
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBound oxygen-atom transfer endows peroxidase-mimic M–N–C with high substrate selectivity†
Xinghua
Chen
a,
Lufang
Zhao
a,
Kaiqing
Wu
a,
Hong
Yang
a,
Qing
Zhou
a,
Yuan
Xu
a,
Yongjun
Zheng
b,
Yanfei
Shen
b,
Songqin
Liu
 a and
Yuanjian
Zhang
*a
a and
Yuanjian
Zhang
*a
aJiangsu Engineering Laboratory of Smart Carbon-Rich Materials and Device, Jiangsu Province Hi-Tech Key Laboratory for Bio-Medical Research, State Key Laboratory of Bioelectronics, School of Chemistry and Chemical Engineering, Southeast University, Nanjing, 211189, China. E-mail: Yuanjian.Zhang@seu.edu.cn
bMedical School, Southeast University, Nanjing, 210009, China
First published on 6th May 2021
Abstract
Advances in nanoscience have stimulated the wide exploration of nanozymes as alternatives to enzymes. Nonetheless, nanozymes often catalyze multiple reactions and are not specialized to a specific substrate, restricting their broad application. Here, we report that the substrate selectivity of the peroxidase-mimic M–N–C can be significantly altered via forming bound intermediates with variable interactions with substrates according to the type of metal. Taking two essential reactions in chemical sensing as an example, Fe–N–C and Co–N–C showed opposite catalytic selectivity for the oxidation of 3,3′,5,5′-tetramethylbenzidine (TMB) and 3-aminophthalhydrazide (luminol), respectively, by factors of up to 200-fold. It was revealed that specific transition metal-N coordination was the origin of the selective activation of H2O2 forming critically bound oxygen intermediates (M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) for oxygen-atom transfer and the consequent oxidization of substrates. Notably, owing to the embedded ligands in the rigid graphitic framework, surprisingly, the selectivity of M–N–C was even superior to that of commonly used horseradish peroxidase (HRP).
O) for oxygen-atom transfer and the consequent oxidization of substrates. Notably, owing to the embedded ligands in the rigid graphitic framework, surprisingly, the selectivity of M–N–C was even superior to that of commonly used horseradish peroxidase (HRP).
Introduction
Substance transformation is involved in many diverse processes, ranging from natural metabolism in living organisms to artificial industrial reactions and chemical sensing.1,2 Despite the various pathways, these interconversions are often accelerated by catalysts. A great example is that of the metabolic enzymes, which precisely drive biological reactions with incredible efficiency thanks to the refined hierarchical structures resulting from millions of years of evolution. Unfortunately, the high cost and environment-dependent activity of these enzymes limits their wide-ranging applications in vitro. Hence, with the increased interest in preparing robust and cost-effective alternatives, many nanomaterials have been reported with intriguing enzyme-like functions and are called nanozymes.3–8 According to their composition, nanozymes can be categorized into carbon, metal oxides, and noble metals/alloys. To date, they have been successfully applied to various applications, such as bio-sensing/-imaging, tumor therapeutics, and anti-bacterial/-inflammation,9–16 demonstrating multiple functions. For instance, owing to the highly reversible affinities of oxygen-related species at surfaces, noble metals and their alloy nanomaterials can catalyze multiple redox reactions, similar to peroxidase, oxidase, catalase, and superoxide dismutation (SOD), depending on the reaction conditions.17,18 In contrast, transition metals in metal oxide-based nanozymes, such as Fe3O4, exhibit strong bonding with O via the d orbitals, enabling the intermediate reactive oxygen species (ROS), for example, ˙OH, to be freely released into the solution and oxidize a diverse range of substrates.19 However, the intrinsic reaction selectivity of the nanozymes is low, which restricts their broad applications.20–23A few efforts have been devoted to addressing this problem, including the early extrinsic integration of the recognition unit by molecular imprinting,14 and very recently, intrinsic engineering of metal complex-based nanozymes.24,25 However, for the former, the selectivity is impeded by the particular types of monomers used for imprinting; for the latter, the general origins that make them stand out from their counterparts are still unclear. Therefore, developing a novel method to improve the reaction selectivity of the nanozymes and obtain the underlying operation principles poses significant challenges.26,27
As a typical non-precious electrocatalyst, Fe–N–C has been widely explored for O2, CO2, and N2 reduction.28–31 The in situ/operando characterization verifies the critical role of Fe–N coordination centers in promoting electrocatalytic activities.32–35 Very recently, under a similar scheme, Fe–N–C was discovered as a kind of single-atom nanozyme with an exceptional high (per)oxidase-like activity.36–43 From a fundamental point of view, coordination between the transition metal and N offers suitable hybrid orbitals that selectively interact and activate the substrates and intermediates, reminiscent of the similar operation principle of metalloporphyrin in natural enzymes.44,45 Indeed, metal ions are ubiquitous in nature, playing structural and/or catalytic roles in almost half of all proteins, thus attracting extensive exploration to understand the involved fundamental operation principle.46 However, for nanozymes following a similar principle, few related studies have been reported to date.
Herein, we report that the peroxidase-like reaction selectivity of the M–N–C nanozymes can be generally regulated by the M–N coordination site (M = Fe, Co, Mn, Ni, and Cu), which has variable affinities for successive H2O2 activation and oxidation of the substrates. The formation of active MO intermediates for the oxygen-atom transfer was disclosed by both practical experiments and density functional theory (DFT) calculations, to determine the origin of the intrinsic reaction selectivity. As two essential reactions in chemical sensing, intriguingly, Fe–N–C and Co–N–C exhibited an opposite selectivity up to 200-fold in the catalytic 3,3′,5,5′-tetramethylbenzidine (TMB) and 3-aminophthalhydrazide (luminol) oxidation, respectively. Owing to the unique embedded ligands in the rigid graphitic framework, surprisingly, the selectivity of M–N–C was even superior to that of commonly used horseradish peroxidase (HRP). Rather than merely copying biologically available enzymes, this work provides an interesting example to learn principles from biology and transfer them to industrial reactions that were not previously accessible to biology, and revealing an outstanding performance.47
Results and discussion
The M–N–C nanozymes were synthesized via high-temperature pyrolysis of poly(o-phenylenediamine) (PoPD) with metal salts loaded on carbon black (CB), followed by acid-etching.28 As a control, N–C without any metals was also synthesized under identical conditions except for the addition of metal salts. The microstructures of M–N–C were first characterized using scanning electron microscopy (SEM). Fig. S1† showed that Fe–N–C mainly consists of nanoparticles with an average size of 40–50 nm. Their size and shape did not significantly rely on the concentration of iron precursor. Similar features were also observed for other M–N–C nanozymes with different transition metals (Fig. S2†). The more detailed texture of Fe–N–C was ascertained using transmission electron microscopy (TEM). Fig. 1a and S3† showed that all the Fe–N–C nanozymes were composed of a CB core (a turbostratic multilayer of graphite, Fig. S4†) and a secondary graphitic shell derived from PoPD during pyrolysis. There were no obvious metal nanoparticles or clusters in the nanocomposites, consistent with the additional powder X-ray diffraction and Raman measurements (Fig. S5 and S6†). Notably, a significantly reduced specific surface area (Table S1†) along with the diminished mesoporous structure of Fe–N–C compared to that of the CB support was further observed using pore size distribution analysis of the N2 sorption isotherms (Fig. S7†). It was associated with the growth of a new layer on the outer surface and the mesopores of the CB support. Other M–N–C, such as Co–N–C, exhibited a similar microstructure according to the characterization results (Fig. S8–S11†).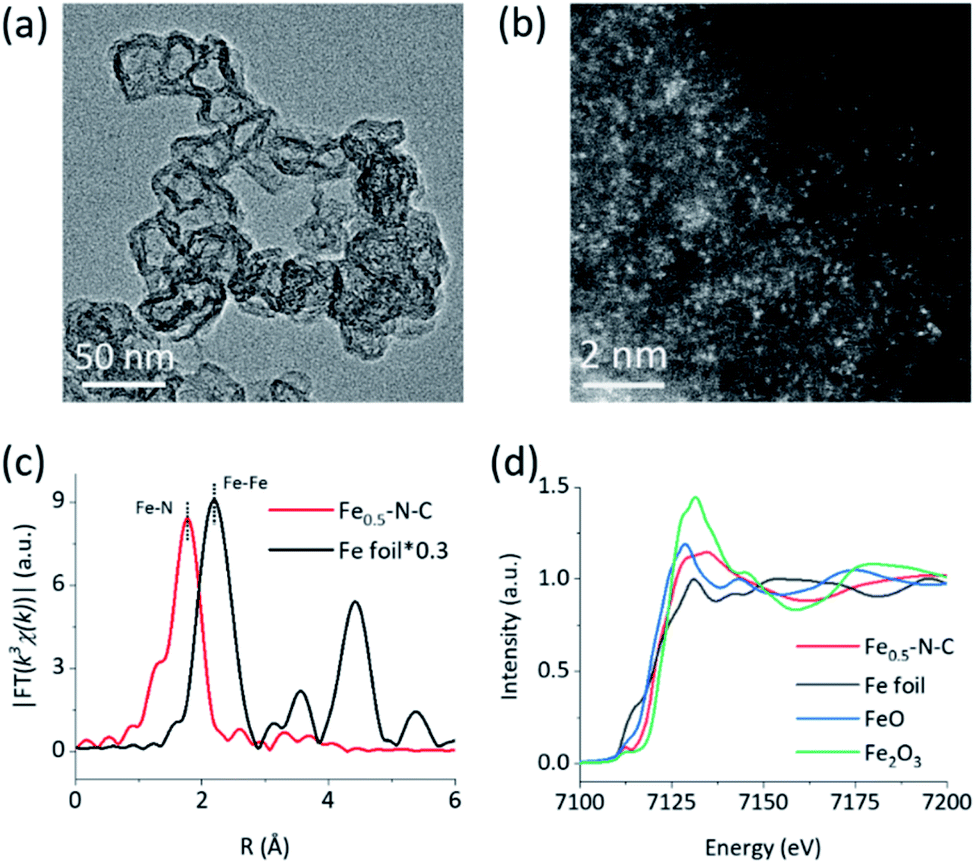 | ||
| Fig. 1 Structural characterization. (a) a TEM image and (b) a HAADF-STEM image of Fe–N–C, (c) Fourier transformed k3-weighted EXAFS spectra of Fe–N–C and Fe foil, and (d) XANES spectra of Fe–N–C and reference samples at the Fe K-edge. | ||
To reveal the specific state of the metals in M–N–C, we then resorted to aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM). The homogeneous, abundant, and isolated single Fe sites of the Fe–N–C nanozyme were identified adequately by synergistically comparing and analyzing the HAADF-STEM image (Fig. 1b) coupled energy-dispersive X-ray spectroscopy (EDS) mapping images (Fig. S12†). The average size of the Fe sites was ca. 1–1.5 Å based on the statistical analysis of the legible bright dots. Owing to the accompanying appearance of the Fe and N phases, it was speculated that effective coordination structures were formed between the Fe and N elements. The affluent Co–N sites of the Co–N–C nanozyme were also confirmed using the same method (Fig. S13†).
The electronic environment and relative content of the doped elements within the near-surface region of the M–N–C nanozymes were analyzed using X-ray photoelectron spectroscopy (XPS). The high-resolution N 1s spectra of Fe–N–C (Fig. S14†) can be deconvoluted into porphyrin-like Fe–N coordinated and/or imine (399.7 eV), as well as pyridinic- (398.3 eV), pyrrolic- (401 eV), graphitic- (402.3 eV), and oxidized- (403.5 eV) N species.48–50 Interestingly, the relative percentage of N centered at 399.7 eV of Fe–N–C (18.49%) was significantly higher than that of N–C (14.45%) and gradually increased with the Fe content (Fig. S14, Table S2†), evidently verifying the formation of Fe–N coordination in Fe–N–C. The very similar M–N coordination structures of the other M–N–C (M = Co, Mn, Ni, and Cu) were also confirmed by deconvoluting the high-resolution N 1s spectra (Fig. S15 and S16†) and the quantification analysis of the N percentage (Table S2†). Detailed analysis of the metal 2p3/2 shake-up photoemission lines offered a more detailed cation state for these metallic species (Fig. S17–S19†).
To obtain the valence state and coordination structure of the Fe in the Fe–N–C nanozyme at the atom-level, X-ray absorption fine structure (XAFS) spectra were further performed at the Fe K-edge. The Fourier-transform-extended X-ray absorption fine structure (FT-EXAFS) curves of Fe–N–C and Fe foil are illustrated in Fig. 1c. An Fe–Fe peak ca. at 2.19 Å for Fe foil was not observed in Fe–N–C, demonstrating the atomic dispersion of Fe in Fe–N–C, which was in accordance with the HAADF-STEM results. The major peak ca. at 1.76 Å corresponding to the Fe–N scattering paths is presented in Fe–N–C. The X-ray absorption near-edge structure (XANES) spectra (Fig. 1d) indicated that the absorption edge position of Fe–N–C was located between that of FeO and Fe2O3, indicating the valence state of Fe was between +2 and +3 in Fe–N–C. The coordination manner of Co in Co–N–C was similar to that in Fe–N–C, except for the metal type and some minor additional Cl(S) ligands (Fig. S20†). It has been suggested that these coordination variations would regulate the selectivity in enzyme-like reactions, owing to their different affinities to substrates.
As two of the essential reactions in chemical sensing, the catalytic oxidation of TMB and luminol with H2O2 were selected as examples to evaluate the selectivity of M–N–C.51,52 In the first set of experiments, the activity of M–N–C for catalyzing the oxidation of TMB with H2O2 in HAc–NaAc buffer solution was assessed (Fig. S21–24†). As shown in Fig. 2a, the typical blue oxidized product of TMB (i.e., TMBox) was observed for most catalysts using the naked eye, but interestingly, Fe–N–C and HRP demonstrated a much darker color. The quantitative evaluation of the characteristic absorption peak at 652 nm of TMBox demonstrated that the initial reaction rates of the oxidation reaction catalyzed by Fe–N–C (6.08 μM s−1) were larger than that by the other M–N–C and N–C by a factor of up to 21-fold (Fig. 2c). Considering the similar particle size, morphology, and carbon crystallinity, the varieties of transition metals in M–N–C should play a crucial role in the selective catalytic activity in TMB oxidation.
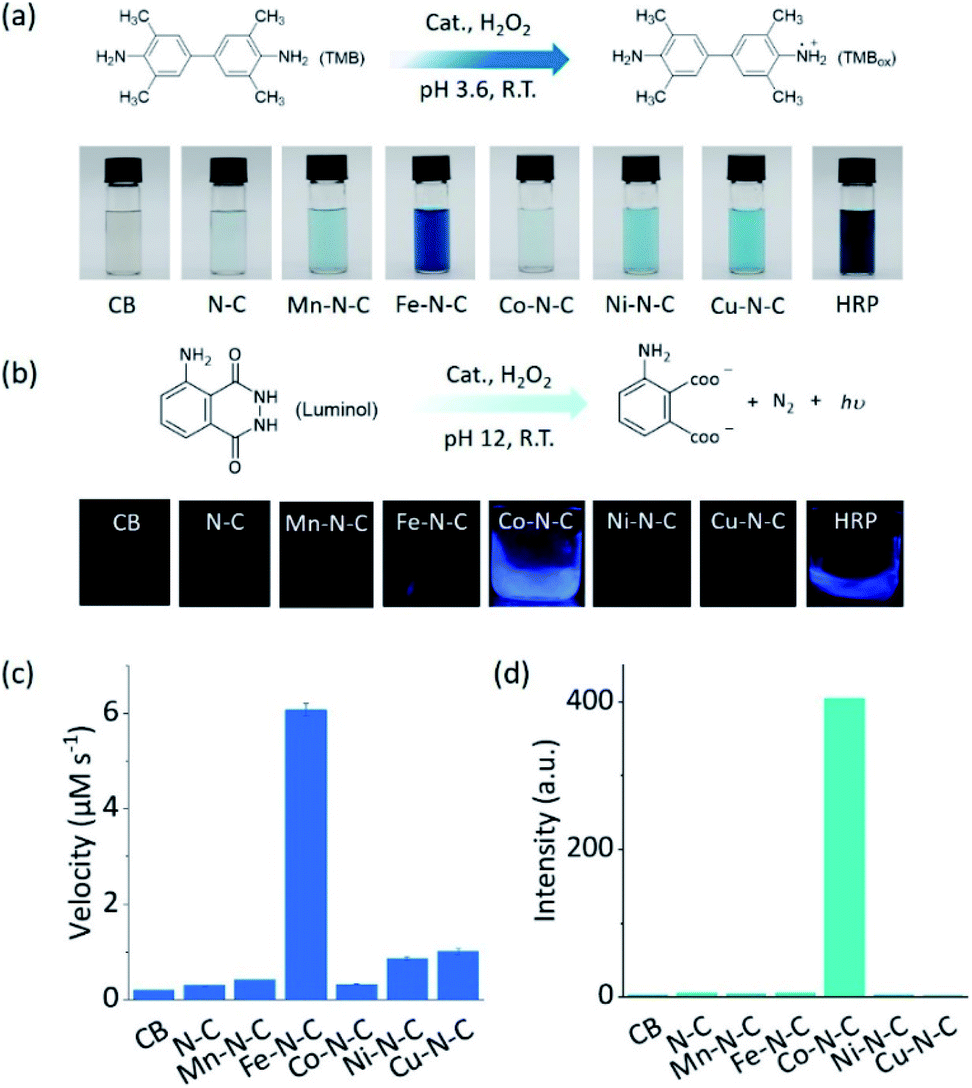 | ||
| Fig. 2 The selective driving of peroxidase-like reactions under different reaction conditions. (a) An equation showing the reaction for TMB oxidation and photos of TMB in solution after the reaction using different catalysts. (b) An equation showing the luminol oxidation reaction and photos of the CL emission in solution during the reaction using different catalysts. (c) The initial velocity of the catalytic oxidation of 1 mM TMB with 100 mM H2O2 in the presence of 20 μg mL−1 CB, N–C, and different M0.5–N–C nanozymes in 0.1 M HAc–NaAc (pH 3.6). (d) The chemiluminescence intensity at 425 nm for monitoring the catalytic oxidation of 2.5 mM luminol with 250 mM H2O2 in the presence of 50 μg mL−1 CB, N–C, and different M–N–C nanozymes in 0.01 M NaOH. | ||
M–N–C activity for catalyzing the oxidation reaction of luminol with H2O2 in an alkaline solution was investigated by detecting the chemiluminescent (CL) emission intensity. In Co–N–C and HRP only,53 the CL light emission was observed by the naked eye (Fig. 2b). The CL intensity-time plots in Fig. S25† showed that the CL was rapidly triggered when luminol was injected into the solution containing Co–N–C and H2O2, and the light intensity attenuated over time. This could be explained by the high catalytic activity of Co–N–C and the gradual consumption of luminol during the reaction. Notably, the catalytic activity of Co–N–C remained almost unaffected in physiological temperatures up to 40 °C (Fig. S26†). In contrast, the other M–N–C (M = Fe, Mn, Ni, Cu), metal-free N–C, and CB did not exhibit any noticeable catalytic activity (up to 200-fold lower, Fig. 2d) using the same method, indicating the nature of the M–N coordination center is the kernel for the catalytic activity.
To understand the opposite catalytic characteristics of Fe–N–C and Co–N–C in H2O2 involved redox reactions, the possible intermediate radical species, for example, free ROS, were first explored using the scavenger trapping technique.36,54 As illustrated in Fig. 3a, neither SOD nor mannitol, which respectively scavenged superoxide and hydroxyl radicals, had significant influences on the oxidation of TMB catalyzed by Fe–N–C. This indicated that the superoxide and hydroxyl radical were not produced owing to the redox of H2O2 in the Fe–N–C catalyzed TMB oxidation. The electron spin resonance (ESR) spectra shown in Fig. 3b and S27† further demonstrated that there was no signal for any ROS-trapping agent adduct during the activation of H2O2 by the Fe–N–C nanozyme, which was similar to the case catalyzed by HRP (Fig. 3c, S27 and S28†).6,55 Directly monitoring the dynamic formation and consumption of FeO species during the reactions would be exciting, but challenging in terms of the experiments;56–58 nonetheless, the ex situ ESR spectra of Fe–N–C in air and degassed conditions showed a significant intensity change for Fe, suggesting the probable formation of FeO species (Fig. 3d).59–61 In contrast, the characteristic peak of the hydroxyl radical-DMPO adducts with a signal intensity of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2:1 appeared during the activation of H2O2 catalyzed by the well-known Fe3O4 nanozyme (Fig. S29†). Taking the metalloporphyrin-like structure of M–N–C into account, we speculate that M–N–C might have a similar catalytic mechanism to HRP via the bound ROS pathway.62,63
:2:2:1 appeared during the activation of H2O2 catalyzed by the well-known Fe3O4 nanozyme (Fig. S29†). Taking the metalloporphyrin-like structure of M–N–C into account, we speculate that M–N–C might have a similar catalytic mechanism to HRP via the bound ROS pathway.62,63
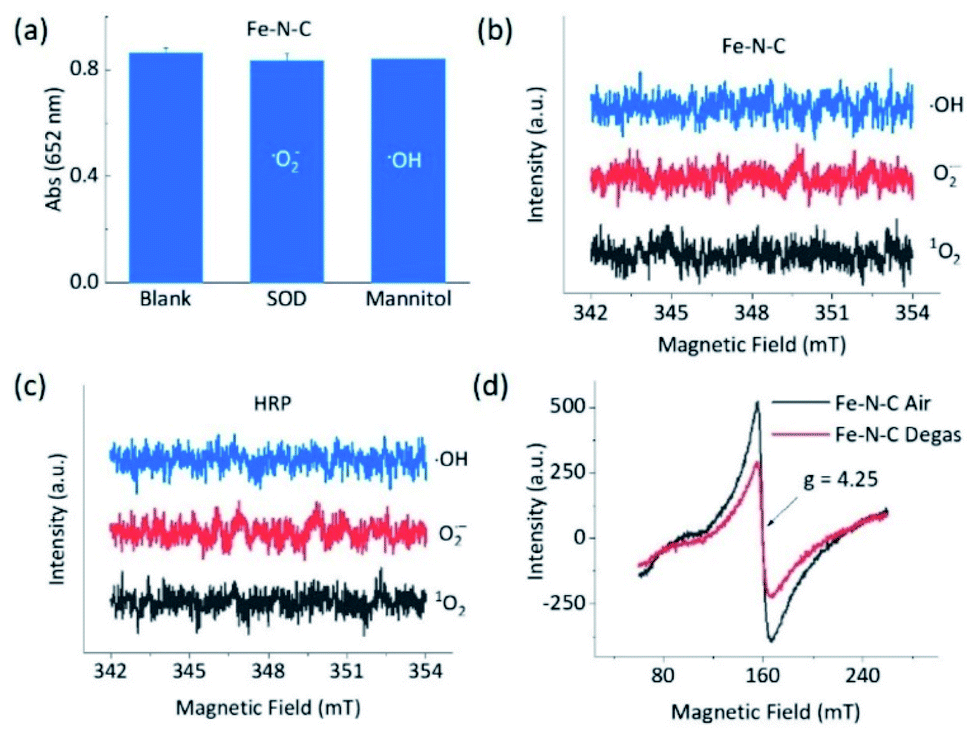 | ||
| Fig. 3 Intermediates in the peroxidase-like reactions. (a) Effects of ROS scavengers on the oxidation of TMB with H2O2 catalyzed by Fe–N–C based on typical absorption at 652 nm. ESR spectra of the spin adduct of the hydroxyl radical, superoxide radical, and singlet oxygen generated during the activation of H2O2 by (b) Fe–N–C and (c) HRP in 0.1 M HAc–NaAc (pH 3.6). (d) EPR spectra at 110 K of Fe–N–C in air and treated via vacuum degassing at 120 °C for 36 h. | ||
The dependence of the reaction rate on the TMB and H2O2 concentration was observed, indicating the Fe–N–C catalyzed oxidation of TMB with H2O2 through a competition between the peroxidase- and catalase-like properties (see further discussion in Fig. S30†), as did the Co–N–C/luminol system. To further investigate the significant difference between Fe–N–C and Co–N–C to catalyze the oxidation of TMB and luminol using H2O2, DFT calculations were carried out, by following a leading postulate that M–N–C primarily has a pyridinic M–N4 ligation environment (see more discussion in Fig. S31†).28,59,64,65 The acid and alkaline systems were modeled by adsorbing the hydrogen atom and hydroxyl on metal atoms, respectively (Fig. S31†).18 As shown in Fig. 4a and S32†, TMB oxidation with H2O2 on M–N–C started with H2O2 adsorption onto the N-coordinated metal site with an adsorption free energy (Eads) of −0.16 and −0.13 eV for Fe–N–C and Co–N–C, respectively. Then, the FeO intermediate state (MS3) was produced by the decomposition of the adsorbed H2O2 into H2O via the transition state (TS1) with an activation barrier (Ea) of 0.08 eV. Compared with the marginal Ea for generating FeO, the formation of CoO required a much larger Ea of 0.94 eV, which was speculated as the crucial factor for the distinctive difference in the catalytic activity between Fe–N–C and Co–N–C. In the next stage, the oxidation of TMB was proceeded through the N–H bond cleavage of TMB and transformed H to MO with an Ea of 0.45 eV (Fig. S32†) on FeO. In contrast, the H2O2 oxidation using the same FeO intermediate, a competitive catalase-like reaction, had a higher Ea of 0.99 eV (Fig. S32†) with respect to the TMB oxidation, rendering it a minor reaction pathway. Notably, although the oxidation of TMB and the second H2O2 (Fig. S33†) on the CoO intermediates were also allowed in terms of thermodynamics, the overall activity of the Co–N–C was practically inert, owing to substantial restriction in the first CoO formation step.
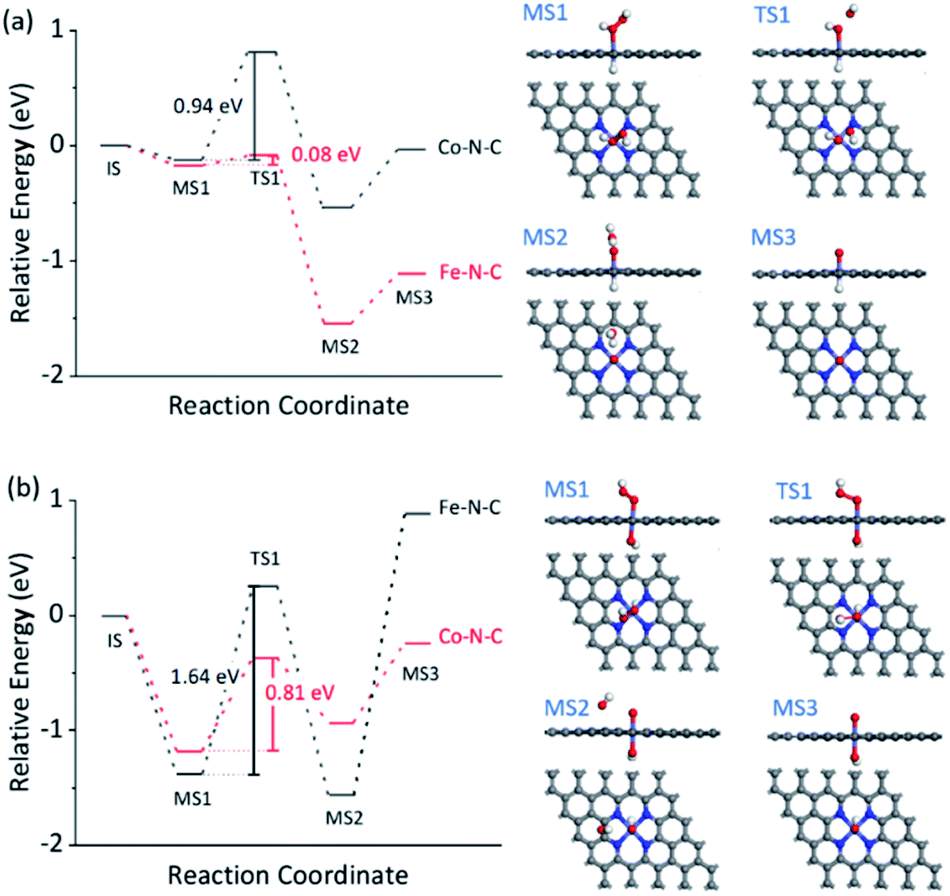 | ||
| Fig. 4 DFT calculations for M–N–C (M = Fe, Co) for selective oxidation with H2O2. The free energy diagram of H2O2 activation for TMB oxidation (a) and HO2− activation for luminol oxidation (b) on Fe–N–C and Co–N–C. The right panels in (a) and (b) show the corresponding structures of the intermediate (MS) and transition (TS) states. The white, grey, blue, red, and cyan balls represent H, C, N, O, and M atoms, respectively. | ||
The reaction mechanism for luminol oxidation with H2O2 on Fe–N–C and Co–N–C was also explored. The Eads values for HO2− and Ea values for MO formation over Fe–N–C/Co–N–C under alkaline conditions were −1.38 and −1.18 eV, and 1.64 and 0.81 eV, respectively (Fig. 4b, S34 and S35†). It was strongly suggested that under alkaline conditions, Co–N–C was more conducive to activating HO2− than Fe–N–C to generate the critically active MO intermediates. The adsorption of luminol on CoO (Fig. S36†) in the next step (Eads = −0.45 eV) was further calculated, which was slightly favored compared to that on the FeO species (Eads = −0.27 eV). The charge density difference of luminol adsorption on Fe–N–C/Co–N–C was calculated to evaluate the electron transfer between luminol and M–N–C. As shown in Fig. S37,† the delocalization of electrons from luminol to CoO was evident, but that to FeO was negligible. Therefore, the lower Ea for the formation of CoO, the greater Eads of luminol on CoO, and the easier the electron transfer between luminol and the CoO intermediates combined to make the catalytic oxidation activity of luminol with H2O2 by Co–N–C much more profound than Fe–N–C.
According to these considerations, a possible mechanism for the reaction selectivity by M–N–C nanozymes was proposed. Briefly, H2O2 was first bound to the N coordinated FeIII in Fe–N–C to form the FeIII-superoxo species (Fig. 5a). Then, O–O cleavage of H2O2 occurred to release a water molecule upon the oxidation of FeIII to FeVO. Whether FeVO was reduced by a second H2O2 to form O2 or TMB to form TMBox was determined by the affinity of TMB on FeVO and the electron-donor ability of TMB. Similarly, in the Co–N–C catalytic redox reaction (Fig. 5b), HO2− first bound to the N coordinated CoII in Co–N–C, creating a CoIII-superoxo species. Then, O–O cleavage of HO2− occurred to release a OH− with the oxidation of CoII to CoIVO. Lastly, whether CoIVO was reduced by a second HO2− to form O2 or luminol to develop an oxidized product relied on the affinity and reducing ability of luminol. In addition, the superoxide radical was experimentally observed (Fig. S38†) and theoretically verified (Fig. 4b) after CoIVO formation in the Co–N–C catalyzed oxidation of luminol with H2O2, consistent with previous reports (Fig. S39†).66 Therefore, in the bound ROS pathway, the ability of M–N to produce active MO intermediates from H2O2 and the reactivity of the substrates at the MO sites jointly determined the selectivity in the oxidation reactions.
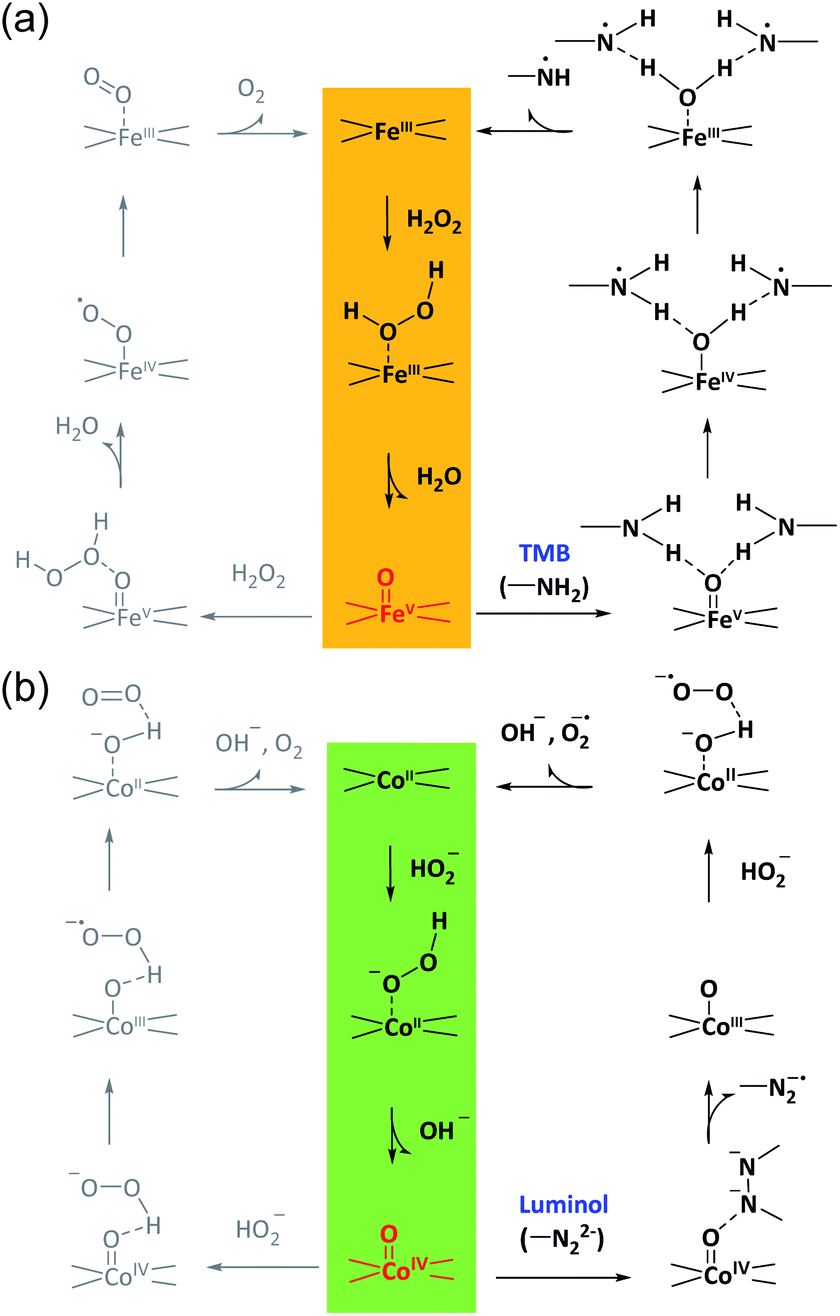 | ||
| Fig. 5 The proposed bound ROS mechanism for M–N–C nanozymes. The proposed reaction pathways for the oxidation of TMB in acid solution and luminol in alkaline solution with H2O2 over Fe–N–C (a) and Co–N–C (b), respectively. | ||
It should be noted that HRP also adopts the bound MO intermediates as the operation principle for the highly selective activation of H2O2.62,63 Nonetheless, because of the oxoiron-(oxidized porphyrin radical) species and Phe41, HRP is not specialized to oxidize a specific substrate; its natural substrates include aromatic amines, phenols, indoles, and sulfonates, owing to millions of years of biological evolution.67 As such, HRP can catalyze the oxidation of both TMB and luminol under acidic and alkaline conditions, respectively. In this sense, owing to the unique embedded ligands in the rigid graphitic framework, the adaptive adjustment of the M–N coordination sites in M–N–C might not occur in a similar manner to that of HRP in a biological system, which in turn, surprisingly enables M–N–C to exhibit a superior reaction selectivity in the catalytic oxidation of TMB and luminol. Moreover, except for MO, for future development, other types of bound intermediates, such as M–H68 and M–COO69 that enable substrates to interact with nanozymes via different absorption and desorption processes, are also applicable. In addition, in this work, to highlight the major role of metals, a series of M–N–C were prepared using following the same recipe. As M–N–C is a large family of doped nanocarbon matters, the M–N centers could be further modulated using an engineered method for more refined structures, such as the specific coordination number and ligands, with an even higher selectivity.
Conclusions
In summary, we propose M–N–C as a peroxidase-mimic platform to improve the substrate selectivity via forming a critically bound MO intermediate with variable interactions with substrates according to the type of metal. As an example, thanks to the unique d orbitals in the transition metal-N interaction, Fe–N–C and Co–N–C demonstrated opposite selectivities of up to 200-fold in catalyzing the oxidation of TMB and luminol with H2O2, respectively. Moreover, the selectivity of M–N–C was even superior to natural HRP, presumably owing to the embedded ligands in the rigid graphitic framework. Both experiments and DFT calculations based on simplified models proved that the M–N center was the origin of the disparate types of peroxidase-like reactions as a result of the intrinsic differences in activating H2O2 and generating essential MO intermediates for oxygen-atom transfer. The consequent interaction of the reductant substrates at the MO active site with different affinities and electron-donating abilities of substrates also significantly determines the overall reaction selectivity. Notably, the route of such bound MO intermediates is completely different from previously reported free ROS intermediate pathways for most peroxidase-like nanozymes. Going beyond conventional free-intermediate pathways, the newly developed bound intermediate pathway for oxygen-atom transfer could open a novel avenue for the development of the next generation of nanozymes with high substrate selectivity for future, more demanding, applications. Learning principles from biology, this work also highlights the significant potential of biomimetic bound-intermediates in endowing nanozymes with high reaction selectivity towards industrial reactions that have previously not been accessible in biology. Work focused on more comprehensive DFT calculations regarding the work functions of carbon/N-doped carbon and full water structural simulations of the reactions is ongoing.
Author contributions
Y. Z. and X. C. conceived and designed the experiments. X. C. performed the synthesis, characterization, activity evaluation, and DFT calculations of the catalysts. L. Z., K. W., Q. Z. and Y. X tested the chemiluminescence. All authors contributed to the analysis and discussion of the results. X. C., H. Y., Y. Z., Y. S., S. L., and Y. Z. co-wrote the manuscript. All authors reviewed the manuscript.Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21775018, 21675022), the Open Funds of the State Key Laboratory of Electroanalytical Chemistry (SKLEAC201909), and the Fundamental Research Funds for the Central Universities. We thank Prof. Haibo Ma and Ms Yaping Wen (Nanjing University) for discussing the reaction mechanism. We also thank Dr Pengfei An (Institute of High Energy Physics, Chinese Academy of Science) for assistance with testing and analyzing the X-ray adsorption fine structure spectra.Notes and references
- J. G. Chen, R. M. Crooks, L. C. Seefeldt, K. L. Bren, R. M. Bullock, M. Y. Darensbourg, P. L. Holland, B. Hoffman, M. J. Janik, A. K. Jones, M. G. Kanatzidis, P. King, K. M. Lancaster, S. V. Lymar, P. Pfromm, W. F. Schneider and R. R. Schrock, Science, 2018, 360, 873 CAS.
- S. Y. Lee, H. U. Kim, T. U. Chae, J. S. Cho, J. W. Kim, J. H. Shin, D. I. Kim, Y. S. Ko, W. D. Jang and Y. S. Jang, Nat. Catal., 2019, 2, 18–33 CrossRef CAS.
- L. Z. Gao, J. Zhuang, L. Nie, J. B. Zhang, Y. Zhang, N. Gu, T. H. Wang, J. Feng, D. L. Yang, S. Perrett and X. Yan, Nat. Nanotechnol., 2007, 2, 577–583 CrossRef CAS PubMed.
- A. Asati, S. Santra, C. Kaittanis, S. Nath and J. M. Perez, Angew. Chem., Int. Ed., 2009, 48, 2308–2312 CrossRef CAS PubMed.
- M. A. Komkova, E. E. Karyakina and A. A. Karyakin, J. Am. Chem. Soc., 2018, 140, 11302–11307 CrossRef CAS PubMed.
- K. L. Fan, J. Q. Xi, L. Fan, P. X. Wang, C. H. Zhu, Y. Tang, X. D. Xu, M. M. Liang, B. Jiang, X. Y. Yan and L. Z. Gao, Nat. Commun., 2018, 9, 1440 CrossRef PubMed.
- N. Singh, M. A. Savanur, S. Srivastava, P. D'Silva and G. Mugesh, Angew. Chem., Int. Ed., 2017, 56, 14267–14271 CrossRef CAS PubMed.
- L. Jiao, H. Y. Yan, Y. Wu, W. L. Gu, C. Z. Zhu, D. Du and Y. H. Lin, Angew. Chem., Int. Ed., 2020, 59, 2565–2576 CrossRef CAS PubMed.
- J. P. Chen, S. Patil, S. Seal and J. F. McGinnis, Nat. Nanotechnol., 2006, 1, 142–150 CrossRef CAS PubMed.
- Y. F. Liu, Y. Cheng, H. Zhang, M. Zhou, Y. J. Yu, S. C. Lin, B. Jiang, X. Z. Zhao, L. Y. Miao, C. W. Wei, Q. Y. Liu, Y. W. Lin, Y. Du, C. J. Butch and H. Wei, Sci. Adv., 2020, 6, eabb2695 CrossRef CAS PubMed.
- B. L. Xu, H. Wang, W. W. Wang, L. Z. Gao, S. S. Li, X. T. Pan, H. Y. Wang, H. L. Yang, X. Q. Meng, Q. W. Wu, L. R. Zheng, S. M. Chen, X. H. Shi, K. L. Fan, X. Y. Yan and H. Y. Liu, Angew. Chem., Int. Ed., 2019, 58, 4911–4916 CrossRef CAS PubMed.
- Y. J. Sang, F. F. Cao, W. Li, L. Zhang, Y. W. You, Q. Q. Deng, K. Dong, J. S. Ren and X. G. Qu, J. Am. Chem. Soc., 2020, 142, 5177–5183 CrossRef CAS PubMed.
- W. Y. Zhen, Y. Liu, W. Wang, M. C. Zhang, W. X. Hu, X. D. Jia, C. Wang and X. E. Jiang, Angew. Chem., Int. Ed., 2020, 59, 9491–9497 CrossRef CAS PubMed.
- Z. J. Zhang, X. H. Zhang, B. W. Liu and J. W. Liu, J. Am. Chem. Soc., 2017, 139, 5412–5419 CrossRef CAS PubMed.
- D. D. Wang, H. H. Wu, S. Z. F. Phua, G. B. Yang, W. Q. Lim, L. Gu, C. Qian, H. B. Wang, Z. Guo, H. Z. Chen and Y. L. Zhao, Nat. Commun., 2020, 11, 357 CrossRef CAS PubMed.
- F. F. Cao, L. Zhang, Y. W. You, L. R. Zheng, J. S. Ren and X. G. Qu, Angew. Chem., Int. Ed., 2020, 59, 5108–5115 CrossRef CAS PubMed.
- X. M. Shen, W. Q. Liu, X. J. Gao, Z. H. Lu, X. C. Wu and X. F. Gao, J. Am. Chem. Soc., 2015, 137, 15882–15891 CrossRef CAS PubMed.
- J. N. Li, W. Q. Liu, X. C. Wu and X. F. Gao, Biomater, 2015, 48, 37–44 CrossRef CAS PubMed.
- L. Z. Gao, K. L. Fan and X. Y. Yan, Theranostics, 2017, 7, 3207–3227 CrossRef CAS PubMed.
- D. W. Jiang, D. L. Ni, Z. T. Rosenkrans, P. Huang, X. Y. Yan and W. B. Cai, Chem. Soc. Rev., 2019, 48, 3683–3704 RSC.
- Y. Y. Huang, J. S. Ren and X. G. Qu, Chem. Rev., 2019, 119, 4357–4412 CrossRef CAS PubMed.
- W. W. Wu, L. Huang, E. K. Wang and S. J. Dong, Chem. Sci., 2020, 11, 9741–9756 RSC.
- H. B. Zhang, X. F. Lu, Z. P. Wu and X. W. D. Lou, ACS Cent. Sci., 2020, 6, 1288–1301 CrossRef CAS PubMed.
- M. H. Li, J. X. Chen, W. W. Wu, Y. X. Fang and S. J. Dong, J. Am. Chem. Soc., 2020, 142, 15569–15574 CrossRef CAS PubMed.
- Y. Wang, G. R. Jia, X. Q. Cui, X. Zhao, Q. H. Zhang, L. Gu, L. R. Zheng, L. H. Li, Q. Wu, D. J. Singh, D. Matsumura, T. Tsuji, Y. T. Cui, J. X. Zhao and W. T. Zheng, Chem, 2021, 7, 436–449 CAS.
- Y. Zhou, H. J. Sun, H. C. Xu, S. Matysiak, J. S. Ren and X. G. Qu, Angew. Chem., Int. Ed., 2018, 57, 16791–16795 CrossRef CAS PubMed.
- M. M. Ma, Z. Q. Liu, N. Gao, Z. F. Pi, X. B. Du, J. S. Ren and X. G. Qu, J. Am. Chem. Soc., 2020, 142, 21702–21711 CrossRef CAS PubMed.
- H. T. Chung, D. A. Cullen, D. Higgins, B. T. Sneed, E. F. Holby, K. L. More and P. Zelenay, Science, 2017, 357, 479–483 CrossRef CAS PubMed.
- J. Gu, C. S. Hsu, L. C. Bai, H. M. Chen and X. L. Hu, Science, 2019, 364, 1091 CrossRef CAS PubMed.
- Y. Wang, X. Q. Cui, J. X. Zhao, G. R. Jia, L. Gu, Q. H. Zhang, L. K. Meng, Z. Shi, L. R. Zheng, C. Y. Wang, Z. W. Zhang and W. T. Zheng, ACS Catal., 2019, 9, 336–344 CrossRef CAS.
- T. Al-Zoubi, Y. Zhou, X. Yin, B. Janicek, C. J. Sun, C. E. Schulz, X. H. Zhang, A. A. Gewirth, P. Huang, P. Zelenay and H. Yang, J. Am. Chem. Soc., 2020, 142, 5477–5481 CrossRef CAS PubMed.
- A. Zitolo, V. Goellner, V. Armel, M. T. Sougrati, T. Mineva, L. Stievano, E. Fonda and F. Jaouen, Nat. Mater., 2015, 14, 937 CrossRef CAS PubMed.
- W. Wang, Q. Y. Jia, S. Mukerjee and S. L. Chen, ACS Catal., 2019, 9, 10126–10141 CrossRef CAS.
- S. Wagner, H. Auerbach, C. E. Tait, I. Martinaiou, S. C. N. Kumar, C. Kubel, I. Sergeev, H. C. Wille, J. Behrends, J. A. Wolny, V. Schunemann and U. I. Kramm, Angew. Chem., Int. Ed., 2019, 58, 10486–10492 CrossRef CAS PubMed.
- N. R. Sahraie, U. I. Kramm, J. Steinberg, Y. J. Zhang, A. Thomas, T. Reier, J. P. Paraknowitsch and P. Strasser, Nat. Commun., 2015, 6, 8618 CrossRef CAS.
- F. He, L. Mi, Y. F. Shen, T. Mori, S. Q. Liu and Y. J. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 35327–35333 CrossRef CAS.
- L. Jiao, W. Q. Xu, H. Y. Yan, Y. Wu, C. R. Liu, D. Du, Y. H. Lin and C. Z. Zhu, Anal. Chem., 2019, 91, 11994–11999 CrossRef CAS PubMed.
- Y. Wang, Z. W. Zhang, G. R. Jia, L. R. Zheng, J. X. Zhao and X. Q. Cui, Chem. Commun., 2019, 55, 5271–5274 RSC.
- L. Huang, J. X. Chen, L. F. Gan, J. Wang and S. J. Dong, Sci. Adv., 2019, 5, eaav5490 CrossRef CAS PubMed.
- M. J. Lu, C. Wang, Y. Q. Ding, M. H. Peng, W. Zhang, K. Li, W. Wei and Y. Q. Lin, Chem. Commun., 2019, 55, 14534–14537 RSC.
- M. S. Kim, J. Lee, H. S. Kim, A. Cho, K. H. Shim, T. N. Le, S. S. A. An, J. W. Han, M. I. Kim and J. Lee, Adv. Funct. Mater., 2020, 30, 1905410 CrossRef CAS.
- Y. Xu, J. Xue, Q. Zhou, Y. J. Zheng, X. H. Chen, S. Q. Liu, Y. F. Shen and Y. J. Zhang, Angew. Chem., Int. Ed., 2020, 59, 14498–14503 CrossRef CAS PubMed.
- C. Zhao, C. Xiong, X. K. Liu, M. Qiao, Z. J. Li, T. W. Yuan, J. Wang, Y. T. Qu, X. Q. Wang, F. Y. Zhou, Q. Xu, S. Q. Wang, M. Chen, W. Y. Wang, Y. F. Li, T. Yao, Y. E. Wu and Y. D. Li, Chem. Commun., 2019, 55, 2285–2288 RSC.
- L. Que and W. B. Tolman, Nature, 2008, 455, 333–340 CrossRef CAS PubMed.
- S. Bang, Y. M. Lee, S. Hong, K. B. Cho, Y. Nishida, M. S. Seo, R. Sarangi, S. Fukuzumi and W. Nam, Nat. Chem., 2014, 6, 934–940 CrossRef CAS PubMed.
- C. E. Valdez, Q. A. Smith, M. R. Nechay and A. N. Alexandrova, Acc. Chem. Res., 2014, 47, 3110–3117 CrossRef CAS PubMed.
- M. Antonietti and P. Fratzl, Macromol. Chem. Phys., 2010, 211, 166–170 CrossRef CAS.
- Y. Y. Sun, L. Silvioli, N. R. Sahraie, W. Ju, J. K. Li, A. Zitolo, S. Li, A. Bagger, L. Arnarson, X. L. Wang, T. Moeller, D. Bernsmeier, J. Rossmeisl, F. Jaouen and P. Strasser, J. Am. Chem. Soc., 2019, 141, 12372–12381 CrossRef CAS PubMed.
- K. Artyushkova, A. Serov, S. Rojas-Carbonell and P. Atanassov, J. Phys. Chem. C, 2015, 119, 25917–25928 CrossRef CAS.
- J. T. Zhang, Z. H. Zhao, Z. H. Xia and L. M. Dai, Nat. Nanotechnol., 2015, 10, 444–452 CrossRef CAS PubMed.
- G. J. Guan, L. Yang, Q. S. Mei, K. Zhang, Z. P. Zhang and M. Y. Han, Anal. Chem., 2012, 84, 9492–9497 CrossRef CAS PubMed.
- B. Jiang, D. M. Duan, L. Z. Gao, M. J. Zhou, K. L. Fan, Y. Tang, J. Q. Xi, Y. H. Bi, Z. Tong, G. F. Gao, N. Xie, A. Tango, G. H. Nie, M. M. Liang and X. Y. Yan, Nat. Protoc., 2018, 13, 1506–1520 CrossRef CAS PubMed.
- G. H. G. Thorpe and L. J. Kricka, Methods Enzymol., 1986, 133, 331–353 CAS.
- C. X. Zhao, H. B. Cui, J. Duan, S. H. Zhang and J. G. Lv, Anal. Chem., 2018, 90, 2201–2209 CrossRef CAS PubMed.
- Y. H. Hu, X. J. J. Gao, Y. Y. Zhu, F. Muhammad, S. H. Tan, W. Cao, S. C. Lin, Z. Jin, X. F. Gao and H. Wei, Chem. Mater., 2018, 30, 6431–6439 CrossRef CAS.
- M. Lucic, D. A. Svistunenko, M. T. Wilson, A. K. Chaplin, B. Davy, A. Ebrahim, D. Axford, T. Tosha, H. Sugimoto, S. Owada, F. S. N. Dworkowski, I. Tews, R. L. Owen, M. A. Hough and J. A. R. Worrall, Angew. Chem., Int. Ed., 2020, 59, 21656–21662 CrossRef CAS PubMed.
- M. Guo, T. Corona, K. Ray and W. Nam, ACS Cent. Sci., 2019, 5, 13–28 CrossRef CAS PubMed.
- E. L. Onderko, A. Silakov, T. H. Yosca and M. T. Green, Nat. Chem., 2017, 9, 623–628 CrossRef CAS PubMed.
- Y. Pan, Y. J. Chen, K. L. Wu, Z. Chen, S. J. Liu, X. Cao, W. O. Cheong, T. Meng, J. Luo, L. R. Zheng, C. G. Liu, D. S. Wang, Q. Peng, J. Li and C. Chen, Nat. Commun., 2019, 10, 4290 CrossRef PubMed.
- W. G. Liu, L. L. Zhang, X. Liu, X. Y. Liu, X. F. Yang, S. Miao, W. T. Wang, A. Q. Wang and T. Zhang, J. Am. Chem. Soc., 2017, 139, 10790–10798 CrossRef CAS PubMed.
- J. Cho, S. Jeon, S. A. Wilson, L. V. Liu, E. A. Kang, J. J. Braymer, M. H. Lim, B. Hedman, K. O. Hodgson, J. S. Valentine, E. I. Solomon and W. Nam, Nature, 2011, 478, 502–505 CrossRef CAS PubMed.
- J. N. Rodriguez-Lopez, D. J. Lowe, J. Hernandez-Ruiz, A. N. P. Hiner, F. Garcia-Canovas and R. N. F. Thorneley, J. Am. Chem. Soc., 2001, 123, 11838–11847 CrossRef CAS PubMed.
- M. Wirstam, M. R. A. Blomberg and P. E. M. Siegbahn, J. Am. Chem. Soc., 1999, 121, 10178–10185 CrossRef CAS.
- T. Marshall-Roth, N. J. Libretto, A. T. Wrobel, K. J. Anderton, M. L. Pegis, N. D. Ricke, T. Van Voorhis, J. T. Miller and Y. Surendranath, Nat. Commun., 2020, 11, 5283 CrossRef CAS PubMed.
- W. Ju, A. Bagger, G. P. Hao, A. S. Varela, I. Sinev, V. Bon, B. Roldan Cuenya, S. Kaskel, J. Rossmeisl and P. Strasser, Nat. Commun., 2017, 8, 944 CrossRef PubMed.
- M. Mayer, S. Takegami, M. Neumeier, S. Rink, A. Jacobi von Wangelin, S. Schulte, M. Vollmer, A. G. Griesbeck, A. Duerkop and A. J. Baeumner, Angew. Chem., Int. Ed., 2018, 57, 408–411 CrossRef CAS PubMed.
- A. D. Ryabov, Adv. Inorg. Chem., 2004, 55, 201–269 CrossRef CAS.
- L. Kuai, Z. Chen, S. J. Liu, E. J. Kan, N. Yu, Y. M. Ren, C. H. Fang, X. Y. Li, Y. D. Li and B. Y. Geng, Nat. Commun., 2020, 11, 48 CrossRef PubMed.
- Z. Li, Y. J. Chen, S. F. Ji, Y. Tang, W. X. Chen, A. Li, J. Zhao, Y. Xiong, Y. E. Wu, Y. Gong, T. Yao, W. Liu, L. R. Zheng, J. C. Dong, Y. Wang, Z. B. Zhuang, W. Xing, C. T. He, C. Peng, W. C. Cheong, Q. H. Li, M. L. Zhang, Z. Chen, N. H. Fu, X. Gao, W. Zhu, J. W. Wan, J. Zhang, L. Gu, S. Q. Wei, P. J. Hu, J. Luo, J. Li, C. Chen, Q. Peng, X. F. Duan, Y. Huang, X. M. Chen, D. S. Wang and Y. D. Li, Nat. Chem., 2020, 12, 764–772 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc02170b |
| This journal is © The Royal Society of Chemistry 2021 |