General and efficient method for direct N-monomethylation of aromatic primary amines with methanol†
Feng
Li
*,
Jianjiang
Xie
,
Haixia
Shan
,
Chunlou
Sun
and
Lin
Chen
School of Chemical Engineering, Nanjing University of Science and Technology, Nanjing 210094, P. R. China. E-mail: fengli@mail.njust.edu.cn; Fax: +86-25-84431939; Tel: +86-25-84317316
First published on 26th July 2012
Abstract
The direct N-monomethylation of aromatic primary amines, including arylamines, arylsulfonamides and amino-azoles, using methanol as a methylating agent has been accomplished in the presence of a [Cp*IrCl2]2/NaOH system. From both synthetic and environmental points of view, the reaction is highly attractive because of low catalyst loading, broad substrate scope and excellent selectivities.
Introduction
The direct N-monomethylation of primary arylamines, one of the most important organic reactions, is widely utilized for the synthesis of numerous natural products, pharmaceuticals and fine chemicals.1 Traditionally, the transformation is performed using highly toxic and hazardous methyl halide or dimethyl sulfate as methylating agents in the presence of bases. In the past several years, dimethyl carbonate (DMC) and other alkyl methyl carbonates were also used as green methylating agents for the N-monomethylation of primary arylamines in the presence of a large amount of zeolites as catalysts (the weight ratio of zeolites and primary amines is over 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2).2 As mentioned above, these procedures inevitably suffer from the generation of N,N-dimethyl amines as side products due to the resulting N-methyl amines being more nucleophilic than the starting primary arylamines in the process of the reactions.
:2).2 As mentioned above, these procedures inevitably suffer from the generation of N,N-dimethyl amines as side products due to the resulting N-methyl amines being more nucleophilic than the starting primary arylamines in the process of the reactions.
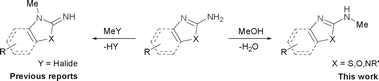
Recently, much attention has been focused on the N-alkylation of amines with alcohols as environmentally benign alkylating agents instead of alkyl halides based on the “hydrogen autotransfer (or hydrogen-borrowing) process”,3 using iridium,4 ruthenium5 or other transition metal catalysts.6 In this process, alcohols are first dehydrogenated to form aldehydes, followed by the condensation of the resulting aldehydes with amines to afford imine intermediates, which are hydrogenated to give the final N-alkylated products. The methodology is attractive because of high atom efficiency and the formation of water as the only side product. As a result, several groups explored transition metal-catalyzed N-methylation of primary arylamines with methanol.7 However, these procedures suffer from a lack of selectivity towards the N-monomethylation7a or are highly restricted in the scope of substrates.7b It was also speculated that a relatively high energy is required for the dehydrogenation of methanol compared with higher alcohols such as ethanol (ΔH = +84 vs. +68 kJ mol−1, respectively).8 Therefore, the development of a simple, efficient and versatile system for the N-monomethylation of primary arylamines with methanol is still an extreme challenge. Very recently, Krische and co-workers reported the direct C–C coupling of allenes with activated methanol catalyzed by an iridium complex.9 We also reported the regioselective N-alkylation of amino-thiazoles10a,b and amino-imidazoles10c with alcohols catalyzed by transition metal/base systems. Encouraged by their research and as part of our continuing interest in exploring selective N-alkylation reactions, we herein wish to describe our efforts towards the direct N-monomethylation of primary arylamines using methanol as a methylating agent catalyzed by a [Cp*IrCl2]2/NaOH system.11 Further, the direct N-monomethylation of arylsulfonamides and regioselective N-monomthylation of amino-azoles with methanol was explored for the first time (Scheme 1).
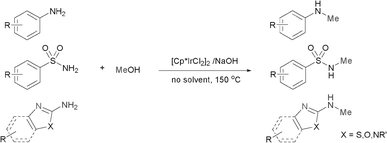 | ||
| Scheme 1 N-Monomethylation of aromatic primary amines. | ||
Results and discussion
The 4-chloroaniline 1a was chosen as a model substrate to explore the feasibility of the reaction. In an initial experiment, the reaction of 1a (2 mmol) with methanol 2 (1 ml) was carried out in the presence of commercially available [Cp*IrCl2]2 (Cp* = pentamethylcyclopentadienyl) (0.1 mol%) at 150 °C for 12h and no conversion was observed (Table 1, entry 1). When K2CO3 or K3PO4 (1.0 equiv.) was used as an additive, reactions afforded the N-monomethylated product 3a with 38% and 46% yields, respectively (entries 2–3). Surprisingly, the product 3a could be obtained with almost quantitative yield in the presence of a strong base such as NaOH, KOH or NaOtBu (entries 4–6).|
|
||||
|---|---|---|---|---|
| Entry | Base | Equiv. | Temp (°C) | Yieldb (%) |
| a Reaction conditions: 2 mmol 1a, 0.1 mol% [Cp*IrCl2]2, x equiv. base, 1 ml methanol, 150 °C, 12 h. b Isolated yield. | ||||
| 1 | — | 1.0 | 150 | 0 |
| 2 | K2CO3 | 1.0 | 150 | 38 |
| 3 | K3PO4 | 1.0 | 150 | 46 |
| 4 | NaOH | 1.0 | 150 | 96 |
| 5 | KOH | 1.0 | 150 | 94 |
| 6 | NaOtBu | 1.0 | 150 | 96 |
| 7 | NaOH | 0.5 | 150 | 72 |
| 8 | NaOH | 0.2 | 150 | 63 |
| 9 | NaOH | 1.0 | 130 | 85 |
| 10 | NaOH | 1.0 | 100 | 56 |
Among them, NaOH was selected as the base for further research. Attempts to decrease the reaction temperature and reduce the amount of NaOH resulted in relatively low yields (entries 7–10). It was also found that no reaction took place in the individual presence of NaOH.
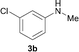
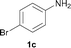
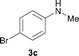
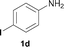
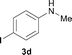
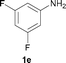
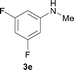
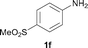
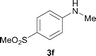
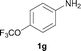
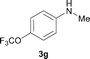
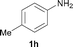
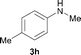
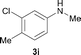
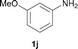
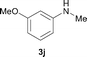
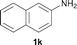
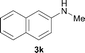
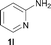
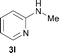
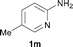
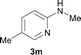
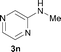
To expand the scope of the reaction, the N-methylation of a variety of arylamines with methanol was examined under the above optimal conditions (Table 1, entry 4) and the results are outlined in Table 2. Reactions of other anilines bearing one or two halide atoms, such as 3-chloro 1b, 4-bromo 1c, 4-iodo 1d and 3,5-difluoro 1e afforded the desired products 3b–e with 73–90% yields (Table 2, entries 1–4). Anilines bearing an electron-withdrawing group, such as 4-methylsulfonyl 1f and 4-trifluoromethoxy 1g, were converted to the corresponding products 3f and 3g with 94% and 96% yields, respectively (entries 5–6). Further, the N-methylation of anilines containing an electron-donating group, such as 4-methyl 1h, 3-chloro-4-methyl 1i, and 3-methoxy 1j, afforded the desired products 3h–3j with 71–96% yields (entries 7–9). The 2-naphthylamine 1k was also proven to be a suitable substrate and the corresponding product 3k was obtained with 88% yield (entry 10). In the case of heteroanilines, such as aminopyridines 1l–m and aminopyrazine 1n, reactions afforded the desired products 3l–n with 90–94% yields (entries 11–13).


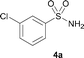
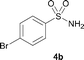
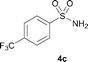
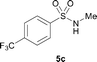
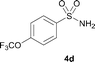
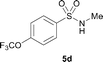
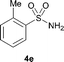
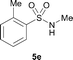
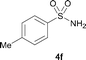
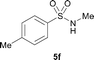
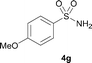
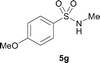
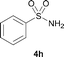
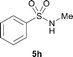
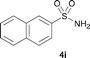
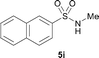
The N-methylation of a series of arylsulfonamides 4 with methanol was then investigated. As shown in Table 3, reactions of benzenesulfonamides bearing a halide atom 4a and 4b afforded the desired products 5a and 5b with 91% and 87% yields, respectively (Table 3, entries 1–2). The N-methylation of benzenesulfonamides bearing an electron-withdrawing group 4c and 4d gave the corresponding products 5c and 5d with 90% and 96% yields, respectively (entries 3–4). Further, the desired products 5e–g were obtained with 92–96% yields when benzenesulfonamides bearing an electron-donating group 4e–g were utilized as substrates (entries 5–7). Transformations of benzenesulfonamide 4h and naphthalenesulfonamide 4i afforded the desired products 5h and 5i with 97% and 87% yields, respectively (entries 8–9).
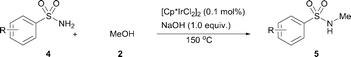

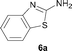
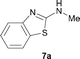
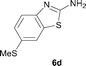
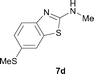
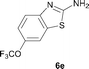
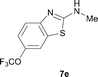
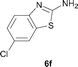
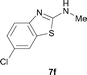
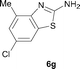
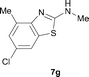
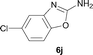
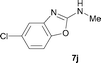
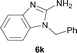
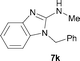
It was well documented that the N-alkylation of amino-azoles with alkyl halides,12 including methyl halides,12e,g–h afforded the N-endosubstituted 3-alkyl-2-iminoazolines as products because the endocyclic nitrogen is more basic than the exocyclic one (Scheme 2, left). However, it was found that only N-exosubstituted products were obtained in the N-methylation of amino-azoles using methanol as the methylating agent (Scheme 2, right). The reaction of 2-aminobenzothiazole 6a with 2 in the presence of [Cp*IrCl2]2 (0.4 mol%) and NaOH (1.0 equiv.) was carried out for 12 h to afford 2-(N-methylamino)benzothiazole 7a with 93% yield (Table 4, entry 1). Similarly, 2-aminobenzothiazoles bearing an electron-donating group 6b–d or electron-withdrawing group 6e were successfully converted to give the corresponding products 7b–e with 88–95% yields (entries 2–5). High yields were also obtained in reactions of 2-aminobenzothiazoles containing a halide atom 6f–g (entries 6–7). Further, reactions of non-benzo-fused 2-aminothiazoles 6h and 6i afforded the desired products 7h and 7i with 90% and 93% yields, respectively (entries 8–9). As amino-oxazole 6j was used as the substrate, the corresponding product 7j was obtained with 87% yield (entry 10). The N-methylation was also applied to amino-imidazoles 6k–m, giving the desired products 7k–m with 90–94% products (entries 11–13).
 | ||
| Scheme 2 Regioselective N-methylation of amino-azoles. | ||

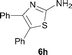
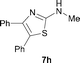
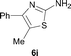
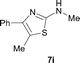
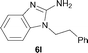
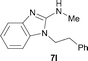
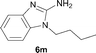
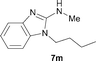
In all cases, apart from the desired N-monomethylated products, no N,N-dimethylated products were detected by 1H NMR of the crude reaction mixture, although an excess amount of methanol was used. In addition, no 3-methyl-2-iminoazoline isomers were observed in the N-methylation of amino-azoles.
To understand the excellent selectivities towards N-monomethylation of this reaction, the N-methylation of the N-methylamine with methanol was then investigated. As shown in Scheme 3, the reaction of 4-chloro-N-methylbenzenamine 3a with 2 was carried out for 12 h and no conversion was detected, indicating that the [Cp*IrCl2]2/NaOH system is only effective for the conversion of aromatic primary amines to the corresponding N-monomethylated products.
 | ||
| Scheme 3 Reaction of N-methylamine 3a with 2. | ||
Based on the experimental results and the “hydrogen autotransfer (or hydrogen-borrowing) process”,3,4 the possible mechanism for the N-monomethylation reaction was proposed in Scheme 4. The methanol was first dehydrogenated to form the formaldehyde coordinated with the iridium hydride species B in the presence of base,13 followed by the condensation of the resulting B with the aromatic primary amine affording the imine coordinated with iridium hydride species C. Further, the addition of iridium hydride into the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond of the imines gave the amido-iridium species D. Finally, the N-monomethylated product was released and the catalytically active alkoxo iridium species A were generated via an amido-alkoxo exchange reaction between D and methanol.14 The regioselective formation of 2-(N-methylamino)azole may be attributed to the exocylic nitrogen being favored over the endocyclic one in the condensation step of the amino-azoles with the resulting formaldehyde.10
N bond of the imines gave the amido-iridium species D. Finally, the N-monomethylated product was released and the catalytically active alkoxo iridium species A were generated via an amido-alkoxo exchange reaction between D and methanol.14 The regioselective formation of 2-(N-methylamino)azole may be attributed to the exocylic nitrogen being favored over the endocyclic one in the condensation step of the amino-azoles with the resulting formaldehyde.10
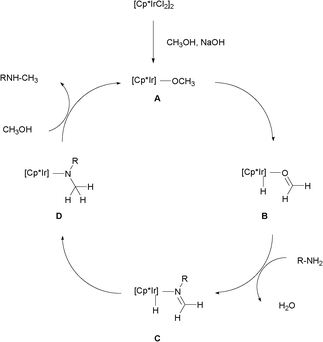 | ||
| Scheme 4 Possible mechanism. | ||
Conclusions
We have demonstrated a simple, efficient and versatile system for the direct N-monomethylation of aromatic primary amines, including arylamines, arylsulfonmides and amino-azoles, with methanol as the methylating agent. From both synthetic and environmental points of view, the iridium-catalyzed reaction is highly attractive because of low catalyst loading, broad substrate scope and excellent selectivities. Further studies to expand the application of the synthetic strategy are currently underway.Experimental section
General experimental details
Infrared spectra were recorded on a Nicolet iS10 FT-IR spectrometer. High-resolution mass spectra (HRMS) were obtained on a HPLC-Q-Tof MS(Micro) spectrometer and are reported as m/z (relative intensity). Accurate masses are reported for the molecular ion [M+H]+. Melting points were measured on a X-6 micro-melting apparatus (Beijing Tech Instrument Co., Ltd). Proton nuclear magnetic resonance (1H NMR) spectra were recorded at 500 MHz using a Bruker Avance 500 spectrometer. Chemical shifts are reported in delta (δ) units, parts per million (ppm) downfield from trimethylsilane or ppm relative to the center of the singlet at 7.26 ppm for CDCl3. Coupling constants as J values are reported in Hertz (Hz), and the splitting patterns were designated as follows: s, singlet; d, doublet; t, triplet; m, multiplet; b, broad. Carbon-13 nuclear magnetic resonance (13C NMR) spectra were recorded at 125 MHz using a Bruker Avance 500 spectrometer. Chemical shifts are reported in delta (δ) units, ppm relative to the center of the triplet at 77.0 ppm for CDCl3. 13C NMR spectra were routinely run with broadband decoupling. Reactions tubes were purchased from Beijing Synthware Glass Inc. All reactions were run under an atmosphere of nitrogen, unless otherwise indicated. Analytical thin-layer chromatography (TLC) was carried out using 0.2 mm commercial silica gel plates.Acknowledgements
Financial support by the National Natural Science Foundation of China (No. 20902046), Natural Science Foundation of Jiangsu Province (No. BK2009384) and Fundamental Research Funds for the Central Universities (No. NUST2011ZDJH19), is greatly appreciated.References
- (a) F. A. Carey and R. J. Sundberg, Advanced Organic Chemistry, 4th ed.; Kluwer Academic: New York, 2001; Part B, Chapter 3.2.5 Search PubMed; (b) R. N. Salvatore, C. H. Yoon and K. W. Jung, Tetrahedron, 2001, 57, 7785 CrossRef CAS.
- (a) M. Selva, A. Bomben and P. Tundo, J. Chem. Soc., Perkin Trans. 1, 1997, 1041 RSC; (b) M. Selva, P. Tundo and A. Perosa, J. Org. Chem., 2001, 66, 677 CrossRef CAS; (c) P. Tundo and M. Selva, Acc. Chem. Res., 2002, 35, 706 CrossRef CAS; (d) M. Selva, P. Tundo and A. Perosa, J. Org. Chem., 2002, 67, 9238 CrossRef CAS; (e) M. Selva, P. Tundo and T. Foccardi, J. Org. Chem., 2005, 70, 2476 CrossRef CAS; (f) M. Selva, A. Perosa and M. Fabris, Green Chem., 2008, 10, 1068 RSC.
- (a) For selected reviews, see: M. H. S. A. Hamid, P. A. Slatford and J. M. J. Williams, Adv. Synth. Catal., 2007, 349, 1555 CrossRef CAS; (b) T. D. Nixon, M. K. Whittlesey and J. M. J. Williams, Dalton Trans., 2009, 753 RSC; (c) G. E. Dobereiner and R. H. Grabtree, Chem. Rev., 2010, 110, 681 CrossRef CAS; (d) G. Guillena, D. Ramon and M. Yus, Chem. Rev., 2010, 110, 1611 CrossRef CAS; (e) T. Suzuki, Chem. Rev., 2011, 111, 1825 CrossRef CAS.
- (a) K. Fujita, K. Yamamoto and R. Yamaguchi, Org. Lett., 2002, 4, 2691 CrossRef CAS; (b) K. Fujita, Z. Li, N. Ozeki and R. Yamaguchi, Tetrahedron Lett., 2003, 44, 2687 CrossRef CAS; (c) K. Fujita, T. Fujii and R. Yamaguchi, Org. Lett., 2004, 6, 3525 CrossRef CAS; (d) S. Whitney, R. Grigg, A. Derrick and A. Keep, Org. Lett., 2007, 9, 3299 CrossRef CAS; (e) K. Fujita, Y. Enoki and R. Yamaguchi, Tetrahedron, 2008, 64, 1943 CrossRef CAS; (f) A. Prades, R. Corberán, M. Poyatos and E. Peris, Chem.–Eur. J., 2008, 14, 11474 CrossRef CAS; (g) R. Yamaguchi, S. Kawagoe, C. Asai and K. Fujita, Org. Lett., 2008, 10, 181 CrossRef CAS; (h) B. Blank, S. Michlik and R. Kempe, Chem.–Eur. J., 2009, 15, 3790 CrossRef CAS; (i) O. Saidi, A. J. Blacker, M. M. Farah, S. P. Marsden and J. M. J. Williams, Chem. Commun., 2010, 46, 1541 RSC; (j) M. Tursky, L. L. R. Lorentz-Petersen, L. B. Olsen and R. Madsen, Org. Biomol. Chem., 2010, 8, 5576 RSC; (k) M. Zhu, K. Fujita and R. Yamaguchi, Org. Lett., 2010, 12, 1336 CrossRef CAS; (l) S. Michlik and R. Kempe, Chem.–Eur. J., 2010, 16, 13193 CrossRef CAS; (m) N. Andrushko, V. Andrushko, P. Roose, K. Moonen and A. Boner, ChemCatChem, 2010, 2, 640 CrossRef CAS; (n) R. Kawahara, K. Fujita and R. Yamaguchi, Adv. Synth. Catal., 2011, 353, 1161 CrossRef CAS; (o) I. Cumpstey, S. Agrawal, K. E. Borbasa and B. Martin-Matute, Chem. Commun., 2011, 47, 7827 RSC; (p) S. Bhat and V. Sridharan, Chem. Commun., 2012, 48, 4701 RSC.
- (a) For selected examples, see: M. H. S. A. Hamid and J. M. J. Williams, Chem. Commun., 2007, 725 RSC; (b) C. Gunanathan and D. Milstein, Angew. Chem., Int. Ed., 2008, 47, 8661 CrossRef CAS; (c) Angew. Chem. 2008, 120, 8789; (d) H. S. A. Hamid, C. L. Allen, G. W. Lamb, A. C. Maxwell, H. C. Maytum, A. J. A. Watson and J. M. J. Williams, J. Am. Chem. Soc., 2009, 131, 1766 CrossRef; (e) S. Bahn, A. Tillack, S. Imm, K. Mevius, D. Michalik, D. Hollmann, L. Neubert and M. Beller, ChemSusChem, 2009, 2, 551 CrossRef; (f) S. Bahn, S. Imm, K. Mevius, L. Neubert, A. Tillack, J. M. J. Williams and M. Beller, Chem.–Eur. J., 2010, 16, 3590 CrossRef; (g) B. Sundararaju, Z. Tang, M. Achard, G. V. M. Sharma, L. Toupet and C. Bruneau, Adv. Synth. Catal., 2010, 352, 3141 CrossRef CAS; (h) S. Imm, S. Bahn, M. Zhang, L. Neubert, H. Neumann, F. Klasovsky, J. Pfeffer, T. Haas and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 7599 CrossRef CAS; (i) M. Zhang, S. Imm, S. Bahn, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 11197 CrossRef CAS; (j) Z. Sahli, B. Sundararaju, M. Achard and C. Bruneau, Org. Lett., 2011, 13, 3964 CrossRef CAS; (k) R. Cano, D. J. Ramón and M. Yus, J. Org. Chem., 2011, 76, 5547 CrossRef CAS; (l) S. Agrawal, M. Lenormand and B. Martın-Matute, Org. Lett., 2012, 14, 1456 CrossRef CAS.
- (a) For selected examples, see; F. Shi, M. K. Tse, X. Cui, D. Gordes, D. Michalik, T. Y. Deng and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 5912 CrossRef CAS; (b) A. Martinez-Asencio, M. Yus and D. J. Ramon, Synthesis, 2011, 22, 3730–3740 Search PubMed; (c) W. He, L. Wang, C. Sun, K. Wu, S. He, J. Chen, P. Wu and Z. Yu, Chem.–Eur. J., 2011, 17, 13308–13317 CrossRef CAS; (d) X. Yu, C. Liu, L. Jiang and Q. Xu, Org. Lett., 2011, 13, 6184–6187 CrossRef CAS.
- (a) K. T. Huh, Y. Tsuji, M. Kobayashi, F. Okuda and Y. Watanabe, Chem. Lett., 1988, 449 CrossRef CAS; (b) S. Naskar and M. Bhattacharjee, Tetrahedron Lett., 2007, 48, 3367 CrossRef CAS.
- (a) M. Qian, M. A. Liauw and G. Emig, Appl. Catal., A, 2003, 238, 211 CrossRef CAS; (b) W. H. Lin and H. F. Chang, Catal. Today, 2004, 97, 181 CrossRef CAS.
- J. Moran, A. Preetz, R. A. Mesch and M. J. Krische, Nat. Chem., 2011, 3, 287 CrossRef CAS.
- (a) F. Li, H. Shan, Q. Kang and L. Chen, Chem. Commun., 2011, 47, 5058 RSC; (b) F. Li, H. Shan, L. Chen, Q. Kang and P. Zou, Chem. Commun., 2012, 48, 603 RSC; (c) F. Li, Q. Kang, H. Shan, L. Chen and J. Xie, Eur. J. Org. Chem. DOI:10.1002/ejoc.201200698.
- Fujita, Yamaguchi and co-workers explored the N-monomethylation of aniline with methanol in the presence of the [Cp*IrCl2]2/K2CO3 system at 110 °C, however, no reaction occurred. See: K. Fujita, Z. Li, N. Ozeki and R. Yamaguchi, Tetrahedron Lett., 2003, 44, 2687 CrossRef CAS , Ref. 8.
- (a) J. V. Metzger, in Thiazoles and their Benzo Derivatives. Comprehensive Heterocyclic Chemistry, Pergamon Press Ltd, New York, 1984, 6, 235–331 Search PubMed; (b) N. Zhang, M. Tomizawa and J. E. Casida, J. Med. Chem., 2002, 45, 2832 CrossRef CAS; (c) X. Zhu, Q. Yu, R. G. Cutler, C. W. Culmsee, H. W. Holloway, D. K. Lahiri, M. P. Mattson and N. H. Greig, J. Med. Chem., 2002, 45, 5090 CrossRef CAS; (d) H. Ramstrom, M. Bourotte, C. Philippe, M. Schmitt, J. Haiech and J. J. Bourguignon, J. Med. Chem., 2004, 47, 2264 CrossRef; (e) S. D. Barchechath, R. I. Tawatao, M. Corr, D. A. Carson and H. B. Cottam, J. Med. Chem., 2005, 48, 6409 CrossRef CAS; (f) J. Coetzee, S. Cronje, L. Dobrzanska, H. G. Raubenheimer, Gisela Joone, M. J. Nell and H. C. Hoppe, Dalton Trans., 2011, 40, 1471 RSC.
- Fujita, Yamaguchi and co-workers revealed the catalytic activity of [Cp*IrCl2]2 towards oxidation of alcohols to the corresponding aldehydes: see: K. Fujita, S. Furukawa and R. Yamaguchi, J. Organomet. Chem., 2002, 649, 289 CrossRef CAS.
- Bergman and co-workers demonstrated the amido-alkoxo exchange reaction for Cp*Ir complexes, see: D. S. Glueck, L. J. N. Winslow and R. G. Bergman, Organometallics, 1991, 10, 1462 CrossRef CAS.
- J. Jiao, X. R. Zhang, N. H. Chang, J. Wang, J. F. Wei, X. Y. Shi and Z. G. Chen, J. Org. Chem., 2011, 76, 1180 CrossRef CAS.
- A. J. Chen, S. T. Wong, C. C. Hwang and C. Y. Mou, ACS Catal., 2011, 1, 786 CrossRef CAS.
- S. Kajigaeshi, T. Kakinami, H. Yamasaki, S. Fujisaki and T. Okamoto, Bull. Chem. Soc. Jpn., 1988, 61, 600 CrossRef CAS.
- S. B. Herzon and J. F. Hartwig, J. Am. Chem. Soc., 2007, 129, 6690 CrossRef CAS.
- B. Flatt, X. H. Gu, R. Martin, R. Mohan, B. Murphy, M. C. Nyman, W. C. Stevens and T. L. Wang,WO2006076202, 2006 Search PubMed.
- G. W. Spears, K. Tsuji, T. Tojo, H. Nishimura and T. Ogino, Synth. Commun., 2000, 30, 565 CrossRef CAS.
- K. C. Majumdar, B. Roy, P. K. Basu and P. Biswas, Indian. J. Chem. B., 2006, 45B, 719 CAS.
- A. Teichert, K. Jantos, K. Harms and A. Studer, Org. Lett., 2004, 6, 3477 CrossRef CAS.
- A. T. Londregan, S. Jennings and L. Wei, Org. Lett., 2010, 12, 5254 CrossRef CAS.
- B. C. Barlaam and R. Ducray, WO2009010794, 2009 Search PubMed.
- A. J. Henderson, M. Hadden, Cheng Guo, Neema Douglas, H. Decornez, M. R. Hellberg, A. Rusinko, M. McLaughlin, N. Sharif, C. Drace and R. Patlib, Bioorg. Med. Chem. Lett., 2010, 20, 1137 CrossRef CAS.
- R. R. Singhaus, R. C. Bernotas, R. Steffan, E. Matelan, E. Quinet, P. Nambi, I. Feingold, C. Huselton, A. Wilhelmsson, A. Goos-Nilsson and J. Wrobel, Bioorg. Med. Chem. Lett., 2010, 20, 521 CrossRef CAS.
- N. M. Sanghavi, V. L. Parab, B. S. Patravale and M. N. Patel, Synth. Commun., 1989, 19, 1499 CrossRef CAS.
- L. A. Xu, H. Shu, Y. Liu, S. H. Zhang and M. L. Trudell, Tetrahedron, 2006, 62, 7902 CrossRef CAS.
- S. Minakata, Y. Morino, Y. Oderaotoshi and M. Komutsu, Org. Lett., 2006, 8, 3335 CrossRef CAS.
- D. W. Slocum and C. A. Jennings, J. Org. Chem., 1976, 41, 3653 CrossRef CAS.
- D. Maclean, R. Hale and M. Chen, Org. Lett., 2001, 3, 2977 CrossRef CAS.
- Y. Kacem, A. Bouraoui, V. Ratovelomanana-Vidal, J. P. Genetc and B. B. Hassine, C. R. Chim., 2002, 5, 611 CrossRef CAS.
- D. Fajkusova and P. Pazdera, Synthesis, 2008, 8, 1297 CrossRef.
- A. D. Jordan, C. Luo and A. B. Reitz, J. Org. Chem., 2003, 68, 8693 CrossRef CAS.
- P. Jimonet and C. Nemecek, Eur. Pat. Appl., 408437, 1991 Search PubMed.
- N. B. Ambati, V. Anand and P. Hanumanthu, Synth. Commun., 1997, 27, 1487 CrossRef CAS.
- G. W. Kabalka and A. R. Mereddy, Tetrahedron Lett., 2006, 47, 5171 CrossRef CAS.
- H. Ogura, S. Mineo and K. Nakagawa, Chem. Pharm. Bull., 1981, 29, 1518 CrossRef CAS.
- A. D. Settimo, A. M. Marini, A. M. Bianucci, G. Primofiore, F. D. Settimo and E. Boldrini, Farmaco, 1994, 49, 829 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and analytical data for N-monomethylated products. See DOI: 10.1039/c2ra21487c |
| This journal is © The Royal Society of Chemistry 2012 |