
Ferromagnetic iron oxide–cellulose nanocomposites prepared by ultrasonication†
Razvan
Rotaru
 ,
Marcela
Savin
,
Nita
Tudorachi
,
Cristian
Peptu
,
Petrisor
Samoila
,
Liviu
Sacarescu
and
Valeria
Harabagiu
*
,
Marcela
Savin
,
Nita
Tudorachi
,
Cristian
Peptu
,
Petrisor
Samoila
,
Liviu
Sacarescu
and
Valeria
Harabagiu
*
“Petru Poni” Institute of Macromolecular Chemistry, Iasi, 700487, Romania. E-mail: hvaleria@icmpp.ro; Fax: +40-232-211299; Tel: +40-232-217454
First published on 27th October 2017
Abstract
This paper highlights the efficiency of ultrasonication, as a clean and energy-saving method for the preparation of cellulose–iron oxide ferromagnetic composites in two steps. Hydroxyl-functionalized maghemite–goethite nanoparticles (MG) having a saturation magnetization of 56.9 emu g−1 at room temperature were first synthesized by a one-pot procedure involving ultrasonication of an aqueous alkaline Fe2+ salt solution. Organic–inorganic nanocomposites were obtained by a second ultrasonication step of an aqueous suspension of micronized cellulose and MG nanoparticles. The cumulative effect of ultrasonication and MG nano-projectiles was found to strongly decrease the degree of crystallinity of cellulose. The microstructural characterisation of the resulting composite evidenced its size polydispersity, with small nanoparticles uniformly attached on the surface of cellulose microfibrils. Vibrating sample magnetic measurement indicated a hysteresis curve specific to ferromagnetic materials and the appearance of a superparamagnetic phenomenon at low temperature (a blocking temperature of 62 K). Cellulose/iron oxide clusters with an average size around 7 nm and characterized by high decomposition temperatures (around 645 °C) were proven to be responsible for the observed superparamagnetic phenomenon. The superiority of the ultrasonication process versus a procedure involving simple mechanical stirring, in terms of composite yield, dispersion uniformity of MG nanoparticles and magnetic properties is discussed.
Introduction
Magnetic cellulose-based nanomaterials have been suggested recently to be of interest as transparent films for magneto-optical applications,1 for recyclable catalysts,2–6 as antibacterial or contrasting materials for magnetic resonance imaging,7 magnetic aerogels,8 magnetically retrievable oil adsorbents9–11 or cellulose composite films.12 Cellulose in combination with magnetic particles was also proposed as a material for magnetic paper13 and other magnetically responsive cellulose composite materials.14,15 Typically, cellulose-based magnetic materials are prepared by incorporating iron oxides into the cellulose matrix.16 The inorganic domains can be either presynthesized and mixed with the cellulose matrix1,17 or synthesized in situ.7,14Besides the above mentioned applications, another area of major interest is concerned with superparamagnetic materials. The phenomenon of superparamagnetism (SP) describes a state of single domain sized grains, when the thermal energy is high enough to overcome the barriers to a magnetization reversal. These barriers arise from magneto-crystalline, magneto-elastic or shape anisotropy, all proportional to the grain volume. When the energy barriers are large with respect to the thermal energy, the magnetization process is stopped and the probability of spontaneous reversal strongly decreases, becoming negligible. If the barriers are low, the thermal excitation can result in the reversal of the magnetization over very short time scales, and a superparamagnetic state is attained. At a given temperature, the volume of a particle that passes from the unblocked state to a blocked one is known as the blocking volume. For a given particle volume, the grain can be blocked by cooling the material below the blocking temperature (the temperature when the reversal magnetization starts).18,19 This type of magnetism that occurs in small ferromagnetic or ferrimagnetic particles implies sizes of a few to a couple of tenth nanometers, depending on the material.20,21 These nanoparticles are single-domain particles. The total magnetic moment of the nanoparticle can be seen as a single giant magnetic moment, composed of all the individual magnetic moments of the atoms forming the nanoparticle.22 However, the size is not the only parameter able to provide the state of superparamagnetism. The surface effects and the crystallinity also affect the magnetic properties, including the SP phenomena.23–26
Ultrasonication assisted processes are more and more studied as unconventional and clean methods in chemical synthesis27 and materials preparation/processing.28 Ultrasound treatments of solutions or suspensions involve multiple effects such as acoustic cavitation (bubbles formation, followed by their growth and implosion generating microcavities), very high local temperatures and pressures, micro-jet and shockwave phenomena.29 Such extreme conditions can accelerate some chemical reactions and have been exploited to prepare amorphous metals, alloys, and oxides, and also to induce new physical properties. Thus, Fang et al. managed to synthesize ferroelectric and ferromagnetic BiFeO3 by intense ultrasonication (ultrasound frequency: 40 kHz) of a bismuth nitrate and iron nitrate mixture.30 Liu et al. produced ultrathin CoPt3 nanowires with ferromagnetic properties at room temperature by dealloying Co99Pt1 nanowires of a larger diameter through ultrasonication.31 Furthermore, high frequency ultrasonication was proved to directly influence the magnetic properties of the treated materials. Thus, the coercive field increases with increasing ultrasound intensity. Also, the saturation magnetization of the magnetic particles synthesized by using ultrasound is higher compared to the values obtained for other synthesis methods.32 These effects of ultrasonication can be explained by an increased internal tension due to strong interactions on the surface (surface effects) and by the reduced size of the crystallites.33
We recently reported the one-pot synthesis of maghemite–goethite nanocomposites possessing high magnetic properties induced by maghemite and hydroxyl groups specific to goethite.11,34 These functional groups are of interest in the preparation of polysaccharide-based composites, since they are able to interact with similar groups of the polymer matrix.35 Thus, hydrophobized viscose-maghemite/goethite composites were prepared and their performances as magnetically recovering oil sorbents were demonstrated.10 Ultrasonication was also used as an efficient method to prepare ferroelectric viscose-barium titanate composites for electromagnetic shielding.11,36
The purpose of this investigation is to provide a clean ultrasonication method for the preparation of micronized cellulose/maghemite–goethite magnetic composite materials and to investigate the ferromagnetic properties of the resulting materials as a function of their microstructure induced by the preparation conditions. The novelty of this approach resides in the absence of an energy consuming thermal treatment. Moreover, ultrasonication alone converts the pristine hydroxyl-functionalized ferromagnetic maghemite–goethite nanoparticles into very small cellulose–iron oxide clusters that show superparamagnetic behaviour. For comparison purpose, a similar composite was prepared by conventional mechanical mixing of the components. The structures and the properties of the composites were compared by means of Fourier Transform infrared spectroscopy (FTIR), scanning (SEM) and transmission electron microscopy (TEM), X-ray diffraction (XRD), small angle X-ray scattering (SAXS), thermogravimetric analysis (TGA) and vibrating sample magnetometer measurements (VSM).
Experimental
Materials
Micronized cellulose powder (C) (20 μm, 98% purity) (Aldrich), iron sulfate heptahydrate (FeSO4·7H2O) (99% purity), sodium hydroxyde (NaOH) (99% purity) and Milli-Q ultrapure distilled water (Merck) were of analytical grade and used without further purification.Preparation of maghemite–goethite nanoparticles (MG)
MG nanoparticles were obtained through a one-pot procedure as previously described.34 1/2 w/w FeSO4 and 1/4 w/w NaOH aqueous solutions were mixed (1/1 w/w) over 60 minutes in an ultrasonic bath, at an ultrasonic frequency of 20 kHz. The resulting suspension was then centrifuged, washed with Milli-Q water, centrifuged again and dried in a Trade Raypa vacuum oven for 24 h at 40 °C to obtain a dark brown powder sample.Preparation of ferromagnetic composites (C-MG)
An aqueous suspension of 4 g micronized cellulose (C) and 1 g maghemite–goethite powders in 50 mL Milli-Q water was mixed by mechanical stirring (50 rotation per min) at 80 °C, for 15 min to give the C-MGst sample. A similar suspension of powder mixture was sonicated for 15 min at 20 kHz ultrasound frequency, when 26.1 kJ of energy was dissipated into the mixture and the temperature reached 80 °C, to provide the C-MGultr sample. After filtration, the samples were dried in a Trade Raypa vacuum oven overnight at 50 °C. The C-MGst sample resulted in 87% yield as a powder of uneven colour (white with black islands inserts), while the C-MGultr composite sample was obtained as a brown powder of uniform colour in 98% yield.Equipment
The ultrasonication experiments were performed in an ultrasonic bath of a Sonics Vibracell ultrasound generator (750 W nominal electric power, 20 kHz ultrasound frequency, provided with a display giving the energy delivered to the end of the probe and sensor for temperature) at an ultrasonic frequency of 20 kHz and an amplitude of 50% from the maximum intensity. The structures of the precursors and composites were investigated by FTIR spectroscopy on potassium bromide pellets by using a Bruker Vertex 70 spectrometer and by X-ray diffraction (XRD) on a Bruker Advance D8 X-ray diffractometer (λ: 1.5405 Å – the wavelength of Cu-Kα radiation, 2θ ranging from 3 to 70°). The surface morphologies were visualized by scanning electron microscopy (SEM) on an ESEM Quanta 200 electronic deflection microscope. Transmission electron microscopy (TEM) analysis was carried out with a Hitachi High-Tech HT7700 instrument, operated in high resolution mode at 100 kV accelerating voltage. Samples were prepared by drop casting from the diluted dispersions of nanoparticles in ethanol, on 300 mesh holey carbon coated copper grids (Ted Pella) and vacuum dried. Small-angle X-ray scattering experiments (SAXS) were performed on a Bruker NanostarU system equipped with a Vantec 2000 detector (diameter of 200 mm) and an X-ray IμS microsource. The wavelength of the incident X-ray beam was λ = 1.54 Å (Cu-Kα). The scattered intensity I(q) was plotted versus the momentum transfer vector q = 4π![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sinθ/λ, where λ is the wavelength of the X-ray beam and θ is half of the scattering angle. The sample-to-detector distance was 107 cm allowing measurements with q values from 0.008 Å−1 to 0.2 Å−1. The angular scale was calibrated by the scattering peaks of silver behenate standard. The samples under study, in the powder form, were measured under vacuum at 25 °C for 20000 seconds. The background was subtracted from the original intensity profiles. The data analysis was done by using the SAXS-NT Bruker integrated software and ATSAS 2.5.1.37
sinθ/λ, where λ is the wavelength of the X-ray beam and θ is half of the scattering angle. The sample-to-detector distance was 107 cm allowing measurements with q values from 0.008 Å−1 to 0.2 Å−1. The angular scale was calibrated by the scattering peaks of silver behenate standard. The samples under study, in the powder form, were measured under vacuum at 25 °C for 20000 seconds. The background was subtracted from the original intensity profiles. The data analysis was done by using the SAXS-NT Bruker integrated software and ATSAS 2.5.1.37
Magnetic measurements were performed on a Quantum Design-PPMSQD-9 vibrating sample magnetometer for the applied magnetic field in the range of −20–20 kOe. The magnetization dependence on temperature was followed by using standard zero field cooling (ZFC) and field cooling (FC) procedures between 10 and 300 K for the applied magnetic field of 200 Oe. The thermal degradation was followed on a thermo balance model STA 449F1 Jupiter (Netzsch, Germany) calibrated with metal standards (Hg, In, Sn, Bi, Zn, Al) from 38.5 °C to 600 °C. Samples of mass ranging from 7 to 10 mg were heated from 25 to 680 °C with a heating rate of 10 °C min−1. High purity He (99.999%) was used as carrier gas at a flow rate of 50 mL min−1, except for sample MG, when the heating rate was 15 °C min−1 and the carrier gas was nitrogen. The protective purging of the thermo balance with the inert gas was performed at a flow rate of 20 mL min−1. The samples were heated in an open Al2O3 crucible, and alumina was used as the reference material. Data collection was carried out with Proteus® software.
Results and discussion
Cellulose–iron oxide composites (C-MG) were obtained by a two-step procedure as depicted in Fig. 1a. Maghemite (γ-Fe2O3)–goethite (α-FeOOH) nanoparticles (MG) were presynthesized by ultrasonication of an alkaline solution of iron sulfate, according to a procedure previously described.11,34 According to the literature, the preparation of maghemite (γ-Fe2O3) nanoparticles is carried out by different procedures,38–42 each of them involving a thermal treatment step at temperatures higher than 300 °C. On the contrary, the less thermally stable goethite (α-FeOOH) can be obtained only at lower temperatures (20–70 °C).43,44 To the best of our knowledge, we are the first to obtain MG nanoparticles through a one-pot synthesis procedure by ultrasonication. The process does not involve any supplementary thermal treatment, thus being more energy-efficient than other processes.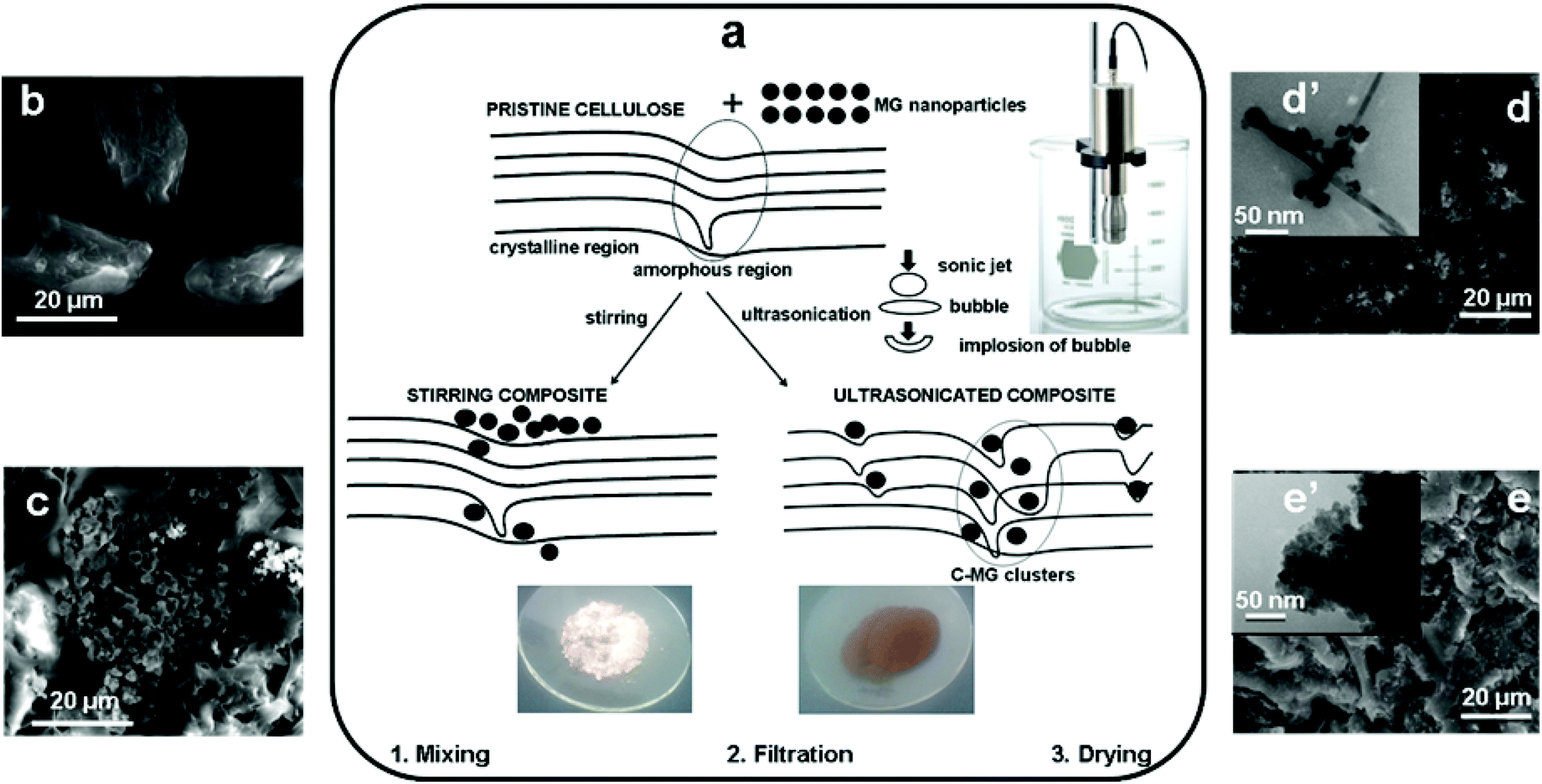 | ||
| Fig. 1 Overview of the preparation of cellulose–maghemite/goethite composites: (a) schematic representation of the preparation methods and of the microstructures of C-MG composites obtained by stirring and ultrasonication; the effect of the combined action of ultrasonication and nanoparticles on the crystalline structure of cellulose is highlighted: SEM images of (b) pristine cellulose, (c) C-MGst (d), MG and (e) C-MGultr; TEM images of (d′) MG and (e′) C-MGultr. | ||
Cellulose–iron oxide composites were obtained in a second step, either by mechanical stirring (C-MGst) or by ultrasonication (C-MGultr). For this purpose, mixtures of MG nanoparticles of a particular shape (acicular goethite particles of more than 200 nm length and diameters lower than 10 nm surrounded by almost spherical agglomerations of maghemite nanoparticles of diameters lower than 50 nm – Fig. 1d′)11,34 and micronized cellulose (C) precursors were used. Ultrasonication was found to be a better homogenization process as proved by the optical microscopy images (insets in Fig. 1a). Thus, the distribution of MG nanoparticles is uneven in the C-MGst sample, where they can be seen as black spots. On the contrary, the C-MGultr sample shows a uniform brown colour. In addition, ultrasonication leads to a higher yield of the C-MGultr composite (98%). The lower yield obtained for the C-MGst sample (87%) is a result of the loss by filtration of a part of MG nanoparticles that were not adhering to cellulose, as will be further demonstrated by FTIR, XRD and TGA analyses.
Structural characterization
 | ||
| Fig. 2 FTIR spectra of the initial components and composites. | ||
Some of the characteristic spectral bands are quite sensitive to the crystalline structure in cellulose materials.46,50 Thus, the bands located at 1431 and 1373 cm−1 and the one at 837 cm−1 correspond to crystalline and amorphous domains, respectively. I1431/I897 and I1373/I2900 ratios (I is the intensity of the absorbance peak ≈ 1 − intensity of the transmittance peak) were used to calculate the empirical crystalline index or lateral order index (LOI) and the total crystalline index (TCI), respectively. TCI is proportional to the crystallinity degree of cellulose and LOI is correlated with the overall degree of order in the cellulose. The ratio of two other infrared bands, I3308/I1330, is known as the hydrogen bond intensity (HBI) closely related to the crystal system and to the degree of intermolecular regularity, considering the chain mobility and bond distance, as well as the amount of bound water. The calculated values of LOI, TCI and HBI for cellulose and its composites are given in Table 1.
The mixing of cellulose with MG nanoparticles by stirring does not change dramatically the crystalline structure of cellulose, as one may see by comparing the LOI, TCI and HBI values of C and C-MGst samples. On the contrary, ultrasonication decreases all these values, less for cellulose ultrasonicated alone (Cultr sample) and more for the C-MGultr sample. Ultrasonic waves are expected to produce a very strong mechanical stress into the treated cellulose containing samples.29 The obtained FTIR data prove that the more rigid crystalline domains are the most affected, while maghemite/goethite nanoparticles increase the amplitude of this phenomenon by playing the role of nano-projectiles thrown by sonic waves into cellulose fibers. As a consequence, the fragmentation of cellulose fibers into smaller entities occurs, as can also be seen in the SEM image of the C-MGultr sample (Fig. 1e compared to Fig. 1b). The effect of MG nanoparticles under ultrasonication on the cellulose microstructure could be depicted as in Fig. 1a.
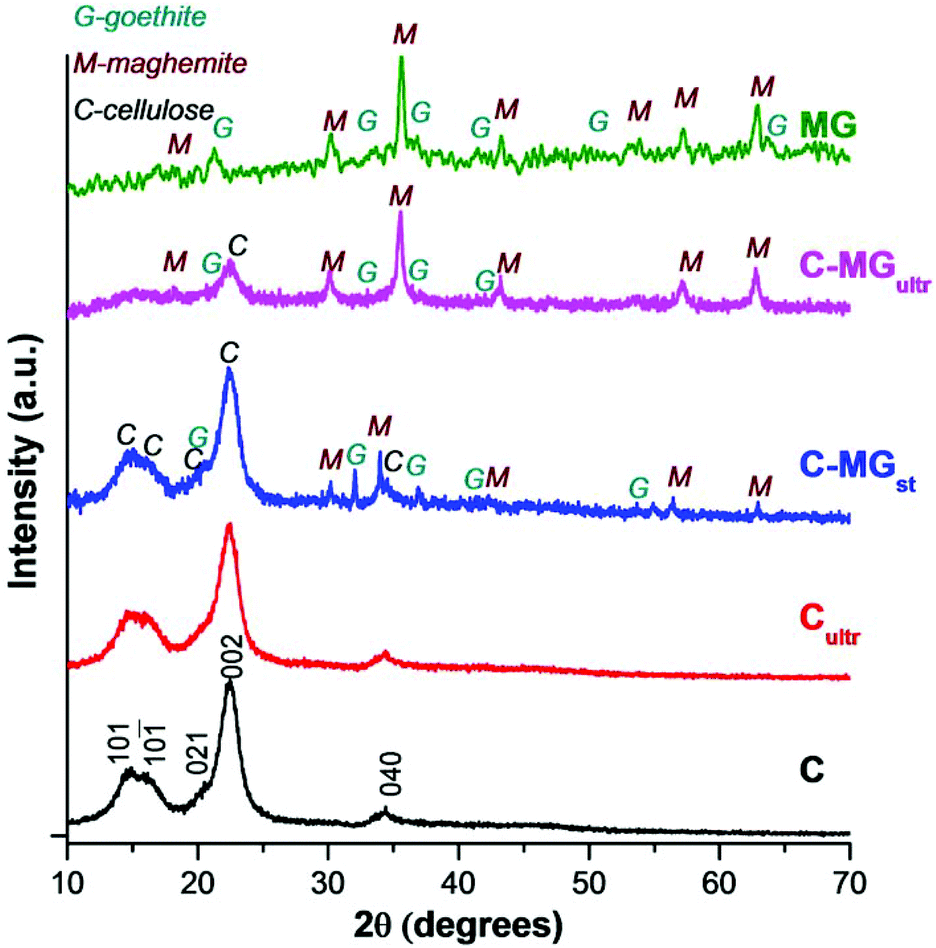 | ||
| Fig. 3 Normalized XRD patterns of pristine cellulose (C), ultrasonicated cellulose (Cultr), maghemite–goethite (MG), and their composites obtained by stirring (C-MGst) and ultrasonication (C-MGultr). | ||
The values of the crystallinity indices for cellulose (Table 1) were calculated by using the deconvolution method (see Fig. S1 in the ESI†), with the following equation:51
| CI (%) = 100 × Sc/St, | (1) |
As one can see in Table 1, similar crystallinity indices were obtained for cellulose, ultrasonicated cellulose and C-MGst sample, and much lower values for the composite sample obtained by ultrasonication, confirming the FTIR data.
Morphology/dimensional characterization
The supramolecular structures of the obtained composites were identified by SEM, TEM and SAXS, compared to those of the pure components. The SEM images of the components and composites are shown in Fig. 1 (larger images are shown in Fig. S2, ESI†). The SEM micrograph of the pristine cellulose sample (Fig. 1b) clearly shows well-separated fibrils of about 30–40 μm length and diameters of around 15 μm. As previously reported, the TEM image of the MG inorganic precursor showed agglomerations of goethite nanorods surrounded by almost spherical maghemite nanoparticles (Fig. 1d′).11,34 The mixing of the components by mechanical stirring gives rise to an uneven distribution of MG between C microfibers (Fig. 1c), while the ultrasonicated sample shows a more uniform distribution of the magnetic nanoparticles and the fragmentation of the cellulose microfibers (Fig. 1e). Optical microscopy images are inserted in Fig. 1a.The C-MGultr sample was also analyzed by TEM. The low magnification images show that the sample contains large aggregates of more than 200 nm size (see Fig. S3 in the ESI†). At higher magnification (Fig. 1e′), the inner structure of these aggregates is resolved, and the component nanoparticles are visible. It is thus possible to notice that the characteristic shape of MG nanoparticles (Fig. 1d′) is no more evidenced and ultrasonication produced almost spherical nanoparticles of a polydisperse size within the 5–30 nm range. These ones are embedded within an organic matrix that contributes to their close packing. No free particles are present outside the polymer matrix indicating the strong connection between the organic and inorganic components.
TEM results were confirmed by SAXS analysis. Fig. 4 shows the dependence of the scattering intensity on the momentum transfer vector (q), obtained by azimuthal integration of the collected 2D patterns to a 1D plot.
 | ||
| Fig. 4 Logarithmic plot of the scattering intensity (I) vs. scattering vector (q) for the C-MGultr sample. | ||
The SAXS scattering curve shows a profile with some specific features. First, there is a low q region, where the scattering intensity obeys a linear monotone decreasing function over several orders of q values. Such an aspect of the curve is characteristic of complex systems with a high degree of disorder. In this case, the results of the scattering measurements should be analyzed by taking into account the fractal geometry concepts. Thus, the intensity of SAXS analysis of fractal objects can be described by the equation:52
| I(q) = Ioqα. | (2) |
The value of the power-law exponent (α) represents the slope of the linear part of the scattering logarithmic plot, while Io is the scattering intensity at q = 0. Using these values, the volume (Dv) and surface (Ds) fractal dimensions were calculated. The fractal dimensions are useful to establish if the scattering object is a volume or surface fractal. The slope calculated according to the linear part of the scattering curve of |α| = 3.5 corresponds to a surface fractal having Ds = 2.5. In other terms, the system contains aggregates with dimensions larger than 100 nm, outside the SAXS range and a medium roughness of the surface.
The second important feature observed in the scattering curve profile is a large shoulder at q = 0.087 Å−1. According to the Bragg law, d = 2α/q, the size of the corresponding objects was estimated to be d = 72 Å. On the other hand, the aspect of the peak suggests a large polydispersity of the scatterers. Therefore, the SAXS analysis reveals that the sample contains nanoparticles having the average size around 7 nm, aggregated in large structures having an irregular shape and a mild roughness, thus confirming the TEM observations.
Thermal properties
The thermal degradation (TG) of the precursors and magnetic composites was followed under an inert gas atmosphere in the 25–680 °C temperature range. TG and DTA curves are shown in Fig. 5. | ||
| Fig. 5 TG (up) and DTA curves (down) of maghemite–goethite (MG), cellulose (C) and of their composites obtained by stirring (C-MGst) and ultrasonication (C-MGultr). | ||
For MG particles, due to the very small values of the mass loss, the highlight of the sample decomposition was possible only at higher heating rates (15 °C min−1) and by using nitrogen as the carrier gas. Four steps of weight loss were found under these conditions, 0.94, 0.83, 0.75 and 1.20%, respectively between 20–165, 165–300, 300–410 and 410–680 °C, while the DTA curve was still not a reliable one. The first decomposition phenomenon can be attributed to the loss of adsorbed water and water linked by hydrogen bonding. According to Liu et al.53 and Chen,54 pure goethite loses the chemically linked hydroxyl groups at temperatures of 200–300 °C and the remnant and partly non stoichiometric OH units at higher temperatures, up to 1000 °C, while pure maghemite shows only a thermal transition to hematite at temperatures of 550–580 °C, depending on particle dimensions. One can thus attribute the second and the third decomposition phenomena of the MG sample to the goethite component dehydroxylation and the transition to hematite, the fourth decomposition step being ascribed to the loss of residual and nonstoichiometric hydroxyl groups in goethite. Since for pure goethite the theoretical mass loss is 10.1% and the mass loss of the MG sample between 165 and 680 °C is 2.78%, a content of 27.5% goethite could be estimated for the MG inorganic particles.
The thermal decomposition for C and C-MGst samples occurs mainly in one degradation step, which runs through endothermic processes with Tpeak at 341.9 °C and 366.8 °C, respectively. The higher Tpeak value for the composite sample can be attributed to the protection of cellulose microfibrils by the more stable inorganic particles on the surface. A high weight loss of 95.6% for C and 90.0% for C-MGst samples was obtained. The C-MGultr sample showed a different thermal stability behavior. Apart from the first decomposition process of a lower weight loss (72.8%) and located at lower Tpeak = 349.7 °C compared to the C-MGst sample, a second degradation occurs at Tpeak = 645.4 °C with a mass loss of 5.0%. The second mass loss can be due to the degradation of cellulose–iron oxide clusters that could appear by chemical bonding between the OH groups of cellulose and those of the MG surface during ultrasonic treatment, as a result of the high energy developed. These structures show higher thermal stability compared to pure cellulose and were also observed in the TEM images of the C-MGultr sample.
The content of the MG ferromagnetic precursor in composites was calculated by considering the residues (ash) at 680 °C for pure cellulose and for C-MG composites (relation (3)).
| MG [%] = Residue for composite − Residue for cellulose | (3) |
Thus, 5.6 and 17.8 wt% MG contents in C-MGst and C-MGultr composites were calculated from the thermogravimetric analysis data. The result confirms the IR data on the higher content for the C-MGultr sample compared to that of C-MGst. Moreover, the MG content of C-MGultr is closer to the value of the initial mixture (20%), proving that sonication represents a more efficient homogenization process of the precursors.
Magnetic properties
The magnetic properties of the maghemite–goethite (MG) and composites (C-MGst, C-MGultr) were monitored by obtaining the hysteresis loops (magnetization curves) and the thermomagnetic curves (magnetization dependence on temperature) in the 10–300 K temperature range. The MG sample shows a hysteresis loop specific for ferromagnetic materials (Fig. 4S in the ESI†). The saturation values of the magnetic moment are 59.6 emu g−1 at 300 K (27 °C) and 75.5 emu g−1 at 10 K (−263 °C).11,34 The found saturation magnetization values for the composites obtained by stirring and ultrasonication at the same temperatures are lower, respectively, higher than the values calculated based on the maghemite–goethite content in the initial mixture (20 wt%) or as resulted from TGA analysis (Table 2).| Sample (MG content from TGA) | Temperature (°C) | M s (emu g−1) | ||
|---|---|---|---|---|
| Calculated based on the MG content in the initial mixture (20 wt%) | Calculated based on the MG content resulted from TGA | Experimental | ||
| C-MGst (5.6%) | 27 | 11.92 | 3.34 | 4.8 |
| −263 | 15.10 | 4.23 | 6.2 | |
| C-MGultr (17.8%) | 27 | 11.92 | 10.51 | 20 |
| −263 | 15.10 | 13.44 | 26 | |
As one may see from Table 2, for the C-MGst sample, the experimental Ms values are closer to those calculated by considering the content of inorganic particles as obtained from TG analysis (5.6%). The result confirms the lower efficiency of the simple stirring procedure that does not promote tight adhering of inorganic MG nanoparticles to cellulose microparticles (see also the comments on FTIR and TG analysis). On the contrary, the C-MGultr sample shows higher experimental Ms values than the calculated ones. A possible explanation is given by the occurrence of surface phenomena between cellulose and inorganic particles leading to an increased coordination of the iron atoms lying at the surface of the ferromagnetic particles and also to a high degree of interparticle interactions, which can lead to increased magnetic properties.18 The emergence of the new hydroxyl groups involved in the coordination with Fe atoms observed in the FTIR spectrum of C-MGultr (Fig. 2, the band at 3119 cm−1) and the presence of cellulose–inorganic particle clusters that showed a thermal decomposition temperature as high as 645 °C (Fig. 5) confirm this supposition.
The ZFC–FC curves representing the magnetic moment dependence on temperature in the presence and absence of an external magnetic field, respectively, are shown in Fig. 6. For the maghemite–goethite and C-MGst composite, ZFC and FC curves evidence characteristic features of ferromagnetic materials: a growth of the magnetic moment with temperature in the absence of an external magnetic field (ZFC curves) and a much lower variation in the presence of an external magnetic field (FC curves). Surprisingly, for the C-Multr composite, the ZFC and FC measurements also show characteristic superparamagnetic features with a blocking temperature (Tb) of 62 K (−211 °C), similar to other reported values. Thus, Cai et al. found a Tb of −163 °C for magnetite nanoparticles obtained in several liquid polyols;55 Rumpf et al. obtained values between −218 and −113 °C for a magnetite–porous silicon composite56 and Correa et al. obtained a Tb of −73 °C for magnetite in cellulose beads.57 In the FC curve of C-MGultr, the magnetic moment increases with decreasing temperatures. As the C-MGultr particles are cooled to low temperatures in the presence of a magnetic field, the magnetization direction of each particle is frozen in the field direction so that the magnetic moment is increasing. As the coercivity value of the sample is different from zero (Fig. 4S in the ESI†), the results support the idea of a polydisperse sample, where the very small cellulose–iron oxide cluster shows superparamagnetic properties, while the entire composite structure shows ferromagnetic properties.
 | ||
| Fig. 6 Thermomagnetic curves (temperature dependence of magnetization measured using the standard zero field cooling (ZFC) and field cooling (FC) procedure) for MG, C-MGst, C-MGultr. | ||
Conclusions
Ferromagnetic composites were obtained by two different mixing procedures of the suspensions of micronized cellulose and goethite–maghemite nanoparticles, i.e. mechanical stirring and ultrasonication. The last mentioned method was proved to yield composites with a higher content of magnetic inorganic particles, more uniformly distributed in the cellulose matrix of a decreased crystallinity induced by the cumulative action of ultrasound and MG nano-projectiles. VSM studies revealed the ferromagnetic character, with saturation magnetization values at room temperature of 4.8 and 20 emu g−1, for the samples obtained by stirring and ultrasonication, respectively. The superparamagnetic phenomenon observed in the ZFC-FC curves of C-MGultr is explained by the size polydispersity of the sample evidenced by TEM and SAXS analyses.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Research funded by Academia Româna (doctoral research program) – Unitatea Executiva pentru Finantarea Invatamantului Superior, a Cercetarii, Dezvoltarii si Inovarii (PN-II-PT-PCCA-2013-4-2210). The authors thank Dr Nicoleta Lupu (National Research and Development Institute for Technical Physics, Iasi, Romania) for VSM measurements and comments on magnetic properties.Notes and references
- Y. Li, H. Zhu, H. Gu, H. Dai, Z. Fang, N. J. Weadock, Z. Guo and L. Hu, J. Mater. Chem. A, 2013, 1(48), 15278–15283 CAS.
- R. Xiong, C. Lu, Y. Wang, Z. Zhou and X. Zhang, J. Mater. Chem. A, 2013, 47, 14910–14918 Search PubMed.
- A. Maleki, A. A. Jafari and S. Yousefia, Carbohydr. Polym., 2017, 175, 409–416 CrossRef CAS PubMed.
- A. Maleki and M. Kamalzare, Catal. Commun., 2014, 53, 67–71 CrossRef CAS.
- A. Maleki, H. Movahed and R. Paydar, RSC Adv., 2016, 6, 13657–13665 RSC.
- A. Maleki and M. Kamalzare, Tetrahedron Lett., 2014, 55, 6931–6934 CrossRef CAS.
- G. Biliuta, L. Sacarescu, V. Socoliuc, M. Iacob, L. Gheorghe, D. Negru and S. Coseri, Macromol. Chem. Phys., 2017, 218 CrossRef CAS , 1700062.
- R. T. Olsson, M. A. S. Azizi Samir, G. Salazar-Alvarez, L. Belova, V. Ström, L. A. Berglund, O. Ikkala, J. Nogues and U. W. Gedde, Nat. Nanotechnol. Lett., 2010, 5, 584–588 CrossRef CAS PubMed.
- S. F. Chin, A. N. Binti Romainor and S. C. Pang, Mater. Lett., 2014, 115, 241–243 CrossRef CAS.
- R. Rotaru, C. Cojocaru, P. Samoila, L. Pricop and V. Harabagiu, Environ. Eng. Manage. J., 2017 Search PubMed , accepted.
- R. Rotaru, PhD thesis, Romanian Academy, 2016.
- S. Liu, J. Zhou and L. Zhang, Cellulose, 2011, 18, 663–673 CrossRef CAS.
- R. H. Marchessault, P. Rioux and L. Raymond, Polymer, 1992, 33, 4024–4028 CrossRef CAS.
- J. A. Carrazana-García, M. A. Lopez-Quintela and J. Rivas-Rey, Colloids Surf., A, 1997, 121, 61–66 CrossRef.
- S. Galland, R. L. Andersson, M. Salajkova, V. Ström, R. T. Olsson and L. A. Berglund, J. Mater. Chem. C, 2013, 1, 7963–7972 RSC.
- W.-B. Wu, Y. Jing, M.-R. Gong, X.-F. Zhou and H.-Q. Dai, BioResources, 2011, 6, 3396–3409 CAS.
- S. Coseri, A. Spatareanu, L. Sacarescu, V. Socoliuc, I. S. Stratulat and V. Harabagiu, J. Appl. Polym. Sci., 2016, 133, 42926–42942 CrossRef.
- J. Bowles, M. Jackson, A. Chen and P. Solheid, IRM Quarterly, 2009, 19, 1–3 Search PubMed.
- R. M. Cornell and U. Schwertmann, The Iron Oxides: Structure, Properties, Reactions and Uses, Wiley-VCH Verlag, Weinheim, 2nd edn, 2003, p. 121 Search PubMed.
- Q. A. Pankhurst, J. Connolly, S. K. Jones and J. Dobson, J. Phys. D: Appl. Phys., 2003, 36, 167–181 CrossRef.
- G. C. Papaefthymiou, Nano Today, 2009, 4, 438–447 CrossRef CAS.
- A. P. Chen, IRM Quarterly, 2009, 19, 4–5 Search PubMed.
- X. Batlle and A. Labarta, J. Phys. D: Appl. Phys., 2002, 35, 15–42 CrossRef.
- C. M. Sorensen, Nanoscale Materials in Chemistry, ed. K. J. Klabunde, Wiley and Sons, New York, 2001, vol. 6, pp. 169–223 Search PubMed.
- N. Feltin and M. P. Pileni, Langmuir, 1997, 13, 3927–3933 CrossRef CAS.
- E. Murad, L. H. Bowen, G. J. Long and T. G. Quin, Clay Miner., 1988, 23, 161–173 CAS.
- (a) T. J. Mason, Chem. Soc. Rev., 1997, 26, 443–451 RSC; (b) B. Banerjee, Ultrason. Sonochem. A, 2017, 35, 1–14 CrossRef CAS PubMed.
- (a) K. S. Suslick and G. J. Price, Annu. Rev. Mater. Sci., 1999, 29, 295–326 CrossRef CAS; (b) D. Chen, S. K. Sharma and A. Mudhoo, Handbook on Applications of Ultrasound: Sonochemistry for Sustainability, CRC Press, Taylor and Francis Group, Boca Raton, London, New York, 2012 Search PubMed.
- H. M. Santos, C. Lodeiro and J.-L. Capelo-Martinez, Ultrasound in Chemistry: Analytical Applications, ed. J.-L. Capelo-Martinez, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2009, ch. 1, pp. 1–15 Search PubMed.
- L. Fang, J. Liu, S. Ju, F. Zheng, W. Dong and M. Shen, Appl. Phys. Lett., 2010, 97, 242501–242503 CrossRef.
- L. Liu, O. Albrecht, E. Pippel, K. Nielsch and U. Gösele, Electrochem. Commun., 2010, 12, 835–838 CrossRef CAS.
- N. O. Dudchenko, A. B. Brik, Y. V. Kardanets and O. E. Grechanivskyy, Proceedings of the International Conference Nanomaterials: Applications and Properties, 2013, vol. 2, CD version Search PubMed.
- V. N. Nikiforov, A. N. Ignatenko and V. Yu. Irkhin, J. Exp. Theor. Phys., 2017, 124, 304–310 CrossRef CAS.
- R. Rotaru, P. Samoila, N. Lupu, M. Grigoras and V. Harabagiu, Rev. Roum. Chim., 2017, 62, 131–138 Search PubMed.
- C. Wan, Y. Jiao, T. Qiang and J. Li, Carbohydr. Polym., 2017, 156, 427–434 CrossRef CAS PubMed.
- R. Rotaru, C. Peptu and V. Harabagiu, Cellul. Chem. Technol., 2016, 50, 621–628 CAS.
- P. V. Konarev, V. V. Volkov, A. V. Sokolova, M. H. J. Koch and D. I. Svergun, J. Appl. Crystallogr., 2003, 36, 1277–1282 CrossRef CAS.
- C. Cannas, A. Ardu, D. Niznansky, D. Peddis, G. Piccaluga and A. Musinu, J. Sol-Gel Sci. Technol., 2011, 60, 266–274 CrossRef CAS.
- J. Hu, I. M. C. Lo and G. H. Chen, Water Res., 2005, 39, 4528–4536 CrossRef CAS PubMed.
- M. Aliahmad and N. Nasiri Moghaddam, Mater. Sci., 2013, 31, 264–268 CAS.
- S. Layek, A. Pandey, A. Pandey and H. C. Verma, Int. J. Eng. Sci. Technol., 2010, 2(8), 33–39 Search PubMed.
- K. Kluchova, R. Zboril, J. Tucek, M. Pecova, L. Zajoncova, I. Safarik, M. Mashlan, I. Markova, D. Jancik, M. Sebela, H. Bartonkova, V. Bellesi, P. Novak and D. Petridis, Biomaterials, 2009, 30, 2855–2863 CrossRef CAS PubMed.
- U. Schwertmann and R. M. Cornell, Iron Oxides in the Laboratory: Preparation and Characterization, WILEY-VCH, Weinheim, New York, Chichester, Brisbane, Singapore, Toronto, 2nd edn, 2000, pp. 67–91 Search PubMed.
- G. Montes-Hernandez, P. Beck, F. Renard, E. Quirico, B. Lanson, R. Chiriac and N. Findling, Cryst. Growth Des., 2011, 11, 2264–2272 CAS.
- M. Fan, D. Dai and B. Huang, Fourier Transform Infrared Spectroscopy for Natural Fibres, ed. S. M. Salih, InTech, Croatia, 2012, ch. 3, pp. 45–68 Search PubMed.
- D. Ciolacu, F. Ciolacu and V. I. Popa, Cellul. Chem. Technol., 2011, 45, 13–21 CAS.
- T. Kondo, Cellulose, 1997, 4, 281–292 CrossRef CAS.
- I. Spiridon, C. A. Teaca and R. Bodirlau, BioResources, 2010, 6, 400–413 Search PubMed.
- D. Dai and M. Fan, Mater. Sci. Appl., 2010, 1, 336–342 CAS.
- S. Y. Oh, I. Y. Dong, Y. Shin, C. K. Hwan, Y. K. Hak, S. C. Yong, H. P. Won and H. Y. Ji, Carbohydr. Res., 2005, 340, 2376–2391 CrossRef CAS PubMed.
- S. Park, J. O. Baker, M. E. Himmel, P. A. Parilla and D. K. Johnson, Biotechnol. Biofuels, 2010, 3, 10 CrossRef PubMed.
- (a) J. E. Martin and A. J. Hurd, J. Appl. Crystallogr., 1987, 20, 61–78 CrossRef CAS; (b) P. W. Schmidt, J. Appl. Crystallogr., 1991, 24, 414–435 CrossRef CAS; (c) J. Teixeira, J. Appl. Crystallogr., 1988, 21, 781–785 CrossRef.
- H. Liu, T. Chen, X. Zou, C. Qing and R. L. Frost, Thermochim. Acta, 2013, 568, 115–121 CrossRef CAS.
- J. H. Chen, J. Alloys Compd., 2013, 553, 194–198 CrossRef.
- W. Cai and J. Wan, J. Colloid Interface Sci., 2007, 305, 366–370 CrossRef CAS PubMed.
- K. Rumpf, P. Granitzer, P. M. Morales, P. Poelt and M. Reissner, Nanoscale Res. Lett., 2012, 7, 445–448 CrossRef PubMed.
- J. R. Correa, E. Bordallo, D. Canetti, V. Leon, L. C. Otero-Diaz, C. Negro, A. Gómez and R. Saez-Puche, Mater. Res. Bull., 2010, 45, 946–953 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7py01587a |
| This journal is © The Royal Society of Chemistry 2018 |