
Nanoscale halide perovskites for photocatalytic CO2 reduction: product selectivity, strategies implemented, and charge-carrier separation
Zhe
Wang
ab,
Chun Hong
Mak
ab,
Jianpei
Feng
ab,
Hsin-Hui
Shen
c,
Bin
Han
d,
Shella Permatasari
Santoso
 j,
Mingjian
Yuan
h,
Fang-Fang
Li
g,
Haisheng
Song
e,
Duu-Jong
Lee
*i,
Juan Carlos
Colmenares
fk and
Hsien-Yi
Hsu
*ab
j,
Mingjian
Yuan
h,
Fang-Fang
Li
g,
Haisheng
Song
e,
Duu-Jong
Lee
*i,
Juan Carlos
Colmenares
fk and
Hsien-Yi
Hsu
*ab
aSchool of Energy and Environment & Department of Materials Science and Engineering & Centre for Functional Photonics (CFP), City University of Hong Kong, Kowloon Tong, Hong Kong, China. E-mail: sam.hyhsu@cityu.edu.hk
bShenzhen Research Institute of City University of Hong Kong, Shenzhen 518057, P. R. China
cDepartment of Materials Science and Engineering, Faculty of Engineering, Monash University, Clayton, Victoria 3800, Australia
dMaterials Institute of Atomic and Molecular Science, Shaanxi University of Science and Technology, Xi'an 710021, China
eWuhan National Laboratory for Optoelectronics (WNLO) and School of Optical and Electronic Information, Huazhong University of Science and Technology, 1037 Luoyu Road, 430074, Wuhan, Hubei, P. R. China
fInstitute of Physical Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, Warsaw 01-224, Poland
gSchool of Materials Science and Engineering, Huazhong University of Science and Technology, Wuhan, Hubei 430074, P. R. China
hKey Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), Renewable Energy Conversion and Storage Center (RECAST), College of Chemistry, Nankai University, Tianjin, 300071, P. R. China
iDepartment of Mechanical Engineering, City University of Hong Kong, Kowloon Tong, Hong Kong, China. E-mail: tuclee@cityu.edu.hk
jChemical Engineering Department, Faculty of Engineering, Widya Mandala Surabaya Catholic University, Surabaya, East Java 60114, Indonesia
kEngineering Research Institute (In3), Universidad Cooperativa de Colombia, Medellín 50031, Colombia
First published on 28th June 2024
Abstract
The over-use of fossil fuels leads to a sharp increase in atmospheric concentrations of carbon dioxide (CO2), which seriously contributes to the energy crisis and climate problems. The direct transformation of CO2 into high-value chemicals through photocatalysis offers an effective way to mitigate these problems. The key to achieving this goal is to discover a cost-effective, highly efficient, and durable photocatalyst. Due to their straightforward synthesis, high light absorption capacity, rapid exciton production efficiency, and long carrier diffusion length, nanoscale halide perovskites (NHPs) have great potential for solar photocatalysis. However, several crucial problems, like poor long-term stability, low product selectivity, and severe charge recombination, have become bottlenecks in the development of NHP photocatalysts. Therefore, this review aims to summarize the principles of the CO2 reduction reaction (CO2RR), the structural features of halide perovskites nanocrystals (NCs) in the system, and the principal approaches to enhancing their photocatalytic activity. Factors that influence the selectivity of CO2RR final products are also discussed. Moreover, this review pays special attention to the techniques for studying photogenerated carrier transport processes and photocatalytic intermediates, which make a significant contribution to the insight into the reaction mechanism of photocatalytic CO2 reduction. Finally, the main challenges and prospects for NHP's further development are also presented. This review will offer instructions for the design of NHP photocatalysts to further enhance the photocatalytic performance and product selectivity for CO2 reduction. It will also offer insights into studying the charge transport process and mechanism for the CO2 photocatalytic reduction reaction.
1. Introduction
The consumption of fossil fuels has drastically increased during the past few decades, resulting in energy shortages as the global population and industry have grown exponentially.1–3 The atmospheric CO2 concentration was 280 parts per million volume (ppmv) before the first industrial revolution, and it has reached 410 ppmv by 2022. Notably, 600 ppmv is expected to be reached by 2100, implying a global temperature increase of 1.9 °C by 2100, which will lead to global warming and other serious climate problems.4 However, fossil fuels will remain the main source of energy for the energy infrastructure for a long time to come. As a result, “carbon neutrality” has gained popularity in recent years. Many countries and regions have committed to reaching this goal by mid-century, such as the European Union, China, and the United Kingdom. The conversion of CO2 and water into carbohydrates at ambient temperature while exposed to light and oxygen is known as photosynthesis, and it is widely regarded as nature's most significant mechanism for preserving the Earth's relative CO2 and O2 balance. This has led to the development of solar-driven photocatalytic conversion of CO2 to useful chemicals as an appealing, practicable, and potentially long-term solution to the problems of energy and the environment.5With a bond energy of up to 750 kJ mol−1, CO2 is a linear, thermodynamically stable molecule.6 Due to the high activation potential, conventional thermocatalytic CO2 conversion usually requires high temperature and high-pressure conditions.7–9 Furthermore, unlike electrocatalysis, which requires additional electrical energy, photocatalytic CO2 conversion can be a green process using solar energy under ambient temperature and pressure conditions. Therefore, it is believed to be a prospective approach in terms of energy efficiency, sustainability, economy and eco-friendliness.10 Photogenerated electrons are stimulated to the conduction band (CB) when a photocatalyst absorbs light with greater or equivalent energy compared to its band gap, leaving holes at the valence band (VB). Subsequently, the electrons and holes are engaged in the reduction reaction of CO2 and the oxidation reaction of sacrificial agents, respectively. Depending on the CB and VB positions of the photocatalyst, CO2 molecules can be reduced to various high value-added chemicals such as CO, CH4,11 CH3OH,12 HCHO,13 HCOOH,14 C2H4,15etc. Therefore, it is crucial to explore photocatalysts with excellent performance and cost effectiveness. Ideal photocatalytic materials should have a wide light absorption range, long carrier lifetime, effective charge separation, excellent stability, abundant active sites, and superb CO2 adsorption.16 The earliest research on CO2 photocatalysis dates back to 1978, when Halmann et al. achieved the photochemical reduction of CO2 by using p-GaAs at a high cathode applied bias.17 Inoue et al. then used a variety of semiconductor particles suspended in aqueous solution for selective photoreduction of CO2.18 Several types of catalyst materials have been used for CO2 photocatalytic reduction, including nitrides (CoN, C3N4, etc.), sulfides (e.g., CdS, CdSe, etc.), metal oxides (TiO2, ZnO, NaNbO3, etc.), MXenes, metal organic frameworks (MOFs), and perovskites (NaNbO3, MAPbI3, CsPbBr3, etc.).19,20 However, most semiconductors are far from satisfactory as photocatalysts due to several constraints such as rapid compounding of photogenerated charge carriers, insufficient charge potential for oxidation and reduction reactions, or activity only under UV irradiation.21 Therefore, the exploration of affordable, reliable, and efficient photocatalysts for CO2 fixation remains a great need.
Nanoscale halide perovskites (NHPs) have recently gained popularity among scientists because of their unique characteristics, including their low cost, simple synthesis, tunable band gap, high defect tolerance, long charge diffusion length, etc.22 Thus, NHPs have been widely used in photovoltaic conversion, H2/O2 evolution, organic degradation, etc.23–28 They have also recently become among the most exciting choices for the photocatalytic reduction of CO2.29 Compared with bulk perovskites, NHPs exhibit fascinating quantum confinement effects and superior microstructural features due to their size being comparable to the exciton Bohr radius, endowing them with a tunable band gap, enhanced optical properties and exciton characteristics, and a larger exposed active surface.30 Besides, uniform size, shape, and surface properties of NHPs hold promise for achieving surface-facet-controlled catalysis for specific reaction substrates. These unique size and quantum effects endow NHPs over bulk perovskites with great application potential in the field of photocatalytic CO2 reduction.31 The practical implementation of NHP in CO2 photoreduction becomes possible under the continuous improvement of synthesis techniques, including ball milling, thermal injection, solvothermal, ultrasonication, ligand-assisted reprecipitation, and microwave assistance.32–36 Currently, extensive research on photocatalytic CO2 reduction for NHPs is focused on solving key problems, like low photocatalytic conversion efficiency, poor product selectivity, and weak photocatalyst stability.37–40 However, review papers related to NHPs for CO2 photoreduction mainly introduced the photocatalysis principle, structure and optical properties of NHPs, synthesis methods, and modification approaches. Noticeably, there is a great demand for researchers to pay attention to the product selectivity and charge transport process of CO2 reduction photocatalysis, which will deepen our understanding of the CO2RR mechanism and guide us to design more efficient and practical photocatalysts. Therefore, this review not only introduces the basic knowledge and modification strategies of NHPs for the CO2RR, but also summarizes the factors affecting the product selectivity and the characterization techniques to identify the photogenerated charge separation and transfer mechanisms. Among the modification strategies, surface engineering, component engineering, heterojunction engineering, and encapsulation engineering are systematically introduced. As for the reduction products of CO2, their selectivity relies on four main factors: photoexcitation properties, band structure, charge carrier separation and catalytically active sites. In general, the production of CO is usually due to rapid charge separation and transfer, while CH4 is mainly produced by changing the B-site perovskites or elements of co-catalysts to provide catalytically active sites. In addition, the characterization techniques for identifying the photogenerated charge transport process and mechanism are presented in four categories according to their functions: charge separation efficiency, charge transfer direction, charge carrier lifetime, and surface reaction intermediate identification. Finally, this review concludes several challenges and prospects on the further study of NHPs for CO2 photocatalytic reduction in terms of environmental impact, stability, photocatalytic efficiency and selectivity, and the catalytic mechanism. This review will offer an overview for the development of efficient NHP NCs in CO2 photocatalytic reduction and contribute to an in-depth understanding of the charge transport processes and reaction mechanism of photocatalytic reactions, which will help to address key questions encountered by the scientific community in the energy and environmental fields including but not restricted to CO2 photoreduction, H2O splitting, and photoelectrochemical reactions.
2. Photocatalytic CO2 reduction
2.1 CO2 photoreduction principle
Understanding the principles of the CO2RR is essential for the development of suitable photocatalysts. Typically, the whole process of the CO2RR mainly involves three key steps: (1) absorption of light at suitable wavelengths by the photocatalyst followed by formation of electron–hole pairs; (2) charge carrier separation and migration from the interior to the surface of the catalyst; and (3) oxidation and reduction reactions between the charge carrier and the reactants on the catalyst surface. When a photocatalyst is irradiated by light with energy equivalent to or higher than its band gap energy (Eg), some of the electrons will be excited to leap to the CB, leaving the holes in the VB, and thus electron–hole pairs are formed. Afterwards, the generated electron–hole pairs are separated and transferred to the surface of the perovskites for reduction and oxidation reactions. Simultaneously, some electrons and holes will inevitably recombine through radiation and non-radiation modes that produce light or heat, respectively. In addition, some photogenerated electrons may also be captured in traps. Fig. 1 shows that the recombination of surface electrons and holes would produce energy through light or heat.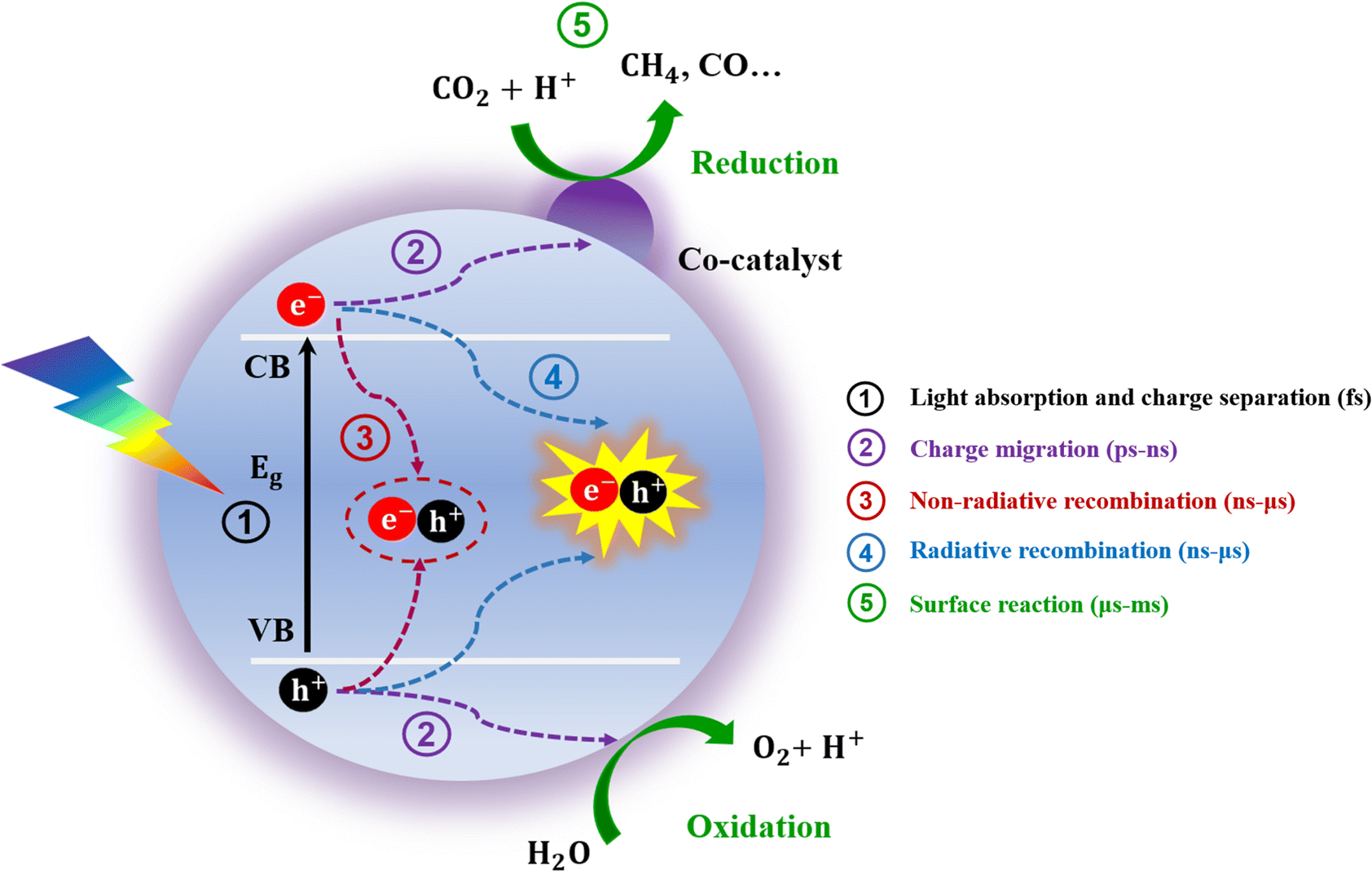 | ||
| Fig. 1 Schematic diagram of the mechanism of the photocatalytic CO2 reduction process and the corresponding timescales.41,42 | ||
Carbon dioxide is a thermodynamically stable molecule composed of two linearly arranged C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bonds, which have a greater bond strength than a variety of chemical bonds including C–H and C–C.6 During the catalytic process, inert CO2 molecules are chemisorbed on the surface of the photocatalyst in a curved structure, through which the carbon dioxide molecules exhibit a lower unoccupied molecular orbital (LUMO) energy level, which makes it easier to receive electrons from the photocatalyst. After accepting the electrons, carbon dioxide will transform into carbonate anion radicals (CO2−) which are more reactive, contributing to further reactions to generate other products.43 As shown in Table 1, by combining different amounts of electrons and hydrogen ions, CO2 can produce a variety of products through different reaction routes.
O double bonds, which have a greater bond strength than a variety of chemical bonds including C–H and C–C.6 During the catalytic process, inert CO2 molecules are chemisorbed on the surface of the photocatalyst in a curved structure, through which the carbon dioxide molecules exhibit a lower unoccupied molecular orbital (LUMO) energy level, which makes it easier to receive electrons from the photocatalyst. After accepting the electrons, carbon dioxide will transform into carbonate anion radicals (CO2−) which are more reactive, contributing to further reactions to generate other products.43 As shown in Table 1, by combining different amounts of electrons and hydrogen ions, CO2 can produce a variety of products through different reaction routes.
| Equation | Products | E 0 [V] |
|---|---|---|
| CO2 + e− → CO2− | Carbonate anion radical | −1.90 |
| CO2 + 2H+ + 2e− → HCOOH(aq) | Formic acid | −0.61 |
| CO2 + 2H+ + 2e− → CO(g) + H2O | Carbon monoxide | −0.53 |
| CO2 + 4H+ + 4e− → HCHO(aq) + H2O | Formaldehyde | −0.48 |
| CO2 + 6H+ + 6e− → CH3OH(aq) + H2O | Methanol | −0.38 |
| 2CO2 + 12H+ + 12e− → C2H4(g) + 4H2O | Ethene | −0.34 |
| 2CO2 + 12H+ + 12e− → C2H5OH(aq) + 3H2O | Ethanol | −0.33 |
| 2CO2 + 8H+ + 8e− → CH3COOH(aq) + 2H2O | Acetic acid | −0.31 |
| 2CO2 + 14H+ + 14e− → C2H6(g) + 4H2O | Ethane | −0.27 |
| CO2 + 8H+ + 8e− → CH4(g) + 2H2O | Methane | −0.24 |
2.2 Reaction systems
CO2 photocatalytic reduction is usually carried out in an air-tight cell, in which the CO2 molecules mixed with water vapor or dissolved in a solvent are reduced by photocatalysts under light. There are two main reaction modes for this reaction, namely, a “solid–liquid system” and “solid–gas system”, as shown in Fig. 2. | ||
| Fig. 2 Schematic diagram of the solid–gas system and solid–liquid system for photocatalytic CO2 reduction. | ||
The solid–liquid system is typically used in the majority of current research, because it only needs to disperse the photocatalyst particles in water or organic solvents to bring the photocatalyst into full contact with the dissolved CO2. In contrast, solid–gas photoreaction systems require uniform loading of the photocatalyst onto the solid substrate by methods such as drip and spin coating to prevent its accumulation or agglomeration into clumps.45 In addition, the solid–gas system requires additional steps to introduce water vapor, such as by heating the water injected in advance or passing CO2 into the reactor along with water vapor. Therefore, the solid–liquid system is simpler and less expensive to operate. However, because of their intrinsic ionic nature, NHPs are particularly vulnerable to moisture and are very unstable in polar solvents.46 CsPbBr3 nanocrystals (NCs), for instance, will rapidly undergo phase changes to CsPb2Br5 in pure water, leading to a significant loss of light absorption capacity.47 Therefore, low-polarity solvents are widely used in solid–liquid photocatalytic systems to mitigate the destruction of NHP structures. By far, the best performing low-polarity solvent is ethyl acetate (EA).48 Some other solvents, such as acetonitrile (ACN), toluene, and benzene, have also been tried for the photocatalytic reduction of CO2.49 Nevertheless, in all these purely low-polar organic solvents, the organic molecules could not provide enough protons/electrons, resulting in very limited photocatalytic activity of the photocatalyst for CO2 reduction. Therefore, researchers subsequently developed a mixture of solvents containing low-polar organic solvents and water that can provide sufficient protons/electrons, such as EA/H2O and ACN/H2O.50 It was found that adding the right amount of water could significantly increase the productivity of photocatalytic CO2 reduction.51 In addition to water, some other alcohol solvents, like methanol, isopropanol, etc., can also be used as electron sacrificial agents to be oxidized to value-added products during the photocatalytic transformation of CO2.52 To avoid the use of expensive and toxic organic reagents, researchers have also started to try to synthesize water-stabilized NHPs and use pure H2O as a dispersant.49,53 Besides, solid–liquid systems have the problems of limited solubility of CO2 in the solvent and hydrogen production, which can limit the yield and selectivity of photocatalytic CO2 reduction, but these two problems are well avoided in solid–gas systems. In terms of product identification, the solid–liquid system can detect liquid-phase products such as CH3OH, HCOOH and gas-phase products (CH4 and CO), while the solid–gas system can only detect gas-phase products. Therefore, it is important to choose the appropriate reaction system according to the need.
2.3 The structure and optical properties of NHPs
 | (1) |
 | (2) |
R A, RB and RX in eqn (1) and (2) correspond to the radius of the A, B, and X-site ions in the ABX3 structure. Their changes directly lead to changes in the lattice parameter and thus affect the crystal structure. As shown in Fig. 3h, theoretically stable NHPs should have a value of [t] from 0.8 to 1.0 and a [μ] value between 0.44 and 0.9, respectively.58 In general, NHPs will exhibit a cubic structure (α-phase) when the [t] value is in the range of 0.9 and 1.0,59 while a perfect cube is formed when [t] = 1.0. α-Phase NHPs are desirable crystal structures for the solar light absorption because of their excellent optical properties. The divalent B-site metal ion radius tends to be large in practice. Therefore, using A-site cations with a greater ionic radius is essential to get the [t] value as close to 1.0 as possible. However, too large A-site cations may also result in serious deformation of the α-phase lattice, which finally leads to crystal structural change.60 This may result in the formation of low-dimensional NHP structures such as two-dimensional sheets, one-dimensional chains, or zero-dimensional clusters.61 Thus, as shown in Fig. 3a–g, in addition to the conventional ABX3 structure, NHPs are also considered to exhibit other structures such as A4BX6, AB2X5, A2BX4, A2BX6, A2B1+B23+X6, and A3B2X9. Among them, the octahedra in the A4BX6 structure dissociate in all ranges and no longer share halide ions between them.62,63 AB2X5 is a type of two-dimensional NHP having a tetragonal phase, which contains Cs+ and [Pb2X5]− layers with alternating Cs+ and [Pb2X5]− layers. A2PbX4 are also 2-dimensional NHPs which have alternating co-angular [PbX6]4−octahedral layers.64 A2BX6 is similar to ABX3 in that the positive 2-valent cation is replaced by a positive 4-valent B-site cation. A2B+B3+X6 is a 3-D structure consisting of two different kinds of B-site cations. Cs3M2X9 is made of isolated clusters, in which each consists of two coplanar octahedra with Cs+ acting as a bridge ion between the clusters.
 | ||
| Fig. 3 Structure schematic diagram of different NHPs. (a) 3D pseudocubic ABX3, (b) 0D A4BX6, (c) 2D AB2X5, (d) 2D A2BX4, (e) 0D A2BX6, (f) 3D A2B+B3+X6, and (g) 2D A3B2X9, and (h) tolerance factors of commonly used ABX3 NHPs. Reproduced from ref. 57 with permission from Royal Society of Chemistry, Copyright (2021). | ||
NHPs with different dimensionalities have different characteristics for photocatalytic applications. Regarding the dimensionality of the NHPs, it can be 3D bulk, 2D nanostructures (nanosheets), 1D nanostructures (nanotubes, nanowires and nanorods) and 0D nanostructures (nanocrystals and quantum dots). In general, the 3D NHP structure usually has a more compact structure and wider light absorption bandwidth, which covers a wider range of the solar spectrum. However, NHPs with low-dimensional shapes (2D, 1D, and 0D) possess optical and electrical properties that are absent in their 3D counterparts as a result of quantum confinement and steric anisotropy effects.65 Since the photogenerated carriers are confined in the three dimensions for the 0D NHPs, they usually possess a high quantum yield and defect-tolerant band gap and, thus, they have great potential for photocatalytic applications. In terms of 1D NHPs, they have one microscopic and two nanoscopic dimensions. The motion of the carrier (electrons and holes) is quantized in two dimensions, which makes them possess a high length-to-width ratio and a well-defined size and morphology. This feature benefits 1D NHPs with efficient transport and propagation of charge carriers and photons along the longitudinal direction. 2D NHPs, with a layered structure, have one nanoscopic and two microscopic dimensions.66 The introduction of large-sized organic molecules as isolation layers between the layers significantly improves the stability of the material. Their band gap can be adjusted by regulating the inter-layer distances, which allows for the fine tuning of the light-absorbing properties. In addition, the 2D structure increases the specific surface area, which is conducive to the separation and transfer of photogenerated carriers at the interface, thus enhancing the photocatalytic activity.67 It is not reasonable to specify which dimension of NHPs is the best in terms of photocatalytic performance, as they all have a disadvantage of their own. For example, 3D NHPs are restricted by their limited surface-to-volume ratio and ineffective carrier transport across whole crystals, 2D NHPs have wider band gaps and cannot fully absorb the visible spectrum, 1D structures are susceptible to stresses and defects and have poorer structural stability, and 0D NHP quantum dots are more sensitive to environmental factors (e.g., water, oxygen, etc.) due to their extremely small scale. Therefore, the more promising approach in reality is to combine various dimensions of NHPs to form different types of heterojunctions, which complement each other to further enhance the photocatalytic performance.
Although using elements like Ge, Sn, Ag and Bi overcomes the problem of high toxicity compared to Pb, their price is over 10 times that of Pb, which still limits their large-scale application. Therefore, researchers started to utilize much cheaper alternatives like Sb, Cu, Ti, and Mg.86,87 For instance, Mu et al. prepared Cs3Sb2Br9 hollow nanospheres (H-Cs3Sb2Br9) using a simple antisolvent method. This structure increases light absorption performance, promotes the separation of photogenerated carriers, and exposes a large number of catalytically active sites.88 Zhou et al. prepared a series of titanium-based halide perovskite materials (Cs2TiX6, X = Cl, Cl0.5Br0.5, Br). These titanium-based perovskite materials have very high stability in harsh environments such as light irradiation and heating. Under 3 hours of light irradiation, Cs2Ti(Cl0.5Br0.5)6 microcrystals catalyzed the production of 176 μmol g−1 of CO and 78.9 μmol g−1 of CH4.89 Zhao et al. investigated the performance of copper-based halide perovskites CsCuCl3 and CsCuCl2Br as photocatalysts for CO2 reduction. Theoretical and experimental analyses revealed that CsCuCl3 has a suitable bandgap (1.92 eV) and conduction band minimum, allowing it to effectively utilize sunlight and drive the reduction of CO2 to CH4 and CO. Partial substitution of bromine ions narrows the bandgap of CsCuCl2Br and facilitates charge carrier transport, resulting in even better photocatalytic performance.90 However, their current photocatalytic performance is still very much lower than that of Ge, Sn, Ag and Bi NHPs. Therefore, some literature combines the low-price metals with Bi, like Cs2NaBiCl6 and Cs3Bi2xSb2−2xI9, which points out a new path for the development of lead-free NHPs.91,92 It is expected that the performance of inexpensive lead-free NHPs could be improved with continuous research in the future.
 | ||
| Fig. 4 Conduction and valence band positions of some commonly used NHP nanocrystals. Reprinted with permission from ref. 43, Copyright 2020, American Chemical Society. | ||
2.4 Selectivity of NHPs for CO2 photoreduction
As mentioned above, CO2 has the highest oxidation state of carbon (+4), so it can be reduced to various lower state products (+2, 0, −2 and −4) by accepting different amounts of protons and electrons. Presently, the principal photoreduction products of CO2 using NHP photocatalysts are CO and CH4, occasionally with some other products like HCOOH.96 However, mixing of multiple products will lead to high cost for separation. Therefore, it is important to improve product selectivity for practical production. Photoexcitation properties, band structure, charge carrier separation and catalytically active sites are four main factors influencing the product selectivity during the photocatalytic CO2 reduction reaction.97Light excitation of NHPs to generate efficient photogenerated electrons and holes is the key to the CO2RR, which is mainly influenced by photon energy and light intensity. Photon energy determines whether the semiconductor can absorb photons to be excited and can thermodynamically affect the product selectivity of the reaction, while light intensity determines the number of photogenerated electrons and holes produced by excitation, which then kinetically affects the reaction rate and product selectivity of a reaction with multiple electrons involved.97 For instance, Wang et al.98 investigated the impact of light intensity on CO2 reduction reactions using Cs3Sb2I9 photocatalysts (Fig. 5a). They found that by varying the light intensity of the full spectrum from 0.5 W cm−2 to 1.5 W cm−2, the production rate of CO increased rapidly with increasing light intensity and the yield of CH4 remained almost constant, leading to a higher CO selectivity.
 | ||
| Fig. 5 (a) CO and CH4 production rates of the Cs3Sb2I9 photocatalyst at different light intensities. Reproduced from ref. 98 with permission from Elsevier, Copyright (2021). (b) Photocatalytic CO2 reduction performance using FAPbBr3 QDs and CsPbBr3 QDs. Reproduced from ref. 99 with permission from Elsevier, Copyright (2020). (c) CO yields of Cs3Bi2Br9, Cs3Bi2Cl9, and Cs3Bi2I9 with time. Reprinted with permission from ref. 100, Copyright 2020, American Chemical Society. (d) Product yields of CsCuCl3 and CsCuCl2Br. Reprinted with permission from ref. 90, Copyright 2022, American Chemical Society. (e) Comparison of photocatalytic performance of g-C3N4, CABB, CABB@C3N4-10% and CABB@C3N4-82%. Reproduced from ref. 101 with permission from Elsevier, Copyright (2020). (f) Effect of rGO in Cs4PbBr6/rGO for photocatalytic CO2 reduction under visible light (>420 nm) irradiation. Reproduced from ref. 102 with permission from Elsevier, Copyright (2020). (g) Photocatalytic CO2 reduction performances of CsPbBr3-Re(x) after 3 h of reaction. Reproduced from ref. 103 with permission from Wiley-VCH, Copyright (2019). (h) Photocatalytic CO2 reduction performance of CABB and the HWO/CABB photocatalyst. Reproduced from ref. 104 with permission from Elsevier, Copyright (2023). (i) Production of gases using various photocatalysts based on pristine CsPbBr3, pristine Ni(tpy), and CsPbBr3-Ni(tpy). Reprinted with permission from ref. 82, Copyright 2020, American Chemical Society. (j) Bar diagram showing relative ratios of CH4 and CO produced using addition of Fe(II) acetate to CsPbBr3, CsPbBr3, and Fe(II)-doped CsPbBr3, only Fe(II) acetate without CsPbBr3 and the control reaction without any additive. Reprinted with permission from ref. 105, Copyright 2019, American Chemical Society. | ||
Additionally, the reduction of CO2 needs photocatalysts with high enough reduction potentials, which are strongly related to their CB positions. The CB positions can be adjusted through changing different cations and anions in NHPs. For instance, Que et al.99 compared the CO2 reduction performance both in FAPbBr3 quantum dots (QDs) and CsPbBr3 QDs (Fig. 5b). They found that in all three reaction mediums (DI, DI + EA, EA), the photocatalytic CO yield rates of FAPbBr3 QDs are higher than that of CsPbBr3 QDs. Specifically, in the DI/EA system, the amount of CO can reach up to 181.25 μmol g−1 h−1 for the FAPbBr3 QDs, while that for CsPbBr3 QDs is 11.23 μmol g−1 h−1. Both QDs exhibited inferior CH4 reduction rates compared to CO. In addition to the A-site, element changes in the X-site can also affect product selectivity. Sheng et al.100 studied the effect of halide elements on the CO2 to CO productivity. As shown in Fig. 5c, Cs3Bi2Br9 exhibits the highest photoreduction performance with 134.76 μmol g−1 of CO yield and a selectivity of 98.7%, which is much better than those of Cs3Bi2Cl9 and Cs3Bi2I9, 83.06 μmol g−1 and 5.78 μmol g−1, respectively. Zhao et al.90 reported that Br-substituted samples (CsCuCl2Br) show higher CH4 selectivity than CsCuCl3 samples (Fig. 5d). CsCuCl2Br microcrystals (MCs) possess the best performance with CO and CH4 yields of 5.61 and 15.36 μmol g−1, respectively, corresponding to a CH4 selectivity of 73.2%. They also found that the morphology of catalysts affects the CO2 reduction performance and that CsCuCl2Br MCs perform better than CsCuCl2Br powders.
Combining NHPs with other materials to construct heterojunctions can change the separation efficiency of photogenerated electrons and holes based on the different energy band structures of the two materials, which also has an impact on the selectivity of the products. Kong et al.103 coupled Re(CO)3Br(dcbpy) with CsPbBr3 NC to enhance photocatalytic performance of CO2 reduction (Fig. 5g). For pure CsPbBr3 NCs, CO and CH4 were both observed, but on coupling with Re(CO)3Br(dcbpy) molecules, CO became the predominant product. The CsPbBr3-Re(600) sample demonstrated the highest performance, yielding 104.37 mol g−1 of CO and 5.64 mol g−1 of H2, with a selectivity for CO of up to 95%. They suggested that the elimination of the CH4 product is because the photogenerated electrons of CsPbBr3 can be transported to Re(CO)3Br(dcbpy) molecules timely and effectively. Wang et al.102 hybridized reduced graphene oxide (rGO) to Cs4PbBr6 for photocatalytic CO2 reduction (Fig. 5f). They discovered that the yield of CO continued to increase while the yield of CH4 decreased as the quantity of rGO in the Cs4PbBr6/rGO composite grew, resulting in better CO selectivity. When the ratio of rGO in the composite of Cs4PbBr6/rGO reaches 10 wt%, the selectivity of CO can be increased to 94.6%. The superior catalytic ability of Cs4PbBr6/rGO is attributed to the fast electron transfer from Cs4PbBr6 to rGO, and the presence of defects on rGO could trap electrons and increase the local charge density. Compared to the Schottky junctions of Re(CO)3Br(dcbpy) and rGO, the photocatalysts in type II heterojunctions promote charge separation more efficiently. Wang et al.101 composed Cs2AgBiBr6@g-C3N4 (CABB@C3N4) for CO2 photoreduction (Fig. 5e). They found that the surface coverage of g-C3N4 on CABB plays an important role in the product selectivity, due to the formation of type-II heterojunctions. The results show that in the CABB@C3N4 composite containing 10% C3N4, electron transfer from the conduction band of g-C3N4 to that of CABB leads to a highly selective conversion of CO2 to CO.
Furthermore, since CO2 reduction is a dynamic multi-step surface catalytic reaction, the types and amounts of catalytically active sites on surface of photocatalysts can significantly affect the adsorption/desorption characteristics of reactants/intermediates, thereby altering the selectivity of the final product. Zhou et al. reported a novel 2D/2D heterojunction H2WO4/Cs2AgBiBr6 (HWO/CABB) photocatalyst, which significantly improves its efficiency and selectivity for photocatalytic CO2 reduction to CH4 by combining the synergistic effects of the 2D heterojunction and surface bromine vacancy defects.104 Specifically, this heterojunction structure facilitates charge transfer and separation, while the surface defects can promote the adsorption and activation of CO2, thereby reducing the formation energy barrier of the CHO* intermediate, resulting in a CH4 selectivity of 86.1%, which is 30 times higher than that of the pure CABB photocatalyst. In addition to vacancies in NHPs, some metals such as Fe, Co, Ni, Cu, etc. can also contribute to product selectivity as catalytically active sites. Shyamal et al.105 found differences in product selectivity between doped and undoped Fe(II) in photocatalysts for the reduction of CO2 (Fig. 5j). For undoped CsPbBr3 NCs, the major product was CO (4.6 μmol g−1 h−1). However, after doping 25% iron ions into the CsPbBr3 NCs, it formed mainly CH4 (6.1 μmol g−1 h−1). They concluded that the active CH4 molecules were desorbed from the catalyst surface faster due to Fe(II) doping, which enhanced the production of CH4. Chen et al.82 coupled CsPbBr3 NCs with the metal complex [Ni(terpy)2]2+ (Ni(tpy)) for photocatalytic CO2 reduction. As seen in Fig. 5i, as the Ni(tpy) content increased, the catalytic activity of CsPbBr3-Ni(tpy) showed remarkable selectivity for CO. The researchers came to the conclusion that Ni(tpy) can serve as an electron sink and can offer catalytic sites, resulting in increased photocatalytic activity for CO2 reduction.
As summarized in Table 2, the selectivity of photocatalytic CO2 reduction products (CO and CH4) using NHPs is mainly influenced by light-excitation, band structure, charge carrier separation and catalytically active sites. In order to enhance the selectivity of CH4, several methods can be taken into consideration: first, by adjusting the types of cations and anions in the perovskite, the position of the conduction band can be tuned to provide the necessary reduction potential for CH4 generation. Selecting perovskites with Bi, Sn or In as the B-site elements can lead to a bandgap structure more favorable for improving CH4 selectivity. Furthermore, constructing heterojunctions between the perovskite and reduced graphene oxide or metal complexes can help enhance the separation and transfer efficiency of photogenerated charge carriers, thereby promoting the multi-electron reduction pathway. Finally, doping the perovskite with metals such as Fe, Co, Ni, Cu, and Zn or coupling it with metal complexes, can introduce catalytically active sites that are beneficial for CH4 formation. In summary, systematically controlling the band structure, charge carrier dynamics, and catalytically active sites can effectively enhance the selectivity towards CH4 in the CO2 reduction reaction using perovskite-based photocatalysts.
| Photocatalysts | Systema | Main product (μmol g−1 h−1) | Selectivity (%) | Influencing factor | Ref. |
|---|---|---|---|---|---|
| a S–L: solid–liquid system and S–V: solid–vapor system. | |||||
| CO selective priority | |||||
| Cs3Bi2I9 | S–V | CO 7.76 | 83.9 | Band structure (Cs+ > Rb+ > MA+) | 106 |
| CH4 1.49 | |||||
| CsPbBr3-Ni(tpy) | S–L | CO 1464 | 84.9 | (a) Charge separation (Schottky scheme) | 82 |
| CH4 260 | (b) Active sites (Ni) | ||||
| Cs3Sb2I9 | S–V | CO 95.7 | 87.8 | (a) Light intensity | 98 |
| CH4 2.9 | |||||
| H2 10.4 | (b) Active sites (defects) | ||||
| FAPbBr3 QDs | S–L | CO 181.25 | 89.9 | Band structure (FA+ > Cs+) | 99 |
| CH4 16.9 | |||||
| H2 3.56 | |||||
| Cs4PbBr6/rGO | S–L | CO 11.4 | 94.6 | (a) Charge separation (Schottky scheme) | 102 |
| (b) Active sites (defects) | |||||
| CsPbBr3-Re(CO)3Br(dcbpy) | S–L | CO 34.79 | 95% | (a) Charge separation (Schottky scheme) | 103 |
| H2 1.88 | (b) Active sites (Re) | ||||
| Mn:CsPb(Br/Cl)3 | S–L | CO 213 | 95.9 | Active sites (Mn) | 107 |
| CH4 9.1 | |||||
| CsPbBr3 QDs/UiO-66(NH2) | S–L | CO 8.21 | 97 | Charge separation (type II scheme) | 108 |
| CH4 0.26 | |||||
| Cs3Bi2Br9 NCs | S–V | CO 26.95 | 98.7 | Band structure (Br > Cl > I) | 100 |
| CsPbBr3 QDs/Cu-TCPP-20 | S–L | CO 71.77 | 99 | Charge separation (type II scheme) | 109 |
| CH4 0.81 | |||||
| Cs2AgBiBr6/Bi2WO6 | S–L | CO 42.19 | 99 | Charge separation (direct Z-scheme) | 110 |
| CH4 0.41 | |||||
| CsPbBr3 NCs/USGO/α-Fe2O3 | S–L | CO 73.8 | 100 | Charge separation (all-solid-state Z-scheme) | 111 |
| FAPbBr3/Bi2WO6 | S–L | CO 170 | 100 | Charge separation (direct Z-scheme) | 112 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||
| CH 4 selective priority | |||||
| Cs2AgInCl6@Ag-2 | S–L | CO 6.59 | 52.3 | Band structure (In3+) | 113 |
| CH4 7.23 | |||||
| Amorphous-TiO2/CsPbBr3 NCs | S–L | CO 11.71 | 55.6 | Charge separation (Schottky scheme) | 114 |
| CH4 20.15 | |||||
| H2 4.38 | |||||
| Fe(II)-doped CsPbBr3 | S–L | CO 3.2 | 66 | Active sites (Fe2+) | 105 |
| CH4 6.1 | |||||
| MAPbI3@PCN221(Fex) | S–L | CO 6.63 | 66 | Active sites (Fe2+) | 115 |
| CH4 12.86 | |||||
| Cs2AgBiBr6@g-C3N4 | S–L | — | ∼70 | Charge separation (direct Z-scheme) | 101 |
| CsCuCl2Br MCs | S–L | CO 1.87 | 73.2 | Band structure (Br + Cl > Cl) | 90 |
| CH4 5.12 | |||||
| α-Fe2O3/amine-RGO/CsPbBr3 NCs | S–V | CO 2.36 | 78.1 | Charge separation (all-solid-state Z-scheme) | 116 |
| CH4 9.45 | |||||
| H2 0.29 | |||||
| CsPbBr3 @ZIF-67 | S–V | ZIF-67 | 82.1, 78.2 | (a) Charge separation (Schottky scheme) | 117 |
| CH4 3.51 | |||||
| CO 0.77 | |||||
| CsPbBr3 @ZIF-8 | ZIF-8 | (b) Active sites (Co2+, Zn2+) | |||
| CH4 1.81 | |||||
| CO 0.51 | |||||
| H2WO4/Cs2AgBiBr6 | S–L | CH4 22.6 | 86.1 | (a) Charge separation (direct Z-scheme) | 104 |
| (b) Br vacancy | |||||
| Cs3Bi2Br9 and Cs2AgBiBr6/mesoporous TiO2 | S–L | CH4 32.9, 24.2 | 88.7, 84.2 | Charge separation (Schottky scheme) | 118 |
| CsPbBr3/ZnPc | S–L | CH4 168 | 89 | (a) Charge separation (direct Z-scheme) | 119 |
| (b) Active sites (Zn) | |||||
| CsCuCl3/Cu | S–L | CH4 58.77 | 92.7 | (a) Charge separation (Schottky scheme) | 120 |
| (b) Active sites (Cu) | |||||
| Cs2AgBiBr6-Cu-RGO | S–V | CO 1.9 | 93 | Active sites (Cu) | 121 |
| CH4 10.7 | |||||
| Cu-RGO-CsPbBr3 NS | S–L | CH4 12.7 | 94.6 | Active sites (Cu) | 122 |
| Cs2SnI6 NCs/SnS2 NS | S–V | CH4 0.61 | 100 | Charge separation (type II scheme) | 123 |
3. Retrofit strategies
Previous research has demonstrated that four essential variables influence the photocatalytic CO2RR performance using NHP as photocatalysts: light absorption, charge separation and transfer, catalytically active sites, and catalyst stability. The light absorption and catalytically active sites depend on the band structure and composition of NHPs, which can be tuned by using a series of methods in surface and component engineering. After absorbing light energy, the catalyst excites photogenerated carriers, which then undergo inside-to-surface transfer. However, large amounts of photogenerated electron–hole pairs are recombined during this complex process, which greatly reduces the efficiency of photogenerated carrier usage. For example, the most common method currently used is to combine NHPs with another catalyst as a photocatalyst, that is to inhibit carrier recombination by forming a heterojunction. Besides, the stability problem is usually solved by performing encapsulation. Herein, the retrofit strategies are divided into four parts as shown in Fig. 6: surface engineering, component engineering, heterojunction engineering and encapsulation engineering. Table 3 summarizes recent literature according to the type of retrofit strategy. | ||
| Fig. 6 Four main categories of retrofit strategies for NHPs. | ||
| Type | Photocatalysts | System | Light source | Products | Stability | Ref. |
|---|---|---|---|---|---|---|
| Surface engineering | ||||||
| Size control | CsPbBr3 QDs | Solid–liquid (EA/H2O) | 300 W Xe-lamp with a standard AM 1.5 filter | CO: 34.1 μmol g−1 (8h) | 8 h | 124 |
| CH4: 12.2 μmol g−1 (8h) | ||||||
| H2: 0.8 μmol g−1 (8h) | ||||||
| Cu-RGO-CsPbBr3 | Solid–liquid (H2O) | Xe-lamp irradiation 400 nm filter | CH4: 12.7 μmol g−1 h−1 | 12 h | 122 | |
| CO: 0.46 μmol g−1 h−1 | ||||||
| H2: 0.27 μmol g−1 h−1 | ||||||
| CsPbBr3 nanosheets | Solid–vapor (H2O vapor) | 300 W Xe lamp (100 mW cm−2) | (a) CsPbBr3 NCs | 30 h | 125 | |
| CO: 5.7 μmol g−1 h−1 (b) 4 nm CsPbBr3 nanosheets | ||||||
| CO: 21.6 μmol g−1 h−1 | ||||||
| Morphology control | 3DOM Au-CPB | Solid–liquid (IPA–EA) | 300 W Xe lamp 420 nm filter | R electron: 38.0 μmol g −1 h −1 | — | 126 |
| NMF/CsPbBr3 nanowires | Solid–liquid (EA/H2O) | 300 W Xe lamp with a 420 nm filter (100 mW cm−2) | CO: 81.0 μmol g −1 h −1 | — | 127 | |
| Cs2AgBiX6 (X = Cl, Br, I) | Solid–liquid (EA) | 405 nm laser diode | (a) Cs2AgBiBr6 NPLs | 9 h | 85 | |
| Total photocatalytic electron consumption = 255.4 μmol g−1 | ||||||
| (b) Cs2AgBiBr6 NCs | ||||||
| Total photocatalytic electron consumption = 30.8 μmol g−1 | ||||||
| CsPbBr3 nanorods | Solid–liquid (EA/H2O) | 450 W Xe lamp (150 mW cm−2) | CO: 40.81 μmol g−1 (2h) | — | 128 | |
| CH4: 74.45 μmol g−1 (2h) | ||||||
| Facet regulation | Cs3Sb2Br9 NCs | Solid–liquid (ODE/H2O) | 300 W Xe-lamp with a AM 1.5 G filter (100 mW cm−2) | (a) Cs3Sb2Br9 NCs | 9 h | 129 |
| CO: 510 μmol g−1 (4 h) | ||||||
| (b) Pristine CsPbBr3 NCs | ||||||
| CO: 50 μmol g−1 (4 h) | ||||||
| CsPbBr3 NCs | Solid–liquid (EA/H2O) | 450 W Xe-lamp (150 mW cm−2) | (a) Cube-shaped CsPbBr3 NCs | 6 h | 130 | |
| CO: 16.4 μmol g−1 (4h) | ||||||
| CH4: 7.6 μmol g−1 (4h) | ||||||
| (b) Hexapod CsPbBr3 NCs | ||||||
| CO: 79.5 μmol g−1 (4h) | ||||||
| CH4: 38.4 μmol g−1 (4h) | ||||||
| (c) Polyhedrons CsPbBr3 NCs | ||||||
| CO: 130.7 μmol g−1 (4h) | ||||||
| CH4: 58.8 μmol g−1 (4h) | ||||||
| Ligand regulation | Surface Pb-rich CsPbCl3 QDs | Solid–liquid (H2O) | AM 1.5 G solar light (150 mW cm−2) | (a) Ni: CsPbCl3 NCs | 3 h | 49 |
| CO: 8.55 μmol g−1 h−1 | ||||||
| (b) Pb-rich Ni: CsPbCl3NCs | ||||||
| CO: 169.37 μmol g−1 h−1 | ||||||
| (c) Pb-rich Mn: CsPbCl3 NCs | ||||||
| CO: 152.49 μmol g−1 h−1 | ||||||
| Cs2AgBiBr6 NCs | Solid–liquid (EA) | AM 1.5 G 100 W Xe-lamp (150 mW cm−2) | (a) Washed Cs2AgBiBr6 NCs | — | 131 | |
| CO: 14.1 μmol g−1 (6h) | ||||||
| CH4: 9.6 μmol g−1 (6h) | ||||||
| (b) As-prepared Cs2AgBiBr6 NCs | ||||||
| CO: 5.5 μmol g−1 (6h) | ||||||
| CH4: 0.65 μmol g−1 (6h) | ||||||
| CsPbBr3-Ni(tpy) | Solid–liquid (EA/H2O) | 300 W Xe-lamp (100 mW cm−2) | CO: 1464 μmol g−1 | 4 h | 82 | |
| CH4: 260 μmol g−1 | ||||||
| Cs2AgInCl6@Ag-2 | Solid–liquid (EA) | 300 W Xe-lamp | CO: 26.4 μmol g−1 (4 h) | 9 h | 113 | |
| CH4: 28.9 μmol g−1 (4 h) | ||||||
| Defect regulation | Cs3Bi2X9 NCs (X = Cl, Br, I) | Solid–vapor (H2O vapor) | 300 W Xe-lamp with a AM 1.5 filter | (a) Cs3Bi2Br9 NCs | 20 h | 100 |
| CO: 134.76 μmol g−1 (5 h) | ||||||
| (b) Cs3Bi2Cl9 NCs | ||||||
| CO: 83.06 μmol g−1 (5 h) | ||||||
| (c) Cs3Bi2I9 NCs | ||||||
| CO: 5.78 μmol g−1 (5 h) | ||||||
| CsPbBr3-BF4/Co | Solid–liquid (EA) | λ > 400 nm | CO: 83.8 μmol g−1 h−1 | 8 h | 132 | |
| CsPbBr3 NCs | Solid–liquid (EA/H2O) | 450 W Xe-lamp (150 mW cm−2) | (a) Cube-shaped CsPbBr3 NCs | 6 h | 130 | |
| CO: 16.4 μmol g−1 (4h) | ||||||
| CH4: 7.6 μmol g−1 (4h) | ||||||
| (b) Hexapod CsPbBr3 NCs | ||||||
| CO: 79.5 μmol g−1 (4h) | ||||||
| CH4: 38.4 μmol g−1 (4h) | ||||||
| (c) Polyhedrons CsPbBr3 NCs | ||||||
| CO: 130.7 μmol g−1 (4h) | ||||||
| CH4: 58.8 μmol g−1 (4h) | ||||||
|
||||||
| Compositional engineering | ||||||
| A-site | A3Bi2I9 (A = Rb+, Cs+ or, MA+) | Solid–vapor (H2O vapor) | UV lamp (32 W, 305 nm, 80.4 μW cm−2) | (a) Cs3Bi2I9 NCs | 12 h | 106 |
| CO: 77.6 μmol g−1 (10 h) | ||||||
| CH4: 14.9 ± 0.8 μmol g−1 (10 h) | ||||||
| (b) Rb3Bi2I9 NCs | ||||||
| CO: 18.2 μmol g−1 (10 h) | ||||||
| CH4: 17.0 ± 1.6 μmol g−1 (10 h) | ||||||
| (c) MA3Bi2I9 NCs | ||||||
| CO: 7.2 μmol g−1 (10 h) | ||||||
| CH4: 9.8 ± 0.6 μmol g−1 (10 h) | ||||||
| FAPbBr3 and CsPbBr3 QDs | Solid–liquid (EA/H2O) | Xenon arc lamp | (a) FAPbBr3 QDs | — | 99 | |
| CO: 181.25 μmol g−1 h−1 | ||||||
| CH4: 16.9 μmol g−1 h−1 | ||||||
| H2: 3.56 μmol g−1 h−1 | ||||||
| (b) CsPbBr3 QDs | ||||||
| CO: 11.23 μmol g−1 h−1 | ||||||
| CH4: 0.45 μmol g−1 h−1 | ||||||
| H2: 0.13 μmol g−1 h−1 | ||||||
| B-site | Cs3Sb2I9 | Solid–vapor (H2O vapor) | Xe-lamp (200 mW cm−2) | CO: 13.2 μmol g−1 h−1 | — | 98 |
| Cs2CuBr4 QDs | Solid–vapor (H2O vapor) | 300 W Xe lamp (1.5 G filter) | Cs2CuBr4 QDs | 20 h | 133 | |
| CH4: 74.81 μmol g−1 (5 h) | ||||||
| CO: 148.98 μmol g−1 (5 h) | ||||||
| Cs2XCl6 (X = Hf, Zr, Te) MCs | Solid–vapor (H2O vapor) | 300 W Xe lamp | (a) Cs2TeCl6 MCs | — | 134 | |
| CO: 284.4 μmol g−1 (3 h) | ||||||
| CH4: 48.96 μmol g−1 (3 h) | ||||||
| (b) Cs2HfCl6 MCs | ||||||
| CO: 204.3 μmol g−1 (3 h) | ||||||
| CH4: 48.66 μmol g−1 (3 h) | ||||||
| (c) Cs2ZrCl6 MCs | ||||||
| CO: 219.2 μmol g−1 (3 h) | ||||||
| CH4: 48.36 μmol g−1 (3 h) | ||||||
| X-site | CsPb(Br0.5/Cl0.5)3 | Solid–liquid (EA) | 300 W Xe-lamp with a AM 1.5 filter | CO: 767 μmol g−1 (9 h) | — | 135 |
| CH4: 108 μmol g−1 (9 h) | ||||||
| Cs2AgBiX6 NCs | Solid–vapor (H2O vapor) | 300 W Xe-lamp with a 420 nm filter | (a) Cs2AgBiI6 NCs | — | 136 | |
| CO: 18.9 μmol g−1 (6 h) | ||||||
| (b) Cs2AgBiCl6 NCs | ||||||
| CO: 13.62 μmol g−1 (6 h) | ||||||
| (c) Cs2AgBi(Br0.5I0.5)6 NCs | ||||||
| CO: 11.88 μmol g−1 (6 h) | ||||||
| Cs3Bi2X9 (X = Cl, Cl0.5Br0.5, Br, Br0.5I0.5, I) | Solid–vapor (H2O vapor) | 300 W Xe-lamp with a 420 nm filter | CO: 54 μmol/g (3h) | 10 h | 137 | |
| CsCuCl2Br MCs | Solid–liquid (EA/IPA) | AM 1.5 G (100 mW cm−2) | (a) CsCuCl3 MCs | 20 h | 90 | |
| CO: 2.79 μmol g−1 (3 h) | ||||||
| CH4: 11.55 μmol g−1 (3 h) | ||||||
| (b) CsCuCl2Br MCs | ||||||
| CO: 5.61 μmol g−1 (3 h) | ||||||
| CH4: 15.36 μmol g−1 (3 h) | ||||||
| Metal doping | Mn:CsPb(Br/Cl)3 | Solid–liquid (EA) | 300 W Xe-lamp with a AM 1.5 filter | (a) Mn:CsPb(Br/Cl)3 | 9 h | 107 |
| CO: 1917 μmol g−1 (9 h) | ||||||
| CH4: 82 μmol g−1 (9 h) | ||||||
| (b) Pristine CsPbBr3 | ||||||
| CO: 135 μmol g−1 (9 h) | ||||||
| CH4: 58.6 μmol g−1 (9 h) | ||||||
| Co-CsPbBr3/Cs4PbBr6 | Solid–liquid (ACN/H2O/MeOH) | 300 W Xe-lamp (100 mW cm−2) | (a) Co 1%@CsPbBr3/Cs4PbBr6 | 15 h | 138 | |
| CO: 1835 μmol g−1 (15 h) | ||||||
| (b) CsPbBr3/Cs4PbBr6 | ||||||
| CO: 678 μmol g−1 (15 h) | ||||||
| Fe(II):CsPbBr3 | Solid–liquid (EA/H2O) | 450 W Xe-lamp (150 mW cm−2) | (a) 3% Fe: CsPbBr3 | 3 h | 105 | |
| CO: 3.2 μmol g−1 h−1 | ||||||
| CH4: 6.1 μmol g−1 h−1 | ||||||
| (b) Pristine CsPbBr3 | ||||||
| CO: 4.6 μmol g−1 h−1 | ||||||
| CH4: 1.9 μmol g−1 h−1 | ||||||
| Pt/CsPbBr3 | Solid–liquid (EA) | 150 W Xe-lamp with a 380 nm cut off filter | CO: 5.6 μmol g−1 h−1 | — | 139 | |
| Ni:CsPbBr2.77Ac0.23 | Solid–vapor (H2O vapor) | 300 W Xe-lamp (100 mW cm−2) | (a) Ni: CsPbBr2.77Ac0.23 | 18 h | 140 | |
| CO: 44.09 μmol g−1 h−1 | ||||||
| (b) Ni: CsPbBr3 | ||||||
| CO: 14.49 μmol g−1 h−1 | ||||||
|
||||||
| Heterojunction engineering | ||||||
| Schottky junction | CsPbBr3 QDs/graphene oxide (GO) | Solid–liquid (EA) | 100 W Xe-lamp with a AM 1.5 filter | CO: 58.7 μmol g−1 (12 h) | 12 h | 48 |
| CH4: 29.6 μmol g−1 (12 h) | ||||||
| H2: 1.58 μmol g−1 (12 h) | ||||||
| CsPbBr3 NC/Pd NS | Solid–vapor (H2O vapor) | 150 W Xe-lamp with a 420 nm filter (150 mW cm−2) | CO:12.63 μmol g−1 (3 h) | 9 h | 141 | |
| CH4: 10.41 μmol g−1 (3 h) | ||||||
| CsPbBr3 NCs/MXene nanosheets | Solid–liquid (EA) | 300 W Xe-lamp with a 420 nm filter | CO: 133.05 μmol g−1 (5 h) | 5 h | 142 | |
| CH4: 33.83 μmol g−1 (5 h) | ||||||
| Cs4PbBr6/rGO | Solid–liquid (EA/H2O) | 300 W Xe lamp with a 420 nm filter (100 mW cm−2) | CO: 11.4 μmol g−1 h−1 | 60 h | 102 | |
| CsPbBr3 NC/BZNW/MRGO | Solid–vapor (H2O vapor) | 150 W Xe-lamp AM 1.5 G with a 420 nm filter (150 mW cm−2) | R electron = 52.02 μmol g−1 h−1 | 12 h | 143 | |
| CsPbBr3 QDs/g-C3N4 | Solid–liquid (ACN/H2O) | 300 W Xe-lamp with a 420 nm cut-off filter | CO: 149 μmol g−1 h−1 | — | 51 | |
| CsPbBr3/g-C3N4 containing TiO species | Solid–liquid (EA/H2O) | Xe-lamp with a 400 nm cut off filter (100 mW cm−2) | CO: 129 μmol g−1 (10 h) | — | 144 | |
| Cs2AgBiBr6/MXene | Solid–vapor (H2O vapor) | Xenon lamp (400 nm) | CO: 11 μmol g−1 h−1 | — | 145 | |
| CH4: 1 μmol g−1 h−1 | ||||||
| H2: 9 μmol g−1 h−1 | ||||||
| CsPbBr3-Re(CO)3Br(dcbpy) | Solid–liquid (toluene/IPA) | AM 1.5 G with a 420 nm filter (150 mW cm−2) | R electron = 73.34 μmol g−1 h−1 | 15 h | 103 | |
| Cs2AgBiBr6-Cu-RGO | Solid–vapor (H2O vapor) | 1 Sun | CO: 1.9 μmol g−1 h−1 | — | 121 | |
| CH4: 10.7 μmol g−1 h−1 | ||||||
| Type II heterojunction | Cs2SnI6 NCs/SnS2 nanosheet | Solid–vapor (H2O/MeOH) | Xe-lamp with a 400 nm filter (150 mW cm−2) | CH4: 6.09 μmol g−1 (10 h) | 9 h | 123 |
| CsPbBr3 NCs/MoS2 NS | Solid–liquid (EA/H2O) | 300 W Xe lamp with a 420 nm filter (200 mW cm−2) | CO: 25.0 μmol g−1 h−1 (3 h) | 30 h | 146 | |
| CH4: 12.8 μmol g−1 h−1 (3 h) | ||||||
| CsPbBr3 QDs/Cu-TCPP-20 | Solid–liquid (ACN) | 300 W Xe-lamp with a 420 nm filter | CO: 287.08 μmol g−1 (4 h) | 16 h | 109 | |
| CH4: 3.25 μmol g−1 (4 h) | ||||||
| CsPbBr3 QDs-PCN | Solid–liquid (ACN/H2O) | 300 W Xe lamp with a 420 nm filter | CO: 148.9 μmol g−1 h−1 | 6 h | 147 | |
| All-solid-state Z-scheme heterojunction | CsPbBr3 NCs/USGO/α-Fe2O3 | Solid–liquid (ACN/H2O) | 300 W Xe-lamp with a 420 nm filter (100 mW cm−2) | CO: 73.8 μmol g−1 h−1 | 16 h | 111 |
| α-Fe2O3/amine-RGO/CsPbBr3 NCs | Solid–vapor (H2O vapor) | AM 1.5 G with a 420 nm filter (150 mW cm−2) | CO: 35.47 μmol g−1 (15 h) | 40 h | 116 | |
| CH4: 141.81 μmol g−1 (15 h) | ||||||
| H2: 4.36 μmol g−1 (15 h) | ||||||
| Direct Z-scheme heterojunction (S-scheme) | CsPbBr3 QDs/Bi2WO6 NS | Solid–liquid (EA/H2O) | 300 W Xe-lamp with a 420 nm filter (100 mW cm−2) | CO + CH4: 503 μmol g−1 (10 h) | 10 h | 148 |
| FAPbBr3/Bi2WO6 | Solid–liquid (trifluorotoluene) | 150 W Xe-lamp with a AM 1.5 G filter (100 mW cm−2) | CO: 170 μmol g−1 h−1 | 20 h | 112 | |
| TiO2/CsPbBr3 | Solid–liquid (ACN/H2O) | 300 W Xe-arc lamp | CO: 9.02 μmol g−1 h−1 | — | 149 | |
| Cs2AgBiBr6@g-C3N4 | Solid–liquid (EA/MeOH) | Xe-lamp (80 mW cm−2) | CO + CH4: 2 μmol g−1 h−1 | — | 101 | |
| Cs2AgBiBr6−xGCN | Solid–liquid (IPA) | 250 W mercury vapor lamp with a wavelength range of 285–700 nm | CO: 12.14 μmol g−1 h−1 | 18 h | 150 | |
| CH4: 8.85 μmol g−1 h−1 | ||||||
| Cs3Bi2I9/Bi2WO6 | Solid–vapor (H2O vapor) | 300 W Xe lamp with a 400 nm filter (100 mW cm−2) | CO: 66 μmol g−1 (9h) | 18 h | 151 | |
| Cs2AgBiBr6/Bi2WO6 | Solid–liquid (EA/IPA) | 300 W Xe lamp with an AM 1.5 G filter (100 mW cm−2) | CO: 42.19 μmol g−1 h−1 | — | 110 | |
| CH4: 0.41 μmol g−1 h−1 | ||||||
| Cs3Bi2I9/CeO2 | Solid–vapor (H2O vapor) | 300 W Xe lamp | CO: 170 μmol g−1 | — | 152 | |
| CH4: 65 μmol g−1 | ||||||
| CsPbBr3/Bi3O4Br | Solid–liquid (H2O) | 300 W Xe lamp with a 420 nm filter | CO: 387.57 μmol g−1 (4h) | 20 h | 153 | |
| CH4: 8.53 μmol g−1 (4h) | ||||||
| MF/WO/CsPbBr3 | Solid–vapor (H2O vapor) | 300 W Xe-lamp | CO: 514.06 μmol g−1 h−1 | 64 h | 154 | |
| CH4: 86.56 μmol g−1 h−1 | ||||||
| CsPbBr3@MTB | Solid–liquid (EA/H2O) | 300 W Xe-lamp AM 1.5 filter | CO: 145.28 μmol g−1 h−1 | 16 h | 155 | |
|
||||||
| Encapsulation engineering | ||||||
| Metal oxide | Amorphous-TiO2/CsPbBr3 NCs | Solid–liquid (EA/IPN) | 150 W Xe-lamp with an AM 1.5 G filter | CO: 11.71 μmol g−1 | 3 h | 114 |
| CH4: 20.15 μmol g−1 | ||||||
| H2: 4.38 μmol g−1 | ||||||
| Cs3Bi2Br9 and Cs2AgBiBr6/mesoporous TiO2 | Solid–liquid (IPA) | 300 W Xe-lamp (70 mW cm−2) | CH4: 32.9 and 24.2 μmol g−1 h−1 | — | 118 | |
| Nonmetallic materials | CsPbBr3 @GDY0.3-Co | Solid–liquid (ACN/H2O) | 300 W Xe lamp with a 400 nm filter (100 mW cm−2) | CO: 27.7 μmol g−1 h−1 | — | 156 |
| C60/CsPbBr3 | Solid–liquid (ACN/H2O) | 300 W Xe lamp with a 420 nm filter (150 mW cm−2) | CO: 71.3 μmol g−1 | — | 157 | |
| CH4: 27.3 μmol g−1 | ||||||
| P3HT/CsPbBr3 | Solid–liquid (ACN/H2O) | 300 W Xe lamp with a 420 nm cutoff filter | CO: 145.45 μmol g−1 h−1 | — | 158 | |
| CH4: 23.05 μmol g−1 h−1 | ||||||
| Cs3Bi2Br9 in MCM-41 | Solid–liquid (H2O) | 300 W Xe-lamp with a 420 nm cut-off filter (350 mW cm−2) | CO: 17.24 μmol g−1 h−1 | — | 159 | |
| H2: 0.527 μmol g−1 h−1 | ||||||
| CsPbIxBr3−x/polyethersulfone (PES) | Solid–vapor (H2O vapor) | AM 1.5 G 1 sun condition solar simulator | (a) CsPbBr3 QDs/PES | 2 h | 160 | |
| CO: 27.22 μmol g−1 h−1 | ||||||
| (b) CsPbI2.6Br0.39 QDs/PES | ||||||
| CO: 32.45 μmol g−1 h−1 | ||||||
| MF/CsPbBr3 | Solid–vapor (H2O vapor) | 300 W Xe-lamp AM 1.5 G | CO: 29.13 μmol g−1 h−1 | 104 h | 161 | |
| CH4: 12.95 μmol g−1 h−1 | ||||||
| MF/CsPbBr3/g-C3N4 | Solid–vapor (H2O vapor) | 300 W Xe-lamp (without a filter) | CO: 872.22 μmol g−1 h−1 | 76 h | 162 | |
| CH4: 103.35 μmol g−1 h−1 | ||||||
| Metal organic frameworks | CsPbBr3 @ZIF-67 CsPbBr3 @ZIF-8 | Solid–vapor (H2O vapor) | 100 W Xe-lamp with a AM 1.5 G filter (150 mW cm−2) | (a) CsPbBr3 @ZIF-67 | 18 h | 117 |
| CH4: 10.537 μmol g−1 (3h) | ||||||
| CO: 2.301 μmol g−1 (3h) | ||||||
| (b) CsPbBr3 @ZIF-8 | ||||||
| CH4: 5.434 μmol g−1 (3h) | ||||||
| CO: 1.515 μmol g−1 (3h) | ||||||
| CsPbBr3 QDs/UiO-66(NH2) | Solid–liquid (EA/H2O) | 300 W Xe-lamp with a 420 nm filter | CO: 98.57 μmol g−1 (12 h) | 36 h | 108 | |
| CH4: 3.08 μmol g−1 (12 h) | ||||||
| Cs2AgBiBr6/Ce-UiO-66-H | Solid–liquid (H2O) | 300 W Xe lamp | CO: 309.01 μmol g−1 h−1 | 10 h | 163 | |
| Cs3Bi2Br9/MOF 525 Co | Solid–vapor (H2O vapor) | 300 W Xe lamp with a AM 1.5 G filter (100 mW cm−2) | CO: 61.2 μmol g−1 h−1 | 20 h | 164 | |
| CH4: 0.3 μmol g−1 h−1 | ||||||
| MAPbI3@PCN221(Fex) | Solid–liquid (EA/H2O) | 300 W Xe lamp with a 400 nm filter (100 mW cm−2) | CO: 530.06 μmol g−1 (80 h) | 80 h | 115 | |
| CH4: 1028.94 μmol g−1 (80 h) | ||||||
3.1 Surface engineering
Extensive research has been reported to modify the surface structure or properties of NHP photocatalysts to improve their photocatalytic activity, which can further be divided into 5 categories: (1) size control, (2) morphology control, (3) facet regulation, (4) ligand regulation, and (5) defect regulation. Different particle sizes and morphologies often result in different surface areas and charge transfer efficiencies of photocatalysts.128 Particle size often affects the clustering, surface area/sites, and charge diffusion pathways of NHP NCs, leading to variations in catalytic activity. Hou's team prepared colloidal CsPbBr3 QDs of different sizes from 3.8 nm to 11.6 nm by adjusting the temperature of the solution phase during synthesis (Fig. 7a).124 Their study showed that the increase in CsPbBr3 QD size is efficient in narrowing the band gap and increasing the utilization of the solar spectrum, where the effect mainly stems from the intrinsic quantum size effect that effectively modulates the band gap and the light absorption of CsPbBr3 NCs in the visible spectral region. Along with NHP QD size, changing the shape of nanoparticles can also enhance mass transfer at the photocatalytic interface and promote CO2 adsorption.166 Wu et al.125 successfully synthesized two-dimensional CsPbBr3 (CPB) nanosheets with different thicknesses (2, 3, 4, and 4.6 nm) and used them for the first time as photocatalysts for CO2 reduction with H2O as the electron donor (Fig. 7b). The nanosheets exhibited significantly higher activity and longer stability in photocatalytic CO2 reduction compared to CsPbBr3 NCs. Besides, Pradhan's group found that the adsorption capacity and product release rate depend on the helical nature of the nanorods (Fig. 7c).128 They controlled the helicity of CsPbBr3 nanorods by manipulating the composition of alkylammonium ions in the reaction system and found that the evolution of CH4 depends on the depth of the helical nanorods. | ||
| Fig. 7 (a) Quantum-size effects in UV/vis absorption spectra and band gap structures of CsPbBr3 QDs with different particle sizes. Reproduced from ref. 124 with permission from Wiley-VCH, Copyright (2017). (b) Schematic illustration of band structures of CsPbBr3 NCs and CsPbBr3 nanosheets derived from the results of the UV-vis absorption spectroscopy and flat-band potential measurements. Reproduced from ref. 125 with permission from Wiley-VCH, Copyright (2021). (c) Comparison of photocatalytic CO2 reduction activity of four CsPbBr3 nanorods. Reprinted with permission from ref. 128, Copyright 2021, American Chemical Society. (d) Histograms showing the formation of CO and CH4 from CO2 reduction reactions after 4 h using noncubes or polyhedra, hexapods, or armed and cube-shaped CsPbBr3 nanostructures as photocatalysts. Reprinted with permission from ref. 165, Copyright 2020, American Chemical Society. (e) Schematic illustration of the regulation of the surface ligand density of the CAIC QDs@Ag-2 composite. Reproduced from ref. 113 with permission from Royal Society of Chemistry, Copyright (2021). (f) Schematic illustration for the surface modification of the CsPbBr3 NCs. Reproduced from ref. 132 with permission from Wiley-VCH, Copyright (2021). | ||
Additionally, the crystalline facets of NHPs influence their photocatalytic performance.167 Different charge separation and transport efficiencies can be induced by the crystal's internal electric field (IEF) along different crystal orientations.168 Photogenerated electrons and holes often cluster in small distinct planes, where those perpendicular to the IEF usually exhibit higher activity in photocatalysis.169 Crystal facets with lower activation potentials and higher adsorption energies are more advantageous for the photocatalytic reduction of CO2.170 For example, Shyamal et al.165 synthesized CsPbBr3 NCs in different crystal shapes, such as conventional six-sided cubic and unique polyhedral non-cubic shapes and six-armed hexapod-shaped CsPbBr3 nanocrystals (Fig. 7d). Assuming comparable surface areas, they claimed that compared to halide rich cubic CsPbBr3 NCs, halide deficient polyhedral shaped CsPbBr3 for CO2 reduction shows better catalytic activity due to the presence of many surface defects, which form lead-rich reaction sites. Moreover, these surface trap states prevent quick charge carrier recombination and offer a chance for effective charge transfer to trigger the photocatalytic reaction.
Organic ligands can also control the shape and size of NCs because they can passivate surface dangling bonds and inhibit particle agglomeration, thus enhancing the stable crystal structure of NHP structures. However, they can also hinder charge transfer between the catalyst and reactants as well as inhibit CO2 uptake at the active site.171 Therefore, an optimal surface ligand density is essential for high performance and good stability. Chen et al.113 controlled the ligand density on Cs2AgInCl6 QDs@Ag-2 (CAIC QDs@Ag-2) by adjusting the volume ratio of hexane/EC washing solvents (1:1, 1:2, 1:3, and1:4) in the purification process, as shown in Fig. 7e. Their results show that the PL intensity of CAIC QDs@Ag-2 composites purified by process II was the lowest compared to the other three purification methods, which implies that reducing the amount of ligands on the catalyst surface is beneficial for charge transfer, but too few ligands will lead to agglomeration of the catalyst and thus reduce the effective separation of the carriers.
In general, the synthesized NHPs inevitably have surface defects, such as the lack of X-site halogen anions or B-site metal cations. These defects usually become charge recombination centers and hinder the effective separation of photogenerated carriers, thus reducing the catalytic performance of photocatalysts. Therefore, it is demanded for researchers to modify the surface defects of photocatalysts. For instance, Wang et al.132 modified the surface defects of CsPbBr3 NCs, i.e., uncoordinated Pb atoms and nanoparticles, by a two-step electrostatic self-assembly method (Fig. 7f). They used NH4BF4 salt as a defect treatment agent to peel off the defective layer on the surface of CsPbBr3. Their experimental results showed that the surface-modified samples (CPB–BF4) exhibited a more than 3 times higher CO2 reduction rate compared with the pristine CsPbBr3 samples, which are 29.8 μmol g−1 h−1 and 8 μmol g−1 h−1, respectively.
In conclusion, the photocatalytic performance of NHPs can be significantly influenced by adjusting their size, morphology, crystal facet orientation, surface ligand density, and defect states. First, tuning the nanoparticle size can substantially modify the quantum confinement effect, thereby regulating the bandgap and light absorption characteristics. Different nanostructures, such as two-dimensional nanosheets and one-dimensional nanorods, can alter the mass transfer process of reactants and the migration channels of photogenerated charge carriers. Additionally, rationally designing the structural morphology can help optimize the entire photocatalytic process. Different crystal facets have distinct internal electric field distributions, which can influence the separation and transfer of photogenerated electron–hole pairs. Meanwhile, appropriate surface ligands can passivate defect sites and suppress charge carrier recombination, but excessive ligands may hinder reactant adsorption and charge transfer. Therefore, the ligand density needs to be controlled within an optimal range. The common surface defects of perovskite nanoparticles, such as halide vacancies and metal ion vacancies, often become centers for charge carrier recombination. Through chemical modification methods, the surface defects can be effectively reduced, enhancing the separation efficiency of photogenerated charge carriers. By comprehensively applying these methods, the performance of perovskite photocatalysts can be significantly enhanced, providing important technical support for realizing efficient photochemical CO2 reduction conversion.
3.2 Component engineering
In addition to the modifications on the NHP surface, changing components of NHPs is another effective approach to improve the photocatalytic performance of NHPs. Specifically, the compositional adjustment mainly includes the changing of organic/inorganic cations and halogen anions, and the doping of metal ions. For example, Bhosale et al.106 synthesized three bismuth-based NHPs with the chemical formulae of A3Bi2I9, where A-cations are Rb+, Cs+ and MA+ (Fig. 8a). In general, the photocatalytic activity of these three photocatalysts for the reduction of CO2 shows a trend Cs3Bi2I9 > Rb3Bi2I9 > MA3Bi2I9. Among them, Cs3Bi2I9 and Rb3Bi2I9 have the greatest yield of CO and CH4, 77.6 and 17.0 μmol g−1, respectively. They concluded that the differences in photocatalytic performance are due to changes in charge transfer properties and catalytic pathways resulting from the use of different cations. Apart from the cation at the A-site, the change in the cation at the B-site also affects the photocatalytic performance of NHPs for CO2. As Fig. 8b shows, with the change of the B-site element from Hf to Zr and finally Te, the CO yield of Cs2BCl6 is enhanced obviously. Fig. 8c illustrates that compared to Pb when the B-site is Cu, the NHP has a stronger adsorption of CO2 molecules due to the synergistic effect with other atoms (Cs and Br). Beyond these, X-site halide replacement can modify the band gap and band edge positions, influencing photon absorption and the quantity of photogenerated carriers, thus significantly affecting the catalytic behavior. Guo et al.135 reported low-cost cubic-phase CsPb(Br0.5/Cl0.5)3 perovskites with different Br and Cl ratios and investigated the effect on photocatalytic CO2 reduction (Fig. 8c). They found that the mixed halide perovskites observed a gradual shift and rapid increase of pristine CsPbBr3 from 517 nm to 413 nm with increasing amount of Cl in the emission spectra, which was attributed to the improved electron separation. With increasing Cl concentration, the formation of CO and CH4 increases. Wu et al.125 changed the I content in NHP 2D nanosheets and synthesized CsPbBr3−xIx (x = 0.3, 0.6, 0.9) (Fig. 8d). The synthesized CsPbBr3−xIx nanosheets exhibited reduced band gaps and red-shifted absorption peaks from 452 nm to 466 nm with increasing I concentration. In comparison to pristine CsPbBr3, all CsPbBr3−xIx nanosheets displayed greater photocatalytic CO2 reduction activity. Additionally, the CsPbBr3−xIx nanosheet surface trapping was boosted by the anion exchange reaction, which decreased the recombination of photogenerated carriers. Hence, this facilitated the CO2 reduction reaction, and enhanced the separation and transfer efficiency of the change carriers. | ||
| Fig. 8 (a) Comparison of production of methane and carbon monoxide during the photochemical reaction for 10 h. Reprinted with permission from ref. 106, Copyright 2019, American Chemical Society. (b) Performance of the photocatalytic CO2 reduction of the as prepared Cs2HfCl6, Cs2ZrCl6 and Cs2TeCl6 microcrystals. Reproduced from ref. 134 with permission from Elsevier, Copyright (2022). (c) Yields of CO2 reduction using CsPb(Brx/Cl1−x)3 (x = 1, 0.7, 0.5, 0.3, 0) nanocrystals as catalysts under 9 h Xe lamp irradiation with an AM 1.5 filter. Reproduced from ref. 135 with permission from Elsevier, Copyright (2018). (d) The schematic illustration of the band structures of CsPbBr3−xIx nanosheets derived from the results of flat-band potential and UV-vis absorption spectroscopy measurements. Reproduced from ref. 125 with permission from Wiley-VCH, Copyright (2021). (e) CO and CH4 evolution rate in EA/IPA over CsPbBr3, a physical mixture of CsPbBr3, and Co2+ cations (CPB/Co), CPB-BF4, and CPB-BF4/Co, respectively. Reproduced from ref. 132 with permission from Wiley-VCH, Copyright (2021). (f) Schematic presentation of Fe-doped CsPbBr3 and undoped CsPbBr3 nanocrystal photocatalysts. Reprinted with permission from ref. 105, Copyright 2019, American Chemical Society. (g) Schematic illustration of the electron spin polarization induced longer photoexcited carrier lifetime under an external magnetic field in Mn-CsPbBr3 NPLs. Reprinted with permission from ref. 172, Copyright 2022, American Chemical Society. | ||
Doping metals can also be used to enhance the charge separation in NHPs by increasing the catalytically active sites and facilitating the adsorption and desorption of intermediates, thus improving the photocatalytic performance of NHPs.173 By using density functional theory (DFT) computations, Tang et al.174 modelled the photocatalytic CO2 RR of CsPbBr3 doped with various metal components, including Co, Fe, Ni, Cu, Ag, Mg, Mn, and Bi in benzene. This study shows that the doping of Co and Fe atoms significantly increased the catalytic activity of CsPbBr3. This conclusion was later confirmed by a couple of teams in their experiments. For example, Wang et al.132 synthesized CsPbBr3-BF4/Co with optimal Co loading by the electrostatic self-assembly methodology. They showed that the photocatalytic activity in EA/isopropanol (IPA) under 100 mW cm−2 light was 83.8 μmol g−1 h−1 at 2.0 μmol (Fig. 8e). Not only did the doped Co metal change the selectivity of the CO2RR product, but it also reduced the formation of photogenerated electron–hole pair complexes, increased photoresponsiveness, and improved the stability of NHPs. According to the report by Shyamal et al.,105 doping Fe(II) accelerates the CsPbBr3 QD photocatalytic reduction of CO2 and modifies the selectivity of the reaction products (Fig. 8f). Besides these, another study by Lin et al. showed that they successfully manipulated the spin-polarized electrons in CsPbBr3 perovskite nanoplates by doping magnetic Mn2+ ions and applying an external magnetic field, which significantly improved the conversion efficiency of photocatalytic CO2 reduction. Specifically, Mn doping increased the number of spin-polarized photoexcited carriers, extended the carrier lifetime, and suppressed carrier recombination, thereby substantially enhancing the photocatalytic performance of Mn-CsPbBr3 nanoplates.172 However, these studies usually use in situ doping techniques to directly inject metal ions inside NHPs, and these metal ions can also serve as carrier recombination sites, thus inhibiting the carrier diffusion. Therefore, post-synthesis cation exchange is being studied as a potential solution to solve this problem by attaching active metal ions to the surface of NHP nanocrystals.175
In summary, modifying the compositional components of NHPs is another effective approach for improving their photocatalytic performance. Substituting different A-site cations or B-site cations can significantly influence the charge transfer characteristics, catalytic pathways, and CO2 adsorption of NHPs, thereby affecting their photocatalytic performance. Replacing the X-site halogens (such as Br, Cl, and I) can modify the bandgap and band-edge positions, impacting light absorption, photogenerated carrier density, and catalytic behavior. Doping with metal dopants (such as Co, Fe, and Mn) can increase the catalytically active sites, promote the adsorption/desorption of intermediates, and enhance carrier separation, thereby improving the photocatalytic activity and selectivity of CO2 reduction products. By adjusting the composition of organic/inorganic cations and halide anions, as well as appropriately adding metal dopants, the charge transfer characteristics, light absorption, and charge carrier separation properties of NHPs can be effectively tuned, thereby significantly enhancing the photocatalytic CO2 reduction activity and selectivity of NHPs, providing a valuable strategy for the development of high efficiency photocatalysts.
3.3 Heterojunction engineering
Numerous studies have demonstrated that one of the major obstacles for improving the photocatalytic performance of NHP materials is the photogenerated carrier spatial separation efficiency. To address the problem of severe carrier recombination leading to low photocatalytic activity, scientists have attempted to integrate pristine NHPs with various functional materials by constructing heterojunctions, such as Schottky junctions, type-II heterojunctions and direct/indirect Z-scheme heterojunctions.177Schottky junctions are usually formed by combining a semiconductor (e.g., NHP NCs) and a conductor (metal, graphene, etc.). The free electrons generated by the NHPs upon photoexcitation are transferred to the conductor, thus suppressing the recombination of electrons and holes. The energy band bending effect at the interface of the two materials leads to the formation of a Schottky barrier, which helps prevent electron migration from the coupled conductor back to the NHP NCs. In 2017, Xu et al.48 prepared CsPbBr3/GO Schottky junctions for photocatalytic CO2 reduction by room temperature antisolvent precipitation (Fig. 9a). The photogenerated electrons in CsPbBr3 can be easily transported to GO because CsPbBr3 has a negative CB edge compared to the Fermi energy level of GO. The electron consumption rate (Relectron) increased by 25.5% for the CsPbBr3/GO composite compared to that of pristine CsPbBr3 NCs in ethyl acetate (EA) solution. Later, Xu's group141 successfully developed another Schottky junction composite by attaching CsPbBr3 NCs to the 2D Pd nanosheet (NS), which can rapidly transfer photogenerated electrons from CsPbBr3 NCs to the Pd NS (Fig. 9b). The Pd NS also offers a number of active sites for CO2 photocatalytic reduction. Thus, the CsPbBr3 NCs/Pd nanosheet composites achieve a high electron consumption rate of 33.79 μmol g−1h−1, which is 2.43 times higher than that of pure CsPbBr3 NCs (9.86 μmol g−1h−1). In addition to these, metal complexes (CsPbBr3 NCs-Ni(tpy))82 and MXenes142 are also regarded as promising candidates for decorating NHPs due to their suitable catalytically active sites and favorable electron receiving capability (Fig. 9c and d).
 | ||
| Fig. 9 (a) Schematic diagram of CO2 photoreduction over the CsPbBr3 QD/GO photocatalyst. Reprinted with permission from ref. 48, Copyright 2017, American Chemical Society. (b) Schematic diagram of CO2 photoreduction over the CsPbBr3 NCs/Pd nanosheet. Reprinted with permission from ref. 141, Copyright 2018, American Chemical Society. (c) Schematic diagram of CO2 photoreduction over CsPbBr3 NCs/MXene. Reprinted with permission from ref. 142, Copyright 2019, American Chemical Society. (d) Schematic diagram of CO2 photoreduction over the CsPbBr3 NCs-Ni(tpy) photocatalyst. Reprinted with permission from ref. 82, Copyright 2020, American Chemical Society. (e) Band structure of the 20 CPB–PCN composite photocatalyst. Reproduced from ref. 51 with permission from Wiley-VCH, Copyright (2018). (f) Schematic illustration of the possible mechanism of photocatalytic CO2 reduction using CsPbBr3 QDs/UiO-66(NH2). Reproduced from ref. 176 with permission from Elsevier, Copyright (2018). (g) Schematic diagram of CO2 photoreduction over the α-Fe2O3/amine-RGO/CsPbBr3 photocatalyst. Reproduced from ref. 116 with permission from Elsevier, Copyright (2020). (h) Schematic illustrations for the CsPbBr3/USGO/α-Fe2O3 photocatalyst. Reproduced from ref. 111 with permission from Wiley-VCH, Copyright (2020). (i) Schematic illustration of perovskite-based heterostructures via type-II to Z-scheme transformation for photocatalytic CO2 reduction through an ohmic contact. Reprinted with permission from ref. 39, Copyright 2024, American Chemical Society. (j) Z-scheme photocatalytic system for CO2 reduction. Reprinted with permission from ref. 148, Copyright 2020, American Chemical Society. (k) Schematic illustration of the TiO2/CsPbBr3 heterojunction. Reproduced from ref. 149 with permission from Springer Nature, Copyright (2020). | ||
Type II heterojunctions are generally considered to exhibit higher photon utilization and catalytic performance compared to Schottky junctions because both NHPs and coupled semiconductors are capable of generating photoexcited carriers. Moreover, the energy bands of the two semiconductors forming the type II heterojunction are interleaved. The photogenerated electrons from the semiconductor with a higher CB will migrate and accumulate on the semiconductor with a lower CB; in contrast, the photogenerated holes will move to the semiconductor with a higher VB. Thus, the oxidation and reduction reactions of the type II heterojunction occur separately on both semiconductors, thus improving the utilization of electrons and holes. Currently, coupled semiconductors with suitable band edge positions (e.g., g-C3N4 (PCN), metal–organic frameworks (MOFs), oxides, and sulfides) have been used to construct type II heterojunctions. For example, Ou et al.51 immobilized CsPbBr3 quantum dots (QDs) onto NHx-rich porous PCN via strong interactions between Br atoms on the surface of CsPbBr3 and N atoms on PCN (Fig. 9e). The separation of broad-generated charges between CsPbBr3 and PCN was facilitated by the formation of type-II heterojunctions, in which photogenerated electrons were transported from CsPbBr3 QDs to PCN for CO2 reduction, while holes are transferred from PCN to CsPbBr3 QDs for H2O oxidation via valence band shifts. Thus, the composite catalyst after forming heterojunctions has a 3-fold higher CO generation rate than that of CsPbBr3 QDs alone. In addition, metal–organic frameworks (MOFs) are a group of crystalline porous materials made up of metal nodes and organic linkers, which are characterized by large surface area and structural tunability.178 By directly combining pre-synthesized MOFs UiO-66(NH2) and CsPbBr3 QDs, Wan et al.176 created composites and obtained high CO2 conversion (CO generation of 98.57 mol g−1) in the EA/water system, which was significantly higher than that of pristine CsPbBr3 QDs and UiO-66(NH2) (Fig. 9f). The superior CO2 reduction photoactivity is attributable to the wide reachable surface area, the better visible light collection capacity, and the improved electron extraction and transfer between two materials owing to the development of type II heterojunctions.
Although it has been demonstrated that type II heterojunctions can effectively encourage space charge separation and subsequently enhance the catalytic performance of CO2 reduction, this improvement comes at the expense of weakening the reduction/oxidation of photogenerated electrons/holes, which is detrimental to the performance of the CO2RR. Therefore, a novel heterojunction structure, Z-scheme heterojunction, has been heavily investigated. Similar to type-II heterojunctions, Z-scheme heterojunctions are usually composed of two semiconductors with interleaved energy bands. However, the difference is that the photogenerated electrons in the semiconductor with a lower CB in the Z-scheme heterojunction combine with the photogenerated holes in the semiconductor with a higher VB. Therefore, the photogenerated electrons left in the semiconductor with a higher CB will reduce CO2 while the holes left in the semiconductor with a lower VB will participate in the oxidation reaction, resulting in a strong redox capability. According to a combination method of photogenerated electrons and holes, Z-scheme heterojunctions are further divided into all-solid-state Z-scheme heterojunctions and direct Z-scheme heterojunctions (S-scheme). An all-solid-state Z-type heterojunction consists of two semiconductors and a conductor that acts as an electron mediator. For effective photocatalytic CO2 reduction, Jiang et al.116 originally presented an all-solid-state Z-scheme composite based on NHPs, α-Fe2O3/amine-rGO/CsPbBr3, where amine-rGO operates as the electron mediator (Fig. 9g). Charge transfer from α-Fe2O3 to CsPbBr3 QD is facilitated by the matching energy band structure, which also lessens the interfacial recombination of photogenerated carriers. With a yield of 181.68 mol g−1 and a selectivity of 93.4%, CH4 was identified as the primary reduction product after 15 hours of nonstop illumination. Within 5 photocatalytic reaction cycles, this Z-composite also demonstrated remarkable stability and nearly consistent photocatalytic performance. Another all-solid-state Z scheme was subsequently introduced by Mu et al.111 CsPbBr3 NCs, rod-like α-Fe2O3, and ultrathin small-sized graphene oxide (USGO) nanosheets were used to construct Z-scheme heterojunctions (Fig. 9h). When combined with α-Fe2O3 and CsPbBr3, USGO functions as an electronic mediator by creating C–O–Fe and Br–O–C bonds, respectively. According to the results, the CO yield of CsPbBr3/USGO/α-Fe2O3 is 19 times higher compared to that of pure CsPbBr3, at 73.8 mol g−1 h−1. Recently, Song et al. explored the transformation from type-II into Z-scheme heterostructures based on perovskite materials to enhance photocatalytic CO2 reduction as shown in Fig. 9i.39 The research showed that by incorporating Au as an electron transport mediator in the CsPbBr3/TiO2 heterostructure, a low-resistance ohmic contact can be formed, thereby transforming the type-II structure into a Z-scheme heterojunction. Compared to pristine CsPbBr3 and CsPbBr3/TiO2, the CsPbBr3/Au/TiO2 Z-scheme heterostructure exhibited a 5.4-fold and 3.0-fold enhancement, respectively, in the electron consumption rate for photocatalytic CO2 reduction.
On the other hand, the presence of electron mediators in an all-solid-state Z-scheme heterojunction may hinder the efficient absorption of light by semiconductor materials. Therefore, a direct Z-scheme heterojunction also called an S-scheme heterojunction without an electron mediator is proposed. It consists of two semiconductors in which one photocatalyst should have both a higher CB position and Fermi energy level than the other one.179 Therefore, at the contact interface of the two semiconductors, the alignment of the Fermi energy levels leads to the formation of an internal electric field (IEF), which not only offers an additional driving force to combine the photogenerated electrons and holes at low CB and VB levels but also prevents the recombination of the carriers at high redox potentials. Wang et al.148 fabricated S-scheme CsPbBr3 QD/Bi2WO6 nanosheet (CPB/BWO) photocatalysts for CO2 reduction (Fig. 9j). The close contact between two semiconductors promotes charge separation and transfer, as the photogenerated electrons on the CB of Bi2WO6 can be transferred and recombined with the photogenerated holes on the VB of CsPbBr3, thus maintaining spatially separated reduced electrons and oxidized holes on the CsPbBr3 QD and Bi2WO6 NS sides, respectively. Compared with pristine CsPbBr3, the CO yield was increased by a factor of 9.5 for a mass ratio of 1:5 of the CsPbBr3:Bi2WO6 heterostructure. Besides, the TiO2/CsPbBr3 S-scheme heterojunction was created by Xu et al. for CO2 photoreduction (Fig. 9k).149 Due to the tight contact between TiO2 and CsPbBr3 in heterojunctions, an IEF pointing from CsPbBr3 to TiO2 is created. The efficiency of photocatalytic CO2 reduction can be increased by preventing charge recombination and the inverse reaction in TiO2/CsPbBr3 heterojunctions using band bending in NHP heterojunctions and related IEFs. Recently, some novel heterojunctions have been fabricated for CO2 photoreduction, such as Cs2AgBiBr6/bismuthine, CsPbBr3@Pd, 3D/2D NiTiO3/Cs3Sb2I9, Cs3Bi2Br9/BiVO4, 0D/2D Cs3Bi2Br9/Bi2WO6, etc.180–184
Overall, the main challenge of NHP photocatalysts is the serious recombination of photogenerated charge carriers, which limits their photocatalytic activity. To solve this problem, researchers have enhanced the separation and transfer of photogenerated charge carriers by constructing heterojunction structures of NHP with conductors, semiconductors, and highly porous materials, such as MOFs. Among them, Schottky junctions and type-II heterojunctions can effectively separate electron–hole pairs and improve the photocatalytic performance of CO2 reduction. However, this enhancement is at the cost of sacrificing reduction/oxidation capability. To maintain strong redox ability, Z-scheme heterojunctions have attracted widespread attention. Both all-solid-state Z-scheme heterojunctions and direct Z-scheme (S-scheme) heterojunctions can better separate and utilize photogenerated charge carriers through optimized band structures and interfacial electric fields. Therefore, Z-scheme heterojunctions are more promising compared to other types for the enhancement of the photocatalytic efficiency of CO2 reduction. The experimental findings do not specify which NHPs are suitable for which type of heterojunction. The application scenarios of different types of heterojunctions always depend on reaction redox potentials that need to be satisfied, the band positions of selected co-catalysts (metals, carbon materials, halide perovskites, etc.), and on the matching of the lattice and Fermi energy levels between the two catalyst materials. Furthermore, when designing MHP heterojunctions, other key factors should also be considered, including interface properties, energy levels, lattice parameters, and the interactions between MHP and its counterparts. In situ growth and atomic sharing strategies can greatly eliminate interface defects to form atomic-level intimate contact interfaces, thereby improving the charge transfer efficiency between the two materials.20
3.4 Encapsulation engineering
Due to their ionic structure, most NHPs have serious stability problems under moisture, oxygen, light and at high temperature, which is not favorable for their practical applications in CO2 photocatalysis.79 Therefore, finding approaches to enhance the stability of NHPs is the main direction for further development. Encapsulates are beneficial for the stability of NHPs by reducing the direct contact between the NHPs and the aqueous solution, and can also be regarded as electron/hole acceptors to promote charge carrier transfer. Currently, many kinds of stable functional materials can be used for NHP encapsulation, such as metal oxides, nonmetallic materials, and MOFs.186 However, considerable research is still needed to reasonably control the type and thickness of the encapsulation layer without significantly weakening the light trapping and reactivity of NHPs.Amorphous TiO2 has been considered as a good encapsulation material for the protection of NHPs, and it can also improve the efficiency of photogenerated charge separation to achieve excellent photocatalytic performance. Xu et al.114 reported novel amorphous TiO2-encapsulated CsPbBr3 NCs for photocatalytic CO2 reduction (Fig. 10a). This effect ultimately increases the photoelectron consumption from 25.7 to 193.3 μmol g−1. In addition, CO2 adsorption and activation are promoted. Based on these synergistic effects, the photoelectron consumption was improved by nearly 6.5 times through the photocatalytic CO2 reduction reaction. Nonmetallic materials have also been applied to encapsulate NHPs. Su et al.156 successfully coated thin graphitic diyne (GDY) in situ on CsPbBr3 NCs and then attached Co2+ on it (Fig. 10b). The coated GDY boosts the stability of CsPbBr3 NCs in an aqueous system and serves as a hole transport layer to speed up the transfer of photogenerated holes into CsPbBr3. Additionally, it offers a platform that is suitable for Co2+ working as catalytically active sites, enabling a photocatalytic activity of up to 27.7 mol g−1 h−1 for the reduction of CO2 to CO. Another attractive possibility for NHP encapsulation is highly conductive C60. In Zhang's work,157 the highly conductive C60 acts as an effective electron acceptor, enhancing the electron transfer process, the photocatalytic cycle stability, and solvent stability of the photocatalyst (Fig. 10c). For the end products, CO and CH4, the C60/CsPbBr3 composites achieved an electron consumption rate of 90.2 μmol g−1 h−1 in the CAN/H2O system. In addition to carbon materials, some organics have been used for the encapsulation of NHPs. Chen et al.161 combined CsPbX3 into a three-dimensional melamine foam (MF), which allowed the composite to exhibit long-term stability in water for 104 h and a CO2 reduction rate of up to 42.08 mol g−1h−1 (Fig. 10d). Moreover, Li et al.158 constructed poly(3-hexylthiophene-2,5-diyl) (P3HT)/CsPbBr3 composites using the P3HT conductive polymer as a protective layer and discovered that it promoted charge separation and improved the stability of CO2 conversion to CO with CH4 yields of 145.45 and 23.05 mol g−1h−1 (Fig. 10e).
 | ||
| Fig. 10 (a) Schematic illustration for the structure of CsPbBr3 encapsulated in amorphous-TiO2. Reproduced from ref. 114 with permission from Wiley-VCH, Copyright (2018). (b) Schematic illustrations for the structure of CsPbBr3 encapsulated in GDY-Co. Reprinted with permission from ref. 156, Copyright 2020, American Chemical Society. (c) Schematic illustration of the CO2 photoreduction process of the C60/CsPbBr3 composite. Reproduced from ref. 157 with permission from Elsevier, Copyright (2021). (d) Schematics of CsPbBr3 attached on MF. Reproduced from ref. 161 with permission from Wiley-VCH, Copyright (2021). (e) Schematic illustration of photoreduction of CO2 over the P3HT/CsPbBr3 composite. Reproduced from ref. 158 with permission from Elsevier, Copyright (2021). (f) Schematic illustrations for the structure of MAPbI3 QDs (large spheres) encapsulated in the pores of PCN-221(Fex). Reproduced from ref. 185 with permission from Wiley-VCH, Copyright (2019). (g) Schematic illustration of the CO2 photoreduction process of CsPbBr3/ZIFs. Reprinted with permission from ref. 117, Copyright 2020, American Chemical Society. | ||
Novel materials such as metal organic frameworks (MOFs) have also been used for NHP encapsulation. MOFs with unique structures and admirable chemical/physical properties have recently attracted much attention in the encapsulation of halide perovskites because of their unique properties such as tunable structure, high surface area, and flexibility. Wu et al.185 encapsulated MAPbI3 QDs in an Fe-based MOF (PCN-211(Fex)) and successfully used it for CO2 photoreduction (Fig. 10f). The tight contact between the MAPbI3 QDs and the Fe catalytic site of PCN-211(Fex) facilitated the transfer efficiency of photogenerated electrons from the NHP QDs to the catalytic sites. The best MAPbI3@PCN-221(Fe0.2) achieved an excellent total yield (CO and CH4) of 1559 μmol g−1 with a CH4 selectivity of 66%. PCN-221(Fex) also prevents MAPbI3 from being hydrolyzed, resulting in extremely high photocatalytic durability (over 80 h). Kong et al.117 reported an in situ growth strategy for the synthesis of zinc/cobalt-based zeolite imidazolyl ester skeletons (ZIF) encapsulated CsPbBr3 QDs (Fig. 10g). The Co2+ center in ZIF can be activated by accepting electrons under light, and further acts as an active center for CO2 activation and reduction. The synergistic effect of CsPbBr3 and ZIF coating not only promotes CO2 capture/activation capacity and charge separation efficiency, but also improves water stability and photocatalytic cycle stability. The ZIF-67 coated NHP with photoabsorption ability has an electron consumption rate of 29.630 μmol g−1 h−1, which is 2.66 times higher than that of the pure CsPbBr3 photocatalyst.
In brief, effective encapsulation of inorganic halide perovskites (NHPs) is a critical stabilization strategy. Various encapsulation materials, such as amorphous TiO2, graphitic diyne, C60, melamine foam, and metal–organic frameworks (MOFs), can not only protect the NHPs but also enhance charge separation efficiency and improve the photocatalytic CO2 reduction performance. The synergistic effects between these encapsulation materials and the NHPs in terms of gas adsorption, charge separation, catalytic activity, and stability are also of great importance. Additionally, these encapsulation materials should also have good electrical conductivity to ensure that the photogenerated charge carriers can reach the surface and react with the substrates. Meanwhile, the challenge of rationally designing the encapsulation layer is to stabilize the NHPs without significantly compromising their light absorption and reactivity, which is crucial for improving the NHPs' photocatalytic CO2 reduction capability.
4. Charge transport process and surface reaction mechanism
In addition to understanding how to improve the performance of halide perovskites on photocatalytic CO2 reduction, it is equally important to explore the separation and charge transfer of photogenerated carriers as a prerequisite to promote the efficiency of the final catalytic reaction, because effectively improving the separation efficiency of charge carriers as well as extending their diffusion distance and lifetime are the keys to enhancing photocatalytic efficiency. A deeper understanding of how photoexcited charge carriers are transported, trapped, and recombined will provide insight into the photocatalytic mechanism and inspire us to design high-performance photocatalysts. Here, we divide the mostly used charge carrier characterization techniques into four categories (Fig. 11), namely, charge separation efficiency, charge transfer direction, charge carrier lifetime, and surface reaction intermediate identification. Finally, techniques in each category together with their features are summarized in Table 4. | ||
| Fig. 11 Characterization techniques of the charge transport process and surface reaction mechanism. | ||
| Category | Techniques | Features |
|---|---|---|
| Charge separation efficiency | Temperature-related photoluminescence | Exciton binding energy |
| Steady-state photoluminescence (PL) | Light intensity | |
| Transient photo-current responses | Photocurrent | |
| Linear scanning voltammetry (LSV) | Photocurrent | |
| Electrochemical impedance spectroscopy (EIS) | Radius of Nyquist plots | |
| Surface photovoltage (SPV) | SPV intensity | |
| Charge transfer direction | In situ irradiation X-ray photoelectron spectroscopy (ISI-XPS) | Electron density changes of various atoms |
| Electron spin resonance/electron paramagnetic resonance (ESR/EPR) | Intensity of DMPO-·O2−/DMPO-·OH signals | |
| Kelvin probe force microscopy (KPFM) | Contact potential difference (CPD) values | |
| Density functional theory (DFT) | Work functions (ϕ) and planar-averaged charge density | |
| Charge carrier lifetime | Time-resolved photoluminescence (TRPL) | Photoluminescence decay |
| Transient absorption spectroscopy (TAS) | Recovery time | |
| Fluorescence imaging (FLIM) | Fluorescence decay | |
| Open circuit photovoltage decay (OCVD) | Photovoltage decay | |
| Transient state photovoltage (TPV) | Photovoltage decay | |
| Intermediate identification | In situ Fourier Transform Infrared (in situ FTIR) | IR peaks |
| Diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) | IR peaks | |
| In situ attenuated total reflectance (in situ ATR-IR) | IR peaks | |
| Density functional theory (DFT) | Gibbs free energy (ΔG) |
4.1 Charge separation efficiency
The absorption of light by the photocatalyst results in the production of excitons, which subsequently separate into electron and hole pairs. The lower binding energy of the exciton means that the carriers can be produced more easily, which increases the carrier separation efficiency. Zhang et al.187 investigated the activation energies of exciton dissociation for pure Cs2AgBiBr6 NCs, and Cs2AgBiBr6/Ti3C2TX heterostructures were investigated using temperature-related photoluminescence (temperature-related PL). Their results (Fig. 12a) showed that as the temperature increases, the PL intensity of both the pristine and heterostructured structures become weaker, leading to activation energies of roughly 55 and 39 meV, respectively. This means that the existence of Ti3C2TX in the heterostructure contributes to a lower exciton binding energy and facilitates the formation of free carriers in the Cs2AgBiBr6 NC. Compared to temperature-related PL, steady-state photoluminescence (PL) measurement has been more commonly utilized to examine the recombination dynamics of photogenerated charge carriers. The weakening of PL intensity indicates a lower recombination rate of the photogenerated holes and electrons. Wu et al.115 used steady-state PL to investigate the photoexcited electron transfer process in the MAPbI3@PCN-221 (Fex) hybrid catalyst. As shown in Fig. 12b, the PL peak intensities of both pristine PCN-221 and composite MAPbI3@PCN-221 decreased dramatically upon the addition of Fe ions. This phenomenon indicates that photoexcited electrons are efficiently transferred from PCN-221 and MAPbI3 QDs to the Fe catalytic sites, which improves the carrier separation efficiency and photocatalytic performance. | ||
| Fig. 12 (a) Pseudocolor map of temperature-dependent PL spectra of Cs2AgBiBr6/Ti3C2Tx heterostructures. Reproduced from ref. 187 with permission from Elsevier, Copyright (2022). (b) Steady-state photoluminescence spectra of PCN-221, PCN-221(Fe0.2), MAPbI3@PCN-221, and MAPbI3@PCN-221(Fe0.2). Reproduced from ref. 115 with permission from Wiley-VCH, Copyright (2019). (c) LSV curves plots of five catalysts. Reproduced from ref. 188 with permission from Elsevier, Copyright (2021). (d) Transient photo-current responses of CBB, ISS and ISS/CBB. Reproduced from ref. 189 with permission from Elsevier, Copyright (2022). (e) EIS Nyquist plots at a bias of 0.25 V Ag/AgCl under irradiation in the EA containing 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6). Reproduced from ref. 190 with permission from Elsevier, Copyright (2022). (f) SPV spectra of CsPbBr3 and CPB/MS samples. Reproduced from ref. 191 with permission from Elsevier, Copyright (2020). | ||
In addition to PL, different photoelectrochemical tests were also carried out to monitor the charge separation properties of the photocatalytic materials. For instance, Zhang et al.188 used linear scanning voltammetry (LSV) curves to study the charge separation in CsPbBr3 (CPB)@Cu-TCPP-x. As shown in Fig. 12c, CPB@Cu-TCPP-x has a much lower overpotential than that of the pristine CPB QD and Cu-TCPP nanosheets, and the CPB@Cu-TCPP-20 composite further reduces the overpotential over the other CPB@TCPP-x composites. The increase in photocurrent density reflects the increased photoinduced charge separation efficiency. In Zhang's work, they investigated the carrier separation behavior of the Cs3Bi2Br9/In4SnS8 (CBB/ISS) heterojunction using photocurrent response under visible light irradiation.189 According to their results (Fig. 12d), the pure CBB and ISS had poor photocurrent responses, but the intensity of the photogenerated current increased greatly for their composite, demonstrating the efficient separation of the photogenerated electron–hole pairs in ISS/CBB. Electrochemical impedance spectroscopy (EIS) is another photoelectrochemical method to evaluate the charge separation efficiency by showing the photogenerated carrier transfer resistance, which can be derived from a Nyquist plot. In general, the small radius of the semicircle in the EIS implies that the electron transport resistance is lower, or that carrier separation is easier. As demonstrated in Fig. 12e, in the Nyquist plots produced by Tian et al.,190 the diameter of the semicircle for the Cs2TeCl6 microcrystals (MCs) was clearly smaller than those for Cs2HfCl6 and Cs2ZrCl6 MCs, which means that the Cs2ZrCl6 MCs possessed a reduced charge-transfer resistance compared to the other two materials.
Another compelling technique to identify charge separation in NHPs is surface photovoltage (SPV) spectroscopy, in which the signal results from variations in surface potential barriers both before and after being exposed to light. The resulting electron–hole pairs can be instantly separated by the built-in electric field, causing a quick SPV response. Normally, greater charge separation efficiency is evidenced by a stronger SPV signal. Fig. 12f displays the SPV spectra of pristine CsPbBr3 and CsPbBr3/MoS2 (CPB/MS) samples measured by Wang et al.191 In contrast to pure CsPbBr3, CPB/MS manifested as a significantly increased SPV response in the 300–550 nm range. It is clearly supported by the strikingly different SPV signals between the CsPbBr3 and CPB/MS composites that the latter's visible light-induced charge separation was more effective and that there was spatial charge accumulation/depletion at the interface.
4.2 Charge transfer direction
The separation and transfer of photogenerated charges is the key point that determines the photocatalytic activity of NHPs during the CO2RR process. Its in-depth study can facilitate the understanding of the charge transfer direction in the semiconductors and the design of better performing photocatalysts. The in situ irradiated X-ray photoelectron spectroscopy (ISI-XPS) technique has been regarded as a competent technique for this because it can detect the density change in electron clouds around different atoms in the ground and excited states, which can directly reveal the direction of charge separation and transfer in heterojunction photocatalysts. Wang et al.192 applied ISI-XPS to verify the type of heterojunction scheme for CABB (Cs2AgBiBr6)@C3N4-82% and CABB@C3N4-10% (Fig. 13a). Under illumination, there is a slight positive shift in the binding energy of Cs 3d and Ag 3d for CABB@C3N4-82% (0.1 eV) and a negative shift in the peak of N 1s (−0.7 eV), indicating a decrease in the electron density on CABB and an increase in the electron density on g-C3N4 in the excited state. It can be concluded that after the binding of CABB to g-C3N4, the electrons on the conduction band of CABB are transferred to the valence band of g-C3N4, which provides direct evidence for the Z-scheme mechanism. The type-II mechanism for CABB@C3N4-10% is supported by the opposing trend of the binding energy shifts. | ||
| Fig. 13 (a) High-resolution XPS of CABB@C3N4-10% and CABB@C3N4-82% in the dark or under 365 nm LED irradiation. Reproduced from ref. 192 with permission from Elsevier, Copyright (2020). (b) KPFM image of CsPbBr3-Au in the dark and under light irradiation at 630 nm. Reproduced from ref. 193 with permission from Elsevier, Copyright (2021). (c) Calculated work functions of CABB and BWO, and the planar-averaged electron density difference Δρ and the charge density difference of CABB/BWO. Reproduced from ref. 110 with permission from Elsevier, Copyright (2022). (d) ESR spectra of DMPO-·O2− and DMPO-·OH in the presence of CsPbBr3 QDs, MTB and the CsPbBr3@MTB hybrid. Reproduced from ref. 155 with permission from Elsevier, Copyright (2021). | ||
Another convincing technique to confirm the electron transfer direction in the heterostructure is electron spin resonance (ESR), also called electron paramagnetic resonance (EPR), which usually uses 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) as a radical trap to capture the oxygen radical (·O2−) and hydroxyl radical (·OH). Dong et al.155 confirmed the carrier transfer direction in CsPbBr3@MTB materials by ESR. As shown on the left of Fig. 13d, the DMPO−·O2− signal intensity of the CsPbBr3@MTB composite is much stronger than that of the pristine CsPbBr3 and MTB, indicating that more photogenerated electrons are accumulated in the CB of CsPbBr3. The right side of Fig. 13d shows that the DMPO−·OH signal intensity of the CsPbBr3@MTB composite is much stronger than that of pure MTB, which implies that the light-generated holes are accumulated on the VB of MTB. Thus, the above two conclusions demonstrate that the photogenerated electrons are transferred to CsPbBr3 and the photogenerated holes are transferred to MTB, thus proving that the CsPbBr3@MTB composite is an S-scheme heterojunction.
Besides, Kelvin probe force microscopy (KPFM) is an interesting improvement to Atomic Force Microscopy (AFM) and SPV, which is capable of imaging surface morphology and surface potential simultaneously with nanoscale spatial resolution and sub-millivolt electrical resolution. Therefore, it can be used to confirm the electron transfer direction. In Liao's work, they compared the contact potential difference (CPD) images between pure CsPbBr3 and the CsPbBr3-Au composite.193 As shown in Fig. 13b, pure CsPbBr3 cannot be excited by light, since there is no significant difference between CPD images collected in the dark and under light. In contrast, the CPD values of the CsPbBr3-Au composites decrease upon light illumination, which suggests the accumulation of electrons on the surface. Through this, they inferred that the hot electrons generated in the Au nanoparticles can be effectively transferred to CsPbBr3.
Aiming to obtain further evidence of the charge separation direction, Wu et al. used density functional theory (DFT) to calculate the work functions (ϕ) of Cs2AgBiBr6 (CABB) and Bi2WO6 (BWO).110 The calculated outcomes (Fig. 13c) demonstrate that CABB has a higher Ef than BWO, which suggests that some electrons will automatically migrate from CABB to BWO at the heterojunction interface until their Ef coincides. Additionally, the generation of heterojunctions will result in the reallocation of electrons at the interface, according to the predicted planar average charge density difference of CABB/BWO. Green represents the electron reduction on the CABB side, whereas yellow represents the electron buildup on the BWO side. As a result, a solid IEF and band bending will form because CABB and BWO will be positively and negatively charged, respectively.
4.3 Charge carrier lifetime
Exploring the interfacial charge transfer of heterojunction photocatalysts is important because it is a prerequisite to show the final catalytic reaction capability, which will provide insight into the photocatalytic mechanism and facilitate the design of high-performance photocatalysts. Since the time scale of charge kinetics in photocatalytic processes varies from femtoseconds to seconds, a series of techniques are applied to investigate the charge transfer kinetics and carrier lifetime inside the photocatalyst as well as at the interface.195Time-resolved photoluminescence (TRPL) spectroscopy is the most widely used technique to investigate excited charge carrier recombination in photocatalysts. After a photocatalyst is excited, electrons and holes will be recombined, leading to photogenerated carrier decay. Carrier decay kinetics can be obtained by extrapolating the carrier density changes directly from the time-resolved PL intensity measured using laser excitation pulses or fast electron techniques. TRPL decay is commonly used in multi-exponential models to extract the lifetimes of different processes. Zhang et al.157 tested the TRPL of pristine CsPbBr3 and the C60/CsPbBr3 composite. The normalized TRPL decay plots confirmed the C60/CsPbBr3 composite's swift charge separation, as seen in Fig. 14a. The decay curves were fitted using triexponential decay kinetics, and the average lifetime for the C60/CsPbBr3 composite was 6.1 ns, a substantial drop from the 15.8 ns of pure CsPbBr3. The C60/CsPbBr3 composite's noticeably faster decay can be attributed to C60's ability to receive electrons, which acted as an electron reservoir and promoted the charge transfer from CsPbBr3 to C60. Different from TRPL, Fluorescence Lifetime Imaging Microscopy (FLIM) is a fluorescence imaging technique. Instead of using the emission spectra of the fluorophores, it bases its contrast on their lifetime. The average time a molecule spends in the excited state before it returns to its ground state and emits a photon is known as the fluorescence lifespan. To determine the PL lifetimes of the excited species, Laishram et al.194 measured FLIM lifetimes on g-C3N4 (CN) and CsPbBr3 NCs with monolayer sheet (CNM) samples. The tri-exponential fitting of the FLIM lifespan curves resulted in the average lifetimes (τave) of the CN and CNM samples being determined to be 1.14 and 1.72 ns, respectively, as shown in Fig. 14b. They attributed the increase in the average lifetime value of CNM to the increase in intrasheet order and the apparent suppression of intrasheet recombination after the conversion of bulk CN into monolayers. It is evident from the increased average lifetime value that transformation of bulk sheets into monolayer sheets increases charge transport on the conjugated CNM sheets, resulting in a more efficient charge separation.
 | ||
| Fig. 14 (a) Time-resolved PL decay spectra of CsPbBr3 NCs and the C60/CsPbBr3 composite. Reproduced from ref. 157 with permission from Elsevier, Copyright (2021). (b) Fluorescence lifetime imaging (FLIM) lifetime decay curve of CN (black) and CNM (blue). Reproduced from ref. 194 with permission from Elsevier, Copyright (2022). (c and d) Voc transient rise/decay obtained during excitation/termination of visible light irradiation and average electron lifetimes (τn) obtained from transient OCVD measurements. Reproduced from ref. 127 with permission from Elsevier, Copyright (2021). (e–g) TA recovery dynamic plots of CsPbBr3 and CsPbBr3@ZIFs. Excitation wavelength: 400 nm. Reprinted with permission from ref. 117, Copyright 2018, American Chemical Society. (h) TPV spectra of CsPbBr3 and CPB/MS samples. Reproduced from ref. 146 with permission from Elsevier, Copyright (2020). | ||
In general, a TRPL instrument is only capable of monitoring emitting materials, which usually does not provide a complete picture of the charge carriers. In contrast, transient absorption spectroscopy (TAS) can monitor both emitting and non-emitting (i.e., trapped) substances, providing more information to model the operation of the photocatalytic system by measuring the change in absorbance or transmittance over time before and after sample excitation. The TAS of CsPbBr3@ZIF-8 and CsPbBr3@ZIF-67 in comparison to pure CsPbBr3 was measured by Kong et al.117 Absorption change (ΔA) curves plotted against wavelength and delay time are shown in Fig. 14e–g. ZIF-8 and ZIF-67 exhibit very little absorption at a pump pulse of 400 nm, which allows CsPbBr3 to be stimulated selectively. The ground state bleaching (GSB) signal of CsPbBr3 is believed to have a unique negative absorption peak at 513 nm that is present in unmodified CsPbBr3. In CsPbBr3@ZIFs, bleaching behavior was more quickly seen, indicating an efficient interfacial electron transport between CsPbBr3 and ZIF shells.
Additionally, photovoltage decay can be used for determining the lifetime of photogenerated electrons. For example, in Xi's work, open circuit photovoltage decay (OCVD) measurement was performed with an electrochemical station under 300 W Xe lamp irradiation.127 The average electron lifetime of CsPbBr3 is noticeably shorter than that of a corresponding Ni-based metal–organic framework (NMF)/CPB, as can be seen in Fig. 14c and d. This suggests that the addition of NMF reduces the recombination of photogenerated charge carriers and lengthens the lifetimes of electrons. In transient photovoltage (TPV), the semiconductor under testing initially remains disconnected from the circuit under steady-state illumination. With the application of an additional short light pulse, the semiconductor is photoexcited to produce carriers. The free electrons migrating to the material surface are collected at the electrode to obtain the photovoltage. The lifetime of the photocarriers can be determined by detecting the decay of the photovoltage. Fig. 14h shows the TPV spectra of Wang's work.146 Both pure CsPbBr3 and CsPbBr3/MoS2 (CPB/MS) samples exhibit a positive TPV response, but the larger response and longer lifetime of CPB/MS suggests that CsPbBr3 is the source of the photogenerated holes that accumulate on the surface of the test electrode. The larger TPV response of CPB/MS suggests a broader space charge zone for the charge diffusion process.
4.4 Surface reaction intermediate identification
Designing effective photocatalysts will benefit from a thorough understanding of the unique active sites and associated reaction mechanisms of NHP NCs. Even though the photocatalytic process of CO2 reduction has been extensively studied, the CO2 reduction reaction pathways on NHP NCs remain unclear. A deeper understanding of the photocatalytic mechanism and the intermediate species involved in the CO2 photoreduction process are required to resolve these challenges. Fourier transform infrared (FTIR) spectroscopy is considered a semi-quantitative tool for studying the molecular structure of materials using infrared radiation. This is due to the different vibrational frequencies of the covalent bonds within molecules, resulting in their ability to absorb different wavelengths of light and thus create different infrared spectra.Several derived characterization methods, such as in situ FTIR, diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS), and in situ Attenuated Total Reflection Infrared (in situ ATR-IR) spectroscopy, have been used to identify intermediates involved in photocatalytic CO2 reduction. DFT theoretical calculations in conjunction with advanced in situ characterization techniques will be more useful in determining the precise photocatalytic mechanism and offer helpful information for designing NHPs for CO2 photocatalytic reduction. For example, Ding et al.163 performed in situ FTIR measurements on the Cs2AgBiBr6/UIO-66 composite in order to detect reaction intermediates in the CO2 photoreduction process. As shown in Fig. 15a, no intermediate peaks were seen prior to the illumination, but as soon as the illumination began, a new set of IR peaks emerged, including CH3O* (1140 cm−1), 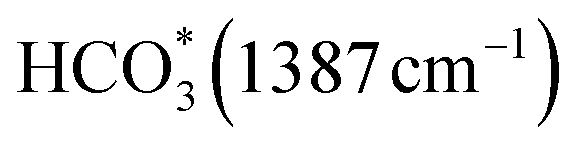 , COOH* (1650 cm−1), and
, COOH* (1650 cm−1), and  , and their intensity increased with time. The results suggest that the primary reaction intermediates in the photocatalytic conversion of CO2 to CO are formate species. In order to detect and monitor adsorbed materials and reaction intermediates on the surface of 10% Bi3O4Br–3% Ni-doped CsPbBr3 (10BC-3N) and CsPbBr3 (CPB) in the dark and under full-spectrum light irradiation, Wang et al. used DRIFTS measurements (Fig. 15b) to identify the surface reactant intermediates.153 They discovered that the –CO2− species matches the wavenumbers that appear at 1308 and 1693 cm−1. The essential intermediate in the multi-step reduction of CO2 to CO or CH4 and the cause of the peak at 1510 cm−1 is COOH*. As indicated by the wave number at 1745 cm−1, some CO may be created during the irradiation process. Additionally, numerous chemical intermediates were discovered in the high wave number region CH3O* (2853 cm−1),
, and their intensity increased with time. The results suggest that the primary reaction intermediates in the photocatalytic conversion of CO2 to CO are formate species. In order to detect and monitor adsorbed materials and reaction intermediates on the surface of 10% Bi3O4Br–3% Ni-doped CsPbBr3 (10BC-3N) and CsPbBr3 (CPB) in the dark and under full-spectrum light irradiation, Wang et al. used DRIFTS measurements (Fig. 15b) to identify the surface reactant intermediates.153 They discovered that the –CO2− species matches the wavenumbers that appear at 1308 and 1693 cm−1. The essential intermediate in the multi-step reduction of CO2 to CO or CH4 and the cause of the peak at 1510 cm−1 is COOH*. As indicated by the wave number at 1745 cm−1, some CO may be created during the irradiation process. Additionally, numerous chemical intermediates were discovered in the high wave number region CH3O* (2853 cm−1),  , and
, and  . For in situ-ATR analysis, Choi et al.196 reported that different characteristics of intermediate formation were evaluated for each catalyst. The photocatalytic reduction process of gaseous CO2 employing perovskite photocatalysts with copper carriers results in IR distinctive peaks (Fig. 15c). At 1532 cm−1 (asymmetric) and 1500 cm−1 (symmetric), significant C2 pathway intermediates *OCCO and *OCCOH were both visible at 3080 cm−1 (olefins), 2987 cm−1 (asymmetric –CH3 alkanes), 2924 cm−1 (asymmetric –CH2 alkanes), and 2876 cm−1 (symmetric –CH3 alkanes). For the synthesis of C2H4, the olefin peak at 3080 cm−1 is particularly significant evidence. At 1495 cm−1, 1414 cm−1, and 1340 cm−1, adsorbed carbonate species derived from pure CO2 gas were noted. In addition to characterization, DFT calculations can also be applied to predict the CO2 photoreduction mechanism. By computing the associated Gibbs free energy (G) at each stage of the CO2 photoreduction process, Sheng's study on Cs2CuBr4 corroborated the hypothesized CO2 photoreduction pathway (Fig. 15d).133 The figure depicts the creation of COOH* by a high energy input internal energy process on the surface of Cs2CuBr4 and CsPbBr3 QDs. As a result, the rate-limiting step is the creation of COOH* intermediates from ˙CO2−. However, Cs2CuBr4 has a stronger photoreduction capacity than CsPbBr3, as evidenced by the fact that it requires less reaction energy (0.48 eV) to convert ˙CO2− to COOH* than CsPbBr3 (1.61 eV) does. The simulations also show that the CsPbBr3-produced CO* intermediate prefers to undergo energetically advantageous desorption because of CO rather than additional hydrogenation to CH4.
. For in situ-ATR analysis, Choi et al.196 reported that different characteristics of intermediate formation were evaluated for each catalyst. The photocatalytic reduction process of gaseous CO2 employing perovskite photocatalysts with copper carriers results in IR distinctive peaks (Fig. 15c). At 1532 cm−1 (asymmetric) and 1500 cm−1 (symmetric), significant C2 pathway intermediates *OCCO and *OCCOH were both visible at 3080 cm−1 (olefins), 2987 cm−1 (asymmetric –CH3 alkanes), 2924 cm−1 (asymmetric –CH2 alkanes), and 2876 cm−1 (symmetric –CH3 alkanes). For the synthesis of C2H4, the olefin peak at 3080 cm−1 is particularly significant evidence. At 1495 cm−1, 1414 cm−1, and 1340 cm−1, adsorbed carbonate species derived from pure CO2 gas were noted. In addition to characterization, DFT calculations can also be applied to predict the CO2 photoreduction mechanism. By computing the associated Gibbs free energy (G) at each stage of the CO2 photoreduction process, Sheng's study on Cs2CuBr4 corroborated the hypothesized CO2 photoreduction pathway (Fig. 15d).133 The figure depicts the creation of COOH* by a high energy input internal energy process on the surface of Cs2CuBr4 and CsPbBr3 QDs. As a result, the rate-limiting step is the creation of COOH* intermediates from ˙CO2−. However, Cs2CuBr4 has a stronger photoreduction capacity than CsPbBr3, as evidenced by the fact that it requires less reaction energy (0.48 eV) to convert ˙CO2− to COOH* than CsPbBr3 (1.61 eV) does. The simulations also show that the CsPbBr3-produced CO* intermediate prefers to undergo energetically advantageous desorption because of CO rather than additional hydrogenation to CH4.
 | ||
| Fig. 15 (a) In situ FTIR spectra of 20CABB/UIO-66 under CO2 and H2O vapor after photo-irradiation for 0, 5, 10, 15, 20, and 25 min. Reproduced from ref. 163 with permission from Elsevier, Copyright (2022). (b) In situ DRIFTS measurements for CO2 and H2O interaction with 10BC-3N and CPB. Reproduced from ref. 153 with permission from Elsevier, Copyright (2022). (c) In situ ATR-IR spectra of photocatalytic CO2 reduction on the CsPbBr3 perovskite photocatalyst embedded in a porous copper scaffold. Reproduced from ref. 196 with permission from Elsevier, Copyright (2021). (d) Calculated free energy of the main reactions in photocatalytic CO2 reduction for Cs2CuBr4 and CsPbBr3 PQDs. Reprinted with permission from ref. 133, Copyright 2018, American Chemical Society. | ||
5. Challenges and prospects
During the past few decades, many scientists have made great progress in the field of photocatalytic CO2 reduction using NHPs. However, it should be realized that the practical applications of NHPs still face several challenges, such as heavy metal (Pb) contamination, stability issues, low catalytic activity and selectivity, and unclear mechanism of photocatalysis. The design of novel efficient NHP photocatalysts is still in progress, and researchers are required to focus on addressing these obstacles.5.1 Environmental impact
Even modest amounts of Pb can cause major environmental and health issues; therefore its toxicity cannot be disregarded. Currently, lead halide perovskites are the most widely used photocatalysts. Its toxicity unquestionably prevents its commercial use and the mass production of NHP photocatalysts. To address this issue, some progress has been made in exploring lead-free NHPs for photocatalytic CO2 reduction, such as various Pb-free transition metals including Ge, Ag, In, Sn, Sb, and Bi.197–199 In addition, bimetallic repetition has been developed due to the synergistic effect of bimetals. Noticeably, most of the current Pb-free NHPs have lower photocatalytic efficiency than Pb-based ones.200 In future studies, lead-free perovskite materials remain a promising strategy to reduce lead leakage by using different optimization strategies as mentioned above such as surface engineering (size, morphology, defects, etc.), component engineering (trying new metal cations or combining different non-lead metal cations) and heterojunction engineering (2D/3D structures) to improve the photocatalytic performance of lead-free perovskite materials. In addition, the physical encapsulation of lead-containing perovskites can slow down the leakage of lead, for example, TiO2, organic materials, and MOFs, but the lead compounds will still leak into the environment after long-term operation. Therefore, lead capture after photocatalytic reactions is an effective way to reduce the leakage of lead into the environment. One of the effective methods is chemisorption, in which some functional materials with multiple functional groups, including thiols, phosphate groups, etc., are bonded to capture the mobile Pb2+. This approach can lead to better protection against Pb leakage because it has superior ability to reduce Pb leakage compared to physical encapsulation.5.2 Stability
The industrialisation of NHP photocatalysts is additionally hampered by stability issues brought on by heat, humidity, UV radiation, self-separation, or repetitive catalytic cycles. The general strategy to increase the stability of NHP is based on effects of chemical or physical bonding. Using encapsulation techniques with a core–shell structure that is almost entirely covered can lessen NHP's direct interaction with the environment. The stability of NHPs can be increased by efficiently preventing polar solvents from coming into direct contact with the NHP core through a densely crystalline protective shell or replacing polar solvents with low-polarity solvents such as ethyl acetate, acetonitrile, and toluene. Besides using encapsulation and low-polarity solvents, the stability of NHPs can also be enhanced by changing the elements, surface ligands and morphology of the NHPs. For example, the intrinsic stability of NHPs can be significantly increased by using mixed cation methods, such as CsxFA1−xPbI3 (ref. 201). The lattice–ion interactions can be improved by B-site metal ion doping, leading to a more stable crystal structure. NHPs can also be made more stable by partially substituting smaller cations (Mn2+ or Zn2+) for Pb2+. Recently, researchers have started using thiocyanate (SCN−) and formate (HCOO−) ions to synthesize stable pseudo-halide perovskite materials for solar cell applications. Compared to halide perovskites, these pseudo-halide perovskites exhibit better stability at room temperature. This is because the SCN− or HCOO− anions occupy the axial positions of the lead halide octahedra, leading to an asymmetric electronic structure that helps maintain the desired structural framework. Furthermore, two-dimensional NHPs can be constructed by introducing large organic cations into the A-site. The hydrophobicity of these organic cations can be leveraged to effectively block external environmental factors, significantly improving the stability of the NHPs.202 In addition, employing specific ligands can also enhance the stability of NHPs. For instance, alkyl phosphinic acid and alkylammonium halide ligands can passivate the damaged surface of NHPs and form a protective layer. These ligands can modify the NHPs and improve their stability during photocatalytic reactions.20 In conclusion, comprehensive material design strategies are key to improving the stability of NHPs. Through the combination of these approaches, the long-term stability of NHP photocatalysts can be achieved, which is crucial for their practical development.5.3 Catalytic efficiency and selectivity
The photocatalytic performance of current NHPs for the photocatalytic reduction of CO2 is not sufficient for future practical applications, so further improvements in photocatalytic efficiency and product selectivity are needed. Numerous features have been thoroughly explored, including how to increase the specific surface area, add additional active sites, encourage the separation of photogenerated electron–hole pairs, and prevent charge recombination. For instance, co-catalysts (such as metals, MXenes, carbon materials, etc.) can be added to speed up the rate of the surface catalytic reaction, and new heterojunctions (such as Schottky junctions, and type II, all-solid-state Z-scheme, and S-scheme heterojunctions) can be built to improve charge separation. Perovskites can also be modified in size and morphology to increase the surface reaction of active sites. Additionally, to enhance catalytic activity, synergistic external field catalysis (such as thermal, electronic, magnetic, and biological enzymes) can be used. In terms of product selectivity, the ideal products for photocatalytic CO2 reduction are chemical feedstocks and fuels (e.g., unsaturated hydrocarbons or alcohols). However, the selectivity of CO2 reduction at present is usually directed towards CO and CH4. Therefore, the design of NHPs with additional economic benefits for multi-carbon hydrocarbon products is the key to addressing the above challenges in the future. As for NHPs, the selectivity of photocatalytic CO2 to CH4 can be improved by tuning the photoexcitation properties, band structure (B-site element: Bi, Sn or In), charge carrier separation, and catalytically active sites (Fe, Co, Zn, and Cu). To improve the selectivity of photocatalytic CO2 reduction towards C2+ products, the following strategies can be adopted to address the limitations of photocatalytic systems: (1) introduce metal co-catalysts such as Au, Ag, and Cu on the surface of NHP photocatalysts to promote C–C coupling reactions; (2) prepare NHP photocatalysts with suitable internal voids or hollow structures to increase the spatial sites for C–C coupling; (3) optimize the crystal phase and facet orientation of the photocatalysts since certain facets are more favorable for C–C bond formation compared to others; (4) optimize reaction conditions like temperature, pH, and light intensity (high photon flux) to influence C–C coupling; (5) construct more effective heterojunctions to regulate the distribution of photogenerated electrons on co-catalysts and enhance the driving force for C–C coupling by minimizing charge recombination;41 (6) doping or surface modification to modulate the electronic structure and surface properties of photocatalysts, e.g., introducing nitrogen-containing functional groups to enhance CO2 adsorption, can also benefit C–C bond formation; (7) integrate NHP photocatalysis with electrochemical processes, such as photoelectrochemical, photothermal, photomagnetic, and photobiosynthetic catalysis, to provide stronger reducing potentials for the generation of C2+ products.203 By comprehensively applying these innovative strategies, the selectivity and efficiency of photocatalytic CO2 reduction towards C2+ products can be further improved.5.4 Catalytic mechanism
A deeper comprehension of the photocatalytic mechanism is necessary for a more logical design of NHP photocatalysts. The chemical pathway of CO2 reduction on NHPs is still unknown, despite substantial research into the photocatalytic mechanism of CO2 reduction on a variety of metals and semiconductors. More fundamental research on the photocatalytic mechanism is required to identify the intermediate species involved in CO2 reduction and provide answers to these problems. DFT calculations allow the calculation of various properties such as crystal structures, band structures, effective masses of electrons and holes, and intermediate products. Therefore, more studies combining theoretical DFT calculations with experimental validation are needed. Moreover, more advanced in situ characterization techniques need to be developed to gain an in-depth understanding of the detailed photocatalytic mechanism and charge carrier separation, as well as provide useful information for the design of NHP photocatalysts for CO2 reduction.In conclusion, using NHPs for photocatalytic CO2RR is a highly effective technique to deal with the problems of the greenhouse effect and the energy crisis at the same time. However, the CO2RR performance of NHP-based catalysts is severely constrained by strong carrier recombination, poor product selectivity, and instability in humid environments. Therefore, our review summarized critical factors that influence the final product selectivity, strategies that have been implemented to enhance the photocatalytic performance of NHPs, and techniques to characterize the charge transport process and CO2RR mechanism. Finally, some challenges and prospects are introduced to indicate the direction of future research. With the rapid development of this fascinating field, we deem that this review could provide crucial guidance for the rational design of efficient photocatalytic systems and conquer the decisive problems encountered in the field of NHP photocatalysts.
Data availability
The data that support the findings of this study are available on request from the corresponding authors, D.-J. Lee and H.-Y. Hsu.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the Research Grants Council of Hong Kong (grant no. CityU 21203518 and F-CityU106/18), Innovation and Technology Commission (grant no. MHP/104/21), Shenzhen Science Technology and Innovation Commission (grant no. JCYJ20210324125612035, R-IND12303 and R-IND12304), City University of Hong Kong (grant no. 9360140, 7005289, 7005580, 7005720, 9667213, 9667229, 9680331 and 9678291), National Natural Science Foundation of China (51901119, 22071070, 61874165, and 21833009), and Major State Basic Research Development Program of China (2019YFB1503401).References
- S. C. Roy, O. K. Varghese, M. Paulose and C. A. Grimes, ACS Nano, 2010, 4, 1259–1278 CrossRef CAS.
- X. Fu, T. He, S. Zhang, X. Lei, Y. Jiang, D. Wang, P. Sun, D. Zhao, H.-Y. Hsu and X. Li, Chem, 2021, 7, 3131–3143 CAS.
- C. Hepburn, E. Adlen, J. Beddington, E. A. Carter, S. Fuss, N. Mac Dowell, J. C. Minx, P. Smith and C. K. Williams, Nature, 2019, 575, 87–97 CrossRef CAS PubMed.
- A. Tabish, A. M. Varghese, M. A. Wahab and G. N. Karanikolos, Catalysts, 2020, 10, 95 CrossRef CAS.
- A. A. Khan and M. Tahir, J. CO2 Util., 2019, 29, 205–239 CrossRef CAS.
- S. Xie, Q. Zhang, G. Liu and Y. Wang, Chem. Commun., 2016, 52, 35–59 RSC.
- G. Zhang, G. Liu, L. Wang and J. T. Irvine, Chem. Soc. Rev., 2016, 45, 5951–5984 RSC.
- S. Rao, X. Zou, S. Wang, T. Shi, Y. Lu, L. Ji, H.-Y. Hsu, Q. Xu and X. Lu, J. Electrochem. Soc., 2019, 166, D427 CrossRef CAS.
- H. Wu, T. H. Tan, R. Liu, H.-Y. Hsu and Y. H. Ng, Sol. RRL, 2021, 5, 2000423 CrossRef CAS.
- A. Saravanan, D.-V. N. Vo, S. Jeevanantham, V. Bhuvaneswari, V. A. Narayanan, P. Yaashikaa, S. Swetha and B. Reshma, Chem. Eng. Sci., 2021, 236, 116515 CrossRef CAS.
- B. S. Kwak, K. Vignesh, N.-K. Park, H.-J. Ryu, J.-I. Baek and M. Kang, Fuel, 2015, 143, 570–576 CrossRef CAS.
- F. Iqbal, B. Abdullah, H. Oladipo, M. Yusuf, F. Alenazey, T. D. Nguyen and M. Ayoub, Nanostructured Photocatalysts, 2021, pp. 519–540 Search PubMed.
- J. Mora-Hernandez, A. M. Huerta-Flores and L. M. Torres-Martinez, J. CO2 Util., 2018, 27, 179–187 CrossRef CAS.
- G. Yin, H. Abe, R. Kodiyath, S. Ueda, N. Srinivasan, A. Yamaguchi and M. Miyauchi, J. Mater. Chem. A, 2017, 5, 12113–12119 RSC.
- W. Gao, S. Li, H. He, X. Li, Z. Cheng, Y. Yang, J. Wang, Q. Shen, X. Wang and Y. Xiong, Nat. Commun., 2021, 12, 4747 CrossRef CAS PubMed.
- X. Chen, C. Li, M. Grätzel, R. Kostecki and S. S. Mao, Chem. Soc. Rev., 2012, 41, 7909–7937 RSC.
- M. Halmann, Nature, 1978, 275, 115–116 CrossRef CAS.
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637–638 CrossRef CAS.
- J. S. Zhao, Y. F. Mu, L. Y. Wu, Z. M. Luo, L. Velasco, M. Sauvan, D. Moonshiram, J. W. Wang, M. Zhang and T. B. Lu, Angew. Chem., 2024, e202401344 CAS.
- J. Wang, Y. Shi, Y. Wang and Z. Li, ACS Energy Lett., 2022, 7, 2043–2059 CrossRef CAS.
- J. L. White, M. F. Baruch, J. E. Pander III, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao, J. Gu and Y. Yan, Chem. Rev., 2015, 115, 12888–12935 CrossRef CAS PubMed.
- Q. A. Akkerman, G. Rainò, M. V. Kovalenko and L. Manna, Nat. Mater., 2018, 17, 394–405 CrossRef CAS.
- X. Song, G. Wei, J. Sun, C. Peng, J. Yin, X. Zhang, Y. Jiang and H. Fei, Nat. Catal., 2020, 3, 1027–1033 CrossRef CAS.
- Y. Wu, P. Wang, X. Zhu, Q. Zhang, Z. Wang, Y. Liu, G. Zou, Y. Dai, M. H. Whangbo and B. Huang, Adv. Mater., 2018, 30, 1704342 CrossRef.
- Y. Tang, C. H. Mak, R. Liu, Z. Wang, L. Ji, H. Song, C. Tan, F. Barrière and H. Y. Hsu, Adv. Funct. Mater., 2020, 2006919 CrossRef CAS.
- C. A. Richard, Z. Pan, H.-Y. Hsu, S. Cekli, K. S. Schanze and J. R. Reynolds, ACS Appl. Mater. Interfaces, 2014, 6, 5221–5227 CrossRef CAS.
- L. Fu, K. Fu, X. Gao, S. Dong, B. Zhang, S. Fu, H.-Y. Hsu and G. Zou, Anal. Chem., 2021, 93, 2160–2165 CrossRef CAS.
- Y. Tang, C. H. Mak, C. Wang, Y. Fu, F.-F. Li, G. Jia, C.-W. Hsieh, H.-H. Shen, J. C. Colmenares, H. Song, M. Yuan, Y. Chen and H.-Y. Hsu, Small Methods, 2022, 6, 2200326 CrossRef CAS.
- L. Liu, H. Huang, Z. Chen, H. Yu, K. Wang, J. Huang, H. Yu and Y. Zhang, Angew. Chem., Int. Ed., 2021, 60, 18303–18308 CrossRef CAS.
- Y. Lin, J. Guo, J. San Martin, C. Han, R. Martinez and Y. Yan, Chem.–Eur. J., 2020, 26, 13118–13136 CrossRef CAS.
- F. Zhong, Y. He, Y. Sun, F. Dong and J. Sheng, J. Mater. Chem. A, 2022, 10, 22915–22928 RSC.
- E. A. Tsiwah, Y. Ding, Z. Li, Z. Zhao, M. Wang, C. Hu, X. Liu, C. Sun, X. Zhao and Y. Xie, CrystEngComm, 2017, 19, 7041–7049 RSC.
- D. M. Jang, D. H. Kim, K. Park, J. Park, J. W. Lee and J. K. Song, J. Mater. Chem. C, 2016, 4, 10625–10629 RSC.
- Z. Long, H. Ren, J. Sun, J. Ouyang and N. Na, Chem. Commun., 2017, 53, 9914–9917 RSC.
- S. Javaid, X. Xu, W. Chen, J. Chen, H.-Y. Hsu, S. Wang, X. Yang, Y. Li, Z. Shao, F. Jones and G. Jia, Nano Energy, 2021, 89, 106463 CrossRef CAS.
- L. Protesescu, S. Yakunin, O. Nazarenko, D. N. Dirin and M. V. Kovalenko, ACS Appl. Nano Mater., 2018, 1, 1300–1308 CrossRef CAS PubMed.
- X. Fan, L. Zhang, M. Wang, W. Huang, Y. Zhou, M. Li, R. Cheng and J. Shi, Appl. Catal., B, 2016, 182, 68–73 CrossRef CAS.
- Y. Wang, J. Wang, M. Zhang, S. Zheng, J. Wu, T. Zheng, G. Jiang and Z. Li, Small, 2023, 19, 2300841 CrossRef CAS.
- W. Song, K. C. Chong, G. Qi, Y. Xiao, G. Chen, B. Li, Y. Tang, X. Zhang, Y. Yao and Z. Lin, J. Am. Chem. Soc., 2024, 146(5), 3303–3314 CrossRef CAS PubMed.
- N. Li, X. Chen, J. Wang, X. Liang, L. Ma, X. Jing, D.-L. Chen and Z. Li, ACS Nano, 2022, 16, 3332–3340 CrossRef CAS PubMed.
- J. Albero, Y. Peng and H. García, ACS Catal., 2020, 10, 5734–5749 CrossRef CAS.
- Z. Wu, H. Tüysüz, F. Besenbacher, Y. Dai and Y. Xiong, Nanoscale, 2023, 15, 5598–5622 RSC.
- S. Shyamal and N. Pradhan, J. Phys. Chem. Lett., 2020, 11, 6921–6934 CrossRef CAS PubMed.
- M. Tahir and N. S. Amin, Energy Convers. Manage., 2013, 76, 194–214 CrossRef CAS.
- R. Cheng, C.-C. Chung, S. Wang, B. Cao, M. Zhang, C. Chen, Z. Wang, M. Chen, S. Shen and S.-P. Feng, Mater. Today Phys., 2021, 17, 100358 CrossRef CAS.
- Y. Wei, Z. Cheng and J. Lin, Chem. Soc. Rev., 2019, 48, 310–350 RSC.
- Q. Chen, Y. Ma, L. Wang, X. Lan and J. Shi, Sol. RRL, 2021, 5, 2000755 CrossRef CAS.
- Y.-F. Xu, M.-Z. Yang, B.-X. Chen, X.-D. Wang, H.-Y. Chen, D.-B. Kuang and C.-Y. Su, J. Am. Chem. Soc., 2017, 139, 5660–5663 CrossRef CAS PubMed.
- J. Zhu, Y. Zhu, J. Huang, L. Hou, J. Shen and C. Li, Nanoscale, 2020, 12, 11842–11846 RSC.
- Z.-C. Kong, H.-H. Zhang, J.-F. Liao, Y.-J. Dong, Y. Jiang, H.-Y. Chen and D.-B. Kuang, Sol. RRL, 2020, 4, 1900365 CrossRef CAS.
- M. Ou, W. Tu, S. Yin, W. Xing, S. Wu, H. Wang, S. Wan, Q. Zhong and R. Xu, Angew. Chem., 2018, 130, 13758–13762 CrossRef.
- H. Huang, J. Zhao, Y. Du, C. Zhou, M. Zhang, Z. Wang, Y. Weng, J. Long, J. Hofkens and J. A. Steele, ACS Nano, 2020, 14, 16689–16697 CrossRef CAS PubMed.
- J. Wang, M. Zhang, Z. Chen, L. Li, G. Jiang and Z. Li, ACS Energy Lett., 2024, 9, 653–661 CrossRef CAS.
- M. A. Peña and J. L. G. Fierro, Chem. Rev., 2001, 101, 1981–2018 CrossRef.
- Y. Zhao and K. Zhu, Chem. Soc. Rev., 2016, 45, 655–689 RSC.
- Z. Yi, N. H. Ladi, X. Shai, H. Li, Y. Shen and M. Wang, Nanoscale Adv., 2019, 1, 1276–1289 RSC.
- M. A. Raza, F. Li, M. Que, L. Zhu and X. Chen, Mater. Adv., 2021, 2, 7187–7209 RSC.
- J. Tian, Q. Xue, Q. Yao, N. Li, C. J. Brabec and H. L. Yip, Adv. Energy Mater., 2020, 10(23), 2000183 CrossRef CAS.
- F. Igbari, Z.-K. Wang and L.-S. Liao, Adv. Energy Mater., 2019, 9, 1803150 CrossRef.
- F. Zhou, Z. Li, H. Chen, Q. Wang, L. Ding and Z. Jin, Nano Energy, 2020, 73, 104757 CrossRef CAS.
- M. I. Saidaminov, O. F. Mohammed and O. M. Bakr, ACS Energy Lett., 2017, 2, 889–896 CrossRef CAS.
- Q. Akkerman, A. Abdelhady and L. Manna, J. Phys. Chem. Lett., 2018, 9(9), 2326–2337 CrossRef CAS.
- R. Liu, C. H. Mak, X. Han, Y. Tang, G. Jia, K.-C. Cheng, H. Qi, X. Zou, G. Zou and H.-Y. Hsu, J. Mater. Chem. A, 2020, 8, 23803–23811 RSC.
- K. A. Huynh, D. L. T. Nguyen, V.-H. Nguyen, D.-V. N. Vo, Q. T. Trinh, T. P. Nguyen, S. Y. Kim and Q. V. Le, J. Chem. Technol. Biotechnol., 2020, 95, 2579–2596 CrossRef CAS.
- A. Feng, X. Jiang, X. Zhang, X. Zheng, W. Zheng, O. F. Mohammed, Z. Chen and O. M. Bakr, Chem. Mater., 2020, 32, 7602–7617 CrossRef CAS.
- S. González-Carrero, R. E. Galian and J. Pérez-Prieto, Part. Part. Syst. Charact., 2015, 32, 709–720 CrossRef.
- F. Temerov, Y. Baghdadi, E. Rattner and S. Eslava, ACS Appl. Energy Mater., 2022, 5, 14605–14637 CrossRef CAS.
- X. Wang, T. Zhang, Y. Lou and Y. Zhao, Mater. Chem. Front., 2019, 3, 365–375 RSC.
- S. P. Santoso, S.-P. Lin, T.-Y. Wang, Y. Ting, C.-W. Hsieh, R.-C. Yu, A. E. Angkawijaya, F. E. Soetaredjo, H.-Y. Hsu and K.-C. Cheng, Int. J. Biol. Macromol., 2021, 175, 526–534 CrossRef CAS PubMed.
- C. H. Mak, Y. Peng, M. H. Chong, L. Yu, M. Du, L. Ji, X. Zou, G. Zou, H.-H. Shen and S. P. Santoso, J. Mater. Chem. C, 2023, 11, 11303–11311 RSC.
- H.-Y. Hsu, H.-H. Hsieh, H.-Y. Tuan and J.-L. Hwang, Sol. Energy Mater. Sol. Cells, 2010, 94, 955–959 CrossRef CAS.
- C.-N. Wu, L.-C. Sun, Y.-L. Chu, R.-C. Yu, C.-W. Hsieh, H.-Y. Hsu, F.-C. Hsu and K.-C. Cheng, Food Chem., 2020, 330, 127244 CrossRef CAS.
- A. Andreas, Z. G. Winata, S. P. Santoso, A. E. Angkawijaya, M. Yuliana, F. E. Soetaredjo, S. Ismadji, H.-Y. Hsu, A. W. Go and Y.-H. Ju, J. Mol. Liq., 2021, 329, 115579 CrossRef CAS.
- X. Yang, J. Yang, M. I. Ullah, Y. Xia, G. Liang, S. Wang, J. Zhang, H.-Y. Hsu, H. Song and J. Tang, ACS Appl. Mater. Interfaces, 2020, 12, 42217–42225 CrossRef CAS PubMed.
- C. H. Mak, X. Han, M. Du, J.-J. Kai, K. F. Tsang, G. Jia, K.-C. Cheng, H.-H. Shen and H.-Y. Hsu, J. Mater. Chem. A, 2021, 9, 4454–4504 RSC.
- E. Luévano-Hipólito, O. L. Quintero-Lizárraga and L. M. Torres-Martínez, Catalysts, 2022, 12(11), 1410 CrossRef.
- C. H. Mak, R. Liu, X. Han, Y. Tang, X. Zou, H. H. Shen, Y. Meng, G. Zou and H. Y. Hsu, Adv. Opt. Mater., 2020, 8, 2001023 CrossRef CAS.
- H.-Y. Hsu, L. Ji, M. Du, J. Zhao, T. Y. Edward and A. J. Bard, Electrochim. Acta, 2016, 220, 205–210 CrossRef CAS.
- H.-Y. Hsu, L. Ji, C. Zhang, C. H. Mak, R. Liu, T. Wang, X. Zou, S.-Y. Leu and E. T. Yu, J. Mater. Chem. C, 2018, 6, 11552–11560 RSC.
- C. H. Mak, X. Huang, R. Liu, Y. Tang, X. Han, L. Ji, X. Zou, G. Zou and H.-Y. Hsu, Nano Energy, 2020, 73, 104752 CrossRef CAS.
- S. Rao, X. Zou, S. Wang, Y. Lu, T. Shi, H.-Y. Hsu, Q. Xu and X. Lu, Mater. Chem. Phys., 2019, 232, 6–15 CrossRef CAS.
- Z. Chen, Y. Hu, J. Wang, Q. Shen, Y. Zhang, C. Ding, Y. Bai, G. Jiang, Z. Li and N. Gaponik, Chem. Mater., 2020, 32, 1517–1525 CrossRef CAS.
- M. A. Raza, F. Li, M. Que, L. Zhu and X. Chen, Mater. Adv., 2021, 2, 7187–7209 RSC.
- C. B. Hiragond, N. S. Powar and S.-I. In, Nanomaterials, 2020, 10, 2569 CrossRef CAS.
- Z. Liu, H. Yang, J. Wang, Y. Yuan, K. Hills-Kimball, T. Cai, P. Wang, A. Tang and O. Chen, Nano Lett., 2021, 21, 1620–1627 CrossRef CAS.
- E. Luévano-Hipólito, M. G. Fabela-Cedillo and L. M. Torres-Martínez, Mater. Lett., 2024, 136066 CrossRef.
- K. Zhang, Y. Zhang, D. Zhou, Y. Yang, Z. Yang, Z. Song, J. Zhang, Q. Wang and J. Qiu, J. Alloys Compd., 2024, 976, 173283 CrossRef CAS.
- Y.-F. Mu, J.-S. Zhao, L.-Y. Wu, K.-Y. Tao, Z.-L. Liu, F.-Q. Bai, D.-C. Zhong, M. Zhang and T.-B. Lu, Appl. Catal., B, 2023, 338, 123024 CrossRef CAS.
- J. Zhou, D. Wu, C. Tian, Z. Liang, H. Ran, B. Gao, Z. Luo, Q. Huang and X. Tang, Small, 2023, 19, 2207915 CrossRef CAS.
- H.-B. Zhao, J.-F. Liao, Y. Teng, H.-Y. Chen and D.-B. Kuang, ACS Appl. Mater. Interfaces, 2022, 14, 43354–43361 CrossRef CAS PubMed.
- G. Chen, P. Wang, Y. Wu, Q. Zhang, Q. Wu, Z. Wang, Z. Zheng, Y. Liu, Y. Dai and B. Huang, Adv. Mater., 2020, 32, 2001344 CrossRef CAS.
- J. Pi, X. Jia, Z. Long, S. Yang, H. Wu, D. Zhou, Q. Wang, H. Zheng, Y. Yang and J. Zhang, Adv. Energy Mater., 2022, 12, 2202074 CrossRef CAS.
- Y. Tang, C. H. Mak, J. Zhang, G. Jia, K.-C. Cheng, H. Song, M. Yuan, S. Zhao, J.-J. Kai, J. C. Colmenares and H.-Y. Hsu, Adv. Mater., 2023, 35, 2207835 CrossRef CAS PubMed.
- C. A. Gueymard, Sol. Energy, 2004, 76, 423–453 CrossRef.
- V. K. Ravi, G. B. Markad and A. Nag, ACS Energy Lett., 2016, 1, 665–671 CrossRef CAS.
- P. Wang, X. Ba, X. Zhang, H. Gao, M. Han, Z. Zhao, X. Chen, L. Wang, X. Diao and G. Wang, Chem. Eng. J., 2023, 457, 141248 CrossRef CAS.
- J. Fu, K. Jiang, X. Qiu, J. Yu and M. Liu, Mater. Today, 2020, 32, 222–243 CrossRef CAS.
- Y. Wang, Q. Zhou, Y. Zhu and D. Xu, Appl. Catal., B, 2021, 294, 120236 CrossRef CAS.
- M. Que, Y. Zhao, L. Pan, Y. Yang, Z. He, H. Yuan, J. Chen and G. Zhu, Mater. Lett., 2021, 282, 128695 CrossRef CAS.
- J. Sheng, Y. He, J. Li, C. Yuan, H. Huang, S. Wang, Y. Sun, Z. Wang and F. Dong, ACS Nano, 2020, 14, 13103–13114 CrossRef CAS PubMed.
- Y. Wang, H. Huang, Z. Zhang, C. Wang, Y. Yang, Q. Li and D. Xu, Appl. Catal., B, 2021, 282, 119570 CrossRef CAS.
- X. Wang, K. Li, J. He, J. Yang, F. Dong, W. Mai and M. Zhu, Nano Energy, 2020, 78, 105388 CrossRef CAS.
- Z.-C. Kong, H.-H. Zhang, J.-F. Liao, Y.-J. Dong, Y. Jiang, H.-Y. Chen and D.-B. Kuang, Sol. RRL, 2020, 4, 1900365 CrossRef CAS.
- B. Zhou, X. Xu, M. Li, L. Wu, S. Xu, L. Yuan, Y. Chong, W. Xie, P. Liu, D. Ye, G. I. N. Waterhouse, Y. Qiu, G. Chen, T. Shi and K. Yan, Chem. Eng. J., 2023, 468, 143754 CrossRef CAS.
- S. Shyamal, S. K. Dutta and N. Pradhan, J. Phys. Chem. Lett., 2019, 10, 7965–7969 CrossRef CAS.
- S. S. Bhosale, A. K. Kharade, E. Jokar, A. Fathi, S.-m. Chang and E. W.-G. Diau, J. Am. Chem. Soc., 2019, 141, 20434–20442 CrossRef CAS.
- Y.-W. Liu, S.-H. Guo, S.-Q. You, C.-Y. Sun, X.-L. Wang, L. Zhao and Z.-M. Su, Nanotechnology, 2020, 31, 215605 CrossRef CAS PubMed.
- S. Wan, M. Ou, Q. Zhong and X. Wang, Chem. Eng. J., 2019, 358, 1287–1295 CrossRef CAS.
- N. Zhang, J.-J. Li, Y. Li, H. Wang, J.-Y. Zhang, Y. Liu, Y.-Z. Fang, Z. Liu and M. Zhou, J. Colloid Interface Sci., 2022, 608, 3192–3203 CrossRef CAS PubMed.
- L. Wu, S. Zheng, H. Lin, S. Zhou, A. Mahmoud Idris, J. Wang, S. Li and Z. Li, J. Colloid Interface Sci., 2023, 629, 233–242 CrossRef CAS.
- Y.-F. Mu, W. Zhang, G.-X. Dong, K. Su, M. Zhang and T.-B. Lu, Small, 2020, 16, 2002140 CrossRef CAS.
- H. Huang, J. Zhao, Y. Du, C. Zhou, M. Zhang, Z. Wang, Y. Weng, J. Long, J. Hofkens, J. A. Steele and M. B. J. Roeffaers, ACS Nano, 2020, 14, 16689–16697 CrossRef CAS.
- T. Chen, M. Zhou, W. Chen, Y. Zhang, S. Ou and Y. Liu, Sustainable Energy Fuels, 2021, 5, 3598–3605 RSC.
- Y.-F. Xu, X.-D. Wang, J.-F. Liao, B.-X. Chen, H.-Y. Chen and D.-B. Kuang, Adv. Mater. Interfaces, 2018, 5, 1801015 CrossRef.
- L.-Y. Wu, Y.-F. Mu, X.-X. Guo, W. Zhang, Z.-M. Zhang, M. Zhang and T.-B. Lu, Angew. Chem., Int. Ed., 2019, 58, 9491–9495 CrossRef CAS PubMed.
- Y. Jiang, J.-F. Liao, H.-Y. Chen, H.-H. Zhang, J.-Y. Li, X.-D. Wang and D.-B. Kuang, Chem, 2020, 6, 766–780 CAS.
- Z.-C. Kong, J.-F. Liao, Y.-J. Dong, Y.-F. Xu, H.-Y. Chen, D.-B. Kuang and C.-Y. Su, ACS Energy Lett., 2018, 3, 2656–2662 CrossRef CAS.
- Q.-M. Sun, J.-J. Xu, F.-F. Tao, W. Ye, C. Zhou, J.-H. He and J.-M. Lu, Angew. Chem., Int. Ed., 2022, 61, e202200872 CrossRef CAS.
- N. Li, J. Wang, G. Zhao, J. Du, Y. Li, Y. Bai, Z. Li and Y. Xiong, ACS Mater. Lett., 2024, 6, 999–1006 CrossRef CAS.
- H.-B. Zhao, J.-N. Huang, Q. Qin, H.-Y. Chen and D.-B. Kuang, Small, 2023, 19, 2302022 CrossRef CAS.
- S. Kumar, I. Hassan, M. Regue, S. Gonzalez-Carrero, E. Rattner, M. A. Isaacs and S. Eslava, J. Mater. Chem. A, 2021, 9, 12179–12187 RSC.
- S. Kumar, M. Regue, M. A. Isaacs, E. Freeman and S. Eslava, ACS Appl. Energy Mater., 2020, 3, 4509–4522 CrossRef CAS.
- X.-D. Wang, Y.-H. Huang, J.-F. Liao, Y. Jiang, L. Zhou, X.-Y. Zhang, H.-Y. Chen and D.-B. Kuang, J. Am. Chem. Soc., 2019, 141, 13434–13441 CrossRef CAS.
- J. Hou, S. Cao, Y. Wu, Z. Gao, F. Liang, Y. Sun, Z. Lin and L. Sun, Chem.–Eur. J., 2017, 23, 9481–9485 CrossRef CAS PubMed.
- L.-Y. Wu, M.-R. Zhang, Y.-X. Feng, W. Zhang, M. Zhang and T.-B. Lu, Sol. RRL, 2021, 5, 2100263 CrossRef CAS.
- R. Tang, H. Sun, Z. Zhang, L. Liu, F. Meng, X. Zhang, W. Yang, Z. Li, Z. Zhao, R. Zheng and J. Huang, Chem. Eng. J., 2022, 429, 132137 CrossRef CAS.
- Y. Xi, X. Zhang, Y. Shen, W. Dong, Z. Fan, K. Wang, S. Zhong and S. Bai, Appl. Catal., B, 2021, 297, 120411 CrossRef CAS.
- S. Bera, S. Shyamal and N. Pradhan, J. Am. Chem. Soc., 2021, 143, 14895–14906 CrossRef CAS.
- C. Lu, D. S. Itanze, A. G. Aragon, X. Ma, H. Li, K. B. Ucer, C. Hewitt, D. L. Carroll, R. T. Williams, Y. Qiu and S. M. Geyer, Nanoscale, 2020, 12, 2987–2991 RSC.
- S. Shyamal, S. K. Dutta, T. Das, S. Sen, S. Chakraborty and N. Pradhan, J. Phys. Chem. Lett., 2020, 11, 3608–3614 CrossRef CAS PubMed.
- L. Zhou, Y.-F. Xu, B.-X. Chen, D.-B. Kuang and C.-Y. Su, Small, 2018, 14, 1703762 CrossRef.
- J.-C. Wang, N. Li, A. M. Idris, J. Wang, X. Du, Z. Pan and Z. Li, Sol. RRL, 2021, 5, 2100154 CrossRef CAS.
- J. Sheng, Y. He, M. Huang, C. Yuan, S. Wang and F. Dong, ACS Catal., 2022, 12, 2915–2926 CrossRef CAS.
- C. Tian, Q. Huang, D. Wu, J. Lai, F. Qi, N. Zhang, W. Zhang and X. Tang, Mater. Today Energy, 2022, 28, 101067 CrossRef CAS.
- S.-H. Guo, J. Zhou, X. Zhao, C.-Y. Sun, S.-Q. You, X.-L. Wang and Z.-M. Su, J. Catal., 2019, 369, 201–208 CrossRef CAS.
- D. Wu, X. Zhao, Y. Huang, J. Lai, H. Li, J. Yang, C. Tian, P. He, Q. Huang and X. Tang, Chem. Mater., 2021, 33, 4971–4976 CrossRef CAS.
- D. Wu, X. Zhao, Y. Huang, J. Lai, J. Yang, C. Tian, P. He, Q. Huang and X. Tang, J. Phys. Chem. C, 2021, 125, 18328–18333 CrossRef CAS.
- G.-X. Dong, W. Zhang, Y.-F. Mu, K. Su, M. Zhang and T.-B. Lu, Chem. Commun., 2020, 56, 4664–4667 RSC.
- Y.-X. Chen, Y.-F. Xu, X.-D. Wang, H.-Y. Chen and D.-B. Kuang, Sustainable Energy Fuels, 2020, 4, 2249–2255 RSC.
- J. Cheng, Y. Mu, L. Wu, Z. Liu, K. Su, G. Dong, M. Zhang and T. Lu, Nano Res., 2022, 15, 1845–1852 CrossRef CAS.
- Y.-F. Xu, M.-Z. Yang, H.-Y. Chen, J.-F. Liao, X.-D. Wang and D.-B. Kuang, ACS Appl. Energy Mater., 2018, 1, 5083–5089 CrossRef CAS.
- A. Pan, X. Ma, S. Huang, Y. Wu, M. Jia, Y. Shi, Y. Liu, P. Wangyang, L. He and Y. Liu, J. Phys. Chem. Lett., 2019, 10, 6590–6597 CrossRef CAS PubMed.
- Y. Jiang, J.-F. Liao, Y.-F. Xu, H.-Y. Chen, X.-D. Wang and D.-B. Kuang, J. Mater. Chem. A, 2019, 7, 13762–13769 RSC.
- X.-X. Guo, S.-F. Tang, Y.-F. Mu, L.-Y. Wu, G.-X. Dong and M. Zhang, RSC Adv., 2019, 9, 34342–34348 RSC.
- Z. Zhang, B. Wang, H.-B. Zhao, J.-F. Liao, Z.-C. Zhou, T. Liu, B. He, Q. Wei, S. Chen, H.-Y. Chen, D.-B. Kuang, Y. Li and G. Xing, Appl. Catal., B, 2022, 312, 121358 CrossRef CAS.
- X. Wang, J. He, L. Mao, X. Cai, C. Sun and M. Zhu, Chem. Eng. J., 2021, 416, 128077 CrossRef CAS.
- M. Ou, W. Tu, S. Yin, W. Xing, S. Wu, H. Wang, S. Wan, Q. Zhong and R. Xu, Angew. Chem., Int. Ed., 2018, 57, 13570–13574 CrossRef CAS PubMed.
- J. Wang, J. Wang, N. Li, X. Du, J. Ma, C. He and Z. Li, ACS Appl. Mater. Interfaces, 2020, 12, 31477–31485 CrossRef CAS PubMed.
- F. Xu, K. Meng, B. Cheng, S. Wang, J. Xu and J. Yu, Nat. Commun., 2020, 11, 4613 CrossRef CAS PubMed.
- S. Purohit, S. Singh, K. L. Yadav, K. K. Pant and S. Satapathi, ACS Appl. Energy Mater., 2023, 6, 5580–5587 CrossRef CAS.
- Z.-L. Liu, R.-R. Liu, Y.-F. Mu, Y.-X. Feng, G.-X. Dong, M. Zhang and T.-B. Lu, Sol. RRL, 2021, 5, 2000691 CrossRef CAS.
- Y.-X. Feng, G.-X. Dong, K. Su, Z.-L. Liu, W. Zhang, M. Zhang and T.-B. Lu, J. Energy Chem., 2022, 69, 348–355 CrossRef CAS.
- X. Wang, Z. Wang, Y. Li, J. Wang and G. Zhang, Appl. Catal., B, 2022, 319, 121895 CrossRef CAS.
- Y. Zhang, L. Shi, H. Yuan, X. Sun, X. Li, L. Duan, Q. Li, Z. Huang, X. Ban and D. Zhang, Chem. Eng. J., 2022, 430, 132820 CrossRef CAS.
- Z. Dong, Z. Zhang, Y. Jiang, Y. Chu and J. Xu, Chem. Eng. J., 2022, 433, 133762 CrossRef CAS.
- K. Su, G.-X. Dong, W. Zhang, Z.-L. Liu, M. Zhang and T.-B. Lu, ACS Appl. Mater. Interfaces, 2020, 12, 50464–50471 CrossRef CAS.
- Z. Zhang, M. Shu, Y. Jiang and J. Xu, Chem. Eng. J., 2021, 414, 128889 CrossRef CAS.
- L. Li, Z. Zhang, C. Ding and J. Xu, Chem. Eng. J., 2021, 419, 129543 CrossRef CAS.
- Z. Cui, P. Wang, Y. Wu, X. Liu, G. Chen, P. Gao, Q. Zhang, Z. Wang, Z. Zheng, H. Cheng, Y. Liu, Y. Dai and B. Huang, Appl. Catal., B, 2022, 310, 121375 CrossRef CAS.
- R. Cheng, C. C. Chung, S. Wang, B. Cao, M. Zhang, C. Chen, Z. Wang, M. Chen, S. Shen and S. P. Feng, Mater. Today Phys., 2021, 17, 100358 CrossRef CAS.
- Q. Chen, Y. Ma, L. Wang, X. Lan and J. Shi, Sol. RRL, 2021, 5, 2000755 CrossRef CAS.
- Q. Chen, X. Lan, Y. Ma, P. Lu, Z. Yuan and J. Shi, Sol. RRL, 2021, 5, 2100186 CrossRef CAS.
- L. Ding, F. Bai, B. Borjigin, Y. Li, H. Li and X. Wang, Chem. Eng. J., 2022, 446, 137102 CrossRef CAS.
- N. Li, X.-P. Zhai, B. Ma, H.-J. Zhang, M.-J. Xiao, Q. Wang and H.-L. Zhang, J. Mater. Chem. A, 2023, 11, 4020–4029 RSC.
- S. Shyamal, S. K. Dutta, T. Das, S. Sen, S. Chakraborty and N. Pradhan, J. Phys. Chem. Lett., 2020, 11(9), 3608–3614 CrossRef CAS PubMed.
- Y. Wang, S. Wang and X. W. Lou, Angew. Chem., Int. Ed., 2019, 58, 17236–17240 CrossRef CAS PubMed.
- J. Yu, J. Low, W. Xiao, P. Zhou and M. Jaroniec, J. Am. Chem. Soc., 2014, 136, 8839–8842 CrossRef CAS.
- L. Li, P. A. Salvador and G. S. Rohrer, Nanoscale, 2014, 6, 24–42 RSC.
- S. Selcuk and A. Selloni, Nat. Mater., 2016, 15, 1107–1112 CrossRef CAS PubMed.
- S. Bai, J. Jiang, Q. Zhang and Y. Xiong, Chem. Soc. Rev., 2015, 44, 2893–2939 RSC.
- J. Yuan, A. Hazarika, Q. Zhao, X. Ling, T. Moot, W. Ma and J. M. Luther, Joule, 2020, 4, 1160–1185 CrossRef CAS.
- C.-C. Lin, T.-R. Liu, S.-R. Lin, K. M. Boopathi, C.-H. Chiang, W.-Y. Tzeng, W.-H. C. Chien, H.-S. Hsu, C.-W. Luo and H.-Y. Tsai, J. Am. Chem. Soc., 2022, 144, 15718–15726 CrossRef CAS PubMed.
- C. Tang, C. Chen, W. Xu and L. Xu, J. Mater. Chem. A, 2019, 7, 6911–6919 RSC.
- C. Tang, C. Chen, W. Xu and L. Xu, J. Mater. Chem. A, 2019, 7, 6911–6919 RSC.
- G. Huang, C. Wang, S. Xu, S. Zong, J. Lu, Z. Wang, C. Lu and Y. Cui, Adv. Mater., 2017, 29, 1700095 CrossRef PubMed.
- S. Wan, M. Ou, Q. Zhong and X. Wang, Chem. Eng. J., 2019, 358, 1287–1295 CrossRef CAS.
- Q. Xu, L. Zhang, B. Cheng, J. Fan and J. Yu, Chem, 2020, 6, 1543–1559 CAS.
- J. W. Maina, C. Pozo-Gonzalo, L. Kong, J. Schütz, M. Hill and L. F. Dumée, Mater. Horiz., 2017, 4, 345–361 RSC.
- L. Zhang, J. Zhang, H. Yu and J. Yu, Adv. Mater., 2022, 34, 2107668 CrossRef CAS.
- M. S. Sena, J. Cui, Y. Baghdadi, E. Rattner, M. Daboczi, A. L. Lopes-Moriyama, A. G. dos Santos and S. Eslava, ACS Appl. Energy Mater., 2023, 6, 10193–10204 CrossRef CAS PubMed.
- H. Xiao, Q. Qian and Z. Zang, Sci. China Mater., 2023, 66, 1810–1819 CrossRef CAS.
- S. Fan, Q. Yang, G. Yin, X. Qi, Y. Feng, J. Ding, Q. Peng, Y. Qu, Q. Wang, Y. Shen, M. Wang and X. Gong, Small, 2024, 2311978 CrossRef CAS PubMed.
- B. Zhou, S. Xu, L. Wu, M. Li, Y. Chong, Y. Qiu, G. Chen, Y. Zhao, C. Feng, D. Ye and K. Yan, Small, 2023, 19, 2302058 CrossRef CAS.
- Y. Feng, D. Chen, Y. Zhong, Z. He, S. Ma, H. Ding, W. Ao, X. Wu and M. Niu, ACS Appl. Mater. Interfaces, 2023, 15, 9221–9230 CrossRef CAS PubMed.
- L. Y. Wu, Y. F. Mu, X. X. Guo, W. Zhang, Z. M. Zhang, M. Zhang and T. B. Lu, Angew. Chem., Int. Ed., 2019, 58, 9491–9495 CrossRef CAS.
- A. Dey, J. Ye, A. De, E. Debroye, S. K. Ha, E. Bladt, A. S. Kshirsagar, Z. Wang, J. Yin, Y. Wang, L. N. Quan, F. Yan, M. Gao, X. Li, J. Shamsi, T. Debnath, M. Cao, M. A. Scheel, S. Kumar, J. A. Steele, M. Gerhard, L. Chouhan, K. Xu, X.-g. Wu, Y. Li, Y. Zhang, A. Dutta, C. Han, I. Vincon, A. L. Rogach, A. Nag, A. Samanta, B. A. Korgel, C.-J. Shih, D. R. Gamelin, D. H. Son, H. Zeng, H. Zhong, H. Sun, H. V. Demir, I. G. Scheblykin, I. Mora-Seró, J. K. Stolarczyk, J. Z. Zhang, J. Feldmann, J. Hofkens, J. M. Luther, J. Pérez-Prieto, L. Li, L. Manna, M. I. Bodnarchuk, M. V. Kovalenko, M. B. J. Roeffaers, N. Pradhan, O. F. Mohammed, O. M. Bakr, P. Yang, P. Müller-Buschbaum, P. V. Kamat, Q. Bao, Q. Zhang, R. Krahne, R. E. Galian, S. D. Stranks, S. Bals, V. Biju, W. A. Tisdale, Y. Yan, R. L. Z. Hoye and L. Polavarapu, ACS Nano, 2021, 15, 10775–10981 CrossRef CAS.
- Z. Zhang, B. Wang, H.-B. Zhao, J.-F. Liao, Z.-C. Zhou, T. Liu, B. He, Q. Wei, S. Chen, H.-Y. Chen, D.-B. Kuang, Y. Li and G. Xing, Appl. Catal., B, 2022, 312, 121358 CrossRef CAS.
- N. Zhang, J.-J. Li, Y. Li, H. Wang, J.-Y. Zhang, Y. Liu, Y.-Z. Fang, Z. Liu and M. Zhou, J. Colloid Interface Sci., 2022, 608, 3192–3203 CrossRef CAS.
- Z. Zhang, M. Wang, Z. Chi, W. Li, H. Yu, N. Yang and H. Yu, Appl. Catal., B, 2022, 313, 121426 CrossRef CAS.
- C. Tian, Q. Huang, D. Wu, J. Lai, F. Qi, N. Zhang, W. Zhang and X. Tang, Mater. Today Energy, 2022, 28, 101067 CrossRef CAS.
- X. Wang, J. He, L. Mao, X. Cai, C. Sun and M. Zhu, Chem. Eng. J., 2021, 416, 128077 CrossRef CAS.
- Y. Wang, H. Huang, Z. Zhang, C. Wang, Y. Yang, Q. Li and D. Xu, Appl. Catal., 2021, 282, 119570 CrossRef CAS.
- J.-F. Liao, Y.-T. Cai, J.-Y. Li, Y. Jiang, X.-D. Wang, H.-Y. Chen and D.-B. Kuang, J. Energy Chem., 2021, 53, 309–315 CrossRef CAS.
- D. Laishram, S. Zeng, K. M. Alam, A. P. Kalra, K. Cui, P. Kumar, R. K. Sharma and K. Shankar, Appl. Surf. Sci., 2022, 592, 153276 CrossRef CAS.
- J. Wang, L. Xiong, Y. Bai, Z. Chen, Q. Zheng, Y. Shi, C. Zhang, G. Jiang and Z. Li, Sol. RRL, 2022, 6, 2200294 CrossRef CAS.
- B. N. Choi, J. Y. Seo, Z. An, P. J. Yoo and C.-H. Chung, Chem. Eng. J., 2022, 430, 132807 CrossRef CAS.
- Y. Tang, C. H. Mak, G. Jia, K.-C. Cheng, J.-J. Kai, C.-W. Hsieh, F. Meng, W. Niu, F.-F. Li, H.-H. Shen, X. Zhu, H. M. Chen and H.-Y. Hsu, J. Mater. Chem. A, 2022, 10, 12296–12316 RSC.
- Z. Chen, H.-Y. Hsu, M. Arca and K. S. Schanze, J. Phys. Chem. B, 2015, 119, 7198–7209 CrossRef CAS.
- F. Meng, Y. Jia, J. Wang, X. Huang, Z. Gui, L. Huang, R. Li, R. Chen, J. Xu, W. Chen, Z. He, H.-Y. Hsu, E. Zhu, G. Che and H.-L. Wang, Sol. RRL, 2019, 3, 1900319 CrossRef CAS.
- S. Ghosh and B. Pradhan, ChemNanoMat, 2019, 5, 300–312 CrossRef CAS.
- L. Protesescu, S. Yakunin, S. Kumar, J. Bär, F. Bertolotti, N. Masciocchi, A. Guagliardi, M. Grotevent, I. Shorubalko, M. I. Bodnarchuk, C.-J. Shih and M. V. Kovalenko, ACS Nano, 2017, 11, 3119–3134 CrossRef CAS PubMed.
- W. Song, G. Qi and B. Liu, J. Mater. Chem. A, 2023, 11, 12482–12498 RSC.
- Y. Peng, C. H. Mak, J.-J. Kai, M. Du, L. Ji, M. Yuan, X. Zou, H.-H. Shen, S. P. Santoso, J. C. Colmenares and H.-Y. Hsu, J. Mater. Chem. A, 2021, 9, 26628–26649 RSC.
| This journal is © The Royal Society of Chemistry 2024 |