
Hyperattenins A–I, bioactive polyprenylated acylphloroglucinols from Hypericum attenuatum Choisy†
Dongyan Li‡
ab,
Yongbo Xue‡a,
Hucheng Zhua,
Yan Lic,
Bin Suna,
Junjun Liua,
Guangmin Yaoa,
Jinwen Zhangb,
Guang Du*b and
Yonghui Zhang*a
aHubei Key Laboratory of Natural Medicinal Chemistry and Resource Evaluation, School of Pharmacy, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, People's Republic of China. E-mail: zhangyh@mails.tjmu.edu.cn; Fax: +86-27-83692762; Tel: +86-27-83692311
bTongji Hospital Affliated to Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, People's Republic of China. E-mail: tjyxb@sina.com; Tel: +86-27-83663643
cState Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650204, People's Republic of China
First published on 12th December 2014
Abstract
Nine new polyprenylated acylphloroglucinols (PPAPs), hyperattenins A–I (1–9), together with thirteen known analogues (10–22), were isolated from the aerial parts of Hypericum attenuatum Choisy. The structures of 1–9 were elucidated by extensive spectroscopic analysis. The absolute configuration of 1 was determined by a single X-ray crystallographic analysis, and the absolute configurations of 2–9 were determined by comparison of experimental and calculated electronic circular dichroism (ECD) spectra. Compound 1 was characterised as a bicyclo[3.3.1]nonane derivative containing an unusual hemiacetal functionality formed by a series of redox reactions on its side chains, which occurs rarely in nature. All new isolates were evaluated for cytotoxic activities against several human cancer cell lines. Compound 9 exhibited significant inhibitory activity against the HL-60 and A-549 cell lines, with IC50 values of 2.04 and 3.26 μM, respectively. Compound 9 also showed low toxicity to Beas-2B cells (IC50 = 14.36 μM), suggesting that it could be a selective anti-tumour agent for leukaemia and lung cancer. Compounds 2–8 were also screened for their anti-HIV-1 activities.
Introduction
The genus Hypericum comprises 484 species worldwide,1 many of which have been used as herbal medicines. H. attenuatum Choisy is one of the most popular medicinal herbs in China, and it is used in the treatment of numerous disorders such as haemostasia, analgesia and stimulation of lactation.2,3 Previous studies on the chemical constituents of H. attenuatum revealed the presence of flavonoids,4 dianthrones,5 and anthraquinones.5 A series of polycyclic polyprenylated acylphloroglucinols (PPAPs) were also isolated according to one recent study by Kong and co-workers.6As a part of an ongoing research program to discover more bioactive secondary metabolites from plants of the genus Hypericum, nine new PPAPs, termed hyperattenins A–I (1–9), were isolated from H. attenuatum Choisy, together with thirteen known analogues called sampsoniones C (11),7 E (12),7 F (13),7 G (14),7 and R (16),8 otogirinins A (15),9 B (17),9 and D (10),9 peroxysampsone B (18),10 furohyperforin (19),11 hyperforin (20),11 hypersampsone K (21),12 and propolone A (22).13 Notably, hyperattenin A (1) is characterised as a bicyclo[3.3.1]nonane derivative that possesses unusual hemiacetal functionality resulting from a series of redox reactions on its side chains, which occurs rarely in nature. Herein, we describe the isolation, structure determination, and cytotoxic and anti-HIV-1 activities of selected compounds.
Results and discussion
Air-dried aerial parts of H. attenuatum Choisy were powdered and extracted with 95% EtOH (4 × 300 L) at room temperature.The extract was concentrated in vacuo to yield brown gum and partitioned successively with petroleum ether, EtOAc, and n-BuOH. The petroleum ether-soluble extract was subjected to a series of column chromatographies, including MCI gel, silica gel, ODS, and Sephadex LH-20, and further purified by semi-preparative high performance liquid chromatography (HPLC) to give nine new PPAPs (1–9) and thirteen known analogues (10–22). The absolute configuration of 1 was determined by a single X-ray crystallographic analysis, and the absolute configurations of 2–9 were determined by comparison of the experimental and calculated electronic circular dichroism (ECD) spectra.
Hyperattenin A (1) was obtained as colourless crystals. The quasi-molecular ion peak at m/z 477.2626 ([M + H]+, calcd as 477.2641) and the 13C nuclear magnetic resonance (NMR) data (Table 2) allowed the assignment of the molecular formula of C30H36O5, corresponding to thirteen degrees of unsaturation. Infrared spectroscopy (IR) spectra showed absorption bands that were indicative of hydroxyl (3427 cm−1) and carbonyl (1729 and 1695 cm−1) groups. The 1H NMR spectrum of 1 (Table 1) indicated the presence of a monosubstituted benzene ring [δH 7.46 (2H, d, J = 7.4 Hz), 7.22 (2H, t, J = 7.6 Hz), and 7.38 (1H, t, J = 7.4 Hz)], two olefinic protons [δH 5.04 (1H, t, J = 7.4 Hz), 4.89 (1H, t, J = 7.2 Hz)], two methine signals [δH 5.97 (1H, brs), 1.48 (1H, m)], four CH2 protons [δH 2.71 (1H, brs) and 1.91 (1H, d, J = 10.2 Hz), 3.06 (1H, dd, J = 14.1, 7.5 Hz) and 2.97 (1H, dd, J = 14.1, 7.4 Hz), 2.20 (2H, m), 2.17 (2H, m)], and six methyl singlets (δH 1.67, 1.59 × 2, 1.53, 1.49, and 1.41). The 13C NMR and DEPT spectra of 1 displayed 30 carbons, resolving into six methyl groups, four CH2 groups, nine methine carbons (including one oxygenated carbon and seven olefinic carbons), and eleven quaternary carbons (including three carbonyls and five olefinic carbons). Based on the aforementioned evidence, 1 was determined to be a PPAP derivative that was closely related to sampsonione L.7
| Position | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| 6 | 5.97 brs | 4.68 t (9.2) | 4.66 dd (11.2, 9.9) | 4.52 d (3.1) | 4.36 dd (10.5, 4.5) |
| 4.49 ddd (11.9, 9.3, 5.7) | |||||
| 7 | 2.71 brs | 2.77 td (12.5, 9.4) | 2.85 dd (14.6, 9.9) | 4.87 d (3.1) | 3.10 dd (13.6, 10.5) |
| 1.91 d (10.2) | 1.90 dd (12.5, 5.6) | 2.80 dd (14.6, 11.2) | 1.83 dd (13.6, 4.5) | ||
| 9 | 2.20 m | 2.29 d (14.2) | 2.24 m | α 2.29 d (14.2) | α 2.87 d (14.7) |
| 2.16 m | β 2.22 dd (14.2, 6.9) | β 2.10 dd (14.7, 7.3) | |||
| 10 | 1.48 m | 1.50 m | 1.52 m | 1.54 m | 1.49 m |
| 15 | 7.46 d (7.4) | 7.47 d (7.6) | 7.59 d (7.5) | 7.52 dd (8.4, 1.1) | 7.69 d (8.0) |
| 16 | 7.22 t (7.6) | 7.22 t (7.6) | 7.26 t (7.8) | 7.23 t (7.8) | 7.37 t (7.7) |
| 17 | 7.38 t (7.4) | 7.38 t (7.3) | 7.40 t (7.4) | 7.38 t (7.4) | 7.51 t (7.3) |
| 18 | 7.22 t (7.6) | 7.22 t (7.6) | 7.26 t (7.8) | 7.23 t (7.8) | 7.38 t (7.7) |
| 19 | 7.46 d (7.4) | 7.47 d (7.6) | 7.59 d (7.5) | 7.52 dd (8.4, 1.1) | 7.70 d (8.0) |
| 20 | 3.06 dd (14.1, 7.5) | 3.03 m | 2.69 dd (13.6, 8.4) | ||
| 2.97 dd (14.1, 7.4) | 2.48 m | ||||
| 21 | 5.04 t (7.4) | 5.05 t (7.3) | 4.97 m | ||
| 23 | 1.59 s | 1.60 s | 1.67 s | ||
| 24 | 1.59 s | 1.60 s | 1.66 s | ||
| 25 | 2.17 m | 2.17 m | 2.30 m | 2.39 m | 2.46 m |
| 1.79 m | 2.33 m | 2.19 m | |||
| 26 | 4.89 t (7.2) | 4.87 t (6.9) | 5.03 t (6.8) | 4.89 t (7.4) | 4.97 m |
| 28 | 1.67 s | 1.69 s | 1.65 s | 1.69 s | 1.70 s |
| 29 | 1.53 s | 1.54 s | 1.55 s | 1.61 s | 1.60 s |
| 30 | 1.49 s | 1.49 s | 1.47 s | 1.51 s | 1.16 s |
| 31 | 1.41 s | 1.42 s | 1.34 s | 1.36 s | 1.03 s |
| 33 | 1.25 s | 1.28 s | 1.19 s | ||
| 34 | 1.41 s | 1.33 s | 1.31 s | ||
| 35 | 3.91 dq (14.0, 7.0) | ||||
| 3.67 dq (14.0, 7.0) | |||||
| 36 | 1.13 t (7.0) | ||||
| 1′ | 2.54 m | 2.58 m | |||
| 2′ | 5.16 t (7.0) | 5.08 m | |||
| 4′ | 2.02 m | 1.97 m | |||
| 5′ | 1.68 s | 1.70 s | |||
| 6′ | 2.08 m | 2.01 m | |||
| 7′ | 5.09 t (6.8) | 5.05 m | |||
| 9′ | 1.67 s | 1.64 s | |||
| 10′ | 1.61 s | 1.56 s |
Comparison of the NMR data of 1 with those of sampsonione L suggested that the most noticeable differences between 1 and sampsonione L were the loss of a 2-hydroxyl-isopropyl and the presence of a hydroxyl at C-6 in 1. This conclusion was evidenced by the 1H–1H COSY correlation from H-6 (δH 5.97) to H2-7 (δH 2.71 and 1.91) and the key HMBC correlations (Fig. 3) from H2-7 to C-4, C-8, C-9, and C-12. The relative configuration of the core structure of 1 was determined on the basis of a NOESY experiment and by comparison with the coupling constants and chemical shifts of 1 with those of sampsonione L.7 However, all of the relative configurations of 1 were determined to be the same as those of sampsonione L, except for the orientation of H-6 owing to the absence of significant interactions in the NOESY spectrum. Fortunately, a single crystal of 1 was obtained from MeOH, and a single X-ray diffraction experiment was performed using Cu Kα radiation (ESI†) (Fig. 2), which clarified the above conclusion and unambiguously confirmed the planar structure and clarified the absolute configuration of 1 as 1S,6R,8R,10S with a probability of 1.000, according to the refined Flack parameter value 0.09(19) and Hooft parameter value 0.06(4) for 2250 Bijvoet pairs.14–16
The molecular formula of hyperattenin B (2) was established as C30H36O4, which was 16 mass units greater than that of 1. The 1H and 13C NMR data (Tables 1 and 2) of 2 were quite similar to those of 1, except that a hemiacetal methine at C-6 in 1 was replaced by an oxygenated methylene group in 2. This deduction was supported by the COSY sequence of H2-6/H2-7 and the HMBC correlations of H2-6 to C-4, C-7, and C-8. The gross structure of 2 was elucidated by careful interpretation of the 2D NMR experiments, and its relative configuration was established by comparison of 2 with 1 in terms of their closely related NMR data including chemical shifts, coupling constants, and NOESY spectroscopic data. The absolute configuration of 2 was determined to be 1S,8R,10S by comparing the experimental ECD spectrum of 2 with that of 1 (Fig. 5a).
| Position | 1 | 2 | 3 | 4 | 5 | Position | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 77.3 | 77.2 | 77.8 | 78.3 | 67.9 | 24 | 17.6 | 17.66 | 18.11 | ||
| 2 | 194.5 | 194.0 | 188.5 | 188.7 | 194.8 | 25 | 29.2 | 28.9 | 30.3 | 30.4 | 28.9 |
| 3 | 115.6 | 115.6 | 118.2 | 119.9 | 118.0 | 26 | 124.3 | 124.4 | 124.8 | 125.5 | 124.9 |
| 4 | 172.3 | 172.9 | 176.9 | 179.1 | 178.1 | 27 | 132.6 | 132.8 | 132.4 | 132.5 | 134.9 |
| 5 | 28 | 25.74 | 25.8 | 25.82 | 25.8 | 26.2 | |||||
| 6 | 103.0 | 72.0 | 93.4 | 99.5 | 93.2 | 29 | 17.8 | 17.68 | 18.0 | 18.1 | 18.08 |
| 7 | 37.3 | 30.6 | 28.0 | 78.1 | 28.8 | 30 | 22.1 | 22.3 | 22.6 | 22.8 | 22.2 |
| 8 | 58.0 | 57.9 | 53.9 | 54.1 | 59.4 | 31 | 26.9 | 27.0 | 26.7 | 27.0 | 26.8 |
| 9 | 37.8 | 35.5 | 39.9 | 39.4 | 39.5 | 32 | 70.7 | 71.0 | 70.7 | ||
| 10 | 47.8 | 47.7 | 48.7 | 48.3 | 46.5 | 33 | 25.0 | 24.1 | 25.2 | ||
| 11 | 48.7 | 49.3 | 49.5 | 49.7 | 46.9 | 34 | 26.9 | 26.0 | 27.1 | ||
| 12 | 205.4 | 205.6 | 207.0 | 207.1 | 205.7 | 35 | 66.6 | ||||
| 13 | 194.0 | 193.6 | 193.4 | 193.2 | 193.1 | 36 | 15.5 | ||||
| 14 | 136.9 | 137.0 | 136.8 | 136.5 | 137.0 | 1′ | 29.6 | 29.6 | |||
| 15 | 128.0 | 128.1 | 128.2 | 128.1 | 129.2 | 2′ | 118.3 | 119.5 | |||
| 16 | 127.9 | 127.9 | 127.9 | 128.0 | 128.3 | 3′ | 138.8 | 138.7 | |||
| 17 | 132.0 | 131.9 | 132.1 | 132.1 | 133.2 | 4′ | 39.9 | 40.0 | |||
| 18 | 127.9 | 127.9 | 127.9 | 128.0 | 128.5 | 5′ | 16.4 | 16.8 | |||
| 19 | 128.0 | 128.1 | 128.2 | 128.1 | 129.4 | 6′ | 26.5 | 26.3 | |||
| 20 | 22.5 | 22.4 | 25.6 | 7′ | 123.9 | 123.7 | |||||
| 21 | 119.3 | 119.5 | 119.6 | 8′ | 131.7 | 131.9 | |||||
| 22 | 132.9 | 132.9 | 132.8 | 9′ | 25.76 | 25.6 | |||||
| 23 | 25.67 | 25.7 | 25.8 | 10′ | 17.8 | 17.7 |
Hyperattenin C (3) was obtained as a colourless oil. The corresponding high-resolution electrospray ionisation mass spectra (HRESIMS) experiment showed a signal at m/z 609.3475 [M + Na]+, which was in accordance with the 13C NMR data, suggesting a molecular formula of C38H50O5. The 1H and 13C NMR data (Tables 1 and 2) of 3 were very similar to those of otogirinin D (10),9 except for the resonances near C-6. Comparing the carbon signals of 3 with those of 10, the chemical shift of C-32 (δC 70.7) was shifted upfield, while C-33 (δC 25.0) and C-34 (δC 26.9) were shifted downfield. These facts revealed that 3 was a C6-epimer of 10. The relative configuration at C-10 of 3 was deduced on the basis of the NOESY correlations of H2-25/Me-30 and of Me-31 with H2-9 observed in the NOESY spectrum of 3 (Fig. 4). These observations implied that the stereochemistry of the isoprenyl moiety at C-10 was in an axial configuration, which was identical to that of 10.9 In addition, NOESY cross-peaks of Me-28/H-6 indicated that H-6 was in the β-orientation. The calculated ECD spectrum of (1S,6S,8S,10S)-3 matched well with the experimental ECD spectrum of 3 (Fig. 5b), which had the structure and absolute configuration of 3 as depicted (Fig. 1).
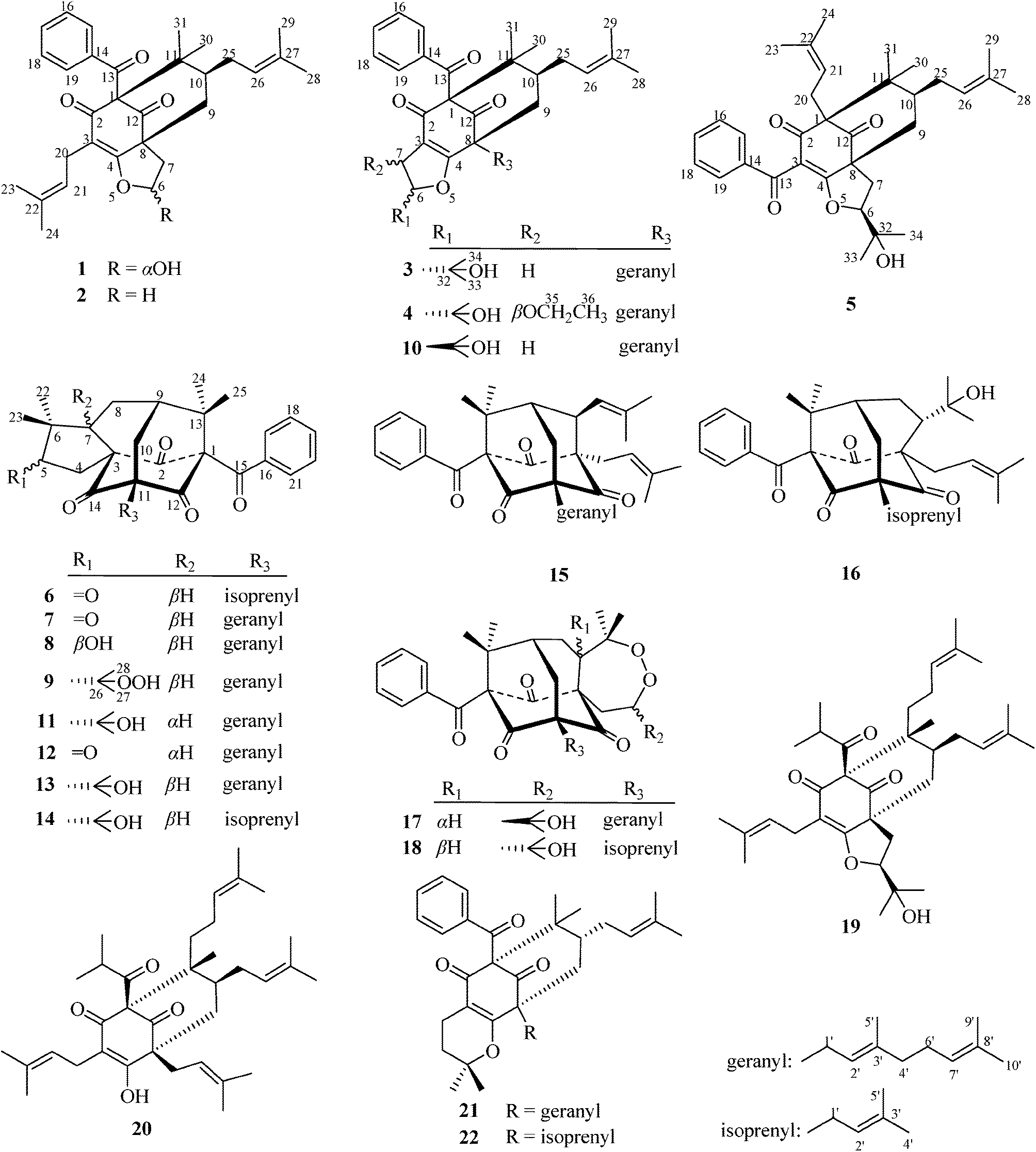 | ||
| Fig. 1 The structures of 1–22. | ||
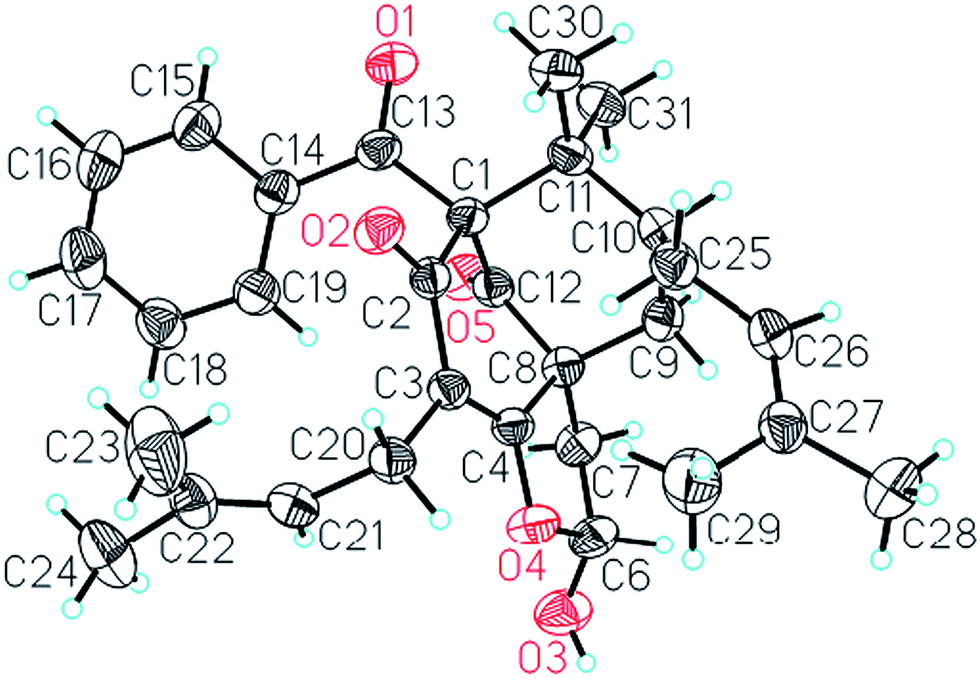 | ||
| Fig. 2 X-ray crystal structure of 1. | ||
Hyperattenin D (4) possessed a molecular formula of C40H54O6 as determined by the 13C NMR data and HRESIMS (m/z 631.3982 [M + H]+). Comparison of the NMR data of 4 with those of 3 revealed structural similarities. The main differences observed in the 13C NMR spectra of 4 and 3 were that the chemical shifts of C-3 (δC 119.9), C-4 (δC 179.1), C-6 (δC 99.5), and C-7 (δC 78.1) were dramatically downshifted, while the chemical shifts of C-33 (δC 24.1) and C-34 (δC 26.0) were upshifted in 4. In the HMBC spectrum, H-7 interacted with C-35, and H2-35 correlated with C-7 and C-36. These findings, together with the multiplicities of H2-35 and Me-36 in the 1H NMR spectrum indicated that an ethoxy group was located at C-7 of 4. The NOESY spectrum exhibited the relative configurations of H-10 and H-6 in 4, which were identical to those of 3. Subsequently, the β-orientation of H-7 was deduced by the cross-peak between H-7 and Me-33 in the NOESY spectrum. Comparing the experimental ECD curves between 4 and 3 (Fig. 5b), the absolute configuration of 4 was determined to be 1R,4S,6S,8S,10S. The ethyl group in 4 was hypothesised to be an artefact formed during isolation, although the de-ethyl precursor of 4 was not obtained in this study.
The HRESIMS and 13C NMR spectra of hyperattenin E (5) suggested the same molecular formula (C33H42O5) as that of sampsonione P, a known compound previously reported from H. sampsonii.8 The 1H NMR and 13C data (Tables 1 and 2) of 5 also closely resembled those of sampsonione P, and the differences were the slight shifts of C-2, C-3, C-4, C-6, C-7, C-8, C-9, C-13, and Me-34. Careful analyses of the 1H–1H COSY, HSQC, and HMBC spectra of 5 indicated the same planar structure as that of sampsonione P.8 The NOESY correlations (Fig. 4) of Me-317/H-9α and H-9β/H2-25 indicated that the isoprenyl group at C-10 was β-oriented. The β-orientation of the 2-(2′-hydroxy)propyl group at C-6 of 5 was determined by the NOESY correlations of H-9β/Me-33. The experimental ECD spectrum (Fig. S105 in the ESI†) was contrary to that of oblongifolin L,17 allowing determination of the absolute configuration of 5 to be 1S,6S,8R,10S.
Hyperattenin F (6) was isolated as yellow oil. Its molecular formula, C30H34O5, was established from the quasi-molecular ion peak at m/z 497.2289 [M + Na]+ (calcd 497.2304, C30H34O5Na) in the positive HRESIMS. The apparent differences between 6 and sampsonione E7 (12) in the 1H and 13C NMR spectra (Tables 3 and 4) indicated that the geranyl side chain at C-11 in 12 was replaced by an isoprenyl group in 6. This elucidation was confirmed by the HMBC correlation (Fig. 3) of H2-1′ (δH 2.64) with C-10 (δC 35.6), C-11 (δC 67.2), C-12 (δC 204.0), C-14 (δC 202.6), C-2′ (δC 118.4), and C-3′ (δC 135.8). Molecular models indicated that the tetracyclic system itself established the relative configurations of the chiral centres of C-1, C-3, C-9, and C-11. The relative configuration of the remaining chiral centre (C-7) was elucidated by analysis of the NOESY spectrum (Fig. 4). NOESY cross-peaks of Me-25/H-10α, H-10β/H-7, and Me-24/H-8α were observed, suggesting that H-7 was β-oriented. To determine the absolute configuration of 6, the ECD experiment and ECD calculation of 6 were conducted. The experimental ECD curve of 6 was in accordance with the calculated ECD curve for (1R,3R,7S,9S,11S)-6 (Fig. 5c), and the structure of 6 was thus established as shown (Fig. 1).
| Position | 6a | 6b | 7a | 7b | 8a | 9a |
|---|---|---|---|---|---|---|
| a Recorded in CDCl3.b Recorded in C5D5N. | ||||||
| 4 | 3.49 d (18.9) | 3.82 d (18.7) | 3.50 d (18.9) | 3.83 d (18.7) | 2.82 dd (14.9, 4.6) | 2.47 m |
| 2.74 d (18.9) | 2.99 d (18.7) | 2.74 d (18.9) | 3.00 d (18.7) | 2.31 m | ||
| 5 | 3.90 d (4.4) | |||||
| 7 | 2.20 m | 2.57 dd (11.4, 8.3) | 2.20 m | 2.58 dd (11.3, 8.3) | 2.31 m | 1.92 m |
| 8 | 2.20 m | 2.13 m | 2.20 m | 2.14 m | 1.98 m | 1.92 m |
| 1.96 m | 1.98 m | 1.96 m | 1.97 m | 1.73 m | 1.68 m | |
| 9 | 2.20 m | 2.21 dd (13.5, 6.1) | 2.20 m | 2.23 m | 2.14 m | 2.10 m |
| 10 | α 2.62 m | 2.70 dd (14.7, 6.8) | 2.63 m | 2.76 dd (14.8, 6.8) | 2.56 dd (14.8, 6.6) | 2.49 dd (14.8, 6.6) |
| β 2.19 m | 2.52 d (14.9) | 2.16 m | 2.50 d (14.8) | 2.23 d (14.8) | 2.20 d (14.8) | |
| 17 | 7.02 d (7.5) | 7.40 d (8.0) | 7.02 d (8.0) | 7.40 d (7.8) | 7.02 dd (8.3, 1.2) | 7.04 d (8.2) |
| 18 | 7.28 t (8.0) | 7.26 t (7.5) | 7.29 t (7.5) | 7.26 t (7.5) | 7.26 t (7.9) | 7.29 t (8.1) |
| 19 | 7.41 t (7.4) | 7.34 t (7.4) | 7.41 t (7.2) | 7.34 t (7.4) | 7.39 t (7.4) | 7.39 t (7.4) |
| 20 | 7.28 t (8.0) | 7.26 t (7.5) | 7.29 t (7.5) | 7.26 t (7.5) | 7.26 t (7.9) | 7.29 t (8.1) |
| 21 | 7.02 d (7.5) | 7.40 d (8.0) | 7.02 d (8.0) | 7.40 d (7.8) | 7.02 dd (8.3, 1.2) | 7.04 d (8.2) |
| 22 | 1.02 s | 1.06 s | 1.02 s | 1.07 s | 0.90 s | 1.11 s |
| 23 | 1.09 s | 1.02 s | 1.09 s | 1.05 s | 1.04 s | 0.95 s |
| 24 | 1.42 s | 1.50 s | 1.42 s | 1.52 s | 1.39 s | 1.42 s |
| 25 | 1.46 s | 1.57 s | 1.47 s | 1.60 s | 1.43 s | 1.384 s |
| 27 | 1.375 s | |||||
| 28 | 1.34 s | |||||
| 1′ | 2.64 m | 2.87 m | 2.65 m | 2.90 d (7.4) | 2.64 d (7.4) | 2.62 m |
| 2′ | 5.26 t (7.6) | 5.53 t (7.3) | 5.29 t (7.3) | 5.59 t (6.9) | 5.30 t (7.5) | 5.30 t (7.5) |
| 4′ | 1.69 s | 1.62 s | 2.07 m | 2.05 m | 2.07 m | 2.07 m |
| 5′ | 1.74 s | 1.65 s | 1.67 s | 1.67 s | 1.67 s | 1.66 s |
| 6′ | 2.07 m | 2.10 m | 2.06 m | 2.08 m | ||
| 7′ | 5.06 t (7.4) | 5.13 t (6.7) | 5.07 t (6.5) | 5.06 t (12.2) | ||
| 9′ | 1.59 s | 1.65 s | 1.66 s | 1.66 s | ||
| 10′ | 1.66 s | 1.54 s | 1.58 s | 1.59 s | ||
| Position | 6a | 6b | 7a | 7b | 8a | 9a | Position | 6a | 6b | 7a | 7b | 8a | 9a |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Recorded in CDCl3.b Recorded in C5D5N. | |||||||||||||
| 1 | 81.4 | 81.4 | 81.4 | 81.5 | 81.0 | 81.2 | 20 | 128.4 | 128.2 | 128.38 | 128.2 | 128.3 | 128.4 |
| 2 | 204.1 | 204.8 | 204.1 | 204.8 | 202.4 | 203.1 | 21 | 128.4 | 128.3 | 128.39 | 128.3 | 128.5 | 128.5 |
| 3 | 68.3 | 68.3 | 68.3 | 68.3 | 74.6 | 70.7 | 22 | 20.8 | 20.2 | 20.8 | 20.2 | 20.0 | 17.3 |
| 4 | 41.0 | 40.9 | 41.0 | 41.0 | 36.5 | 30.4 | 23 | 26.2 | 25.3 | 26.2 | 25.4 | 22.2 | 29.2 |
| 5 | 217.3 | 216.5 | 217.3 | 216.4 | 82.2 | 56.0 | 24 | 22.5 | 22.0 | 22.5 | 22.0 | 22.4 | 22.5 |
| 6 | 49.3 | 48.7 | 49.3 | 48.7 | 48.8 | 46.2 | 25 | 25.07 | 24.4 | 25.0 | 24.4 | 25.3 | 25.3 |
| 7 | 50.1 | 49.4 | 50.2 | 49.6 | 51.9 | 56.2 | 26 | 85.0 | |||||
| 8 | 25.09 | 24.5 | 25.1 | 24.5 | 22.8 | 22.7 | 27 | 24.0 | |||||
| 9 | 42.3 | 41.7 | 42.3 | 41.8 | 42.3 | 42.2 | 28 | 24.7 | |||||
| 10 | 35.6 | 35.3 | 35.4 | 35.2 | 35.4 | 35.1 | 1′ | 29.2 | 29.1 | 29.0 | 29.0 | 28.8 | 28.9 |
| 11 | 67.2 | 67.1 | 67.2 | 67.1 | 67.3 | 67.6 | 2′ | 118.4 | 118.8 | 118.5 | 118.8 | 118.6 | 118.9 |
| 12 | 204.0 | 204.6 | 204.0 | 204.6 | 204.3 | 204.6 | 3′ | 135.8 | 134.9 | 139.5 | 138.5 | 139.3 | 139.0 |
| 13 | 47.8 | 47.7 | 47.7 | 47.6 | 47.8 | 47.8 | 4′ | 18.1 | 17.3 | 40.0 | 39.6 | 40.0 | 40.0 |
| 14 | 202.6 | 202.1 | 202.6 | 202.1 | 206.0 | 203.6 | 5′ | 26.1 | 25.4 | 16.4 | 15.7 | 16.4 | 16.3 |
| 15 | 192.4 | 192.7 | 192.4 | 192.7 | 192.6 | 192.8 | 6′ | 26.5 | 26.2 | 26.6 | 26.6 | ||
| 16 | 134.8 | 134.7 | 134.8 | 134.9 | 134.8 | 134.9 | 7′ | 124.1 | 124.0 | 124.1 | 124.2 | ||
| 17 | 128.4 | 128.3 | 128.39 | 128.3 | 128.5 | 128.5 | 8′ | 131.5 | 130.7 | 131.5 | 131.4 | ||
| 18 | 128.4 | 128.2 | 128.38 | 128.2 | 128.3 | 128.4 | 9′ | 17.7 | 17.0 | 17.7 | 17.7 | ||
| 19 | 132.5 | 132.2 | 132.5 | 132.2 | 132.3 | 132.3 | 10′ | 25.8 | 25.1 | 25.8 | 25.8 | ||
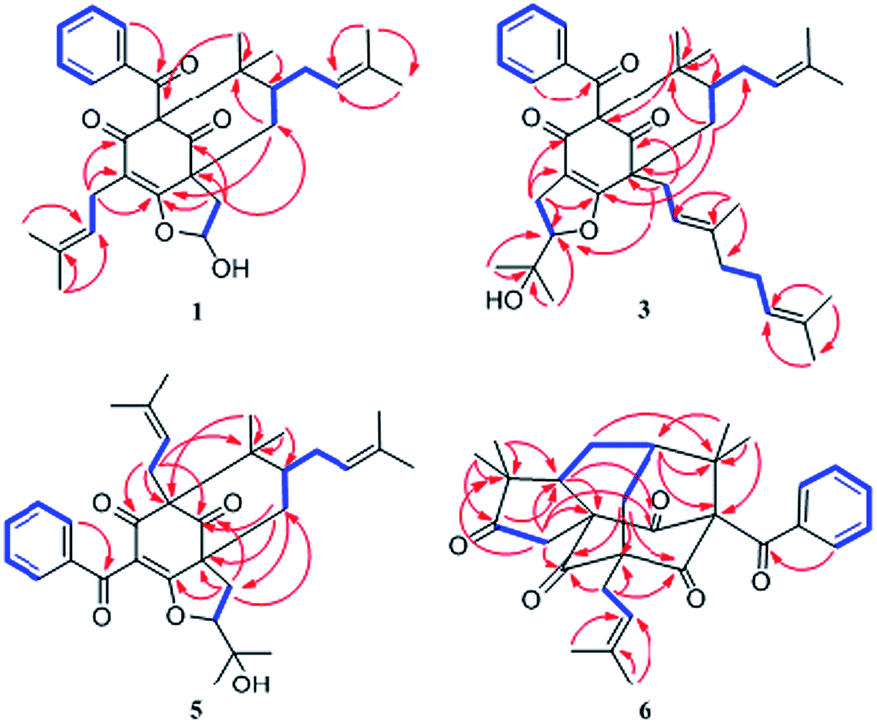 | ||
| Fig. 3 1H–1H COSY (bold) and key correlations observed in the HMBC (arrow) spectrum of 1, 3, 5, 6. | ||
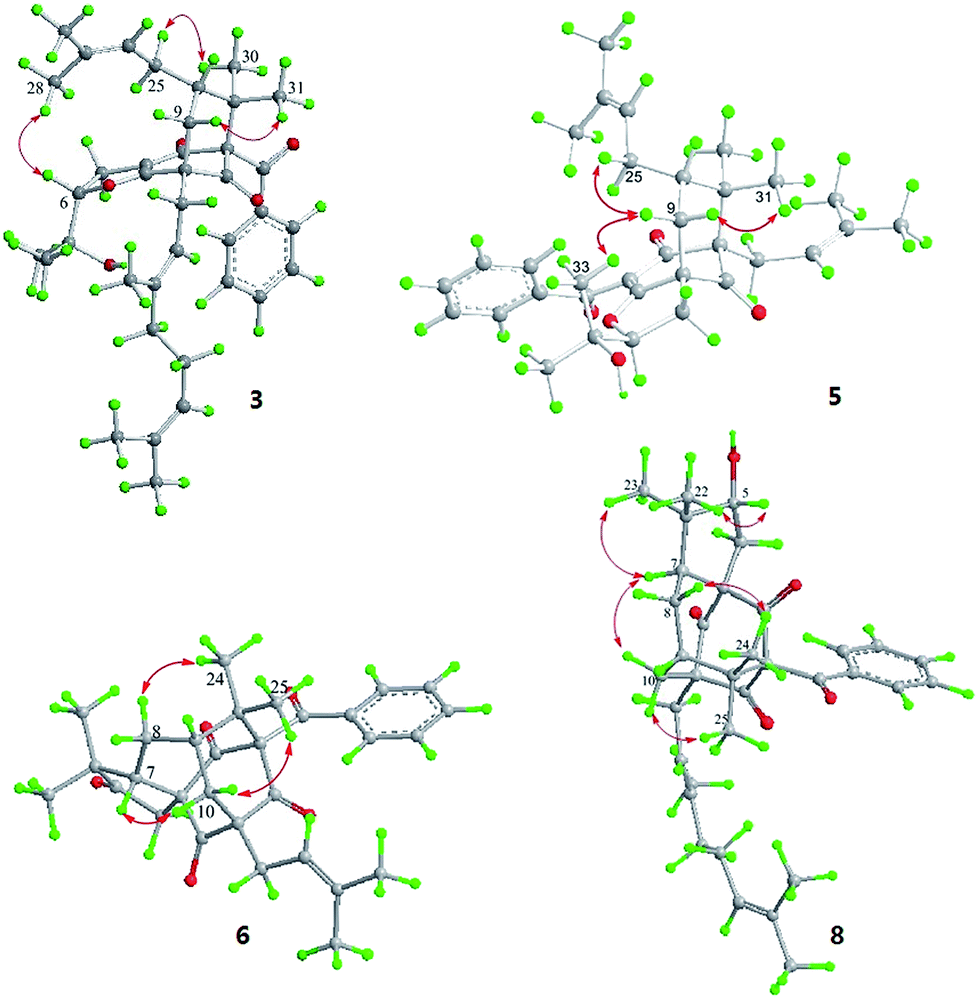 | ||
| Fig. 4 Key NOESY correlations of 3, 5, 6, and 8. | ||
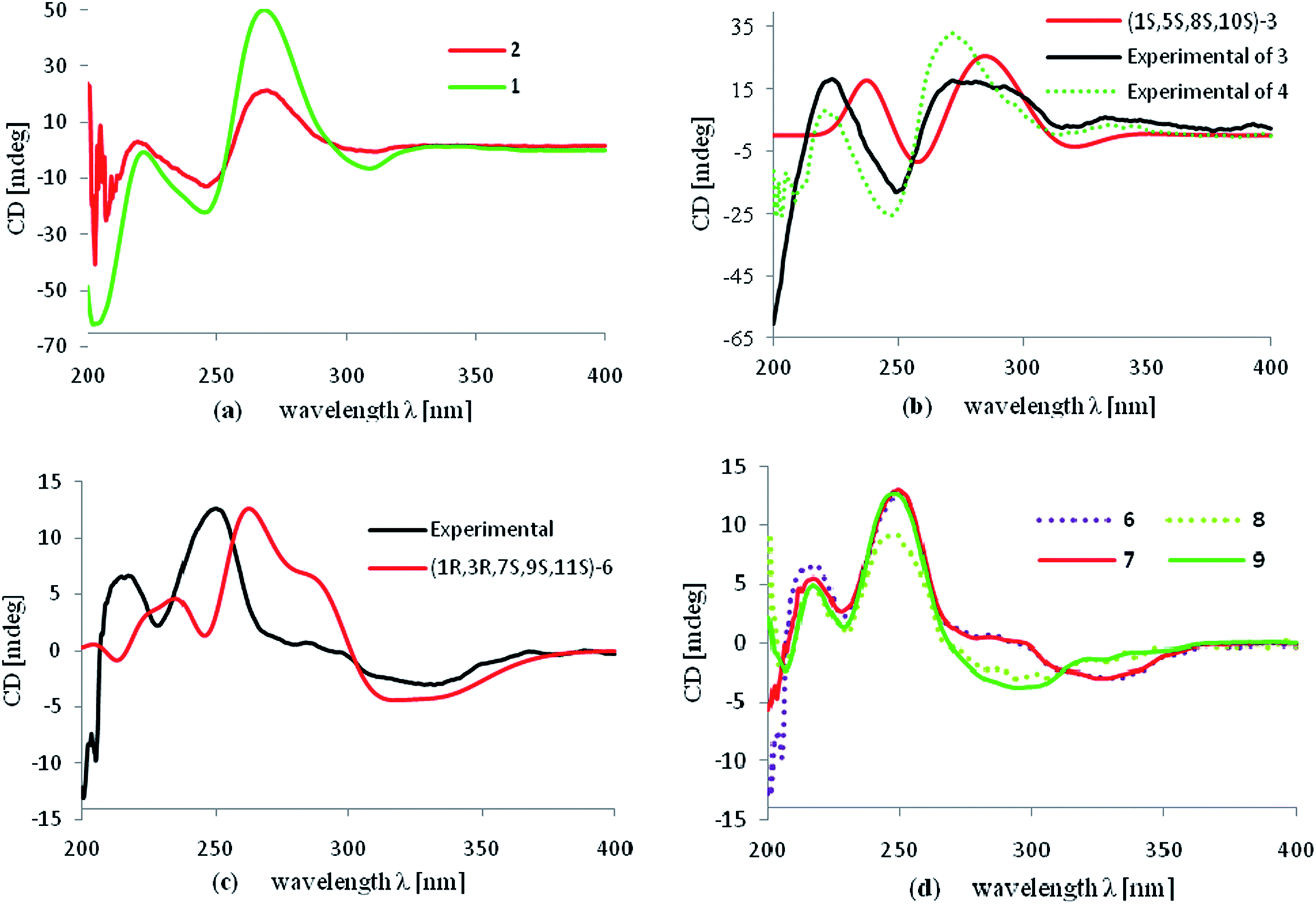 | ||
| Fig. 5 (a) Experimental ECD spectra of 1 and 2; (b) calculated ECD spectrum of 3 and the experimental ECD curves of 3 and 4; (c) calculated and experimental ECD spectrum of 6; (d) experimental ECD spectra of 6–9. | ||
Hyperattenin G (7) was assigned the molecular formula of C35H42O5, corresponding to [M + Na]+ (565.2913, calcd 565.2930) in the HRESIMS and 13C NMR spectra. Comparison of the NMR spectroscopic data of 7 (Tables 3 and 4) with those of 6 revealed that the only difference between 7 and 6 was the substitution at C-11. The isoprenyl group at C-11 in 6 was replaced by a geranyl moiety at C-11 in 7, which was evidenced by the HMBC correlations (Fig. 3) from H2-1′ to C-10, C-11, C-12, and C-14. A detailed analysis of the NOESY spectrum of 7 revealed that the relative configurations of all the chiral centres in 7 were identical to those of 6. Furthermore, the experimental ECD spectrum of 7 was in good agreement with that of 6 (Fig. 5d), which indicated that the absolute configuration of 7 was 1R,3R,7S,9S,11S.
Hyperattenin H (8) was obtained as colourless oil, and the HRESIMS of 8 indicated a molecular ion at m/z 567.3066 ([M + Na]+, calcd 567.3086), suggesting a molecular formula of C35H44O5. The 1H and 13C NMR spectroscopic data of 8 (Tables 3 and 4) were quite similar to those of 7, except for the absence of signal for a free carbonyl group (δC 217.3) and the appearance of oxygenated methine functionality (δC 82.2) in 8. The β-orientation of H-7 was established by analysis of the NOESY correlations of 8 (Fig. 4). In addition, the NOESY interactions of H-7/Me-23 and Me-22/H-5 implied the β-orientation of OH-5 in the molecule. The experimental ECD spectrum of 8 was in good agreement with that of 6 (Fig. 5d), which revealed that the absolute configuration of 8 was 1R,3R,5R,7S,9S,11S.
Hyperattenin I (9), a colourless oil, had a molecular formula of C38H50O6, based on the HRESIMS data (m/z 625.3488 [M + Na]+, calcd for C38H50O6Na, 625.3505) and its 13C NMR spectrum. The 1H and 13C NMR spectra of 9 (Tables 3 and 4) were very close to those of attenuatumione D,6 except for the noticeable differences in the chemical shifts of C-5 (δC 56.0), C-26 (δC 85.0), C-27 (δC 24.0), and C-28 (δC 24.7) in 9. These observations, along with the HRESIMS data of 9, implied the presence of a peroxide group at C-26 of the molecule. The relative configurations of the chiral centres at C-1, C-3, C-7, C-9, and C-11 in 9 were determined to be the same as those of 6, according to the NOESY spectra. H-5 was determined to be β-oriented by the comparable 13C NMR and NOESY spectra of 9 and attenuatumione D.6 The absolute configuration of 9 was thus determined to be (1R,3R,5S,7S,9S,11S)-9 by comparison of the experimental ECD spectra of 9 and 6.
The known compounds 10–22 (Fig. 1) were identified by NMR spectroscopy and mass spectrometry, as well as by comparison of physical and spectroscopic data with compounds reported in the literature.7–13
All new isolates were screened for cytotoxic activities against five human cancer cell lines, HL-60, A-549, SMMC-7721, MCF-7, and SW-480, as well as the immortalised non-cancerous Beas-2B human bronchial epithelial cell line. The results (Table 5) showed that compounds 1–5 and 8–9 possessed cytotoxic activities against a set of human cancer cell lines, while compounds 6 and 7 were found to be inactive (IC50 > 40 μM). Compound 9 exhibited significant inhibitory activities against the HL-60 and A-549 cell lines, with IC50 values of 2.04 and 3.26 μM, respectively. Notably, compound 9 could be a selective anti-tumour agent for leukaemia and lung cancer. In addition, compounds 2–8 were evaluated for anti-HIV-1 activity. However, all the tested compounds were found to be inactive (Table 6).
| Compound | HL-60 | SMMC-7721 | A-549 | MCF-7 | SW480 | BEAS-2B |
|---|---|---|---|---|---|---|
| a DDP (cis-platin) and paclitaxel were used as positive controls. | ||||||
| 1 | 9.62 | 9.89 | 12.40 | 12.34 | 28.96 | 16.38 |
| 2 | 15.26 | 23.15 | 21.50 | 16.79 | >40 | >40 |
| 3 | 18.03 | 35.34 | 16.20 | 19.04 | >40 | 18.12 |
| 4 | >40 | 26.86 | 30.36 | 19.31 | >40 | 22.13 |
| 5 | 31.60 | >40 | 30.89 | 31.38 | >40 | >40 |
| 6 | >40 | >40 | >40 | >40 | >40 | >40 |
| 7 | >40 | >40 | >40 | >40 | >40 | >40 |
| 8 | >40 | 20.51 | >40 | >40 | >40 | >40 |
| 9 | 2.04 | 11.32 | 3.26 | 10.06 | 15.88 | 14.36 |
| DDPa | 2.12 | 7.47 | 9.01 | 15.23 | 13.10 | 11.46 |
| Paclitaxela | <0.008 | <0.008 | <0.008 | <0.008 | <0.008 | 1.03 |
| Compound | CC50a (μM) | EC50a (μM) | SIb |
|---|---|---|---|
| a Cytotoxicity (CC50) and antiviral activity (EC50) were determined using an MTT assay on C8166 cells.b Selectivity index (SI) is the ratio of CC50 to EC50.c NT not tested. | |||
| 2 | 9.63 | 9.93 | 0.97 |
| 3 | 13.40 | 8.11 | 1.65 |
| 4 | 4.58 | 11.67 | 0.39 |
| 5 | 0.64 | 13.93 | 0.05 |
| 6 | 17.21 | 15.02 | 1.15 |
| 7 | 87.87 | NTc | NT |
| 8 | 13.10 | >50 | NT |
| AZT | >0.25 | 0.0032 | >78.13 |
Conclusions
In summary, twenty-two PPAPs were isolated from H. attenuatum Choisy, including nine novel and thirteen known analogues. Hyperattenins A–I (1–9) along with eleven known compounds (12–22) were reported from H. attenuatum Choisy for the first time. Interestingly, hyperattenin A (1) was characterised as a bicyclo[3.3.1]nonane derivative containing unusual hemiacetal functionality, which occurs very rarely in nature.18 Usually, natural PPAPs are obtained as oil or gum because the molecules contain polyprenyl substitutions; thus, it is difficult to crystallise these metabolites and determine their absolute configurations.19 Fortunately, the single crystals of 1 were obtained from MeOH, allowing us to establish its absolute configuration using single X-ray diffraction. Furthermore, the absolute configurations of 2–9 were also elucidated by comparison of their experimental ECD curves with the calculated ECD spectra. Hyperattenin I (9) exhibited significant cytotoxicity against the HL-60 and A-549 cell lines and low toxicity to Beas-2B cells, suggesting that it could be of interest in the search for effective new anti-tumour agents for leukaemia and lung cancer.Experimental section
General experimental procedures
Melting points were measured using a Beijing Tech X-5 micro-melting point apparatus without correction. 1D and 2D NMR spectra were acquired on a Bruker AM-400 spectrometer, and the 1H and 13C NMR chemical shifts were referenced with respect to the solvent or solvent impurity peaks for CDCl3 at δH 7.26 and δC 77.0. Optical rotations were obtained on a Perkin-Elmer 341 polarimeter equipped with a sodium lamp (589 nm) and a 1 dm microcell. UV spectra were determined on a Varian Cary 50 instrument. ECD spectra were obtained with a JASCO J-810 spectrometer. IR spectra were recorded on a Bruker Vertex 70 FT-IR spectrophotometer. HRESIMS were acquired in the positive-ion mode on a Thermo Fisher LC-LTQ-Orbitrap XL spectrometer. The crystallographic data were obtained on a Bruker SMART APEX-II CCD diffractometer equipped with graphite-monochromatised Cu Kα radiation (λ = 1.54178 Å). Semi-preparative HPLC was carried out on a Dionex quaternary system with a diode array detector at a flow rate of 2.5 mL min−1 using a reversed-phased C18 column (5 μm, 10 × 250 mm, YMC-pack ODS-A). Silica gel (Qingdao Marine Chemical Inc., China), ODS (50 μm, YMC Co. Ltd., Japan), and Sephadex LH-20 (GE Healthcare Bio-Sciences AB, Sweden) were applied for column chromatography. Thin-layer chromatography (TLC) was conducted with silica gel 60 F254 (Yantai Chemical Industry Research Institute) and RP-C18 F254 plates (Merck, Germany).Plant material
The aerial parts of Hypericum attenuatum Choisy were collected in Qichun County, Hubei province, People's Republic of China, in October 2011. The plants were identified by Prof. J. P. Wang at the School of Pharmacy, Tongji Medical College, Huazhong University of Science and Technology. A voucher specimen (no. 20111001) was deposited in the herbarium of Hubei Key Laboratory of Natural Medicinal Chemistry and Resource Evaluation, Tongji Medical College, Huazhong University of Science and Technology.Extraction and isolation
The dried plants (79 kg) were powdered and extracted with 95% EtOH (4 × 300 L) at room temperature. The 95% EtOH extracts were filtered, combined, and concentrated in vacuo to form a residue (5.1 kg). The residue was suspended in H2O and fractionated successively with petroleum ether, EtOAc, and n-BuOH to yield a petroleum ether-soluble fraction (500 g), EtOAc-soluble fraction (430 g), and n-BuOH-soluble fraction (700 g), respectively. The petroleum ether-soluble fraction (500 g) was fractionated to seven fractions (A–G) via silica gel column chromatography (CC), and the fractions were eluted with petroleum ether–acetone (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 1:1). Fraction C (50 g) was subjected to a RP C-18 column (MeOH–H2O, 60:40 to 100:0) to yield four subfractions, C1–C4. Subfraction C2 (11.2 g) was further fractionated over Sephadex LH-20 CC (CH2Cl2–MeOH, 1:1) to yield five fractions (C2a–C2e). Compound 15 (4.3 mg) was isolated from fraction C2b by repeated semi-preparative HPLC (MeOH–H2O, 88:12). Similarly, compounds 16 (11.6 mg), 21 (7.7 mg) and 22 (4.1 mg) were purified from fraction C2c by semi-preparative HPLC (MeOH–H2O, 85:15). Fraction D (75 g) was purified over MCI gel CC (MeOH–H2O, 8:2 to 10:0) to yield two subfractions (D1–D2). Fraction D1 (51.8 g) was loaded on a silica gel CC eluted with petroleum ether–acetone (1:0 to 0:1) to yield five fractions (D1a–D1e). Fraction D1b (35.0 g) was further purified on an RP-C18 column (MeOH–H2O, 50:50 to 100:0) to yield four subfractions (D1b1–D1b4). Fraction D1b1 (7.1 g) was fractionated into seven fractions (D1b1a–D1b1g) over Sephadex LH-20 CC eluted with MeOH. Fraction D1b1a was purified by a RP-C18 column (MeOH–H2O, 89:11) to yield compound 4 (5.4 mg). Fraction D1b1c was purified by an RP-C18 column (MeOH–H2O, 88:12) to yield compound 7 (5.5 mg). Fraction D1b1d was subjected to semi-preparative HPLC (CH3CN–H2O, 85:15) to yield compounds 8 (7.1 mg), 13 (21.2 mg) and 12 (3.3 mg). Fraction D1b2 (7.6 g) was chromatographed on a Sephadex LH-20 CC (MeOH) and an ODS column (MeOH–H2O, (7:3) and purified by a RP C-18 column (CH3CN–H2O, 75:25) to yield compounds 6 (16.1 mg) and 14 (14.9 mg). Fraction D1b3 was subjected to a Sephadex LH-20 CC (MeOH) to obtain three subfractions, D1b3a–D1b3c. Separation of fraction D1b3a was conducted by a RP-C18 column eluted with a gradient system of MeOH–H2O (50:50 to 100:0) to yield three main subfractions, D1b3a1–D1b3a3. Subfraction D1b3a1 was separated using semi-preparative HPLC (CH3CN–H2O, 82:18) to obtain compounds 19 (6.1 mg) and 20 (4.4 mg). Fraction E (78 g) was subjected to an MCI gel CC (MeOH–H2O, 85:15 to 100:0) to yield two fractions (E1–E2). Fraction E1 (58 g) was chromatographed on a silica gel eluted with petroleum ether–EtOAc (20:1 to 1:1) to yield seven subfractions (E1a–E1g). Fraction E1c (29.5 g) was chromatographed over an ODS column to yield three fractions (E1c1–E1c3). Fraction E1c1 was subjected to Sephadex LH-20 CC (CH2Cl2–MeOH, 1:1) to yield three fractions E1c1a–E1c1c. Compounds 1 (35.1 mg) and 2 (4.4 mg) were obtained by repeated silica gel CC, eluted with a petroleum ether–acetone gradient, and then followed by semi-preparative HPLC (MeOH–H2O, 80:20). Fraction E1c2 was separated into two fractions (E1c2a–E1c2b) by Sephadex LH-20 CC (CH2Cl2–MeOH, 1:1). Compounds 5 (4.8 mg) and 18 (5.0 mg) were obtained from E1c2a by semi-preparative HPLC (MeCN–H2O, 85:15). Fraction E1c3 (17.0 g) was separated into five fractions (E1c3a–E1c3e) by a Sephadex LH-20 CC with CH2Cl2–MeOH (1:1) and a silica gel CC eluting with petroleum ether–EtOAc (20:1 to 1:1). Fraction E1c3a was subjected to a Sephadex LH-20 CC (MeOH) and further purified by a RP-C18 column (MeOH–H2O, 85:15) to obtain compound 9 (3.5 mg). Compounds 3 (14.7 mg), 10 (21.1 mg), 11 (25.4 mg), and 17 (42.8 mg) were isolated from E1c3b by semi-preparative HPLC (MeOH–H2O, 85:15).
ε) 278 (3.99), 248 (4.10), 204 (4.36) nm; ECD (c 11.62 × 10−4 M, CH3CN) λmax nm (Δε) 203 (−16.15), 245 (−5.77), 268 (+13.04), 309 (−1.70); IR (KBr) vmax 3427, 2968, 2924, 1729, 1695 cm−1; 1H and 13C NMR data, see Tables 1 and 2; positive HRESIMS: m/z 477.2626 [M + H]+ (calcd for C30H37O5, 477.2641).ε) 276 (3.97), 247 (4.07), 204 (4.33) nm; ECD (c 11.84 × 10−4 M, CH3CN) λmax nm (Δε) 203 (−10.42), 245 (−3.30), 269 (+5.51), 310 (−0.14); IR (KBr) vmax 3437, 2967, 2925, 1727, 1696 cm−1; 1H and 13C NMR data, see Tables 1 and 2; positive HRESIMS: m/z 461.2692 [M + H]+ (calcd for C30H37O4, 461.2692).ε) 286 (3.90), 248 (3.97), 204 (4.38) nm; ECD (c 8.96 × 10−4 M, MeOH) λmax nm (Δε) 223 (+6.05), 249 (−6.23), 272 (+5.94), 333 (+1.96); IR (KBr) vmax 3449, 2973, 2923, 1724, 1699 cm−1; 1H and 13C NMR data, see Tables 1 and 2; positive HRESIMS: m/z 609.3475 [M + Na]+ (calcd for C38H50O5Na, 609.3556).ε) 278 (2.88), 247 (3.01), 205 (3.37) nm; ECD (c 9.90 × 10−4 M, MeOH) λmax nm (Δε) 221 (+2.39), 248 (−7.77), 272 (+10.05), 336 (+1.02); IR (KBr) vmax 3520, 2972, 2918, 1725, 1698 cm−1; 1H and 13C NMR data, see Tables 1 and 2; positive HRESIMS: m/z 631.3982 [M + H]+ (calcd for C40H55O6, 631.3999).ε) 252 (4.30), 205 (4.44) nm; ECD (c 19.68 × 10−4 M, MeOH) λmax nm (Δε) 222 (−6.00), 250 (+5.38), 263 (+5.18), 299 (−3.13); IR (KBr) vmax 3450, 2972, 2928, 1732, 1713 cm−1; 1H and 13C NMR data, see Tables 1 and 2; positive HRESIMS: m/z 519.3090 [M + H]+ (calcd for C33H43O5, 519.3111).ε) 244 (4.17), 205 (4.59) nm; ECD (c 10.86 × 10−4 M, MeOH) λmax nm (Δε) 217 (+1.85), 250 (+3.51), 328 (−0.85); IR (KBr) vmax 2960, 2926, 2856, 1740, 1705 cm−1; 1H and 13C NMR data, see Tables 3 and 4; positive HRESIMS: m/z 497.2289 [M + Na]+ (calcd for C30H34O5Na, 497.2304).ε) 247 (4.02), 205 (4.49) nm; ECD (c 5.86 × 10−4 M, MeOH) λmax nm (Δε) 217 (+2.85), 249 (+6.73), 325 (−1.55); IR (KBr) vmax 2962, 2924, 2855, 1743, 1703 cm−1; 1H and 13C NMR data, see Tables 3 and 4; positive HRESIMS: m/z 565.2913 [M + Na]+ (calcd for C35H42O5Na, 565.2930).ε) 245 (4.16), 204 (4.57) nm; ECD (c 3.97 × 10−4 M, MeOH) λmax nm (Δε) 217 (+3.78), 249 (+7.11), 294 (−2.29); IR (KBr) vmax 3542, 2963, 2914, 1736, 1699 cm−1; 1H and 13C NMR data, see Tables 3 and 4; positive HRESIMS: m/z 567.3066 [M + Na]+ (calcd for C35H44O5Na, 567.3086).ε) 246 (4.01), 203 (4.40) nm; ECD (c 5.71 × 10−4 M, MeOH) λmax nm (Δε) 217 (+2.57), 248 (+6.73), 295 (−2.06); IR (KBr) vmax 3435, 2979, 2926, 1736, 1699 cm−1; 1H and 13C NMR data, see Tables 3 and 4; positive HRESIMS: m/z 625.3488 [M + Na]+ (calcd for C38H50O6Na, 625.3505).571 unique reflections (θ = 3.79 to 67.49°) were collected using graphite monochromatised Cu Kα radiation (λ = 1.54178 Å) on a Bruker APEX-II diffractometer. The structure was solved using direct methods (SHELXTL program package) and refined with Full-matrix least-squares on F2. Hydrogen atoms were located by the geometric calculation method. Final R indicates R1 = 0.0410, wR2 = 0.1247. The goodness of fit on F2 was 1.059. Flack parameter = 0.09(19). The Hooft parameter is 0.06(4) for 2250 Bijvoet pairs.†
:1 to 1:1). Fraction C (50 g) was subjected to a RP C-18 column (MeOH–H2O, 60:40 to 100:0) to yield four subfractions, C1–C4. Subfraction C2 (11.2 g) was further fractionated over Sephadex LH-20 CC (CH2Cl2–MeOH, 1:1) to yield five fractions (C2a–C2e). Compound 15 (4.3 mg) was isolated from fraction C2b by repeated semi-preparative HPLC (MeOH–H2O, 88:12). Similarly, compounds 16 (11.6 mg), 21 (7.7 mg) and 22 (4.1 mg) were purified from fraction C2c by semi-preparative HPLC (MeOH–H2O, 85:15). Fraction D (75 g) was purified over MCI gel CC (MeOH–H2O, 8:2 to 10:0) to yield two subfractions (D1–D2). Fraction D1 (51.8 g) was loaded on a silica gel CC eluted with petroleum ether–acetone (1:0 to 0:1) to yield five fractions (D1a–D1e). Fraction D1b (35.0 g) was further purified on an RP-C18 column (MeOH–H2O, 50:50 to 100:0) to yield four subfractions (D1b1–D1b4). Fraction D1b1 (7.1 g) was fractionated into seven fractions (D1b1a–D1b1g) over Sephadex LH-20 CC eluted with MeOH. Fraction D1b1a was purified by a RP-C18 column (MeOH–H2O, 89:11) to yield compound 4 (5.4 mg). Fraction D1b1c was purified by an RP-C18 column (MeOH–H2O, 88:12) to yield compound 7 (5.5 mg). Fraction D1b1d was subjected to semi-preparative HPLC (CH3CN–H2O, 85:15) to yield compounds 8 (7.1 mg), 13 (21.2 mg) and 12 (3.3 mg). Fraction D1b2 (7.6 g) was chromatographed on a Sephadex LH-20 CC (MeOH) and an ODS column (MeOH–H2O, (7:3) and purified by a RP C-18 column (CH3CN–H2O, 75:25) to yield compounds 6 (16.1 mg) and 14 (14.9 mg). Fraction D1b3 was subjected to a Sephadex LH-20 CC (MeOH) to obtain three subfractions, D1b3a–D1b3c. Separation of fraction D1b3a was conducted by a RP-C18 column eluted with a gradient system of MeOH–H2O (50:50 to 100:0) to yield three main subfractions, D1b3a1–D1b3a3. Subfraction D1b3a1 was separated using semi-preparative HPLC (CH3CN–H2O, 82:18) to obtain compounds 19 (6.1 mg) and 20 (4.4 mg). Fraction E (78 g) was subjected to an MCI gel CC (MeOH–H2O, 85:15 to 100:0) to yield two fractions (E1–E2). Fraction E1 (58 g) was chromatographed on a silica gel eluted with petroleum ether–EtOAc (20:1 to 1:1) to yield seven subfractions (E1a–E1g). Fraction E1c (29.5 g) was chromatographed over an ODS column to yield three fractions (E1c1–E1c3). Fraction E1c1 was subjected to Sephadex LH-20 CC (CH2Cl2–MeOH, 1:1) to yield three fractions E1c1a–E1c1c. Compounds 1 (35.1 mg) and 2 (4.4 mg) were obtained by repeated silica gel CC, eluted with a petroleum ether–acetone gradient, and then followed by semi-preparative HPLC (MeOH–H2O, 80:20). Fraction E1c2 was separated into two fractions (E1c2a–E1c2b) by Sephadex LH-20 CC (CH2Cl2–MeOH, 1:1). Compounds 5 (4.8 mg) and 18 (5.0 mg) were obtained from E1c2a by semi-preparative HPLC (MeCN–H2O, 85:15). Fraction E1c3 (17.0 g) was separated into five fractions (E1c3a–E1c3e) by a Sephadex LH-20 CC with CH2Cl2–MeOH (1:1) and a silica gel CC eluting with petroleum ether–EtOAc (20:1 to 1:1). Fraction E1c3a was subjected to a Sephadex LH-20 CC (MeOH) and further purified by a RP-C18 column (MeOH–H2O, 85:15) to obtain compound 9 (3.5 mg). Compounds 3 (14.7 mg), 10 (21.1 mg), 11 (25.4 mg), and 17 (42.8 mg) were isolated from E1c3b by semi-preparative HPLC (MeOH–H2O, 85:15).
ε) 278 (3.99), 248 (4.10), 204 (4.36) nm; ECD (c 11.62 × 10−4 M, CH3CN) λmax nm (Δε) 203 (−16.15), 245 (−5.77), 268 (+13.04), 309 (−1.70); IR (KBr) vmax 3427, 2968, 2924, 1729, 1695 cm−1; 1H and 13C NMR data, see Tables 1 and 2; positive HRESIMS: m/z 477.2626 [M + H]+ (calcd for C30H37O5, 477.2641).ε) 276 (3.97), 247 (4.07), 204 (4.33) nm; ECD (c 11.84 × 10−4 M, CH3CN) λmax nm (Δε) 203 (−10.42), 245 (−3.30), 269 (+5.51), 310 (−0.14); IR (KBr) vmax 3437, 2967, 2925, 1727, 1696 cm−1; 1H and 13C NMR data, see Tables 1 and 2; positive HRESIMS: m/z 461.2692 [M + H]+ (calcd for C30H37O4, 461.2692).ε) 286 (3.90), 248 (3.97), 204 (4.38) nm; ECD (c 8.96 × 10−4 M, MeOH) λmax nm (Δε) 223 (+6.05), 249 (−6.23), 272 (+5.94), 333 (+1.96); IR (KBr) vmax 3449, 2973, 2923, 1724, 1699 cm−1; 1H and 13C NMR data, see Tables 1 and 2; positive HRESIMS: m/z 609.3475 [M + Na]+ (calcd for C38H50O5Na, 609.3556).ε) 278 (2.88), 247 (3.01), 205 (3.37) nm; ECD (c 9.90 × 10−4 M, MeOH) λmax nm (Δε) 221 (+2.39), 248 (−7.77), 272 (+10.05), 336 (+1.02); IR (KBr) vmax 3520, 2972, 2918, 1725, 1698 cm−1; 1H and 13C NMR data, see Tables 1 and 2; positive HRESIMS: m/z 631.3982 [M + H]+ (calcd for C40H55O6, 631.3999).ε) 252 (4.30), 205 (4.44) nm; ECD (c 19.68 × 10−4 M, MeOH) λmax nm (Δε) 222 (−6.00), 250 (+5.38), 263 (+5.18), 299 (−3.13); IR (KBr) vmax 3450, 2972, 2928, 1732, 1713 cm−1; 1H and 13C NMR data, see Tables 1 and 2; positive HRESIMS: m/z 519.3090 [M + H]+ (calcd for C33H43O5, 519.3111).ε) 244 (4.17), 205 (4.59) nm; ECD (c 10.86 × 10−4 M, MeOH) λmax nm (Δε) 217 (+1.85), 250 (+3.51), 328 (−0.85); IR (KBr) vmax 2960, 2926, 2856, 1740, 1705 cm−1; 1H and 13C NMR data, see Tables 3 and 4; positive HRESIMS: m/z 497.2289 [M + Na]+ (calcd for C30H34O5Na, 497.2304).ε) 247 (4.02), 205 (4.49) nm; ECD (c 5.86 × 10−4 M, MeOH) λmax nm (Δε) 217 (+2.85), 249 (+6.73), 325 (−1.55); IR (KBr) vmax 2962, 2924, 2855, 1743, 1703 cm−1; 1H and 13C NMR data, see Tables 3 and 4; positive HRESIMS: m/z 565.2913 [M + Na]+ (calcd for C35H42O5Na, 565.2930).ε) 245 (4.16), 204 (4.57) nm; ECD (c 3.97 × 10−4 M, MeOH) λmax nm (Δε) 217 (+3.78), 249 (+7.11), 294 (−2.29); IR (KBr) vmax 3542, 2963, 2914, 1736, 1699 cm−1; 1H and 13C NMR data, see Tables 3 and 4; positive HRESIMS: m/z 567.3066 [M + Na]+ (calcd for C35H44O5Na, 567.3086).ε) 246 (4.01), 203 (4.40) nm; ECD (c 5.71 × 10−4 M, MeOH) λmax nm (Δε) 217 (+2.57), 248 (+6.73), 295 (−2.06); IR (KBr) vmax 3435, 2979, 2926, 1736, 1699 cm−1; 1H and 13C NMR data, see Tables 3 and 4; positive HRESIMS: m/z 625.3488 [M + Na]+ (calcd for C38H50O6Na, 625.3505).571 unique reflections (θ = 3.79 to 67.49°) were collected using graphite monochromatised Cu Kα radiation (λ = 1.54178 Å) on a Bruker APEX-II diffractometer. The structure was solved using direct methods (SHELXTL program package) and refined with Full-matrix least-squares on F2. Hydrogen atoms were located by the geometric calculation method. Final R indicates R1 = 0.0410, wR2 = 0.1247. The goodness of fit on F2 was 1.059. Flack parameter = 0.09(19). The Hooft parameter is 0.06(4) for 2250 Bijvoet pairs.†Computational methods
The conformational analyses were carried out for compounds 3 and 6 using BALLOON11 and confab12 programs. The BALLOON program searches conformational space with a generic algorithm, whereas the confab program systematically generates diverse low energy conformations that are supposed to be close to crystal structures. The conformations generated by both programs were grouped together by removing the duplicated conformations in which the root mean square (RMS) distance was less than 0.5 Å. Semi-empirical PM3 quantum mechanical geometry optimisations were performed on the conformations using the Gaussian 0913 program. The duplicated conformations after geometry optimisation were then identified and removed. The remaining conformations were further optimised at the B3LYP/6-31G* level of theory in methanol solvent with the IEFPCM314 solvation model using the Gaussian 09 program, and the duplicated conformations emerging after these calculations were removed according to the same RMS criteria indicated above. The harmonic vibrational frequencies were performed to confirm the stability of the obtained conformers (Fig. S106 and S107†). The oscillator strengths and rotational strengths of the 20 weakest electronic excitations of each conformer were calculated using the TDDFT methodology at the B3LYP/6-311++G** level of theory with methanol as the solvent by the IEFPCM solvation model implemented in the Gaussian 09 program. The ECD spectra for each conformer were then simulated using a Gaussian function with a bandwidth σ of 0.45 eV. The calculated spectra for each conformation were combined after Boltzmann weighting according to their population contribution (Tables S1 and S2†).Cytotoxicity assay
The cytotoxicity of the new PPAPs was determined by MTS assays, as reported previously.20–22 Five human cancer cell lines, myeloid leukaemia cell line HL-60, hepatocellular carcinoma cell line SMMC-7721, lung cancer cell line A-549, breast cancer cell line MCF-7, and colon cancer cell line SW480, together with one noncancerous human pulmonary epithelial cell line, BEAS-2B, were used. All cells were grown in RPMI-1640 or in Dulbecco's Modified Eagle Medium (Hyclone, Logan, UT, USA) and supplemented with 10% foetal bovine serum (Hyclone, Logan, UT, USA) in 5% CO2 at 37 °C. A volume of 100 μL of adherent cells was seeded into each well of the 96-well culture plates at an initial density of 1 × 105 cells per mL, and cells were allowed to adhere for 12 h before adding test compounds. Each cancer cell line was exposed to test compounds at concentrations of 0.064, 0.32, 1.6, 8, and 40 μM in triplicate for 48 h in the incubator, and DDP (cis-platin, Sigma, St. Louis, MO, USA) and paclitaxel (Sigma, St. Louis, MO, USA) were used as positive controls. Then, 100 μL of medium and 20 μL of MTS (Sigma, St. Louis, MO, USA) were added to each well for 4 h. Later, the cell viability was measured using a 96-well microtitre plate (Bio-Rad 680) at λ = 595 nm, and a cell growth curve was graphed. The IC50 value of each test compound was calculated by Reed and Muench's method.23Anti-HIV assay
Anti-HIV assays include cytotoxicity and HIV-1 replication inhibition assays. The cytotoxicity was measured using the MTT method as previously described.24 HIV-infected C8166 cells (1 × 104 per well) were seeded into a 96-well microtitre plate in the absence or presence of various concentrations of test compounds in triplicate and incubated at 37 °C in a humidified atmosphere of 5% CO2. After a 5 day incubation, cell viability was measured. The concentration that caused the reduction of viable cells by 50% (CC50) was determined. In parallel with the MTT assay, the HIV-1 replication inhibition assay was conducted using p24 antigen capture enzyme-linked immunosorbent assay (ELISA).25 C8166 cells were exposed to HIV-1LAI (MOI = 0.01) at 37 °C for 1.5 h, washed with phosphate buffered saline (PBS) to remove free viruses, and then seeded into a 96-well microtitre plate at 2 × 104 cells per well in the absence or presence of test compounds (zidovudine was used as the positive control). Then, the supernatant was collected and inactivated by 0.5% Triton X-100. The supernatant was diluted three times, added to the plate coated with anti-p24 McAb (provided by Dr Bin Yan, Wuhan Institute of Virology, China), and incubated at 37 °C for 1 h. After washing five times with phosphate buffered saline with Tween-20 (PBST), the horseradish peroxidase labelled anti-p24 antibody (provided by Dr Bin Yan, Wuhan Institute of Virology, China) was added and incubated at 37 °C for 1 h. Then, the plate was washed 5 times with PBST, followed by adding OPD to the reaction mixture. The assay plate was read at 490 nm using a microplate reader within 30 min. The inhibition rate and EC50 based on p24 antigen expression level were calculated, and the selective index was calculated as CC50/EC50.Acknowledgements
We are grateful to Prof. J. P. Wang at Huazhong University of Science and Technology for the authentication of this plant material. This project was financially supported by the National Natural Science Foundation of China (31370372, 31200258, and 81202423), Program for New Century Excellent Talents in University, State Education Ministry of China (NCET-2008-0224), and National Science and Technology Project of China (nos 2011ZX09102-004).Notes and references
- S. I. Crockett and N. K. B. Robson, Plant Sci. Biotechnol., 2011, 5, 1–12 CrossRef.
- Flora of China Editorial Committee of Chinese Academy of Sciences, in Flora of China, Science Press, Beijing, 1990, vol. 50, p. 69 Search PubMed.
- Jiangsu New Medicine College, in Dictionary of Traditional Chinese Medicine, Shanghai Science and Technology Press, Shanghai, 1986, p. 1823 Search PubMed.
- J. Y. Dong and Z. J. Jia, Chin. Pharm. J., 2005, 40, 897–899 CAS.
- O. M. ka, Ukr. Biokhim. Zh., 2000, 72, 110–113 Search PubMed.
- Z. B. Zhou, Y. M. Zhang, K. Pan, J. G. Luo and L. Y. Kong, Fitoterapia, 2014, 95, 1–7 CrossRef CAS PubMed.
- L. H. Hu and K. Y. Sim, Tetrahedron, 2000, 56, 1379–1386 CrossRef CAS.
- Z. Y. Xiao, Q. Mu, W. K. P. Shiu, Y. H. Zeng and S. Gibbons, J. Nat. Prod., 2007, 70, 1779–1782 CrossRef CAS PubMed.
- Y. Ishida, O. Shirota, S. Sekita, K. Someya, F. Tokita, T. Nakane and M. Kuroyanagi, Chem. Pharm. Bull., 2010, 58, 336–343 CrossRef CAS.
- Z. Y. Xiao, Y. H. Zeng, Q. Mu, W. K. P. Shiu and S. Gibbons, Chem. Biodiversity, 2010, 7, 953–958 CAS.
- L. Verotta, G. Appendino, E. Belloro, J. Jakupovic and E. Bombardelli, J. Nat. Prod., 1999, 62, 770–772 CrossRef CAS PubMed.
- Y. H. Zeng, K. Osman, Z. Y. Xiao, S. Gibbons and Q. Mu, Phytochem. Lett., 2012, 5, 200–205 CrossRef CAS PubMed.
- O. C. Rubio, A. Cuellar, N. Rojas, H. V. Castro, L. Rastrelli and R. Aquino, J. Nat. Prod., 1999, 62, 1013–1015 CrossRef CAS PubMed.
- H. D. Flack, Acta Crystallogr., Sect. A: Found. Crystallogr., 1983, 39, 876–881 CrossRef.
- H. D. Flack, Chirality, 2008, 20, 681–690 CrossRef CAS PubMed.
- R. W. W. Hooft, L. H. Straver and A. L. Spek, J. Appl. Crystallogr., 2008, 41, 96–103 CrossRef CAS PubMed.
- H. Zhang, L. Tao, W. W. Fu, S. Liang, Y. F. Yang, Q. H. Yuan, D. J. Yang, A. P. Lu and H. X. Xu, J. Nat. Prod., 2014, 77, 1037–1046 CrossRef CAS PubMed.
- S. Trifunovic, V. Vajs, S. Macura, N. Juranic, Z. Djarmati, R. Jankov and S. Milosavljevic, Phytochemistry, 1998, 49, 1305–1310 CrossRef CAS.
- X. W. Yang, X. Deng, X. Liu, C. Y. Wu, X. N. Li, B. Wu, H. R. Luo, Y. Li, H. X. Xu, Q. S. Zhao and G. Xu, Chem. Commun., 2012, 48, 5998–6000 RSC.
- H. Zheng, Q. Chen, M. Zhang, Y. Lai, L. Lei, P. Shu, J. Zhang, Y. Xue, Z. Luo, Y. Li, G. Yao and Y. Zhang, J. Nat. Prod., 2013, 76, 2253–2262 CrossRef CAS PubMed.
- A. H. Cory, T. C. Owen, J. A. Barltrop and J. G. Cory, Cancer Commun., 1991, 3, 207–212 CAS.
- Z. Luo, F. Wang, J. Zhang, X. Li, M. Zhang, X. Hao, Y. Xue, Y. Li, F. D. Horgen, G. Yao and Y. Zhang, J. Nat. Prod., 2012, 75, 2113–2120 CrossRef CAS PubMed.
- L. J. Reed and H. Muench, Am. J. Hyg., 1938, 27, 493–497 Search PubMed.
- Y. T. Zheng, W. F. Zhang, K. L. Ben and J. H. Wang, Immunopharmacol. Immunotoxicol., 1995, 17, 69–79 CrossRef CAS PubMed.
- G. Zhang, H. Q. Wang, J. J. Chen, X. M. Zhang, S. C. Tam and Y. T. Zheng, Biochem. Biophys. Res. Commun., 2005, 334, 336–812 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: UV, IR, HRESIMS, and NMR spectra for compounds 1–9 and X-ray crystal structure of compound 1. CCDC 1023874, as well as ECD computational details. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra11675e |
| ‡ These authors contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |