. Jiang, Y.-W. Yang, S. Niu, B. Sun, J. Wu and J.-S. Hu, ACS Appl. Mater. Interfaces, 2017, 9, 28473–28477 CAS.
- R. Díaz, M. G. Orcajo, J. A. Botas, G. Calleja and J. Palma, Mater. Lett., 2012, 68, 126–128 CrossRef.
- Y. Yang and D. S. Sholl, J. Mater. Chem. A, 2022, 10, 4242–4253 RSC.
- K. Ren, X.-F. Guo, Y.-J. Tang, B.-H. Huang and H. Wang, Analyst, 2020, 145, 7349–7356 RSC.
- B. An, Z. Li, Y. Song, J. Zhang, L. Zeng, C. Wang and W. Lin, Nat. Catal., 2019, 2, 709–717 CrossRef CAS.
- B. Li, Z. Ju, M. Zhou, K. Su and D. Yuan, Angew. Chem., Int. Ed., 2019, 58, 7687–7691 CrossRef CAS.
- R. Chen, L. Cheng, J. Liu, Y. Wang, W. Ge, C. Xiao, H. Jiang, Y. Li and C. Li, Small, 2022, 18, 2200720 CrossRef CAS.
- Y. Zhang, Q. Zhou, Z. F. Qiu, X. Y. Zhang, J. Q. Chen, Y. Zhao, F. Gong and W. Y. Sun, Adv. Funct. Mater., 2022, 32, 2203677 CrossRef CAS.
- M. Wen, N. Sun, L. Jiao, S. Q. Zang and H. L. Jiang, Angew. Chem., Int. Ed., 2024, 63, e202318338 CrossRef CAS.
- L. Jiao, J. Zhu, Y. Zhang, W. Yang, S. Zhou, A. Li, C. Xie, X. Zheng, W. Zhou and S.-H. Yu, J. Am. Chem. Soc., 2021, 143, 19417–19424 CrossRef CAS PubMed.
- Z. Jin, D. Jiao, Y. Dong, L. Liu, J. Fan, M. Gong, X. Ma, Y. Wang, W. Zhang and L. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202318246 CrossRef CAS.
- Y. Zhang, L. Jiao, W. Yang, C. Xie and H. L. Jiang, Angew. Chem., Int. Ed., 2021, 60, 7607–7611 CrossRef CAS PubMed.
- L. L. Zhuo, P. Chen, K. Zheng, X. W. Zhang, J. X. Wu, D. Y. Lin, S. Y. Liu, Z. S. Wang, J. Y. Liu and D. D. Zhou, Angew. Chem., Int. Ed., 2022, 61, e202204967 CrossRef CAS PubMed.
- R. Iqbal, M. B. Akbar, A. Ahmad, A. Hussain, N. Altaf, S. Ibraheem, G. Yasin, M. A. Khan, M. Tabish and A. Kumar, Adv. Mater. Interfaces, 2022, 9, 2101505 CrossRef CAS.
- Z. H. Zhu, Z. L. Liang, Z. H. Jiao, X. L. Jiang, Y. Xie, H. Xu and B. Zhao, Angew. Chem., Int. Ed., 2022, 61, e202214243 CrossRef CAS PubMed.
- P. Lamagni, M. Miola, J. Catalano, M. S. Hvid, M. A. H. Mamakhel, M. Christensen, M. R. Madsen, H. S. Jeppesen, X. M. Hu and K. Daasbjerg, Adv. Funct. Mater., 2020, 30, 1910408 CrossRef CAS.
- Q. Mo, S. Li, C. Chen, H. Song, Q. Gao and L. Zhang, ACS Sustainable Chem. Eng., 2024, 12, 6093–6101 CrossRef CAS.
- T. Zhang, X. Han, H. Liu, M. Biset-Peiró, J. Li, X. Zhang, P. Tang, B. Yang, L. Zheng and J. R. Morante, Adv. Funct. Mater., 2022, 32, 2111446 CrossRef CAS.
- Y. Zhang, Y.-Y. Xing, C. Wang, R. Pang, W.-W. Ren, S. Wang, Z.-M. Li, L. Yang, W.-C. Tong and Q.-Y. Wang, ACS Appl. Mater. Interfaces, 2022, 14, 23909–23915 CrossRef CAS PubMed.
- T. Zhao, H. Wu, X. Wen, J. Zhang, H. Tang, Y. Deng, S. Liao and X. Tian, Coord. Chem. Rev., 2022, 468, 214642 CrossRef CAS.
- X. Wang, Z. Chen, X. Zhao, T. Yao, W. Chen, R. You, C. Zhao, G. Wu, J. Wang and W. Huang, Angew. Chem., Int. Ed., 2018, 57, 1944–1948 CrossRef CAS.
- Y. Zhang, Q. Zhou, Z. F. Qiu, X. Y. Zhang, J. Q. Chen, Y. Zhao, F. Gong and W. Y. Sun, Adv. Funct. Mater., 2022, 32, 2203677 CrossRef CAS.
- J. Li, G. Chen, Y. Zhu, Z. Liang, A. Pei, C.-L. Wu, H. Wang, H. R. Lee, K. Liu and S. Chu, Nat. Catal., 2018, 1, 592–600 CrossRef CAS.
- C. Genovese, M. E. Schuster, E. K. Gibson, D. Gianolio, V. Posligua, R. Grau-Crespo, G. Cibin, P. P. Wells, D. Garai and V. Solokha, Nat. Commun., 2018, 9, 935 CrossRef PubMed.
- A. R. Woldu, Z. Huang, P. Zhao, L. Hu and D. Astruc, Coord. Chem. Rev., 2022, 454, 214340 CrossRef CAS.
- J. Yu, J. Wang, Y. Ma, J. Zhou, Y. Wang, P. Lu, J. Yin, R. Ye, Z. Zhu and Z. Fan, Adv. Funct. Mater., 2021, 31, 2102151 CrossRef CAS.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- R. K. Aparna, V. Surendran, D. Roy, B. Pathak, M. M. Shaijumon and S. Mandal, ACS Appl. Energy Mater., 2023, 6, 4072–4078 CrossRef CAS.
- Y. Wang, B. J. Park, V. K. Paidi, R. Huang, Y. Lee, K.-J. Noh, K.-S. Lee and J. W. Han, ACS Energy Lett., 2022, 7, 640–649 CrossRef CAS.
- X. Jiang, H. Li, J. Xiao, D. Gao, R. Si, F. Yang, Y. Li, G. Wang and X. Bao, Nano Energy, 2018, 52, 345–350 CrossRef CAS.
- Z. Meng, J. Luo, W. Li and K. A. Mirica, J. Am. Chem. Soc., 2020, 142, 21656–21669 CrossRef CAS PubMed.
- H. Zhong, M. Ghorbani-Asl, K. H. Ly, J. Zhang, J. Ge, M. Wang, Z. Liao, D. Makarov, E. Zschech and E. Brunner, Nat. Commun., 2020, 11, 1409 CrossRef CAS PubMed.
- J. H. Cho, C. Lee, S. H. Hong, H. Y. Jang, S. Back, M. g. Seo, M. Lee, H. K. Min, Y. Choi and Y. J. Jang, Adv. Mater., 2023, 35, 2208224 CrossRef CAS.
- T. Wen, M. Liu, S. Chen, Q. Li, Y. Du, T. Zhou, C. Ritchie and J. Zhang, Small, 2020, 16, 1907669 CrossRef CAS PubMed.
- S. Dou, J. Song, S. Xi, Y. Du, J. Wang, Z. F. Huang, Z. J. Xu and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 4041–4045 CrossRef CAS PubMed.
- R. Shimoni, Z. Shi, S. Binyamin, Y. Yang, I. Liberman, R. Ifraemov, S. Mukhopadhyay, L. Zhang and I. Hod, Angew. Chem., Int. Ed., 2022, 61, e202206085 CrossRef CAS PubMed.
- Z. Zhang, J. Zhu, S. Chen, W. Sun and D. Wang, Angew. Chem., Int. Ed., 2023, 62, e202215136 CrossRef CAS PubMed.
- H. Li, X. Liu, S. Chen, D. Yang, Q. Zhang, L. Song, H. Xiao, Q. Zhang, L. Gu and X. Wang, Adv. Energy Mater., 2019, 9, 1900072 CrossRef.
- F. Pan, H. Zhang, K. Liu, D. Cullen, K. More, M. Wang, Z. Feng, G. Wang, G. Wu and Y. Li, ACS Catal., 2018, 8, 3116–3122 CrossRef CAS.
- Z. Li, J. Jiang, X. Liu, Z. Zhu, J. Wang, Q. He, Q. Kong, X. Niu, J. S. Chen and J. Wang, Small, 2022, 18, 2203495 CrossRef CAS.
- D.-H. Nam, O. Shekhah, G. Lee, A. Mallick, H. Jiang, F. Li, B. Chen, J. Wicks, M. Eddaoudi and E. H. Sargent, J. Am. Chem. Soc., 2020, 142, 21513–21521 CrossRef CAS PubMed.
- T. A. Al-Attas, N. N. Marei, X. Yong, N. G. Yasri, V. Thangadurai, G. Shimizu, S. Siahrostami and M. G. Kibria, ACS Catal., 2021, 11, 7350–7357 CrossRef CAS.
- X. Wang, Z. Chen, X. Zhao, T. Yao, W. Chen, R. You, C. Zhao, G. Wu, J. Wang and W. Huang, Angew. Chem., Int. Ed., 2018, 130, 1962–1966 CrossRef.
- X. Yang, J. Cheng, H. Lv, X. Yang, L. Ding, Y. Xu, K. Zhang, W. Sun and J. Zhou, Chem. Eng. J., 2022, 450, 137950 CrossRef CAS.
- X. Zhang, Y. Zhang, Q. Li, X. Zhou, Q. Li, J. Yi, Y. Liu and J. Zhang, J. Mater. Chem. A, 2020, 8, 9776–9787 RSC.
- Y. Y. Liu, J. R. Huang, H. L. Zhu, P. Q. Liao and X. M. Chen, Angew. Chem., Int. Ed., 2023, 62, e202311265 CrossRef CAS.
- P. Song, B. Hu, D. Zhao, J. Fu, X. Su, W. Feng, K. Yu, S. Liu, J. Zhang and C. Chen, ACS Nano, 2023, 17, 4619–4628 CrossRef CAS PubMed.
- Z. Xin, Y.-R. Wang, Y. Chen, W.-L. Li, L.-Z. Dong and Y.-Q. Lan, Nano Energy, 2020, 67, 104233 CrossRef CAS.
- R. Yun, R. Xu, C. Shi, B. Zhang, T. Li, L. He, T. Sheng and Z. Chen, Nano Res., 2023, 16, 1–7 CrossRef.
- Y. Li, W. Shan, M. J. Zachman, M. Wang, S. Hwang, H. Tabassum, J. Yang, X. Yang, S. Karakalos and Z. Feng, Angew. Chem., Int. Ed., 2022, 61, e202205632 CrossRef CAS.
- R. Yun, F. Zhan, X. Wang, B. Zhang, T. Sheng, Z. Xin, J. Mao, S. Liu and B. Zheng, Small, 2021, 17, 2006951 CrossRef CAS.
- Y. Yang, L. Chen, Z. Guo, S. Liu, P.-d. Wu, Z. Fang, K. Zhang and H. Li, J. Mater. Chem. A, 2024, 12, 8991–9001 RSC.
- J. W. Lim, D. H. Choo, J. H. Cho, J. Kim, W. S. Cho, O. F. N. Okello, K. Kim, S. Lee, J. Son and S.-Y. Choi, J. Mater. Chem. A, 2024, 12, 11090–11100 RSC.
- J. D. Yi, D. H. Si, R. Xie, Q. Yin, M. D. Zhang, Q. Wu, G. L. Chai, Y. B. Huang and R. Cao, Angew. Chem., Int. Ed., 2021, 133, 17108–17114 CrossRef.
- L. L. Ling, L. Jiao, X. Liu, Y. Dong, W. Yang, H. Zhang, B. Ye, J. Chen and H. L. Jiang, Adv. Mater., 2022, 34, 2205933 CrossRef CAS.
- M. Feng, X. Wu, H. Cheng, Z. Fan, X. Li, F. Cui, S. Fan, Y. Dai, G. Lei and G. He, J. Mater. Chem. A, 2021, 9, 23817–23827 RSC.
- H. Cheng, X. Wu, M. Feng, X. Li, G. Lei, Z. Fan, D. Pan, F. Cui and G. He, ACS Catal., 2021, 11, 12673–12681 CrossRef CAS.
- F. Mao, Y.-H. Jin, P. F. Liu, P. Yang, L. Zhang, L. Chen, X.-M. Cao, J. Gu and H. G. Yang, J. Mater. Chem. A, 2019, 7, 23055–23063 RSC.
- C. Hu, W. Yao, X. Yang, K. Shen, L. Chen and Y. Li, Adv. Sci., 2024, 11, 2306095 CrossRef CAS.
- C. Hu, Y. Zhang, A. Hu, Y. Wang, X. Wei, K. Shen, L. Chen and Y. Li, Adv. Mater., 2023, 35, 2209298 CrossRef CAS.
- Y. Takaoka, J. T. Song, A. Takagaki, M. Watanabe and T. Ishihara, Appl. Catal., B, 2023, 326, 122400 CrossRef CAS.
- S.-Z. Hou, X.-D. Zhang, W.-W. Yuan, Y.-X. Li and Z.-Y. Gu, Inorg. Chem., 2020, 59, 11298–11304 CrossRef CAS.
- Y. Zhou, S. Liu, Y. Gu, G.-H. Wen, J. Ma, J.-L. Zuo and M. Ding, J. Am. Chem. Soc., 2021, 143, 14071–14076 CrossRef CAS.
- Z.-H. Zhao, J.-R. Huang, D.-S. Huang, H.-L. Zhu, P.-Q. Liao and X.-M. Chen, J. Am. Chem. Soc., 2024, 146, 14349–14356 CrossRef CAS PubMed.
