
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBenchmark selectivity p-xylene separation by a non-porous molecular solid through liquid or vapor extraction†
Na
Sun
a,
Shi-Qiang
Wang
 c,
Ruqiang
Zou
a,
Wen-Gang
Cui
b,
Anqi
Zhang
a,
Tianzhen
Zhang
a,
Qi
Li
b,
Zhan-Zhong
Zhuang
a,
Ying-Hui
Zhang
b,
Jialiang
Xu
*b,
Michael J.
Zaworotko
c and
Xian-He
Bu
*ab
c,
Ruqiang
Zou
a,
Wen-Gang
Cui
b,
Anqi
Zhang
a,
Tianzhen
Zhang
a,
Qi
Li
b,
Zhan-Zhong
Zhuang
a,
Ying-Hui
Zhang
b,
Jialiang
Xu
*b,
Michael J.
Zaworotko
c and
Xian-He
Bu
*ab
aState Key Laboratory of Elemento-Organic Chemistry, College of Chemistry, Nankai University, Tianjin 300071, China. E-mail: buxh@nankai.edu.cn
bSchool of Materials Science and Engineering, National Institute for Advanced Materials, TKL of Metal and Molecule-Based Material Chemistry, Nankai University, Tianjin 300350, China. E-mail: jialiang.xu@nankai.edu.cn
cDepartment of Chemical Sciences and Bernal Institute, University of Limerick, Limerick, Republic of Ireland
First published on 6th August 2019
Abstract
Solid–liquid separation of similarly sized organic molecules utilizing sorbents offers the potential for new energy-efficient approaches to a number of important industrial separations such as xylenes (C8) separations. Research on selective C8 sorption has tended to focus upon rigid porous materials such as zeolites and MOFs but has revealed generally weak selectivity that is inconsistent across the range of C8 molecules. Nevertheless, there are a few recent examples of non-porous molecular materials that exhibit relatively high selectivity for p-xylene (pX) from pX/oX, approaching that of the current benchmark pX sorbent, the zeolite H/ZSM-5. Herein, we report that a L-shaped Ag(I) complex, AgLClO4 (M), which crystallizes as a non-porous molecular solid material, offering exceptional performance for pX selectivity across the range of C8 isomers with liquid extraction selectivity values of 24.0, 10.4 and 6.2 vs. oX, eB and mX, respectively. The pX selectivities over oX and eB are among the highest yet reported. Moreover, M also exhibits strong vapor extraction selectivity and can be regenerated by exposure to vacuum drying.
Introduction
Molecular separations are important in the production of clean water, specialty chemicals, commodities, and fuels.1,2 Separation of the C8 isomers (para-xylene, pX, ortho-xylene, oX, meta-xylene, mX, ethylbenzene, eB) is critical as their end use includes polymers, plastics, resins, pigments, and fungicides.3–5 Indeed, separation of xylene isomers has been described as “one of the seven chemical separations to change the world”6 because of (i) the commercial value; and (ii) its difficulty due to their inherently similar structures and physical properties (boiling points, kinetic diameters, etc., Table S1†).7–11 Approaches such as fractional crystallization,12 adsorption,13 sieving,14 complexation,15 and isomerization16 have been employed to separate xylene isomers. With respect to adsorption, there is of great potential to reduce the energy footprint of C8 separations, but most sorbents exhibit low selectivity for the aforementioned reasons (Table S2†). With respect to the current benchmark sorbents, it is interesting to note that, whereas rigid 3D porous materials such as MOFs are the most widely studied, they are not prominent amongst the leading materials for performance in terms of pX or oX selectivity. For example, the 0D non-porous coordination complex [Ni(NCS)2(ppp)4]17 and the 2D layered coordination network sql-1-Co-NCS18 are the most selective sorbents for oX. The leading pX favoring sorbents are the 0D organic molecular EtP6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 19 and the zeolite H/ZSM-5.13
19 and the zeolite H/ZSM-5.13
Whereas it is perhaps counter-intuitive to study non-porous molecular solids for gas and vapor separations, their performance to date for xylenes prompted us to study other classes of molecular solids that can form host–guest complexes or inclusion compounds upon contact with organic molecules.20 Such stimulus-induced phase transformations of host molecules has been exemplified in studies reported by the Atwood and Barbour groups.17,21 Recently, Huang et al. have developed a family of nonporous adaptive crystals, which exhibited superior performances for molecular adsorption and separation.22,23 With respect to C8 separations, Báthori,24 Kawahata,12 Zhang & Moore25 and Nassimbeni26 all demonstrated the potential for C8 isomer separation by “0D” molecular solids is promising. The nonporous adaptive crystals have been also demonstrated to be an ideal separation platform for C8 compounds.19,27,28 These contributions prompted us to investigate a very different class of nonporous adaptive crystals based on metal–organic coordination compounds, as detailed herein.
Results
The non-porous molecular complex M was synthesized by solvothermal reaction of AgClO4 with the ligand 2,3-bis[3-(pyridin-2-yl)-1H-pyrazol-1-yl·methyl]quinoxaline29,30 (see full details in ESI†). M offers π binding sites that are readily accessible thanks to its L-shaped geometry (Fig. S1†). As we report herein, although M forms a non-porous molecular crystal, it switches to C8 loaded phases when exposed to C8 isomers and readily reverts to M upon exposure to vacuum drying (Fig. 1). Single crystals of M were obtained from water/methanol under solvothermal conditions. Single crystal X-ray diffraction (SCXRD) results reveal that the Ag+ cations adopt a slightly distorted square planar coordination mode to four nitrogen atoms of two 3-(2-pyridyl)pyrazole species. The L-shaped cations feature strong C–H⋯π (2.79 Å and 3.32 Å) and C–H⋯N (2.69 Å) interactions (Fig. S1†), and form into a non-porous framework as indicated by Platon calculations (∼2.6%).31 | ||
| Fig. 1 Schematic illustration of the adsorption behavior of the non-porous molecular crystal M. M readily forms host–guest complexes upon exposure to xylene isomers in liquid, vapor or solution phases and can be recovered upon exposure to vacuum drying. | ||
Solvothermal reactions in the presence of each of the pure xylene isomers (ESI, Experimental section†) afforded crystals of the respective inclusion compounds with varying host:guest ratios, n, for pX (n = 4:5), mX (n = 2:1) and oX (n = 1:1) (Fig. 2, S2–S8 and Table S3†). The corresponding reaction with eB under the same conditions did not result in an inclusion compound. The crystal structures reveal that the formation of the inclusion compounds is driven by the rearrangement of M, two L-shaped building blocks, into a supramolecular box-like cage. This cage enclathrates the respective guests with one xylene molecule per cage. The cage dimensions are independent of the guest: 10.28 × 7.03 × 10.07 Å3 for pX; 10.18 × 6.98 × 10.04 Å3 for mX; 10.26 × 7.06 × 10.02 Å3 for oX. However, the interstitial spaces formed between the cages are different (pX: 3.43, 3.61 Å; mX: 3.47, 3.06 Å; oX: 4.65, 3.55 Å). It is the chemical environments between the cages that result in different host:guest ratios. For pX@M, there are four pX chemical environments. Two of them are inside the cage: one with ordered pX and the other with disordered pX. The other pX molecules lie between cages: one located between adjacent cages and the other lying in the gap of cages. For mX@M, all mX molecules lie in the center of cages and there are none out of the cages. For oX@M, one oX is inside the cage and the other lies between the cages (Fig. S3–S8†).
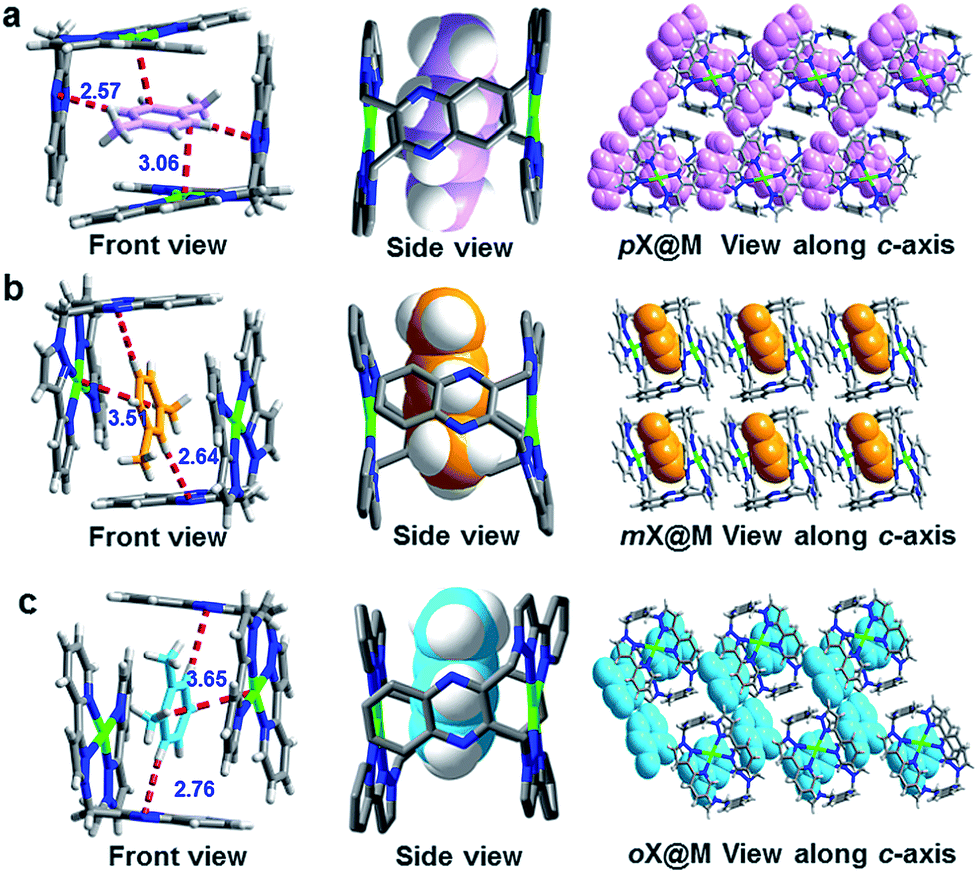 | ||
| Fig. 2 Single crystal structures of the host–guest complexes formed between M and (a) pX, (b) oX and (c) mX. | ||
Xylene selectivity studies in liquid
To study the xylene selectivity of M, solid–liquid sorption experiments using a 1:1:1 volumetric ratio of oX:mX:pX were conducted using various conditions (the solubility of the host in xylenes is negligible, Fig. S9†). Because of slow kinetics, a solvothermal method (Experimental section in ESI†) was ultimately preferred. The resulting material, pX/mX/oX@M, afforded a PXRD pattern resembling the calculated PXRD pattern of pX@M (Fig. 3a). 1H NMR measurements revealed that pX was the predominant isomer included in pX/mX/oX@M (Fig. 3b and Table S4, ESI†).
 | ||
| Fig. 3 Separation performance of M towards xylenes. (a) The PXRD pattern of pX/mX/oX@Mvs. the calculated PXRD patterns of oX@M, mX@M, pX@M. (b) The 1H NMR spectra (DMSO) of M after soaking in xylenes vs. reference extractions. pX is the predominant isomer: (I) mX; (II) mX@M; (III) oX; (IV) oX@M; (V) pX; (VI) pX@M; (VII) xylenes@M. (c) Kinetics of sorption for pX/oX at 383 K as determined by GC. (d) Relative amount of pX and oX extracted by M as measured by GC (average value of five extractions). | ||
The kinetics of solid–liquid sorption for M in a 1:1 mixture of pX and oX at 383 K was monitored by gas chromatography (GC) (Table S5†). As shown in Fig. 3c, the uptake of pX in M increased over time and after 2 h the mass percentage of pX reached ∼12.3% vs. ∼0.35% for oX. This result indicated a high selectivity of M towards pX (about 30:1 for pX & oX). Based upon this kinetic study, we conducted five replicates of this experiment (2 h at 383 K), and the average pX/oX selectivity was found to be 24.0. GC was then used to quantitatively determine the selectivity of M towards other pairs of xylene isomers following the same procedure used for pX/oX. Selectivity values of 24.0, 6.19, 10.36 and 3.93 were determined for pX/oX, pX/mX, pX/eB and mX/oX, respectively (Fig. S10–S13, Tables S6 and S7†). The pX/oX, pX/eB, mX/oX and pX/mX selectivity values demonstrated herein are among the highest values yet reported.32–37
Commercial grade xylenes are usually produced by methylation of toluene and benzene and contains pX, mX, oX and ethylbenzene (eB).1 A commercial grade xylenes C8 mixture (oX:mX:pX:eB = 20:20:20:40, v/v) was also tested using the same procedure. The results suggest that M strongly favors pX over the other C8 isomers with selectivity values consistent with those obtained from the 1:1 extractions (Fig. S14, ESI†).
To study the recyclability of M, we conducted variable temperature X-ray diffraction (VT-XRD) and thermogravimetric analysis (TGA) experiments. The TGA suggest the materials thermal stability until 573 K (Fig. 4a). The VT-XRD data suggest that increasing temperature causes pX@M to gradually transform back to M (Fig. 4b). 1H NMR experiments were conducted after vacuum drying (Experimental section in ESI†) of a sample of pX@M for 12 h and verified the presence of M (Fig. 4c). Recycled samples of M can be used for pX selection experiments at least 5 times without any performance decrease (Fig. 4d).
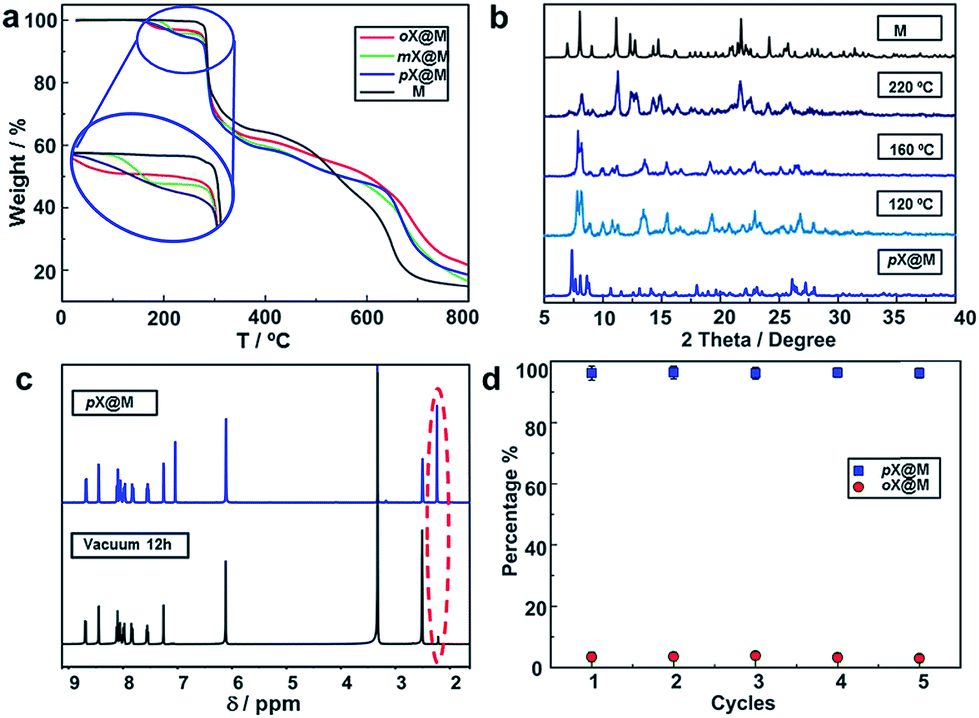 | ||
| Fig. 4 Recyclability of M. (a) The TGA profiles for M, oX@M, mX@M, pX@M. Loss of weight: pX@M > mX@M > oX@M. (b) VT-XRD experiments for pX@M. With the temperature increasing, the pattern of PXRD transformed from pX@M into M. (c) The 1H NMR spectra of pX@M and the sample after vacuum drying. (d) Relative uptake of pX and oX in M after M is recycled for 5 times. | ||
Xylene selectivity studies in vapor
Vapor sorption experiments were conducted to determine if C8 vapors can also induce switching from closed to open phases. The kinetics of xylene vapour sorption on M was studied at 298 K (Fig. 5a). It reveals that, under the same conditions, pX vapor can be adsorbed much more efficiently and sufficiently than mX or oX. This could be attributed to the smaller kinetic diameter of pX (Table S1†), which benefits the diffusion of pX in the structure of M for faster occupation of the “box-like” cage. It was observed that, whereas M switches in the presence of pX vapor, there are differences in terms of uptake and structure vs. the solution experiments. As shown in Fig. 5b, the PXRD of M after pX vapor sorption sample is consistent with that of M after contact with mX solution. The mass uptake and PXRD are consistent with n = 2:1 and pX molecules can be removed in vacuo drying to regenerate M (Fig. S15†).
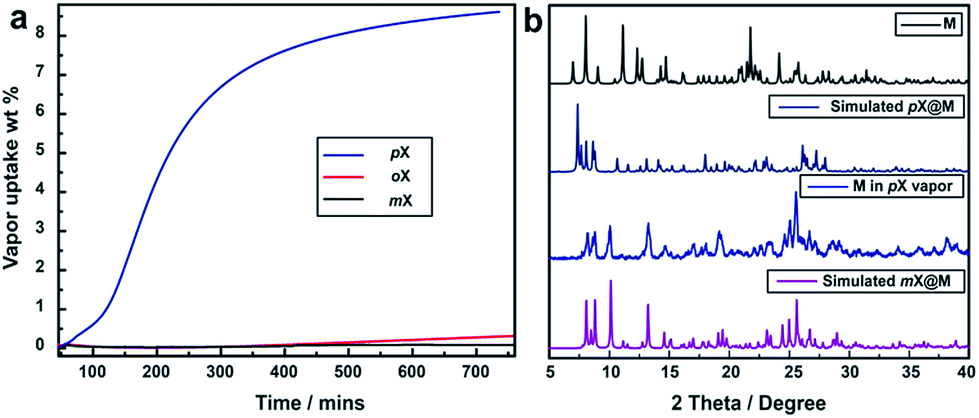 | ||
| Fig. 5 Vapor sorption of M. (a) Kinetics of pX (blue), mX (black), oX (red) vapour sorption for M at 298 K, respectively. (b) The PXRD of M in pX vapor sorption sample is most consistent with the mX@M in liquid. | ||
Vapour-phase binary mixture separation experiments were conducted on M and selectivities were determined by GC. Selectivity values were found to be 20.3, 5.4, 8.0 and 1.15 for pX/oX, pX/mX, pX/eB and mX/oX, respectively (Fig. S16–S20, Tables S8 and S9†). The hierarchy of the selectivity (pX > mX > oX) is consistent with that observed during the liquid sorption experiments. The pX/oX selectivity of 20.3 is to our knowledge among the highest value yet reported for vapor extractions (Table S2†).
Discussion
To understand the mechanism behind the observed selectivity of M for pX, we analyzed the crystal structures with particular emphasis upon host–guest interactions. For guests within the cage (Fig. 2 and S3–S8†), the shortest distance between pX molecules and M results from C–H⋯N interactions formed by aromatic C–H moieties and the quinoxaline rings. This distance is shortest for pX: 2.57 vs. 2.64 vs. 2.76 Å for pX, mX and oX, respectively. The interaction distances between the benzene ring of each xylene and Ag+ cations in the coordination complex are also shortest for pX (3.06 vs. 3.51 vs. 3.65 Å for pX, mX and oX, respectively). These differences in host–guest interactions can be attributed to shape differences of the C8 aromatics with pX enabling closer interactions, presumably because the methyl groups can orient towards the windows of the cage. This could be helps to explain why M exhibits high pX selectivity (Table S2†). Moreover, DFT calculations also proved that the binding energy of pX@M is higher than that of oX@M and mX@M (Table S10†). This is in consistent with the kinetics studies (Fig. 3c and 5a) and could be the reason why M has a high selectivity for pX.We note that, because M forms a distinct cage-like structure that serves as a host for xylenes, it exhibits a different binding phenomenon from that observed in other molecular compounds that exhibit such high selectivity, EtP6 (ref. 19) (pX selective) and [Ni(NCS)2(ppp)4]17 (oX selective, Table S2†). Further, the shape of the supramolecular cage formed by M enables us to gain insight into the selectivities observed herein, which for pX/oX and pX/eB are among the highest yet reported. The behavior observed herein is therefore most closely related to clathration approaches driven by weak van der Waals interactions reported by Atwood and Barbour et al.21
Conclusion
In summary, the L-shaped non-porous molecular material M exhibits new benchmarks for pX selectivity as it preferentially forms host–guest complexes with pX from liquid or vapor C8 mixtures. We attribute the performance of M to its ability to form a cage-like structure that is well-suited for the size and shape of pX. Although the approach taken herein, the use of a non-porous solid to serve as a selective sorbent, is perhaps counter-intuitive, it offers superior performance vs. rigid porous materials such as zeolites and MOFs. Moreover, as is likely to be the case for molecular materials in general, M is readily recyclable. Whereas the promise for non-porous molecular materials to serve as selective sorbents is high based upon this and other recent studies, slow kinetics and low uptake remain unsolved challenges for molecular materials. Future work will focus upon overcoming such problems.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (91256124, 21531005 and 21773168) and “111 Project” of China (B18030). We also gratefully acknowledge the support of Science Foundation Ireland (13/RP/B3529 and 16/IA/4624).Notes and references
-
J. Fabri, U. Graeser and T. A. Simo, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, Germany, 2000 Search PubMed
.
-
Materials for Separation Technologies, Energy and Emission Reduction Opportunities, Oak Ridge National Laboratory, Oak Ridge, TN, USA, 2005 Search PubMed
- M. Minceva and A. E. Rodrigues, AIChE J., 2007, 53, 138 CrossRef CAS
-
J. Scheirs and T. E. Long, Industrial Modern Polyesters: chemistry and technology of polyesters and copolyesters, Wiley, Chichester, 2003 Search PubMed
-
J. L. Pellegrino, Energy and Environmental Profile of the Chemicals Industry, U.S. Department of Energy, 2000 Search PubMed
- D. S. Sholl and R. P. Lively, Nature, 2016, 532, 435 CrossRef PubMed
- R. Krishna, Phys. Chem. Chem. Phys., 2015, 17, 39 RSC
- D. Peralta, G. Chaplais, J.-L. Paillaud, A. Simon-Masseron, K. Barthelet and G. D. Pirngruber, Microporous Mesoporous Mater., 2013, 173, 1 CrossRef CAS
-
A. Méthivier, in Zeolites for Cleaner Technologies, Catalytic Science Series, Imperial College Press, London, 2002, vol. 3, pp. 209–221 Search PubMed
-
R. Szostak, Handbook of Molecular Sieves: Structure, Springer, 1992 Search PubMed
- P. S. Barcia, D. Guimaraes, P. A. P. Mendes, J. A. C. Silva, V. Guillerm, H. Chevreau, C. Serre and A. E. Rodrigues, Microporous Mesoporous Mater., 2011, 139, 67 CrossRef CAS
- M. Kawahata, T. Hyodo, M. Tominaga and K. Yamaguchi, CrystEngComm, 2018, 20, 5667 RSC
- M. Rasouli, N. Yaghobi, S. Chitsazan and M. H. Sayyar, Microporous Mesoporous Mater., 2012, 150, 47 CrossRef CAS
- D.-Y. Koh, B. A. McCool, H. W. Deckman and R. P. Lively, Science, 2016, 353, 804 CrossRef CAS PubMed
- W.-Y. Zhang, Y.-J. Lin, Y.-F. Han and G.-X. Jin, J. Am. Chem. Soc., 2016, 138, 10700 CrossRef CAS PubMed
- J. Jarvis, P. He, A. Wang and H. Song, Fuel, 2019, 236, 1301 CrossRef CAS
- M. Lusi and L. J. Barbour, Angew. Chem., Int. Ed., 2012, 51, 3928 CrossRef CAS PubMed
- S.-Q. Wang, S. Mukherjee, E. Patyk-Kazmierczak, S. Darwish, A. Bajpai, Q.-Y. Yang and M. J. Zaworotko, Angew. Chem., Int. Ed., 2019, 58, 6630 CrossRef CAS PubMed
- K. Jie, M. Liu, Y. Zhou, M. A. Little, A. Pulido, S. Y. Chong, A. Stephenson, A. R. Hughes, F. Sakakibara, T. Ogoshi, F. Blanc, G. M. Day, F. Huang and A. I. Cooper, J. Am. Chem. Soc., 2018, 140, 6921 CrossRef CAS PubMed
- H.-N. Zhang, Y. Lu, W.-X. Gao, Y.-J. Lin and G.-X. Jin, Chem.–Eur. J., 2018, 24, 18913 CrossRef CAS PubMed
- J. L. Atwood, L. J. Barbour, A. Jerga and B. L. Schottel, Science, 2002, 298, 1000 CrossRef CAS PubMed
- K. C. Jie, Y. J. Zhou, E. R. Li, Z. T. Li, R. Zhao and F. H. Huang, J. Am. Chem. Soc., 2017, 139, 15320 CrossRef CAS PubMed
- K. C. Jie, Y. J. Zhou, E. R. Li and F. H. Huang, Acc. Chem. Res., 2018, 51, 2064 CrossRef CAS PubMed
- M. M. Wicht, L. R. Nassimbeni and N. B. Báthori, Polyhedron, 2019, 163, 7 CrossRef CAS
- D. Venkataraman, S. Lee, J. Zhang and J. S. Moore, Nature, 1994, 371, 591 CrossRef CAS
- L. R. Nassimbeni, Acc. Chem. Res., 2003, 36, 631 CrossRef CAS PubMed
- K. C. Jie, M. Liu, Y. J. Zhou, M. A. Little, S. Bonakala, S. Y. Chong, A. Stephenson, L. J. Chen, F. H. Huang and A. I. Cooper, J. Am. Chem. Soc., 2017, 139, 2908 CrossRef CAS PubMed
- B. Gao, L. L. Tan, N. Song, K. Li and Y. W. Yang, Chem. Commun., 2016, 52, 5804 RSC
- R.-Q. Zou, C.-S. Liu, Z. Huang, T.-L. Hu and X.-H. Bu, Cryst. Growth Des., 2006, 6, 99 CrossRef CAS
- F. Li, J. K. Clegg, L. Goux-Capes, G. Chastanet, D. M. D'Alessandro, J.-F. Letard and C. J. Kepert, Angew. Chem., Int. Ed., 2011, 50, 2820 CrossRef CAS PubMed
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9 CrossRef CAS PubMed
- Z.-Y. Gu, D.-Q. Jiang, H.-F. Wang, X.-Y. Cui and X.-P. Yan, J. Phys. Chem. C, 2010, 114, 311 CrossRef CAS
- J.-M. Lin, C.-T. He, P.-Q. Liao, R.-B. Lin and J.-P. Zhang, Sci. Rep., 2015, 5, 11537 CrossRef CAS PubMed
- J. E. Warren, C. G. Perkins, K. E. Jelfs, P. Boldrin, P. A. Chater, G. J. Miller, T. D. Manning, M. E. Briggs, K. C. Stylianou, J. B. Claridge and M. J. Rosseinsky, Angew. Chem., Int. Ed., 2014, 53, 4592 CrossRef CAS PubMed
- M. Rasouli, N. Yaghobi, F. Allahgholipour and H. Atashi, Chem. Eng. Res. Des., 2014, 92, 1192 CrossRef CAS
- M. Rasouli, N. Yaghobi, S. Z. M. Gilani, H. Atashi and M. Rasouli, Chin. J. Chem. Eng., 2015, 23, 64 CrossRef CAS
- F. Vermoortele, M. Maes, P. Z. Moghadam, M. J. Lennox, F. Ragon, M. Boulhout, S. Biswas, K. G. M. Laurier, I. Beurroies, R. Denoyel, M. Roeffaers, N. Stock, T. Düren, C. Serre and D. E. De Vos, J. Am. Chem. Soc., 2011, 133, 18526 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1889509–1889512. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc02621e |
| This journal is © The Royal Society of Chemistry 2019 |