
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning the structure, reactivity and magnetic communication of nitride-bridged uranium complexes with the ancillary ligands†
Chad T.
Palumbo
 a,
Luciano
Barluzzi
a,
Rosario
Scopelliti
a,
Ivica
Zivkovic
b,
Alberto
Fabrizio
a,
Clémence
Corminboeuf
*a and
Marinella
Mazzanti
*a
a,
Luciano
Barluzzi
a,
Rosario
Scopelliti
a,
Ivica
Zivkovic
b,
Alberto
Fabrizio
a,
Clémence
Corminboeuf
*a and
Marinella
Mazzanti
*a
aInstitut des Sciences et Ingénierie Chimiques, Ecole Polytechnique Fédérale de Lausanne (EPFL), 1015 Lausanne, Switzerland. E-mail: marinella.mazzanti@epfl.ch
bLaboratory for Quantum Magnetism, Institute of Physics, Ecole Polytechnique Fédérale de Lausanne (EPFL), CH-1015 Lausanne, Switzerland
First published on 13th August 2019
Abstract
Molecular uranium nitride complexes were prepared to relate their small molecule reactivity to the nature of the U![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NU bonding imposed by the supporting ligand. The U4+–U4+ nitride complexes, [NBu4][{((tBuO)3SiO)3U}2(μ-N)], [NBu4]-1, and [NBu4][((Me3Si)2N)3U}2(μ-N)], 2, were synthesised by reacting NBu4N3 with the U3+ complexes, [U(OSi(OtBu)3)2(μ-OSi(OtBu)3)]2 and [U(N(SiMe3)2)3], respectively. Oxidation of 2 with AgBPh4 gave the U4+–U5+ analogue, [((Me3Si)2N)3U}2(μ-N)], 4. The previously reported methylene-bridged U4+–U4+ nitride [Na(dme)3][((Me3Si)2)2U(μ-N)(μ-κ2-C,N-CH2SiMe2NSiMe3)U(N(SiMe3)2)2] (dme = 1,2-dimethoxyethane), [Na(dme)3]-3, provided a versatile precursor for the synthesis of the mixed-ligand U4+–U4+ nitride complex, [Na(dme)3][((Me3Si)2N)3U(μ-N)U(N(SiMe3)2)(OSi(OtBu)3)], 5. The reactivity of the 1–5 complexes was assessed with CO2, CO, and H2. Complex [NBu4]-1 displays similar reactivity to the previously reported heterobimetallic complex, [Cs{((tBuO)3SiO)3U}2(μ-N)], [Cs]-1, whereas the amide complexes 2 and 4 are unreactive with these substrates. The mixed-ligand complexes 3 and 5 react with CO and CO2 but not H2. The nitride complexes [NBu4]-1, 2, 4, and 5 along with their small molecule activation products were structurally characterized. Magnetic data measured for the all-siloxide complexes [NBu4]-1 and [Cs]-1 show uncoupled uranium centers, while strong antiferromagnetic coupling was found in complexes containing amide ligands, namely 2 and 5 (with maxima in the χ versus T plot of 90 K and 55 K). Computational analysis indicates that the U(μ-N) bond order decreases with the introduction of oxygen-based ligands effectively increasing the nucleophilicity of the bridging nitride.
NU bonding imposed by the supporting ligand. The U4+–U4+ nitride complexes, [NBu4][{((tBuO)3SiO)3U}2(μ-N)], [NBu4]-1, and [NBu4][((Me3Si)2N)3U}2(μ-N)], 2, were synthesised by reacting NBu4N3 with the U3+ complexes, [U(OSi(OtBu)3)2(μ-OSi(OtBu)3)]2 and [U(N(SiMe3)2)3], respectively. Oxidation of 2 with AgBPh4 gave the U4+–U5+ analogue, [((Me3Si)2N)3U}2(μ-N)], 4. The previously reported methylene-bridged U4+–U4+ nitride [Na(dme)3][((Me3Si)2)2U(μ-N)(μ-κ2-C,N-CH2SiMe2NSiMe3)U(N(SiMe3)2)2] (dme = 1,2-dimethoxyethane), [Na(dme)3]-3, provided a versatile precursor for the synthesis of the mixed-ligand U4+–U4+ nitride complex, [Na(dme)3][((Me3Si)2N)3U(μ-N)U(N(SiMe3)2)(OSi(OtBu)3)], 5. The reactivity of the 1–5 complexes was assessed with CO2, CO, and H2. Complex [NBu4]-1 displays similar reactivity to the previously reported heterobimetallic complex, [Cs{((tBuO)3SiO)3U}2(μ-N)], [Cs]-1, whereas the amide complexes 2 and 4 are unreactive with these substrates. The mixed-ligand complexes 3 and 5 react with CO and CO2 but not H2. The nitride complexes [NBu4]-1, 2, 4, and 5 along with their small molecule activation products were structurally characterized. Magnetic data measured for the all-siloxide complexes [NBu4]-1 and [Cs]-1 show uncoupled uranium centers, while strong antiferromagnetic coupling was found in complexes containing amide ligands, namely 2 and 5 (with maxima in the χ versus T plot of 90 K and 55 K). Computational analysis indicates that the U(μ-N) bond order decreases with the introduction of oxygen-based ligands effectively increasing the nucleophilicity of the bridging nitride.
Introduction
Uranium nitride compounds are of high interest because of their recently discovered ability to promote the stoichiometric transformation of small molecules such as CO2, CO and N2,1 which are widely available C1 and N feedstocks for the synthesis of high-value organic compounds.2 Notably, examples of small molecule activation by nitride compounds remain scarce3 both in d-block4 and in f-block chemistry. Moreover, uranium nitrides are driving experimental and computational studies because they are well suited to probe the nature of bonding in actinide compounds5,6 and to promote magnetic communication between uranium centers in various oxidation states.1i,7 | ||
| Chart 1 Previously characterized diuranium (U(IV)) nitride-bridged complexes supported by silylamide and siloxide ligands. | ||
The number of isolated terminal and bridging uranium nitrides continues to increase,1a,1e,1f,1i,5b-5d,7b,8 but their synthesis remains far from trivial in part because of their high reactivity that may result in solvent deprotonation, C–H activation8i,9 or C–O cleavage1d of the ancillary ligand to afford amido or imido complexes. Moreover, studies of the reactivity of uranium nitrides have so far been limited to three ligand systems,1c,1d,1f-1i namely, complexes of the tris(tertbutoxy)siloxide ligand OSi(OtBu)3, the U5+–U5+ nitride [(U(N[t-Bu]Ar)3)2(μ-N)][B(ArF)4] with amide ligands, and lastly terminal nitrides supported by the tripodal tris-amido ligand TrenTIPS (TrenTIPS = N(CH2CH2NSiiPr3)3). The nitride in complexes of the siloxide and TrenTIPS ligands acts as a strong nucleophile and reacts with CO2, CO, and CS2 to give products of N–C bond formation.1c,1d,1f-1i The siloxide supported complexes also effect the cleavage of H2 leading to N–H formation.1c,1d,1f-1i In contrast, the aforementioned [(U(N[t-Bu]Ar)3)2(μ-N)][B(ArF)4] was reported unreactive with CO but showed electrophilic reactivity towards cyanide resulting in the formation of a cyanoimide bridged U4+–U4+ complex.5c
The geometric and electronic parameters leading to the observed differences in reactivity remain unclear. Moreover, the effects of the inner-sphere cation on the reactivity of the siloxide supported complex [Cs{(LO)3U}2(μ-N)] (LO = (tBuO)3SiO), [Cs]-1 (Chart 1),8g,1b was not elucidated. It occurred to us that the bulky silylamide ligand (LN = N(SiMe3)2) is well suited for determining ligand effects on the reactivity and magnetic properties of bridging nitrides because it is more electron-donating than the siloxide ligand LO with only a slightly higher steric demand.10 Earlier room temperature studies from the Hayton group8f with the LN ligand led to the synthesis of the methylene-bridged U4+–U4+ nitride complex [Na(dme)3][(LN)2U(μ-N)(μ-κ2-C,N-CH2SiMe2NSiMe3)-U(LN)2] (dme = 1,2-dimethoxyethane), 3 (Chart 1).
Here we report the synthesis of three new diuranium U4+–U4+ nitride complexes, namely the all siloxide [NBu4][{(LO)3U}2(μ-N)], [NBu4]-1, the all-amide [NBu4][{(LN)3U}2(μ-N)] (LN = N(SiMe3)2), 2, and the mixed-ligand [Na(dme)3][{(LN)3U}{(LO)(LN)2U}(μ-N)], 5, which allow a straightforward analysis of the effects of cation and ancillary ligand on nitride reactivity. Comparative reactivity studies with small molecules, CO, CO2, and H2 showed dramatic differences between the nitride complexes depending on the supporting ligands. No reactivity was shown by the all-amide complex 2, but high nucleophilic reactivity of the nitride was observed with CO, CO2, and H2 for the all-siloxide complex [NBu4]-1. Moreover, the presence of only one siloxide ligand out of six supporting ligands is sufficient to enable CO, CO2 activation by complex 5. Computational studies inferred that the different behavior can be interpreted in terms of differences in the UNU bonding between the all-siloxide and the all-silylamide ligand. The increased orbital overlap between the UNU centers found in the all-silylamide complex results in stronger magnetic coupling between the two uranium centers and reduced nucleophilic character of the bridging nitride compared to the all-siloxide one.
Results and discussion
Preparation of the nitride complexes
The U(III) complexes [U(LN)3]11 and [U(LO)2(μ-LO)]2 (ref. 12) react with NBu4N3 to produce [NBu4][{(LN)3U}2(μ-N)] (L = LO = OSi(OtBu)3, [NBu4]-1; L = LN = N(SiMe3)2, 2), Scheme 1.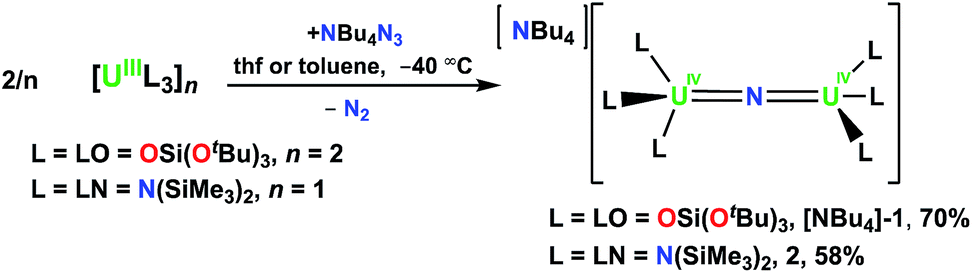 | ||
| Scheme 1 Synthesis of [NBu4][{(L)3U}2(μ-N)] (L = LO = OSi(OtBu)3, [NBu4]-1; L = LN = N(SiMe3)2, 2). | ||
Compound [NBu4]-1 is obtained in 70% yield using a method similar to that used to prepare the previously reported Cs analogue [Cs]-18g in which reduction of azide by [U(LO)2(LO)]2 in thf is done at low temperature (−40 °C). Single-crystals of [NBu4]-1 were obtained after storage of an 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 toluene/hexane solution at −40 °C. The 1H NMR spectrum of [NBu4]-1 shows resonances for the NBu4+ cation and a single resonance for the OSi(OtBu)3 in thf-d8 and in toluene-d8, indicating fluxional behavior of the siloxide ligands. Similar fluxional behavior had been observed for [Cs]-1 both in thf and toluene solution. Complex 2 can also be synthesized in thf, but in toluene precipitates as a brown powder in 58% yield. On several occasions while investigating the low temperature synthesis of 2 in toluene-d8, single crystals of 2 grew in the NMR tube, which allowed its structural characterization. The 1H NMR spectrum of 2 at −60 °C in thf-d8 shows two narrow and four broad resonances attributed to the SiMe3 groups, suggesting a rigid structure under these conditions. As the temperature is elevated to room temperature, the resonances become increasingly broadened until they are indistinguishable from the baseline; at room temperature, only those of the [NBu4]+ cation are observed.
:1 toluene/hexane solution at −40 °C. The 1H NMR spectrum of [NBu4]-1 shows resonances for the NBu4+ cation and a single resonance for the OSi(OtBu)3 in thf-d8 and in toluene-d8, indicating fluxional behavior of the siloxide ligands. Similar fluxional behavior had been observed for [Cs]-1 both in thf and toluene solution. Complex 2 can also be synthesized in thf, but in toluene precipitates as a brown powder in 58% yield. On several occasions while investigating the low temperature synthesis of 2 in toluene-d8, single crystals of 2 grew in the NMR tube, which allowed its structural characterization. The 1H NMR spectrum of 2 at −60 °C in thf-d8 shows two narrow and four broad resonances attributed to the SiMe3 groups, suggesting a rigid structure under these conditions. As the temperature is elevated to room temperature, the resonances become increasingly broadened until they are indistinguishable from the baseline; at room temperature, only those of the [NBu4]+ cation are observed.
Previously, it was shown that [Cs]-1 decomposes slowly at room temperature and more rapidly at 80 °C to afford the tert-butyl imide complex, [Cs{(LO)3U(μ-NtBu)(μ-O2Si(OtBu)2)U(LO)2}],1d due to the high nucleophilic character of the bridging nitride in [Cs]-1. Monitoring the 1H NMR spectrum of [NBu4]-1 at room temperature or at 80 °C demonstrates that its thermal stability is similar to that of [Cs]-1, but an imide product could not be identified in the decomposition mixture.
1H NMR studies showed that 2 decomposes slowly (over six days) at room temperature and more rapidly at 65 °C (2 h) in thf-d8 to afford a mixture of species. Among the products, the proton resonances of the cyclometalate uranium nitride anion, [(LN)2U(μ-N)(μ-κ2-C,N-CH2SiMe2NSiMe3)U(LN)2]−, were identified.8f Thus, a NBu4+ analogue of the U(IV) nitride bridged complex [Na(dme)3][(LN)2U(μ-N)(μ-κ2-C,N-CH2SiMe2NSiMe3)U(LN)2] (dme = 1,2-dimethoxyethane), 3, reported previously by Hayton and coworkers,8f is formed during the decomposition. These results indicate that the cyclometallation side-reaction observed by Hayton and coworkers during the reaction of the [U(LN)3] precursor with azide is prevented when the reaction is performed at low temperature.
In order to investigate the effect of the uranium oxidation state on the spectroscopic features and reactivity of the N(SiMe3)2 nitride complexes, a U4+–U5+ analogue of 2, namely [{(LN)3U}2(μ-N)], 4, was also prepared. Complex 4 was obtained in 60% yield by oxidation of 2 with 1 equiv. of AgBPh4, Scheme 2. The 1H NMR spectrum of 4 is similar to that of 2 in that it consists of two narrow and four broad resonances at −60 °C and at room temperature, the resonances broaden and become indistinguishable from the baseline. Single crystals of 4 characterizable by X-ray crystallography were obtained by cooling a concentrated toluene solution to −40 °C.
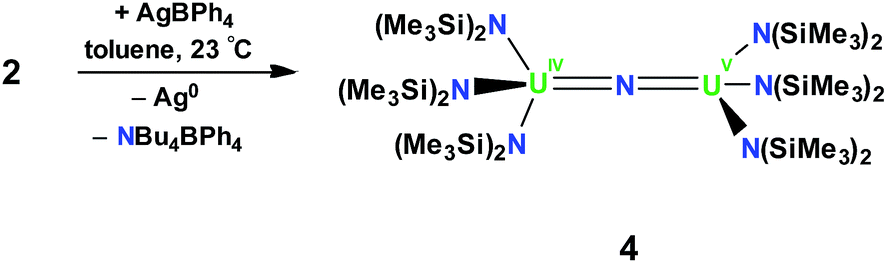 | ||
| Scheme 2 Synthesis of [{(LN)3U}2(μ-N)], 4. | ||
In order to probe further ligand effects on reactivity and bonding, the synthesis of OSi(OtBu)3/N(SiMe3)2 mixed-ligand uranium nitride complexes was pursued. We first explored protonolysis reactions and attempted to synthesize [NBu4]-1 by reacting 2 with 6 equiv. of HOSi(OtBu)3, but 1H NMR spectroscopy indicated that no reaction occurred between these two reagents at room temperature. This suggests that the U–N bonds in complex 2 are significantly more resistant to protonolysis compared to those in the U(III) complex [U(LN)3]11 that reacts rapidly with HOSi(OtBu)3 to afford the protonolysis product [U(LO)2(μ-LO)]2.12
However, the cyclometalate U4+ nitride complex, [Na(dme)3][(LN)2U(μ-N)(μ-κ2-C,N-CH2SiMe2NSiMe3)U(LN)2], 3,8f reacts immediately with HOSi(OtBu)3, affording a mixed-ligand nitride of N(SiMe3)2 and OSi(OtBu)3 in 70% yield, [Na(dme)3][(LN)3U(μ-N)U(LN)2(LO)], 5, Scheme 3. The room temperature 1H NMR spectrum of 5 in thf-d8 displays three resonances; two resonances at 1.08 ppm and −0.02 ppm are attributed to the N(SiMe3)2 ligands and another broad resonance at −5.40 ppm has been assigned to OSi(OtBu)3. Single crystals of 5 were grown by diffusion of hexane into an Et2O solution of 5 in the presence of dme.
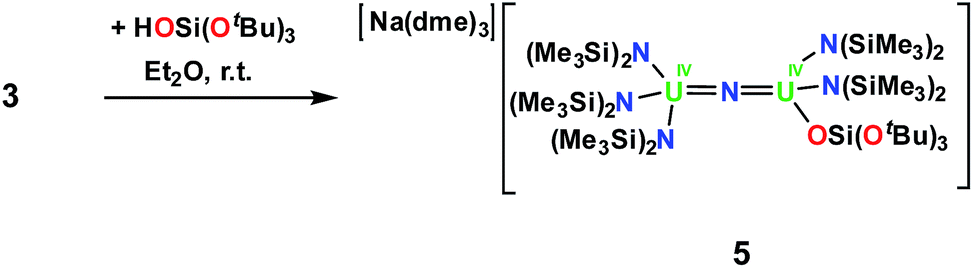 | ||
| Scheme 3 Synthesis of [Na(dme)3][(LN)3U(μ-N)U(LN)2LO], 5. | ||
Structural characterization of the nitride-bridged complexes
The solid-state structures of complexes [NBu4]-1, 2, 4 and 5 were determined by X-ray diffraction studies. The metrical parameters are presented in Table 1, including those previously reported for complex [Cs]-18g and [Na(dme)2(tmeda)]-3 (ref. 8f) for comparison. The molecular structure of 4 (Fig. S39†) and a discussion of the metrical parameters of 4 are presented in the ESI.†| Complex | [Cs]-1 | [NBu4]-1 | 5 | [Na(dme)2(tmeda)]-3 (ref. 8f) | 2 | 4 |
|---|---|---|---|---|---|---|
| a Taken as an average of 1.5 molecules in the unit cell of [NBu4][{(LN)3U}2(μ-N)], 2. b Defined as the average angle between the two closest adjacent planes defined by Nnitride, U, and Osiloxide or Namide atoms. | ||||||
| U–Nnitride | 2.058(5), 2.079(5) | 2.032(2), 2.067(2) | 2.055(4), 2.066(4) | 1.95(1), 2.12(1) | 2.076(5), 2.083(5), 2.075(2) | 2.080(5), 2.150(5) |
| U–(Osiloxide)avg | 2.19(1) | 2.2(1) | 2.208(4) | — | — | — |
| U–(Namide)avg | — | — | 2.35(1) | 2.35(1) | 2.35(1) | 2.274(5) |
| U–N–Ua | 170.3(3) | 172.2(2), 174(1), 177(1) | 168.4(3) | 123.5(5) | 179(1) | 179.4(3) |
| Twist angleb | 24(11) | 6(2) | 15(1) | — | 1.7(4) | 18(1) |
Complex [NBu4]-1 crystallizes in the I2 space group and its molecular structure (Fig. 1) shows the presence of a U4+–U4+ complex bridged by a nitride ligand. In the solid-state structure of [NBu4]-1, the U4+ ions of [NBu4]-1 are disordered between two positions that were refined with 50% occupancy. Analysis of the X-ray data of [NBu4]-1 reveals significant differences in the overall structure with respect to the previously reported [Cs]-1, but the disorder of the U–N–U core is such that its parameters are not reliable. In [NBu4]-1, each U(IV) ion is four-coordinate and in a pseudo tetrahedral coordination environment bound by three κ1-OSi(OtBu)3 ligands and one μ-nitride ligand, which bridges the two [(LO)3U]+ units. The OSi(OtBu)3 ligands are in a staggered orientation with respect to those of the adjacent [(LO)3U]+ moiety as reflected in the 6(2)° twist angle. In the previously reported8g structure of [Cs]-1, the binding situation is more complicated. There are one κ1-OSi(OtBu)3, one κ2-OSi(OtBu)3, and four μ-κ2-O,OtBu-OSi(OtBu)3 ligands which bridge U(IV) and Cs+ cations. The inner-sphere Cs+ is located at the apical position of the nitride ligand. The metal–siloxide bond distances/angles are very similar in [NBu4]-1 and [Cs]-1. The disorder in the uranium atoms gives three distinct U–N–U angles (U1A–N–U1A = 177(1)°, U1B–N–U1B = 174(1)° and U1A–N–U1B = 172.2(2)°) and suggests many possibilities of U–N–U angle in the solid state. This could indicate the presence of two molecules with a UNU angle of 172.2(2)° or four molecules with different UNU angles (two at 172.2(2)°, one at 177(1)°, and one at 174(1)°).
 | ||
| Fig. 1 Molecular structure of (a) [NBu4][{(LO)3U}2(μ-N)], [NBu4]-1, and (b) [NBu4][{(LN)3U}2(μ-N)], 2, with thermal ellipsoids drawn at the 50% probability level. Hydrogen atoms, the methyl groups of the tert-butyl moieties and the NBu4+ cation have been omitted for clarity. | ||
 | ||
| Fig. 2 Molecular structure of [Na(dme)3][(LN)3U(μ-N)U(LN)2LO], 5, with thermal ellipsoids drawn at the 50% probability level. Hydrogen atoms and a [Na(dme)3]+ cation are omitted for clarity. | ||
The structure of the [{(LN)3U}2(μ-N)]− anion (Fig. 1b) in 2 shows the presence of a dinuclear U(IV) complex with two [(LN)3U]+ units bridged by a nitride ligand. The coordination geometry of each four-coordinate uranium ion is pseudo tetrahedral and the arrangement of the N(SiMe3)2 ligands in the two [((LN)3U]+ units is staggered as reflected by the 1.7(4)° twist angle. The 178.7(2)° and 180.0° U–N–U angles found in 2 are larger than that found in the siloxide complex Cs-1 and are very close to linearity. For comparison, the angle in the previously reported [NBu4][{((3,5-Me2C6H3)(tBu)N)3U}2(μ-N)]5c complex containing the (3,5-Me2C6H3)(tBu)N amido ligand is 175.1(2)°. The other metrical parameters in [NBu4][{((3,5-Me2C6H3)(tBu)N)3U}2(μ-N)] are similar to 2 (see Table S2 in the ESI†), but changes between the U4+–U4+ and U4+–U5+ complexes differ in the two classes of compounds. The U–Nnitride distances in the U4+–U5+ compound 4 are different suggesting the presence of localized valence. A detailed structural comparison of 2 and 4 with [NBu4][{((3,5-Me2C6H3)(tBu)N)3U}2(μ-N)] and [{((3,5-Me2C6H3)(tBu)N)3U}2(μ-N)] is presented in the ESI.†
The molecular structure of the mixed-ligand complex 5 shows a dinuclear nitride-bridged U(IV) complex with an outer sphere cation, [Na(dme)3]+, (Fig. 2). The [(LN)2(LO)U(μ-N)U(LN)3]− anion shows two different ligand environments for the two U(IV) centers; one U(IV) is bound by two N(SiMe3)2 amide ligands and one κ1-OSi(OtBu)3 siloxide ligand while the second U(IV) is ligated by three amides. Both U(IV) ions are four-coordinate in a pseudotetrahedral coordination environment and the ligands are gauche with respect to those of the adjacent U(IV) ion. A comparison of the structure of 5 with those of [Cs]-1 and 2 shows they have similar U–Nnitride and U–Osiloxide and U–Namide bond distances. Interestingly, the presence of only one siloxide ligand results in a significant deviation of the U–N–U angle from linearity. The value of the U–N–U angle in 5 (168.4(3)°) is close to that found in Cs-1 (170.3(3)°) containing six siloxide ligands. These structural data indicate that the ligands significantly affect the solid-state metrical parameters of the UNU core.
We assessed the small molecule reactivity of the complexes to explore the effect of ancillary ligands on the reactivity.
Small molecule reactivity
The reactivity of the newly prepared nitride-bridged complexes with CO, CO2, and H2 was investigated and compared with that of [Cs]-11b-1d,1h with the objective of relating differences in reactivity to structure. The reactivity of 5–10 mM solutions of the complexes was monitored by 1H and 13C NMR spectroscopies and X-ray crystallography when single crystals were obtainable. The NMR spectra and additional X-ray characterization data are presented in the ESI.†In our previous work, computational studies had pointed out the importance of cooperative binding of CO by the multimetallic U–N–Cs nitride fragment. Therefore, we investigated the reaction of the [NBu4]-1 that does not contain a bound Cs+ cation. The reaction of [NBu4]-1 with 13CO in both toluene-d8 and thf-d8 also showed conversion of 13CO to 13CN−. In toluene-d8, the 1H NMR spectrum of the reaction mixture is more complicated than for the [Cs]-11c case and, among the products, the resonances of the known μ-oxo complex, [{(LO)3U}2(μ-O)],7a and of the tetrakis(siloxide) complex, [U(LO)4],13 were identified, and indicated ligand scrambling reactions. The tetrakis(siloxide) product of ligand scrambling was observed for [Cs]-1 only in trace amounts or only after heating solutions of [Cs{(LO)3U}2(μ-CN)(μ-O)] to 66 °C. The difference in behavior could be explained by an increased stability that the cesium cation provides to [Cs{(LO)3U}2(μ-CN)(μ-O)]. Notably, the inner sphere cesium cation binds four siloxide oxygen atoms and the bridging oxo, which could hold together the overall structure and prevent scrambling. In the thf-d8 reaction of [NBu4]-1 with 13CO, only a single product was observed in the 1H NMR spectrum that resonates at 2.66 ppm. In the 13C spectrum of the reaction mixture in thf-d8, we identified a resonance at 634.7 ppm, which we have assigned to a uranium bound 13CN− ligand. In both the toluene-d8 and thf-d8 reactions, when the reaction residue is hydrolysed with pD = 12 D2O, 13CN− is observed as the only product by 13C NMR spectroscopy. The spectra are shown in the ESI (Fig. S15–S19†).
A solution of the diuranium(IV) amide complex 2 was monitored by 1H NMR spectroscopy after exposure to 1 atm of CO. After 5 h, only minor resonances consistent with decomposition were observed. The U4+–U5+ amide complex 4 also showed no reactivity when exposed to 1 atm of CO.
The 13CO reactivity of the mixed-ligand 5 was explored in thf-d8 (Fig. S23–S26†) but not in toluene-d8 due to its insolubility. Complex 5 reacts with 1–2 equiv. of 13CO, but more slowly than the [Cs]-1 and [NBu4]-1 complexes. Full consumption of 5 was observed only after ca. 24 h and the 1H NMR spectrum appeared complicated. Analysis of the reaction mixture by 13C NMR spectroscopy in thf-d8 did not allow an assignment of a resonance for a bound 13CN− ligand. However, after removal of the volatiles and hydrolysis with pD = 12 D2O, 13CN− was identified by 13C NMR spectroscopy. Attempts to grow single crystals from reactions with 1–2 equiv. of natural isotope abundance CO produced only amorphous powders.
To test whether a U(IV) nitride complex without a OSi(OtBu)3 ligand would react with CO, we examined 3 (Fig. S20–S22†). We report only its reactivity in thf-d8 due to its limited solubility in toluene-d8. The 1H NMR spectra in thf-d8 from reactions of 3 with either 1 atm of CO or 1–2 equiv. of 13CO appear identical (see Fig. S24 and S25 in the ESI†) suggesting that the product distribution is not affected by the stoichiometry of CO. In the 1–2 equiv. of 13CO reaction, complete consumption of 3 was observed after ca. 9 h. Similar to the 5 result, we did not identify a resonance attributable to a bound 13CN− ligand in the thf-d813C NMR spectrum, but did observe 13CN− when the reaction residue was hydrolysed with pD = 12 D2O. From the reactions of 3 with 1 atm of CO, crystals of several U(IV) oxo products were characterized by X-ray crystallography, namely the trimeric bis(amide) oxo, [(LN)2U(O)]3, 6, and an oxo product where CO had inserted into the uranium–methylene bond, [(LN)2U(μ-O)U(μ-κ2-O,N-OC(C)SiMe2NSiMe3)U(LN)2], 7, Fig. 3 (see ESI† for structural description). The products obtained from the reaction of 3 with 1 atm of CO are illustrated in Scheme 4.
 | ||
| Fig. 3 Molecular structure of (a) [(LN)2U(O)]3, 6, and (b) [(LN)2U(μ-O)U(μ-κ2-O,N-OC(C)SiMe2NSiMe3)U(LN)2], 7, with thermal ellipsoids drawn at the 50% probability level. Hydrogen atoms have been omitted for clarity. The asymmetric unit contains 2 independent molecules. Only 1 is shown for sake of clarity. Selected bond lengths and angles of [(LN)2U(O)]3: U–(Namide)avg, 2.28(1) Å; U–(Ooxo)avg, 2.10(2) Å; U–O–U, 144(1)°. Selected bond lengths and angles of [(LN)2U(μ-O)U(μ-κ2-O,N-OC(C)SiMe2NSiMe3)U(LN)2]: U1–(Namide)avg, 2.27(4) Å; U2–(Namide)avg, 2.26(1) Å; U1–O1, 2.082(15) Å; U2–O1, 2.130(15) Å; U1–N3, 2.256(16) Å; U2–O2, 2.074(13) Å; C36–C37, 1.32(3); U1–O1–U2, 150.3(7). | ||
 | ||
| Scheme 4 Reaction of [Na(dme)3][(LN)2U(μ-N)(μ-κ2-C,N-CH2SiMe2NSiMe3)U(LN)2], 3, with CO. | ||
The CO insertion chemistry has been observed previously in mononuclear uranium(IV) metallacycles,14 but these results demonstrate its extension to uranium nitride complexes.
The reactivity of the all-siloxide complexes [Cs]-1 and [NBu4]-1, which effect the cleavage of CO under ambient conditions, differs significantly from that of the all-amide complexes 2 and 4, which do not react with CO despite differences in their oxidation states. The CO cleavage reactivity is reinstated in the mixed ligand amide siloxide 5 and methanide 3 complexes, although in the case of 3, CO insertion into the U–C bond also occurs concomitantly with CO cleavage. These results suggest reduced nucleophilic character of the nitride in the all-amide complexes compared to those with siloxide or methanide ligands.
:1 toluene/hexane mixture at −40 °C, Fig. 4. The 1H NMR spectrum generated from crystals of 8 in toluene-d8 shows a broad resonance ranging 2–0 ppm assigned to OSi(OtBu)3 of 8. The 13C NMR spectrum in toluene-d8 shows a broad resonance at 141.5 ppm assigned to the bound dicarbamate ligand. Hydrolysis of crystals of 8 with pD = 12 D2O and analysis by 13NMR spectroscopy showed only D13CO3−. Similar results were obtained when the reaction was performed in thf and hydrolysis of the reaction residue similarly showed only D13CO3−, Fig. S30.†
 | ||
| Scheme 5 Reaction of [NBu4][{(LO)3U}2(μ-N)], [NBu4]-1, with CO2. | ||
 | ||
| Fig. 4 Molecular structure of [NBu4][{(LO)3U}2(μ-NC2O4)], 8 with thermal ellipsoids drawn at the 50% probability level. Hydrogen atoms and the methyl groups of the tert-butyl moieties have been omitted for clarity. U1–(Osiloxide)avg = 2.14(2) Å, U2–(Osiloxide)avg = 2.13(2) Å, U1–O2 = 2.320(6) Å, U1–N1 = 2.460(7) Å, U2–O3 = 2.222(6) Å, U2–O1 = 2.267(6) Å, N1–C1 = 1.344(12) Å, O2–C1 = 1.279(12) Å, O1–C1 = 1.285(12) Å, O3–C2 = 1.323(11) Å, O4–C2 = 1.225(12) Å, N1–C1–O2 = 112.7(1)°, torsion angles: O2–C1–N1–C2 = 176.5(8)°, O4–C2–N1–C1 = −174.5(9)°. | ||
The structure of the anion in 8 can be described as a dinuclear complex in which an asymmetric μ-κ2-O,O-:κ2-O,N-N(CO2)23− dicarbamate ligand bridges two U(IV) cations in two [(LO)3U]+ units. One uranium cation is bound by the nitrogen atom and a carboxylate oxygen atom, while the second one is bound by two oxygen atoms from the two different carboxylate units. The U2NC2O4 core comprises two fused rings, with one six membered ring (UOCNCO) and one four membered ring (UNCO) which share the C–N bond and are coplanar. A similar binding mode is found for the Cs analogue [Cs{(LO)3U}2(μ-NC2O4)]1d except for the fact that the dicarbamate moiety also binds a Cs cation.
We explored the CO2 reactivity of the amide complexes 2 and 4 with CO2 but found that exposure of thf-d8 solutions of these complexes to 1 atm of CO2 does not produce any change in their respective 1H NMR spectra indicating no reactivity.
The bridging nitrides in the heteroleptic amide methanide, 3, and amide siloxide, 5, both react with 2 equiv. or 1 atm CO2 in thf-d8 to produce a complicated mixture of products. The products could not be isolated, but hydrolysis with pD = 12 D2O of the reaction residues after reactions of 3 or 5 with 2–6 equiv. 13CO2 showed N13CO− as the only product originating of the nitride; evidence for a dicarbamate product was not obtained in either case. The formation of cyanate indicates that the reaction of 3 and 5 with excess CO2 proceeds with deoxygenation and N–C bond formation. Similar reactivity leading to the formation of oxo cyanate complexes had been reported for [Cs]-1 at substoichiometric ratios of CO2 (ref. 1d) and for terminal U(V) nitrides.1g
These results show that the replacement of one amide ligand (out of six ligands) by one siloxide is sufficient to promote nitride transfer to CO2. The different reactivity of 3 and 5 compared to [NBu4]-1 indicates that bridging nitrides in complexes supported by amide ligands are less nucleophilic and react preferentially with one CO2 molecule with the rate of (NCO2)3− cleavage to yield NCO− and O2− being faster than the addition of a second CO2 molecule. All of the spectra are shown in Fig. S27–S35 in the ESI.†
:1 toluene/hexane mixture, Scheme 6 (Fig. 5).
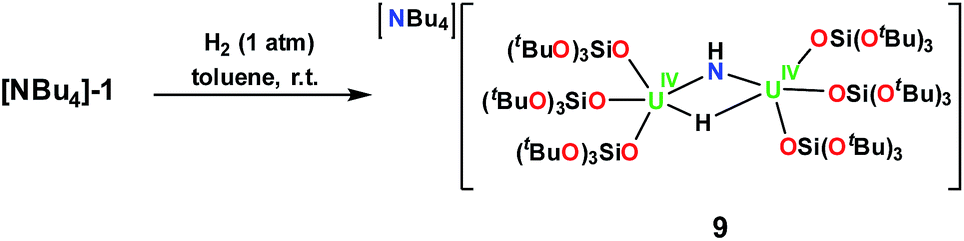 | ||
| Scheme 6 Reaction of [NBu4][{(LO)3U}2(μ-NH)(μ-H)] with H2,9. | ||
Complex 9 is the NBu4+ analogue of the [Cs{U(LO)3}2(μ-H)(μ-NH)] complex previously obtained when [Cs]-1 was reacted with H2 under similar conditions.1h The NH and H resonances of [NBu4][{(LO)3U}2(μ-NH)(μ-H)] were identified at 188.1 and 720.2 ppm, respectively, in toluene-d8. Consistent with this, when D2 is used the resonances are no longer observable. The [NBu4][{(LO)3U}2(μ-NH)(μ-H)] complex is stable to dynamic vacuum and shows no evidence of reversible binding of H2. In contrast, H2 addition to the [Cs]-1 complex was found to be reversible.1h The reversibility of H2 addition to [Cs]-1 and the different behavior observed for [NBu4]-1 can be rationalized in the terms of the differences in the solid-state structures of the two imide hydride complexes. Complex 9 can be described as a dinuclear complex in which μ-NH and a μ-H ligands bridge two 5-coordinate U(IV) ions bound as two [(LO)3U]+ units. The U–N bond distances (2.222(3) Å and 2.209(3) Å) are significantly longer than those found in the parent compound, [NBu4]-1 (2.032(2) Å and 2.067(2) Å), and are similar to those found in the imido bridged diuranium(IV) complexes [Cs{U(LO)3}2(μ-H)(μ-NH)],1h (2.231(7) Å and 2.288(8) Å), and [K2{[U(LO)3]2(μ-NH)2}] (2.192(3) Å and 2.273(3) Å).1i The bridging hydride was located in the Fourier difference map and the U–H distances (2.31(4) Å and 2.23(4) Å) are similar to those found in the Cs analogue (2.18(6) and 2.36(6) Å),1h and those determined by neutron diffraction studies for the complex [U(C5Me5)2(μ-H)2]2 of 2.134(9) Å.15 In the cesium complex, the cesium cation lies at the apical position of the hydride ligand with a Cs–H distance of 2.73(6) Å. The Cs–H interaction is probably rendering more labile the U–H binding and facilitating the H2 elimination when the H2 headspace is removed. In contrast, the absence of the coordinated cesium cation evidently reduces its reactivity towards H2 elimination.
 | ||
| Fig. 5 Molecular structure of [NBu4][{(LO)3U}2(μ-NH)(μ-H)], 9, with thermal ellipsoids drawn at the 50% probability level. Hydrogen atoms of the siloxide ligands, and a toluene molecule have been omitted for clarity. Selected structural parameters: U–(Osiloxide)avg = 2.17(1) Å, U–Navg = 2.216(2) Å, U–Havg = 2.27(3) Å, U1–N1–U2 = 116.71(14)°, U1–H1–U2 = 113 (2)°. | ||
H2 cleavage was not observed for the heteroleptic complexes, 3 and 5, nor for the all-amide complexes 2 and 4 at ambient conditions. This can be explained in term of their reduced nucleophilic character.
Overall, the correlations between structural parameters and nucleophilic reactivity are not obvious except that the all-amide complexes with the linear UNU angle are the least reactive. The different reactivity with H2 of the mixed-ligand complex 5 and the all-siloxide complexes 1 suggests a reduced nucleophilicity of the nitride in complex 5 in spite of the presence of a bent UNU angle. This suggests that the nucleophilicity of the nitride is affected by ligand electronics which do not always result in obvious structural differences.
In order to further investigate how differences in bonding could explain the observed difference in reactivity we turned to other methods.
Magnetic properties
Magnetic communication between uranium centers can provide important information on the nature of bonding5b,7a,8f,16 and lead to attractive magnetic properties.17 Examples of unambiguous magnetic coupling are rare in uranium chemistry and mostly limited to antiferromagnetic coupling between uranium(V) centers with values of χ maximum in the χ versus T plots ranging from 5 to 77 K.7a,16b,18 Stronger antiferromagnetic coupling with the highest value of χ maximum at 110 K was reported by Cummins and Diaconescu for an arene-bridged U(III) dimer.19 In contrast, two examples of unambiguous magnetic coupling between two U(IV) ions were reported for diuranium(IV) chalcogenide complexes with χ maxima at 3 and 20 K.20 Strong antiferromagnetic coupling has been also suggested from the ∼60 K maximum in the χ versus T plot of an amide supported nitride-bridged U4+–U4+ also presenting a linear UNU core, complex.7b
The χ versus T plot for complex 2 (Fig. 6) shows the magnetic behavior of an antiferromagnetically coupled dinuclear complex with a maximum in the χ versus T plot of approximately 90 K. Below 20 K, an increase of the magnetic susceptibility is observed which we attribute to small amounts of paramagnetic impurities. Thus, the linear U(IV)NU(IV) unit in the all amide complex exhibits a strong coupling between the two f2 ions higher than for any other U4+–U4+ complex.
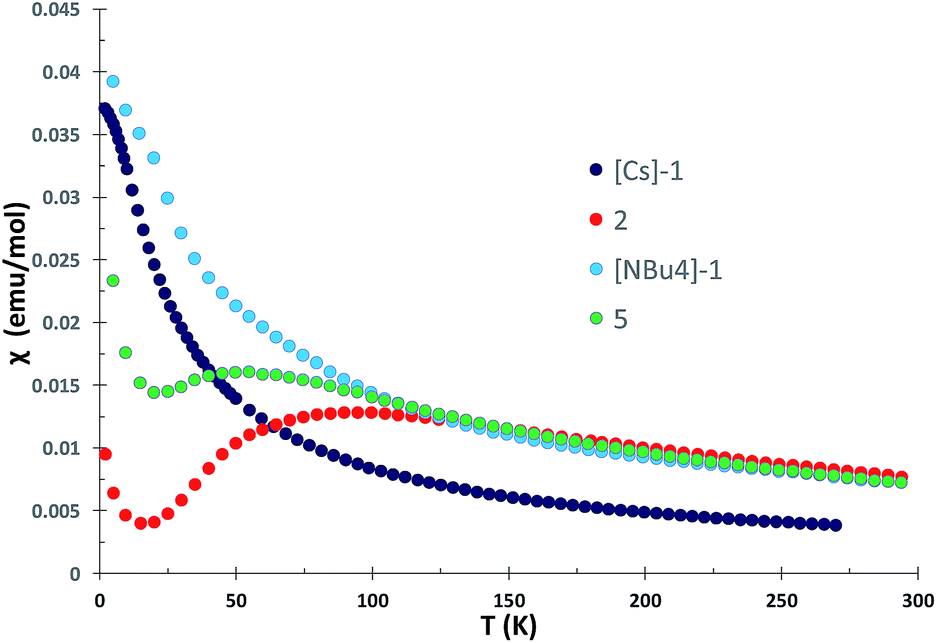 | ||
| Fig. 6 Plot of χ versus temperature data for the all-siloxide complexes [Cs]-1 and [NBu4]-1 and the all-amide complex 2 measured in 1 T field. | ||
For the all-siloxide complexes [NBu4]-1 and [Cs]-1 (Fig. 6), and for the all-amide U4+–U5+ complex 4 (Fig. S43†), the χ versus T plot shows a continuous increase and for [NBu4]-1 and [Cs]-1 approaches a temperature-independent plateau at low temperature. This behavior is characteristic of magnetically isolated uranium ions in complexes.8e,18f,20b,21 A similar behavior was also reported for the amide methanide complex 3, which was interpreted in terms of a localized UN bonding interactions of the bridging nitrido ligand (as suggested by the inequivalent U–Nnitride distances).8f A shorter U–U distance is found in the solid-state structure of the all-siloxide complexes [NBu4]-1 (4.107(2) Å) and Cs-1 (4.1214(4) Å) compared to the amide complex 2 (4.15 Å), but coupling is observed only in the amide complex. The χ versus T plot for the mixed siloxide-amide complex 5 (Fig. 6) shows the magnetic behavior of an antiferromagnetically coupled U4+–U4+ complex with a maximum in the χ versus T at 55 K, significantly lower than for the all-amide complex 2. In this complex, the U–N–U angle (168°) is similar to what found in all siloxide complex [Cs]-1 suggesting that there is no a relation between this structural parameter and the magnetic properties. The dramatic difference in the magnetic properties of complexes 2 and 5 compared to complexes [NBu4]-1 and [Cs]-1 are likely to arise from the electronic properties of the supporting ligands, which evidently lead to increased orbital overlap between the nitride and the two uranium(IV) centers in the amide supported complex, enhancing the electronic communication between the two uranium centers. Such communication is reduced when one amide out of six is replaced by one siloxide. This is in line with the reactivity the nitride ligand in complex 5 which is increased compared to the all-amide complexes, but still lower than for all-siloxide complexes. In order to assess more thoroughly the U–N–U bonding scheme, we have computed the electronic structures of the three complexes.
Computational studies
Depending on the arrangement of the two 5f electrons on each UIV center, the synthesized complexes are characterized either by a quintet, triplet, or singlet ground-state. Computations on the all-siloxide 1, the all-amide 2 and the mixed-ligand 5 complexes for each of these spin-states were performed at the DFT level (B3LYP/Def2-SVP/Def-SDD and M06L(-D3)/Def2-SVP/Def-SDD (see ESI†)). The two formally degenerate states (triplet and singlet) were treated within a broken-symmetry (BS) formalism, starting from a spin-flip ansatz from the non-degenerate high-spin quintet state. According to both density functionals, the all-amide is characterized by a singlet anti-ferromagnetic ground-state (exchange couplings: JB3LYP (UU) = −15.02 cm−1 and JM06L (UU) = −50.15 cm−1), while the quintet state is the most stable for the all-siloxide. Assigning the multiplicity of the mixed-ligand compound is more delicate, since this compound is more sensitive to the choice of the density functional, which has to be able to capture simultaneously the asymmetry in the ligand environment between the two uranium centers and mimic the effects of state degeneracies (see ESI†). This complex results in a quintet with M06L and in a singlet anti-ferromagnetic state (exchange coupling: JB3LYP (UU) = −9.44 cm−1) with B3LYP. Although the results with B3LYP are in line with experimental magnetic properties for all the complexes, the relatively small energy differences (see ESI†) between spin-states has to be taken with care.Beside the differences in magnetic properties, Mayer bond order (MBO) analysis22 on the three complexes reveals a correlation between the increased reactivity of the complexes and a decreased bond-order between the uranium centers and the bridging nitrogen, despite the relatively small magnitude of the bond-order differences (Fig. 7 top panel). An opposite trend is found when considering the bond order between the uranium centers and the non-bridging ligands, suggesting that the modulation of the reactivity is linked to the relative strength of the O–U bond, compared to the N–U (Fig. 7 bottom panel). The overall reduced bond order between the bridging nitrogen and the uranium centers in the presence of oxygen-based ligands indicate an increased localization of the electron density on the bridging nitrogen and a consequent increase of its nucleophilic character.
 | ||
| Fig. 7 (Top) Mayer Bond Order for U–N bond in function of the complexes ordered per increasing reactivity. (Bottom) Mayer Bond Order between uranium centers and non-bridging ligands in function of the complexes ordered per increasing reactivity. L1 indicates the position of the siloxide ligand in the mixed complex. | ||
Conclusions
In summary, four new examples of diuranium nitride bridged complexes have been prepared and structurally characterized, the siloxide supported [NBu4]-1, the amide supported 2 and 4, and the amide siloxide 5. The previously reported amide methanide complex 38f demonstrated to be a useful precursor for the preparation of the mixed-ligand amide siloxide complex 5. The small molecule reactivity of the nitride group in the [NBu4]-1, 2, 3 and 5 has been investigated with CO, CO2, and H2. Although [Cs]-1 and [NBu4]-1 show differences in structure, their reactivity is similar. Both complexes convert CO to CN−, react with CO2 to give dicarbamate (N(CO2)2)3−, and react with H2 to yield imide hydride complexes. The only observed effect of the presence of an inner sphere cesium cation is that reversible H2 cleavage is observed in the Cs+ case versus NBu4+ where it is irreversible.The overall reactivity of the described nitride complexes can be summarized according to the ligand; the all-amide complexes 2 and 4 are unreactive with CO, CO2, and H2 whereas the all-OSi(OtBu)3 complexes 1 react with all of these substrates. The heteroleptic complexes 3 and 5 react with CO and CO2 but not H2. Complexes 1, 3, and 5 convert of CO to CN− but their reactivity with CO2 is different; the all-OSi(OtBu)31 convert CO2 to dicarbamate, whereas the mixed ligand complexes 3 and 5 favor NCO− formation. Finally, only the all-OSi(OtBu)3 complexes 1 react with H2 and give imide/hydride complexes.
These results demonstrate that the all-OSi(OtBu)3 ligand environment in U4+–U4+ nitride complexes allows reactivity that is not observed when amide ligands are used, (e.g., dicarbamate formation and reversible and irreversible H2 heterolysis), and highlights the unique chemistry that can be obtained in this coordination environment. The all-OSi(OtBu)3 ligand environment provides nitrides that are highly nucleophilic and more reactive than those with amide ligands. In contrast, the all-silylamide ligand promotes magnetic exchange coupling but gives less reactive complexes. Computational analysis at the DFT level shows that increasing reactivity of the complexes correlates with a decrease in bond order of the bridging nitride, as well as with an increase in the bond order of the ancillary ligands.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge support from the Swiss National Science Foundation grant number 200021_162430, 200021_178793 and 200021_169699 and the Ecole Polytechnique Fédérale de Lausanne (EPFL). We thank Dr Euro Solari for carrying out the elemental analyses, Farzaneh Fadaei-Tirani for important contributions to the X-ray single crystal structure analyses.Notes and references
- (a) M. Falcone, L. Chatelain, R. Scopelliti, I. Zivkovic and M. Mazzanti, Nature, 2017, 547, 332–335 CrossRef CAS PubMed; (b) M. Falcone, L. Chatelain, R. Scopelliti and M. Mazzanti, Chimia, 2017, 71, 209–212 CrossRef CAS PubMed; (c) M. Falcone, C. E. Kefalidis, R. Scopelliti, L. Maron and M. Mazzanti, Angew. Chem., Int. Ed. Engl., 2016, 55, 12290–12294 CrossRef CAS PubMed; (d) M. Falcone, L. Chatelain and M. Mazzanti, Angew. Chem., Int. Ed. Engl., 2016, 55, 4074–4078 CrossRef CAS PubMed; (e) D. M. King and S. T. Liddle, Coord. Chem. Rev., 2014, 266, 2–15 CrossRef; (f) P. A. Cleaves, D. M. King, C. E. Kefalidis, L. Maron, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed. Engl., 2014, 53, 10412–10415 CrossRef CAS PubMed; (g) P. A. Cleaves, C. E. Kefalidis, B. M. Gardner, F. Tuna, E. J. L. McInnes, W. Lewis, L. Maron and S. T. Liddle, Chem.–Eur. J., 2017, 23, 2950–2959 CrossRef CAS PubMed; (h) M. Falcone, L. N. Poon, F. F. Tirani and M. Mazzanti, Angew. Chem., Int. Ed. Engl., 2018, 57, 3697–3700 CrossRef CAS PubMed; (i) L. Barluzzi, L. Chatelain, F. Fadaei-Tirani, I. Zivkovic and M. Mazzanti, Chem. Sci., 2019, 10, 3543–3555 RSC.
- (a) L. M. Duman and L. R. Sita, J. Am. Chem. Soc., 2017, 139, 17241–17244 CrossRef CAS PubMed; (b) D. J. Knobloch, E. Lobkovsky and P. J. Chirik, J. Am. Chem. Soc., 2010, 132, 10553–10564 CrossRef CAS PubMed; (c) M. Aresta, Carbon Dioxide as a Chemical Feedstock, Wiley VCH, 2010 CrossRef; (d) I. Klopsch, M. Kinauer, M. Finger, C. Wurtele and S. Schneider, Angew. Chem., Int. Ed. Engl., 2016, 55, 4786–4789 CrossRef CAS PubMed; (e) A. J. Keane, W. S. Farrell, B. L. Yonke, P. Y. Zavalij and L. R. Sita, Angew. Chem., Int. Ed. Engl., 2015, 54, 10220–10224 CrossRef CAS PubMed; (f) R. J. Burford and M. D. Fryzuk, Nat. Rev. Chem., 2017, 1, 0026 CrossRef CAS.
- (a) J. S. Silvia and C. C. Cummins, J. Am. Chem. Soc., 2009, 131, 446–447 CrossRef CAS PubMed; (b) J. A. Buss, C. Cheng and T. Agapie, Angew. Chem., Int. Ed. Engl., 2018, 57, 9670–9674 CrossRef CAS PubMed; (c) B. Askevold, J. T. Nieto, S. Tussupbayev, M. Diefenbach, E. Herdtweck, M. C. Holthausen and S. Schneider, Nat. Chem., 2011, 3, 532–537 CrossRef CAS PubMed; (d) Y. Ishida and H. Kawaguchi, J. Am. Chem. Soc., 2014, 136, 16990–16993 CrossRef CAS PubMed; (e) B. L. Tran, M. Pink, X. F. Gao, H. Park and D. J. Mindiola, J. Am. Chem. Soc., 2010, 132, 1458–1459 CrossRef CAS PubMed; (f) J. J. Scepaniak, R. P. Bontchev, D. L. Johnson and J. M. Smith, Angew. Chem., Int. Ed. Engl., 2011, 50, 6630–6633 CrossRef CAS PubMed; (g) S. D. Brown and J. C. Peters, J. Am. Chem. Soc., 2005, 127, 1913–1923 CrossRef CAS PubMed; (h) A. F. Cozzolino, J. S. Silvia, N. Lopez and C. C. Cummins, J. Chem. Soc., Dalton Trans., 2014, 43, 4639–4652 RSC; (i) J. K. Brask, V. Dura-Vila, P. L. Diaconescu and C. C. Cummins, Chem. Commun., 2002, 902–903 RSC; (j) J. S. Silvia and C. C. Cummins, J. Am. Chem. Soc., 2010, 132, 2169–2170 CrossRef CAS PubMed; (k) S. P. Semproni and P. J. Chirik, Angew. Chem., Int. Ed. Engl., 2013, 52, 12965–12969 CrossRef CAS PubMed; (l) S. P. Semproni and P. J. Chirik, J. Am. Chem. Soc., 2013, 135, 11373–11383 CrossRef CAS PubMed.
- (a) J. M. Smith, Prog. Inorg. Chem., 2014, 58, 417–470 CAS; (b) R. A. Eikey and M. M. Abu-Omar, Coord. Chem. Rev., 2003, 243, 83–124 CrossRef CAS.
- (a) M. B. Jones and A. J. Gaunt, Chem. Rev., 2013, 113, 1137–1198 CrossRef CAS PubMed; (b) D. M. King, P. A. Cleaves, A. J. Wooles, B. M. Gardner, N. F. Chilton, F. Tuna, W. Lewis, E. J. L. McInnes and S. T. Liddle, Nat. Commun., 2016, 7, 13773 CrossRef CAS PubMed; (c) A. R. Fox, P. L. Arnold and C. C. Cummins, J. Am. Chem. Soc., 2010, 132, 3250–3251 CrossRef CAS PubMed; (d) T. W. Hayton, Chem. Commun., 2013, 49, 2956–2973 RSC; (e) J. Su, E. R. Batista, K. S. Boland, S. E. Bone, J. A. Bradley, S. K. Cary, D. L. Clark, S. D. Conradson, A. S. Ditter, N. Kaltsoyannis, J. M. Keith, A. Kerridge, S. A. Kozimor, M. W. Loble, R. L. Martin, S. G. Minasian, V. Mocko, H. S. La Pierre, G. T. Seidler, D. K. Shuh, M. P. Wilkerson, L. E. Wolfsberg and P. Yang, J. Am. Chem. Soc., 2018, 140, 17977–17984 CrossRef CAS PubMed; (f) E. Lu, S. Sajjad, V. E. J. Berryman, A. J. Wooles, N. Kaltsoyannis and S. T. Liddle, Nat. Commun., 2019, 10, 634 CrossRef PubMed.
- (a) N. H. Anderson, S. O. Odoh, Y. Y. Yao, U. J. Williams, B. A. Schaefer, J. J. Kiernicki, A. J. Lewis, M. D. Goshert, P. E. Fanwick, E. J. Schelter, J. R. Walensky, L. Gagliardi and S. C. Bart, Nat. Chem., 2014, 6, 919–926 CrossRef CAS PubMed; (b) N. Kaltsoyannis, Inorg. Chem., 2013, 52, 3407–3413 CrossRef CAS PubMed.
- (a) M. Falcone, L. Barluzzi, J. Andrez, F. F. Tirani, I. Zivkovic, A. Fabrizio, C. Corminboeuf, K. Severin and M. Mazzanti, Nat. Chem., 2019, 11, 154–160 CrossRef CAS PubMed; (b) J. Du, D. M. King, L. Chatelain, E. Lu, F. Tuna, E. J. L. McInnes, A. J. Wooles, L. Maron and S. T. Liddle, Chem. Sci., 2019, 10, 3738–3745 RSC.
- (a) A. R. Fox and C. C. Cummins, J. Am. Chem. Soc., 2009, 131, 5716–5717 CrossRef CAS PubMed; (b) W. J. Evans, S. A. Kozimor and J. W. Ziller, Science, 2005, 309, 1835–1838 CrossRef CAS PubMed; (c) I. Korobkov, S. Gambarotta and G. P. A. Yap, Angew. Chem., Int. Ed. Engl., 2002, 41, 3433–3436 CrossRef CAS; (d) T. K. Todorova, L. Gagliardi, J. R. Walensky, K. A. Miller and W. J. Evans, J. Am. Chem. Soc., 2010, 132, 12397–12403 CrossRef CAS PubMed; (e) G. Nocton, J. Pecaut and M. Mazzanti, Angew. Chem., Int. Ed. Engl., 2008, 47, 3040–3042 CrossRef CAS PubMed; (f) S. Fortier, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2010, 132, 6888–6889 CrossRef CAS PubMed; (g) C. Camp, J. Pecaut and M. Mazzanti, J. Am. Chem. Soc., 2013, 135, 12101–12111 CrossRef CAS PubMed; (h) L. Maria, I. C. Santos, V. R. Sousa and J. Marcalo, Inorg. Chem., 2015, 54, 9115–9126 CrossRef CAS PubMed; (i) D. M. King, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Nat. Chem., 2013, 15, 482–488 CrossRef PubMed; (j) D. M. King, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Science, 2012, 337, 717–720 CrossRef CAS PubMed; (k) D. M. King, J. McMaster, F. Tuna, E. J. L. McInnes, W. Lewis, A. J. Blake and S. T. Liddle, J. Am. Chem. Soc., 2014, 136, 5619–5622 CrossRef CAS PubMed; (l) L. Chatelain, R. Scopelliti and M. Mazzanti, J. Am. Chem. Soc., 2016, 138, 1784–1787 CrossRef CAS PubMed; (m) N. Tsoureas, A. F. R. Kilpatrick, C. J. Inman and F. G. N. Cloke, Chem. Sci., 2016, 7, 4624–4632 RSC.
- (a) R. K. Thomson, T. Cantat, B. L. Scott, D. E. Morris, E. R. Batista and J. L. Kiplinger, Nat. Chem., 2010, 2, 723–729 CrossRef CAS PubMed; (b) K. C. Mullane, H. Ryu, T. Cheisson, L. N. Grant, J. Y. Park, B. C. Manor, P. J. Carroll, M. H. Baik, D. J. Mindiola and E. J. Schelter, J. Am. Chem. Soc., 2018, 140, 11335–11340 CrossRef CAS PubMed.
- S. M. Mansell, J. H. Farnaby, A. I. Germeroth and P. L. Arnold, Organometallics, 2013, 32, 4214–4222 CrossRef CAS.
- (a) D. L. Clark, A. P. Sattelberger, S. G. Bott and R. N. Vrtis, Inorg. Chem., 1989, 28, 1771–1773 CrossRef CAS; (b) J. L. Stewart and R. A. Andersen, Polyhedron, 1998, 17, 953–958 CrossRef CAS.
- V. Mougel, C. Camp, J. Pecaut, C. Coperet, L. Maron, C. E. Kefalidis and M. Mazzanti, Angew. Chem., Int. Ed. Engl., 2012, 51, 12280–12284 CrossRef CAS PubMed.
- O. Cooper, C. Camp, J. Pécaut, C. E. Kefalidis, L. Maron, S. Gambarelli and M. Mazzanti, J. Am. Chem. Soc., 2014, 136, 6716–6723 CrossRef CAS PubMed.
- (a) O. Benaud, J. C. Berthet, P. Thuery and M. Ephritikhine, Inorg. Chem., 2010, 49, 8117–8130 CrossRef CAS PubMed; (b) S. J. Simpson and R. A. Andersen, J. Am. Chem. Soc., 1981, 103, 4063–4066 CrossRef CAS; (c) P. L. Arnold, Z. R. Turner, A. I. Germeroth, I. J. Casely, G. S. Nichol, R. Bellabarba and R. P. Tooze, J. Chem. Soc., Dalton Trans., 2013, 42, 1333–1337 RSC.
- D. Grant, T. J. Stewart, R. Bau, K. A. Miller, S. A. Mason, M. Gutmann, G. J. McIntyre, L. Gagliardi and W. J. Evans, Inorg. Chem., 2012, 51, 3613–3624 CrossRef CAS PubMed.
- (a) B. M. Gardner, D. M. King, F. Tuna, A. J. Wooles, N. F. Chilton and S. T. Liddle, Chem. Sci., 2017, 8, 6207–6217 RSC; (b) L. P. Spencer, E. J. Schelter, P. Yang, R. L. Gdula, B. L. Scott, J. D. Thompson, J. L. Kiplinger, E. R. Batista and J. M. Boncella, Angew. Chem., Int. Ed. Engl., 2009, 48, 3795–3798 CrossRef CAS PubMed.
- (a) D. P. Mills, F. Moro, J. McMaster, J. van Slageren, W. Lewis, A. J. Blake and S. T. Liddle, Nat. Chem., 2011, 3, 454–460 CrossRef CAS PubMed; (b) V. Mougel, L. Chatelain, J. Pecaut, R. Caciuffo, E. Colineau, J. C. Griveau and M. Mazzanti, Nat. Chem., 2012, 4, 1011–1017 CrossRef CAS PubMed.
- (a) R. K. Rosen, R. A. Andersen and N. M. Edelstein, J. Am. Chem. Soc., 1990, 112, 4588–4590 CrossRef CAS; (b) G. Nocton, P. Horeglad, J. Pécaut and M. Mazzanti, J. Am. Chem. Soc., 2008, 130, 16633–16645 CrossRef CAS PubMed; (c) V. Mougel, P. Horeglad, G. Nocton, J. Pecaut and M. Mazzanti, Angew. Chem., Int. Ed. Engl., 2009, 48, 8477–8480 CrossRef CAS PubMed; (d) P. L. Arnold, G. M. Jones, S. O. Odoh, G. Schreckenbach, N. Magnani and J. B. Love, Nat. Chem., 2012, 4, 221–227 CrossRef CAS PubMed; (e) L. Chatelain, V. Mougel, J. Pécaut and M. Mazzanti, Chem. Sci., 2012, 3, 1075–1079 RSC; (f) A.-C. Schmidt, F. W. Heinemann, W. W. Lukens Jr and K. Meyer, J. Am. Chem. Soc., 2014, 136, 11980–11993 CrossRef CAS PubMed.
- (a) P. L. Diaconescu, P. L. Arnold, T. A. Baker, D. J. Mindiola and C. C. Cummins, J. Am. Chem. Soc., 2000, 122, 6108–6109 CrossRef CAS; (b) B. Vlaisayljevich, P. L. Diaconescu, W. L. Lukens Jr, L. Gagliardi and C. C. Cummins, Organometallics, 2013, 32, 1341–1352 CrossRef.
- (a) B. M. Gardner, J. C. Stewart, A. L. Davis, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9265–9270 CrossRef CAS PubMed; (b) O. P. Lam, F. W. Heinemann and K. Meyer, Chem. Sci., 2011, 2, 1538–1547 RSC.
- E. J. Schelter, P. Yang, B. L. Scott, J. D. Thompson, R. L. Martin, P. J. Hay, D. E. Morris and J. L. Kiplinger, Inorg. Chem., 2007, 46, 7477–7488 CrossRef CAS PubMed.
- A. J. Bridgeman, G. Cavigliasso, L. R. Ireland and J. Rothery, J. Chem. Soc., Dalton Trans., 2001, 2095–2108 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic methods, NMR spectra, crystallographic data, computational details. CCDC 1913257–1913264. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc02149c |
| This journal is © The Royal Society of Chemistry 2019 |