- Y. Yang, J. Huang, Y. Zou, Y. Li, T. Zhan, L. Huang, X. Ma, Z. Zhang and S. Xiang, Appl. Surf. Sci., 2023, 618, 156664 CrossRef CAS.
- Z. Jiang, M. Zhang, X. Chen, B. Wang, W. Fan, C. Yang, X. Yang, Z. Zhang, X. Yang and C. Li, Angew. Chem., Int. Ed., 2023, 62, e202311223 CrossRef CAS PubMed.
- R. Yang, Q. Huang, X. Sha, B. Gao and J. Peng, Int. J. Mol. Sci., 2023, 24, 13838 CrossRef CAS.
- D. Yao, C. Tang, A. Vasileff, X. Zhi, Y. Jiao and S. Z. Qiao, Angew. Chem., Int. Ed., 2021, 60, 18178–18184 CrossRef CAS.
- S. Ma, K. Wu, S. Fan, Y. Li, Q. Xie, J. Ma and L. Yang, Sep. Purif. Technol., 2024, 339, 126520 CrossRef CAS.
- Z. Gao, M. Hou, Y. Shi, L. Li, Q. Sun, S. Yang, Z. Jiang, W. Yang, Z. Zhang and W. Hu, Chem. Sci., 2023, 14, 6860–6866 RSC.
- L. Mi, B. Chen, X. Xu, S. Cai, Y. He, Y. Wei, Y. Jiang, C. Zheng, S. Zhong and W. Hu, J. Alloys Compd., 2024, 978, 173516 CrossRef CAS.
- Q. Huang, X. Sha, R. Yang, H. Li and J. Peng, ACS Appl. Mater. Interfaces, 2024, 16, 13882–13892 CrossRef CAS.
- Y. Zhang, S. Liu, N. Ji, L. Wei, Q. Liang, J. Li, Z. Tian, J. Su and Q. Chen, J. Mater. Chem. A, 2024, 12, 7528–7535 RSC.
- C. Qiu, K. Qian, J. Yu, M. Sun, S. Cao, J. Gao, R. Yu, L. Fang, Y. Yao and X. Lu, Nano-Micro Lett., 2022, 14, 167 CrossRef CAS.
- C. Cao, D. D. Ma, J. F. Gu, X. Xie, G. Zeng, X. Li, S. G. Han, Q. L. Zhu, X. T. Wu and Q. Xu, Angew. Chem., Int. Ed., 2020, 59, 15014–15020 CrossRef CAS PubMed.
- Y. Zhang, Q. Zhou, P. Wang, Y. Zhao, F. Gong and W. Y. Sun, ChemSusChem, 2022, 15, e202102528 CrossRef CAS.
- G. Liu, Q. T. Trinh, H. Wang, S. Wu, J. M. Arce-Ramos, M. B. Sullivan, M. Kraft, J. W. Ager, J. Zhang and R. Xu, Small, 2023, 2301379 CrossRef CAS PubMed.
- Y.-Y. Liu, H.-L. Zhu, Z.-H. Zhao, N.-Y. Huang, P.-Q. Liao and X.-M. Chen, ACS Catal., 2022, 12, 2749–2755 CrossRef CAS.
- X. Tan, C. Yu, C. Zhao, H. Huang, X. Yao, X. Han, W. Guo, S. Cui, H. Huang and J. Qiu, ACS Appl. Mater. Interfaces, 2019, 11, 9904–9910 CrossRef CAS PubMed.
- J. Lv, W. Li, J. Li, Z. Zhu, A. Dong, H. Lv, P. Li and B. Wang, Angew. Chem., Int. Ed., 2023, 62, e202217958 CrossRef CAS PubMed.
- Y. L. Yang, Y. R. Wang, L. Z. Dong, Q. Li, L. Zhang, J. Zhou, S. N. Sun, H. M. Ding, Y. Chen and S. L. Li, Adv. Mater., 2022, 34, 2206706 CrossRef CAS PubMed.
- Y. Liu, S. Li, L. Dai, J. Li, J. Lv, Z. Zhu, A. Yin, P. Li and B. Wang, Angew. Chem., Int. Ed., 2021, 60, 16409–16415 CrossRef CAS.
- Y. Zhang, X.-Y. Zhang and W.-Y. Sun, ACS Catal., 2023, 13, 1545–1553 CrossRef CAS.
- S. Chen, W. H. Li, W. Jiang, J. Yang, J. Zhu, L. Wang, H. Ou, Z. Zhuang, M. Chen and X. Sun, Angew. Chem., Int. Ed., 2022, 61, e202114450 CrossRef CAS.
- H.-L. Zhu, J.-R. Huang, X.-W. Zhang, C. Wang, N.-Y. Huang, P.-Q. Liao and X.-M. Chen, ACS Catal., 2021, 11, 11786–11792 CrossRef CAS.
- P. Shao, W. Zhou, Q. L. Hong, L. Yi, L. Zheng, W. Wang, H. X. Zhang, H. Zhang and J. Zhang, Angew. Chem., Int. Ed., 2021, 60, 16687–16692 CrossRef CAS.
- T. Yan, J.-H. Guo, Z.-Q. Liu and W.-Y. Sun, ACS Appl. Mater. Interfaces, 2021, 13, 25937–25945 CrossRef CAS.
- X.-D. Zhang, J.-M. Huang, X. Zhu, C. Liu, Y. Yin, J.-Y. Huang, Y. Li and Z.-Y. Gu, Chin. Chem. Lett., 2024, 109937 CrossRef.
- T. Yan, P. Wang and W. Y. Sun, Small, 2023, 19, 2206070 CrossRef CAS PubMed.
- D. H. Nam, O. Shekhah, A. Ozden, C. McCallum, F. Li, X. Wang, Y. Lum, T. Lee, J. Li and J. Wicks, Adv. Mater., 2022, 34, 2207088 CrossRef CAS.
- X.-F. Qiu, H.-L. Zhu, J.-R. Huang, P.-Q. Liao and X.-M. Chen, J. Am. Chem. Soc., 2021, 143, 7242–7246 CrossRef CAS.
- D.-S. Huang, H.-L. Zhu, Z.-H. Zhao, J.-R. Huang, P.-Q. Liao and X.-M. Chen, ACS Catal., 2022, 12, 8444–8450 CrossRef CAS.
- Z.-H. Zhao, H.-L. Zhu, J.-R. Huang, P.-Q. Liao and X.-M. Chen, ACS Catal., 2022, 12, 7986–7993 CrossRef CAS.
- J. Wang, J. Liu, Y. Song, S. Geng, Z. Peng, J. Yu, F. Liu, Y. Wang, S. Xi and Z. Zhang, ACS Mater. Lett., 2023, 5, 2121–2130 CrossRef CAS.
- J. Feng, W. Zhang, D. Shi, Y. Jia, Y. Tang, Y. Meng and Q. Gao, Chem. Sci., 2024, 15, 9173–9182 RSC.
- C. F. Wen, M. Zhou, P. F. Liu, Y. Liu, X. Wu, F. Mao, S. Dai, B. Xu, X. L. Wang and Z. Jiang, Angew. Chem., Int. Ed., 2022, 61, e202111700 CrossRef CAS.
- H. Sun, L. Chen, L. Xiong, K. Feng, Y. Chen, X. Zhang, X. Yuan, B. Yang, Z. Deng and Y. Liu, Nat. Commun., 2021, 12, 6823 CrossRef CAS.
- C. Liu, X.-D. Zhang, J.-M. Huang, M.-X. Guan, M. Xu and Z.-Y. Gu, ACS Catal., 2022, 12, 15230–15240 CrossRef CAS.
- M. Kempasiddaiah, R. Samanta, S. Panigrahi, R. K. Trivedi, B. Chakraborty and S. Barman, Nanoscale, 2024, 16, 10458–10473 RSC.
- K. Zhao, Y. Liu, X. Quan, S. Chen and H. Yu, ACS Appl. Mater. Interfaces, 2017, 9, 5302–5311 CrossRef CAS PubMed.
- X. Yang, J. Cheng, X. Yang, Y. Xu, W. Sun and J. Zhou, Chem. Eng. J., 2022, 431, 134171 CrossRef CAS.
- J. Liu, D. Yang, Y. Zhou, G. Zhang, G. Xing, Y. Liu, Y. Ma, O. Terasaki, S. Yang and L. Chen, Angew. Chem., Int. Ed., 2021, 60, 14473–14479 CrossRef CAS PubMed.
- C. Hu, Z. Jiang, Q. Wu, S. Cao, Q. Li, C. Chen, L. Yuan, Y. Wang, W. Yang and J. Yang, Nat. Commun., 2023, 14, 4767 CrossRef CAS.
- P. Lu, J. Lv, Y. Chen, Y. Ma, Y. Wang, W. Lyu, J. Yu, J. Zhou, J. Yin and Y. Xiong, Nano Lett., 2024, 24, 1553–1562 CrossRef CAS PubMed.
- S. Li, J. Yu, S. Zhang, W. Qiu, X. Tang, Z. Lin, R. Cai, Y. Fang, S. Yang and X. Cai, Adv. Funct. Mater., 2024, 34, 2311989 CrossRef CAS.
- W. Su, W. Guo and Y. Fan, Chem. Eng. J., 2023, 477, 147204 CrossRef CAS.
- Z.-H. Zhao, J.-R. Huang, P.-Q. Liao and X.-M. Chen, J. Am. Chem. Soc., 2023, 149, 26783–26790 CrossRef.
- J. Liu, Y. Wang, P. Mo, F. Yang, K. Jiang, Z. Cheng, Y. Liu, Z. Sun, Z. Liu and Y. Zhang, Nano Res., 2024, 17, 3888–3894 CrossRef CAS.
- Y. Zhang, K. Li, M. Chen, J. Wang, J. Liu and Y. Zhang, ACS Appl. Nano Mater., 2019, 3, 257–263 CrossRef.
- K. Zhao, X. Nie, H. Wang, S. Chen, X. Quan, H. Yu, W. Choi, G. Zhang, B. Kim and J. G. Chen, Nat. Commun., 2020, 11, 2455 CrossRef CAS.
- S. Payra, S. Kanungo and S. Roy, Nanoscale, 2022, 14, 13352–13361 RSC.
- Z. Tang, W. He, Y. Wang, Y. Wei, X. Yu, J. Xiong, X. Wang, X. Zhang, Z. Zhao and J. Liu, Appl. Catal., B, 2022, 311, 121371 CrossRef CAS.
- W. Su, W. Guo and Y. Fan, Chem. Eng. J., 2023, 147, 147204 CrossRef.
- Z.-H. Gao, K. Wei, T. Wu, J. Dong, D.-e. Jiang, S. Sun and L.-S. Wang, J. Am. Chem. Soc., 2022, 144, 5258–5262 CrossRef CAS PubMed.
- Y. Pan, R. Lin, Y. Chen, S. Liu, W. Zhu, X. Cao, W. Chen, K. Wu, W.-C. Cheong and Y. Wang, J. Am. Chem. Soc., 2018, 140, 4218–4221 CrossRef CAS.
- W. Xie, T. Yang, N. Tian, X.-H. Liu and J. Chen, Energy Fuels, 2022, 36, 958–964 CrossRef CAS.
- H. Kim, D. Shin, W. Yang, D. H. Won, H.-S. Oh, M. W. Chung, D. Jeong, S. H. Kim, K. H. Chae and J. Y. Ryu, J. Am. Chem. Soc., 2021, 143, 925–933 CrossRef CAS PubMed.
- P. Wang, S. Meng, B. Zhang, M. He, P. Li, C. Yang, G. Li and Z. Li, J. Am. Chem. Soc., 2023, 145, 26133–26143 CrossRef CAS PubMed.
- R. Wang, J. Liu, L.-Z. Dong, J. Zhou, Q. Huang, Y.-R. Wang, J.-W. Shi and Y.-Q. Lan, CCS Chem., 2023, 5, 2237–2250 CrossRef CAS.
- Q. Lei, L. Huang, J. Yin, B. Davaasuren, Y. Yuan, X. Dong, Z.-P. Wu, X. Wang, K. X. Yao and X. Lu, Nat. Commun., 2022, 13, 4857 CrossRef CAS PubMed.
- Z. Jiang, T. Wang, J. Pei, H. Shang, D. Zhou, H. Li, J. Dong, Y. Wang, R. Cao and Z. Zhuang, Energy Environ. Sci., 2020, 13, 2856–2863 RSC.
- Y. Zhao, L. Zheng, D. Jiang, W. Xia, X. Xu, Y. Yamauchi, J. Ge and J. Tang, Small, 2021, 17, 2006590 CrossRef CAS.
- C. Cui, Q. Cao, H. Jing, Z. Zhao, D. Wang and Y. J. S. Li, Surf. Interfaces, 2024, 48, 104353 CrossRef CAS.
- X. Peng, L. Zeng, D. Wang, Z. Liu, Y. Li, Z. Li, B. Yang, L. Lei, L. Dai and Y. Hou, Chem. Soc. Rev., 2023, 52, 2193–2237 RSC.
- B. Zhang, Y. Jiang, M. Gao, T. Ma, W. Sun and H. Pan, Nano Energy, 2021, 80, 105504 CrossRef CAS.
- K. A. Adegoke and N. W. Maxakato, Coord. Chem. Rev., 2022, 457, 214389 CrossRef CAS.
- H. Meng, Y. Han, C. Zhou, Q. Jiang, X. Shi, C. Zhan and R. Zhang, Small Methods, 2020, 4, 2000396 CrossRef CAS.
- C. Li, L. Zhang, J. Chen, X. Li, J. Sun, J. Zhu, X. Wang and Y. Fu, Nanoscale, 2021, 13, 485–509 RSC.
- S. Mandal, S. Natarajan, P. Mani and A. Pankajakshan, Adv. Funct. Mater., 2021, 31, 2006291 CrossRef CAS.
- D.-W. Lim and H. Kitagawa, Chem. Soc. Rev., 2021, 50, 6349–6368 RSC.
- J. Ye, J. Yan, Y. Peng, F. Li and J. Sun, Catal. Today, 2023, 410, 68–84 CrossRef CAS.
- M. Tang, H. Shen and Q. Sun, J. Phys. Chem. C, 2019, 123, 26460–26466 CrossRef CAS.
- Q. Cui, G. Qin, W. Wang, K. Geethalakshmi, A. Du and Q. Sun, Appl. Surf. Sci., 2020, 500, 143993 CrossRef CAS.
- Y. Tian, Y. Wang, L. Yan, J. Zhao and Z. Su, Appl. Surf. Sci., 2019, 467, 98–103 CrossRef.
- G. Xing, L. Cheng, K. Li, Y. Gao, H. Tang, Y. Wang and Z. Wu, New J. Chem., 2020, 44, 12299–12306 RSC.
- Y. Tian, C. Zhu, L. Yan, J. Zhao and Z. Su, J. Mater. Chem. A, 2019, 7, 15341–15346 RSC.
- H. Zhong, M. Wang, G. Chen, R. Dong and X. Feng, ACS Nano, 2022, 16, 1759–1780 CrossRef CAS PubMed.
- L. Majidi, A. Ahmadiparidari, N. Shan, S. N. Misal, K. Kumar, Z. Huang, S. Rastegar, Z. Hemmat, X. Zou and P. Zapol, Adv. Mater., 2021, 33, 2004393 CrossRef CAS.
- J. Liu, D. Yang, Y. Zhou, G. Zhang, G. Xing, Y. Liu, Y. Ma, O. Terasaki, S. Yang and L. Chen, Angew. Chem., Int. Ed., 2021, 60, 14473–14479 CrossRef CAS PubMed.
- L. Huang, Z. Liu, G. Gao, C. Chen, Y. Xue, J. Zhao, Q. Lei, M. Jin, C. Zhu and Y. Han, J. Am. Chem. Soc., 2023, 145, 26444–26451 CrossRef CAS PubMed.
- M. G. Lee, S. Kandambeth, X.-Y. Li, O. Shekhah, A. Ozden, J. Wicks, P. Ou, S. Wang, R. Dorakhan and S. Park, J. Am. Chem. Soc., 2024, 146, 14267–14277 CrossRef CAS.
- Z.-Z. Wu, F.-Y. Gao and M.-R. Gao, Energy Environ. Sci., 2021, 14, 1121–1139 RSC.
- Q. Fan, X. Zhang, X. Ge, L. Bai, D. He, Y. Qu, C. Kong, J. Bi, D. Ding and Y. Cao, Adv. Energy Mater., 2021, 11, 2101424 CrossRef CAS.
- S. Li, A. V. Nagarajan, Y. Li, D. R. Kauffman, G. Mpourmpakis and R. Jin, Nanoscale, 2021, 13, 2333–2337 RSC.
- X.-L. Lv, L. Feng, L.-H. Xie, T. He, W. Wu, K.-Y. Wang, G. Si, B. Wang, J.-R. Li and H.-C. Zhou, J. Am. Chem. Soc., 2021, 143, 2784–2791 CrossRef CAS PubMed.
- X. Cheng, J. Zhang, X. Tan, L. Zheng, D. Tan, L. Liu, G. Chen, Q. Wan, B. Zhang and F. Zhang, Chem. Commun., 2020, 56, 7637–7640 RSC.
- X. Mao, W. Gong, Y. Fu, J. Li, X. Wang, A. P. O'Mullane, Y. Xiong and A. Du, J. Am. Chem. Soc., 2023, 145, 21442–21453 CrossRef CAS.
- A. K. Singh, L. Gu, A. Dutta Chowdhury and A. Indra, Inorg. Chem., 2023, 62, 8803–8811 CrossRef CAS.
- T. Al-Attas, S. K. Nabil, A. S. Zeraati, H. S. Shiran, T. Alkayyali, M. Zargartalebi, T. Tran, N. N. Marei, M. A. Al Bari and H. Lin, ACS Energy Lett., 2022, 8, 107–115 CrossRef.
- A. Bencini and V. Lippolis, Coord. Chem. Rev., 2010, 254, 2096–2180 CrossRef CAS.
- J. Wang, X. Huang, S. Xi, H. Xu and X. Wang, Angew. Chem., Int. Ed., 2020, 59, 19162–19167 CrossRef CAS.
- X. Xie, X. Zhang, M. Xie, L. Xiong, H. Sun, Y. Lu, Q. Mu, M. H. Rummeli, J. Xu, S. Li, J. Zhong, Z. Deng, B. Ma, T. Cheng, W. A. Goddard and Y. Peng, Nat. Commun., 2022, 13, 63 CrossRef CAS.
- Z. Xin, J. Liu, X. Wang, K. Shen, Z. Yuan, Y. Chen and Y.-Q. Lan, ACS Appl. Mater. Interfaces, 2021, 13, 54959–54966 CrossRef CAS PubMed.
- J. Deng, L. Qiu, M. Xin, W. He, W. Zhao, J. Dong and G. Xu, Small, 2024, 2311060 CrossRef CAS.
- C.-Y. Yuan, L. Feng, X.-T. Qin, J.-X. Liu, X. Li, X.-C. Sun, X.-X. Chang, B.-J. Xu, W.-X. Li and D. Ma, Angew. Chem., Int. Ed., 2024, e202405255 CAS.
- F. Chang, K. Zhu, C. Liu, J. Wei, S. Yang, Q. Zhang, L. Yang, X. Wang and Z. Bai, Adv. Funct. Mater., 2024, 2400893 CrossRef.
- C. Jia, Y. Zhao, S. Song, Q. Sun, Q. Meyer, S. Liu, Y. Shen and C. Zhao, Adv. Energy Mater., 2023, 13, 2302007 CrossRef CAS.
- P. Su, K. Iwase, S. Nakanishi, K. Hashimoto and K. Kamiya, Small, 2016, 12, 6083–6089 CrossRef CAS PubMed.
- Z. Qi, Y. Zhou, R. Guan, Y. Fu and J. B. Baek, Adv. Mater., 2023, 35, 2210575 CrossRef CAS.
- B. Pan, X. Zhu, Y. Wu, T. Liu, X. Bi, K. Feng, N. Han, J. Zhong, J. Lu and Y. Li, Adv. Sci., 2020, 7, 2001002 CrossRef CAS PubMed.
- M. Qu, Z. Chen, Z. Sun, D. Zhou, W. Xu, H. Tang, H. Gu, T. Liang, P. Hu and G. J. N. R. Li, Nano Res., 2023, 16, 2170–2176 CrossRef CAS.
- S. Liu, L. Wang, H. Yang, S. Gao, Y. Liu, S. Zhang, Y. Chen, X. Liu and J. Luo, Small, 2022, 18, 2104965 CrossRef CAS.
- X. Zhang, X. Sun, S.-X. Guo, A. M. Bond and J. Zhang, Energy Environ. Sci., 2019, 12, 1334–1340 RSC.
- N. Han, Y. Wang, H. Yang, J. Deng, J. Wu, Y. Li and Y. Li, Nat. Commun., 2018, 9, 1320 CrossRef PubMed.
- J. P. Jones, G. S. Prakash and G. A. Olah, Isr. J. Chem., 2014, 54, 1451–1466 CrossRef CAS.
- Q. Huang, X. Sha, R. Yang, H. Li and J. Peng, ACS Appl. Mater. Interfaces, 2024, 16, 13882–13892 CrossRef CAS PubMed.
- W. He, I. Liberman, I. Rozenberg, R. Ifraemov and I. Hod, Angew. Chem., Int. Ed., 2020, 59, 8262–8269 CrossRef CAS PubMed.
- W. Zhang, C. Huang, J. Zhu, Q. Zhou, R. Yu, Y. Wang, P. An, J. Zhang, M. Qiu and L. Zhou, Angew. Chem., Int. Ed., 2022, 61, e202112116 CrossRef CAS PubMed.
- W. Zhang, Y. Yang, Y. Tang and Q. Gao, J. Energy Chem., 2022, 70, 414–436 CrossRef CAS.
- P. Liu, P. Jing, X. Xu, B. Liu and J. Zhang, ACS Appl. Energy Mater., 2021, 4, 12128–12136 CrossRef CAS.
- C. Bressler and M. Chergui, Chem. Rev., 2004, 104, 1781–1812 CrossRef CAS PubMed.
- K. Adarsh, N. Chandrasekaran and V. Chakrapani, Front. Chem., 2020, 8, 137 CrossRef CAS PubMed.
- S. Liu, L. Song, R. Liu, L. Li, D. Yang, S. Yuan and X. Dai, Small, 2023, 19, 2304808 CrossRef CAS.
- C. Farber, J. Li, E. Hager, R. Chemelewski, J. Mullet, A. Y. Rogachev and D. Kurouski, ACS Omega, 2019, 4, 3700–3707 CrossRef CAS.
- M. R. Smith, C. B. Martin, S. Arumuganainar, A. Gilman, B. E. Koel and M. L. Sarazen, Angew. Chem., Int. Ed., 2023, 62, e202218208 CrossRef CAS PubMed.
- G. Hai, X. Xue, S. Feng, Y. Ma and X. Huang, ACS Catal., 2022, 12, 15271–15281 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |