
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvometallurgical recovery of cobalt from lithium-ion battery cathode materials using deep-eutectic solvents†
Nand
Peeters
 ,
Koen
Binnemans
and
Sofía
Riaño
*
,
Koen
Binnemans
and
Sofía
Riaño
*
KU Leuven, Department of Chemistry, Celestijnenlaan 200F, P.O. box 2404, B-3001 Leuven, Belgium. E-mail: Sofia.Riano@kuleuven.be
First published on 9th June 2020
Abstract
Recycling of cobalt from end-of-life lithium-ion batteries (LIBs) is gaining interest because they are increasingly used in commercial applications such as electrical vehicles. A common LIB cathode material is lithium cobalt oxide (LiCoO2). Besides the cathode, LIBs contain other components, such as metallic aluminium and copper as current collectors, which are often separated at initial hydrometallurgical recycling stages. Leaching of cobalt from LiCoO2 is mainly driven by reducing cobalt(III) in LiCoO2 to cobalt(II) via adding reducing agents. In this work, a green, cheap and safe approach is proposed by using a choline chloride–citric acid deep-eutectic solvent (DES) as lixiviant. Aluminium and copper were evaluated as reducing agents for cobalt(III). After optimisation, lithium and cobalt were quantitatively leached from LiCoO2 in the presence of aluminium and copper. Copper was the most effective reducing agent for cobalt(III), so that no additional reducing agents or a pre-separation step were required. A speciation study of the pregnant leach solution (PLS) confirmed the dominance of chloro complexes. DES leaching was compared with conventional hydrochloric acid leaching, whereby the DES avoided the formation of toxic chlorine gas. Finally, the DES PLS was used as the more polar phase in a non-aqueous solvent extraction process. This process consisted of a copper(I/II) extraction step with the extractant LIX 984, followed by selective extraction of cobalt(II) with the extractant Aliquat 336. Both metals were completely stripped from the loaded organic phases by oxalic acid. The total recovery yield of cobalt was 81%, as a 99.9% pure oxalate precipitate.
Introduction
Lithium-ion batteries (LIBs) are on the market since the early 1990s.1,2 The use of LIBs in electric devices has been increasing sharply during the least 20 years due to the advantages of LIBs compared to other rechargeable batteries, such as nickel metal hydride batteries (NMH batteries).3–7 End-of-life LIBs are worth being recycled because of the presence of aluminium and copper as current collectors, but especially because of the cobalt in the cathode materials.2 Furthermore, apart from economic perspectives, recycling of LIBs is also important for environmental reasons. This is because the releasement of certain metals and electrolytes that are present in the LIBs could be environmentally harmful.2,7 Popular cobalt-containing cathode materials are lithium cobalt oxide (LiCoO2) and mixed nickel manganese cobalt oxide.2,5,8–10 Since cobalt is a critical metal, development of efficient recycling processes for recovery of cobalt from end-of-life LIBs becomes important and could decrease the environmental impact of mining and dependence of geopolitical unstable countries.11–15Hydrometallurgical recycling routes comprise dismantling of the LIBs and separation of the main components as the first processing steps, followed by thermal treatment and/or dissolution steps to recover cobalt and other valuable metals.16–18 Other industrial processes use first a thermal treatment step, followed by mechanical separation. This is cheaper, but less pure cathode materials are obtained.19,20 In both types of recycling processes, the obtained pretreated material is leached, usually by mineral acids, to dissolve the metals and cobalt is recovered by solvent extraction (SX) or precipitation.1 Efficient leaching of cobalt from cathode materials such as LiCoO2 is achieved by reducing cobalt(III) to the more soluble cobalt(II) oxidation state.21–23 Often used lixiviants are (1) sulfuric acid in the presence of hydrogen peroxide as reducing agent or (2) hydrochloric acid.2,5,16,24 Whereby leaching with hydrochloric acid releases toxic chlorine gas. Huang et al.1 and Chagnes et al.25 compared leaching of LIB cathode materials with different mineral acids and reducing agents. Chen et al.26 described the use of organic acids and organic reducing agents as milder lixiviant systems. The use of LIB current collectors as reducing agents during leaching of cathode materials is less investigated. Joulié et al.10 illustrated that aluminium or copper separately are suitable reducing agents for cobalt(III), without describing the combined effect of both metals on LIB leaching. Peng et al.27 included the combined effect since it mimics a more realistic scenario due to the fact that industrial LIB recycling routes usually cannot obtain cathode materials that solely contain aluminium or copper. Nevertheless, these two studies describe conventional hydrometallurgical leaching based on aqueous solutions.10,27
Another possibility for recovery of cobalt from LIB cathode materials is applying solvometallurgy, where the aqueous phase used in hydrometallurgical processes is replaced by organic solvents.28 Solvometallurgical routes produce less aqueous waste and are often more selective than hydrometallurgical processes.28–31 Often used green solvents in solvometallurgy are deep-eutectic solvents (DESs).32–34 DESs are low-melting mixtures with a melting point much lower than those of its individual components. They are composed of an organic salt such as choline chloride and a hydrogen bond donor such as citric acid.35–37 Synthesis of DESs is generally straightforward and advantageous properties like biodegradability, safety, greenness and low price can be obtained by a proper selection of the DES components.35,38–40 Moreover, DESs can be potentially used as lixiviants in leaching processes and polar organic phases in non-aqueous solvent extraction processes.28 The DES could then avoid emission of toxic and environmentally harmful gasses such as chlorine gas, nitrogen- or sulfur oxides, which are commonly produced during leaching by mineral acids.22,26,28
For example, Zürner et al.41 used a choline chloride–oxalic acid DES for the leaching and selective extraction of indium(III) and tin(II) from metallurgical waste. Foreman developed a DES-based process for recovery of metals from NMH batteries.42 Riaño et al.43 applied a choline chloride–lactic acid DES for NdFeB magnets recycling, utilizing leaching and non-aqueous solvent extraction. In this paper, a model study is done to recover cobalt from LiCoO2 by solvometallurgy, utilizing a water-diluted choline chloride–citric acid DES (ChCl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CA, molar ratio 1:2, diluted with 35 wt% water). Choline chloride is an inexpensive chicken feed additive, produced at a large scale.40,44,45 Citric acid is a renewable, non-toxic and relatively cheap organic acid, produced at large scale as well.46,47 The choline chloride–citric acid DES is used as a lixiviant and more polar phase in non-aqueous solvent extraction (non-aqueous SX). Leaching by this DES is compared with leaching by hydrochloric acid, whereby the DES avoids the emission of toxic chlorine gas, which is produced when hydrochloric acid is used as lixiviant. Furthermore, the current collectors aluminium and copper are used in a combined way as reducing agents for cobalt(III) during leaching, in order to avoid additional reducing agents and thus to simplify the leaching process. The chosen citric acid and choline chloride content in the DES caused that no pH or chloride concentration adjustments were necessary in the downstream non-aqueous SX process. These adjustments are usually done in a conventional solvent extraction process when a pregnant leach solution (PLS) (after leaching by mineral acids) is extracted by LIX 984 and Aliquat 336 (A336) respectively.
:CA, molar ratio 1:2, diluted with 35 wt% water). Choline chloride is an inexpensive chicken feed additive, produced at a large scale.40,44,45 Citric acid is a renewable, non-toxic and relatively cheap organic acid, produced at large scale as well.46,47 The choline chloride–citric acid DES is used as a lixiviant and more polar phase in non-aqueous solvent extraction (non-aqueous SX). Leaching by this DES is compared with leaching by hydrochloric acid, whereby the DES avoids the emission of toxic chlorine gas, which is produced when hydrochloric acid is used as lixiviant. Furthermore, the current collectors aluminium and copper are used in a combined way as reducing agents for cobalt(III) during leaching, in order to avoid additional reducing agents and thus to simplify the leaching process. The chosen citric acid and choline chloride content in the DES caused that no pH or chloride concentration adjustments were necessary in the downstream non-aqueous SX process. These adjustments are usually done in a conventional solvent extraction process when a pregnant leach solution (PLS) (after leaching by mineral acids) is extracted by LIX 984 and Aliquat 336 (A336) respectively.
Experimental
Products
Choline chloride (99%), L(+)-ascorbic acid (99%), aluminium powder (0.075 mm, 99%), cobalt(II) chloride anhydrous (97%), ethylene glycol (99.5%), L(−)-malic acid (99%), copper(I) chloride (97%) and copper(II) chloride (anhydrous, 95%) and ethanol (99%) were purchased from Acros Organics (Geel, Belgium). Aliquat® 336 (a mixture of quaternary ammonium chlorides, with 88.2–90.6% quaternary ammonium content) and copper powder (0.075 mm, 99%) were obtained from Sigma-Aldrich (Overijse, Belgium). Citric acid (anhydrous, 99.5%) was purchased from Carl Roth® (Karlsruhe, Germany). Hydrochloric acid (37%) was purchased from VWR Chemicals (Heverlee, Belgium). Lithium cobalt oxide (LiCoO2, 0.005 mm, 97%) was purchased from Alfa Aesar (Kandel, Germany). Nitric acid (65%) was obtained from Chemlab-Analytical (Zedelgem, Belgium). Oxalic acid (99%) was purchased from J&K Scientific (Pelt, Belgium). The aliphatic diluent Shell GTL GS190 (C10-C13 aliphatic hydrocarbon diluent) was obtained from Shell (Rotterdam, Netherlands). LIX® 984 (a mixture of 5-dodecylsalicylaldoxime and 2-hydroxy-5-nonylacetophenone oxime) was provided by BASF (Antwerp, Belgium). Malonic acid (99%) and ICP standards of aluminium, cobalt, lithium and scandium were obtained from Merck (Overijse, Belgium). All the chemicals were used as received without any further purification. Ultrapure water (18.2 MΩ cm, reference A+ Milli-Q water purification system) was prepared by a Milli-Q® Academic ultrapure water system.Instrumentation
Metal concentrations in the heavy DES phase were determined by inductively coupled plasma-optical emission spectroscopy (ICP-OES) using an Optima 8300 spectrometer equipped with an axial (AX)/radial (RAD) dual plasma view, a GemTip Cross-Flow II nebuliser, a Scott double pass with inert Ryton spray chamber and a demountable one-piece Hybrid XLT ceramic torch with a 2.0 mm internal diameter sapphire injector. Dilutions were done with 2 vol% nitric acid solutions and all ICP-OES sample analyses were performed in triplicate. Samples were diluted 2000 times and scandium was used as internal standard. Metal concentrations in the light phase were determined by using a total X-ray reflection fluorescence spectrometer (TXRF; Bruker S2 Picofox), equipped with a molybdenum X-ray source and operated at a voltage of 50 kV. The quartz glass sample carriers were first heated for half an hour at 60 °C in a hot air oven. Sample preparation was done by mixing 20 μL of loaded light phase together with 50 μL of gallium ICP standard and 930 μL of ethanol. Analysis was done by adding 3 μL of this prepared sample on the preheated carriers followed by drying 30 min at the same temperature. Qualitative and quantitative analysis of the PLS was done by UV/VIS absorption spectroscopy with a Agilent Cary 6000i spectrophotometer and Cary WinUV software. A Nemus Life Thermo Shaker TMS-200 was used for shaking the glass vials during the extraction experiments. A Heraeus Labofuge 200 centrifuge was used to accelerate phase separation after equilibration of the two phases.48 Detection of chlorine gas formed during leaching was done by a GasAlert extreme Cl2 0–50 ppm GAXT-C-DL chlorine detector.Synthesis deep-eutectic solvents
The choline chloride based DESs were prepared by adding choline chloride and an organic compound at a specific molar ratio (n:n) to a glass beaker. Subsequently, a certain mass percentage of water was added to the beaker, followed by addition of a magnetic stirring bar. The mixture was stirred at 30 °C on a heating plate, equipped with a thermocouple to control the temperature, until a homogeneous solution was obtained.
Leaching by deep-eutectic solvents
Leaching experiments were started by adding certain amounts of LiCO2 and/or aluminium and/or copper powders into a glass vial. The added amount of aluminium and copper metal was based on their average mass ratios in LIBs and kept constant, which was Al:LIB = 4 wt% (e.g. Al:LiCoO2 = 12 wt%) and Cu:LIB = 8 wt% (e.g. Cu:LiCoO2 = 24 wt%).5,49,50 This was accompanied by addition of a certain volume of DES, according to the desired solid-to-liquid ratio (S:L) and a magnetic stirring bar was added. The vial was sealed and then placed in a sand bath on a heating plate, equipped with a thermocouple for temperature control, at a stirring speed of 900 rpm. If necessary, intermediate samples of 350 μL were taken at certain time intervals and filtered with syringe filters (Chromafil® pore size 0.45 μm, diameter 25 mm). A constant DES volume of 0.04 L (maximum capacity of the vials used) and a varying amount of LiCoO2, Al and Cu according the chosen S:L were used for all tests where these intermediate samples were taken. This relatively high volume of DES was chosen to minimize the effect of volume losses during the intermediate sampling as much as possible. The PLS was separated from the solid residue via the same filtering procedure. All experiments were performed in duplicate. The percentage leaching (%L) was calculated via the following equation:14,27,51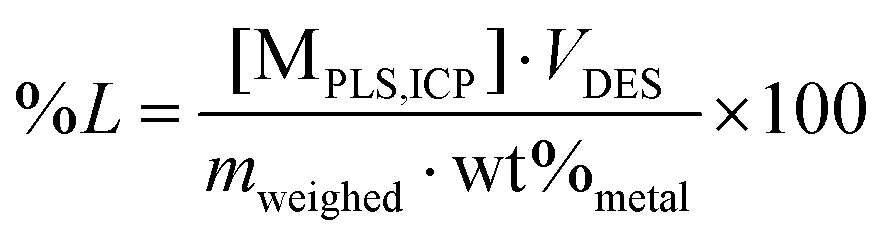 | (1) |
Non-aqueous solvent extraction
The PLS was obtained at optimised conditions and contacted with an organic light phase at an equal volume ratio (VLP:VDES) (unless stated otherwise) in a 4 mL glass vial and shaken at 2250 rpm during 0.5 h at room temperature. The vial was then centrifuged at 5000 rpm for 5 min and the metal content was analysed for both phases. ICP-OES is suitable for analysing water soluble samples and TXRF can be used to measure organic samples. Therefore, the DES phase was used for ICP-OES analysis and the loaded organic phases for TXRF analysis. Scrubbing and/or stripping experiments were both executed via similar procedure. All experiments were performed in duplicate. The percentage extraction (%E) was calculated via the following equation: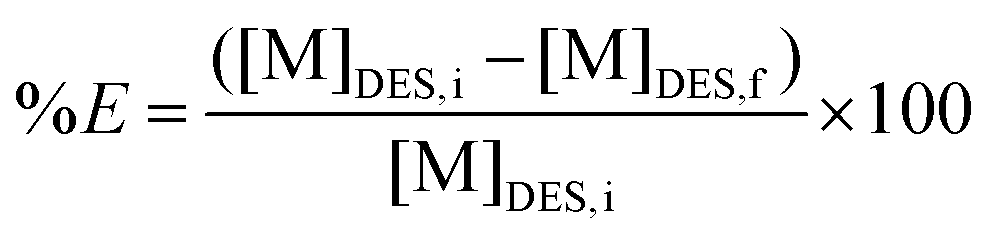 | (2) |
With [M]DES,i and [M]DES,f the respective initial and final metal concentration in the DES (heavy phase), both determined by ICP-OES.
Since stripping by forming a precipitate was used for metal recovery, metal stripping efficiencies were determined by calculating the precipitation efficiency (%PE). This was done via the following equation:
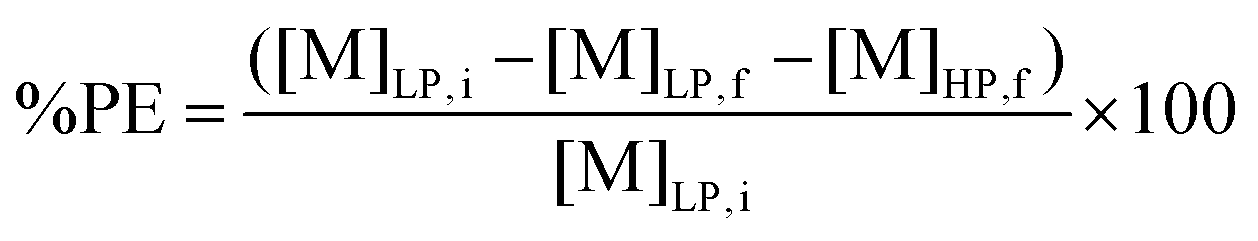 | (3) |
[M]LP,i and [M]LP,f are the metal concentrations in the light organic phase after extraction and after stripping, respectively. Both metal concentrations were determined by TXRF. [M]HP,f is the metal concentration of the aqueous supernatant (e.g. heavy phase) after stripping, and was determined by ICP-OES.
UV/VIS analysis
Copper(I)-, copper(II) and cobalt(II) chloride salts were dissolved in the DES with similar metal concentration as in the PLS, followed by recording the absorption spectra. These were hereafter compared with the recorded PLS spectrum for qualitative analysis. A calibration curve was constructed by dissolving different amounts of cobalt(II) chloride in a 35 wt% water-diluted choline chloride solution for cobalt(II) chloride quantification in the DES. The cobalt(II) concentration ranged between 0.005 and 0.625 g L−1.Results and discussion
Choice of lixiviant
Several deep-eutectic solvents (DESs) were synthesised by combining choline chloride with organic compounds such as citric acid, ethylene glycol, malic acid, malonic acid and oxalic acid in 1:1 molar ratios. The viscosity of these DESs was reduced by addition of 20 wt% water. The capacity of the DESs for leaching of cobalt from lithium cobalt oxide (LiCoO2), in presence of aluminium and copper metal, was evaluated (Table 1).
| DES (1:1 molar ratio) |
%L Co |
|---|---|
|
a Leaching parameters: 60 °C, S:L = 20 g L−1, Al:LiCoO2 = 12 wt%, Cu:LiCoO2 = 24 wt%, 4 h at 900 rpm. All DESs: 1:1 molar ratio and diluted with 20 wt% H2O.
b Formation of precipitate observed.
|
|
| Choline chloride–citric acid | 99.6 |
| Choline chloride–ethylene glycol | 2.1 |
| Choline chloride–malic acid | 81.2 |
| Choline chloride–malonic acidb | 24.4 |
| Choline chloride–oxalic acidb | 19.6 |
Only DESs comprising relatively strong organic acids combined with choline chloride were able to leach cobalt. Ethylene glycol, being the weakest acid considered in this study, could not leach cobalt.52 However, although malonic acid and oxalic acid have the highest acidity of the acids shown in Table 1, their conjugated bases form poorly soluble salts with cobalt(II).4,53–55 These acids form poorly soluble copper(II) salts as well, which coprecipitate with the cobalt(II) salts, making separation of cobalt from copper difficult.4,53,56,57 The choline chloride–citric acid DES (ChCl:CA) leached more cobalt(II) compared to the choline chloride–malic acid DES, due to the higher acidity of citric acid.52,54,55 Moreover, citric acid is also cheaper and more readily available than malic acid.47,58 However, bulky molecules, like citric acid, lead to a DES with higher viscosities, when combined with choline chloride.36 This undesired increase in viscosity was mitigated by changing the choline chloride:citric acid molar ratio from 1:1 to 2:1, combined with addition of 35 wt% of water. Hammond et al. confirmed that most DESs retain their typical network of hydrogen bonds up to a water content of ca. 42 wt%.59,60 Thus, a water content of 35 wt% still ensured that the mixture has the typical behaviour of a DES, and not of a diluted aqueous solution.59,60 The choline chloride–citric acid DES (ChCl:CA 2:1 +35 wt% H2O) could still leach cobalt quantitatively and was therefore considered as being the most suited for leaching of lithium cobalt oxide.
Time and temperature effect on leaching
First insights in the leaching mechanism were obtained by plotting the percentage of leaching (%L) as a function of time for different temperatures, during leaching of lithium cobalt oxide with the choline chloride–citric acid DES, in the presence of aluminium and copper metal (Fig. 1). | ||
| Fig. 1 Leaching efficiencies (%L) of LiCoO2 + Al + Cu mixtures in choline chloride–citric acid DES as a function of time at different temperatures. Temperatures: room temperature (left above, a), 40 °C (right above, b), 60 °C (left below, c) and 80 °C (right below, d). DES: ChCl:CA 2:1 +35 wt% H2O. Leaching parameters: S:L = 20 g L−1, Al:LiCoO2 = 12 wt%, Cu:LiCoO2 = 24 wt% at 900 rpm. | ||
Fig. 1 indicates that leaching of the metals is in general faster at higher temperatures. Cobalt, copper and lithium show a very similar behaviour, while dissolution of aluminium is slower. The efficiency of copper dissolution decreases over time at 80 °C, because of the cementation reaction with aluminium.61 Aluminium reduces copper(I/II) during cementation and gets oxidised to aluminium(III), and the reaction rate increased with increasing temperatures, as shown in Fig. 1(d).61,62 Although quantitative cobalt leaching could be achieved at room temperature after 4 h, the condition of 1 hour at 40 °C was chosen for further study. These conditions leached 38% aluminium(III), 98% cobalt(II), 94% copper(I/II) and 93% lithium(I) and ensured a good balance between selectivity, reaction time and energy consumption. The remaining solid residue after leaching thus contained an amount of aluminium metal with LiCoO2 and copper in low concentrations as impurities. This residue could be further processed by selectively leaching aluminium(III) with sodium hydroxide.63 Another approach is to leach the residue with a mineral acid such as hydrochloric acid, followed by quantitative and selective extraction of aluminium(III), cobalt(II), copper(I/II) and lithium(I) by acid extractants like D2EHPA or Cyanex 272 (C272).64,65 However, due to the very low amounts of metals present in the residue, the cost-effectiveness of this secondary waste stream should be taken into account.
Solid-to-liquid ratio optimisation
The PLS metal content influences the extraction behaviour in the subsequent non-aqueous SX step. Therefore, the solid-to-liquid ratio was optimised (Fig. 2).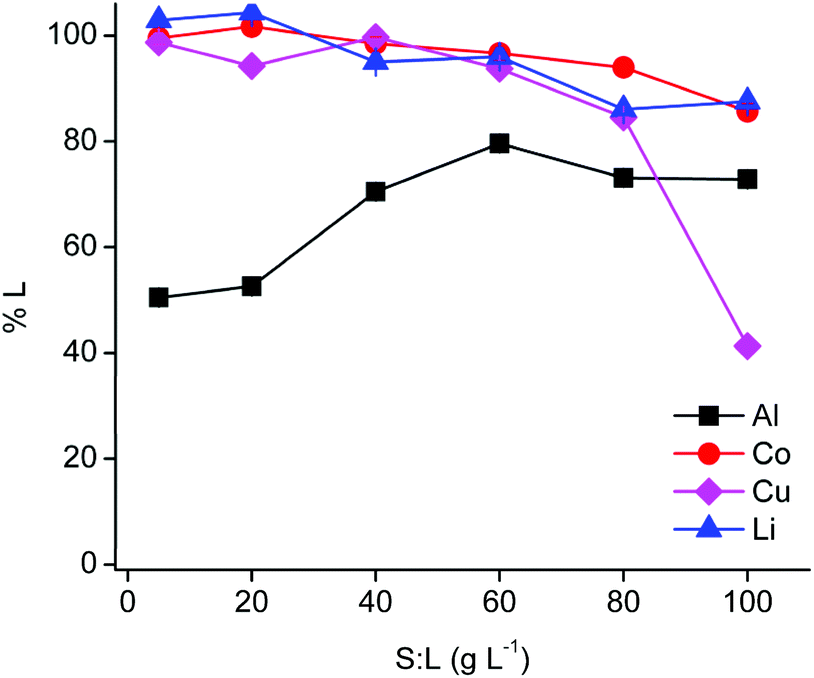 | ||
| Fig. 2 Variation of the solid-to-liquid ratio (S:L) during leaching by choline chloride–citric acid DES. Composition of the DES: ChCl:CA 2:1 +35 wt% H2O. Leaching parameters: 1 h at 40 °C, Al:LiCoO2 = 12 wt%, Cu:LiCoO2 = 24 wt% at 900 rpm. | ||
The efficiency of cobalt(II) and lithium(I) leaching decreased slightly with increasing solid-to-liquid ratios until a leaching efficiency of ca. 90% at 100 g L−1, while copper(I/II) leaching decreased dramatically at a solid-to-liquid ratio of 100 g L−1 due to precipitation. Aluminium(III) leaching increased up to a solid-to-liquid ratio of 60 g L−1 and remained constant for higher ratios. Cementation of copper(I/II) by aluminium metal is favoured at higher copper concentrations, which is most likely occurring at a solid-to-liquid ratio of 100 g L−1.62 Furthermore, a solid-to-liquid ratio of 20 g L−1 was selected as a suitable solid-to-liquid ratio because this ratio provides a higher selectivity and a reasonably high cobalt(II) concentration in the PLS of about 12 g L−1. Nevertheless, it should kept in mind that the DES is able to leach efficiently at higher solid-to-liquid ratios and that it is possible to obtain a PLS containing about 55 g L−1 cobalt(II). Unfortunately, these more extreme conditions cause a drastic increase in viscosity due to the high loading of the DES with metals.
Effect of current collector metals on leaching
The effect of aluminium and copper metal was examined by comparing four different leaching systems (Fig. 3). Lithium cobalt oxide was leached under the following conditions: (a) without addition of a reducing agent, (b) in the presence of aluminium metal, (c) in the presence of copper metal, (d) in the presence of both aluminium and copper metal. Condition (a) describes conventional leaching without addition of a reducing agent, which can in a general and simplified way described by the following equation:1,66,67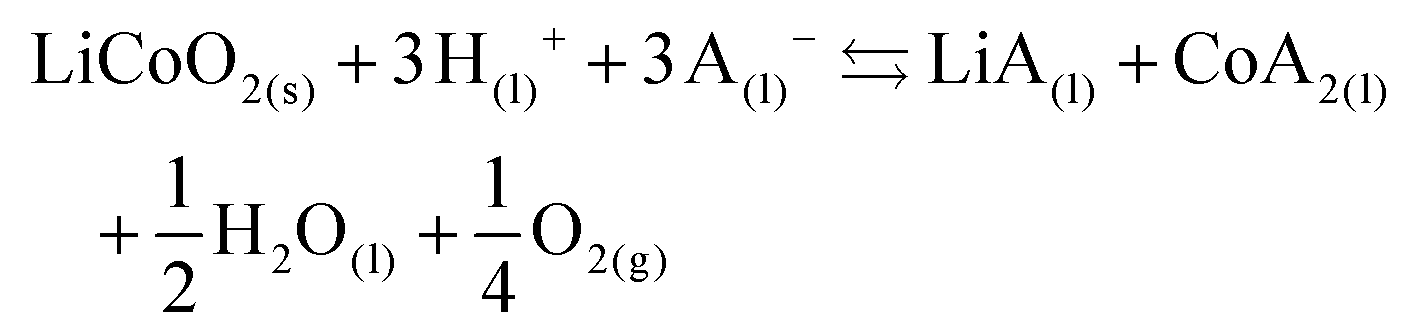 | (4) |
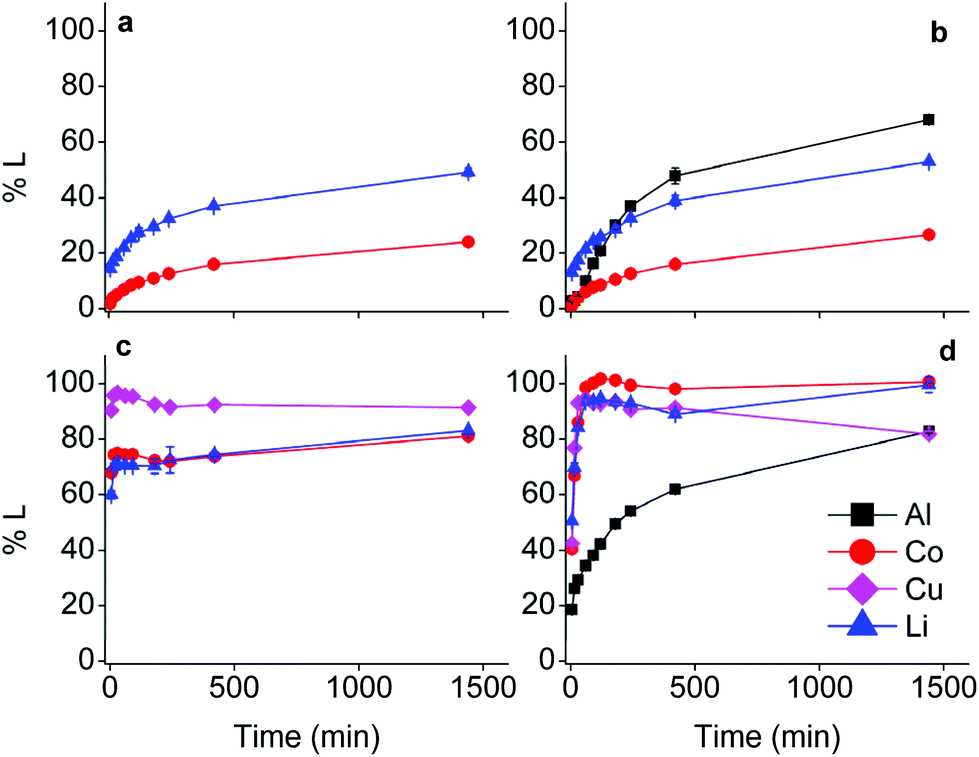 | ||
| Fig. 3 Comparison of the effects of current collector metals on the leaching of lithium cobalt oxide by choline chloride–citric acid DES. Conditions: Only LiCoO2 (left above, a), LiCoO2 with Al (right above, b), LiCoO2 with Cu (left below, c), LiCoO2 with Al + Cu (right below, d). DES: ChCl:CA 2:1 +35 wt% H2O. Leaching parameters: S:L = 20 g L−1, 40 °C, Al:LiCoO2 = 12 wt%, Cu:LiCoO2 = 24 wt% at 900 rpm. | ||
Citric acid provides the protons and chlorides are the ligands A− in eqn (4) due to the high concentrations of these species in the DES.68 Although eqn (4) is simplified description of the chemical reaction occurring during leaching in the sense that the exact stoichiometry of the complexes is not known, the formation of oxygen gas has been reported in the literature during the dissolution of LiCoO2.1,22,69 In the absence of reducing agents, reduction of cobalt(III) is accomplished by oxidising the oxide ions present in lithium cobalt oxide to oxygen gas. This redox reaction is kinetically unfavoured and requires higher temperatures together with unpractically long leaching times, explaining the low cobalt leaching efficiency seen in Fig. 3(a).22 Condition (b) shows a slightly higher cobalt leaching efficiency compared to condition (a) and thus indicates small effect of the presence of aluminium metal on the reduction of cobalt(III). Reduction of cobalt(III) by aluminium is favoured by thermodynamics, but aluminium is more prone to side reactions such as reduction of water and protons, both causing formation of hydrogen gas.8,10,61,70,71 This was evidenced by formation of plenty of gas bubbles and a pressure build-up in the leaching vials, but the gas could not be identified. Joulié et al. reported that a 750% excess of aluminium metal is required to reduce cobalt(III) efficiently during leaching of LIB cathode materials by sulphuric acid.10 Condition (c) shows clearly that copper is an effective reducing agent for cobalt(III) during leaching, in agreement with the results reported by Joulié et al. and Peng et al.10,27 Nevertheless, quantitative leaching of cobalt could not be achieved due to the presence of less than stoichiometric amounts of copper metal. This was mitigated in condition (d), where quantitative cobalt leaching was achieved in the presence of both aluminium and copper metal. Aluminium metal reduces the copper(I) and copper(II) that is formed due to reduction of cobalt(III), back to copper metal and this copper metal is then available again to reduce more cobalt(III).61,62 Thus, copper metal is the decisive reducing agent for cobalt(III), while aluminium is inefficient for reduction of cobalt(III) due to the competitive reduction of protons, copper(I) and copper(II).
Although the focus of this study was mainly on cobalt, lithium was quantitatively leached in condition (d) as well. Lithium(I) is present in the interstices of the in the LiCoO2 lattice.46,72 Attack by acids partially opens the crystal structure and lithium(I) leaches more easily than cobalt(III) since no redox reaction is required. Furthermore, increase in cobalt leaching causes more distortion of the LiCoO2 crystal lattice and results in more lithium leaching as well.73,74 This is supported by the similar trends of cobalt and lithium leaching in conditions (c) and (d).
The powder characteristics such as the particle size and morphology are an important factor that influences the redox activity of copper or aluminium.75–78 The particle sizes of the copper and aluminium powders used were kept constant during these experiments (0.075 mm for both powders). However, industrial processes usually do not give this fine particle sizes after crushing and sieving the spent LIBs. For example, common industrially obtained particle sizes range from 6.0 to 0.5 mm.19,27,79 Thus, investigating the effect of larger particle sizes would be essential when it is the intention to industrially apply this process. Furthermore, real industrial wastes would contain much more metals than the ones discussed in this work.19,24,27 Since present work is a fundamental model study rather than an industrial application, particle size investigations were not executed.
Metal complex identification during DES leaching
Counter anions and ligands of metals influence the solvent extraction behaviour and partially assist the choice of a suitable extractant. Thus, identification of the complexes present in the PLS is essential for understanding of the reaction mechanism and to develop an efficient process. Aluminium(III), cobalt(II), copper(I), copper(II) and lithium(I) chloride salts were dissolved in the choline chloride–citric acid DES. The cobalt(II), copper(I) and copper(II) complexes were coloured and could be investigated by UV-VIS absorption spectroscopy, whereas the aluminium(III) and lithium(I) complexes are spectroscopically silent. Therefore, speciation of the complexes in the PLS was based on cobalt and copper. Fig. 4 compares the absorption spectra of the dissolved copper(I)- and copper(II) chloride salts with the spectrum of dissolved copper metal. The peak shapes and the wavelengths of maximum absorbance of copper(I) chloride and copper metal are quite similar, indicating that copper(I) chloride complexes are probably predominantly formed during leaching of LiCoO2 with aluminium and copper by the choline chloride–citric acid DES. The dissolution of copper metal in a choline chloride–ethylene glycol DES is described by Green et al.80 These authors describe the comproportionation/disproportionation equilibrium between copper(I)- and copper(II) chlorides, with the former being the dominant species. This is supported by the smaller absorption band at about 405 nm, which is characteristic for the tetrachlorocuprate(II) complex.48,80,81 The intensity of this absorption band decreases when copper(I) chloride is dissolved but is still slightly present, indicating the copper(I)–copper(II) equilibrium.80 Moreover, the copper(II) oxidation state is stable in aqueous media, even at low chloride concentrations whereas monovalent copper(I) is only stable at higher chloride concentrations (≥1 mol L−1,82–84 like in the DES). This supports the fact that copper(I) chlorides can be formed in the DES. These high chloride concentrations cause copper corrosion by surface adsorption of the chloride ions, followed by a dissolution reaction.80,85,86 Thus, copper is most likely dissolved by the DES via a combination of corrosive chloride attack and a comproportionation/disproportionation equilibrium described as:80,85,86| Cu + [CuCl4]2− ⇆ 2[CuCl2]− | (5) |
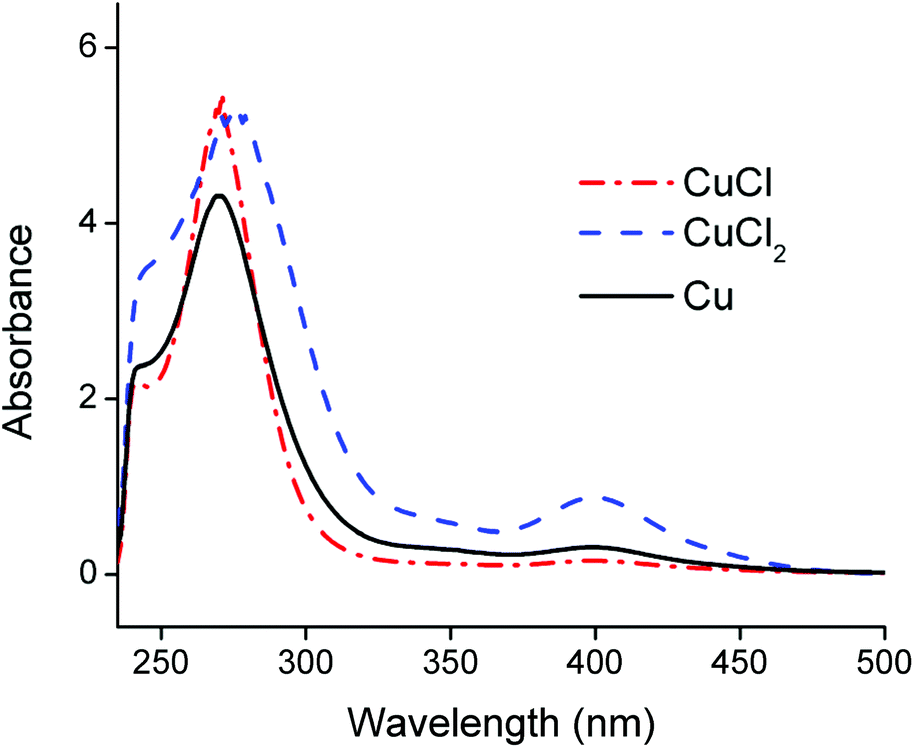 | ||
| Fig. 4 Optical absorption spectra of CuCl (red curve) and CuCl2 (blue curve) dissolved in the choline chloride–citric acid DES. The black lines is the absorption spectrum of the copper species formed by dissolution of copper metal in the DES. DES: ChCl:CA 2:1 +35 wt% H2O. Concentrations of the dissolved copper ions in the DES are ca. 1 g L−1. | ||
Fig. 5 compares spectra of cobalt(II) and copper(I) chloride salts with the spectra of the PLS. The latter spectra showed the typical absorption bands of the tetrachlorocobaltate(II) complex at 590–735 nm, a copper(II) chloride band at 405 nm and a copper(I) chloride band at 275 nm, as well as a peak at 240 nm with unknown origin.48 Quantification of tetrachlorocobaltate(II) in the PLS via UV-VIS absorption spectroscopy was done by first dissolving different amounts of CoCl2 in a 35 wt% water diluted choline chloride solution to exclude formation of citrate complexes, followed by calibrating the 590–735 nm spectral region and quantification (the calibration curve together with spectrum can be found in the ESI Fig. S1†). This result was compared with the quantification of cobalt in the PLS by ICP-OES. The quantification by UV-VIS and ICP-OES resulted in a cobalt(II) concentration of 12.5 g L−1 and 12.7 g L−1 respectively. Comparison of these two values confirms that cobalt(II) is nearly quantitatively present as tetrachlorocobaltate(II) in the PLS. The dominance of the latter complexes was also described by Tran et al., in a choline chloride–ethylene glycol based DES.87 Furthermore, this also shows that the peak at 240 nm in Fig. 5 is most likely not related with cobalt complexation, but is probably associated with the absorption by organic compounds such as citric acid.
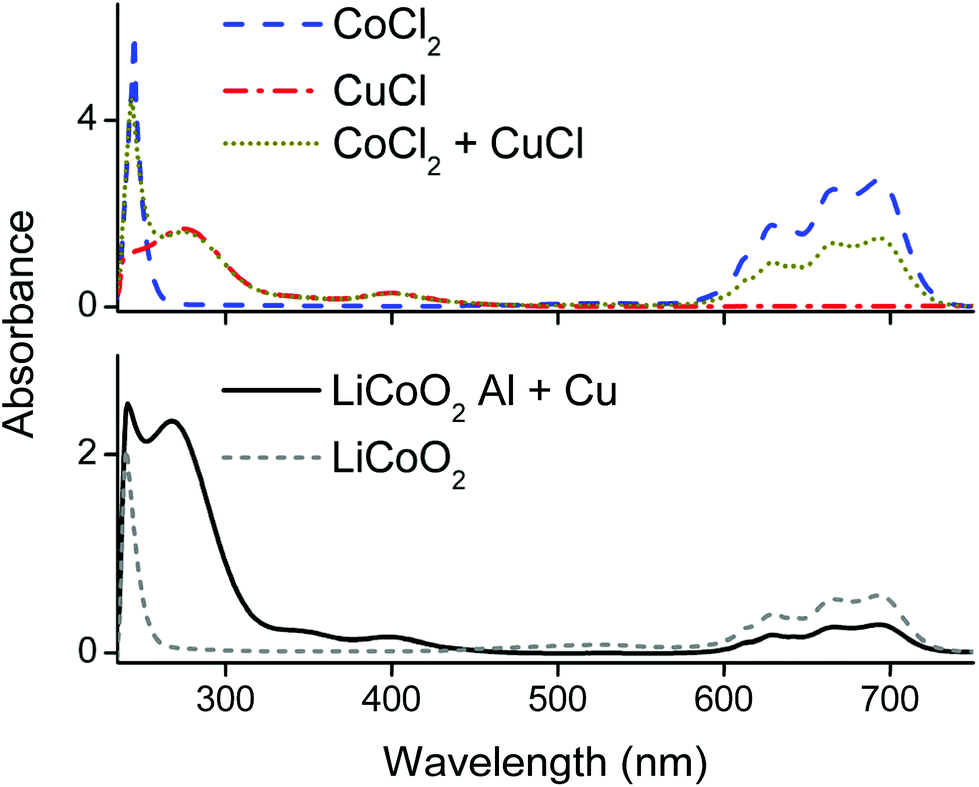 | ||
| Fig. 5 Optical absorption spectra of the DES pregnant leach solution. DES: ChCl:CA 2:1 +35 wt% H2O. Input concentrations CuCl, CoCl2ca. 2 g L−1, the PLS was diluted 40 times. Metal chloride salts (above) compared with the spectra of the PLS (below) with and without the Al and Cu. | ||
Comparison of DES and conventional HCl leaching
Fig. S2† indicates that choline chloride or citric acid are poor lixiviants on their own, and these results show that both protons and chloride anions are necessary for efficient dissolution of LiCoO2. Therefore, hydrochloric acid (HCl) is a commonly used as lixiviant for LIB.5,88–90 The leaching behaviour of hydrochloric acid at similar optimised conditions was compared with that of the choline chloride–citric acid DES (Fig. 6). Although the citric acid concentration in the DES is about 3.6 mol L−1, a lower HCl concentration of 1 mol L−1 was selected for the experiments, because higher HCl concentrations resulted in very fast leaching, accompanied with vigorous gas formation and splashing of the solution, making accurate data collection difficult (see Fig. S3†). Fig. 6 shows that hydrochloric acid is less selective; aluminium metal is dissolved almost instantly. Moreover, comparison of the first data points (after ca. 1 min of leaching) shows that about 45% of the cobalt is leached in both cases. However, the leaching by the DES shows clearly that leaching is enabled by cobalt(III)/copper redox reactions, while this is not the case for leaching with hydrochloric acid. In the latter case, cobalt(III) is most likely reduced to cobalt(II) by chloride anions, which are oxidised to chlorine gas.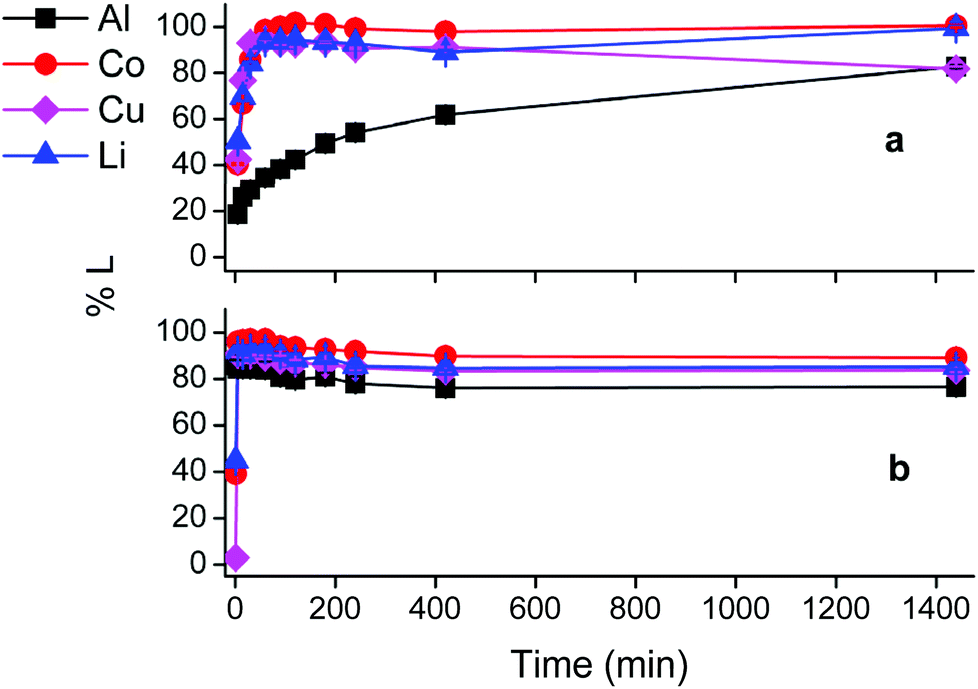 | ||
| Fig. 6 Comparison between leaching with leaching of LiCoO2 by DES and HCl solution. Top: DES leaching (ChCl:CA 2:1 +35 wt% H2O). Bottom: Leaching with HCl 1 mol L−1. Leaching parameters: S:L = 20 g L−1, 40 °C, Al:LiCoO2 = 12 wt%, Cu:LiCoO2 = 24 wt% at a stirring rate of 900 rpm. | ||
Both series of experiments were performed in open vials with a chlorine gas detector positioned above them. Chlorine gas was detected only during hydrochloric acid leaching. This supports the hypothesis of a redox reaction between cobalt(III) and chloride ions. Aluminium dissolves much faster in hydrochloric acid than in the DES because it favours reduction of protons, this tendency is higher due to the higher acidity of hydrochloric acid compared with the DES.54,71,91 Fig. S4† confirms these assumptions. Thus, hydrochloric acid leaches faster than the DES, but is less selective and is more hazardous due to the chlorine gas formation. Although chloride anions are present in the DES as well, chlorine gas formation in the DES is unlikely because these anions are retained by hydrogen bonds.42 The chloride anions of the DES stabilise cobalt(II) cations and have no significant reducing power. The presence of copper metal is the decisive parameter during leaching by the DES because it reduces cobalt(III), while the latter is mainly done by chloride anions along hydrochloric acid leaching. This comparison shows that the role of the metallic current collectors (copper and aluminium) in the leaching process is different for DES and hydrochloric acid leaching. These metals dissolve LiCoO2 in the DES by reducing cobalt(III), with copper metal being the more effective reducing agent, while their dissolution in hydrochloric acid is mainly controlled by acidity. Hence, leaching with DES is a more selective, safer and milder approach than conventional leaching with hydrochloric acid. However, leaching of LIB in aqueous solutions is still industrially favoured due to the higher possible S:L ratios, lower viscosities and lower energy consumption compared with DES systems.
Mechanism of leaching of LiCoO2 by DES in the presence of copper and aluminium
After parameter optimisation and identification of the metal complexes, the mechanism of leaching of LiCoO2 by the choline chloride–citric acid DES can be formulated (Fig. 7). Protons of citric acid and chloride anions of choline chloride in the DES attack the crystal structure of LiCoO2. The leaching is accelerated by copper metal, which reduces cobalt(III) to cobalt(II) and gets oxidised to mainly copper(I), which is in equilibrium with copper(II) and is stabilised by chloride anions. The copper(I/II) complexes formed during the dissolution reaction are reduced to metallic copper by metallic aluminium via cementation. This partially regenerates copper and copper becomes available for further cobalt(III) reduction until complete dissolution of LiCoO2. Aluminium metal reduces protons as a side reaction, causing hydrogen gas formation. Furthermore, chloride anions stabilise cobalt(II) by forming chloro complexes, with tetrachlorocobaltate(II) being the predominant species. Although aluminium(III) and lithium(I) chloro complexes could not be identified by absorption spectroscopy because they do not absorb in the UV-VS spectral region, the formation of chloro complexes with these metal ions can be excluded because of their strong affinity for water molecules.92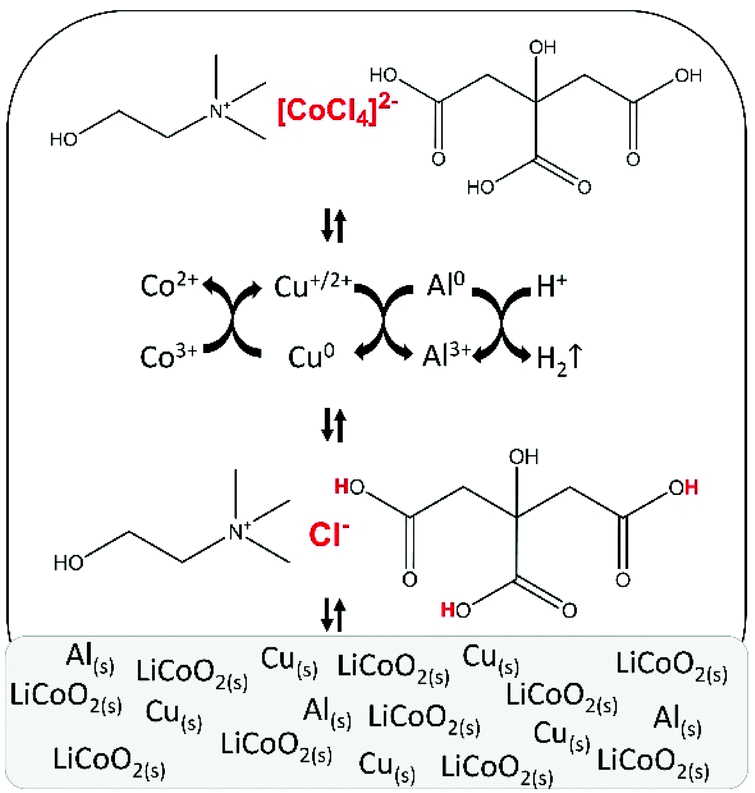 | ||
| Fig. 7 Schematic representation of leaching of lithium cobalt oxide (LiCoO2) by a choline chloride–citric acid DES, in the presence of aluminium and copper metal. | ||
Non-aqueous solvent extraction process for cobalt(II) recovery
Because the relatively high chloride concentration in the DES caused the dominant formation of anionic tetrachlorocobaltate(II) complexes, the anion-exchanging extractant A336 is an obvious choice to extract cobalt from the PLS.93,94 Notice that we use here direct solvent extraction from a DES with a water content lower than 50 vol%, thus not from an aqueous phase, hence the extraction process can be labelled as non-aqueous SX. It is common practice to use aromatic diluents for extractions by A336 because the solubilities of A336 and the extracted metals complexes in toxic aliphatic diluents are too low. To avoid the use of these aromatic diluents, we decided to use undiluted A336 as extracting phase. Prior to use, A336 was saturated with water to reduce its viscosity. The efficiency for cobalt(II) extraction is high, but copper is unfortunately co-extracted as well. Undiluted A336 extracted 82% of cobalt(II) and 41% of copper(I/II) from the PLS that contained 1.0 g L−1 aluminium(III), 12.1 g L−1 cobalt(II), 4.7 g L−1 copper(I/II) and 1.1 g L−1 lithium(I). Co-extraction of aluminium(III) and lithium(I) was insignificant, since their quantities were below the ICP-OES detection limit. To avoid this co-extraction of cobalt and copper, copper(I/II) was firstly extracted from the PLS using the chelating extractant LIX 984. LIX 984 is a commercially oxime-based mixture which is widely used as copper extractant.95,96 Since most pure extractants are viscous, they are in general diluted. Therefore, LIX 984 was diluted in an aliphatic diluent (Shell GS190) and optimisation of its concentration is shown in Fig. 8. Shell GTL diluents are biodegradable and show low ecotoxicity.28,97Fig. 8 indicates that 60 vol% LIX 984 in an aliphatic diluent extracts about 92% copper(I/II) with co-extraction of the other metals below 7%. This concentration was used in further experiments, LIX 984 extracts copper(I/II) via chelation and proton exchange. Protons of the citric acid in the DES are consumed during leaching. The proton exchange reaction during the solvent extraction step transfers protons to the DES, enabling to regenerate the DES so that it can be reused in the new leaching step. It was not confirmed that LIX 984 extracts copper(I) or copper(II) species, because ICP-OES cannot distinguish metal oxidation states. The purpose of this purification step is to remove copper ions quantitatively, regardless of their oxidation states. Hence, this first extraction step with LIX 984 showed to be an essential step in the non-aqueous solvent extraction flow sheet. Variation of the (VLIX984:VPLS) phase ratios showed that a LIX 984 (60 vol%) to PLS volume ratio of 1.5 was an optimum condition for complete copper removal (≥99.9 %E). In a next step, 85% of the cobalt(II) was extracted from the PLS raffinate with undiluted water-presaturated A336, with co-extraction of insignificant amounts of aluminium(III) and lithium(I). Selective extractions by LIX 984 or A336 are usually established by controlling the pH or the chloride concentration of the PLS respectively. The former is often done by adding alkaline compounds (when a PLS is obtained after leaching by mineral acids), the latter by dissolving a chloride salt or by adding hydrochloric acid.98,99 As mentioned, these additional adjustments were avoided by preparing the DES at a choline chloride:citric acid ratio of 2:1. Hereby the suitable range of pH and chloride concentration were obtained for copper and cobalt extraction by LIX 984 and A336 respectively. However, it is important to mention that higher extraction efficiencies could be obtained via further increasing the phase ratio (Vorg:VPLS) or by simulating both extractions in mixer-settlers via a continuous multistage counter – current circuit. This latter approach is known to increase both the selectivity and efficiency of extraction processes.98,100,101 Furthermore, since increasing the phase ratio would dilute the metal of interest in further downstream processes, a counter – current extraction operation would be the most suitable approach.
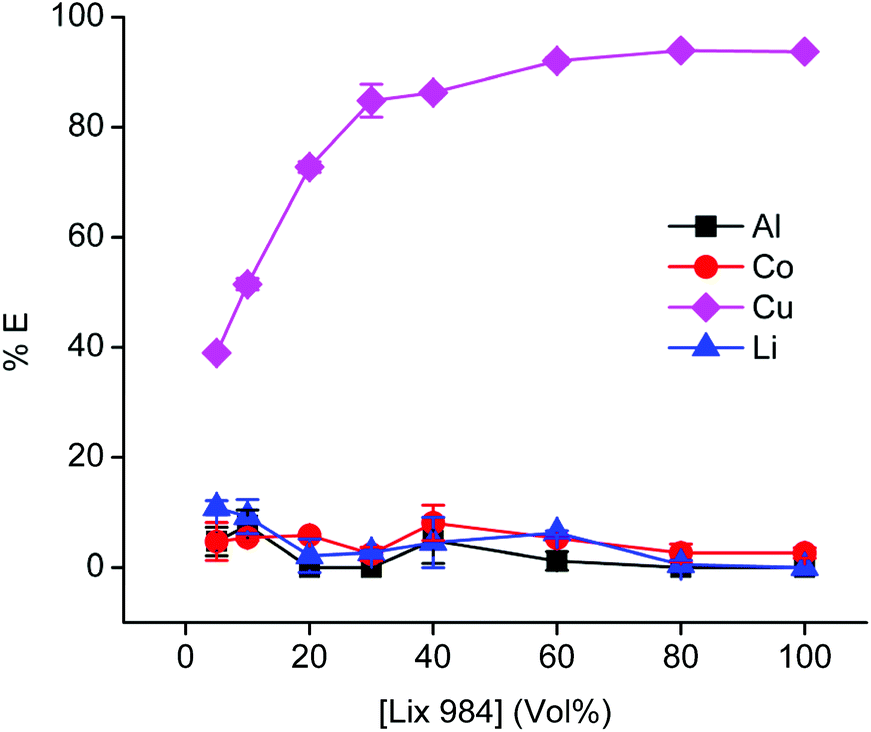 | ||
| Fig. 8 Optimisation of the LIX 984 concentration in the aliphatic diluent during extraction of metals from the PLS. Initial metal concentrations in the PLS: Al(III) 1.0 g L−1, Co(II) 12.1 g L−1, Cu(I/II) 4.7 g L−1 and Li(I) 1.1 g L−1. Equal phase ratio was used. Equilibration time = 30 min, shaking speed = 2250 rpm, room temperature. | ||
As mentioned above, copper(I/II)- and cobalt(II) oxalate precipitates are easily formed.4,53,57 Therefore, oxalic acid was used to strip and precipitate copper from the loaded LIX 984 phase and cobalt from the loaded A336 phase.102Fig. 9 shows that 0.25 mol L−1 oxalic acid is efficient to precipitate copper(I/II) and cobalt(II). TXRF analysis of the light organic phases after oxalic acid stripping showed negligible concentrations of copper and cobalt. Thus, both metals could by quantitatively stripped and precipitated by a 0.25 mol L−1 oxalic acid solution, causing the regeneration of their corresponding light phases. The solubilities of both aluminium(III) and lithium(I) oxalate precipitates are higher than cobalt(II) and copper(I/II) oxalate precipitates.54,103 Moreover, ICP-OES analysis confirmed that aluminium(III) and lithium(I) were not extracted by A336. Therefore, the purity of the cobalt(II) oxalate precipitate was determined by measuring the metal content of the supernatant after stripping of A336 with ICP-OES and assuming that only cobalt(II) was precipitated (since all copper(I/II) was previously removed), which resulted in a purity of 99.9%. However, XRD analysis of the isolated precipitate would be a more straightforward approach. The aqueous waste that is generated after filtering these precipitates would only contain free oxalic acid molecules, due to the high precipitation efficiencies and the low cobalt(II)- and copper(I/II) oxalate solubilities.54 These free oxalic acid molecules were not consumed to produce the oxalate precipitates. So in principle, the aqueous waste residue after stripping copper(I/II) from LIX 984 and filtering the precipitate could be used to strip cobalt(II) from A336 and vice versa. However, it is important to optimize this recycling approach in order to estimate the amount of oxalic acid that should be added after a certain number of cycles to compensate for the loss that is made during the formation of these precipitates, and thus ensure an efficient recycling. Oxalate precipitates can easily be converted to their oxide forms by a calcination step. The latter is very effective for cobalt(II) oxalate, since cobalt(II) oxide is a common lithium cobalt oxide precursor.4,88 The final DES raffinate contained ca. 1.0 g L−1 aluminium(III) and lithium(I), which could both be recovered via non-aqueous SX and/or precipitation. Aluminium(III) could be selectively extracted from the final raffinate by acidic extractants such as D2EPHA, C272 or Versatic 10.65,104 This is because the acidity of the DES is approximately in the preferred pH range for Al(III) extraction by these extractants, while lithium(I) is extracted at much higher pH values and will therefore stay in the DES raffinate.52,105,106 Lithium(I) could then be recovered in the next step by adding sodium carbonate in order to precipitate lithium(I) carbonate.4,103,105 Another approach would be to extract lithium(I) first from the final raffinate by alcohols such as n-butanol or 2-ethylhexanol, which are known to extract lithium(I) selectively in the presence of aluminium(III).107,108 Hereafter, aluminium(III) could be extracted by acidic extractants as mentioned above. These secondary recovery routes were not further investigated, since the focus of this manuscript is on cobalt(II). However, the removal of both metals from the final raffinate is crucial for the recycling of the DES as lixiviant for the next leaching cycle. Furthermore, this manuscript confirmed that, apart from the reducing agents, the leaching of cobalt(II) from LiCoO2 is also influenced by chloride anions and protons. As mentioned, the former is given by choline chloride and the latter by citric acid in the DES. So it is important to investigate the amount of these compounds that are lost during the entire process and take this into account for the recycling. For example, acidic protons are consumed during leaching, this is partially compensated by LIX 984 that donates protons back during extraction. However, a major drawback of non-aqueous SX is the mutual solubility between the used heavy- and the light phases.28 Determining the solubility of choline chloride and citric acid in 60 vol% LIX 984 and pre-saturated A336 would result in the estimation of the lost amounts of these DES components. The latter will provide the amount of choline chloride and citric acid that should be added each cycle to ensure an efficient recycling.
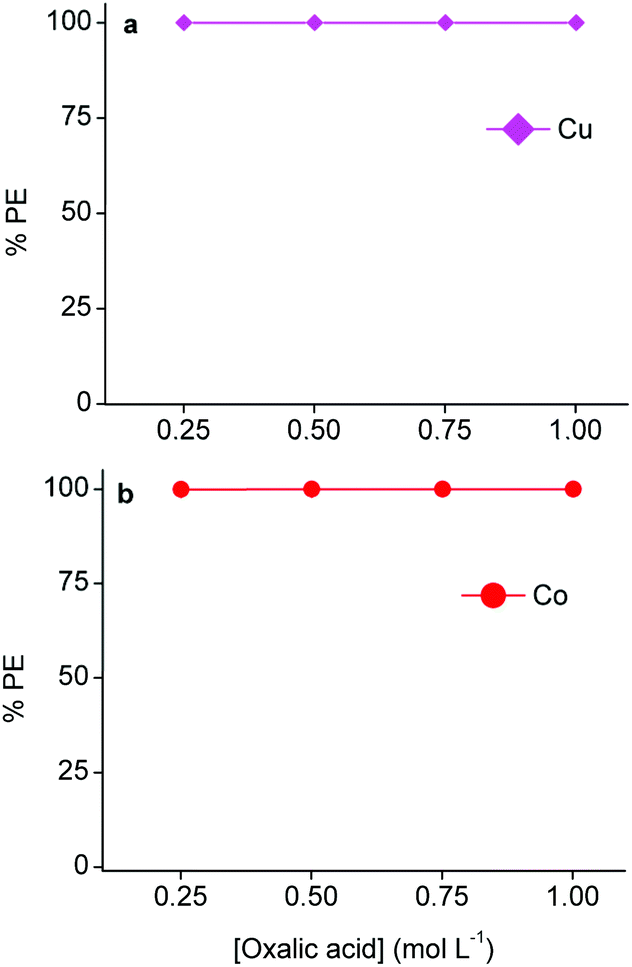 | ||
| Fig. 9 Efficiencies of stripping precipitation of metal oxalates at different oxalic acid concentrations. (a) Cu(I/II) oxalate formation by stripping from loaded 60 vol% LIX 984 in an aliphatic diluent and (b) cobalt(II) oxalate formation by stripping from loaded undiluted water-presaturated A336. The loaded LIX 894 and A336 contained 4.7 g L−1 Cu(I/II) and 9.8 g L−1 Co(II). Phase ratio = 1:1, equilibration = 30 min, shaking speed = 2250 rpm, room temperature. | ||
Conclusion
A choline chloride–citric acid DES, prepared at a 2:1 molar ratio and diluted with 35 wt% water, was found to be an efficient lixiviant for lithium cobalt oxide (LiCoO2) in the presence of metallic aluminium and copper. The optimised leaching conditions were 60 min of leaching at 40 °C with a solid-to-liquid ratio of 20, which ensured to leach more than 98% cobalt(II) of the LiCoO2. The effect of current collectors aluminium and copper on the leaching efficiency was investigated, confirming that copper is the decisive reducing agent for cobalt(III). Aluminium is not as effective as copper for reduction of cobalt(III) because it mainly reacts with protons to form hydrogen gas. Speciation studies showed that the dominant species in the pregnant leach solution (PLS) were chloro complexes of copper(I/II) and cobalt(II). Study of the leaching mechanism show that the necessary components for leaching of LiCoO2 are: (1) a reducing agent to reduce cobalt(III) to cobalt(II), (2) chloride anions to form metal chloro complexes and (3) protons to react with oxide ions to form water molecules. In the choline chloride–citric acid DES, choline chloride acts as chloride source and citric acid as proton source. Leaching of LiCoO2 with the DES and hydrochloric acid were compared. The advantage of leaching with DES is that no chlorine gas evolves, but leaching is slower than when hydrochloric acid is used. Reduction of cobalt(III) in the DES is predominantly controlled by copper metal, while part of the reduction of cobalt(III) is achieved by oxidation of chloride anions to toxic chlorine gas in hydrochloric acid. Copper and cobalt could be recovered from the PLS by non-aqueous solvent extraction by LIX 984 and A336 respectively; without using additional components to control the pH and chloride concentration of the PLS. The metals could be stripped from the loaded organic phase by an oxalic acid solution. Schematic overviews of entire process and the recovery yields of all the involved metals during the entire flowsheet are shown in Fig. 10 and Fig. S5,† respectively. The choline chloride–citric acid DES leaches 38% aluminium(III), 98% cobalt(II), 94% copper(I/II) and 93% lithium(I). Subsequently, copper(I/II) is quantitatively extracted by 60 vol% LIX 984 with 5% co-extraction of the other three metals. A336 extracted 85% cobalt(II) from the remaining raffinate with no co-extraction of aluminium(III) and lithium(I). Oxalic acid quantitatively precipitated both cobalt(II) and copper(I/II) from their corresponding loaded organic phases, resulting in a total recovery yield of 81% cobalt(II) and 94% copper(I/II) as oxalate precipitates, with a cobalt(II) oxalate purity of 99.9%. Approximately 95% of aluminium(III) and lithium(I) remained in the final DES raffinate.
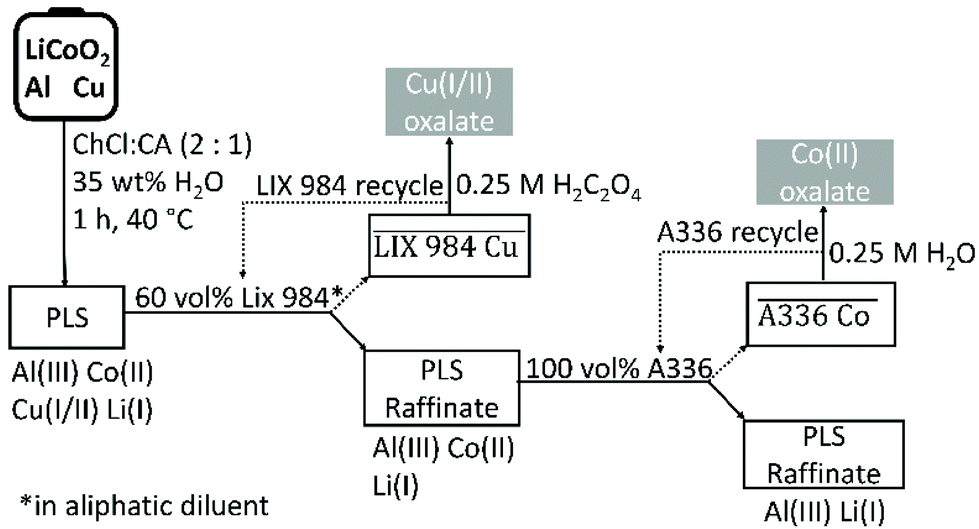 | ||
| Fig. 10 Conceptual flow sheet for the solvometallurgical recovery of cobalt(II) from LiCoO2 by a choline chloride–citric acid DES. | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research project has received funding from the European Union's EU Framework Programme for Research and Innovation Horizon 2020 under Grant Agreement No 776473 (CROCODILE). This work reflects only the author's view, exempting the Community from any liability. Website: https://h2020-crocodile.eu/.Notes and references
- B. Huang, Z. Pan, X. Su and L. An, J. Power Sources, 2018, 399, 274–286 CrossRef CAS.
- H. Zou, E. Gratz, D. Apelian and Y. Wang, Green Chem., 2013, 15, 1183–1191 RSC.
- L. Li, E. Fan, Y. Guan, X. Zhang, Q. Xue, L. Wei, F. Wu and R. Chen, ACS Sustainable Chem. Eng., 2017, 5, 5224–5233 CrossRef CAS.
- J. Kang, J. Sohn, H. Chang, G. Senanayake and S. M. Shin, Adv. Powder Technol., 2010, 21, 175–179 CrossRef CAS.
- M. Petániková, A. Miskufová, Z. Takácová and A. Pehkonen, in 18th International Student's Day of Metallurgy, Leoben, 2011.
- G. P. Nayaka, K. V. Pai, G. Santhosh and J. Manjanna, Hydrometallurgy, 2016, 161, 54–57 CrossRef CAS.
- H. Nie, L. Xu, D. Song, J. Song, X. Shi, X. Wang, L. Zhang and Z. Yuan, Green Chem., 2015, 17, 1276–1280 RSC.
- X. Zeng, J. Li and B. Shen, J. Hazard. Mater., 2015, 295, 112–118 CrossRef CAS PubMed.
- Z. Peng, D. Gregurek, C. Wenzl and J. F. White, JOM, 2016, 68, 2313–2315 CrossRef.
- M. Joulié, E. Billy, R. Laucournet and D. Meyer, Hydrometallurgy, 2017, 169, 426–432 CrossRef.
- B. Buijs, H. Sievers and L. A. T. Espinoza, Proc. Inst. Civ. Eng.: Waste Resour. Manage., 2012, 165, 201–208 Search PubMed.
- M. Buchert, A. Manhart, D. Bleher and D. Pingel, Recycling critical raw materials from waste electronic equipment, Öko-Institut e.V., Darmstadt, 2012 Search PubMed.
- A. H. Tkaczyk, A. Bartl, A. Amato, V. Lapkovskis and M. Petranikova, J. Phys. D: Appl. Phys., 2018, 51, 1–26 CrossRef.
- Y. Yang, X. Meng, H. Cao, X. Lin, C. Liu, Y. Sun, Y. Zhang and Z. Sun, Green Chem., 2018, 20, 3121–3133 RSC.
- C. Banza Lubaba Nkulu, L. Casas, V. Haufroid, T. De Putter, N. D. Saenen, T. Kayembe-Kitenge, P. Musa Obadia, D. Kyanika Wa Mukoma, J. M. Lunda Ilunga, T. S. Nawrot, O. Luboya Numbi, E. Smolders and B. Nemery, Nat. Sustainable, 2018, 1, 495–504 CrossRef PubMed.
- P. Meshram, B. D. Pandey and T. R. Mankhand, Chem. Eng. J., 2015, 281, 418–427 CrossRef CAS.
- H. Ku, Y. Jung, M. Jo, S. Park, S. Kim, D. Yang, K. Rhee, E. M. An, J. Sohn and K. Kwon, J. Hazard. Mater., 2016, 313, 138–146 CrossRef CAS PubMed.
- G. P. Nayaka, K. V. Pai, G. Santhosh and J. Manjanna, J. Environ. Chem. Eng., 2016, 4, 2378–2383 CrossRef CAS.
- C. Peng, J. Hamuyuni, B. P. Wilson and M. Lundström, Waste Manag., 2018, 76, 582–590 CrossRef CAS PubMed.
- A. Sonoc, J. Jeswiet and V. K. Soo, Procedia CIRP, 2015, 29, 752–757 CrossRef.
- Q. Meng, Y. Zhang and P. Dong, Waste Manag., 2017, 64, 214–218 CrossRef CAS PubMed.
- L. Sun and K. Qiu, Waste Manag., 2012, 32, 1575–1582 CrossRef CAS PubMed.
- R. Golmohammadzadeh, F. Rashchi and E. Vahidi, Waste Manag., 2017, 64, 244–254 CrossRef CAS PubMed.
- X. Chen, J. Li, D. Kang, T. Zhou and H. Ma, Green Chem., 2019, 21, 6342–6352 RSC.
- A. Chagnes and B. Pospiech, J. Chem. Technol. Biotechnol., 2013, 88, 1191–1199 CrossRef CAS.
- X. Chen, C. Guo, H. Ma, J. Li, T. Zhou, L. Cao and D. Kang, Waste Manag., 2018, 75, 459–468 CrossRef CAS PubMed.
- C. Peng, F. Liu, A. T. Aji, B. P. Wilson and M. Lundström, Waste Manag., 2019, 95, 604–611 CrossRef CAS PubMed.
- K. Binnemans and P. T. Jones, J. Sustainable Metall., 2017, 3, 570–600 CrossRef.
- Z. Li, X. Li, S. Raiguel and K. Binnemans, Sep. Purif. Technol., 2018, 201, 318–326 CrossRef CAS.
- N. K. Batchu, T. Vander Hoogerstraete, D. Banerjee and K. Binnemans, Sep. Purif. Technol., 2017, 174, 544–553 CrossRef CAS.
- X. Li, W. Monnens, Z. Li, J. Fransaer and K. Binnemans, Green Chem., 2020, 22, 417–426 RSC.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 CrossRef CAS PubMed.
- A. P. Abbott, G. Frisch, S. J. Gurman, A. R. Hillman, J. Hartley, F. Holyoak and K. S. Ryder, Chem. Commun., 2011, 47, 10031–10033 RSC.
- A. P. Abbott, G. Frisch, J. Hartley and K. S. Ryder, Green Chem., 2011, 13, 471–481 RSC.
- M. A. R. Martins, S. P. Pinho and J. A. P. Coutinho, J. Solution Chem., 2018, 48, 962–982 CrossRef.
- L. P. Silva, L. Fernandez and A. P. Coutinho, ACS Sustainable Chem. Eng., 2018, 6, 10724–10734 CrossRef CAS.
- H. Cruz, N. Jordão and L. C. Branco, Green Chem., 2017, 19, 1653–1658 RSC.
- Q. Zhang, K. De Oliveira Vigier, S. Royer and F. Jérôme, Chem. Soc. Rev., 2012, 41, 7108–7146 RSC.
- G. R. T. Jenkin, A. Z. M. Al-Bassam, R. C. Harris, A. P. Abbott, D. J. Smith, D. A. Holwell, R. J. Chapman and C. J. Stanley, Miner. Eng., 2016, 87, 18–24 CrossRef CAS.
- S. Khandelwal, Y. K. Tailor and M. Kumar, J. Mol. Liq., 2016, 215, 345–386 CrossRef CAS.
- P. Zürner and G. Frisch, ACS Sustainable Chem. Eng., 2019, 7, 5300–5308 CrossRef.
- M. R. S. Foreman, Cogent Chem., 2016, 2, 1–11 Search PubMed.
- S. Riaño, M. Petranikova, B. Onghena, T. Vander Hoogerstraete, D. Banerjee, M. R. S. Foreman, C. Ekberg and K. Binnemans, RSC Adv., 2017, 7, 32100–32113 RSC.
- G. R. T. Jenkin, A. Z. M. Al-Bassam, R. C. Harris, A. P. Abbott, D. J. Smith, D. A. Holwell, R. J. Chapman and C. J. Stanley, Miner. Eng., 2016, 87, 18–24 CrossRef CAS.
- F. J. Albler, K. Bica, M. R. S. J. Foreman, S. Holgersson and M. S. Tyumentsev, J. Cleaner Prod., 2018, 167, 806–814 CrossRef.
- I. L. Santana, T. F. M. Moreira, M. F. F. Lelis and M. B. J. G. Freitas, Mater. Chem. Phys., 2017, 190, 38–44 CrossRef CAS.
- C. R. Soccol, L. P. S. Vandenberghe, C. Rodrigues and A. Pandey, Food Technol. Biotechnol., 2006, 44, 141–149 CAS.
- R. Lommelen, T. Vander Hoogerstraete, B. Onghena, I. Billard and K. Binnemans, Inorg. Chem., 2019, 58, 12289–12301 CrossRef CAS PubMed.
- M. J. Lain, J. Power Sources, 2001, 97–98, 736–738 CrossRef CAS.
- T. Georgi-Maschler, B. Friedrich, R. Weyhe, H. Heegn and M. Rutz, J. Power Sources, 2012, 207, 173–182 CrossRef CAS.
- C. Sun, L. Xu, X. Chen, T. Qiu and T. Zhou, Waste Manag. Res., 2018, 36, 113–120 CrossRef CAS PubMed.
- A. Skulcova, A. Russ, M. Jablonsky and J. Sima, BioResources, 2018, 13, 5042–5051 CAS.
- F. Wang, R. Sun, J. Xu, Z. Chen and M. Kang, RSC Adv., 2016, 6, 85303–85311 RSC.
- J. A. Dean, Lange's handbook of chemistry, McGraw-Hill, New York, 1990, vol. 5 Search PubMed.
- D. R. Lide, Handbook of Chemistry and Physics, CRC Press, Cleveland, 1977 Search PubMed.
- A. M. Donia and D. Dollimore, Thermochim. Acta, 1997, 290, 139–147 CrossRef CAS.
- A. T. Royappa, A. D. Royappa, R. F. Moral, A. L. Rheingold, R. J. Papoular, D. M. Blum, T. Q. Duong, J. R. Stepherson, O. D. Vu, B. Chen, M. R. Suchomel, J. A. Golen, G. André, N. Kourkoumelis, A. D. Mercer, A. M. Pekarek and D. C. Kelly, Polyhedron, 2016, 119, 563–574 CrossRef CAS.
- D. Sortwell and A. Woo, Improving the Flavor of Fruit Products with Acidulants, presented at Expotechnoalimentaria '96, New Mexico, 1996 Search PubMed.
- O. S. Hammond, D. T. Bowron, A. J. Jackson, T. Arnold, A. Sanchez-Fernandez, N. Tsapatsaris, V. Garcia Sakai and K. J. Edler, J. Phys. Chem. B, 2017, 121, 7473–7483 CrossRef CAS PubMed.
- O. S. Hammond, D. T. Bowron and K. J. Edler, Angew. Chem., Int. Ed., 2017, 56, 9782–9785 CrossRef CAS PubMed.
- D. J. MacKinnon and T. R. Ingraham, Can. Metall. Q., 2014, 10, 197–201 CrossRef.
- N. Demirkran and A. Künkül, Trans. Nonferrous Met. Soc. China, 2011, 21, 2778–2782 CrossRef.
- D. A. Ferreira, L. M. Z. Prados, D. Majuste and M. B. Mansur, J. Power Sources, 2009, 187, 238–246 CrossRef CAS.
- V. Branzoi, F. Golgovici and F. Branzoi, Mater. Chem. Phys., 2003, 78, 122–131 CrossRef.
- P. E. Tsakiridis and S. Agatzini-Leonardou, Hydrometallurgy, 2005, 80, 90–97 CrossRef CAS.
- P. Meshram, B. D. Pandey and T. R. Mankhand, Waste Manag., 2015, 45, 306–313 CrossRef CAS PubMed.
- X. Zheng, Z. Zhu, X. Lin, Y. Zhang, Y. He, H. Cao and Z. Sun, Engineering, 2018, 4, 361–370 CrossRef CAS.
- E. L. Smith, Trans. Inst. Met. Finish., 2013, 91, 241–248 CrossRef CAS.
- M. K. Jha, A. Kumari, A. K. Jha, V. Kumar, J. Hait and B. D. Pandey, Waste Manag., 2013, 33, 1890–1897 CrossRef CAS PubMed.
- J. V. Petrocelli, J. Electrochem. Soc., 2007, 97, 1–10 Search PubMed.
- A. O. Dudoladov, O. A. Buryakovskaya, M. S. Vlaskin, A. Z. Zhuk and E. I. Shkolnikov, Int. J. Hydrogen Energy, 2016, 41, 2230–2237 CrossRef CAS.
- F. Xiong, H. J. Yan, Y. Chen, B. Xu, J. X. Le and C. Y. Ouyang, Int. J. Electrochem. Sci., 2012, 7, 9390–9400 CAS.
- G. P. Nayaka, J. Manjanna, K. V. Pai, R. Vadavi, S. J. Keny and V. S. Tripathi, Hydrometallurgy, 2015, 151, 73–77 CrossRef CAS.
- G. Dorella and M. B. Mansur, J. Power Sources, 2007, 170, 210–215 CrossRef CAS.
- J. Kiwi and M. Graetzel, J. Am. Chem. Soc., 1979, 101, 7214–7217 CrossRef CAS.
- S. Mrowec and A. Stokłosa, Oxid. Met., 1971, 3, 291–311 CrossRef CAS.
- B. Rufino, F. Boulc'h, M. V. Coulet, G. Lacroix and R. Denoyel, Acta Mater., 2007, 55, 2815–2827 CrossRef CAS.
- A. V. Parmuzina and O. V. Kravchenko, Int. J. Hydrogen Energy, 2008, 33, 3073–3076 CrossRef CAS.
- X. Wang, G. Gaustad and C. W. Babbitt, Waste Manag., 2016, 51, 204–213 CrossRef CAS PubMed.
- T. A. Green, P. Valverde and S. Roy, J. Electrochem. Soc., 2018, 165, D313–D320 CrossRef CAS.
- P. De Vreese, N. R. Brooks, K. Van Hecke, L. Van Meervelt, E. Matthijs, K. Binnemans and R. Van Deun, Inorg. Chem., 2012, 51, 4972–4981 CrossRef CAS PubMed.
- Y. Meng and A. J. Bard, Anal. Chem., 2015, 87, 3498–3504 CrossRef CAS PubMed.
- K. Nobe, J. Electrochem. Soc., 1986, 133, 2035–2043 CrossRef.
- J. Brugger, B. Etschmann, W. Liu, D. Testemale, J. L. Hazemann, H. Emerich, W. van Beek and O. Proux, Geochim. Cosmochim. Acta, 2007, 71, 4920–4941 CrossRef CAS.
- M. Scendo, Corros. Sci., 2008, 50, 1584–1592 CrossRef CAS.
- N. Habbache, N. Alane, S. Djerad and L. Tifouti, Chem. Eng. J., 2009, 152, 503–508 CrossRef CAS.
- M. K. Tran, M. T. F. Rodrigues, K. Kato, G. Babu and P. M. Ajayan, Nat. Energy, 2019, 4, 339–345 CrossRef CAS.
- L. Chen, X. Tang, Y. Zhang, L. Li, Z. Zeng and Y. Zhang, Hydrometallurgy, 2011, 108, 80–86 CrossRef CAS.
- P. Zhang, T. Yokoyama, O. Itabashi, Y. Wakui, T. M. Suzuki and K. Inoue, Hydrometallurgy, 1998, 50, 61–75 CrossRef CAS.
- S. Gabriele and M. C. Caria, Ospedali d'Italia, 1989, 24, 204–208 Search PubMed.
- S. M. Abd El Haleem, S. Abd El Wanees, E. E. Abd El Aal and A. Farouk, Corros. Sci., 2013, 68, 1–13 CrossRef CAS.
- D. Richens, The Chemistry Of Aqua ions, Wiley, New York, 1997 Search PubMed.
- S. Sobekova Foltova, T. Vander Hoogerstraete, D. Banerjee and K. Binnemans, Sep. Purif. Technol., 2019, 210, 209–218 CrossRef CAS.
- K. Larsson and K. Binnemans, Green Chem., 2014, 16, 4595–4603 RSC.
- E. Rodríguez De San Miguel, J. C. Aguilar, J. P. Bernal, M. L. Ballinas, M. T. J. Rodríguez, J. De Gyves and K. Schimmel, Hydrometallurgy, 1997, 47, 19–30 CrossRef.
- V. Sridhar, J. K. Verma and S. A. Kumar, Hydrometallurgy, 2009, 99, 124–126 CrossRef CAS.
- Shell, Solvents: GTL fluids and solvents, http://www.shell.com/business-customers/chemicals/our-products/solvents-gtl-solvents-and-fluids.html, (accessed 1 May 2018).
- M. Jun, R. R. Srivastava, J. Jeong, J. C. Lee and M. S. Kim, Green Chem., 2016, 18, 3823–3834 RSC.
- C. Deferm, M. Van De Voorde, J. Luyten, H. Oosterhof, J. Fransaer and K. Binnemans, Green Chem., 2016, 18, 4116–4127 RSC.
- F. Xie, T. A. Zhang, D. Dreisinger and F. Doyle, Miner. Eng., 2014, 56, 10–28 CrossRef CAS.
- K. Larsson, C. Ekberg and A. Ødegaard-Jensen, Hydrometallurgy, 2013, 133, 168–175 CrossRef CAS.
- S. Riaño and K. Binnemans, Green Chem., 2015, 17, 2931–2942 RSC.
- B. Swain, Sep. Purif. Technol., 2017, 172, 388–403 CrossRef CAS.
- M. M. J. Correa, F. P. C. Silvas, P. Aliprandini, V. T. de Moraes, D. Dreisinger and D. C. R. Espinosa, Braz. J. Chem. Eng., 2018, 35, 919–930 CrossRef CAS.
- Y. Yang, S. Xu and Y. He, Waste Manag., 2017, 64, 219–227 CrossRef CAS PubMed.
- V. T. Nguyen, J. C. Lee, J. Jeong, B. S. Kim and B. D. Pandey, Met. Mater. Int., 2014, 20, 357–365 CrossRef CAS.
- J. A. Epstein, E. M. Feist, J. Zmora and J. Marcus, Hydrometallurgy, 1981, 6, 269–275 CrossRef CAS.
- G. G. Gabra and A. E. Torma, Hydrometallurgy, 1978, 3, 23–33 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Quantification of [CoCl4]2− in the PLS by UV-VIS absorption spectroscopy, comparison of leaching with solely citric acid and leaching with solely choline chloride, leaching with 3.6 mol L−1 HCl, influence of aluminium on leaching with DES and hydrochloric acid, summary of the recovery values for all metals during the entire flowsheet. See DOI: 10.1039/d0gc00940g |
| This journal is © The Royal Society of Chemistry 2020 |