
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Single atom catalysts for water electrolysis: from catalyst-coated substrate to catalyst-coated membrane
Sol A
Lee†
ab,
Sang Eon
Jun†
ac,
Sun Hwa
Park
c,
Ki Chang
Kwon
c,
Jong Hun
Kang
*d,
Min Sang
Kwon
 *a and
Ho Won
Jang
*ae
*a and
Ho Won
Jang
*ae
aDepartment of Materials Science and Engineering, Research Institute of Advanced Materials, Seoul National University, Seoul 08826, Korea. E-mail: hwjang@snu.ac.kr
bLiquid Sunlight Alliance (LiSA), Department of Applied Physics and Materials Science, California Institute of Technology, Pasadena, California 91125, USA
cInterdisciplinary Materials Measurement Institute, Korea Research Institute of Standards and Science, Daejeon 34113, Republic of Korea
dSchool of Chemical and Biological Engineering, and Institute of Chemical Process, Seoul National University, Seoul 08826, Republic of Korea
eAdvanced Institute of Convergence Technology, Seoul National University, Suwon, 16229, Republic of Korea
First published on 5th October 2023
Abstract
Green hydrogen production through water electrolysis is considered the next-generation technology capable of industrial-scale hydrogen production to achieve carbon neutrality. The core of constructing a water electrolyzer lies in designing the membrane electrode assembly (MEA) with optimal integration of the membrane, electrocatalysts, and gas diffusion layer. Among the two representative MEA fabrication methods, catalyst-coated substrates (CCS) and catalyst-coated membranes (CCM), CCM shows great promise due to its catalyst layer/membrane interface contact and scalability. The key factor in the CCM method is the effective application of the powdered catalyst onto the membrane. In this respect, the utilization of single-atom catalysts (SACs) has emerged as a noteworthy focus due to their unprecedented catalytic activity resulting from unique electronic/atomic configurations and high atomic utilization efficiency. Incorporating SACs into CCM–MEA has the potential to be a cutting-edge water electrolysis technology. However, it is still in its infancy due to the instability of the components (SACs, membranes, ionomers, supports) and degradation during the SACs–CCM–MEA fabrication and cell operation. Herein, we outline the representative fabrication method of MEA and provide a comprehensive analysis of SACs applicable to MEA. Then, we discuss the advantages of SACs–CCM–MEA and the challenges for industrial hydrogen production. Finally, this review concludes with future perspectives on the development of single-atom catalyst-coated membranes and the expected achievements.
 Sol A Lee | Sol A. Lee received her PhD from the Department of Materials Science and Engineering of Seoul National University under the supervision of Prof. Ho Won Jang in 2021. She is currently a postdoctoral fellow at the California Institute of Technology under the supervision of Prof. Harry Atwater. Her research focuses on the design of tandem structures for water splitting and CO2 reduction. |
 Sang Eon Jun | Sang Eon Jun is currently a PhD candidate under the supervision of Prof. Ho Won Jang in the Department of Materials Science and Engineering at Seoul National University (SNU). He received his BS degree from the School of Integrative Engineering, Chung-Ang University, in 2019. His research focuses on the synthesis and characterization of 2-dimensional materials and single atom catalysts for photoelectrochemical energy conversion reactions. |
 Ho Won Jang | Ho Won Jang is a full professor in Department of Materials Science and Engineering at Seoul National University. He earned his PhD from the Department of Materials Science and Engineering of Pohang University of Science and Technology in 2004. He worked as a research associate at University of Madison-Wisconsin from 2006 to 2009. He is a member of Young Korean Academy of Science and Technology and is serving as an associate editor for journals, Exploration and Eco Energy. His research interests include materials synthesis and device fabrication for solar fuel generation, chemical sensing, nonvolatile data storage, neuromorphic computing, plasmonics, ferroelectronics, and metal–insulator transition. |
Broader contextHydrogen is considered as the most promising energy source due to its high energy density, non-toxicity, and zero CO2 emission. Electrochemical water electrolysis, including hydrogen evolution reaction (HER) and oxygen evolution reaction (OER), is one of the promising methods to obtain pure hydrogen. For a highly efficient water electrolysis, it is necessary to design the membrane electrode assembly (MEA) with optimal combination of membrane, electrocatalysts, and gas diffusion layer. In this respect, the catalyst-coated membrane (CCM) method has been developed for low ohmic losses, efficient mass transport, and scalability. Additionally, single atom catalysts, demonstrating exceptional catalytic activity and atomic utilization efficiency, attracted much attention. In this review, we introduce the integration of SACs and CCM–MEA, which enables to exhibit high utilization of SACs, facile ion transport, low interfacial resistance, and excellent scalability. |
1. Introduction
In recent decades, the use of fossil fuels has led to a significant increase in carbon dioxide emissions, resulting in severe environmental pollution and global warming. Therefore, the development of a new energy economic system based on renewable energy has become an urgent and imperative task worldwide.1–4 Hydrogen, as an energy carrier, has emerged as a prime interest as a substitute for fossil fuels toward carbon neutrality.5–8 Despite its potential as a clean energy source, mature hydrogen production technologies to date are using fossil fuels; reforming technologies (steam reforming process, partial oxidation, autothermal reforming, etc.), pyrolysis of hydrocarbons, and gasification of coals. The representative steam reforming reaction is the thermochemical process under high pressure and temperature by reacting hydrocarbons and steam to produce hydrogen and carbon dioxide represented by the following equations:| CH4 + H2O ⇌ CO + 3H2 | (1) |
| CO + 3H2 ⇌ CO2 + H2 | (2) |
First, methane reacts with steam to form carbon monoxide and hydrogen with the aid of heat. Then, carbon monoxide and steam are reacted to generate carbon dioxide and hydrogen called a water–gas shift reaction. The pure hydrogen is finally separated from the mixed gas and impurities by pressure swing adsorption. However, current hydrogen production technologies use confined resources and generate carbon products, which are inadequate for carbon neutralization.9,10 Therefore, the production of so-called “green” hydrogen, which emits no CO2, is gaining substantial attention by utilizing renewable energy sources.11–14 One promising approach to accomplish renewable H2 production involves integrating solar energy conversion technology with water electrolysis such as photovoltaic-electrolysis and photoelectrolysis.15–18
Water electrolysis technology has been considered a sustainable way for large-scale hydrogen production.19–22 To enhance cost competitiveness compared to conventional hydrogen production methods, a highly efficient water electrolysis system should be developed.23–25 In particular, the development of this technology should not only focus on improving the performance of individual functional components such as electrodes and catalysts,26–29 but also on the operation of a merged cell with optimized performance.30 From this perspective, alkaline water electrolysis is the most mature technology for hydrogen production.31–33 However, it faces challenges such as low current density, gas crossover, and limitations in operating under high-pressure conditions.31,34 Therefore, novel water electrolysis technologies using ion-exchange membranes have been developed, providing advantages in terms of improved reaction kinetics with high current density.35 Depending on the type of membrane used, the water electrolyzer (WE) can be classified into two categories: proton exchange membrane water electrolysis (PEMWE) and anion exchange membrane water electrolysis (AEMWE). In an ion-exchange membrane water electrolysis system, a key component is the membrane electrode assembly (MEA), which comprises an ion exchange membrane, catalyst layer, and liquid/gas diffusion layer (GDL).36 The early stage of MEA fabrication involved the catalyst-coated substrates (CCS) method, where catalyst-coated supports were sandwiched with membranes.37 Subsequently, recognizing the importance of the catalyst/membrane interface, the catalyst-coated membrane (CCM) method was developed, allowing the direct introduction of catalysts onto the membrane.38–40 This method demonstrates several advantages that position itself as a compelling option for water electrolysis, including low ohmic losses, efficient mass transport, and scalability. Recent studies have highlighted the significant impact of the MEA fabrication method on cell performance and scalability.41,42
Taking advantage of the CCM method, we propose the incorporation of recently spotlighted single-atom catalysts (SACs) into CCM–MEA. In recent years, the field of SACs has witnessed significant advancements in various electrochemical reactions such as the hydrogen evolution reaction (HER),43–46 the oxygen evolution reaction (OER),29,47–49 the carbon dioxide reduction reaction (CO2RR),50–55 the oxygen reduction reaction (ORR),56–58 the nitrogen reduction reaction (NRR),59–61 and the chlorine evolution reaction (ClER).62,63 In addition, SACs have been developed in the field of photoelectrochemical,47,64–73 and photocatalytic74–81 water splitting, and organic synthesis.82–86 Atomically dispersed metals offer several advantages over conventional nanoparticulate counterparts, including high atomic utilization efficiency, enhanced adsorption of reactants, improved charge separation, and selective reactions.29,72 These advantages arise from the unique electronic configurations and environmental coordination of single atoms.87 Thus, SACs have demonstrated exceptional catalytic performance by serving highly active catalytic sites, which facilitates interactions between reactants and catalysts.88,89 Recent studies have demonstrated that high loading of SACs and delicate tuning of the metal–support interaction could accelerate surface reaction rates in water electrolysis and enhance the mass activity.90–92 By combining the advantages of SACs and CCM–MEA, it is possible to achieve the ultimate water electrolysis system for efficient and sustainable hydrogen production.
Fig. 1 shows the schematic illustration of the SACs-incorporated MEA water electrolyzer and developments in both ion exchange membrane water electrolyzer and single atom catalysts. For decades, MEA-based water electrolyzers including PEMWE and AEMWE have been developed a lot in respect of stacking structure, electrolyte, catalyst deposition, and efficiency.93–97 Meanwhile, the synthesis and application of SACs for electrochemical reactions have been in the spotlight and advanced recently.98–104
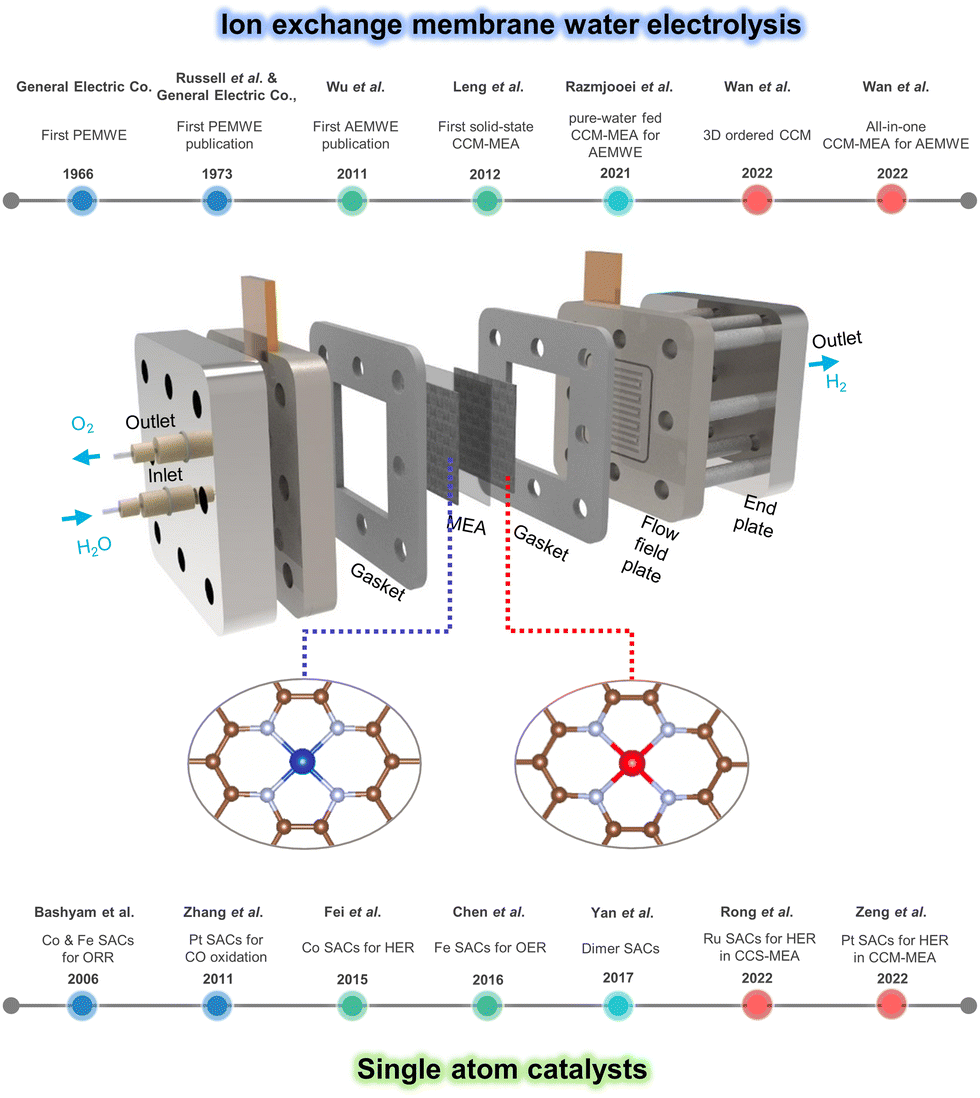 | ||
| Fig. 1 Schematic illustration of SACs-incorporated MEA water electrolyzer and timelines of developments in ion exchange membrane water electrolyzer and single atom catalysts. | ||
In this review, we focus on the integration of SACs and CCM architecture, which possesses great potential for highly efficient, durable, and cost-effective water electrolysis. The combination of SACs and CCM represents a promising approach that harnesses the individual strengths of both technologies, leading to synergistic effects and enhanced water electrolysis cell performance. It is expected that SACs–CCM–MEA will exhibit high utilization of SACs, facile ion transport to single metal atoms, low interfacial resistance between the SACs and the membrane, and scalability. Specifically, the uniformly dispersed catalyst layer would ensure complete contact between the SACs and the membrane, eliminating any inactive and empty reaction sites. Additionally, it might provide a direct and shorter path not only for H+ or OH− ions to move from the membrane to the reaction sites, but also for charges to efficiently transfer at the electrode–electrolyte interface. Moreover, large-scale fabrication of MEA with high performance can be achieved through SACs–CCM–MEA for the industrialization of hydrogen production. This novel approach to integrating individual high-level techniques has the potential to overcome the limitations of conventional electrolysis systems.
2. Catalyst-coated substrate and catalyst-coated membrane for the membrane electrode assembly
Water electrolysis techniques have undergone development, progressing from alkaline water electrolysis to proton/anion exchange membrane water electrolysis, aiming at efficient and cost-effective generation of hydrogen in a large-scale. The MEA-based water electrolyzer is considered a promising technology for industrial hydrogen production due to its improved structure providing low ohmic resistance and enhanced mass transfer properties.105Fig. 1 illustrates the components of the MEA-based WE, including an MEA, gaskets, flow field plates/current collectors, and end plates; an electrochemical reaction occurs at MEA. Gaskets serve the important function of preventing both electrolyte leakages and electrical contact with the flow field plates/current collectors, except for the active region. The electrolytes circulate through the flow field plate with a carved flow channel, while the end plates are employed for the assembly purpose. Since the performance of water electrolysis significantly relies on the MEA, studies have focused on enhancing interfacial interactions within the MEA by developing various components (catalysts, membranes, supports), as well as MEA fabrication methods.106,1072.1 Membrane electrode assembly
The MEA represents a core component of the WE where the electrochemical reactions occur. The MEA has a sandwiched structure comprising ion exchange membrane and a pair of OER/HER electrodes, which consist of catalysts and supports. The ion exchange membrane selectively allows the ion transport, depending on the type of membrane utilized such as anion exchange membrane or proton exchange membrane. The GDL serves as a catalyst support, facilitating the transport of liquid and gas products, as well as charge carriers. The performance of MEA is significantly influenced by the employed fabrication method, as it determines the resistance, ion movement, and gas diffusion properties of the MEA. Contingent upon the fabrication method, the MEA can be categorized as a CCS if the catalysts are loaded onto the substrates, or as a CCM if the catalysts are coated on the membrane surface.2.2 Catalyst-coated substrate
In the early stage, the MEA was mostly prepared with CCS by taking advantage of its simple and easy fabrication process.108,109 In the CCS fabrication process, the catalysts are coated on supports or substrates, and then the membrane and catalyst/support are hot-pressed to form the MEA. Due to its independent fabrication process of membrane and electrode, it enables the manufacture of bulk CCS-type MEAs (denoted as CCS–MEAs). Spray coating is one of the widely employed technologies for CCS fabrication, wherein the catalyst ink, catalysts and ionomers dispersed in a solvent are sprayed onto the gas diffusion electrodes (GDE), as illustrated in Fig. 2a. The composition of catalyst ink is a pivotal factor in achieving a uniform catalyst layer on substrates. Thus, optimization of the ionomer content, catalysts loading, and the solvent system is necessary. Numerous studies have reported on the effects of ionomer contents and catalyst loading on the performances of water electrolysis.110,111 | ||
| Fig. 2 (a) Schematic of spray coating. SEM images of GDL (b) before catalyst loading and (c) after catalyst loading. Reproduced with permission from ref. 36. Copyright 2017, Elsevier. | ||
To deposit powder catalysts on substrates, the popular and simple method is hand spraying, which involves using a spray gun to apply catalyst ink onto the substrates. After spraying the catalyst inks, the solvent is evaporated, and the process is repeated several times to load a desired amount of catalysts. As a selective example, Vincent et al. prepared catalyst inks consisting of deionized water (DI), isopropyl alcohol (IPA), alkaline ionomer, and commercial catalysts (Acta 3030 (CuCoOx)).36 The catalysts were coated using the hand spraying method on microporous carbon paper with an air spray gun and dried at 80 °C for 2 hours. Fig. 2b and c show that the catalyst layer was formed on substrates without voids or cracks. Despite being a straightforward technique for electrode preparation, hand spraying lacks precise control over the uniformity of the catalyst layer due to its reliance on manual operation. As an advanced technique, sonicated spraying provides even finer control over catalyst distribution and loading by atomizing catalyst inks into a fine mist through the ultrasonic nozzle. Bühler et al. used an ExactaCoat device (SonoTek) to spray coat Ir-based catalysts inks on a titanium fiber anodic porous transport layer.112 They investigated the influence of IrO2 loadings and the amount of ionomer content (Nafion) on the behavior of PEMWE. The same method was applied to prepare a bipolar MEA, where IrO2 was uniformly spray coated between the AEM and PEM by Mayerhöfer et al.108
Despite the mature techniques for synthesizing powder catalyst, the loading of such catalysts requires the use of organic binders (ionomer) which can poison the catalytic active sites and impede the diffusion of generated gas species. An alternative approach that offers an advantage in terms of catalyst–support adhesion and a binder-free reaction environment is the direct growth of catalysts on substrates. This direct growth can be accomplished through physical vapor deposition methods or solution-based methods.
The physical deposition of catalysts onto substrates provides advantages in reproducibility and scalability. One method that utilizes the physical deposition is the magnetron sputtering in an oblique angle deposition (MS-OAD) technique, which has been employed to manufacture nanostructured anodes for AEMWE. López-Fernández et al. showed the preparation of Ni, NiFe, and CuxCo3−xO4 catalysts on carbon paper as an anode using MS-OAD.113 They achieved a high WE performance by leveraging the porous nanostructure of the catalyst layer, while using a minimal load of catalysts.
Solution-based methods include sol–gel methods, electrodeposition, chemical bath deposition, etc.114–119 Ahn et al. demonstrated the preparation of Ni/carbon paper (CP) electrodes through electrodeposition, serving as both the anode and cathode for AEMWE.120 They highlighted the promising aspect of direct growth of Ni on CP using electrodeposition, particularly in terms of catalyst loading on substrates. When compared to the bulk electrodes with high catalyst loading of up to a few mg, the fabricated MEA showed a current density of 150 mA cm−2 at a cell voltage of 1.9 V, operating at a temperature of 50 °C, with a low Ni amount of 17.0 μgNi cm−2 on CP. In another study, Lee et al. fabricated a NiFeV layered double hydroxide (LDH) OER electrode by surface corrosion of Ni foam in a chemical bath containing Fe3+ and V3+.121 The AEMWE using the NiFeV LDH anode showed comparable performance compared to a WE utilizing commercial IrO2 and Pt/C catalysts.
Various CCS fabrication methods have been developed for ion exchange membrane water electrolysis toward large-scale hydrogen production. With the development of a catalyst coating system, it is possible to fabricate CCS over 100 cm2 scale.122 One such method for ion exchange membrane water electrolysis is CCS–MEA, which involves the hot-pressing of a membrane and catalyst/supports. However, fabricating large-scale CCS–MEA is challenging due to the mechanical contact between the membrane and the catalyst/support, resulting in a performance loss caused by ohmic resistance at the catalyst/membrane interface. Additionally, the low catalyst utilization on supports (e.g., GDE) poses a bottleneck in terms of cost-effectiveness. Consequently, the need to directly coat catalysts on the membrane has prompted the development of the catalyst-coated membrane fabrication method.
2.3 Catalyst-coated membrane
The CCM refers to one of the methods used for MEA fabrication, wherein catalysts are directly deposited on the membrane surface. In comparison to the CCS method, the CCM method provides a facile catalyst–membrane interlayer, resulting in low interfacial resistance during the electrochemical reaction. Additionally, it enables the increase of mass efficiency with low catalyst loading, thereby leading to a high-performance MEA water electrolyzer. Park et al. conducted a comparative study on the AEMWE performance of CCS–MEA with and without hot-pressing, along with CCM–MEA. The result indicated that the CCM–MEA showed the better performance with good repeatability.123 However, MEA fabrication using the CCM method faced the challenges such as membrane swelling and catalyst detachment.124 Therefore, the development of an appropriate preparation method has become significant for the fabrication of stable CCM–MEAs. Several CCM fabrication methods, such as spray coating, decal transfer coating, doctor blade coating, brushing, inkjet printing, layer-by-layer deposition, bar coating, and solvothermal method have been developed.97,125,126Similar to CCS fabrication methods, direct spray coating is widely employed for CCM. Klose et al. demonstrated the use of all-hydrocarbon MEAs, utilizing a sulfonated poly(phenylene sulfone) (sPPS) membrane onto which the catalysts were spray-coated using catalysts inks.127 IrO2 with a loading of 1.5 mg cm−2 and 0.5 mg cm−2 of Pt was deposited as the OER and HER catalyst, respectively. As shown in Fig. 3a and b, the OER/HER catalyst layers were directly formed on sPPS and Nafion N115 membranes, each with a thickness of ca. 7 μm. The CCM–MEA using sPPS showed lower gas cross-over compared to the MEA using Nafion and exhibited a current density of 3.5 A cm−2 at 1.8 V.
 | ||
| Fig. 3 Cross-sections of (a) sulfonated poly(phenylene sulfone) (sPPS)-MEA and (b) Nafion-MEA by spray coating. Reproduced with permission from ref. 127. Copyright 2020, WILEIY-VCH. | ||
The decal transfer method is another extensively used approach for CCM preparation, which is considered a feasible method for mass production. It provides the advantage of low resistance between the catalyst layer and polymer electrolyte membrane, as well as reduced mass transfer resistance with the thin catalyst layer.128–130 The conventional decal transfer process consists of multiple steps.130,131 First, a catalyst slurry or ink is cast onto the substrates. Subsequently, the catalyst layer is transferred to the polymer membrane through hot-pressing. This hot-pressing step is performed above the glass transition temperature (Tg) of the polymer used as a membrane. Above Tg, the polymer membrane softens, allowing for interpenetration of the polymer across the interface. After hot-pressing, the substrate is removed. Park et al. employed the decal transfer method to prepare an IrO2 inverse-opal MEA for polymer exchange membrane water electrolysis in order to reduce the catalyst loading.130Fig. 4 shows the process of preparing the inverse-opal MEA. The decal transfer method involves the following steps: (1) preparation of the IrO2 inverse-opal electrode via pulse electrodeposition, (2) hot-pressing of electrodes onto the membrane, (3) removal of the sacrificing layer, and (4) spray coating of the cathode catalyst layer on the opposite side of the membrane. The interconnected pores and increased surface area of the inverse-opal structure enable enhanced catalyst utilization, leading to high mass activity for PEMWE. Bernt et al. utilized the decal transfer method to fabricate 5 cm2 MEAs for PEMWE, employing Pt/C and IrO2/TiO2 as the HER and OER catalysts, respectively.132 The catalyst inks were prepared by mixing solvent, catalysts, DI water, and Nafion ionomer solution. These catalysts inks were then coated on decal substrates such as ethylene-tetrafluoroethylene (ETFE) and polytetrafluoroethylene (PTFE). After drying, the electrodes were hot-pressed onto a Nafion membrane. They showed that optimizing the electrode thickness and catalyst loadings can enhance the interface resistance and catalyst utilization.
 | ||
| Fig. 4 Photographs of preparation steps for inverse-opal MEA in the decal-transfer method. Reproduced with permission from ref. 130. Copyright 2019, Elsevier. | ||
The hot-pressing process can significantly affect the performance of the CCM–MEA because it influences the physical characteristics of the interface contacts among MEA components. Yazdanpour et al. demonstrated the direct coating of ink-jet printed Pt/MWCNT on Nafion membranes for polymer electrolyte membrane fuel cells (PEMFC).133 They optimized the hot-pressing conditions (temperature, pressure, and duration) to fabricate a CCM–MEA and compared its performance with that of CCM created based on the decal transfer method. They demonstrated a high-quality and dense CCM–MEA fabricated by hot-pressing compared to the CCM method by decal transfer. The typical method for catalyst layer formation involves the use of catalyst inks. However, the use of catalyst inks can result in a dense catalyst layer, which impedes the mass transfer of liquid/gas and reduces catalyst utilization. To address this issue, researchers have developed some direct catalyst layer deposition techniques.
Wan et al. designed an all-in-one MEA by fabricating a 3D-ordered catalyst layer on a porous polypropylene (PP) membrane using hydrothermal synthesis.97 First, the CoNi LDHs were synthesized on a porous PP membrane through the solvothermal method and converted to CoNiS by a hydrothermal process. The as-prepared CCM was subsequently pressed to fabricate the CCM–MEA at 70 °C for 3 minutes at 0.1 MPa. The fabricated all-in-one MEA showed a current density of 1 A cm−2 at 1.57 V benefiting from its 3D-ordered catalyst layer structure, which facilitates liquid/gas mass transfer, provides a high catalytic active sites density, and accelerates ionic transport at the integrated catalyst/membrane interface.
Koch et al. developed the bar coating method to directly deposit the catalyst for CCM fabrication.126 The catalyst layers were bar-coated onto a membrane, allowing for control over the catalyst loading by introducing adhesive foil to protect against swelling and wrinkling. Bar-coated CCM showed comparable cell efficiency (1 A cm−2 at 1.8 V) to the spray-coated CCM–MEA. The uniformity of catalyst coating, dual side coating, repeatability of the coating, and durability are required for electrolysis coating applications using large-scale CCM. Efforts have been made to fabricate large-scale CCM for water electrolysis up to a few hundred cm2 scales with the aid of catalyst coating equipment such as an ultrasonic coating system. For PEMWE, the fabrication of CCM has been developed for industrial application, however, low catalyst utilization and dissolution of platinum group catalysts during operation are the main challenges toward large-scale CCM–MEA. In the case of AEMWE, the durability of the membrane/electrocatalysts and deformation during the fabrication process make it difficult to design CCM–MEA, limiting the practical application. Therefore, adequate strategies for designing integrated CCM–MEA are required for securing long lifetime and high performance.
3. Single atom catalysts (SACs) for MEA in water electrolysis
3.1 Why SACs in water electrolysis?
Numerous researchers have endeavored to improve the catalytic activity and sustainability of electrocatalysts for highly efficient hydrogen production, while simultaneously reducing the cost involved in the synthetic process.134–137 In line with this objective, a groundbreaking advancement in electrocatalysts has emerged: the utilization of single atom catalysts. This cutting-edge approach possesses tremendous potential to revolutionize the field of catalysis. Unlike conventional catalysts consisting of nanoparticles or bulk materials, atomically dispersed catalysts possess unique electronic and geometric characteristics that render them exceptionally efficient in promoting both HER and OER.138 Depending on the support matrix, these properties can be elaborately modulated, thereby influencing the surface redox chemistry during water electrolysis.139The utilization of single atom catalysts in water electrolysis provides several advantages which can be categorized into the following two categories: atomically confined active sites optimized for both water reduction and oxidation, and high atomic utilization efficiency. Firstly, the atomically confined active sites of SACs enhance the intrinsic catalytic activity by increasing the opportunities for the target reactions to occur.140,141 The catalytic activity of SACs can be significantly influenced by the support matrix, which is the material that provides an environment for dispersing and anchoring the single metal atoms. This influence is attributed to the variation of the oxidation state and coordination environment of SACs depending on the supports, leading to the modulated adsorption and activation of reactant molecules. Secondly, the high atomic utilization efficiency in SACs provides significant resource efficiency.142,143 Due to the extremely high surface-to-volume ratio of SACs, all the individual metal atoms actively participate in the surface reaction of water electrolysis. As a result, the usage of SACs can reduce the consumption of noble or rare metals required for the synthesis of electrocatalysts. This characteristic makes SACs more economically and environmentally sustainable by minimizing the use of scarce resources. These advantages of SACs are applicable to both HER and OER.
Jiang et al. introduced single-atom Ru catalysts anchored on a nanoporous MoS2 (np-MoS2) and investigated the synergistic effect between Ru sites and sulfur vacancies in alkaline HER.144 By tuning the ligament size of np-MoS2, the curvature-induced strain can be introduced into sulfur vacancies (Fig. 5a). Through the DFT calculations, they explained that H2O molecules are easily adsorbed on the Ru sites of Ru/MoS2 and subsequently dissociated into H and OH species due to a lower water adsorption energy of −0.516 eV and a low energy barrier for the Volmer step, respectively. Furthermore, the H–H coupling can be readily accomplished by Ru sites on Ru/MoS2 due to the small hydrogen adsorption free energy (Fig. 5b). In Fig. 5c, the absorption edge of Ru K-edge X-ray absorption near edge structure (XANES) spectrum of Ru/np-MoS2 is located between those of RuCl3 and RuO2, indicating the oxidation state of +3 to +4 after doping in MoS2. In Fig. 5d, the Fourier-transformed extended X-ray absorption fine structure (FT-EXAFS) spectrum of Ru/np-MoS2 exhibits a prominent peak at ca. 1.6 Å, corresponding to the scattering path between Ru and S. This implies that Ru element exists in isolation without Ru-Ru bonding in Ru/np-MoS2. As shown in Fig. 5e, the catalytic activity of Ru/np-MoS2 was compared with those of nanoporous MoS2 (np-MoS2), Ru single atoms on plane MoS2 (Ru/P-MoS2), and Ru single atoms on nanoporous MoS2 with larger ligament (Ru/Lnp-MoS2). Compared to the other samples, Ru/np-MoS2 shows a much lower overpotential of 30 mV to achieve a current density of 10 mA cm−2 and a Tafel slope of 31 mV dec−1. In Fig. 5f, the electrochemically active surface area (ECSA)-normalized current density of Ru/np-MoS2 at an overpotential of 100 mV is larger than those of Ru/Lnp-MoS2 and Ru/P-MoS2. This indicates that the intrinsic catalytic activity in HER significantly depends on the magnitude of strain, as higher strain induces more variation in the electronic states and atomic structure of Ru sites and sulfur vacancies.
 | ||
| Fig. 5 (a) Schematic illustration of the construction of Ru/np-MoS2. (b) Free energy diagrams at difference sites in HER. (c) XANES and (d) FT-EXAFS spectra of Ru/np-MoS2, Ru foil, RuCl3, and RuO2. (e) Polarization curves of Ru/np-MoS2 as compared with np-MoS2, Ru/P-MoS2, Ru/Lnp-MoS2, Ru/C, and Pt/C. (f) ECSA-normalized current density at an overpotential of 100 mV for Ru/P-MoS2, Ru/Lnp-MoS2, and Ru/np-MoS2. Reproduced with permission from ref. 144. Copyright 2021. Springer Nature. | ||
Zhang et al. selectively anchored Ir single atoms onto the three-fold hollow sites and oxygen vacancy sites of defective CoOOH to explore how the catalytic activity in OER is controlled by anchoring sites.49 Defective CoOOH offers three possible sites for immobilizing the metal species as support for single atom anchoring. These sites include the three-fold face-centered cubic (fcc) hollow sites of oxygen, the three-fold hexagonal close-packed (hcp) hollow sites of oxygen, and the oxygen vacancy sites (Fig. 6a). In this research, singly-dispersed iridium cations were anchored to the three-fold hollow sites of surface oxygen (Ir1/TO-CoOOH), while isolated iridium anions were located at oxygen vacancies (Ir1/VO-CoOOH) via cathodic and anodic electrochemical deposition, respectively. In Fig. 6b, the white line intensity implies that the valence state of Ir in Ir1/TO-CoOOH ranges from +3 to +4, while in Ir1/VO-CoOOH, it is slightly higher than +4. This difference can be attributed to the charge transfer under reductive and oxidative environments during the synthesis. In Fig. 6c, no dominant peak at 2.71 Å was observed in either sample, indicating the absence of Ir–Ir interaction. As shown in Fig. 6d, Ir1/VO-CoOOH showed dramatically enhanced catalytic activity compared to Ir1/TO-CoOOH and the original CoOOH with an overpotential of 200 mV to attain a current density of 10 mA cm−2. In Fig. 6e, Ir1/VO-CoOOH exhibited a specific activity of 17.3 mA cm−2 at an overpotential of 300 mV, which was 5.75 times higher than that of Ir1/TO-CoOOH. Fig. 6f shows the free energy diagrams of CoOOH, Ir1/TO-CoOOH, and Ir1/VO-CoOOH, with the rate-determining step energy barrier of Ir1/VO-CoOOH showing the lowest value of 1.63 eV.
