
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A surface passivated fluorinated polymer nanocomposite for carbon monoxide resistant plasmonic hydrogen sensing†
I.
Östergren
a,
I.
Darmadi
 bc,
S.
Lerch
a,
R. R.
da Silva
a,
M.
Craighero
a,
S. H. K.
Paleti
a,
K.
Moth-Poulsen
adef,
C.
Langhammer
*b and
C.
Müller
*a
bc,
S.
Lerch
a,
R. R.
da Silva
a,
M.
Craighero
a,
S. H. K.
Paleti
a,
K.
Moth-Poulsen
adef,
C.
Langhammer
*b and
C.
Müller
*a
aDepartment of Chemistry and Chemical Engineering, Chalmers University of Technology, 412 96 Göteborg, Sweden. E-mail: christian.muller@chalmers.se
bDepartment of Physics, Chalmers University of Technology, 412 96 Göteborg, Sweden. E-mail: clangham@chalmers.se
cResearch Center for Photonics, National Research and Innovation Agency, BJ Habibie Science and Technology Park, 15314 South Tangerang, Indonesia
dInstitute of Materials Science of Barcelona, ICMAB-CSIC, 08193, Bellaterra, Barcelona, Spain
eCatalan Institution for Research and Advanced Studies ICREA, Pg. Lluís Companys 23, Barcelona, Spain
fDepartment of Chemical Engineering, Universitat Politècnica de Catalunya, EEBE, Eduard Maristany 10–14, 08019 Barcelona, Spain
First published on 29th February 2024
Abstract
Plasmonic hydrogen sensors are promising safety monitoring devices for the emerging hydrogen economy provided a fast response time and poisoning resistance can be achieved. Nanocomposites composed of palladium nanoparticles embedded in a polymer matrix facilitate rapid hydrogen diffusion if a fluorinated polymer is used, while a denser polymer such as atactic poly(methyl methacrylate) (PMMA) facilitates a high degree of gas selectivity. However, nanocomposites that combine a fast response with poisoning resistance have not yet been realized. Here, these two properties are achieved simultaneously by modifying the surface of a fluorinated polymer nanocomposite with a thin PMMA coating, which functions as a molecular sieve that effectively blocks carbon monoxide. The resulting surface passivated nanocomposite shows a high degree of poisoning resistance without compromising a fast sensing response of 2–3 seconds upon exposure to 100 mbar of hydrogen. The sensor signal and response are preserved over 55 cycles of synthetic air containing 5% hydrogen and 500 ppm of carbon monoxide, indicating that nanocomposites are a viable approach for the realization of robust hydrogen sensors.
Introduction
Hydrogen is important for a wide range of industrial processes, such as fossil-free steel production and chemical synthesis. Furthermore, when produced using, for example, green electricity, hydrogen is a carbon-free energy source that can either be combusted or used in fuel cells creating little to zero emissions other than water.1 However, hydrogen needs to be handled with caution since it has a low ignition energy and a wide flammability range of 4–74% in air.2 Furthermore, hydrogen has been identified as a potential ozone precursor in the atmosphere, which would make it an indirect greenhouse gas if leaked in large amounts.3 Early detection and prevention of leakage is therefore of high importance for addressing both safety and environmental concerns. Thus, hydrogen sensors that can detect leaks and ease process monitoring are needed to ensure safety along the entire value chain that ranges from hydrogen production to storage and end use. Consequently, various industries and national governments have set specific performance criteria for hydrogen sensors in general and leakage detection sensors in particular. Here, fast response times ranging from 1 to 30 s depending on the specific application4,5 and a lifetime of 10 years in ambient air, which also contains air pollutants such as CO, NOx and SOx,6 are two of the most important but at the same time challenging performance targets.To date, multiple hydrogen sensing technologies exist that encompass (1) catalytic sensors, which monitor the temperature or resistance change caused by the catalytic combustion of hydrogen with oxygen,7,8 (2) electrical sensors, that exploit changes in resistance induced by hydrogen absorption into a metal, typically Pd or a Pd-based alloy,9,10 (3) mechanical sensors, where hydrogen absorption strains a host metal (again typically Pd-based) due to the induced volume expansion,11 and (4) optical sensors that use changes in the optical contrast of thin metal films or metal nanoparticles induced by hydrogen absorption.12–14 Among optical sensors, so-called plasmonic hydrogen sensors based on Pd or Pd-alloy nanoparticles are of particular interest since they offer a selective and ultrafast sub-second response to hydrogen (see Fig. 1)15,16 with limits of detection down to a few hundred parts per billion,17 paired with optical fiber compatibility, which enables remote sensing in confined spaces without any risk for spark generation by the sensor electronics.18
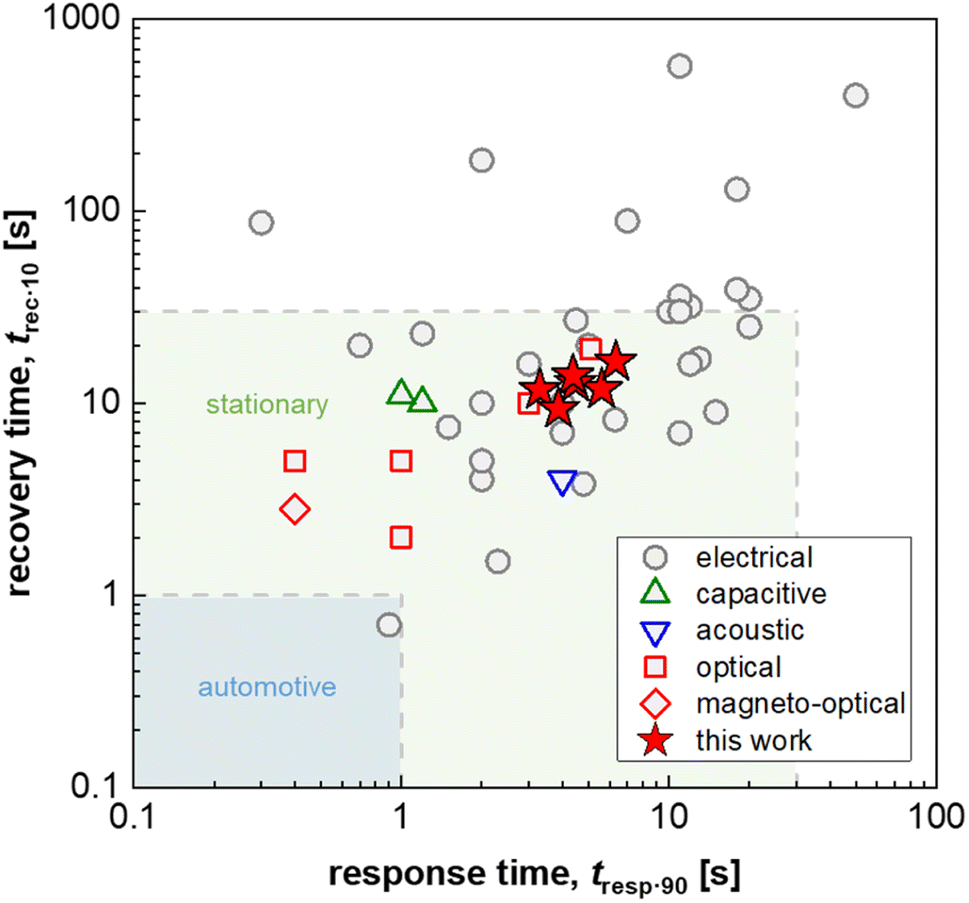 | ||
| Fig. 1 Response and recovery time, t90·resp and t10·rec, i.e. the time to reach 90% of the sensor response and the time to lose all but 10% of the sensor response, respectively, of fast H2 sensors with an electrical, capacitive, acoustic, optical or magneto-optical transducer platform (1 mbar H2 or t90·resp ≤ 5 s; data from ref. 16 and 25–29) as well as Δt90·resp = t90·resp − t10·resp and Δt10·rec = t10·rec − t90·rec values for PMMA coated Teflon AF:Pd nanoparticle composites obtained in this work (100 mbar H2); for automotive and stationary industrial applications a H2 sensor response/recovery time of not more than 1 s and 30 s, respectively, are required.16 | ||
At the same time, a drawback of Pd-based hydrogen sensors in general, and of Pd-based plasmonic sensors in particular, is the susceptibility of Pd surfaces to CO poisoning at ambient conditions, e.g., due to the presence of CO molecules in urban air.6 Mechanistically, CO blocks the ability for hydrogen molecules to dissociate on a Pd surface and therefore hinders hydrogen absorption into the Pd lattice.19 To address this challenge multiple strategies have been developed, including alloying of Pd with Cu to weaken the CO–sensor surface interaction to eliminate poisoning20,21 or the creation of a protective diffusion barrier for CO, as well as other molecular species that include NO2, CO2 and CH4, by applying, e.g., a poly(methyl methacrylate) (PMMA) thin film onto the sensor surface.17,22
Inspired by the effectiveness of polymer coatings to protect Pd-based hydrogen sensors from chemical deactivation/poisoning, polymer–metal nanoparticle nanocomposites – plasmonic plastics – have recently emerged as an intriguing alternative that allows to augment the processing toolbox that can be used to fabricate plasmonic devices in general with scalable techniques common in the plastics industry such as melt compounding, extrusion, and fused deposition modeling (FDM) 3D printing, and to prepare 3D printed plasmonic hydrogen sensors.23–25 Specifically, and as a key advantage, such polymer nanocomposites enable one step processing of the active sensing elements and the protective polymer coating material, as we have demonstrated recently using colloidal Pd or PdAu alloy nanoparticles as plasmonic hydrogen sensing elements, and PMMA or the fluorinated polymer Teflon AF as the polymer matrix (see Fig. 2 for chemical structures).23–25
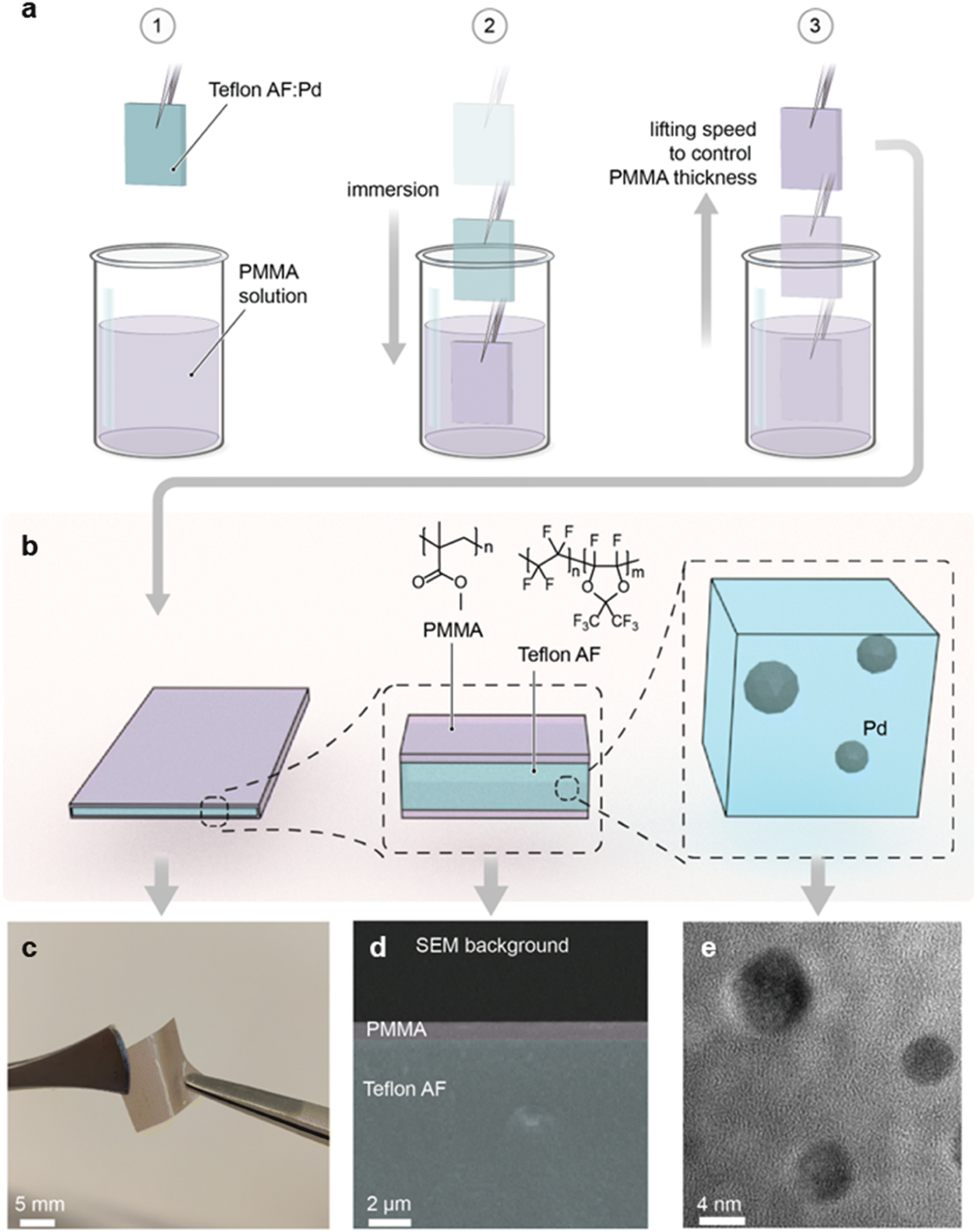 | ||
| Fig. 2 Preparation and characterization of Teflon AF:Pd – PMMA core:shell structures. (a) Schematic of the dip coating process. (b) Architecture of Teflon AF:Pd films coated with PMMA, including the chemical structures of PMMA and Teflon AF. (c) Photograph of a Teflon AF:Pd film coated with PMMA. (d) False-colored SEM images of the cross section of a Teflon AF:Pd film dip coated with PMMA at 5 mm s−1. (e) TEM image of Pd nanoparticles in Teflon AF. | ||
The choice of matrix polymer critically influences both the response time and poisoning/deactivation resistance. With regard to the response time, semicrystalline polymers such as poly(vinylidene difluoride) (PVDF) would result in too slow sensors. Instead, the use of amorphous atactic PMMA allowed the fabrication of reasonably fast sensors with a response time of down to 12 s.24 The selection of an amorphous fluoropolymer with high H2 permeability, Teflon AF, improved the response time down to only a couple of seconds for a pressure step from vacuum to 100 mbar,25 which is similarly fast compared to other state-of-the-art H2 sensors based on an electrical, capacitive, acoustical optical or magneto-optical transducer platform and approaches the sensing speed required for stationary applications (Fig. 1).16 Mechanistically, the fast response can be understood on the basis of the significantly larger fractional free volume of Teflon AF compared to atactic PMMA, which significantly enhances the diffusion rate of molecular hydrogen through the polymer matrix to the dispersed Pd nanoparticle sensing elements.25 However, the larger fractional free volume, which enhances H2 diffusion, also allows larger molecules, such as CO, to diffuse through the polymer more efficiently, which in turn means that the molecular filtering function is reduced, and that CO poisoning is no longer suppressed.
Here, we demonstrate that this trade-off between response time and poisoning resistance can be resolved by applying a selective coating to a polymer nanocomposite-based plasmonic hydrogen sensor. We use colloidal Pd nanoparticles as sensing elements, which we combine with the two polymers Teflon AF and PMMA in a core:shell-type fashion. As a result, bulk-processed polymer nanocomposite plasmonic hydrogen sensors can be realized, which feature both, a fast response time that is at a par with those of other reported sensor platforms (Fig. 1), and resistance against CO poisoning.
Results and discussion
As the first step, we prepared a Teflon AF:Pd nanocomposite with 0.037 wt% Pd nanoparticle loading according to an earlier reported procedure.25 The colloidal Pd nanoparticles were made using a continuous flow synthesis method,30 which provides cubic nanoparticles stabilized with poly(vinylpyrrolidone) (PVP) and a size of 9–12 nm (ESI Fig. S1†). PVP was chosen as the stabilizing agent since it has a positive impact on the sensing kinetics compared to halide-cationic stabilizers31 and because it improves the compatibility of Pd nanoparticles with the Teflon AF matrix.25 The 0.037 wt% Pd nanoparticle concentration in the composite was selected because it generates a large optical signal and enables a low detection limit, as we have shown earlier.25 To prepare the composite we compounded the Pd nanoparticles and the Teflon AF powder, extruded the mixture, and finally melt pressed the extruded filaments into 100 μm thick nanocomposite films at 250 °C. This thickness was selected because it facilitates a response time of 2–3 s.25To coat the Teflon AF:Pd nanocomposite films with a PMMA barrier layer, we H2 plasma treated samples for 30 s to promote adhesion between Teflon AF and PMMA. Subsequently, we dip coated the melt pressed films in a 10 g L−1 solution of PMMA in anisole for 10 s with an Ossila dip coater, followed by removal of the samples at speeds ranging from 0.1 to 10 mm s−1, which allowed us to investigate the impact of the coating speed on PMMA coverage (Fig. 2a). Finally, the dip coated films were annealed at 110 °C for 5 min in an oven to yield highly flexible Teflon AF:Pd nanocomposite films with a thin PMMA coating on both sides (Fig. 2b and c).
To confirm the presence of the PMMA layer, we recorded cross-sectional scanning electron microscopy (SEM) images of cryofractured samples (Fig. 2d and ESI Fig. S2†). Energy dispersive X-ray spectroscopy (EDX) revealed a strong fluorine peak, assigned to Teflon AF, when the interior is investigated while the same signal is absent in the EDX spectra of the surface coating (ESI Fig. S3†). Finally, to confirm both the structural integrity and dispersion of the Pd nanoparticles in the composite, we acquired transmission electron microscopy (TEM) images of cryo-microtomed melt pressed films, which revealed well-dispersed individual Pd nanoparticles that have undergone slight reshaping from their as-synthesized cubic shape due to heating to 250 °C during the compounding process (Fig. 2e and ESI Fig. S4†). This observation is in agreement with restructuring of bare Pd nanoparticles at 150 to 220 °C, which was previously reported by Pekkari et al.30 Reassuringly, we have previously found that restructuring of Pd nanocubes upon melt compounding of Teflon AF:Pd nanoparticle composites at 250 °C has no adverse effect on the ability to sense H2.25
To investigate the impact of the dip coating speed on the obtained PMMA film thickness, we employed Fourier transform infrared (FTIR) microscopy. FTIR spectra of PMMA feature a characteristic absorption peak at 1734 cm−1 due to the stretch vibration of its carbonyl groups, while Teflon AF does not absorb in this region (ESI Fig. S5a†). Evidently, the intensity of the 1734 cm−1 absorption peak increases with the dip coating speed (Fig. 3a and ESI Fig. S5b†), indicating the formation of a thicker PMMA layer compared to films dip coated at slower speeds. This is in agreement with literature stating that the film thickness is depended on the ratio between the viscous drag and the gravitational forces, which increases with higher withdrawal speeds.32,33 The PMMA layer thickness deduced from cross sectional SEM images (Fig. 2d and ESI Fig. S2†) (for dip coating speed of 1, 5 and 10 mm s−1) scales with the intensity of the FTIR absorption peak at 1734 cm−1 (Fig. 3b), which allowed us to use FTIR to determine the thickness of the thinnest PMMA layers (dip coating speed of 0.5 and 0.1 mm s−1) that cannot be resolved by SEM. We find that dip coating of Teflon AF:Pd nanoparticle films resulted in PMMA coatings with an average thickness ranging from 90 to 720 nm (Fig. 3b). We also performed FTIR mapping of larger areas with a step size of 25 μm, which indicated a continuous PMMA coating but also a gradual variation in coating thickness (ESI Fig. S6†).
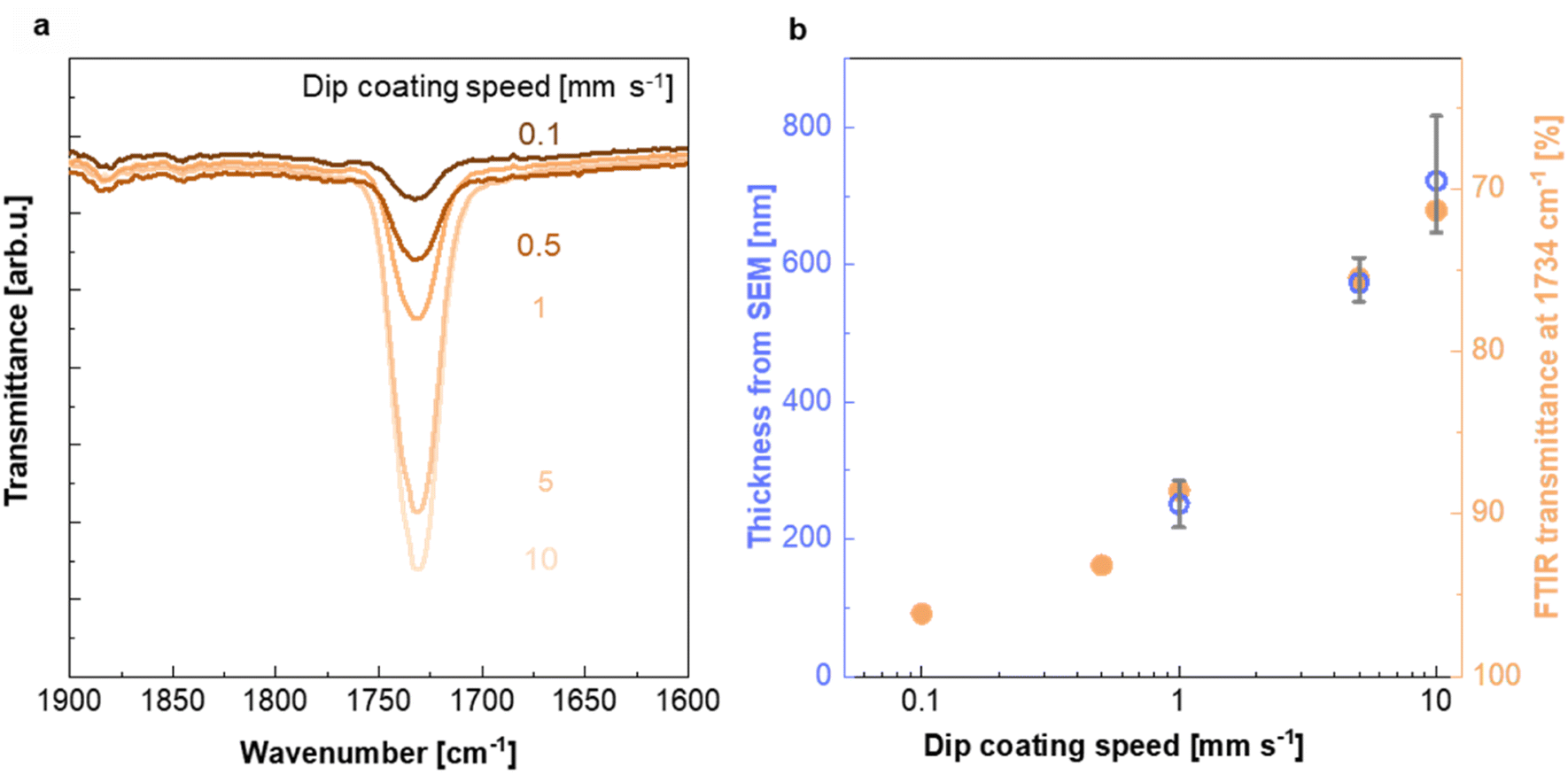 | ||
| Fig. 3 Characterization of the PMMA coating. (a) FTIR absorption peak at 1734 cm−1 recorded for Teflon AF:Pd films coated with PMMA at different dip coating speeds (see ESI Fig. S5† for full FTIR spectra). (b) PMMA layer thickness from SEM and baseline-corrected FTIR transmittance at 1734 cm−1 as a function of dip coating speed; thickness from SEM images (blue circles; mean values and max–min errors of three samples) and FTIR transmittance at 1734 cm−1 (orange circles). | ||
Tensile deformation and nanoindentation were used to investigate the mechanical properties of the PMMA coated Teflon AF:Pd nanoparticle films. For neat Teflon AF:Pd nanocomposite films (100 μm thick) we measured a Young's modulus E = 490 ± 44 MPa and εbreak = 29 ± 4% (ESI Fig. S7†). After dip-coating with PMMA dissolved in anisole, the samples became stiffer with a higher E = 641 ± 123 MPa but lower εbreak = 10 ± 1% (570 nm thick PMMA coating; ESI Fig. S7†). Considering that the PMMA coating is much thinner than the nanocomposite film (note that for PMMA a value of E ≈ 3 GPa is typical), we argue that the observed changes in mechanical properties are mostly the result of the dip coating process. PMMA coated films were also subjected to nanoindentation, which revealed a gradual decrease in reduced modulus with maximum indentation depth hmax from Er ≈ 3.1 GPa for a sample with a 570 nm PMMA coating (hmax = 50 nm; E = (1 − ν2)Er = 2.6 GPa assuming a Poisson's ratio of ν = 0.4) to 1.5 GPa (hmax ≈ 1 μm; ESI Fig. S8†). The latter value is similar to the Er ≈ 1.3 GPa measured for a neat Teflon AF:Pd nanoparticle composite film (hmax ≈ 1.25 μm). We explain the gradual decrease in Er with an increasing contribution from the nanocomposite film as the nanoindenter tip penetrates deeper into the coated sample.
To evaluate the H2 sensing ability of Teflon AF:Pd films coated with PMMA we used a vacuum chamber with optical access for transmittance measurements24 that allows recording of optical extinction spectra as a function of applied hydrogen pressure in real time (Fig. 4a). The sensing steps begin with H2 diffusion through the PMMA coating and the Teflon matrix. Afterwards, the H2 is dissociated on the Pd nanoparticle surface and absorbed by the Pd lattice to form palladium hydride (PdHx).24 The transformation of Pd to PdHx causes a shift in the plasmonic resonance of the nanoparticles. The shift also manifests itself in the extinction spectrum change that is proportional to the amount of hydrogen being absorbed by Pd.34 The corresponding optical response of the coated nanocomposites was recorded in the form of self-referenced optical extinction spectra, ![[small epsilon, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0c6.gif) , defined as the difference between the extinction prior to and after H2 exposure, εPd(λ) and εPdHx(λ, t), normalized by εPd(λ) (Fig. 4b):
, defined as the difference between the extinction prior to and after H2 exposure, εPd(λ) and εPdHx(λ, t), normalized by εPd(λ) (Fig. 4b):
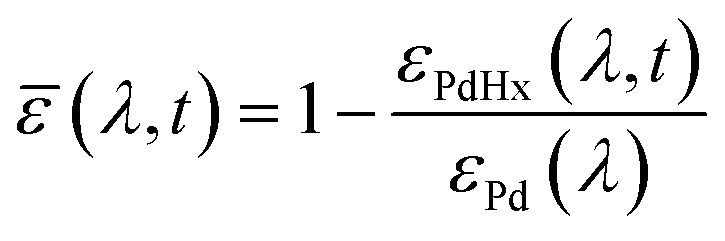 | (1) |
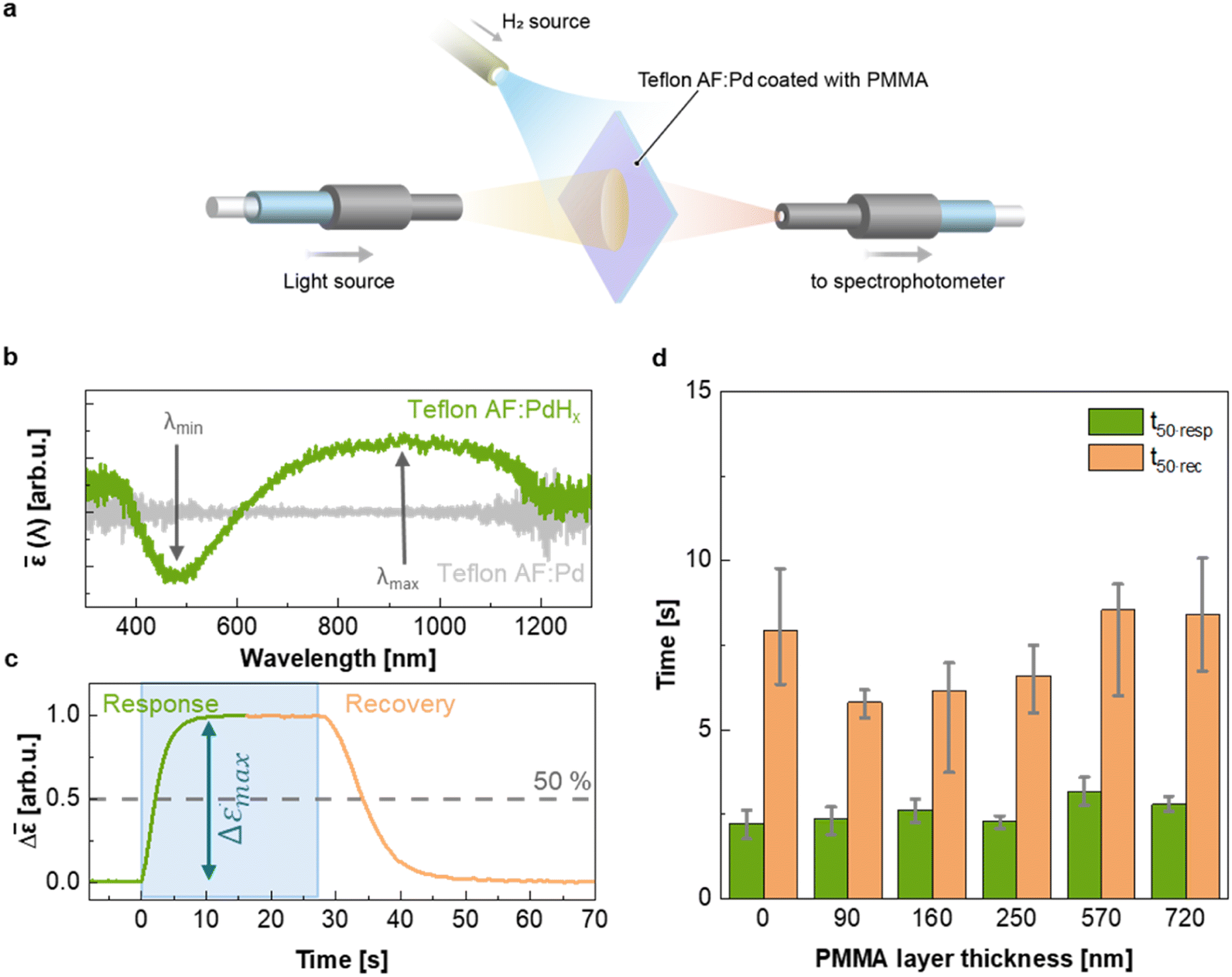 | ||
| Fig. 4 Plasmonic hydrogen sensing of PMMA coated Teflon AF:Pd. (a) Schematic of optical H2 detection via measurements of extinction spectra of a melt pressed nanocomposite film. (b) Self-referenced extinction spectra (λ, t) of Teflon AF:Pd coated with 250 nm of PMMA prior to H2 exposure (grey) and fully hydrated upon exposure to 100 mbar H2 at t = 54 s (green). (c) Temporal evolution of the difference between maximum and minimum extinction Δ(t) of Teflon AF:Pd coated with 250 nm of PMMA, upon a stepwise increase in H2 pressure from 0 to 100 mbar H2 followed by a stepwise decrease from 100 to 0 mbar H2. The blue region marks the exposure to 100 mbar H2. (d) H2 response and recovery time, t50·resp and t50·rec, of the coated Teflon AF:Pd films as a function of PMMA layer thickness (error bars denote the max–min value of three measurements). | ||
Self-referenced extinction spectra of the hydrogenated films feature a maximum and minimum at (λmax, t) and (λmin, t) (Fig. 4b). Here, we used the difference between (λmax, t) and (λmin, t) as the sensor response given by:
| Δ(t) = (λmax, t) − (λmin, t). | (2) |
By monitoring Δ at different H2 pressures at 30 °C, we constructed an optical pressure-composition isotherm, which exhibits the characteristic shape of the palladium-hydrogen system35,36 with an α-phase at low pressure, a β-phase at high pressure and a two-phase coexistence plateau during the first-order phase transformation (ESI Fig. S9†).36
To evaluate the temporal response as a function of PMMA coating thickness, we exposed samples to hydrogen by stepwise increasing the pressure from 0 to 100 mbar followed by a stepwise decrease from 100 to 0 mbar (Fig. 4c and ESI Fig. S10†). We then extracted the response and recovery time, t50·resp and t50·rec, which correspond to the time it takes to reach 50% of the total signal change (Fig. 4c). Neat Teflon AF:Pd as well as all PMMA coated Teflon AF:Pd films feature a response time t50·resp ≈ 2–3 s and a slightly longer recovery time t50·rec ≈ 6–8 s (Fig. 4d). Evidently, the sensing kinetics were not significantly affected by the addition of a PMMA coating layer with a thickness of up to 720 nm (see Fig. 3b). We also evaluated the response and recovery of the sensor by determining the difference between the time it takes to reach 10% and 90% of the total signal (ESI Fig. S11†), which also indicated that the PMMA coating did not affect the sensing kinetics. We conclude that a thin layer of PMMA does not hinder the diffusion of H2 into the nanocomposite film and therefore the fast response time of neat Teflon AF:Pd is preserved.
In a further set of experiments, executed in a flow reactor with optical access and operating at atmospheric pressure, we evaluated the ability of PMMA to protect the sensing material from CO poisoning and thereby enabling sensor operation in a CO-rich environment. The samples coated with five different PMMA film thicknesses were exposed to five cycles of synthetic air containing 5% of H2 and five cycles of synthetic air containing 5% H2 and 500 ppm CO while continuously monitoring Δ (Fig. 5a). Evidently, the presence of a PMMA coating with a thickness ranging from 90 nm to 720 nm does not alter the magnitude of the Δ response compared to the neat Teflon AF:Pd control (cf. Δ step height in Fig. 5a) and eliminates a decrease in the speed of the hydrogen desorption kinetics in the presence of CO (Δt90·rec = t10·rec − t90·rec = 100 s upon CO exposure of uncoated samples vs. Δt90·rec = 27–33 s for PMMA coated samples; ESI Fig. S12†). We argue that the PMMA coating is unlikely to affect the absorption and desorption kinetics as long as the characteristic diffusion time CDT = d2/6D is much less than t50·resp ≈ 2–3 s. Using a diffusion coefficient of H2 in PMMA of D = 6.6 × 10−7 cm2 s−1 (ref. 25) and CDT = 0.1 × t50·resp we obtain a minimum PMMA coating thickness of d ≈ 9–11 μm that would be needed to significantly affect the sensor response. Evidently, the here explored coating thicknesses of 90 to 720 nm are too thin to impact the sensor response as indeed observed (cf.Fig. 4d and ESI Fig. S11 and S12†).
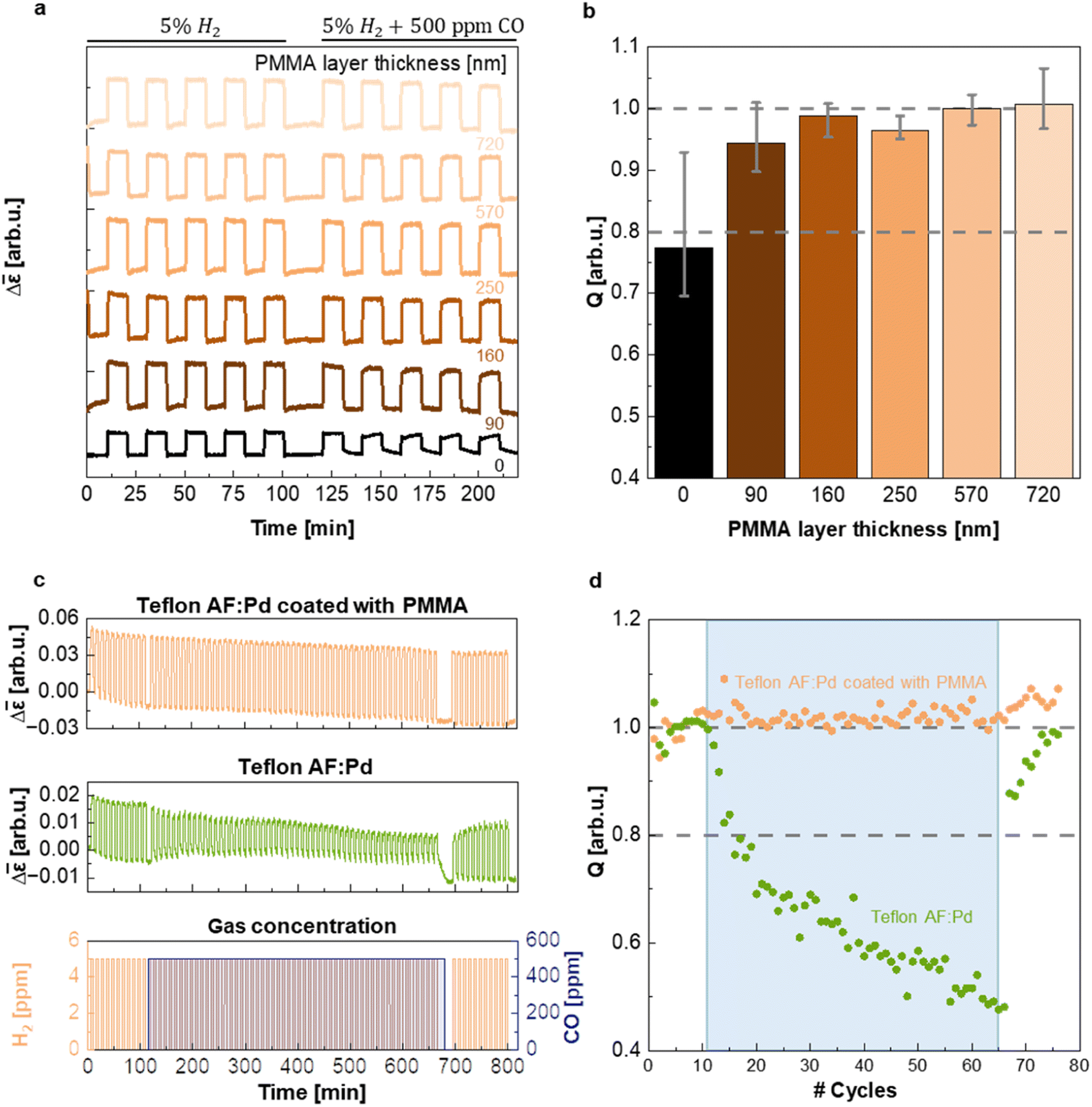 | ||
| Fig. 5 (a) Sensor response, Δ, for Teflon AF:Pd coated with PMMA (layer thickness as indicated), during five cycles of exposure to synthetic air containing 5% H2 and five cycles of synthetic air containing 5% H2 + 500 ppm CO. (b) Quality of the sensor for Teflon AF:Pd coated with PMMA from sensor tests shown in (a); error bars denote max–min values of five measurements. (c) Long-term stability test of Teflon AF:Pd coated with 720 nm of PMMA (top) and neat Teflon AF:Pd (middle) upon changes in gas concentration in the atmosphere with cyclic exposure to 5% H2 and a constant level of 500 ppm CO (bottom). (d) Quality of the sensor for Teflon AF:Pd coated with 720 nm of PMMA (orange) and neat Teflon AF:Pd (green) for each cycle during the long-term stability test shown in (c). The blue region marks the presence of 500 ppm CO. | ||
To evaluate the influence of CO on the sensor response of films with different PMMA layer thickness in more detail, we calculated the ratio of the maximum sensor response Δmax in pure H2 and in H2 with CO background (Fig. 5b), i.e., the quality factor, Q, of the sensor:
 | (3) |
Evidently, after five cycles containing CO the Q-factor of the neat Teflon AF:Pd sensor is reduced to 0.77, i.e. significantly smaller than 1, which indicates sizable poisoning. This decrease in Q-factor also exceeds the limit of ±20% deviation of the sensor response set by the performance standard for stationary H2 sensors for usage at, e.g., refueling stations,5 which would disqualify the neat Teflon AF nanocomposite for real applications. Gratifyingly, however, the presence of a 90 nm thin PMMA layer already increased Q to 0.94 and thus positions the corresponding sensor well within the limits set by the performance standard. Intriguingly, the resistance to CO poisoning could be further improved by increasing the thickness of the PMMA coating, with Q values approaching 1 for PMMA coatings with a thickness of 160 to 720 nm (given the error in the determined Q values we deem the differences between samples with a 160 to 720 nm thick PMMA coating as not statistically significant; see Fig. 5b). The hydrogen desorption kinetics drastically slowed down for neat Teflon AF:Pd under CO exposure, as indicated by the increase in recovery time, Δt90·rec (ESI Fig. S12†). Instead, Teflon AF:Pd coated with PMMA maintained its recovery time (ESI Fig. S12†). Interestingly, the response times were not notably affected by exposure to CO, with both neat Teflon AF:Pd and coated films showing comparable values (ESI Fig. S12†). We conclude that a sub-micrometer thin PMMA coating is sufficient to create a protective barrier against CO, such that the sensors not only retain their overall response but also a fast response and recovery time in the presence of CO.
The superior selectivity of PMMA can be understood by considering the Robeson upper bound limit, i.e. a more selective gas membrane is characterized by a lower permeability.37 Teflon AF has a large fractional free volume38 resulting in a more than two orders of magnitude higher permeability of 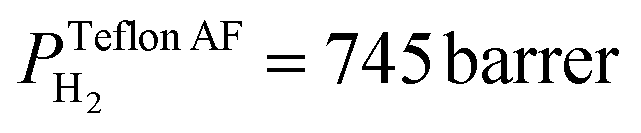 at 30 °C compared with PMMA
at 30 °C compared with PMMA 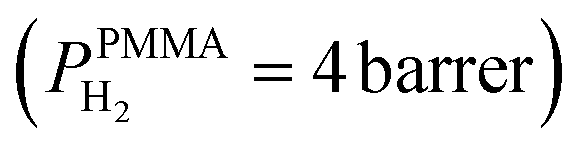 .25 Hence, Teflon AF is a suitable matrix polymer for Pd nanoparticles, while a PMMA coating with a lower permeability can provide gas selectivity. The selectivity of two gases is given by the ratio of their permeability:37
.25 Hence, Teflon AF is a suitable matrix polymer for Pd nanoparticles, while a PMMA coating with a lower permeability can provide gas selectivity. The selectivity of two gases is given by the ratio of their permeability:37
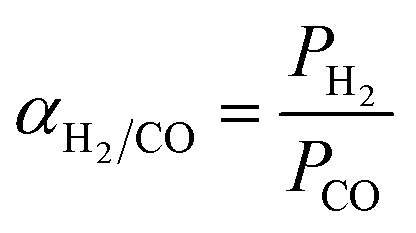 | (4) |
The upper bound limit is given by:37
| PH2 = k × αH2/COn | (5) |
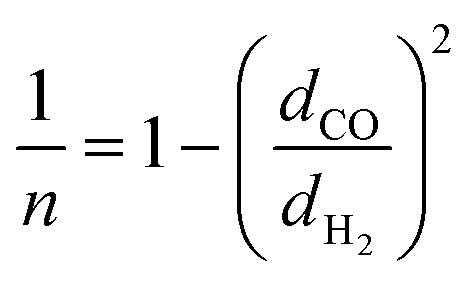 | (6) |
Using values of dCO = 3.2 Å and dH2 = 2.2 Å,39 we obtain n ≈ −1.12 and hence a selectivity ratio of 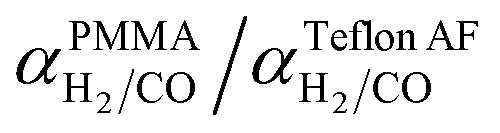 ≈ 106, i.e. the permeability ratio is up to 106 times larger in case of PMMA. Hence, PMMA is the more suitable barrier layer.
≈ 106, i.e. the permeability ratio is up to 106 times larger in case of PMMA. Hence, PMMA is the more suitable barrier layer.
As the final step of our sensing performance evaluation, we chose to investigate the long-term stability of Teflon AF:Pd films coated with a 720 nm PMMA layer. We chose this thickness because it offered the best protection against CO with Q ≈ 1 (Fig. 5b). For this purpose, we exposed the sensor to 77 cycles of synthetic air containing 5% H2 (Fig. 5c) where 55 of those cycles also contained 500 ppm of CO (blue shaded area in Fig. 5c). The PMMA coated sensor exhibited excellent stability and CO protection throughout all cycles. Instead, neat Teflon AF:Pd was gradually deactivated (Fig. 5c). For neat Teflon AF:Pd a value of Q = 0.8 was reached after only 6 cycles of H2 exposure with a concomitant constant exposure to CO, further decreasing to Q = 0.48 after 55 cycles with a CO background (Fig. 5d). Interestingly, the Q-factor gradually recovers after the CO is removed from the test which highlights that the CO poisoning is not permanent. This test also shows that it takes 10 H2 flushes to recover from the CO poisoning.
Conclusions
We have prepared a plasmonic plastic core:shell architecture by coating a Teflon AF:Pd nanocomposite with a thin PMMA layer. The addition of a thin PMMA coating provided poisoning resistance, where a PMMA coating as thin as 90 nm maintained a sensor quality of 94% upon introduction of 500 ppm CO. Gratifyingly, a PMMA coating of 720 nm resulted in a preserved sensor quality during the entire length of a stability test comprising 55 cycles of H2 and constant CO exposure. The sensing kinetics were not affected by the addition of a sub-micrometer PMMA layer and thus the fast response time of 2–3 s that is characteristic for neat Teflon AF:Pd was retained. We have demonstrated that by using a core:shell sensor design, a plasmonic plastic hydrogen sensor that features both a fast sensor response and a promising degree of CO protection can be realized.Experimental section
Synthesis of Pd nanocubes
All chemicals used were of analytical grade (purity > 99%) and used as received from Sigma-Aldrich. All solutions were prepared with MiliQ-water with resistivity of 18.2 MΩ cm−1. The automated segmented flow synthesis was performed using a flow system featuring two peristaltic pumps, an air-heated reaction zone and full automation control using the connected computer with integrated software (Vapourtec E-series). Reagents were pumped in high purity grade perfluoroalkoxy (PFA) tubes (i.d. 1 mm). An aqueous solution of sodium tetrachloropalladate (Na2PdCl4) (63.3 mM) was pumped with a peristaltic pump at a rate of 0.091 mL min−1 and was interfaced in a (ethylene tetrafluoroethene (EFTE), o.d. 1.57 mm) T-junction with a mixture of potassium chloride 307.08 mM, potassium bromide 5.41 mM, polyvinylpyrrolidone 118.23 mM, and ascorbic acid 42.71 mM pumped at a rate of 0.242 mL min−1. The outlet of the T-junction was connected to a coiled flow reactor (10 mL; high purity grade perfluoroalkoxy (PFA) tubes with an inner diameter of 1 mm) which was kept at constant temperature (80 °C) by hot air (Vapourtec system) and collected in a vial. For medium exchange from water to isopropanol, the Pd suspension was mixed with acetone in the ratio 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (acetone:aqueous suspension of Pd nanocubes). For instance, 10 mL of aqueous suspension of Pd nanocubes was placed in a 50 mL centrifuge tube and 30 mL of acetone was added. The mixture was centrifuged at speed of 3000 rpm for 5 min. The supernatant was discarded, and 10 mL of isopropanol was added. The suspension of nanoparticles in isopropanol was placed in an ultrasound bath for 5 min to improve the colloidal stability.
:1 (acetone:aqueous suspension of Pd nanocubes). For instance, 10 mL of aqueous suspension of Pd nanocubes was placed in a 50 mL centrifuge tube and 30 mL of acetone was added. The mixture was centrifuged at speed of 3000 rpm for 5 min. The supernatant was discarded, and 10 mL of isopropanol was added. The suspension of nanoparticles in isopropanol was placed in an ultrasound bath for 5 min to improve the colloidal stability.
Polymer nanocomposite compounding
Poly[4,5-difluoro-2,2-bis-(trifluoromethyl)-1,3-dioxole-co-tetrafluoroethylene] (Teflon AF 1600, here called Teflon AF) with a dioxole content of 65 mol% and a density of 1.78 g cm−3 was obtained from Sigma-Aldrich. 3 mL of the Pd nanocube dispersion was added to 4 mL of Teflon AF powder and was kept at 50 °C over night for iso-propanol removal. The dry mixture was compounded at 250 °C for 5 min in an Xplore Microcompounder MC5.Melt pressing
The Teflon AF/Pd composite was melt pressed into films with a hot press from AB Nike Hydralik at 240 °C and 50 kN for 3 min followed by 100 kN for 2 min. Metal spacers were used to control the film thickness.Plasma treatment
The Teflon AF:Pd films were H2 plasma treated on each side with the following recipe: 30 s exposure of 100 W, 250 mTorr, and 40 sccm H2. The plasma treatment was carried out in a BatchTop m/95 PlasmaTherm Reactive Ion Etcher (RIE).Dip coating
Poly(methyl methacrylate) (PMMA) powder with a weight-average molecular weight of Mw = 75 kg mol−1, a polydispersity index PDI of 2.8 and a density of 1.2 g cm−3 was obtained from Polysciences Inc. PMMA was dissolved in anisole (99% extra pure from Acros Organics) with a concentration of 10 g L−1 by heating it to 60 °C under stirring. 100 μm thick Teflon AF:Pd films where dipped with a Ossila dip coater into the PMMA solution with an immersion time of 10 s and withdrawal speeds varying from 0.1 to 10 mm s−1. The coated films were soft baked in an oven at 110 °C for 5 min.Fourier transform infrared spectroscopy-attenuated total reflectance (FTIR-ATR)
Infrared transmittance measurements on the PMMA coated Teflon AF:Pd films were performed with a PerkinElmer FT-IR Spectrometer ‘Frontier’. The mapping of PMMA coated Teflon AF:Pd films was performed with a Bruker Hyperion 3000 microscope coupled to a Bruker Vertex 70v spectrometer. The atmosphere was flushed with nitrogen for 20–30 minutes before the measurement. 100 × 100 μm or 125 × 125 μm areas were mapped with a step size of 25 μm.Transmission electron microscopy (TEM)
The Teflon AF:Pd films were embedded in Epoxy Hard plus Resin-812 (EMS, Hatfield, PA) and polymerized in the oven for 16 h at 60 °C. Ultrathin sections of approximately 70 nm thickness were cut with a LEICA EM UC6 microtome (Leica, Germany) and collected on lacey copper grids. TEM was performed using an FEI Titan with a field emission gun (FEG), monochromator and US1000 CCD, operated at 300 kV. Images were obtained at a variety of magnifications ranging from 35k to 480k.Elemental analysis
Elemental analysis was carried out by Mikrolab Kolbe, Germany.Scanning electron microscopy with energy dispersive X-ray analysis (SEM-EDX)
The samples for SEM were cryofractured in liquid nitrogen. Samples were sputter coated with a 10 nm layer of gold with a Leica EM ACE600 Gold and Chrome Sputter. The SEM analysis was performed with a Zeiss Ultra 55 FEG SEM equipped with a secondary electron detector set at 3 kV. EDX data was collected at 10 kV with an Oxford instruments EDX detector and INCA Energy Software.Mechanical characterization
Tensile deformation was carried out with a Q850 dynamic mechanical analyzer from TA instruments at room temperature in controlled force mode with a force rate of 0.5 N min−1, a preload force of 0.005 N and gauge length of 5 to 7 mm (sample width of 1.8 to 2 mm and thickness of 0.1 mm). Nanoindentation was carried out with a Bruker TI Premier nanoindenter at room temperature in controlled force mode with a force rate of 5 to 500 μN s−1 and using a Berkovich diamond tip, calibrated with a fused quartz film. The indentation depth was varied by changing the maximum load from 25 to 2500 μN while keeping the drift at less than 0.1 nm s−1.Hydrogen sensing measurements
The hydrogen sensing tests were carried out using two different setups: (i) a vacuum chamber for the hydrogen sensing response/recovery time test and (ii) a flow reactor with optical access operated at atmospheric pressure for the simulated hydrogen sensing in synthetic air and CO background. The schematics of both setups are available in ref. 32.40 Both setups are equipped with a polychromatic halogen light source (Avantes Avalight-Hal) and a spectrophotometer (Avantes SensLine AvaSpec-2048XL), which enable monitoring of the optical response in transmission mode. The light source and spectrophotometer were connected using a fiber optic cable (Avantes) and a collimating lens (Thorlabs) towards the sensor. The temperature was maintained in a feedback loop manner using a system consisting of a resistive coil wrapped around the chamber, a DC power supply and a PID temperature controller (Euroterm 3216). All measurements were performed at 30 °C.Vacuum chamber: The sensor response/recovery time tests were performed by exposing the sensor to a stepwise change in absolute H2 pressure: vacuum to 100 mbar (for response time) and 100 mbar to vacuum (for recovery time). The stepwise pressure change was executed using manual valves (Nupro), which separate the chamber from the 100% H2 source and the vacuum turbo pump (minimum vacuum pressure of 10 μbar). The chamber pressure was monitored using a capacitance manometer (MKS Barathron 626C). The sensor spectra were monitored using a custom Matlab program.
Flow reactor: The relative gas concentration at atmospheric pressure was controlled by adjusting each gas flow rate using mass flow controllers (Bronkhorst El-Flow series). The total gas flow rate was kept constant at 200 mL min−1. The feed gases consisted of synthetic air (20% O2, 80% N2), 10% CO (diluted in Ar), and 25% H2 (diluted in Ar) provided by Strandmøllen. The sensor spectra were monitored in real-time using the Insplorer® software from Insplorion AB, Göteborg, Sweden.
Conflicts of interest
C. Langhammer is a scientific advisor at Insplorion AB that markets plasmonic hydrogen sensors. S. Lerch and R. R. da Silva are today employed at Nanoscientifica AB.Acknowledgements
The authors acknowledge financial support from the Swedish Foundation for Strategic Research project RMA15-0052, the Knut and Alice Wallenberg Foundation for a Wallenberg Fellow Prolongation Grant and the European Union’s Horizon 2020 research and innovation programme through the Marie Skłodowska-Curie grant agreement no. 955837 (HORATES). Part of this work was carried out at the Chalmers Materials Analysis Laboratory (CMAL). The authors also acknowledge the Centre for Cellular Imaging at the University of Gothenburg and the National Microscopy Infrastructure, NMI (VR-RFI 2019-00022), for providing assistance with microtome sectioning. The authors thank Anders Mårtensson for his assistance with SEM-EDX measurements and Insplorion AB for providing the Ossila dip coater.References
- I. E. Agency, The Future of Hydrogen, 2019 Search PubMed.
- W. J. Buttner, M. B. Post, R. Burgess and C. Rivkin, Int. J. Hydrogen Energy, 2011, 36, 2462–2470 CrossRef CAS.
- R. Derwent, P. Simmonds, S. O'Doherty, A. Manning, W. Collins and D. Stevenson, Int. J. Nucl. Hydrogen Prod. Appl., 2006, 1, 57–67 Search PubMed.
- U.S Department of Energy, Energy Efficiency and Renewable Energy (EERE), Multi-Year Research, Development, and Demonstration Plan, 2011-2020, Section 3.7 Hydrogen Safety, Codes and Standards, 2015 Search PubMed.
- ISO 26142:2010 – Hydrogen Detection Apparatus – Stationary Applications, 2010 Search PubMed.
- U.S. Environmental Protection Agency, Criteria Air Pollutants, https://www.epa.gov/criteria-air-pollutants, accessed October 2023 Search PubMed.
- M. G. Jones and T. G. Nevell, Sens. Actuators, 1989, 16, 215–224 CrossRef CAS.
- C.-H. Han, D.-W. Hong, I.-J. Kim, J. Gwak, S.-D. Han and K. C. Singh, Sens. Actuators, B, 2007, 128, 320–325 CrossRef CAS.
- R. K. Pippara, P. S. Chauhan, A. Yadav, V. Kishnani and A. Gupta, Micro Nano Eng., 2021, 12, 100086 CrossRef CAS.
- D. Zhang, Y. e. Sun, C. Jiang and Y. Zhang, Sens. Actuators, B, 2017, 242, 15–24 CrossRef CAS.
- H. Yamazaki, Y. Hayashi, K. Masunishi, D. Ono and T. Ikehashi, J. Manuf. Syst., 2018, 28, 094001 Search PubMed.
- R. J. Westerwaal, J. S. A. Rooijmans, L. Leclercq, D. G. Gheorghe, T. Radeva, L. Mooij, T. Mak, L. Polak, M. Slaman, B. Dam and T. Rasing, Int. J. Hydrogen Energy, 2013, 38, 4201–4212 CrossRef CAS.
- L. Bannenberg, H. Schreuders and B. Dam, Adv. Funct. Mater., 2021, 31, 2010483 CrossRef CAS.
- J. Villatoro and D. Monzón-Hernández, Opt. Express, 2005, 13, 5087–5092 CrossRef CAS PubMed.
- C. Wadell, S. Syrenova and C. Langhammer, ACS Nano, 2014, 8, 11925–11940 CrossRef CAS PubMed.
- I. Darmadi, F. A. A. Nugroho and C. Langhammer, ACS Sens., 2020, 5, 3306–3327 CrossRef CAS PubMed.
- F. A. A. Nugroho, I. Darmadi, L. Cusinato, A. Susarrey-Arce, H. Schreuders, L. J. Bannenberg, A. B. da Silva Fanta, S. Kadkhodazadeh, J. B. Wagner, T. J. Antosiewicz, A. Hellman, V. P. Zhdanov, B. Dam and C. Langhammer, Nat. Mater., 2019, 18, 489–495 CrossRef CAS PubMed.
- M. Ando, TrAC, Trends Anal. Chem., 2006, 25, 937–948 CrossRef CAS.
- H. Amandusson, L. G. Ekedahl and H. Dannetun, Appl. Surf. Sci., 2000, 153, 259–267 CrossRef CAS.
- I. Darmadi, S. Z. Khairunnisa, D. Tomeček and C. Langhammer, ACS Appl. Nano Mater., 2021, 4, 8716–8722 CrossRef CAS.
- I. Darmadi, F. A. A. Nugroho, S. Kadkhodazadeh, J. B. Wagner and C. Langhammer, ACS Sens., 2019, 4, 1424–1432 CrossRef CAS PubMed.
- J. Hong, S. Lee, J. Seo, S. Pyo, J. Kim and T. Lee, ACS Appl. Mater. Interfaces, 2015, 7, 3554–3561 CrossRef CAS PubMed.
- I. Darmadi, I. Östergren, S. Lerch, A. Lund, K. Moth-Poulsen, C. Müller and C. Langhammer, Acc. Chem. Res., 2023, 56, 1850–1861 CrossRef CAS PubMed.
- I. Darmadi, A. Stolaś, I. Östergren, B. Berke, F. A. A. Nugroho, M. Minelli, S. Lerch, I. Tanyeli, A. Lund, O. Andersson, V. P. Zhdanov, M. Liebi, K. Moth-Poulsen, C. Müller and C. Langhammer, ACS Appl. Nano Mater., 2020, 3, 8438–8445 CrossRef CAS.
- I. Östergren, A. M. Pourrahimi, I. Darmadi, R. da Silva, A. Stolas, S. Lerch, B. Berke, M. Guizar-Sicairos, M. Liebi, G. Foli, V. Palermo, M. Minelli, K. Moth-Poulsen, C. Langhammer and C. Müller, ACS Appl. Mater. Interfaces, 2021, 13, 21724–21732 CrossRef PubMed.
- Q. Li, S. Yang, X. Y. Lu, T. Q. Wang, X. M. Zhang, Y. Fu and W. Qi, Small, 2022, 18, 2106874 CrossRef CAS PubMed.
- A. Kumar, T. Thundat and M. T. Swihart, ACS Appl. Nano Mater., 2022, 5, 5895–5905 CrossRef CAS.
- M.-S. Jo, K.-H. Kim, J.-S. Lee, S.-H. Kim, J.-Y. Yoo, K.-W. Choi, B.-J. Kim, D.-S. Kwon, I. Yoo, J.-S. Yang, M.-K. Chung, S.-Y. Park, M.-H. Seo and J.-B. Yoon, ACS Nano, 2023, 17, 23649–23658 CrossRef CAS PubMed.
- H. M. Luong, T. A. Ngo, M. T. Pham, Y. P. Zhao, G. K. Larsen, T. Q. Nguyen and T. D. Nguyen, Nano Energy, 2023, 109, 108332 CrossRef CAS.
- A. Pekkari, Z. Say, A. Susarrey-Arce, C. Langhammer, H. Härelind, V. Sebastian and K. Moth-Poulsen, ACS Appl. Mater. Interfaces, 2019, 11, 36196–36204 CrossRef CAS PubMed.
- A. Stolaś, I. Darmadi, F. A. A. Nugroho, K. Moth-Poulsen and C. Langhammer, ACS Appl. Nano Mater., 2020, 3, 2647–2653 CrossRef.
- D. Grosso, J. Mater. Chem., 2011, 21, 17033–17038 RSC.
- P. Yimsiri and M. R. Mackley, Chem. Eng. Sci., 2006, 61, 3496–3505 CrossRef CAS.
- I. Zorić, E. M. Larsson, B. Kasemo and C. Langhammer, Adv. Mater., 2010, 22, 4628–4633 CrossRef PubMed.
- R. Bardhan, L. O. Hedges, C. L. Pint, A. Javey, S. Whitelam and J. J. Urban, Nat. Mater., 2013, 12, 905–912 CrossRef CAS PubMed.
- S. Syrenova, C. Wadell, F. A. A. Nugroho, T. A. Gschneidtner, Y. A. Diaz Fernandez, G. Nalin, D. Świtlik, F. Westerlund, T. J. Antosiewicz, V. P. Zhdanov, K. Moth-Poulsen and C. Langhammer, Nat. Mater., 2015, 14, 1236–1244 CrossRef CAS PubMed.
- L. M. Robeson, J. Membr. Sci., 2008, 320, 390–400 CrossRef CAS.
- M. Minelli and G. C. Sarti, J. Membr. Sci., 2017, 521, 73–83 CrossRef CAS.
- J. Wang, Y. Zhang, W. Wang, L. Yin, M. Xie, J. Y. Lee, H. Shi and H. Liu, J. Phys. Chem. A, 2023, 127, 517–526 CrossRef CAS PubMed.
- I. Darmadi, Polymer-Nanoparticle Hybrid Materials for Plasmonic Hydrogen Detection, PhD thesis, Chalmers University of Technology, 2021.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta00055b |
| This journal is © The Royal Society of Chemistry 2024 |