
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTowards clustered carbonyl cations [M3(CO)14]2+ (M = Ru, Os): the need for innocent deelectronation†
Malte
Sellin
 a,
Christian
Friedmann
a,
Maximilian
Mayländer
b,
Sabine
Richert
b and
Ingo
Krossing
*a
a,
Christian
Friedmann
a,
Maximilian
Mayländer
b,
Sabine
Richert
b and
Ingo
Krossing
*a
aInstitut für Anorganische und Analytische Chemie, Albert-Ludwigs-Universität Freiburg, Albertstr. 21, 79104 Freiburg, Germany. E-mail: krossing@uni-freiburg.de
bInstitut für Physikalische Chemie, Albert-Ludwigs-Universität Freiburg, Albertstr. 21, 79104 Freiburg, Germany
First published on 7th July 2022
Abstract
To access the hitherto almost unknown class of clustered transition metal carbonyl cations, the trimetal dodecacarbonyls M3(CO)12 (M = Ru, Os) were reacted with the oxidant Ag+[WCA]−, but yielded the silver complexes [Ag{M3(CO)12}2]+[WCA]− (WCA = [Al(ORF)4]−, [F{Al(ORF)3}2]−; RF = –OC(CF3)3). Addition of further diiodine I2 to increase the redox potential led for M = Ru non-specifically to divalent mixed iodo-RuII-carbonyl cations. With [NO]+, even the N–O bond was cleaved and led to the butterfly carbonyl complex cation [Ru4N(CO)13]+ in low yield. Obviously, ionization of M3(CO)12 with retention of its pseudo-binary composition including only M and CO is difficult and the inorganic reagents did react non-innocently. Yet, the radical cation of the commercially available perhalogenated anthracene derivative 9,10-dichlorooctafluoroanthracene (anthraceneHal) is a straightforward accessible innocent deelectronator with a half-wave potential E1/2 of 1.42 V vs. Fc0/+. It deelectronates M3(CO)12 under a CO atmosphere and leads to the structurally characterized cluster salts [M3(CO)14]2+([WCA]−)2 including a linear M3 chain. The structural characterization as well as vibrational and NMR spectroscopies indicate the presence of three electronically independent sets of carbonyl ligands, which almost mimic M(CO)5, free CO and even [M(CO)6]2+ in one and the same cation.
Introduction
The carbon monoxide ligand is omnipresent in organometallic chemistry and of high interest for both fundamental chemistry as well as industrial applications.1,2 The combination of the σ-bonding and π-backbonding interactions with transition metals enables the stabilization of transition metal carbonyls (TMCs) in formal oxidation states between −IV and +III.3 There is no other neutral ligand class, with the exception of the closely related alkylisocyanides (−II to +V),4 which allows the stabilization of homoleptic transition metal complexes over such a huge range of eight formal oxidation states.Closer analysis of the bonding within the transition metal carbonyls has led to the classification of two different kinds of carbonyl complexes by S. H. Strauss et al. in 1994:5 the first class is addressed as being “classical”, in which the π-backdonation from the d-electrons of the metal centre towards the π*-orbital of the ligand is the primary interaction, leading to a weakening of the carbon–oxygen bond and a red shift of the ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/char_e0e1.gif) CO stretching vibrations compared to free carbon monoxide. The second class is addressed as “nonclassical” carbonyl complexes, where the σ-donation of the carbonyl ligand towards the metal centre is the dominant interaction, apparently leading to a strengthening of the carbon–oxygen bond and a blueshift of the CO stretch relative to free carbon monoxide.6 Alternatively, Aubke and Willner addressed them as predominantly σ-bonded carbonyl cations or σ-carbonyls.7
CO stretching vibrations compared to free carbon monoxide. The second class is addressed as “nonclassical” carbonyl complexes, where the σ-donation of the carbonyl ligand towards the metal centre is the dominant interaction, apparently leading to a strengthening of the carbon–oxygen bond and a blueshift of the CO stretch relative to free carbon monoxide.6 Alternatively, Aubke and Willner addressed them as predominantly σ-bonded carbonyl cations or σ-carbonyls.7
The prediction of the nature of the carbonyl ligand within homoleptic transition metal carbonyl cations (TMCCs) is not intuitive: while [Ag(CO)1–2]+ complexes are nonclassical, other monocationic homoleptic TMCCs like [Mn(CO)6]+ are classical.8 The prediction is even less straightforward for heteroleptic TMCCs: an illustrative example is [(Cp*)2Fe–CO]2+, which bears iron in the formal oxidation state +IV, but is a classical carbonyl complex.9,10 Hence, the investigation of novel homoleptic TMCCs remains an interesting field with potential for new discoveries, despite the fact that meanwhile the monometallic TMCCs of many transitions metals were already isolated in condensed phases.2,7,11
Still, the weak interaction of carbon monoxide with metal cations requires the strict absence of other nucleophiles, which implies the use of both weakly coordinating anions (WCAs) and weakly coordinating solvents/media. Hence, super acids are a common medium to stabilize transition metal complexes in unusually high oxidation states in general.12 Likewise, reductive carbonylation in super acidic mixtures like fluoroantimonic acid (HF/SbF5) or “magic acid” (HSO3F/SbF5) was utilized by Willner and Aubke to yield several diamagnetic 14, 16 & 18 valence electron (VE) TMCCs containing mono-to trivalent metals.7 Yet, this system is limited by the coordination ability of the [SbF6]− anion, which is incompatible with the strong fluorophilicity of early transition metals13,14 as well as the strong acidity, which obstructs Brønsted basic TMCs as starting materials.15 While perfluorinated alkoxyaluminate based WCAs have a higher affinity to protons than hexafluoroantimonate,16 transition metal complexes are much more weakly coordinated with this class of anions due to the poor polarizability of the C–F bonds and the delocalization of the negative charge over a much larger volume.17 The combination of this type of WCA, paired with inorganic oxidative cations in fluorinated benzene derivatives by our group has enabled the access to paramagnetic 17 VE TMCCs like [M(CO)6]+˙ (M = Cr, Mo, W),18,19, [Fe(CO)5]+˙20 and [Ni(CO)4]+˙21 from their respective neutral TMCs as well as the isolation of the first metal heptacarbonyls [M(CO)7]+ (M = Nb, Ta) (Fig. 1).22
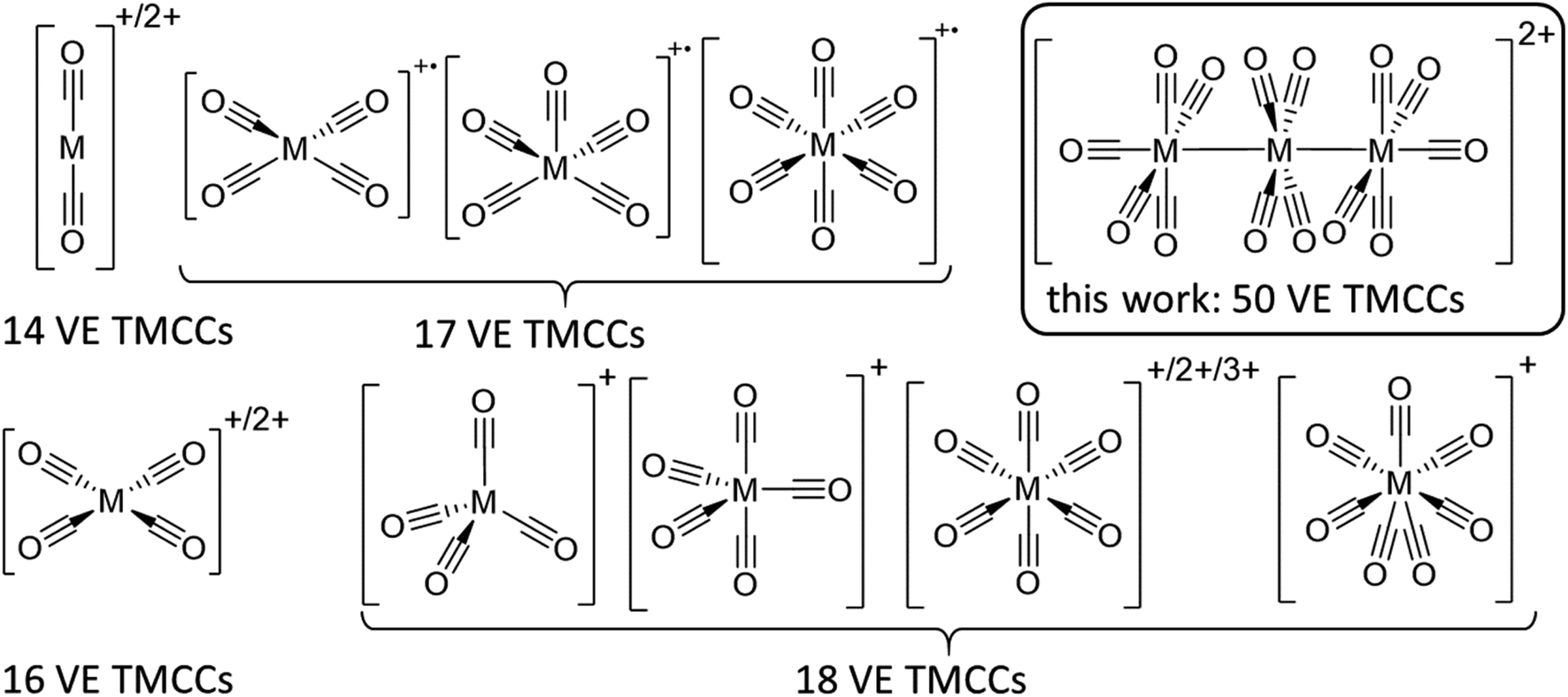 | ||
| Fig. 1 Known structurally characterized homoleptic TMCCs without strong contacts to counterions. 14 VE: [M(CO)2]+/2+ Ag+, Hg2+; 16 VE: [M(CO)4]+/2+ Rh+, Pd2+, Pt2+; 17 VE: [M(CO)4]+˙ Ni+; [M(CO)5]+˙ Fe+ [M(CO)6]+˙ Cr+, Mo+, W+; 18 VE: [M(CO)4]+ Cu+; [M(CO)5]+ Co+; [M(CO)6]+/2+/3 + Mn+, Tc+, Re+, Fe2+, Ru2+, Os2+, Ir3+; [M(CO)7]+ Nb+, Ta+; 50 VE: [M3(CO)14]2+ {Ru3}2+, {Os3}2+. | ||
By contrast, clustered TMCCs are still very scarce and in their infancy: apart from mass spectrometric investigations,23 there are only two reports of homodinuclear transition metal carbonyl cations available in the condensed phase: [Hg2(CO)2]2+ and [Pt2(CO)6]2+. Yet, the instability of these complexes has not allowed full characterisation including single crystal X-ray diffraction (scXRD).24 Therefore, we turned our attention to the preparation of clustered TMCCs. With the thermodynamic instability of their respective monometallic pentacarbonyl complexes M(CO)5 ref. 25,26 (M = Ru, Os) and their manifold of known cluster chemistry,27 triruthenium and triosmium dodecacarbonyl M3(CO)12 seemed to be good candidates for the deelectronation to the first homotrimetallic TMCCs.
Results and discussion
To convert the trimetal dodecacarbonyls to their respective TMCCs, an oxidation agent is needed which retains the pseudo-binary composition of the complexes. Previously we showed, e.g. for the group 6 hexacarbonyls,19,28 that the typical inorganic oxidants Ag+, Ag+/0.5 X2 and NO+ act in several unwanted non-innocent side reactions (eqn (1b), (2b) and (3b)) instead of the desired deelectronation (eqn (1a), (2a) and (3a)). Note, that we use electronation and deelectronation in their strict sense, i.e. removal/addition of electrons in innocent reactions, hence without non-innocent complications. For reasoning see29,30 and the literature included with.29,31 In this work we even observed the cleavage of the N–O bond in [NO]+ in eqn (3c). Hence, the strongest bond known to condensed phase chemistry.32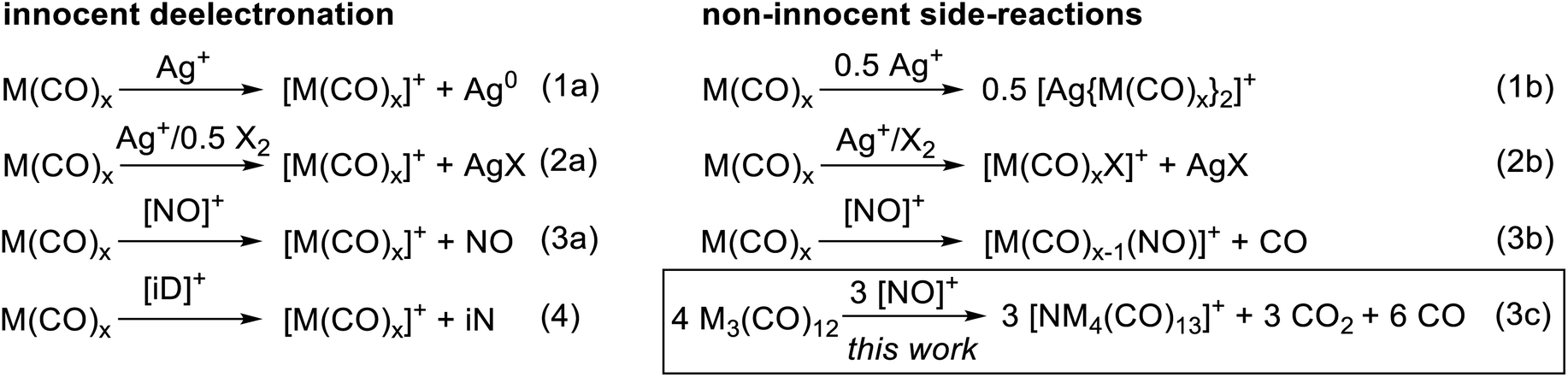
Accordingly, we learned that the use of innocent deelectronators ([iD]+), that do not perform side reactions with Lewis basic transition metal complexes, are superior for an innocent deelectronation towards homoleptic TMCCs as in eqn (4). In the following, we first introduce the reactions of M3(CO)12 with the non-innocent inorganic oxidants before turning to a novel [iD]+ deelectronator salt accessible from a commercial innocent neutral (iN) and describing its structural demands and reactions according to eqn (4) in separate sections.
Syntheses with inorganic oxidant cations
At first, we reacted the commercially available carbonyl complexes Ru3(CO)12 and Os3(CO)12 with the inorganic oxidant cations [ox]+ ([NO]+, Ag+, Ag+/0.5 I2) partnered with the weakly coordinating perfluorinated alkoxyaluminate anion [Al(ORF)4]− (RF = OC(CF3)3). All reactions were performed in 1,2,3,4-tetrafluorobenzene (TFB) as a polar, but very weakly coordinating and non-basic solvent. Fig. 2 includes an overview of the outcome of the reactions with triruthenium dodecacarbonyl (1a).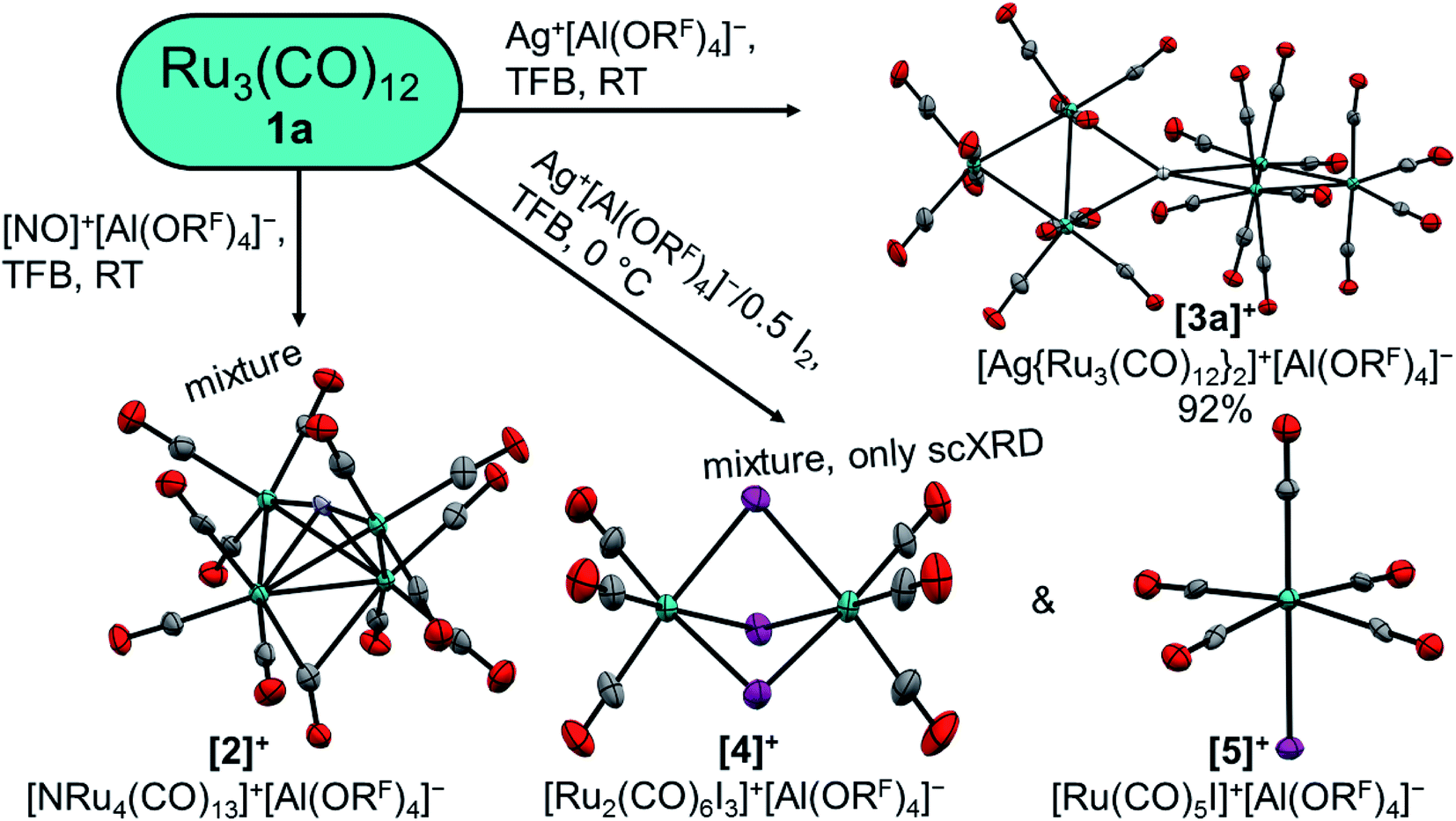 | ||
| Fig. 2 Reactions of 1a with typical inorganic oxidant cations. Molecular structures of the cations are drawn with thermal ellipsoids set at 50% probability; WCAs and co-crystallized solvent molecules were omitted for clarity. Colour code: carbon – grey, nitrogen – light blue, oxygen – red, ruthenium – turquoise, silver – white, iodine – purple. | ||
 | (5) |
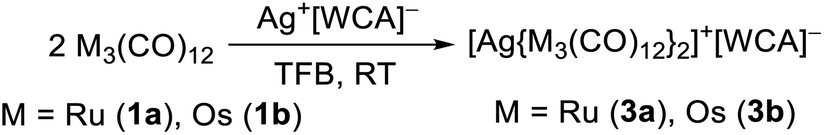
While the structure of [3a]+[Al(ORF)4]−·C6F4H2 is well resolved, the structure of the osmium analogue has severe disorders within the structure of the cluster cation. Therefore, the reaction was performed analogously with Ag+[F{Al(ORF)3}2]−, which yielded the salt [3b]+[F{Al(ORF)3}2]−˙C6F4H2, in which the cluster cation is well ordered. In both structures, the silver cation is coordinated by the M–M bonds of two trimetal dodecarbarbonyls in a D2d coordination geometry (τ = 73°). In addition to the strong interactions of the metal atoms with the silver cation, the proximal carbonyl ligands coordinate weakly to the silver(I) cation, which results in reduced M–C–O angles of ca. 171° and weakly bonding Ag–C distances between 2.69 and 2.77 Å (cf. sum of vdW radii Ag + C = 3.42 Å).37 All aspects of the structures are well in line with the DFT calculated optimized structures (B3LYP(D3BJ)/def2-TZVPP).
The formation of adducts of TMCs with silver(I) cations has already been observed by our group28,38 with recent additions from the Dias group.39,40 These complexes can be seen as metal-only Lewis pairs (MOLPs).41 While zero valent and pentacoordinated group 8 complexes are known for having Lewis-basic behaviour and silver cations are known to be Lewis acidic, their heterodinuclear complexes are scarce.41,42 In contrast to the previously known cationic group 8 → group 11 MOLPs, the primary interaction in [3a]+ and [3b]+ is the donation of electron density from a metal–metal σ-bond instead of a single metal donor orbital. This donation leads to an elongation of the tangential metal–metal bond by 0.13 Å,43 while the other metal does not experience significant shifts.
In addition, we investigated the bonding situation between the {M3(CO)12}2 (M = Ru, Os) and the Ag+ fragments with the EDA-NOCV method.44 The assignments of the orbital interactions are closely related to those observed in [Ag{M(CO)6}2]+ (M = Cr, Mo, W).28 Therefore they are omitted here and shown in the ESI.† The interaction energies of the silver cation with {M3(CO)12}2 are relatively strong (−118.5/−123.7 kcal mol−1 Ru/Os) and even exceed the values of Ag+ with the group 6 hexacarbonyls {M(CO)6}2 −79.8/−86.1/−86.7 (Cr/Mo/W) kcal mol−1 28 and iron pentacarbonyl {Fe(CO)5}2 (−97.6 kcal mol−1).39 The deformation density plot of the [(OC)12M3]→Ag+←[M3(CO)12] σ-donation explains the elongation of the tangential metal–metal bond and the bending of the carbonyl ligands proximal to the silver(I) cation very well (Fig. 3).
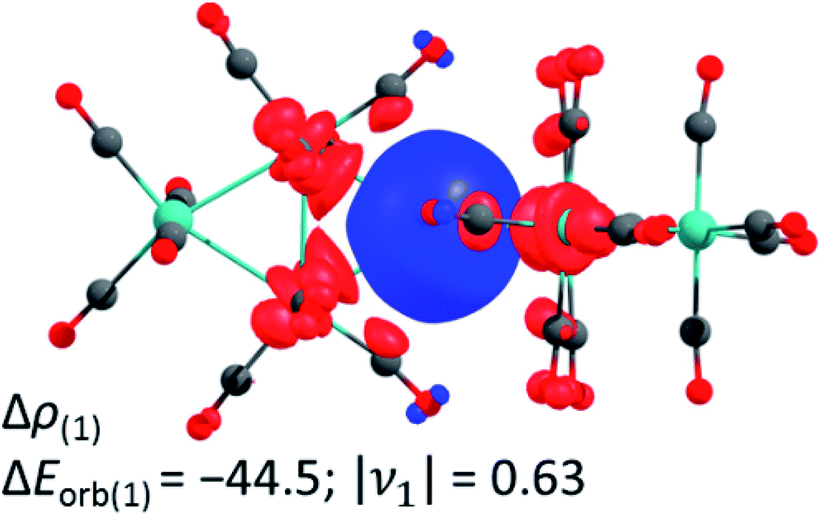 | ||
| Fig. 3 The shape of the deformation density of Δρ(1), which is associated with ΔEorb(1) in kcal mol−1 calculated at the BP86(D3BJ)/TZ2P level; relativistic effects were considered with ZORA. It presents mainly the donation of electron density from the M–M σ-bond to the empty 5s orbital of Ag+. The eigenvalue ν1 gives the value of the charge migration. The direction of the charge flow is from red to blue. Colour code: carbon – grey, oxygen – red, ruthenium – turquoise. | ||
As a potential candidate, the simplest perfluorinated arene, perfluorobenzene (benzeneF), needs extreme reagents like iridium hexafluoride for the deelectronation to its radical cation.48 Additionally, the benzeneF radical cation is highly Lewis acidic and causes the slow decomposition of the hexafluorometalate WCAs [MF6]− (M = Ir, Sb) at room temperature. Fortunately, the ionization energies of the fluorinated arenes decrease with the number of linearly fused rings. Seppelt et al. investigated the deelectronation of larger fluorinated arenes like perfluoronaphthalene (naphthaleneF) and 9,10-dichlorooctafluoroantracene (anthraceneHal).49 While the first was deelectronated and crystallographically characterized as [naphthaleneF]+˙[As2F11]− naphthaleneF, the latter, under reaction conditions, undergoes a halogen exchange to perfluoroanthracene (anthraceneF), which was deelectronated twice to give [anthraceneF]2+ ([Sb2F11]−)2. By contrast, the larger fluorinated arenes like perfluorotetracene (tetraceneF) and perfluoropentacene (pentaceneF) have surprisingly low half-wave potentials of just 1.02 V and 0.79 V vs. Fc0/+ in 1,2-dichlorobenzene.50 Yet, tetraceneF and pentaceneF can be only accessed by multi-step syntheses, while anthraceneHal is commercially available. Cyclovoltammetry of anthraceneHal (6) shows an electrochemically reversible half-wave potential E1/2 at 1.42 V vs. Fc0/+ in both oDFB and TFB, which is 130 mV higher than that of the previously used “phenazineF” (Fig. 4B).10 When 6 is reacted with [NO]+[F{Al(ORF)3}2]− in TFB (but not in oDFB), a dark green solution and evolution of gas is observed, indicating the generation of [6]+˙ and nitrogen monoxide (eqn (6)).
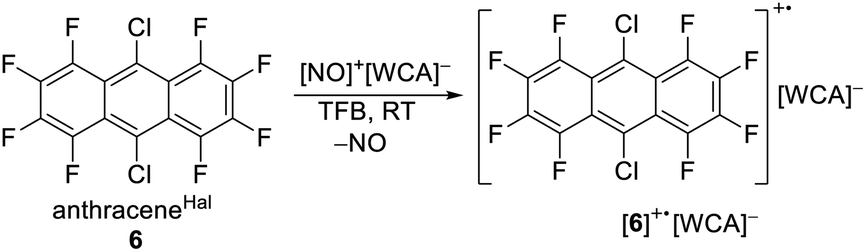 | (6) |
Dark green crystalline blocks suitable for scXRD could be obtained by layering the solution with n-pentane in 80% yield. The product was further analysed with pXRD, as well as IR, Raman, NMR and EPR spectroscopy (Fig. 4). The comparison of the solid state structure of [6]+˙[F{Al(ORF)3}2]− with its innocent neutral (6)51 shows a shortening of all the carbon halogen bonds and an alternation of shortening and elongation of the carbon carbon bond lengths of the aromatic core, this fits the DFT calculated (B3LYP(D3BJ)/def2-TZVPP) SOMO of [6]+˙ (see ESI Chapter 3†). The EPR spectrum shows a delocalization of the spin density across all the substituents (Fig. 4E). The hyperfine coupling constants of the fluorine atoms on the α-position are bigger than those for the fluorine atoms in the β-position, which is in line with the smaller spin density at the β-fluorine atoms (Fig. 4D). The spin in [6]+˙ is therefore similarly delocalised as in the [iD]+ [“phenazineF”]+˙. Favourably, 6 has a higher availability, a smaller molecular weight and a higher deelectronation potential as phenazineF.
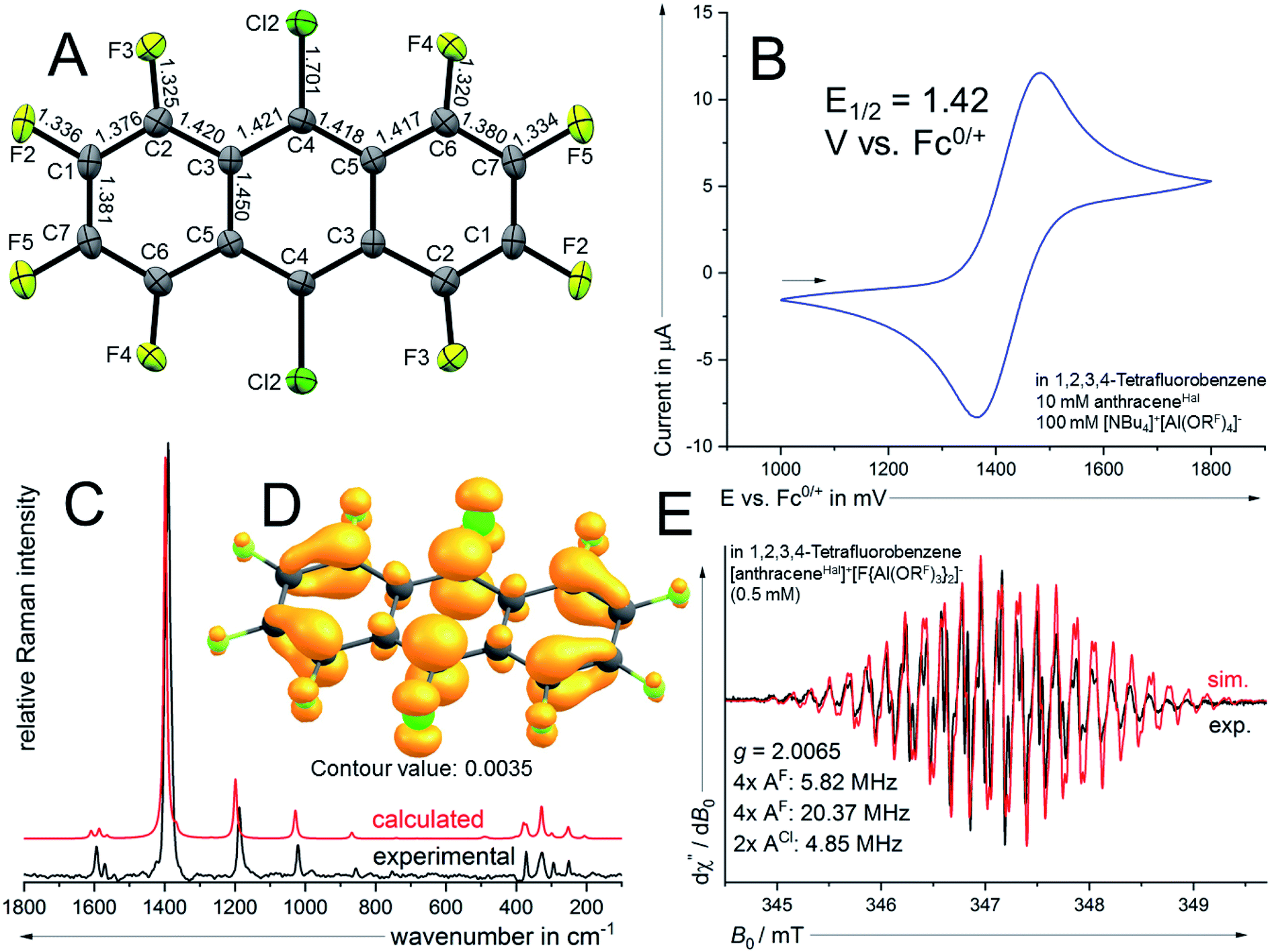 | ||
| Fig. 4 (A) Molecular structure of the radical cation in [6]+˙[F{Al(ORF)3}2]− (ellipsoids set at 50% probability, distances in Å and standard deviations of the C–C bonds in the range of 0.003 Å). Colour code: carbon – grey, fluorine – light green, chlorine – dark green. (B) Cyclovoltammetry (2nd cycle; 100 mV s−1) of 6. (C) calculated (B3LYP(D3BJ)/def2-TZVPP) Raman spectrum of [6]+˙ and experimental Raman spectrum of crystalline [6]+˙[F{Al(ORF)3}2]−. (D) Spin density plot (B3LYP(D3BJ)/def2-TZVPP) of [6a]+˙. (E) EPR spectrum of a 0.5 mM solution of [6]+˙[F{Al(ORF)3}2]− in TFB and simulated EPR spectrum using the hyperfine coupling constants indicated in the figure. | ||
Synthesis and characterisation of the trimetal tetradecarbonyl dications
To test the novel [iD]+ reagent, we reacted two equivalents of the in situ generated [6]+˙[Al(ORF)4]− with the trimetal dodecacarbonyls 1a and 1b under CO pressure according to eqn (3). The intense green colour vanishes after a few minutes leading to brownish solutions, which were layered with n-pentane yielding clear colourless crystals of [7a]2+([Al(ORF)4]−)2·2C6F4H2 and [7b]2+([Al(ORF)4]−)2·2C6F4H2 suitable for scXRD and in 88% (Ru)/72% (Os) yield.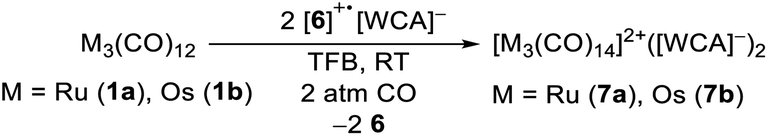 | (3) |
The products were additionally analysed in the bulk by pXRD and by NMR, IR and Raman spectroscopy. The reaction works analogously with [6]+˙[F{Al(ORF)3}2]−, which yields [7a]2+([F{Al(ORF)3}2]−)2·C6F4H2 and [7b]2+([F{Al(ORF)3}2]−)2·C6F4H2.
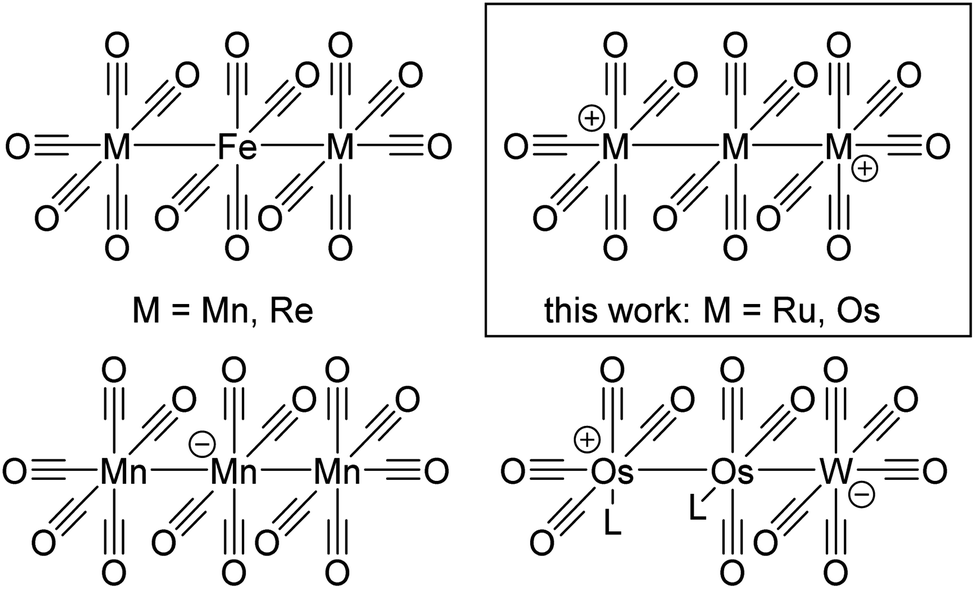 | ||
| Fig. 5 Overview of known and new trimetal tetradecacarbonyls and their relatives. | ||
Trimetal tetradecacarbonyls with 50 valence electrons (VE) and their heteroleptic derivatives can be seen as a combination of two terminal -M(CO)5 fragments (17 VE → pseudohalogen)55 and an inserted -M(CO)4- fragment (16 VE → pseudochalcogen), which are connected by metal metal single bonds. The Lewis-structure shows a positive formal-charge on each terminal metal atom at [7a]2+ and [7b]2+. A quantum theory of atoms in molecules (QTAIM) analysis of [7a]2+ (B3LYP(D3BJ)/def2-TZVPP) is well in line with this Lewis structure and assigns a +0.875 charge on each terminal -Ru(CO)5 fragment and only a charge of +0.25 on the central -Ru(CO)4- fragment. The charge-distribution in [7b]2+ is even more unequal with a +0.17 charge on the central -Os(CO)4- fragment.
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and are isomorphous. The linear homotrimetallic core is coordinated by fourteen carbonyl ligands resulting in a square pyramidal coordination geometry for the terminal metal atoms and a square planar coordination geometry for the middle metal atom always referring to the carbonyl ligands. The torsion angles between the equatorial carbonyl ligands of two neighbouring metals are close to 45° leading to an ecliptic arrangement of the terminal -M(CO)5 fragments in an overall D4h symmetry. Through minor interactions of the complex dication with the WCA and solvent, the ideal D4h geometry is slightly distorted in [M3(CO)14]2+ to the crystallographic Ci symmetry. The co-crystallized solvent molecules have weak interactions with the complex salt leading to the degradation of the crystalline phase upon drying of the crystals in vacuo (see ESI†). It is noticeable, that, in all the four crystal structures of [7a]2+ and [7b]2+, the metal carbon bond lengths elongate from M–COcen,eq over M–COout,eq to M–COout,ax, while the opposite trend is observed for the carbon–oxygen bond lengths. This can be explained by a more pronounced π-backbonding in the central fragment. The span of the bond lengths is remarkable, because the bond lengths of M–C
and are isomorphous. The linear homotrimetallic core is coordinated by fourteen carbonyl ligands resulting in a square pyramidal coordination geometry for the terminal metal atoms and a square planar coordination geometry for the middle metal atom always referring to the carbonyl ligands. The torsion angles between the equatorial carbonyl ligands of two neighbouring metals are close to 45° leading to an ecliptic arrangement of the terminal -M(CO)5 fragments in an overall D4h symmetry. Through minor interactions of the complex dication with the WCA and solvent, the ideal D4h geometry is slightly distorted in [M3(CO)14]2+ to the crystallographic Ci symmetry. The co-crystallized solvent molecules have weak interactions with the complex salt leading to the degradation of the crystalline phase upon drying of the crystals in vacuo (see ESI†). It is noticeable, that, in all the four crystal structures of [7a]2+ and [7b]2+, the metal carbon bond lengths elongate from M–COcen,eq over M–COout,eq to M–COout,ax, while the opposite trend is observed for the carbon–oxygen bond lengths. This can be explained by a more pronounced π-backbonding in the central fragment. The span of the bond lengths is remarkable, because the bond lengths of M–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Ocen,eq are similar to those observed in the respective neutral metal pentacarbonyls M(CO)5 (M = Ru, Os),56 while the bond lengths of M–COout,ax are close to those observed in the superelectrophilic [M(CO)6]2+ dications.57
(CO) modes spread over 136 cm−1 in [7a]2+([Al(ORF)4]−)2 (Fig. 6) and 158 cm−1 in [7b]2+([Al(ORF)4]−)2. The average carbonyl stretching vibrational frequencies weighed over the degeneracies59 of (CO)av = 2120/2117 (Ru/Os) cm−1 are slightly below that of free CO(g) at 2143 cm−1 and lay between that of the respective metal pentacarbonyls M(CO)5 (2038/2032 cm−1) and the known metal hexacarbonyl dications [M(CO)6]2+ (2220/2216 cm−1).25,57 This shows the presence of some π-interactions between the carbonyl ligands and the metal centres, classifying the complexes as still being “classical carbonyl complexes”.
Ocen,eq are similar to those observed in the respective neutral metal pentacarbonyls M(CO)5 (M = Ru, Os),56 while the bond lengths of M–COout,ax are close to those observed in the superelectrophilic [M(CO)6]2+ dications.57
(CO) modes spread over 136 cm−1 in [7a]2+([Al(ORF)4]−)2 (Fig. 6) and 158 cm−1 in [7b]2+([Al(ORF)4]−)2. The average carbonyl stretching vibrational frequencies weighed over the degeneracies59 of (CO)av = 2120/2117 (Ru/Os) cm−1 are slightly below that of free CO(g) at 2143 cm−1 and lay between that of the respective metal pentacarbonyls M(CO)5 (2038/2032 cm−1) and the known metal hexacarbonyl dications [M(CO)6]2+ (2220/2216 cm−1).25,57 This shows the presence of some π-interactions between the carbonyl ligands and the metal centres, classifying the complexes as still being “classical carbonyl complexes”.
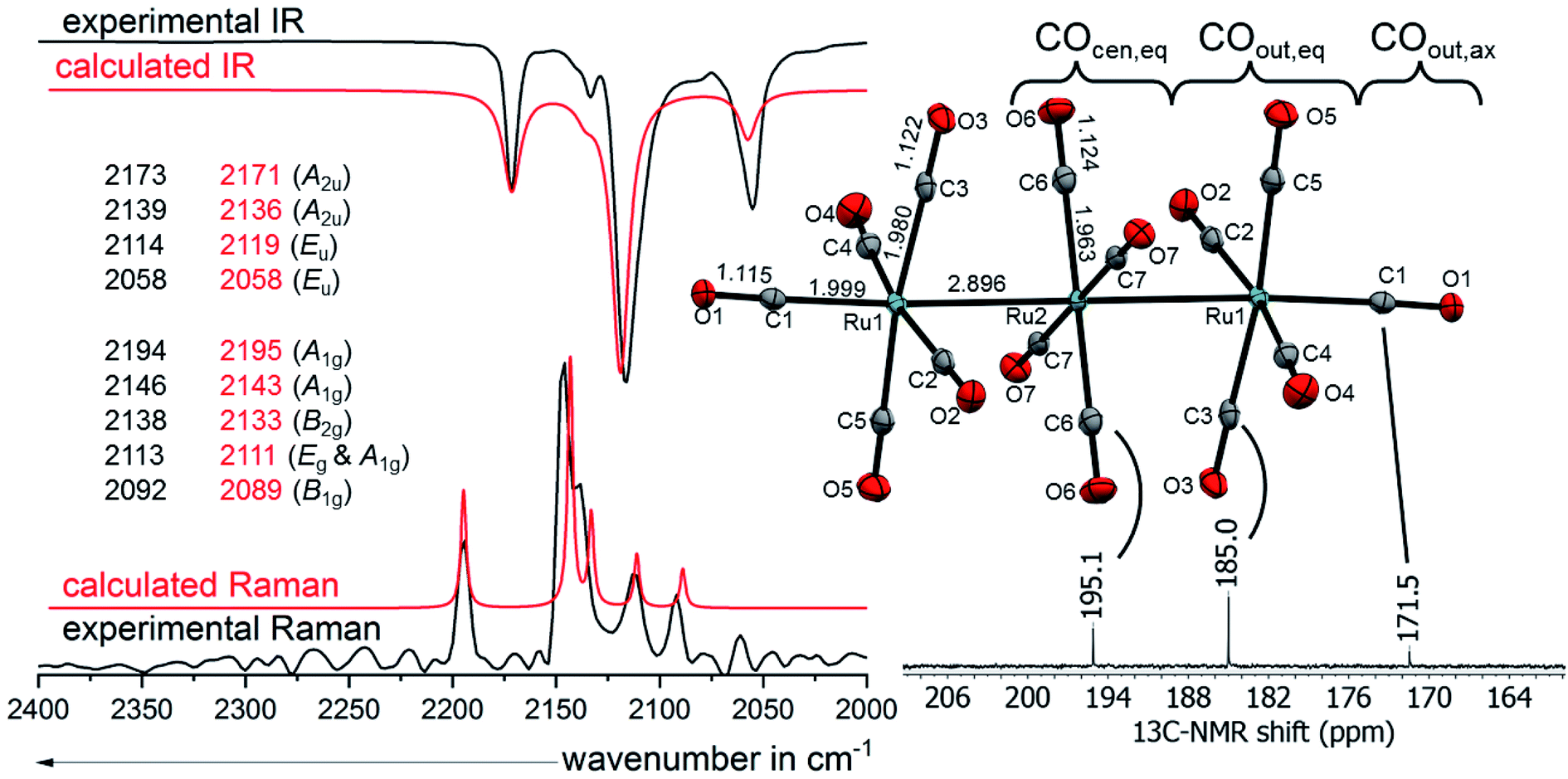 | ||
| Fig. 6 Molecular structure of the dication [7a]2+ in [7a]2+([Al(ORF)4]−)2·2C6F4H2 (ellipsoids set at 50% probability, average distances in Å and standard deviations of the C–O bonds in the range of 0.002 Å) colour code: carbon – grey, oxygen – red, ruthenium – turquoise with the experimental and calculated IR and Raman spectra calculated at B3LYP(D3BJ)/def2-TZVPP level and scaled by 0.968 according to M. A. Duncan et al.58 and 13C-NMR from [7a]2+([Al(ORF)4]−)2 in TFB. | ||
Upon closer look to the vibrational modes, however, not all different carbonyl ligands are active in all vibrational modes (Fig. 7). Therefore, we calculated the respective (CO)av for the symmetry inequivalent carbonyl ligands. While this method does not “unmix” the vibrational modes, the trend from the scXRD can be reproduced, showing a more prominent π-interaction in the M–COcen,eq modes (COcen,eq)av = 2103/2095 cm−1 in comparison to a “nonclassical” interaction in the M–COout,ax modes (COout,ax)av = 2153/2150 cm−1.
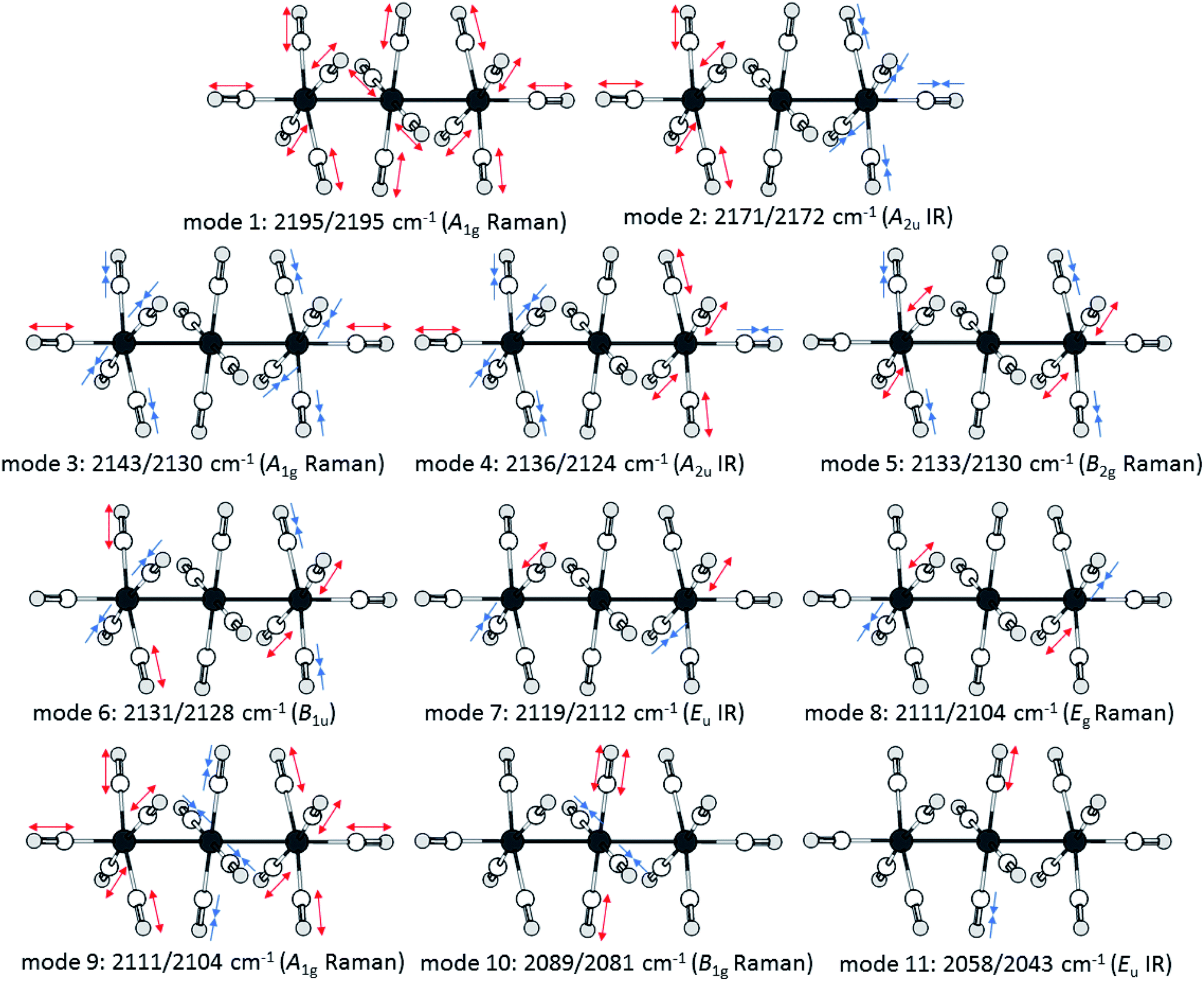 | ||
| Fig. 7 Visualisation of the movements of the vibrational modes of the dications [7a]2+ and [7b]2+ according to calculations at the B3LYP(D3BJ)/def2-TZVPP level and scaled by 0.968 as suggested by M. A. Duncan et al.58 | ||
The force constants of the carbon–oxygen bonds can be estimated with the Cotton–Kraihanzel approximation (see ESI chapter 9 for detailed formulae†).60 Through this approximation, the force constants of all the symmetrical inequivalent carbonyl ligands can be compared separately with free CO(g), which has a force constant of 1856 N m−1.61 The B1g symmetric mode 10 gives the force constant of the carbonyl ligands in the central –M(CO)4– fragment kcen,eq = 1767/1753 N m−1, which is just a little stronger than those for Ru(CO)5 (keq = 1653 N m−1 and kax = 1728 N m−1).62 The same procedure could be used to determine kout,eq from B1u mode 6. However, this mode is neither IR nor Raman active, but it has a close relative: the Raman active B2g mode 5, in which the two –M(CO)5 fragments vibrate in a symmetric instead of an antisymmetric fashion. The vibrational coupling between the two fragments is expected to be negligible and the DFT calculations just show a subliminal spit of 2 cm−1 between the modes. Therefore, the experimentally observed mode 5 was used as an approximation for mode 6, which yields kout,eq = 1846/1842 N m−1. This force constant of the interaction between the cis-standing carbonyl ligands in the –M(CO)5 fragment is determined from the previously calculated kout,eq and mode 8, which yields kout-out = 21/23 N m−1. The A2u symmetric mode 2 yields finally with the previously calculated kout,eq and kout-out the force constant for the axial carbonyl ligands as kout,ax = 1981/2039 N m−1, which are near the force constants of the respective [M(CO)6]2+ dications of k = 2048/2059 N m−1.57
The large difference of kout,ax between [7a]2+ and [7b]2+ (1981/2039 N m−1) is surprising at the first glance, especially given the relatively similar kout,eq and kcen,eq values between the ruthenium and osmium analogues. This can be also directly read out of the experimental vibrational spectra: the complex [7b]2+ has generally slightly lower frequencies than [7a]2+ except for the two most blue shifted experimental bands, which are located at higher frequencies. These two modes, 1 and 2, deviate the most from the DFT calculated values in [7b]2+ with 7 cm−1, while these modes are well predicted for [7a]2+ with a deviation of lower than 2 cm−1. The strengthening of kout,ax relative to kout,eq is known for group 7 dimetal decacarbonyls. While the axial carbonyl ligands have weaker C–O bonds than the equatorial carbonyl ligands in Mn2(CO)10, the relative bond strengths are inverse in Re2(CO)10.63 A possible explanation could be the increasing trans effect on the axial CO-ligand from the –M(CO)5 or the –M(CO)4–(CO)5 fragments, which increases with the metal metal bond strength from 3d over 4d to 5d metals.
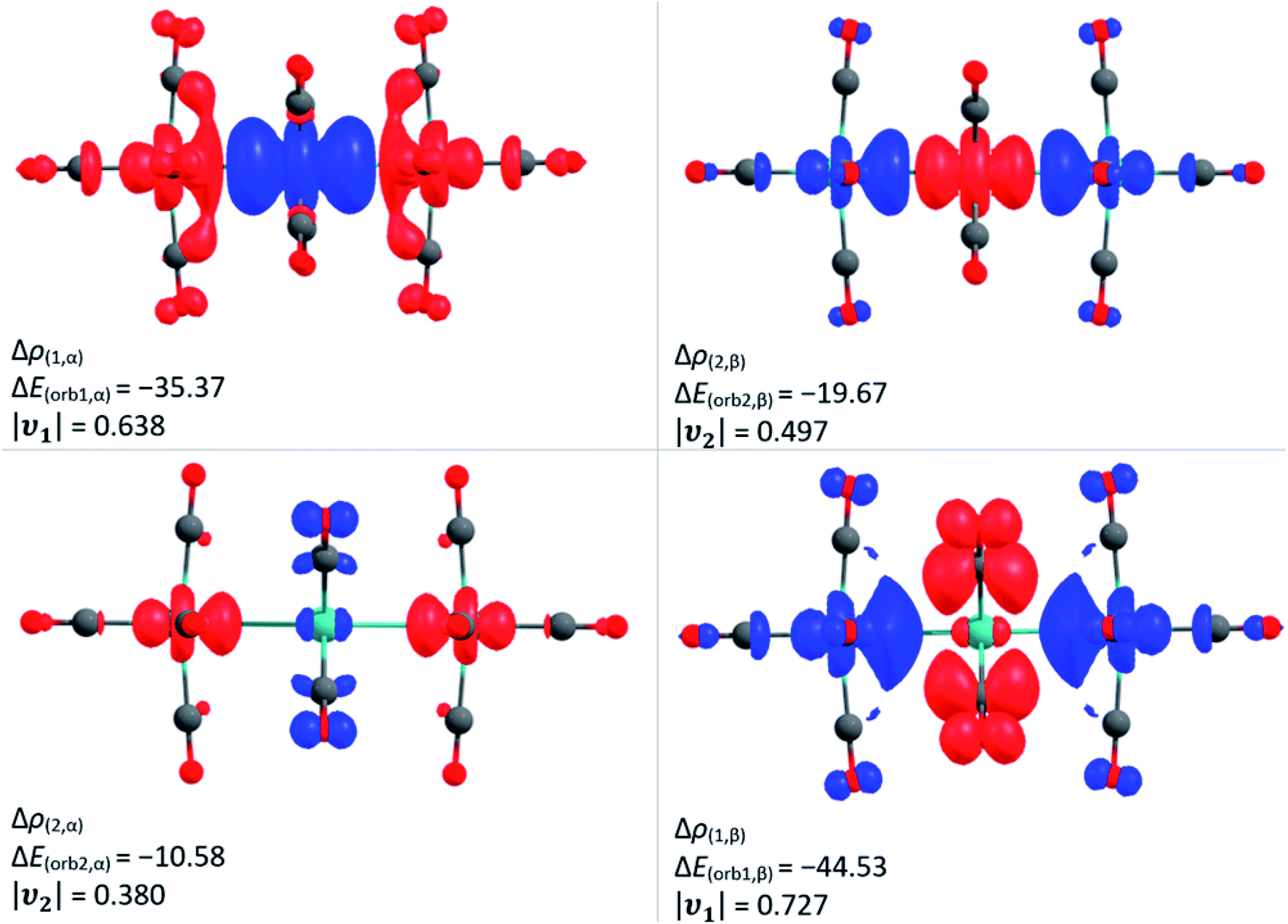 | ||
| Fig. 8 The shape of the deformation densities Δρ of [7a]2+, which are associated with ΔEorb calculated at the BP86(D3BJ)/TZ2P level; relativistic effects were considered with ZORA. The eigenvalue ν gives the value of the charge migration. The direction of the charge flow is from red to blue. Colour code: carbon – grey, oxygen – red, ruthenium – turquoise. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:1. The 13C NMR peak of the central –Ru(CO)4– fragment COcen,eq (195.1 ppm) is little upfield shifted relative to binary ruthenium carbonyl complexes e.g. Ru(CO)5 (200.4 ppm)25 and Ru3(CO)12 (199.7 ppm).66 This indicates, that the central ruthenium atom is indeed only slightly influenced by the dicationic charge of the cluster. The 13C NMR peaks of the equatorial ligands of the –Ru(CO)5 fragment COout,eq on the other hand are more high-field shifted towards 185.0 ppm. The shielding of the 13COout,ax at 171.5 ppm is even more pronounced, being only a few ppm upfield from the superelectrophilic [Ru(CO)6]2+ (168.8 ppm).57 The 13C NMR spectrum of [7b]2+([Al(ORF)4]−)2 is very similar, except for a 15–20 ppm upfield shift, which is also observed for the (pseudo)binary osmium carbonyls compared to their respective ruthenium analogues.57,66 The peak of 13COout,ax in [7b]2+ is unfortunately not visible, probably because it overlaps with the large TFB solvent signal expected to occur in the same spectral range.
:4:1. The 13C NMR peak of the central –Ru(CO)4– fragment COcen,eq (195.1 ppm) is little upfield shifted relative to binary ruthenium carbonyl complexes e.g. Ru(CO)5 (200.4 ppm)25 and Ru3(CO)12 (199.7 ppm).66 This indicates, that the central ruthenium atom is indeed only slightly influenced by the dicationic charge of the cluster. The 13C NMR peaks of the equatorial ligands of the –Ru(CO)5 fragment COout,eq on the other hand are more high-field shifted towards 185.0 ppm. The shielding of the 13COout,ax at 171.5 ppm is even more pronounced, being only a few ppm upfield from the superelectrophilic [Ru(CO)6]2+ (168.8 ppm).57 The 13C NMR spectrum of [7b]2+([Al(ORF)4]−)2 is very similar, except for a 15–20 ppm upfield shift, which is also observed for the (pseudo)binary osmium carbonyls compared to their respective ruthenium analogues.57,66 The peak of 13COout,ax in [7b]2+ is unfortunately not visible, probably because it overlaps with the large TFB solvent signal expected to occur in the same spectral range.
| Ru(CO)5/Os(CO)5 | [7a]2+/[7b]2+ | [Ru(CO)6]2+/[Os(CO)6]2+ | ||||
|---|---|---|---|---|---|---|
| COcen,eq | COaverage | COout,eq | COout,ax | |||
| d C–O/Å | 1.126(2)/1.130(4) | 1.126(2)/1.126(6) | 1.122(2)/1.121(6) | 1.121(2)/1.123(6) | 1.115(2)/1.105(5) | 1.100(8)/1.109(11) |
| d M–C/Å | 1.941(13)ax& 1.961(9)eq/1.982(20)ax & 1.937(19)eq | 1.964(1)/1.963(4) | 1.979(2)/1.981(5) | 1.981(2)/1.988(5) | 1.999(1)/1.987(4) | 2.023(3)/2.020(8) |
| δ 13CCO/ppm | 200.4/182.6 | 195.1/176.8 | 186.0/ | 185.0/163.8 | 171.5/ | 168.8/150.6 |
| (CO)av/cm−11 |
-/- | 2103/2095 | 2120/2115 | 2135/2132 | 2153/2150 | 2220/2216 |
| Δ(COfree)/cm− |
−40/−48 | −23/−27 | −8/−11 | +10/+7 | +77/+73 | |
| k CO/N m−1 | 1728ax & 1653eq/- | 1767/1754 | 1843/1845 | 1846/1842 | 1981/2039 | 2048/2059 |
| q CO (QTAIM) | −0.157/−0.180 | −0.090/−0.125 | −0.022/−0.046 | −0.008/−0.026 | +0.055/+0.030 | +0.150/+0.128 |
| q M (QTAIM) | 0.787/0.900 | M cen = 0.610/0.668 | M av = 0.771/0.883 | M out = 0.851/0.990 | 1.099/1.231 | |
Conclusion
In this report we investigated the reactions of the trimetal dodecacarbonyls 1a and 1b with the inorganic oxidants [NO]+, Ag+ and Ag+/0.5 I2. The reaction of 1a with [NO]+ leads to a mixture, which contains the cationic butterfly complex [2]+ and remarkably breaks the N–O bond in [NO]+. The treatment of 1a and 1b with silver cations yields the respective metal-only Lewis pairs [3a]+ and [3b]+ quantitatively. However, upon increase of the oxidation power of the silver cation through formation of the elemental diiodine complex [Ag2I2]2+, this only led to a mixture of [4]+ and [5]+, which is the result of iodonium additions, formally as in [Ag–I–I]+ → AgI + I+. Consequently, we looked for an innocent deelectronator able to ionize the carbonyls without competing side reactions. Because the only known innocent deelectronator, perfluorinated “phenazineF”,10 has a tedious and time consuming synthesis, we developed an alternative, which can be directly generated from a commercially available chemical. The anthracene derivative 6 has a half-wave potential of 1.42 V vs. Fc0/+ and can be deelectronated by [NO]+[WCA]− in TFB to its radical cation salt [6]+˙[WCA]−. The easy access to the innocent deelectronator [6]+˙[WCA]− will ease the access to known and unknown TMCCs, but also other cations in the future (cf. our work on the organic cations in67). Two equivalents of this arenium cation are able to deelectronate the trimetal dodecacarbonyls 1a and 1b twice under CO pressure, which yields the first homotrimetallic TMCCs [7a]2+ and [7b]2+, respectively. The trimetal tetradecacarbonyl dications have been analysed by NMR as well as vibrational spectroscopy and show very different binding interactions between the carbonyl ligands and the metal centre within the complexes: while the carbonyl ligands in the central –M(CO)4– fragment COcen,eq are closely related to the respective zero-valent metal pentacarbonyls M(CO)5, the equatorial carbonyl ligands of the terminal fragments COout,eq have a similar force constant to free carbon monoxide. The axial carbonyl ligands, on the other hand, are predominantly σ-bound to the metal centres.Data availability
Data available on request.Author contributions
MS performed most of the syntheses, characterizations, performed the DFT calculations and analyses and wrote together with IK the manuscript. CF helped actively with all electrochemical measurements. MM and SR performed the EPR measurements/simulations and wrote and reviewed the respective passages for the manuscript. IK supervised and conceptionally devised the project.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors would like to thank Dr T. Ludwig and Dr M. Daub for the measurements of the pXRD, Dr Harald Scherer and Fadime Bitgül for the NMR measurements and Paul A. Albrecht for the determination of the force constants. The authors acknowledge support by the state of Baden-Württemberg through bwHPC and the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) through grant no INST 40/467-1 and 575-1 FUGG (JUSTUS1 and 2 cluster) and project number 417643975 (S.R.). I.K. acknowledges the DFG for support though grants in the normal procedure. M. S. is grateful for a Ph D fellowship from the “Fonds der Chemischen Industrie FCI”.References
- (a) E. W. Abel and F. G. A. Stone, Q. Rev., Chem. Soc., 1970, 24, 498 RSC; (b) A. J. Lupinetti, S. H. Strauss and G. Frenking, in Progress in Inorganic Chemistry, 2001, pp. 1–112 Search PubMed; (c) G. Frenking, I. Fernández, N. Holzmann, S. Pan, I. Krossing and M. Zhou, JACS Au, 2021, 1, 623 CrossRef CAS PubMed.
- Q. Xu, Coord. Chem. Rev., 2002, 231, 83 CrossRef CAS.
- (a) C. Bach, H. Willner, F. Aubke, C. Wang, S. J. Rettig and J. Trotter, Angew. Chem., Int. Ed., 1996, 35, 1974 CrossRef CAS; (b) J. T. Lin, G. P. Hagen and J. E. Ellis, J. Am. Chem. Soc., 1983, 105, 2296 CrossRef CAS.
- (a) M. Sellin, S. M. Rupf, U. Abram and M. Malischewski, Inorg. Chem., 2021, 60, 5917 CrossRef CAS PubMed; (b) J. A. Corella, R. L. Thompson and N. J. Cooper, Angew. Chem., Int. Ed., 1992, 31, 83 CrossRef.
- P. Schützenberger, Ann. Chim. Phys., 1868, 15, 100 Search PubMed.
- S. C. C. van der Lubbe, P. Vermeeren, C. Fonseca Guerra and F. M. Bickelhaupt, Chem. – Eur. J., 2020, 26, 15690 CrossRef CAS PubMed.
- H. Willner and F. Aubke, Chem. – Eur. J., 2003, 9, 1668 CrossRef CAS PubMed.
- E. O. Fischer, K. Fichtel and K. Öfele, Chem. Ber., 1962, 95, 249 CrossRef CAS.
- M. Malischewski, K. Seppelt, J. Sutter, D. Munz and K. Meyer, Angew. Chem., Int. Ed., 2018, 57, 14597 CrossRef CAS PubMed.
- M. Schorpp, T. Heizmann, M. Schmucker, S. Rein, S. Weber and I. Krossing, Angew. Chem., 2020, 132, 9540 CrossRef.
- H. Willner and F. Aubke, Angew. Chem., 1997, 109, 2506 CrossRef.
- (a) M. Malischewski, in Reference Module in Chemistry, Molecular Sciences and Chemical Engineering, Elsevier, 2021 Search PubMed; (b) H. F. T. Klare and M. Oestreich, J. Am. Chem. Soc., 2021, 143, 15490 CrossRef CAS PubMed; (c) M. Sellin, V. Marvaud and M. Malischewski, Angew. Chem., Int. Ed., 2020, 59, 10519 CrossRef CAS PubMed.
- R. Bröchler, D. Freidank, M. Bodenbinder, I. H. T. Sham, H. Willner, S. J. Rettig, J. Trotter and F. Aubke, Inorg. Chem., 1999, 38, 3684 CrossRef PubMed.
- R. Bröchler, I. H. Sham, M. Bodenbinder, V. Schmitz, S. J. Rettig, J. Trotter, H. Willner and F. Aubke, Inorg. Chem., 2000, 39, 2172 CrossRef PubMed.
- M. S. Foster and J. L. Beauchamp, J. Am. Chem. Soc., 1975, 97, 4808 CrossRef CAS.
- A. Kraft, J. Beck, G. Steinfeld, H. Scherer, D. Himmel and I. Krossing, Organometallics, 2012, 31, 7485 CrossRef CAS.
- (a) T. S. Cameron, A. Decken, I. Dionne, M. Fang, I. Krossing and J. Passmore, Chem. – Eur. J., 2002, 8, 3386 CrossRef CAS PubMed; (b) I. Krossing and I. Raabe, Chem. – Eur. J., 2004, 10, 5017 CrossRef CAS PubMed; (c) I. Krossing and I. Raabe, Angew. Chem. Int. Ed., 2004, 43, 2066 CrossRef CAS PubMed.
- J. Bohnenberger, W. Feuerstein, D. Himmel, M. Daub, F. Breher and I. Krossing, Nat. Commun., 2019, 10, 624 CrossRef CAS PubMed.
- J. Bohnenberger, M. Schmitt, W. Feuerstein, I. Krummenacher, B. Butschke, J. Czajka, P. J. Malinowski, F. Breher and I. Krossing, Chem. Sci., 2020, 11, 3592 RSC.
- J. M. Rall, M. Schorpp, M. Keilwerth, M. Mayländer, C. Friedmann, S. Richert and I. Krossing, 2022, DOI:10.1002/anie.202204080.
- M. Schmitt, M. Mayländer, J. Goost, S. Richert and I. Krossing, Angew. Chem., Int. Ed., 2021, 60, 14800 CrossRef CAS PubMed.
- W. Unkrig, M. Schmitt, D. Kratzert, D. Himmel and I. Krossing, Nat. Chem., 2020, 12, 647 CrossRef CAS PubMed.
- (a) G. Wang, J. Cui, C. Chi, X. Zhou, Z. H. Li, X. Xing and M. Zhou, Chem. Sci., 2012, 3, 3272 RSC; (b) E. M. Markin and K. Sugawara, J. Phys. Chem. A, 2000, 104, 1416 CrossRef CAS; (c) J. Cui, X. Zhou, G. Wang, C. Chi, Z. H. Li and M. Zhou, J. Phys. Chem. A, 2014, 118, 2719 CrossRef CAS PubMed; (d) C. Chi, J. Cui, X. Xing, G. Wang, Z.-P. Liu and M. Zhou, Chem. Phys. Lett., 2012, 542, 33 CrossRef CAS.
- (a) M. Bodenbinder, G. Balzer-Jöllenbeck, H. Willner, R. J. Batchelor, F. W. B. Einstein, C. Wang and F. Aubke, Inorg. Chem., 1996, 35, 82 CrossRef CAS PubMed; (b) Q. Xu, B. T. Heaton, C. Jacob, K. Mogi, Y. Ichihashi, Y. Souma, K. Kanamori and T. Eguchi, J. Am. Chem. Soc., 2000, 122, 6862 CrossRef CAS.
- P. Rushman, G. N. van Buuren, M. Shiralian and R. K. Pomeroy, Organometallics, 1983, 2, 693 CrossRef CAS.
- J. Washington, R. McDonald, J. Takats, N. Menashe, D. Reshef and Y. Shvo, Organometallics, 1995, 14, 3996 CrossRef CAS.
- (a) C. R. Eady, B. Johnson and J. Lewis, J. Organomet. Chem., 1972, 37, C39–C40 CrossRef CAS; (b) D. H. Farrar, B. F. G. Johnson, J. Lewis, P. R. Raithby and M. J. Rosales, J. Chem. Soc., Dalton Trans., 1982, 2051 RSC; (c) V. J. Johnston, F. W. B. Einstein and R. K. Pomeroy, J. Am. Chem. Soc., 1987, 109, 8111 CrossRef CAS; (d) V. J. Johnston, F. W. B. Einstein and R. K. Pomeroy, J. Am. Chem. Soc., 1987, 109, 7220 CrossRef CAS.
- J. Bohnenberger, D. Kratzert, S. Pan, G. Frenking and I. Krossing, Chem. – Eur. J., 2020, 26, 17203 CrossRef CAS PubMed.
- V. Radtke, D. Himmel, K. Pütz, S. K. Goll and I. Krossing, Chem. – Eur. J., 2014, 20, 4194 CrossRef CAS PubMed.
- We use the elementary steps electronation and deelectronation in their strict sense, i.e., addition or removal of e–. Thus, a classically termed ‘oxidant’ is addressed as a ‘deelectronator’ and a ‘reductant’ as an ‘electronator’, if only a single electron transfer as the elementary step takes place. This particle-based terminology is related to the acid-base picture, where the terms deprotonation and protonation describe the transfer of a proton between two partners, i.e., deelectronation is the electron-based equivalent to a deprotonation. This terminology has been discussed in ref. 29 and ref. 31 from our work on the protoelectric potential map and is transferred to describe these types of reactions more accurately.
- D. Himmel, V. Radtke, B. Butschke and I. Krossing, Angew. Chem., Int. Ed., 2018, 57, 4386 CrossRef CAS PubMed.
- R. Kalescky, E. Kraka and D. Cremer, J. Phys. Chem. A, 2013, 117, 8981 CrossRef CAS PubMed.
- S. Harris, M. L. Blohm and W. L. Gladfelter, Inorg. Chem., 1989, 28, 2290 CrossRef CAS.
- J. P. Attard, B. F. G. Johnson, J. Lewis, J. M. Mace and P. R. Raithby, J. Chem. Soc., Chem. Commun., 1985, 1526 RSC.
- J. Bohnenberger and I. Krossing, Angew. Chem., Int. Ed., 2020, 59, 5581 CrossRef CAS PubMed.
- S. C. Meier, D. Himmel and I. Krossing, Chem. – Eur. J., 2018, 24, 19348 CrossRef CAS PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef CAS.
- P. J. Malinowski and I. Krossing, Angew. Chem., Int. Ed., 2014, 53, 13460 CrossRef CAS PubMed.
- S. Pan, S. M. N. V. T. Gorantla, D. Parasar, H. V. R. Dias and G. Frenking, Chem. – Eur. J., 2021, 27, 6936 CrossRef CAS PubMed.
- (a) G. Wang, Y. S. Ceylan, T. R. Cundari and H. V. R. Dias, J. Am. Chem. Soc., 2017, 139, 14292 CrossRef CAS PubMed; (b) G. Wang, T. T. Ponduru, Q. Wang, L. Zhao, G. Frenking and H. V. R. Dias, Chem. – Eur. J., 2017, 23, 17222 CrossRef CAS PubMed; (c) T. T. Ponduru, G. Wang, S. Manoj, S. Pan, L. Zhao, G. Frenking and H. V. R. Dias, Dalton Trans., 2020, 49, 8566 RSC; (d) G. Wang, A. Noonikara-Poyil, I. Fernández and H. V. R. Dias, Chem. Commun., 2022, 58, 3222 RSC.
- J. Bauer, H. Braunschweig and R. D. Dewhurst, Chem. Rev., 2012, 112, 4329 CrossRef CAS PubMed.
- (a) L. Song and W. C. Trogler, Angew. Chem., Int. Ed., 1992, 31, 770 CrossRef; (b) K. Koessler and B. Butschke, in Encyclopedia of Inorganic and Bioinorganic Chemistry, John Wiley & Sons, Ltd, 2021, pp. 1–31 Search PubMed.
- L. J. Farrugia and H. M. Senn, J. Phys. Chem. A, 2012, 116, 738 CrossRef CAS PubMed.
- (a) L. Zhao, M. Hermann, W. H. E. Schwarz and G. Frenking, Nat. Rev. Chem., 2019, 3, 48 CrossRef CAS; (b) M. P. Mitoraj, A. Michalak and T. Ziegler, J. Chem. Theory Comput., 2009, 5, 962 CrossRef CAS PubMed.
- P. J. Malinowski, D. Himmel and I. Krossing, Angew. Chem., Int. Ed., 2016, 55, 9259 CrossRef CAS PubMed.
- (a) V. Zhuravlev and P. J. Malinowski, Angew. Chem., 2018, 130, 11871 CrossRef; (b) P. J. Malinowski, D. Himmel and I. Krossing, Angew. Chem. Int. Ed., 2016, 55, 9262 CrossRef CAS PubMed.
- (a) H. K. Nagra, R. J. Batchelor, A. J. Bennet, F. W. B. Einstein, E. C. Lathioor, R. K. Pomeroy and W. Wang, J. Am. Chem. Soc., 1996, 118, 1207 CrossRef CAS; (b) H. K. Sanati, A. Becalska, A. K. Ma and R. K. Pomeroy, J. Chem. Soc., Chem. Commun., 1990, 197 Search PubMed.
- T. J. Richardson, F. L. Tanzella and N. Bartlett, J. Am. Chem. Soc., 1986, 108, 4937 CrossRef CAS.
- M. A. Khanfar and K. Seppelt, J. Fluorine Chem., 2015, 179, 193 CrossRef CAS.
- (a) Y. Sakamoto, T. Suzuki, M. Kobayashi, Y. Gao, Y. Inoue and S. Tokito, Mol. Cryst. Liq. Cryst., 2006, 444, 225 CrossRef CAS; (b) Y. Sakamoto, T. Suzuki, M. Kobayashi, Y. Gao, Y. Fukai, Y. Inoue, F. Sato and S. Tokito, J. Am. Chem. Soc., 2004, 126, 8138 CrossRef CAS PubMed.
- J. F. Tannaci, M. Noji, J. McBee and T. D. Tilley, J. Org. Chem., 2007, 72, 5567 CrossRef CAS PubMed.
- R. Bau, S. W. Kirtley, T. N. Sorrell and S. Winarko, J. Am. Chem. Soc., 1974, 96, 988 CrossRef CAS.
- L. S. Crocker, B. M. Mattson, D. M. Heinekey and G. K. Schulte, Inorg. Chem., 1988, 27, 3722 CrossRef CAS.
- (a) R. J. Batchelor, F. W. B. Einstein, R. K. Pomeroy and J. A. Shipley, Inorg. Chem., 1992, 31, 3155 CrossRef CAS; (b) R. J. Batchelor, H. B. Davis, F. W. B. Einstein and R. K. Pomeroy, J. Am. Chem. Soc., 1990, 112, 2036 CrossRef CAS.
- R. Hoffmann, Angew. Chem., Int. Ed., 1982, 21, 711 CrossRef.
- (a) J. Huang, K. Hedberg and R. K. Pomeroy, Organometallics, 1988, 7, 2049 CrossRef CAS; (b) J. Huang, K. Hedberg, H. B. Davis and R. K. Pomeroy, Inorg. Chem., 1990, 29, 3923 CrossRef CAS.
- E. Bernhardt, C. Bach, B. Bley, R. Wartchow, U. Westphal, I. H. T. Sham, B. von Ahsen, C. Wang, H. Willner, R. C. Thompson and F. Aubke, Inorg. Chem., 2005, 44, 4189 CrossRef CAS PubMed.
- M. K. Assefa, J. L. Devera, A. D. Brathwaite, J. D. Mosley and M. A. Duncan, Chem. Phys. Lett., 2015, 640, 175 CrossRef CAS.
- A. J. Lupinetti, G. Frenking and S. H. Strauss, Angew. Chem., Int. Ed., 1998, 37, 2113 CrossRef CAS PubMed.
- F. A. Cotton and C. S. Kraihanzel, J. Am. Chem. Soc., 1962, 84, 4432 CrossRef CAS.
- A. S. Goldman and K. Krogh-Jespersen, J. Am. Chem. Soc., 1996, 118, 12159 CrossRef CAS.
- M. F. Gregory, M. Poliakoff and J. J. Turner, J. Mol. Struct., 1985, 127, 247 CrossRef CAS.
- G. Bor and G. Sbrignadello, J. Chem. Soc., Dalton Trans., 1974, 440 RSC.
- (a) A. Veillard and M.-M. Rohmer, Int. J. Quantum Chem., 1992, 42, 965 CrossRef CAS; (b) M. Martin, B. Rees and A. Mitschler, Acta Cryst B, 1982, 38, 6 CrossRef.
- (a) B. von Ahsen, M. Berkei, G. Henkel, H. Willner and F. Aubke, J. Am. Chem. Soc., 2002, 124, 8371 CrossRef CAS PubMed; (b) H. Willner, M. Bodenbinder, R. Bröchler, G. Hwang, S. J. Rettig, J. Trotter, B. von Ahsen, U. Westphal, V. Jonas, W. Thiel and F. Aubke, J. Am. Chem. Soc., 2001, 123, 588 CrossRef CAS PubMed.
- A. Forster, B. F. G. Johnson, J. Lewis, T. W. Matheson, B. H. Robinson and W. G. Jackson, J. Chem. Soc., Chem. Commun., 1974, 1042 RSC.
- M. Hermann, T. Böttcher, M. Schorpp, S. Richert, D. Wassy, I. Krossing and B. Esser, Chem. – Eur. J., 2021, 27, 4964 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2150901, 2150925, 2151155, 2151474, 2151510, 2151836, 2151840, 2151852, 2153977, 2159967. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc02358j |
| This journal is © The Royal Society of Chemistry 2022 |