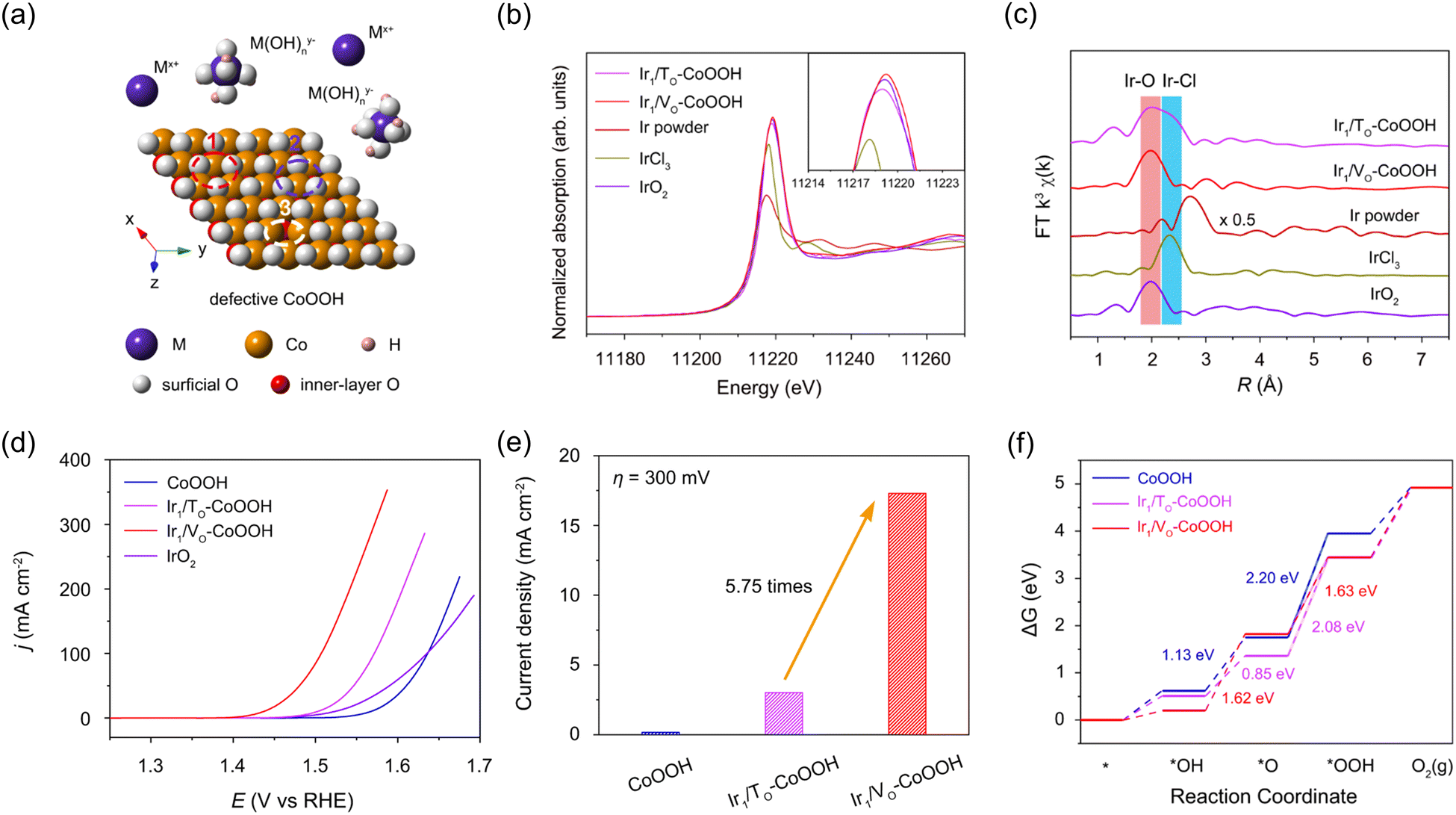 | ||
| Fig. 6 (a) Schematic illustration of oxygen-terminated (001) surface of defective CoOOH with the single atom anchoring sites. (b) XANES and (c) FT-EXAFS spectra of Ir1/TO-CoOOH, and Ir1/VO-CoOOH, Ir powder, IrCl3, and IrO2. (d) Polarization curves of CoOOH, Ir1/TO-CoOOH, and Ir1/VO-CoOOH. (e) Specific activities of samples normalized by ECSA at an overpotential of 300 mV. (f) Free energy diagram of CoOOH, Ir1/TO-CoOOH, and Ir1/VO-CoOOH in OER. Reproduced with permission from ref. 49. Copyright 2022. Springer Nature. | ||
Until now, various types of SACs have been reported for highly efficient HER and OER including noble metal-based SACs, non-noble metal-based SACs, single atom alloys, and dual metal SACs. In HER, elements such as Pt,145–147 Ru,148–150 Ni,151–153 Co,154–156 Fe,157–159 and V160–162 have been selected to promote atomically confined hydrogen adsorption and desorption. For OER, Ir,163–166 Ru,141,167,168 Au,169,170 Fe,171–173 Ni,174,175 and Co176–178 single atoms were developed. Recently, multi-component SACs have been explored to outperform the performance of single-component SACs. For instance, Pt–Ru and W–Mo dual atom catalysts have shown superior performance in HER and OER, respectively.179–182
3.2 Application of SACs into MEA
Single atom catalysts can be applied to both CCS and CCM methods. As can be seen in Fig. 7a, the CCS method involves the preparation, coating, and drying of catalyst inks containing SACs. Separate substrates, acting as gas diffusion layers, are employed to coat the catalyst inks prepared for both HER and OER. Then, the MEA is fabricated by integrating two catalyst-coated substrates and a membrane (Fig. 7b). This fabrication process typically involves sandwiching the two substrates between an anion-exchange membrane or a proton exchange membrane by hot pressing. On the other hand, in the case of the CCM method, the catalyst ink with SACs is directly coated onto both sides of the membrane, which is the key distinction compared to the CCS method (Fig. 7c). There are several coating techniques such as spray coating, spin coating, inkjet printing, doctor blade coating, and decal transfer method. Subsequently, the gas diffusion layers and the catalyst-coated membrane are assembled by hot pressing, resulting in a CCM–MEA with SACs (Fig. 7d). In both cases, the actual reactions occur at the interface between the membrane and the catalyst layer. Therefore, it is necessary to significantly increase the quantity of SACs at the interface to achieve high performance with the smallest amount of SACs.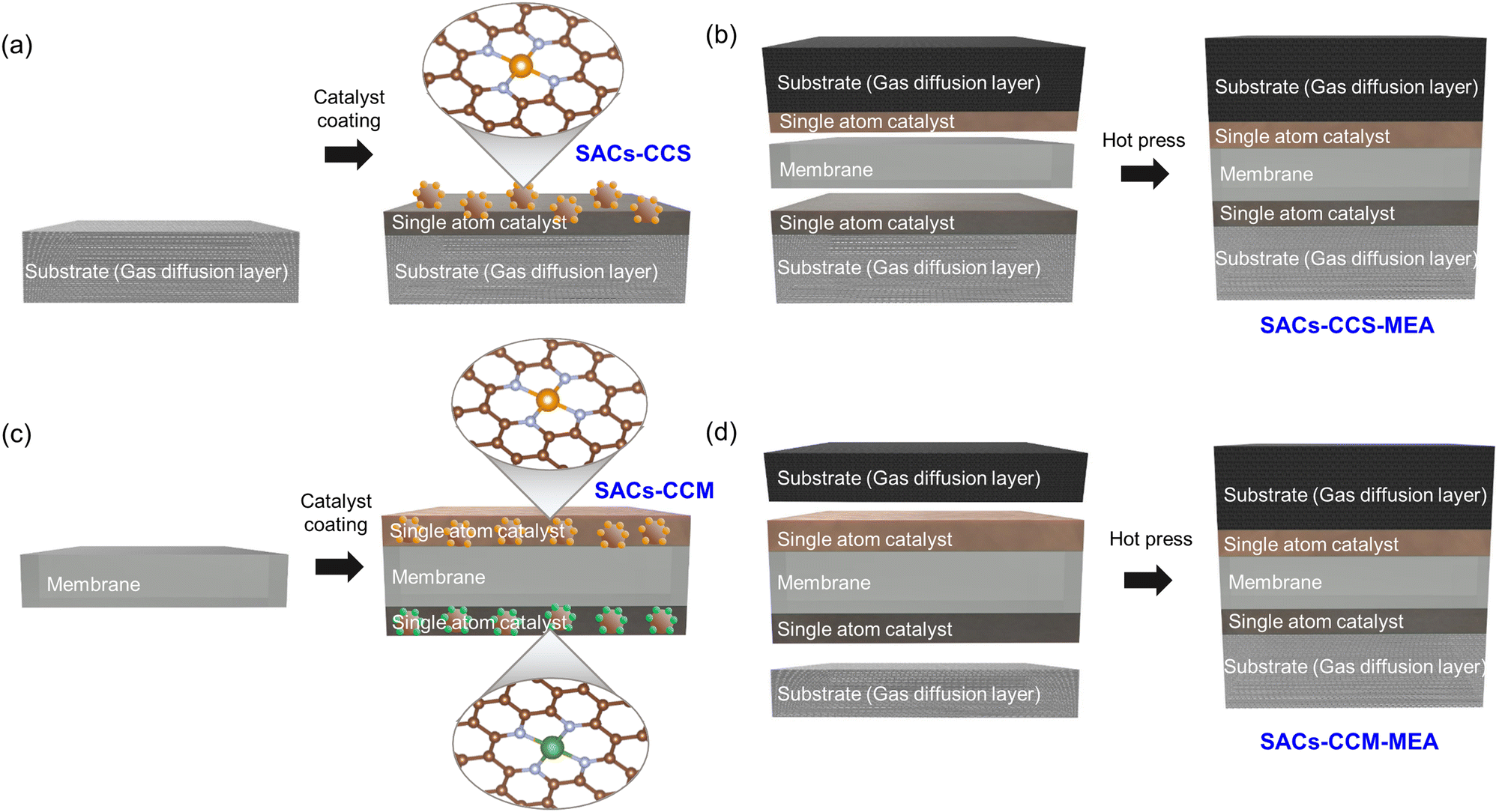 | ||
| Fig. 7 Schematic of (a) SAC-CCS–MEA and (b) MEA fabrication by hot pressing. (c) Schematic of the SAC–CCM–MEA and (d) MEA fabrication. | ||
3.3 Binder-free SACs with bottom-up method
Atomic layer deposition. Atomic layer deposition (ALD) is a cyclic process that relies on sequential self-limiting reactions between gas phase chemical precursors and a solid substrate surface.183,184 This technique enables the formation of atomically dispersed metals as well as the uniform and conformal thin films, within structures with high aspect ratios and porous materials.185
Sun's group successfully prepared one-to-one bimetallic Pt–Ru dimer single atom catalysts through an ALD process (Fig. 8a).179 Firstly, for the deposition of Pt on NCNTs, they utilized trimethyl(methylcyclopentadienyl)-platinum(IV) (MeCpPtMe3) as the precursor and the process was conducted at 250 °C. The high-purity N2 was used for both the purging and carrier gas. The container for MeCpPtMe3 was maintained at 65 °C to ensure a steady-state flux of Pt to the chamber. The Pt precursor shows a tendency to react with the N atoms of NCNTs, establishing a strong metal–support interaction through the chemical bonding between atomically distributed Pt metals and N sites. Then, Ru metal was selectively anchored beside Pt single atoms using bis(ethylcyclopentadienyl)ruthenium(II) precursors (Ru(C2H5C5H4)2). The ratio of dimer structure is around 70%, which indicates that most of the single atoms exist in dimers. Recently, Zhang et al. synthesized Co single atom-modified Pt nanoparticles on NCNTs via the ALD process.186 After the formation of Pt NPs, Co single atoms were selectively deposited using bis(ethylcyclopentadienyl)cobalt(II) (Co(C5H5)2) as a precursor and high-purity N2 as both purging and carrier gas.
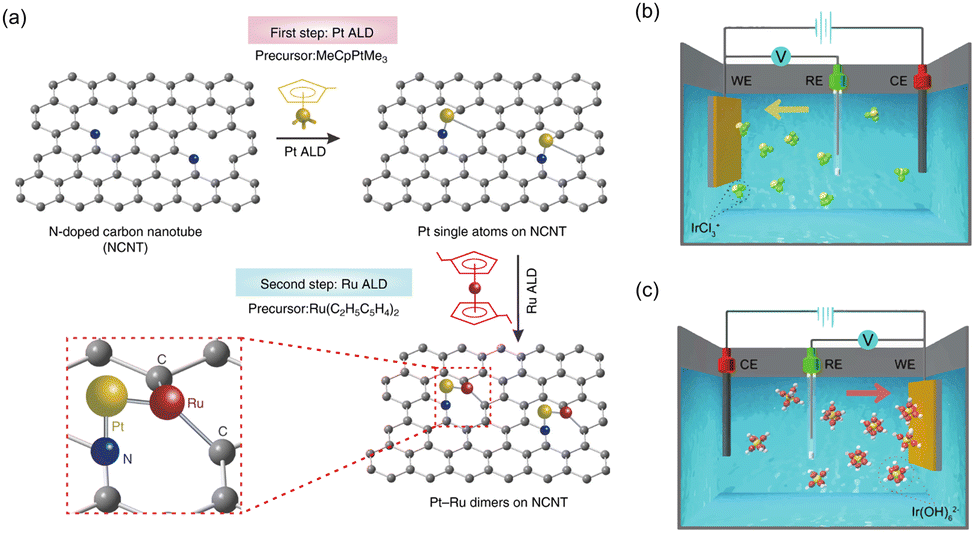 | ||
| Fig. 8 (a) Schematic of synthesis process of the Pt–Ru dimer SACs using ALD process. Reproduced with permission from ref. 179. Copyright 2019, Springer Nature. Schematics of (b) cathodic and (c) anodic deposition of Ir species using three-electrode system. Reproduced with permission from ref. 187. Copyright 2020. Springer Nature. | ||
Electrochemical deposition. Zeng's group reported a universal electrochemical deposition method which is applicable to various metal elements and supports for the formation of SACs.187 The depositions occurred on both the cathode and anode, resulting in the SACs acquiring unique electronic states due to the various redox reactions. Representatively, Ir single atoms were anchored on Co(OH)2 nanosheets using a standard three-electrode system. A low concentration (100 μM) of IrCl4 was added to a 1 M KOH electrolyte and the cathodic deposition was performed with a depositing potential ranging from 0.10 to −0.40 V. Conversely, the anodic deposition utilized a potential range of 1.10 to 1.80 V. A scanning rate of 5 mV s−1 was employed for one scanning cycle, which was repeated ten times to obtain C–Ir1/Co(OH)2 from the cathode and three times to obtain A–Ir1/Co(OH)2 from the anode. In Fig. 8b, iridium cations were moving toward the cathode during the cathodic deposition. Based on the coordination number (CN) of 3.1 for the Ir–Cl contribution, the cations were supposed to be IrCl3+. Furthermore, considering the CN of 3.3 for the Ir–O contribution, they proposed that the IrCl3+ cations coordinated with three oxygen atoms on Co(OH)2. Fig. 8c shows the movement of iridium-complex anions toward the anode during the anodic deposition. The UV-vis adsorption spectroscopy indicated the adsorption of Ir(OH)62− species, suggesting that Ir(OH)62− anions serve as the depositing species during the anodic deposition. This observation aligns with the EXAFS result obtained for A–Ir1/Co(OH)2, which showed a coordination number of 5.8 from the Ir–O contribution.
3.4 Powder-type SACs with top-down method
Pyrolysis of spatially confined element. Chen et al. proposed a two-step pyrolysis method derived from metal–organic frameworks to obtain Co SACs bound with N, P, and S on a hollow carbon polyhedron support (Fig. 9a).192 The initial step involved the pyrolysis of highly cross-linked poly(cyclotriphospazene-co-4,4′-sulfonyldiphenol) (PZS) coated zeolitic imidazolate framework (ZIF-8) core–shell composites (ZIF-8@PZS) at 650 °C under an Ar atmosphere. This process resulted in the formation of nitrogen, phosphorus, and sulfur co-doped hollow carbon polyhedron (NPS-HC-650). The formation of the hollow structure followed the Kirkendall effect, in which the interdiffusion occurred between the S2− ion from the PZS shell and the Zn2+ ion from the ZIF-8 core. The smaller ionic radius of the Zn2+ ion compared to the S2− ion led to faster outward transport of Zn2+ ions compared to the inward transport of S2− ions. The continuous unequal interdiffusion created Kirkendall voids, resulting in the generation of a hollow structure. Similar to the S species, the P species from the PZS shell diffused into the hollow NPS-HC-650 architecture and dispersed uniformly. Subsequently, the cobalt precursor was added, which could be absorbed onto the hierarchical pore structure of NPS-HC-650, forming Co/NPS-HC-650. Finally, Co1-N3PS/HC catalyst was obtained by the pyrolysis of Co/NPS-HC-650 at 950 °C under an Ar atmosphere.
 | ||
| Fig. 9 (a) Schematic illustration of synthetic process of Co1-N3PS/HC. Reproduced with permission from ref. 192. Copyright 2020. Wiley-VCH. (b) TEM images of AgNP/MnO2 at different temperature of 50, 150, 200, and 300 °C. Reproduced with permission from ref. 193. Copyright 2020. Wiley-VCH. (c) Schematic illustration of the ball-milling process to obtain single atoms. Reproduced with permission from ref. 194. Copyright 2021. Wiley-VCH. | ||
Thermal transformation. Li's group reported the synthesis of Ag single atom catalysts using the thermal transformation of Ag nanoparticles.193 The Ag NPs were prepared by dissolving AgNO3 into ammonium hydroxide and DI water, followed by the addition of the solution to the MnO2 suspension. To observe the transformation of Ag NPs, in situ environmental transmission electron microscopy (ETEM) was utilized. As can be seen in Fig. 9b, both the number and diameter of particles were significantly decreased as the temperature was increased to 300 °C. At 350 °C, the Ag NPs vanished in the ETEM image. From aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC-HAADF-STEM) image, it was confirmed that both small Ag clusters and single atoms exist on thermally transformed AgNP/MnO2-350 °C.
Ball milling. Wang et al. introduced a ball-milling method for the large-scale synthesis of Co single atom catalysts surrounded by sulfur and nitrogen atoms (Fig. 9c).194 The surface atoms existing in Co powders are easily dissociated and captured by sulfur and nitrogen co-doped carbon (SNC) due to the stronger binding between Co and N atoms in CoN4 than that between Co and Co atoms in bulk Co powders. For the synthesis, SNC support and Co powder were combined and placed into an iron tank. Subsequently, 50 agate balls with a diameter of 3 mm were added to the tank. The iron tank was then inserted into a ball milling machine and milled at a power of 100 W for a duration of 4 hours. After the completion of the ball milling process, the agate balls were separated, and the resulting powder was stirred in a mixed solution of hydrochloric acid (3 M) and hydrofluoric acid (1 M) for 2 hours. The XANES spectra showed that the Co absorption edge of Co-SNC is far from that of Co foil, indicating the positive electronic states of the Co in Co-SNC catalyst. Moreover, FT-EXAFS spectrum of Co-SNC exhibits only one distinct peak at 1.44 Å, corresponding to the first coordination shell of Co–N bonding. This result suggests that Co atoms in Co-SNC are fully dispersed via ball-milling techniques and N atoms possess a stronger binding energy with Co atoms compared to S atoms.
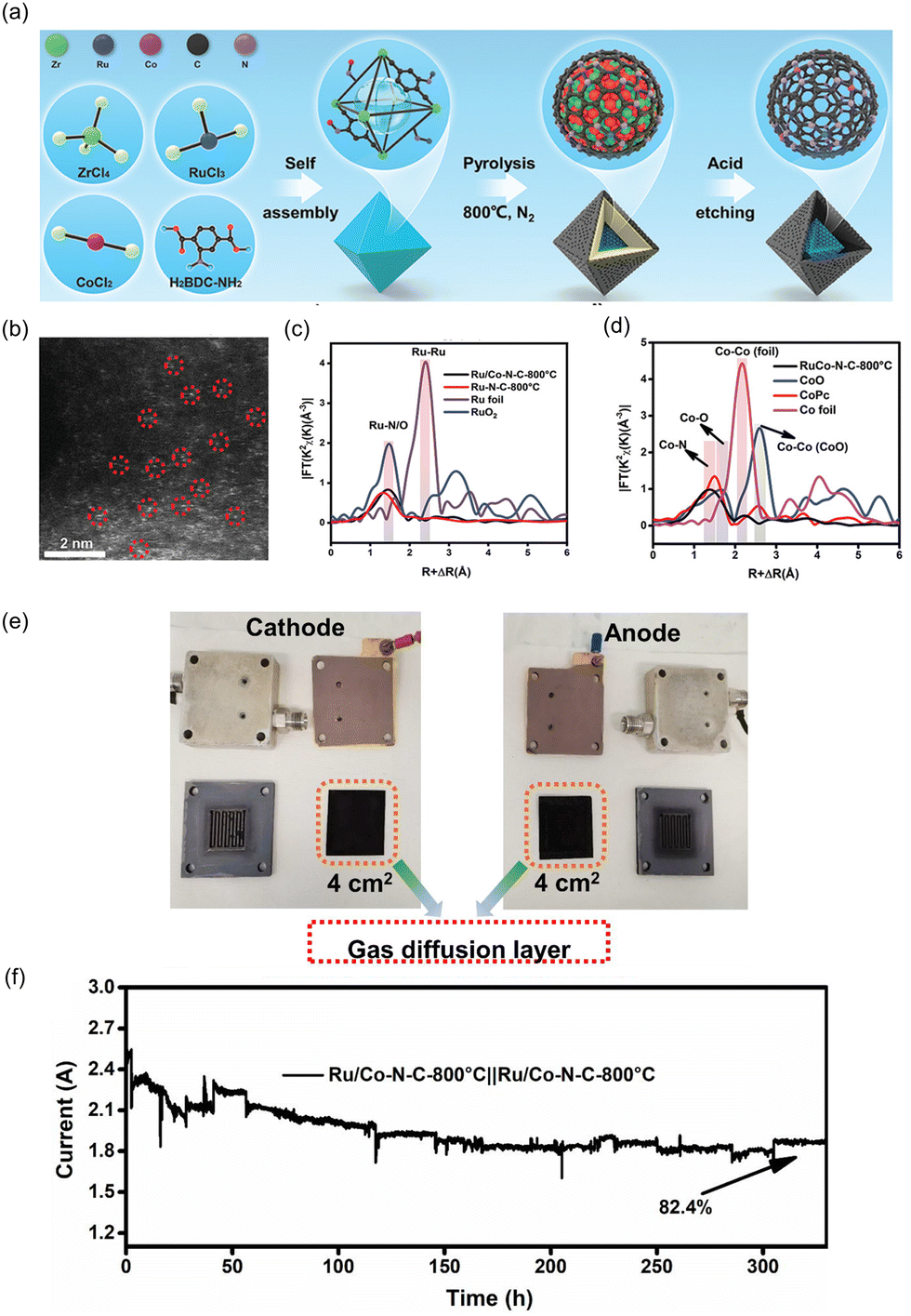 | ||
| Fig. 10 (a) Schematic illustration of synthetic process of Ru/Co–N–C-800 °C. (b) HAADF-STEM image of Ru/Co–N–C-800 °C showing the isolated Ru and Co single atoms. The normalized EXAFS spectra of Co foil, CoPc, CoO, and Ru/Co–N–C-800 °C for (c) Co element and (d) Ru element. (e) Digital photograph of components in PEMWE device. (f) The current–time stability of Ru/Co–N–C-800 °C as both anode and cathode in acidic PEM. Reproduced with permission from ref. 103. Copyright 2022. Wiley-VCH. | ||
Wu et al. investigated a Ni-incorporated RuO2 catalyst to improve the stabilization of the RuO2 lattice under acidic OER conditions.195 The powder-type Ni–RuO2 catalyst was applied to the anode in PEMWE. In Fig. 11a, a schematic illustration of the synthetic process of Ni–RuO2 is provided. Firstly, metal precursors were applied onto a carbon black support by a wet impregnation process. The resultant mixture was annealed in an H2/Ar atmosphere, leading to the formation of Ru3Ni nanoparticles. Secondly, Ru3Ni nanoparticles were converted into Ru3NiOx, while the carbon support was simultaneously eliminated by air annealing. Finally, the resulting Ru3NiOx catalyst underwent an acid-leaching process, which effectively removed unstable Ni species and yielded the desired Ni–RuO2 catalyst. In Fig. 11b, the normalized Ni K-edge XANES spectrum of Ni–RuO2 indicates that the oxidation state of Ni in Ni–RuO2 is closer to that of NiO. Fig. 11c depicts the FT-EXAFS spectrum of Ni-incorporated RuO2, which exhibits a dominant peak at 1.6 Å corresponding to the scattering path between Ni and O. As-synthesized Ni–RuO2 electrocatalyst showed significantly improved OER activity and stability in acidic conditions compared to bare RuO2. This improvement was attributed to the lowered limiting potential and reduced Ru demetallation. This powder-type Ni-incorporated RuO2 catalyst was applied to construct the PEMWE, with Pt/C and Nafion 117 as the cathode and membrane, respectively (Fig. 11d and e). For the fabrication of the CCS, 3.1 mg cm−2 of Ni–RuO2 powder mixed with 20 wt.% PTFE binder was drop-casted onto a platinized titanium fiber felt electrode. The electrode was then dried and pressed under a pressure of less than 0.5 MPa using a hot-press machine. Finally, it was annealed in air for 30 minutes at 350 °C, followed by the circulation of a 0.1 M HClO4 solution. In Fig. 11f, the current–voltage curves exhibit that the device with Ni–RuO2 shows higher water electrolysis activity compared to those with RuO2 and commercial RuO2. Specifically, at room temperature, it requires only 1.78 V, 1.95 V, and 2.10 V to achieve current densities of 500 mA cm−2, 1000 mA cm−2, and 1500 mA cm−2, respectively.
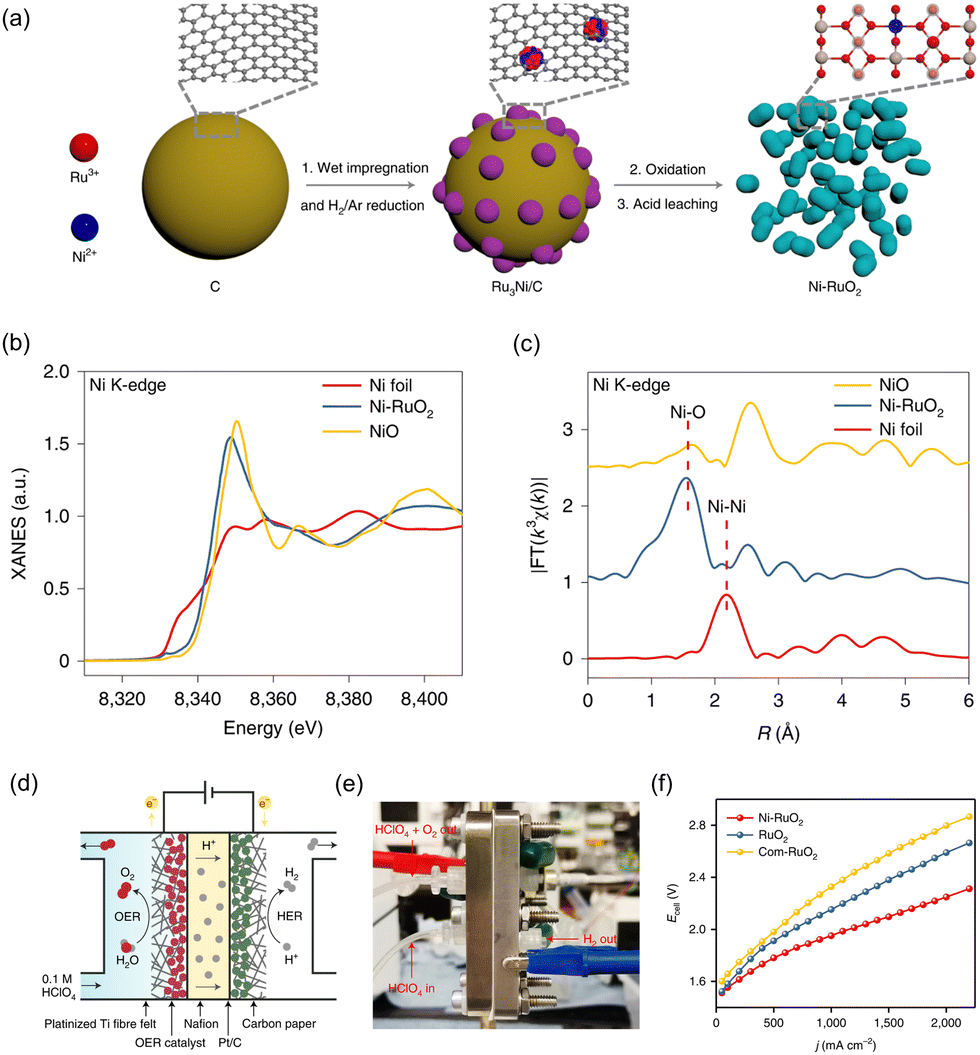 | ||
| Fig. 11 (a) Schematic illustration of synthetic process of Ni–RuO2. The normalized (b) XANES and (c) EXAFS spectra of NiO, Ni foil, and Ni–RuO2. (d) Schematic and (e) digital photograph of PEMWE device. (f) I–V curves of PEMWE device using Ni–RuO2, RuO2, and Com-RuO2 as OER catalyst and commercial Pt/C as HER catalyst. Reproduced with permission from ref. 195. Copyright 2023. Springer Nature. | ||
Wang et al. introduced a novel NiHO/Ni5P4 heterostructure with both Ru nanoclusters and single atoms as a highly active HER catalyst in alkaline conditions.196Fig. 12a shows a HAADF-STEM image revealing the uniform dispersion of both Ru nanoclusters and single atoms on the surface of amorphous NiHO. As shown in Fig. 12b, slight changes in the bonding distances were observed for the NS-Ru@NiHO/Ni5P4 in comparison to the reference Ru foil and RuO2. The distance between Ru atoms decreased from 2.37 to 2.35 Å, while the distance between Ru and O atoms in the Ru–O bonding increased from 1.52 to 1.56 Å. These changes can be attributed to the interface coupling between the Ru nanoclusters and the NiHO substrate, as well as the unsaturated coordination of the Ru surface atoms with oxygen atoms from the NiHO substrate. In Fig. 12c, NS-Ru@NiHO/Ni5P4 shows the lowest overpotential of 16 mV to achieve a current density of 10 mA cm−2 for the HER in an alkaline electrolyte. They suggest that the alkaline HER pathway begins with the adsorption of a water molecule near the surface Ru single atoms, followed by the formation of a bridged H intermediate configuration, which effectively reduces the energy barrier for breaking the O–H bond in water. During the water dissociation process, the bridged H atom is captured by the adjacent atomically-layered Ru NCs, while the resulting OH-species diffuses into the electrolyte. Simultaneously, another water molecule is adsorbed by the Ru surface atoms and undergoes dissociation with the collaborative action of the nanoclusters and surface atoms. Based on the high catalytic activity and stability demonstrated by the powder-type NS-Ru@NiHO/Ni5P4 in an alkaline environment, an AEMWE system was constructed using IrO2-plated nickel foam as the anode and FAA-3-PK-30 (Fumasep) as the membrane. The measurement was conducted at 50 °C with a flow of 1.0 M KOH electrolyte. In Fig. 12d, the device with the NS-Ru@NiHO/Ni5P4‖IrO2 system showed outstanding performance in water electrolysis, achieving a low cell potential of 1.7 V to obtain a current density of 1.0 A cm−2; the catalytic performance to obtain this level of current density was retained for over 150 hours, as shown in Fig. 12e.
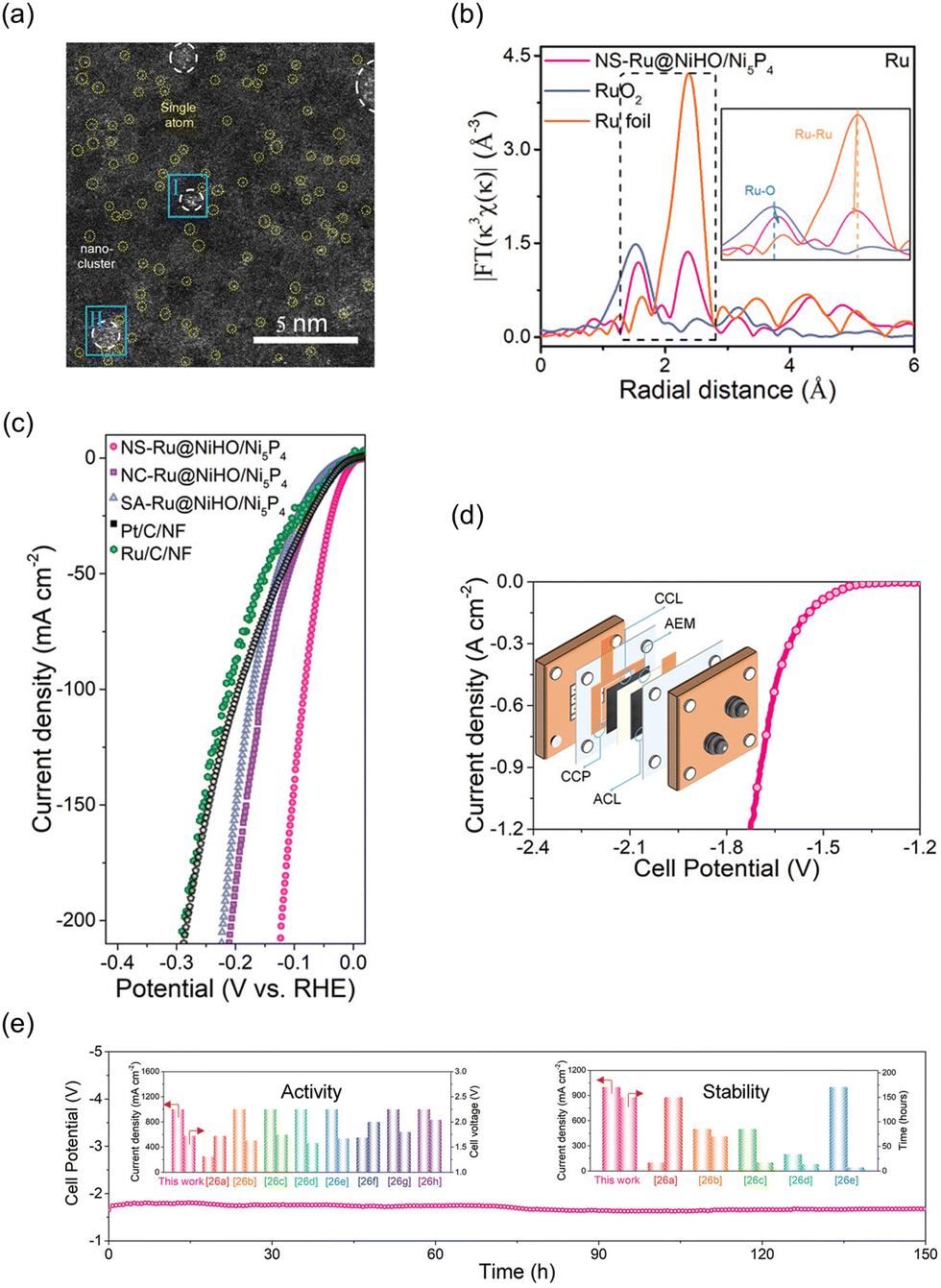 | ||
| Fig. 12 (a) HAADF-STEM image of NS-Ru@NiHO/Ni5P4 showing the isolated Ru single atoms. The normalized EXAFS spectra of Ru foil, RuO2, and CoO, and NS-Ru@NiHO/Ni5P4. (c) Polarization curves of the HER catalysts. (d) I–V curves of AEMWE device using NS-Ru@NiHO/Ni5P4 and IrO2. (e) Chronopotentiometry curve of NS-Ru‖IrO2 AEMWE at 1.0 A cm−2, inset: comparison of activity (left) and stability (right) for NS-Ru‖IrO2 and other previously reported AEMWEs. Reproduced with permission from ref. 196. Copyright 2023. Wiley-VCH. | ||
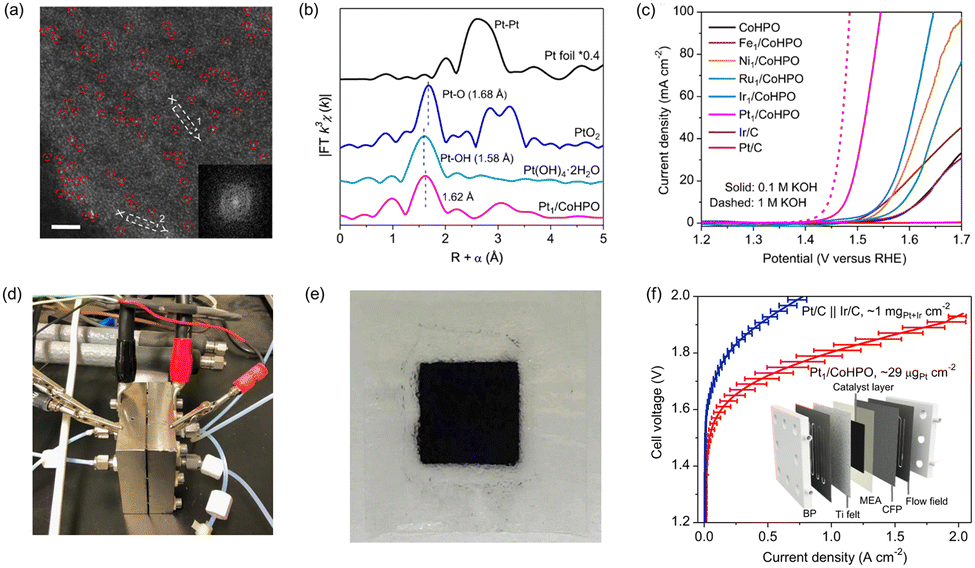 | ||
| Fig. 13 (a) HAADF-STEM image of Pt1/CoHPO showing the isolated Pt single atoms. The normalized EXAFS spectra of Pt foil, PtO2, Pt(OH4)·2H2O and Pt1/CoHPO. (c) Polarization curves of the OER catalysts. Digital photograph of (d) AEMWE device and (e) CCM-type MEA. (f) I–V curves of AEMWE device using benchmark Pt/C‖Ir/C and Pt1/CoHPO. Reproduced with permission from ref. 104. Copyright 2022. Springer Nature. | ||
4. Advantages of SACs–CCM–MEA
Two-types of MEA fabrication methods were introduced to apply SACs in water electrolysis cells, SACs–CCS–MEA and SACs–CCM–MEA. Compared to SACs–CCS–MEA, SACs with CCM-type MEA have several advantages regarding cell performance (Fig. 14).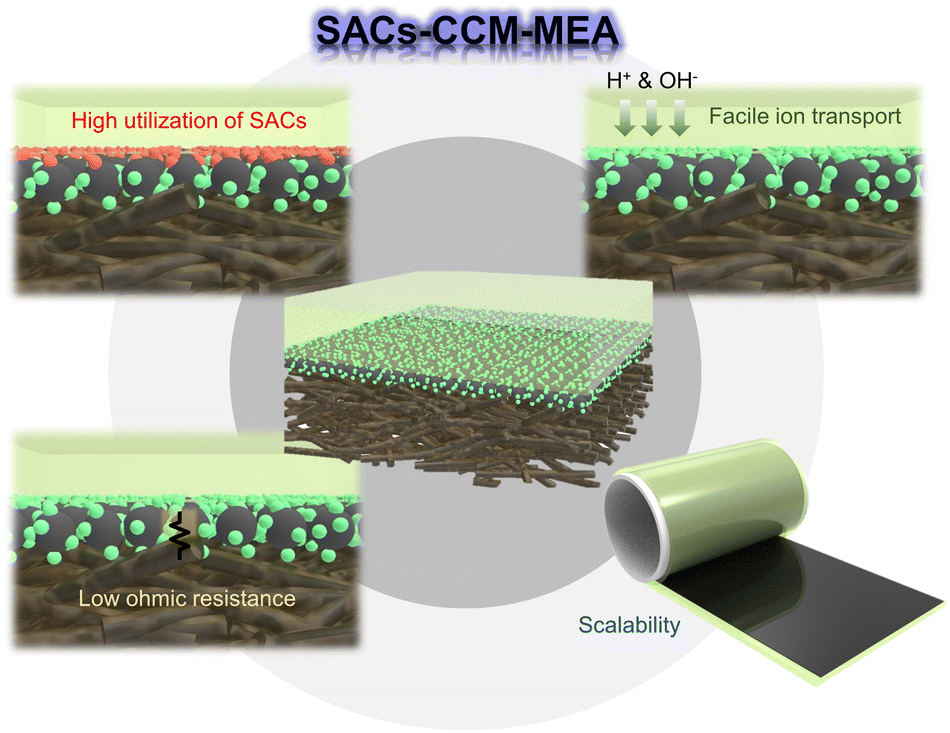 | ||
| Fig. 14 Schematic of the advantages of CCM–MEA with single atom catalysts. | ||
Firstly, the SACs–CCM–MEA facilitates the high utilization efficiency of SACs. This approach involves directly applying the catalyst ink onto the surface of the membrane, ensuring that it does not remain inside the GDL and therefore remains fully utilized. This method enables more efficient utilization of the catalyst. Additionally, a majority of the metal single atoms, anchored on the support matrix, are uniformly dispersed on the membrane. This uniform dispersion ensures complete contact between the SACs and the membrane, eliminating any inactive and empty reaction sites.
Secondly, the SACs–CCM–MEA offers an advantage in ion transport by shortening the diffusion path for ions (H+ for PEM and OH− for AEM). It secures a direct and shorter path for ions to travel between the membrane and the reaction sites of the catalyst, as proximity is ensured. Thus, the overall water electrolysis process is more efficient, enabling higher current densities.
Thirdly, the SACs–CCM–MEA minimizes interfacial resistance between the catalyst layer and the membrane. The direct contact between SACs and the membrane enables efficient charge transfer and reduces the resistance at the electrode–electrolyte interface. By reducing interfacial resistance, it is possible to lower the overall ohmic resistance, resulting in improved water electrolysis performance. Moreover, this approach facilitates uniform current distribution across the interface in the MEA, reducing the risk of localized high-resistance areas. In the case of SACs–CCS–MEA, the SACs are in sparse contact with the membrane, although they are well distributed along the support, resulting in the localization of highly resistive sites.
Lastly, the SACs–CCM–MEA method offers excellent scalability for MEA production. Scalability is crucial for meeting increasing demand and achieving cost-effective production of MEA on a larger scale. Many researchers have developed state-of-the-art synthetic methods for large amounts and high metal loadings of SAC powders. For improved mass production and reduced complexity of MEA, the CCM method is suitable since the continuous and automated manufacturing process is possible. In addition, the catalyst loading can be precisely controlled by adjusting the amount of catalyst ink applied to the membrane.
Several advantages of SACs–CCM–MEA were introduced, including high utilization of SACs, facile ion transport, low ohmic resistance, and scalability. Despite its remarkable superiority, some potential challenges related to SACs, membrane, and the assembly of SACs–CCM–MEA hinder practical applications. Thus, it is essential to address the limitations of integrating SACs and the CCM architecture for highly efficient and cost-effective hydrogen production.
5. Challenges
Although SACs–CCM–MEA is promising for green hydrogen production by taking advantage of a zero-gap structure with a favorable catalyst/membrane interface, there is still a long way to go toward achieving commercial hydrogen production. Fig. 15 displays the challenges of SACs–CCM–MEA, focusing on two main components: SACs and CCM, as well as the fabrication of MEA using SACs and CCM.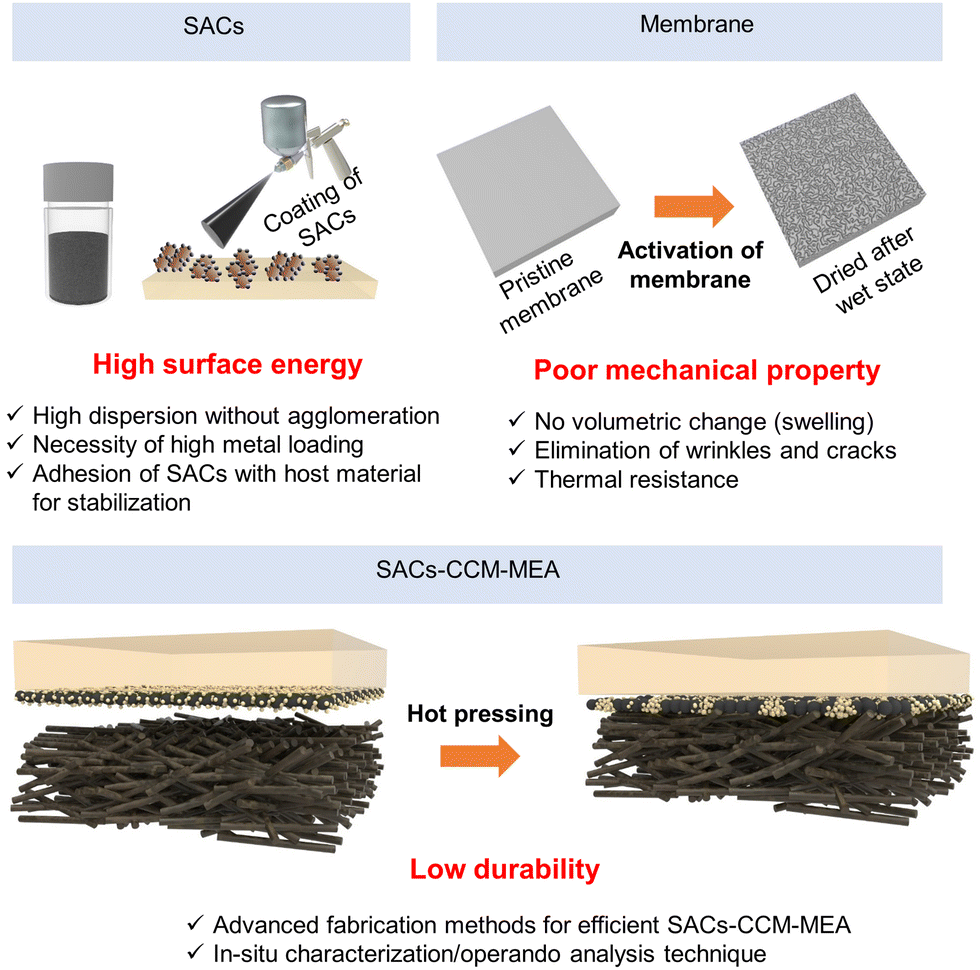 | ||
| Fig. 15 Schematic of challenges of SACs–CCM–MEA for water electrolysis. | ||
5.1 Challenges of SACs
Introducing SACs with high atomic utilization and unique properties has shown a promising pathway toward water electrolysis with high current density. Although remarkable advance has been achieved in the use of SACs in water electrolysis, significant efforts should be devoted to developing fabrication methods and stability.To fabricate a CCM–MEA for large-scale hydrogen production, the SACs should be synthesized on an industrial scale. The SACs can be synthesized as powder-type or directly formed on the substrates through various techniques. In the case of indirect coating of SACs for CCM–MEA, the catalyst ink with SACs poses a challenge in terms of its poor dispersibility leading to agglomeration during the coating process. It should be carefully prepared with the optimized mixture of solvents. Additionally, scaling up SACs poses another challenge in achieving commercial-scale production for higher levels of technological maturity. Increasing metal-atom densities in SACs is challenging, typically exceeding 5 wt%. Xia et al. reported the synthesis of SACs with high metal loadings up to 40 wt% based on the crosslinking and self-assembly of graphene quantum dots.197 Achieving high metal atom density with sufficient spacing can prevent the aggregation of SACs and lead to increased catalytic activity.
In addition, developing SACs with excellent stability is imperative but still a challenge due to the high surface-free energy and low coordination number of SACs under synthetic and reaction conditions. They are easily aggregated and dissolved if there is a poor chemical interaction between metal atoms and support matrix. Therefore, a thorough comprehension of metal–support interaction is necessary to achieve highly stable SACs. Until now, strong chemical and structural interactions have been reported using various substrates such as metal oxides, graphene, g-C3N4, and metal–organic frameworks. For example, Wu et al. synthesized highly stable Ni-incorporated RuO2 for acidic OER and Ni atoms embedded into the lattice of RuO2 with Ni–O interactions not only boosting the catalytic activity but also stabilizing the catalysts.195 There are other strong interactions between metal atoms and surrounding atoms including Pt–O,145 Pt–N2C2,198 and Ni–C4.199 Thus, for future work, it is necessary to find the unique chemical bondings and focus on enhancing the interactions.
Furthermore, catalyst layers containing SACs are easily detached or peeled off from membranes under large current density due to the low structural stability at the interface. In this respect, improving the quality of catalyst ink dispersion is necessary to ensure a highly robust MEA. The crucial consideration in controlling the dispersion of catalyst ink is its composition. Lots of studies have revealed the importance of ionomer to carbon ratio.200,201 One step further, in SACs–CCM–MEA, it is necessary to find an optimized ratio between SACs and carbon or carbon with SACs and ionomer exhibiting the highest structural stability without detachment.
Moreover, similar to the micro/nano electrocatalysts, SACs are usually prepared on carbon-based supports. However, SACs on carbon-based support can be damaged during anodic reaction due to the oxidation of carbon-based supports, resulting in physical damage. Therefore, proper SAC support materials with good adhesion are required for water electrolysis.
5.2 Challenges of membrane
The ion exchange membrane, composed of polymer backbones as the main chains and cationic/anionic groups as the secondary functional groups, enables water molecule transport and prevents gas permeation. Ion exchange membranes are classified into two types, CEMs and AEMs, based on their selective permeability to specific ions, which is determined by their inherent charges. The main properties for evaluating ion exchange membranes in water electrolysis are as follows: (1) ion exchange conductivity that refers to the number of exchangeable ions per membrane dry weight, (2) swelling ratio, which measures the linear expansion of the membrane when exposed to water, calculated as the difference between the membrane length in a dry state and a wet state, (3) water uptake, indicating the degree of change in membrane mass change when exposed to water, (4) gas permeation that quantifies the extent of gas cross-over, (5) mechanical stability of the membrane in the electrolytes, which is one of the most important properties. For practical CCM–MEA applications, the membrane should exhibit excellent ionic conductivity or low area resistance, long-term operational stability, and high mechanical stability while maintaining low cost.The consensus regarding the most challenging aspects of the membranes lies in the stability issues associated with mechanical deformation under operation conditions. Compared to the commercial cation exchange membrane (e.g., Nafion), ensuring excellent stability in an AEM poses a challenge due to the polymer backbone degradation caused by OH− ions. Under the KOH-fed condition of AEMWE, various degradation mechanisms of commercial AEMs have been reported.202 Cationic groups, such as quaternary ammonium (QA) groups, pyridinium, and imidazolium, can cause AEM degradation through the bimolecular nucleophilic substitution (SN2), Hofmann elimination, phenyl oxidation, radical oxygen reaction, and other degradation mechanisms.203 The polymer backbone chains can also be broken through dehydrofluorination, hydrolysis, and other chain-cutting processes. As selected examples, Parrondo et al. conducted 1H-NMR analysis to examine the degradation of polysulfone-based AEMs using post-mortem samples and reported backbone degradation during long WE operation.204 Chen et al. prepared AEMs with a polymer with a poly(fluorenyl ether ketone sulfone) backbone and imidazolium- and quaternary ammonium (QA)-functionalized groups.205 They compared various membrane properties, including water uptake, OH− conductivity, and alkaline/thermal stability. After immersing the membrane in 1 M NaOH, the QA-functionalized membrane was broken into small pieces, indicating a decrease in mechanical stability. The ion exchange capacity (IEC) of both membranes was reduced at elevated temperatures, suggesting degradation of the cationic groups and polymer backbone under an alkaline environment. Arges et al. addressed the durability issue of polysulfone (PSF) AEMs in alkaline solutions using 2D NMR techniques, including correlation spectroscopy (COSY) and heteronuclear multiple quantum correlation spectroscopy (HMQC). The hydrolysis of the PSF-based AEM backbone was confirmed through the 2D NMR spectra, leading to the formation of phenyl alcohol.
In summary, ensuring the stability of membranes for CCM–MEA in the operating environment is imperative. However, still, it remains challenging to scrutinize the degradation mechanism during operation. The majority of the research has focused on reporting the degradation of membranes using post-mortem samples, while the difficulty of unveiling the in situ deformation of membranes poses a bottleneck to designing durable membranes.
5.3 Challenges of SACs–CCM–MEA
Although the SACs and CCM are both promising for water electrolysis, the design of SACs–CCM–MEA is still in its infancy with only a few reports available on water electrolysis. The main challenges faced by SACs–CCM–MEAs are their instability originating from the components such as SACs, membranes, ionomers, supports, as well as the fabrication methods and degradation that occurs during operation.First, there are several ways to fabricate CCM–MEA including direct/indirect methods. Compared to CCS–MEA, adjusting the in situ catalyst deposition technique is difficult due to the swelling and nonconducting characteristics of the polymer membrane. Since SACs are typically prepared on supporting materials, a simple and plausible method for fabricating SACs–CCM–MEA is using catalyst ink or slurry to form a catalyst layer on the membrane. However, the membrane can undergo swelling and shrinkage during the solvent absorption and drying process.206 These results in severely wrinkled and unstable catalyst layers, posing significant challenges in mass production. The catalytically active single atomic sites can be hampered by the ionomer or binder. Consequently, it is essential to develop an adequate fabrication technique to obtain durable SACs–CCM–MEA.126
Second, the CCM fabrication method generally includes hot-pressing of the catalyst/membrane and GDL to ensure favorable contact at the interfaces. The hot-pressing temperature is determined by considering the glass transition temperature (Tg) of the membrane and ionomer. Several studies have reported the effects of hot-pressing CCM–MEA with micro/nano-catalysts on water electrolysis performance. For example, Siracusano et al. investigated the effect of hot-pressing of MEA on PEMWE performance.207 They controlled the hot-pressing temperature (180 °C, 200 °C) and time (1.5 min, 3 min) and tested the cell at an operating current density of 1 A cm−2 at 55 °C. Depending on the hot-pressing conditions, the MEAs showed different stability due to the varied extent of adhesion of the catalyst layer to the membrane. Optimal hot-pressing conditions are prerequisites for fabricating CCM–MEA since the interface contact significantly impacts the MEA performance. Despite the several advantages of hot-pressed CCM–MEAs, hot-pressing can result in the migration and agglomeration of SACs, which are major issues associated with SACs.208 Various strategies have been implemented to atomically disperse SACs and inhibit their movement on the supports. However, the heat and compression process can provide sufficient energy to reconstruct SACs at the interface. Recent studies have demonstrated hot-pressing of CCM–MEA at lower temperatures, which can inflict less damage to the membrane and catalyst layer.
Finally, the establishment of standardized testing criteria for CCM–MEA is crucial to facilitate reliable performance comparisons. Numerous MEAs undergo testing under diverse operating conditions, including variations in temperature, type of electrolytes, and catalyst loading level, which poses challenges to compare the MEA performance. For example, the SAC loading level can lead to significant disparities in WE performance, thereby necessitating the presentation of data under a consensus setup. Additionally, the operating temperature and electrolytes can have significant influences on several membrane properties, such as durability and ion exchange capacity. Therefore, it is imperative to establish a standardized and consensus testing condition for assessing and reporting the WE performance of MEAs.
6. Conclusion and perspectives
Water electrolysis is considered one of the most promising technologies for producing green hydrogen to accomplish carbon neutrality. Water electrolysis has been developed from alkaline water electrolysis, a mature technology, using the diaphragm to separate generated hydrogen and oxygen. However, the low energy efficiency (especially low current density) and gas crossover are the main challenges. Since General Electric Co. first used PEM for electrolysis in 1967, dedicated efforts to develop ion exchange membrane water electrolysis technologies have led to achieving competitiveness for industrial-scale hydrogen production. The core of ion exchange membrane water electrolysis is the zero-gap MEA, which consists of a catalyst/support and a membrane. Studies on SACs have demonstrated their potential as promising electrocatalysts for WE over the past few decades, due to their maximum atomic efficiency and extraordinary catalytic activity. In the context of ion exchange membrane water electrolyzer, studies on MEA preparations have revealed that MEA fabrication significantly impacts resultant WE performances. Two major methods for MEA fabrication are the CCS and CCM methods. While the CCS method was developed early on due to its simplicity, the non-uniform catalyst/membrane interface resulted in elevated high-frequency resistance. On the other hand, the CCM method shows promise as it enables direct contact between the catalyst and membrane, thus reducing the ohmic resistance at the interface. Therefore, a combination of SACs and CCM–MEA has the potential to be a cutting-edge technology for green hydrogen production for the following reasons.Fig. 16a shows the promising aspects of designing SACs–CCM–MEA in terms of cell voltage. Initially, the MEA was developed in the form of CCS–MEA employing micro/nano-catalysts because of its simple and easy fabrication process. The development of the CCS fabrication method subsequently led to the incorporation of nanostructured catalysts on support/substrates, thus enhancing mass transport through structural effects. The CCM–MEA with the nanostructured catalysts can provide the advantage of reduced operating voltage by minimizing ohmic resistance at the catalyst/membrane interface. Additionally, nanostructured catalysts play a role in increasing catalytic active sites and facilitating gas/liquid transfer. By introducing SACs into the CCM–MEA, it is possible to reduce cell voltage through high mass activity and increased catalytic active sites. Fig. 16b shows the typical polarization curve of MEAs, illustrating the kinetic, ohmic, and mass transfer regions as rate-determining steps. Compared to conventional CCS–MEA with micro/nano electrocatalysts, SACs–CCM–MEAs can enhance cell performance due to the introduction of catalytic active SACs (kinetic region), close contact between catalyst/membrane (ohmic region), and high utilization of SACs with low catalyst loading (mass transfer region). Consequently, the energy efficiency of hydrogen production can be significantly enhanced under high current operation.
 | ||
| Fig. 16 (a) Effects of different types of MEA on operating cell voltage. (b) Expected J–V curves of MEAs. (c) Comparison of CCS–MEA with micro/nano-catalysts and CCM–MEA with SACs. | ||
Fig. 16c shows the overall comparison between CCS–MEA with micro/nano-catalysts and SACs–CCM–MEA. CCS has a less uniform catalyst/membrane interface than CCM, resulting in an increase in ohmic resistance. The presence of layers of micro/nano catalysts layer with ionomer can cause ionomer aggregation and hinder gas diffusion and ion transport. These phenomena can ultimately result in mass transfer limitations in MEA cells, particularly in high current density regions. In contrast, SACs–CCM forms a uniform catalyst/membrane interface, expediting ion transport and lowering ohmic resistance. Since the utilization of SACs can significantly reduce the catalyst loading, gas/liquid diffusion can be accelerated. Furthermore, the thickness of the catalyst layer at the atomic scale can mitigate the issues associated with ionomer aggregation.
However, current technologies are facing several challenges in the context of SACs–CCM–MEA for water electrolysis toward green hydrogen production. In terms of catalysts, comparing the activities of SACs in MEA is difficult because of different experimental conditions. The operating temperature, extent of the compression, and catalyst loading pose obstacles to direct comparison among different WE devices. Furthermore, SACs can migrate and undergo reconstruction during operation, similar to other electrocatalysts. Thus, it is necessary to develop operando characterization techniques to understand the degradation mechanism of SACs. Strategies to ensure the stability of SACs are imperative to prevent the dissolution or aggregation of SACs. In the case of an ion exchange membrane, the degradation mechanism during operation should be unveiled to design a durable membrane. Current techniques rely on post-mortem membranes, which provide limited information on polymer degradation. Particularly, the stability of the AEM in an alkaline environment should be improved for durable AEMWE.
The development of SACs–CCM is more challenging than current CCM fabrication using nanoparticles mainly due to the difficulty of synthesizing large amounts of SACs. Although current technologies enable scalable CCM up to a few hundred cm2, preparing ample SACs ink without agglomeration is a prerequisite for practical CCM fabrication. Optimized fabrication methods of SACs–CCM–MEA should be developed to enable large-scale hydrogen production with high efficiency. Although various approaches exist for designing CCM–MEA, the WE performance is significantly affected by the CCM preparation methods, wherein catalyst distribution, catalyst/membrane contact, and membrane deformation should be considered.
Introducing SACs into CCM–MEA has a long road ahead. Nevertheless, we believe that SACs–CCM–MEA, coupled with advanced fabrication methods, will shine the light on the path toward cost-effectiveness and enhanced cell performance in industrial green hydrogen production for carbon neutrality. The feasibility of SACs–CCM–MEA can also be demonstrated in various practical applications, such as CO2 electroreduction and fuel cells, which require high operating current density with scalability at a low-cost since MEAs and SACs for these applications also have been widely investigated. We anticipate that this review will provide valuable insights into the design of state-of-the-art technology for MEA water electrolyzers, contributing to green hydrogen production.
Author contributions
SA Lee and SE Jun contributed equally to this work. They wrote the manuscript under the supervision of JH Kang, MS Kwon, and HW Jang. All authors contributed to the general discussion.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. A. Lee and S. E. Jun contributed equally to this work. This work was supported by the Korea Research Institute of Standards and Science (KRISS) MPI Lab. Program and the National Research Foundation of Korea (NRF) granted by the Korea Government MSIT (grant no. 2021M3H4A1A03057403, 2021R1A4A3027878, and 2021R1C12006142). The Inter-University Semiconductor Research Center and Institute of Engineering Research at Seoul National University provided research facilities for this work.References
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS
.
- J. A. Turner, Science, 2004, 305, 972–974 CrossRef CAS PubMed
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS
- S. A. Lee, S. Choi, C. Kim, J. W. Yang, S. Y. Kim and H. W. Jang, ACS Mater. Lett., 2020, 2, 107–126 CrossRef CAS
- X. Li, L. Zhao, J. Yu, X. Liu, X. Zhang, H. Liu and W. Zhou, Nano-Micro Lett., 2020, 12, 131 CrossRef CAS
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef
- S. Chu, Y. Cui and N. Liu, Nat. Mater., 2017, 16, 16–22 CrossRef
- S. A. Lee, M. G. Lee and H. W. Jang, Sci. China Mater., 2022, 65, 3334–3352 CrossRef
- G. Venugopalan, D. Bhattacharya, E. Andrews, L. Briceno-Mena, J. Romagnoli, J. Flake and C. G. Arges, ACS Energy Lett., 2022, 7, 1322–1329 CrossRef CAS
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef CAS
- B. M. Hunter, H. B. Gray and A. M. Müller, Chem. Rev., 2016, 116, 14120–14136 CrossRef CAS
- S. A. Lee, J. W. Yang, T. H. Lee, I. J. Park, C. Kim, S. H. Hong, H. Lee, S. Choi, J. Moon, S. Y. Kim, J. Y. Kim and H. W. Jang, Appl. Catal., B, 2022, 317, 121765 CrossRef CAS
- S. A. Lee, I. J. Park, J. W. Yang, J. Park, T. H. Lee, C. Kim, J. Moon, J. Y. Kim and H. W. Jang, Cell Rep., 2020, 1, 100219 CAS
- S. E. Jun, J. K. Lee and H. W. Jang, Energy Adv., 2023, 2, 34–53 RSC
- S. E. Jun, S. Choi, S. Choi, T. H. Lee, C. Kim, J. W. Yang, W.-O. Choe, I.-H. Im, C.-J. Kim and H. W. Jang, Nano-Micro Lett., 2021, 13, 81 CrossRef CAS
- J. Park, S. Lee, T. H. Lee, C. Kim, S. E. Jun, J. H. Baek, J. Y. Kim, M. G. Lee, S. H. Ahn and H. W. Jang, Nano Convergence, 2022, 9, 33 CrossRef CAS PubMed
- S. S. M. Bhat, S. E. Jun, S. A. Lee, T. H. Lee and H. W. Jang, Energies, 2020, 13, 974 CrossRef CAS
- J. Chi and H. Yu, Chin. J. Catal., 2018, 39, 390–394 CrossRef CAS
- S. A. Grigoriev, V. N. Fateev, D. G. Bessarabov and P. Millet, Int. J. Hydrogen Energy, 2020, 45, 26036–26058 CrossRef CAS
- M. Chatenet, B. G. Pollet, D. R. Dekel, F. Dionigi, J. Deseure, P. Millet, R. D. Braatz, M. Z. Bazant, M. Eikerling, I. Staffell, P. Balcombe, Y. Shao-Horn and H. Schäfer, Chem. Soc. Rev., 2022, 51, 4583–4762 RSC
- A. J. Shih, M. C. O. Monteiro, F. Dattila, D. Pavesi, M. Philips, A. H. M. da Silva, R. E. Vos, K. Ojha, S. Park, O. van der Heijden, G. Marcandalli, A. Goyal, M. Villalba, X. Chen, G. T. K. K. Gunasooriya, I. McCrum, R. Mom, N. López and M. T. M. Koper, Nat. Rev. Methods Primers, 2022, 2, 84 CrossRef CAS
- O. J. Guerra, J. Eichman, J. Kurtz and B.-M. Hodge, Joule, 2019, 3, 2425–2443 CrossRef
- F. Mueller-Langer, E. Tzimas, M. Kaltschmitt and S. Peteves, Int. J. Hydrogen Energy, 2007, 32, 3797–3810 CrossRef CAS
- S. A. Lee, J. Bu, J. Lee and H. W. Jang, Small Sci., 2023, 3, 2200109 CrossRef CAS
- H. R. Kwon, H. Park, S. E. Jun, S. Choi and H. W. Jang, Chem. Commun., 2022, 58, 7874–7889 RSC
- S. Choi, C. Kim, J. Y. Lee, T. H. Lee, K. C. Kwon, S. Kang, S. A. Lee, K. S. Choi, J. M. Suh, K. Hong, S. E. Jun, W. K. Kim, S. H. Ahn, S. Han, S. Y. Kim, C.-H. Lee and H. W. Jang, Chem. Eng. J., 2021, 418, 129369 CrossRef CAS
- Z. Yu, X. Rui and Y. Yu, EES Catal., 2023, 1, 695–703 RSC
- X. Zheng, J. Yang and D. Wang, EES Catal., 2023, 1, 665–676 RSC
- L. Wan, Z. Xu, Q. Xu, M. Pang, D. Lin, J. Liu and B. Wang, Energy Environ. Sci., 2023, 16, 1384–1430 RSC
- M. Carmo, D. L. Fritz, J. Mergel and D. Stolten, Int. J. Hydrogen Energy, 2013, 38, 4901–4934 CrossRef CAS
- S. E. Jun, S.-P. Hong, S. Choi, C. Kim, S. G. Ji, I. J. Park, S. A. Lee, J. W. Yang, T. H. Lee, W. Sohn, J. Y. Kim and H. W. Jang, Small, 2021, 17, 2103457 CrossRef CAS PubMed
- G. Lee, S. E. Jun, Y. Kim, I.-H. Park, H. W. Jang, S. H. Park and K. C. Kwon, Materials, 2023, 16, 3280 CrossRef CAS PubMed
- S. Sebbahi, N. Nabil, A. Alaoui-Belghiti, S. Laasri, S. Rachidi and A. Hajjaji, Mater. Today: Proc., 2022, 66, 140–145 CAS
- H. A. Miller, K. Bouzek, J. Hnat, S. Loos, C. I. Bernäcker, T. Weißgärber, L. Röntzsch and J. Meier-Haack, Sustainable Energy Fuels, 2020, 4, 2114–2133 RSC
- I. Vincent, A. Kruger and D. Bessarabov, Int. J. Hydrogen Energy, 2017, 42, 10752–10761 CrossRef CAS
- C. C. Pavel, F. Cecconi, C. Emiliani, S. Santiccioli, A. Scaffidi, S. Catanorchi and M. Comotti, Angew. Chem., Int. Ed., 2014, 53, 1378–1381 CrossRef CAS PubMed
- S. Koch, L. Metzler, S. K. Kilian, P. A. Heizmann, F. Lombeck, M. Breitwieser and S. Vierrath, Adv. Sustainable Syst., 2023, 7, 2200332 CrossRef CAS
- J. Hnát, M. Plevova, R. A. Tufa, J. Zitka, M. Paidar and K. Bouzek, Int. J. Hydrogen Energy, 2019, 44, 17493–17504 CrossRef
- R. R. Raja Sulaiman, W. Y. Wong and K. S. Loh, Int. J. Energy Res., 2022, 46, 2241–2276 CrossRef CAS
- B. Chen, P. Mardle and S. Holdcroft, J. Power Sources, 2022, 550, 232134 CrossRef CAS
- T. Y. Yoo, J. Lee, S. Kim, M. Her, S.-Y. Kim, Y.-H. Lee, H. Shin, H. Jeong, A. K. Sinha, S.-P. Cho, Y.-E. Sung and T. Hyeon, Energy Environ. Sci., 2023, 16, 1146–1154 RSC
- J. Yang, Y. Huang, H. Qi, C. Zeng, Q. Jiang, Y. Cui, Y. Su, X. Du, X. Pan, X. Liu, W. Li, B. Qiao, A. Wang and T. Zhang, Nat. Commun., 2022, 13, 4244 CrossRef CAS
- J. Wei, K. Xiao, Y. Chen, X.-P. Guo, B. Huang and Z.-Q. Liu, Energy Environ. Sci., 2022, 15, 4592–4600 RSC
- Y. Wu, Q. Wu, Q. Zhang, Z. Lou, K. Liu, Y. Ma, Z. Wang, Z. Zheng, H. Cheng, Y. Liu, Y. Dai, B. Huang and P. Wang, Energy Environ. Sci., 2022, 15, 1271–1281 RSC
- J. Yu, Y. Qian, Q. Wang, C. Su, H. Lee, L. Shang and T. Zhang, EES Catal., 2023, 1, 571–579 RSC
- S. E. Jun, Y.-H. Kim, J. Kim, W. S. Cheon, S. Choi, J. Yang, H. Park, H. Lee, S. H. Park, K. C. Kwon, J. Moon, S.-H. Kim and H. W. Jang, Nat. Commun., 2023, 14, 609 CrossRef CAS
- Z. Lei, W. Cai, Y. Rao, K. Wang, Y. Jiang, Y. Liu, X. Jin, J. Li, Z. Lv, S. Jiao, W. Zhang, P. Yan, S. Zhang and R. Cao, Nat. Commun., 2022, 13, 24 CrossRef CAS PubMed
- Z. Zhang, C. Feng, D. Wang, S. Zhou, R. Wang, S. Hu, H. Li, M. Zuo, Y. Kong, J. Bao and J. Zeng, Nat. Commun., 2022, 13, 2473 CrossRef CAS
- G. Shi, Y. Xie, L. Du, X. Fu, X. Chen, W. Xie, T.-B. Lu, M. Yuan and M. Wang, Angew. Chem., Int. Ed., 2022, 61, e202203569 CrossRef CAS PubMed
- J. Chen, T. Wang, X. Wang, B. Yang, X. Sang, S. Zheng, S. Yao, Z. Li, Q. Zhang, L. Lei, J. Xu, L. Dai and Y. Hou, Adv. Funct. Mater., 2022, 32, 2110174 CrossRef CAS
- J. Wang, M. Zheng, X. Zhao and W. Fan, ACS Catal., 2022, 12, 5441–5454 CrossRef CAS
- W. Huang, J. Zhu, S. Liu, W. Zhang, L. Zhou and L. Mai, EES Catal., 2023, 1, 434–458 RSC
- M. K. Aslam, K. Yang, S. Chen, Q. Li and J. Duan, EES Catal., 2023, 1, 179–229 RSC
- S. M. Lee, W. S. Cheon, M. G. Lee and H. W. Jang, Small Struct., 2023, 4, 2200236 CrossRef CAS
- F. Liu, C. Shi, L. Pan, Z.-F. Huang, X. Zhang and J.-J. Zou, EES Catal., 2023, 1, 562–570 RSC
- M. Liu, L. Wang, K. Zhao, S. Shi, Q. Shao, L. Zhang, X. Sun, Y. Zhao and J. Zhang, Energy Environ. Sci., 2019, 12, 2890–2923 RSC
- C. Wan, X. Duan and Y. Huang, Adv. Energy Mater., 2020, 10, 1903815 CrossRef CAS
- G. Kour, X. Mao and A. Du, J. Mater. Chem. A, 2022, 10, 6204–6215 RSC
- D. Chen, M. Luo, S. Ning, J. Lan, W. Peng, Y.-R. Lu, T.-S. Chan and Y. Tan, Small, 2022, 18, 2104043 CrossRef CAS
- Y. Kong, Y. Li, X. Sang, B. Yang, Z. Li, S. Zheng, Q. Zhang, S. Yao, X. Yang, L. Lei, S. Zhou, G. Wu and Y. Hou, Adv. Mater., 2022, 34, 2103548 CrossRef CAS PubMed
- Y. Yao, L. Zhao, J. Dai, J. Wang, C. Fang, G. Zhan, Q. Zheng, W. Hou and L. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202208215 CrossRef CAS PubMed
- T. Lim, G. Y. Jung, J. H. Kim, S. O. Park, J. Park, Y.-T. Kim, S. J. Kang, H. Y. Jeong, S. K. Kwak and S. H. Joo, Nat. Commun., 2020, 11, 412 CrossRef
- L. Mao, Y.-C. Huang, H. Deng, F. Meng, Y. Fu, Y. Wang, M. Li, Q. Zhang, C.-L. Dong, L. Gu and S. Shen, Small, 2023, 19, 2203838 CrossRef CAS PubMed
- G. Yang, Y. Li, H. Lin, X. Ren, D. Philo, Q. Wang, Y. He, F. Ichihara, S. Luo, S. Wang and J. Ye, Small Methods, 2020, 4, 2000577 CrossRef CAS
- S.-Y. Yuan, L.-W. Jiang, J.-S. Hu, H. Liu and J.-J. Wang, Nano Lett., 2023, 23, 2354–2361 CrossRef CAS
- S. Tong, B. Fu, L. Gan and Z. Zhang, J. Mater. Chem. A, 2021, 9, 10731–10738 RSC
- Q. Guo, Q. Zhao, R. Crespo-Otero, D. Di Tommaso, J. Tang, S. D. Dimitrov, M.-M. Titirici, X. Li and A. B. Jorge Sobrido, J. Am. Chem. Soc., 2023, 145, 1686–1695 CrossRef CAS
- X. Zhang, P. Zhai, Y. Zhang, Y. Wu, C. Wang, L. Ran, J. Gao, Z. Li, B. Zhang, Z. Fan, L. Sun and J. Hou, J. Am. Chem. Soc., 2021, 143, 20657–20669 CrossRef CAS PubMed
- Y. Miao, J. Liu, L. Chen, H. Sun, R. Zhang, J. Guo and M. Shao, Chem. Eng. J., 2022, 427, 131011 CrossRef CAS
- Y. Zhao, K. R. Yang, Z. Wang, X. Yan, S. Cao, Y. Ye, Q. Dong, X. Zhang, J. E. Thorne, L. Jin, K. L. Materna, A. Trimpalis, H. Bai, S. C. Fakra, X. Zhong, P. Wang, X. Pan, J. Guo, M. Flytzani-Stephanopoulos, G. W. Brudvig, V. S. Batista and D. Wang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 2902–2907 CrossRef CAS PubMed
- D. Liu, X. Wan, T. Kong, W. Han and Y. Xiong, J. Mater. Chem. A, 2022, 10, 5878–5888 RSC
- E. Zhao, K. Du, P.-F. Yin, J. Ran, J. Mao, T. Ling and S.-Z. Qiao, Adv. Sci., 2022, 9, 2104363 CrossRef CAS
- Z.-H. Xue, D. Luan, H. Zhang and X. W. Lou, Joule, 2022, 6, 92–133 CrossRef CAS
- J. Sui, H. Liu, S. Hu, K. Sun, G. Wan, H. Zhou, X. Zheng and H.-L. Jiang, Adv. Mater., 2022, 34, 2109203 CrossRef CAS
- B. Wang, H. Cai and S. Shen, Small Methods, 2019, 3, 1800447 CrossRef
- B. Yan, H. Song and G. Yang, Chem. Eng. J., 2022, 427, 131795 CrossRef CAS
- B.-H. Lee, S. Park, M. Kim, A. K. Sinha, S. C. Lee, E. Jung, W. J. Chang, K.-S. Lee, J. H. Kim, S.-P. Cho, H. Kim, K. T. Nam and T. Hyeon, Nat. Mater., 2019, 18, 620–626 CrossRef CAS PubMed
- Z. Yu, Y. Li, A. Torres-Pinto, A. P. LaGrow, V. M. Diaconescu, L. Simonelli, M. J. Sampaio, O. Bondarchuk, I. Amorim, A. Araujo, A. M. T. Silva, C. G. Silva, J. L. Faria and L. Liu, Appl. Catal., B, 2022, 310, 121318 CrossRef CAS
- M. Ren, X. Zhang, Y. Liu, G. Yang, L. Qin, J. Meng, Y. Guo and Y. Yang, ACS Catal., 2022, 12, 5077–5093 CrossRef CAS
- P. Wang, S. Fan, X. Li, J. Wang, Z. Liu, Z. Niu, M. O. Tadé and S. Liu, Nano Energy, 2022, 95, 107045 CrossRef CAS
- W. Liu, H. Feng, Y. Yang, Y. Niu, L. Wang, P. Yin, S. Hong, B. Zhang, X. Zhang and M. Wei, Nat. Commun., 2022, 13, 3188 CrossRef CAS PubMed
- T.-N. Ye, Z. Xiao, J. Li, Y. Gong, H. Abe, Y. Niwa, M. Sasase, M. Kitano and H. Hosono, Nat. Commun., 2020, 11, 1020 CrossRef CAS
- Y. Zhang, Y. Cheng, X. Wang, Q. Sun, X. He and H. Ji, ACS Catal., 2022, 12, 15091–15096 CrossRef CAS
- W.-H. Li, J. Yang, H. Jing, J. Zhang, Y. Wang, J. Li, J. Zhao, D. Wang and Y. Li, J. Am. Chem. Soc., 2021, 143, 15453–15461 CrossRef CAS
- G. Liu, A. W. Robertson, M. M.-J. Li, W. C. H. Kuo, M. T. Darby, M. H. Muhieddine, Y.-C. Lin, K. Suenaga, M. Stamatakis, J. H. Warner and S. C. E. Tsang, Nat. Chem., 2017, 9, 810–816 CrossRef CAS
- J. Kim, S. Choi, J. Cho, S. Y. Kim and H. W. Jang, ACS Mater. Au, 2022, 2, 1–20 CrossRef CAS
- B. Singh, V. Sharma, R. P. Gaikwad, P. Fornasiero, R. Zbořil and M. B. Gawande, Small, 2021, 17, 2006473 CrossRef CAS
- T. Sun, S. Mitchell, J. Li, P. Lyu, X. Wu, J. Pérez-Ramírez and J. Lu, Adv. Mater., 2021, 33, 2003075 CrossRef CAS
- H. Yang, L. Shang, Q. Zhang, R. Shi, G. I. N. Waterhouse, L. Gu and T. Zhang, Nat. Commun., 2019, 10, 4585 CrossRef
- J. Wang, Z. Li, Y. Wu and Y. Li, Adv. Mater., 2018, 30, 1801649 CrossRef
- Y. Zhou, X. Tao, G. Chen, R. Lu, D. Wang, M.-X. Chen, E. Jin, J. Yang, H.-W. Liang, Y. Zhao, X. Feng, A. Narita and K. Müllen, Nat. Commun., 2020, 11, 5892 CrossRef CAS
- A. G. Rocha, R. Ferreira, D. Falcão and A. M. F. R. Pinto, Energies, 2022, 15, 7937 CrossRef CAS
- Y. Leng, G. Chen, A. J. Mendoza, T. B. Tighe, M. A. Hickner and C.-Y. Wang, J. Am. Chem. Soc., 2012, 134, 9054–9057 CrossRef CAS PubMed
- F. Razmjooei, T. Morawietz, E. Taghizadeh, E. Hadjixenophontos, L. Mues, M. Gerle, B. D. Wood, C. Harms, A. S. Gago, S. A. Ansar and K. A. Friedrich, Joule, 2021, 5, 1776–1799 CrossRef CAS
- L. Wan, Z. Xu, Q. Xu, P. Wang and B. Wang, Energy Environ. Sci., 2022, 15, 1882–1892 RSC
- L. Wan, M. Pang, J. Le, Z. Xu, H. Zhou, Q. Xu and B. Wang, Nat. Commun., 2022, 13, 7956 CrossRef CAS PubMed
- R. Bashyam and P. Zelenay, Nature, 2006, 443, 63–66 CrossRef CAS PubMed
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed
- H. Fei, J. Dong, M. J. Arellano-Jiménez, G. Ye, N. Dong Kim, E. L. G. Samuel, Z. Peng, Z. Zhu, F. Qin, J. Bao, M. J. Yacaman, P. M. Ajayan, D. Chen and J. M. Tour, Nat. Commun., 2015, 6, 8668 CrossRef CAS
- P. Chen, T. Zhou, L. Xing, K. Xu, Y. Tong, H. Xie, L. Zhang, W. Yan, W. Chu, C. Wu and Y. Xie, Angew. Chem., 2017, 129, 625–629 CrossRef
- H. Yan, Y. Lin, H. Wu, W. Zhang, Z. Sun, H. Cheng, W. Liu, C. Wang, J. Li, X. Huang, T. Yao, J. Yang, S. Wei and J. Lu, Nat. Commun., 2017, 8, 1070 CrossRef
- C. Rong, X. Shen, Y. Wang, L. Thomsen, T. Zhao, Y. Li, X. Lu, R. Amal and C. Zhao, Adv. Mater., 2022, 34, 2110103 CrossRef CAS
- L. Zeng, Z. Zhao, F. Lv, Z. Xia, S.-Y. Lu, J. Li, K. Sun, K. Wang, Y. Sun, Q. Huang, Y. Chen, Q. Zhang, L. Gu, G. Lu and S. Guo, Nat. Commun., 2022, 13, 3822 CrossRef CAS
- M. Mandal, ChemElectroChem, 2021, 8, 36–45 CrossRef CAS
- P. Chen and X. Hu, Adv. Energy Mater., 2020, 10, 2002285 CrossRef CAS
- J. R. Varcoe, P. Atanassov, D. R. Dekel, A. M. Herring, M. A. Hickner, P. A. Kohl, A. R. Kucernak, W. E. Mustain, K. Nijmeijer, K. Scott, T. Xu and L. Zhuang, Energy Environ. Sci., 2014, 7, 3135–3191 RSC
- B. Mayerhöfer, D. McLaughlin, T. Böhm, M. Hegelheimer, D. Seeberger and S. Thiele, ACS Appl. Energy Mater., 2020, 3, 9635–9644 CrossRef
- Q. Xu, L. Zhang, J. Zhang, J. Wang, Y. Hu, H. Jiang and C. Li, EnergyChem, 2022, 4, 100087 CrossRef CAS
- J. Xie, F. Xu, D. L. Wood, K. L. More, T. A. Zawodzinski and W. H. Smith, Electrochim. Acta, 2010, 55, 7404–7412 CrossRef CAS
- C. Li, K. Yu, A. Bird, F. Guo, J. Ilavsky, Y. Liu, D. A. Cullen, A. Kusoglu, A. Z. Weber, P. J. Ferreira and J. Xie, Energy Environ. Sci., 2023, 16, 2977–2990 RSC
- M. Bühler, F. Hegge, P. Holzapfel, M. Bierling, M. Suermann, S. Vierrath and S. Thiele, J. Mater. Chem. A, 2019, 7, 26984–26995 RSC
- E. López-Fernández, C. Gómez-Sacedón, J. Gil-Rostra, J. P. Espinós, A. R. González-Elipe, F. Yubero and A. de Lucas-Consuegra, Chem. Eng. J., 2022, 433, 133774 CrossRef
- S. A. Lee, T. H. Lee, C. Kim, M. G. Lee, M.-J. Choi, H. Park, S. Choi, J. Oh and H. W. Jang, ACS Catal., 2018, 8, 7261–7269 CrossRef CAS
- Y. Kim, S. E. Jun, G. Lee, S. Nam, H. W. Jang, S. H. Park and K. C. Kwon, Materials, 2023, 16, 3044 CrossRef CAS
- S. A. Lee, T. H. Lee, C. Kim, M.-J. Choi, H. Park, S. Choi, J. Lee, J. Oh, S. Y. Kim and H. W. Jang, ACS Catal., 2020, 10, 420–429 CrossRef CAS
- S. A. Lee, J. W. Yang, S. Choi and H. W. Jang, Exploration, 2021, 1, 20210012 CrossRef
- N. K. Oh, J. Seo, S. Lee, H.-J. Kim, U. Kim, J. Lee, Y.-K. Han and H. Park, Nat. Commun., 2021, 12, 4606 CrossRef CAS PubMed
- D. H. Kweon, M. S. Okyay, S.-J. Kim, J.-P. Jeon, H.-J. Noh, N. Park, J. Mahmood and J.-B. Baek, Nat. Commun., 2020, 11, 1278 CrossRef CAS
- S. H. Ahn, B.-S. Lee, I. Choi, S. J. Yoo, H.-J. Kim, E. Cho, D. Henkensmeier, S. W. Nam, S.-K. Kim and J. H. Jang, Appl. Catal., B, 2014, 154–155, 197–205 CrossRef CAS
- J. Lee, H. Jung, Y. S. Park, S. Woo, J. Yang, M. J. Jang, J. Jeong, N. Kwon, B. Lim, J. W. Han and S. M. Choi, Small, 2021, 17, e2100639 CrossRef PubMed
- J. Lee, H. Jung, Y. S. Park, N. Kwon, S. Woo, N. C. S. Selvam, G. S. Han, H. S. Jung, P. J. Yoo, S. M. Choi, J. W. Han and B. Lim, Appl. Catal., B, 2021, 294, 120246 CrossRef CAS
- J. E. Park, S. Y. Kang, S.-H. Oh, J. K. Kim, M. S. Lim, C.-Y. Ahn, Y.-H. Cho and Y.-E. Sung, Electrochim. Acta, 2019, 295, 99–106 CrossRef CAS
- B. H. Lim, E. H. Majlan, A. Tajuddin, T. Husaini, W. R. Wan Daud, N. A. Mohd Radzuan and M. A. Haque, Chin. J. Chem. Eng., 2021, 33, 1–16 CrossRef CAS
- Y. Shi, Z. Lu, L. Guo and C. Yan, Int. J. Hydrogen Energy, 2017, 42, 26183–26191 CrossRef CAS
- S. Koch, L. Metzler, S. K. Kilian, P. A. Heizmann, F. Lombeck, M. Breitwieser and S. Vierrath, Adv. Sustainable Syst., 2022, 7, 2200332 CrossRef
- C. Klose, T. Saatkamp, A. Münchinger, L. Bohn, G. Titvinidze, M. Breitwieser, K. D. Kreuer and S. Vierrath, Adv. Energy Mater., 2020, 10, 1903995 CrossRef CAS
- M. S. Saha, D. K. Paul, B. A. Peppley and K. Karan, Electrochem. Commun., 2010, 12, 410–413 CrossRef CAS
- G. Wei, Y. Wang, C. Huang, Q. Gao, Z. Wang and L. Xu, Int. J. Hydrogen Energy, 2010, 35, 3951–3957 CrossRef CAS
- J. E. Park, S. Kim, O.-H. Kim, C.-Y. Ahn, M.-J. Kim, S. Y. Kang, T. I. Jeon, J.-G. Shim, D. W. Lee, J. H. Lee, Y.-H. Cho and Y.-E. Sung, Nano Energy, 2019, 58, 158–166 CrossRef CAS
- J. H. Montoya, L. C. Seitz, P. Chakthranont, A. Vojvodic, T. F. Jaramillo and J. K. Norskov, Nat. Mater., 2016, 16, 70–81 CrossRef PubMed
- M. Bernt, A. Siebel and H. A. Gasteiger, J. Electrochem. Soc., 2018, 165, F305–F314 CrossRef CAS
- M. Yazdanpour, A. Esmaeilifar and S. Rowshanzamir, Int. J. Hydrogen Energy, 2012, 37, 11290–11298 CrossRef CAS
- H. Park, J. W. Bae, T. H. Lee, I. J. Park, C. Kim, M. G. Lee, S. A. Lee, J. W. Yang, M.-J. Choi, S. H. Hong, S. Y. Kim, S. H. Ahn, J. Y. Kim, H. S. Kim and H. W. Jang, Small, 2022, 18, 2105611 CrossRef CAS
- H. Kim, J. Kim, W. Guo, G. H. Han, S. Hong, J. Park, H. W. Jang, S. Y. Kim and S. H. Ahn, Int. J. Hydrogen Energy, 2022, 47, 2093–2102 CrossRef CAS
- M. G. Lee, J. W. Yang, H. R. Kwon and H. W. Jang, CrystEngComm, 2022, 24, 5838–5864 RSC
- M. K. Lee, M. Shokouhimehr, S. Y. Kim and H. W. Jang, Adv. Energy Mater., 2022, 12, 2003990 CrossRef CAS
- J. Yang, W. Li, D. Wang and Y. Li, Adv. Mater., 2020, 32, 2003300 CrossRef CAS PubMed
- F. Zhang, Y. Zhu, Q. Lin, L. Zhang, X. Zhang and H. Wang, Energy Environ. Sci., 2021, 14, 2954–3009 RSC
- X. Mu, X. Gu, S. Dai, J. Chen, Y. Cui, Q. Chen, M. Yu, C. Chen, S. Liu and S. Mu, Energy Environ. Sci., 2022, 15, 4048–4057 RSC
- P. Zhai, M. Xia, Y. Wu, G. Zhang, J. Gao, B. Zhang, S. Cao, Y. Zhang, Z. Li, Z. Fan, C. Wang, X. Zhang, J. T. Miller, L. Sun and J. Hou, Nat. Commun., 2021, 12, 4587 CrossRef CAS PubMed
- S. Liang, C. Hao and Y. Shi, ChemCatChem, 2015, 7, 2559–2567 CrossRef CAS
- L. Zhang, K. Doyle-Davis and X. Sun, Energy Environ. Sci., 2019, 12, 492–517 RSC
- K. Jiang, M. Luo, Z. Liu, M. Peng, D. Chen, Y.-R. Lu, T.-S. Chan, F. M. F. de Groot and Y. Tan, Nat. Commun., 2021, 12, 1687 CrossRef CAS PubMed
- T. Ma, H. Cao, S. Li, S. Cao, Z. Zhao, Z. Wu, R. Yan, C. Yang, Y. Wang, P. A. van Aken, L. Qiu, Y.-G. Wang and C. Cheng, Adv. Mater., 2022, 34, 2206368 CrossRef CAS PubMed
- S. Ye, F. Luo, Q. Zhang, P. Zhang, T. Xu, Q. Wang, D. He, L. Guo, Y. Zhang, C. He, X. Ouyang, M. Gu, J. Liu and X. Sun, Energy Environ. Sci., 2019, 12, 1000–1007 RSC
- K. L. Zhou, C. Wang, Z. Wang, C. B. Han, Q. Zhang, X. Ke, J. Liu and H. Wang, Energy Environ. Sci., 2020, 13, 3082–3092 RSC
- Y. Sun, Z. Xue, Q. Liu, Y. Jia, Y. Li, K. Liu, Y. Lin, M. Liu, G. Li and C.-Y. Su, Nat. Commun., 2021, 12, 1369 CrossRef CAS PubMed
- Y. Liu, X. Liu, A. R. Jadhav, T. Yang, Y. Hwang, H. Wang, L. Wang, Y. Luo, A. Kumar, J. Lee, H. T. D. Bui, M. Gyu Kim and H. Lee, Angew. Chem., Int. Ed., 2022, 61, e202114160 CrossRef CAS PubMed
- T. Luo, J. Huang, Y. Hu, C. Yuan, J. Chen, L. Cao, K. Kajiyoshi, Y. Liu, Y. Zhao, Z. Li and Y. Feng, Adv. Funct. Mater., 2023, 33, 2213058 CrossRef CAS
- P. Li, G. Zhao, P. Cui, N. Cheng, M. Lao, X. Xu, S. X. Dou and W. Sun, Nano Energy, 2021, 83, 105850 CrossRef CAS
- Q. Wang, Z. L. Zhao, S. Dong, D. He, M. J. Lawrence, S. Han, C. Cai, S. Xiang, P. Rodriguez, B. Xiang, Z. Wang, Y. Liang and M. Gu, Nano Energy, 2018, 53, 458–467 CrossRef CAS
- Y. Xue, B. Huang, Y. Yi, Y. Guo, Z. Zuo, Y. Li, Z. Jia, H. Liu and Y. Li, Nat. Commun., 2018, 9, 1460 CrossRef
- S. Yang, Z. Si, G. Li, P. Zhan, C. Liu, L. Lu, B. Han, H. Xie and P. Qin, Small, 2023, 19, 2207651 CrossRef CAS
- Y. Zhao, P. V. Kumar, X. Tan, X. Lu, X. Zhu, J. Jiang, J. Pan, S. Xi, H. Y. Yang, Z. Ma, T. Wan, D. Chu, W. Jiang, S. C. Smith, R. Amal, Z. Han and X. Lu, Nat. Commun., 2022, 13, 2430 CrossRef CAS
- X. Liu, Y. Deng, L. Zheng, M. R. Kesama, C. Tang and Y. Zhu, ACS Catal., 2022, 12, 5517–5526 CrossRef CAS
- Y. Pan, S. Liu, K. Sun, X. Chen, B. Wang, K. Wu, X. Cao, W.-C. Cheong, R. Shen, A. Han, Z. Chen, L. Zheng, J. Luo, Y. Lin, Y. Liu, D. Wang, Q. Peng, Q. Zhang, C. Chen and Y. Li, Angew. Chem., Int. Ed., 2018, 57, 8614–8618 CrossRef CAS PubMed
- C. Hu, E. Song, M. Wang, W. Chen, F. Huang, Z. Feng, J. Liu and J. Wang, Adv. Sci., 2021, 8, 2001881 CrossRef CAS
- L. Wang, X. Liu, L. Cao, W. Zhang, T. Chen, Y. Lin, H. Wang, Y. Wang and T. Yao, J. Phys. Chem. Lett., 2020, 11, 6691–6696 CrossRef CAS
- A. Han, X. Zhou, X. Wang, S. Liu, Q. Xiong, Q. Zhang, L. Gu, Z. Zhuang, W. Zhang, F. Li, D. Wang, L.-J. Li and Y. Li, Nat. Commun., 2021, 12, 709 CrossRef CAS PubMed
- K. Wang, L. Jiang, T. Xin, Y. Li, X. Wu and G. Zhang, Chem. Eng. J., 2022, 429, 132229 CrossRef CAS
- G.-L. Hou, T. Yang, M. Li, J. Vanbuel, O. V. Lushchikova, P. Ferrari, J. M. Bakker and E. Janssens, Angew. Chem., Int. Ed., 2021, 60, 27095–27101 CrossRef CAS PubMed
- X. Zheng, J. Tang, A. Gallo, J. A. Garrido Torres, X. Yu, C. J. Athanitis, E. M. Been, P. Ercius, H. Mao, S. C. Fakra, C. Song, R. C. Davis, J. A. Reimer, J. Vinson, M. Bajdich and Y. Cui, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2101817118 CrossRef CAS PubMed
- Q. Wang, Z. Zhang, C. Cai, M. Wang, Z. L. Zhao, M. Li, X. Huang, S. Han, H. Zhou, Z. Feng, L. Li, J. Li, H. Xu, J. S. Francisco and M. Gu, J. Am. Chem. Soc., 2021, 143, 13605–13615 CrossRef CAS
- Q. Wang, X. Huang, Z. L. Zhao, M. Wang, B. Xiang, J. Li, Z. Feng, H. Xu and M. Gu, J. Am. Chem. Soc., 2020, 142, 7425–7433 CrossRef CAS
- J. Yin, J. Jin, M. Lu, B. Huang, H. Zhang, Y. Peng, P. Xi and C.-H. Yan, J. Am. Chem. Soc., 2020, 142, 18378–18386 CrossRef CAS PubMed
- X. Duan, P. Li, D. Zhou, S. Wang, H. Liu, Z. Wang, X. Zhang, G. Yang, Z. Zhang, G. Tan, Y. Li, L. Xu, W. Liu, Z. Xing, Y. Kuang and X. Sun, Chem. Eng. J., 2022, 446, 136962 CrossRef CAS
- Y. Hu, G. Luo, L. Wang, X. Liu, Y. Qu, Y. Zhou, F. Zhou, Z. Li, Y. Li, T. Yao, C. Xiong, B. Yang, Z. Yu and Y. Wu, Adv. Energy Mater., 2021, 11, 2002816 CrossRef CAS
- J. Zhang, J. Liu, L. Xi, Y. Yu, N. Chen, S. Sun, W. Wang, K. M. Lange and B. Zhang, J. Am. Chem. Soc., 2018, 140, 3876–3879 CrossRef CAS PubMed
- S. Choi, S. A. Lee, J. W. Yang, W. Sohn, J. Kim, W. S. Cheon, J. Park, J. H. Cho, C. W. Lee, S. E. Jun, S. H. Park, J. Moon, S. Y. Kim and H. W. Jang, J. Mater. Chem. A, 2023, 11, 17503–17513 RSC
- C. Chen, M. Sun, F. Zhang, H. Li, M. Sun, P. Fang, T. Song, W. Chen, J. Dong, B. Rosen, P. Chen, B. Huang and Y. Li, Energy Environ. Sci., 2023, 16, 1685–1696 RSC
- X. Wang, L. Sun, W. Zhou, L. Yang, G. Ren, H. Wu and W.-Q. Deng, Cell Rep., 2022, 3, 100804 CAS
- Z. Zhang, C. Feng, X. Li, C. Liu, D. Wang, R. Si, J. Yang, S. Zhou and J. Zeng, Nano Lett., 2021, 21, 4795–4801 CrossRef CAS
- Y. Li, Z.-S. Wu, P. Lu, X. Wang, W. Liu, Z. Liu, J. Ma, W. Ren, Z. Jiang and X. Bao, Adv. Sci., 2020, 7, 1903089 CrossRef CAS PubMed
- S. E. Jun, S. Choi, J. Kim, K. C. Kwon, S. H. Park and H. W. Jang, Chin. J. Catal., 2023, 50, 195–214 CrossRef
- P. Kumar, K. Kannimuthu, A. S. Zeraati, S. Roy, X. Wang, X. Wang, S. Samanta, K. A. Miller, M. Molina, D. Trivedi, J. Abed, M. A. Campos Mata, H. Al-Mahayni, J. Baltrusaitis, G. Shimizu, Y. A. Wu, A. Seifitokaldani, E. H. Sargent, P. M. Ajayan, J. Hu and M. G. Kibria, J. Am. Chem. Soc., 2023, 145, 8052–8063 CrossRef CAS PubMed
- Z. Li, Z. Wang, S. Xi, X. Zhao, T. Sun, J. Li, W. Yu, H. Xu, T. S. Herng, X. Hai, P. Lyu, M. Zhao, S. J. Pennycook, J. Ding, H. Xiao and J. Lu, ACS Nano, 2021, 15, 7105–7113 CrossRef CAS PubMed
- Y. Liu, S. Zhang, C. Jiao, H. Chen, G. Wang, W. Wu, Z. Zhuo and J. Mao, Adv. Sci., 2023, 10, 2206107 CrossRef CAS PubMed
- L. Zhang, R. Si, H. Liu, N. Chen, Q. Wang, K. Adair, Z. Wang, J. Chen, Z. Song, J. Li, M. N. Banis, R. Li, T.-K. Sham, M. Gu, L.-M. Liu, G. A. Botton and X. Sun, Nat. Commun., 2019, 10, 4936 CrossRef PubMed
- M. Luo, J. Cai, J. Zou, Z. Jiang, G. Wang and X. Kang, J. Mater. Chem. A, 2021, 9, 14941–14947 RSC
- R. Li and D. Wang, Adv. Energy Mater., 2022, 12, 2103564 CrossRef CAS
- Y. Yang, Y. Qian, H. Li, Z. Zhang, Y. Mu, D. Do, B. Zhou, J. Dong, W. Yan, Y. Qin, L. Fang, R. Feng, J. Zhou, P. Zhang, J. Dong, G. Yu, Y. Liu, X. Zhang and X. Fan, Sci. Adv., 2020, 6, eaba6586 CrossRef CAS PubMed
- S. M. George, Chem. Rev., 2010, 110, 111–131 CrossRef CAS PubMed
- B. J. O’Neill, D. H. K. Jackson, J. Lee, C. Canlas, P. C. Stair, C. L. Marshall, J. W. Elam, T. F. Kuech, J. A. Dumesic and G. W. Huber, ACS Catal., 2015, 5, 1804–1825 CrossRef
- J. Fonseca and J. Lu, ACS Catal., 2021, 11, 7018–7059 CrossRef CAS
- L. Zhang, Q. Wang, L. Li, M. N. Banis, J. Li, K. Adair, Y. Sun, R. Li, Z.-J. Zhao, M. Gu and X. Sun, Nano Energy, 2022, 93, 106813 CrossRef CAS
- Z. Zhang, C. Feng, C. Liu, M. Zuo, L. Qin, X. Yan, Y. Xing, H. Li, R. Si, S. Zhou and J. Zeng, Nat. Commun., 2020, 11, 1215 CrossRef CAS PubMed
- T. Gan, Q. He, H. Zhang, H. Xiao, Y. Liu, Y. Zhang, X. He and H. Ji, Chem. Eng. J., 2020, 389, 124490 CrossRef CAS
- P. Rao, D. Wu, Y.-Y. Qin, J. Luo, J. Li, C. Jia, P. Deng, W. Huang, Y. Su, Y. Shen and X. Tian, J. Mater. Chem. A, 2022, 10, 6531–6537 RSC
- S. Kang, Y. K. Jeong, S. Mhin, J. H. Ryu, G. Ali, K. Lee, M. Akbar, K. Y. Chung, H. Han and K. M. Kim, ACS Nano, 2021, 15, 4416–4428 CrossRef CAS PubMed
- N. Fu, X. Liang, X. Wang, T. Gan, C. Ye, Z. Li, J.-C. Liu and Y. Li, J. Am. Chem. Soc., 2023, 145, 9540–9547 CrossRef CAS PubMed
- Y. Chen, R. Gao, S. Ji, H. Li, K. Tang, P. Jiang, H. Hu, Z. Zhang, H. Hao, Q. Qu, X. Liang, W. Chen, J. Dong, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 3212–3221 CrossRef CAS PubMed
- N. Zhang, X. Zhang, L. Tao, P. Jiang, C. Ye, R. Lin, Z. Huang, A. Li, D. Pang, H. Yan, Y. Wang, P. Xu, S. An, Q. Zhang, L. Liu, S. Du, X. Han, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 6170–6176 CrossRef CAS PubMed
- H. Wang, X. Wang, J. Pan, L. Zhang, M. Zhao, J. Xu, B. Liu, W. Shi, S. Song and H. Zhang, Angew. Chem., 2021, 133, 23338–23342 CrossRef
- Z.-Y. Wu, F.-Y. Chen, B. Li, S.-W. Yu, Y. Z. Finfrock, D. M. Meira, Q.-Q. Yan, P. Zhu, M.-X. Chen, T.-W. Song, Z. Yin, H.-W. Liang, S. Zhang, G. Wang and H. Wang, Nat. Mater., 2023, 22, 100–108 CrossRef CAS PubMed
- K. Wang, J. Cao, X. Yang, X. Sang, S. Yao, R. Xiang, B. Yang, Z. Li, T. O'Carroll, Q. Zhang, L. Lei, G. Wu and Y. Hou, Adv. Funct. Mater., 2023, 33, 2212321 CrossRef CAS
- C. Xia, Y. Qiu, Y. Xia, P. Zhu, G. King, X. Zhang, Z. Wu, J. Y. Kim, D. A. Cullen, D. Zheng, P. Li, M. Shakouri, E. Heredia, P. Cui, H. N. Alshareef, Y. Hu and H. Wang, Nat. Chem., 2021, 13, 887–894 CrossRef CAS PubMed
- H. Jin, S. Sultan, M. Ha, J. N. Tiwari, M. G. Kim and K. S. Kim, Adv. Funct. Mater., 2020, 30, 2000531 CrossRef CAS
- L. Zhang, Y. Jia, G. Gao, X. Yan, N. Chen, J. Chen, M. T. Soo, B. Wood, D. Yang, A. Du and X. Yao, Chem, 2018, 4, 285–297 CAS
- S. Khandavalli, J. H. Park, N. N. Kariuki, D. J. Myers, J. J. Stickel, K. Hurst, K. C. Neyerlin, M. Ulsh and S. A. Mauger, ACS Appl. Mater. Interfaces, 2018, 10, 43610–43622 CrossRef CAS PubMed
- S. Shukla, S. Bhattacharjee, A. Weber and M. Secanell, J. Electrochem. Soc., 2017, 164, F600 CrossRef CAS
- G. A. Lindquist, Q. Xu, S. Z. Oener and S. W. Boettcher, Joule, 2020, 4, 2549–2561 CrossRef CAS
- S. A. Lee, J. Kim, K. C. Kwon, S. H. Park and H. W. Jang, Carbon Neutralization, 2022, 1, 26–48 CrossRef
- J. Parrondo, C. G. Arges, M. Niedzwiecki, E. B. Anderson, K. E. Ayers and V. Ramani, RSC Adv., 2014, 4, 9875 RSC
- D. Chen and M. A. Hickner, ACS Appl. Mater. Interfaces, 2012, 4, 5775–5781 CrossRef CAS PubMed
- N. Du, C. Roy, R. Peach, M. Turnbull, S. Thiele and C. Bock, Chem. Rev., 2022, 122, 11830–11895 CrossRef CAS PubMed
- S. Siracusano, N. Van Dijk, R. Backhouse, L. Merlo, V. Baglio and A. S. Aricò, Renewable Energy, 2018, 123, 52–57 CrossRef CAS
- Y. Chen, S. Ji, C. Chen, Q. Peng, D. Wang and Y. Li, Joule, 2018, 2, 1242–1264 CrossRef CAS
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |