
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceC–H functionalization of 2-alkyl tryptamines: direct assembly of azepino[4,5-b]indoles and total synthesis of ngouniensines†
Kejing Xie‡
a,
Zeyuan Shen‡b,
Peng Chenga,
Haoxiang Donga,
Zhi-Xiang Yu *b and
Liansuo Zu*a
*b and
Liansuo Zu*a
aSchool of Pharmaceutical Sciences, Key Laboratory of Bioorganic Phosphorus Chemistry & Chemical Biology (Ministry of Education), Beijing Frontier Research Center for Biological Structure, Tsinghua University, Beijing 100084, China. E-mail: zuliansuo@tsinghua.edu.cn
bBeijing National Laboratory for Molecular Sciences (BNLMS), Key Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education, College of Chemistry, Peking University, Beijing 100871, China. E-mail: yuzx@pku.edu.cn
First published on 5th July 2024
Abstract
The Pictet–Spengler type condensation of tryptamine derivatives and aldehydes or ketones is a classic reaction, and has been previously applied to assemble indole-annulated 5-, 6- and 8-membered heterocyclic rings. In this work, we further expand the synthetic scope of this reaction to the 7-membered azepino[4,5-b]indole skeleton through the direct C–H functionalization of 2-alkyl tryptamines, in which the non-activated methylene group participates in a 7-membered ring formation with aldehydes. By combining this unprecedented ring-forming process with a second C–H olefination at the same carbon, the concise total synthesis of natural products ngouniensines is achieved, demonstrating the synthetic potential of the developed chemistry in simplifying retrosynthetic disconnections.
Introduction
The Pictet–Spengler type condensation of tryptamine derivatives and aldehydes or ketones is a classic reaction, and represents a direct and convergent strategy for the synthesis of indole-annulated heterocyclic structures.1–4 Depending on whether there is a substituent at the C2-position, two types of cyclization occurring at C2 or C3 have been well known, leading to the formation of 6-membered tetrahydro-b-carboline (I) and 5-membered spiroindolenine (II) skeletons, respectively (Fig. 1). In addition, with a 2-vinyl substituent, the vinylogous Pictet–Spengler type cyclization is also feasible, affording the 8-membered tetrahydroazocinoindole (III) ring system.5 The wide presence of these skeletons (I, II, III) in natural products and biologically important molecules has prompted the growth of this class of reactions in popularity. The further development of novel reaction modes for the Pictet–Spengler type condensation would be highly desirable to expand the synthetic scope of this class of reactions. | ||
| Fig. 1 Azepino[4,5-b]indole skeleton and the proposed C–H functionalization based strategy. | ||
The 7-membered azepino[4,5-b]indole skeleton (IV, Fig. 1) is characteristic of a variety of indole alkaloids, as exemplified by the structures of ngouniensines (1a, 1b), ibogaine and many others.6–16 The significance of the azepino[4,5-b]indole skeleton as an essential therapeutic pharmacophore has been recently highlighted by the structural simplification of ibogaine, leading to the development of tabernanthalog (Fig. 1) as a safer and non-hallucinogenic psychedelic analog with therapeutic potential.17 In contrast to the structural and biological importance of the azepino[4,5-b]indole skeleton (IV), relatively few synthetic methods have been developed for its synthesis, which mainly have relied on the inherent reactivity of functional groups toward ring forming processes.18–31 While a variety of azepino[4,5-b]indoles with varied substitution patterns could be thus generated, the resulting structural diversity could not fully meet the demand for the synthesis of natural products and diversified synthetic analogs. Herein, we report an unprecedented approach for the synthesis of the azepino[4,5-b]indole skeleton by the direct C–H functionalization of 2-alkyl tryptamines, and showcase the synthetic utility in the concise total synthesis of ngouniensines.
The application of C–H functionalization logic in chemical synthesis has become a vibrant research area due to the admirable ability in simplifying retrosynthetic disconnections by avoiding the preinstallation of requisite functional groups.32 The indole alkaloids ngouniensines (1a, 1b)33,34 structurally feature the azepino[4,5-b]indole core and an exocyclic conjugated alkene, which were both constructed by the manipulation of preinstalled functional groups in the previous synthesis.6 We surmised to develop a C–H functionalization based approach, not only to offer a concise entry to ngouniensines, but also to facilitate the preparation of azepino[4,5-b]indole containing molecules in general. Specifically, as depicted in Fig. 1, we envisioned to construct the azepino[4,5-b]indole skeleton by the direct C–H functionalization of 2-alkyl tryptamines 2, in which the non-activated methylene at C2 would participate in a 7-membered ring formation with aldehydes 3. The direct coupling of 2 and 3 could be regarded as a homologated Pictet–Spengler condensation, which, if realized, would further expand the synthetic scope of this classic reaction beyond the indole-annulated 5-, 6- and 8-membered heterocyclic skeletons (I–III, Fig. 1). The exocyclic conjugated alkene in ngouniensines could be introduced by a second C–H functionalization of the same carbon with formaldehyde (R1 = H). Based on the above hypothesis, the chemical synthesis of ngouniensines could be attempted with unconventional bond disconnection tactics.
Results and discussion
Synthesis of azepino[4,5-b]indoles
In continuation with our interest in developing skeletal rearrangements within the indole system,35–40 we became interested in the possible fate of certain spiroindolenine intermediates toward subsequent transformations. As depicted in the model reaction of 2a and 3a (Table 1), we envisioned that the tautomerization of spiroindolenine B (generated by the Pictet–Spengler reaction) to the exocyclic enamine C would set the basis for the C–H functionalization of the non-activated methyl group, and the 7-membered azepino[4,5-b]indole skeleton would be subsequently formed via the intermediate D through either the concerted [3,3]-sigmatropic rearrangement or stepwise Mannich/retro Mannich pathway. Based on such a proposal, different Brønsted acids were screened with this model reaction. Using AcOH as the acid in chloroform, we were pleased to observe the formation of the desired azepino[4,5-b]indole 4a, albeit in only 11% yield (Table 1, entry 1). The structure of 4a was unambiguously confirmed by single crystal X-ray diffraction. Camphorsulfonic acid (CSA, entry 2), p-toluene sulfonic acid (p-TsOH, entry 3) and diphenyl phosphate (entry 4) were not successful promotors for the formation of 4a. A promising result was obtained using trifluoroacetic acid (TFA), delivering 4a in 52% yield (entry 5). The equivalent of TFA was then found to be a key factor (entries 6–8). With 4.0 equivalent of TFA as the acid, 4a could be isolated in 85% yield (entry 7). The reaction also proceeded in other solvents: similar yield using CH2Cl2, albeit with lower yield using toluene.
|
|||
|---|---|---|---|
| Entry | Acid | Solvent | Yieldb (%) |
| a A mixture of 2a (0.2 mmol) and 3a (0.4 mmol) in the specified solvent (0.2 M) was stirred at rt in the presence of the specified acid.b Isolated yield. rt = room temperature. TFA = trifluoroacetic acid. CSA = camphorsulfonic acid. p-TsOH = p-toluenesulfonic acid. | |||
| 1 | AcOH (2.0 equiv.) | CHCl3 | 11 |
| 2 | CSA (2.0 equiv.) | CHCl3 | Trace |
| 3 | p-TsOH (2.0 equiv.) | CHCl3 | Trace |
| 4 | (PhO)2POOH (2.0 equiv.) | CHCl3 | Trace |
| 5 | TFA (2.0 equiv.) | CHCl3 | 52 |
| 6 | TFA (1.0 equiv.) | CHCl3 | Trace |
| 7 | TFA (4.0 equiv.) | CHCl3 | 85 |
| 8 | TFA (6.0 equiv.) | CHCl3 | 60 |
| 9 | TFA (4.0 equiv.) | CH2Cl2 | 80 |
| 10 | TFA (4.0 equiv.) | Toluene | 56 |

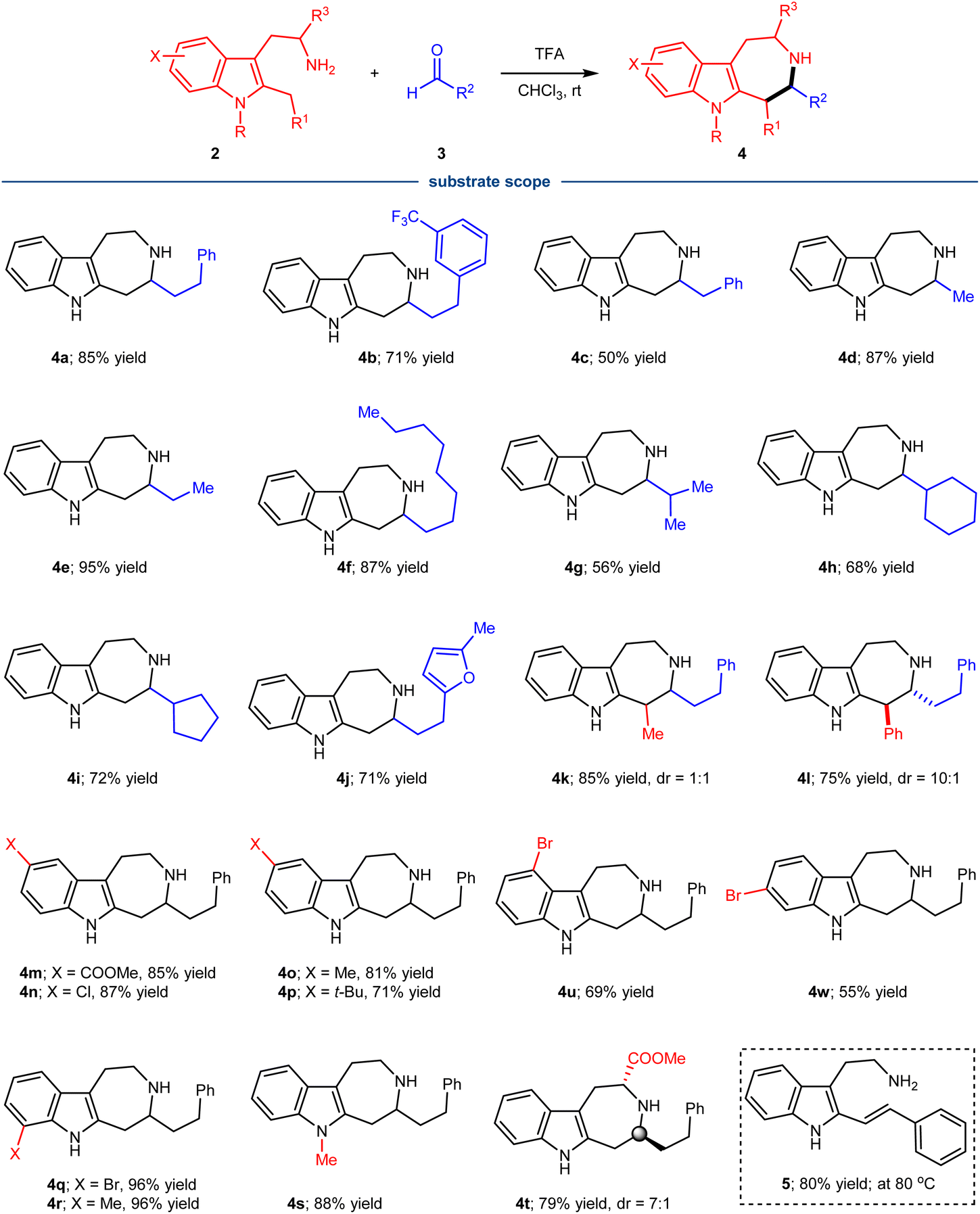
With the reaction conditions identified, the substrate scope of the condensation of tryptamine derivatives 2 with different aliphatic aldehydes 3 was investigated (Scheme 1). It turned out that the reaction represented a general approach for the synthesis of azepino[4,5-b]indoles 4, tolerating significant structural variations of both partners. A variety of linear aldehydes of different size were successful substrates, generating the corresponding products with good to excellent yield (4a–f). The steric more hindered branched aldehydes containing isopropyl-, cyclopentyl- and cyclohexyl-groups also proved to be efficient substrates (4g–i). Of note, the reaction could tolerate the presence of heterocyclic furan (4j), which is also a good nucleophile for the Pictet–Spengler type reaction and could potentially interact with the related active intermediates. When R1 was a methyl (4k) or phenyl group (4l), both reactions proceeded well with good yields, but the diastereoselectivity was different. The poor diastereoselectivity of 4k and excellent diastereoselectivity of 4l indicated the importance of steric effect of R1 in governing the stereochemistry. The relative stereochemistry of 4l was determined by single crystal X-ray diffraction of a derivative. Substituents (X) on the benzenoid part of 2 were also well tolerated, including both electron donating and withdrawing groups at the varied positions (4m–4r, 4u, 4w). Finally, the substrate with a methyl group on indole-N also participated in the condensation process with good yield (4s). While attempts to make the process catalytically enantioselective using chiral Brønsted acids were not successful at this stage, the direct condensation for 7-membered ring formation could be rendered asymmetric by using a chiral tryptophan derivative, affording product 4t with 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereoselectivity. The relative stereochemistry of 4t was determined by single crystal X-ray diffraction of a derivative. Of note, when aromatic benzaldehyde was allowed for the condensation with tryptamine derivatives 2a, the reaction did not lead to the formation of the related 7-membered ring, instead the generation of alkene 5 was observed under slightly varied conditions.
:1 diastereoselectivity. The relative stereochemistry of 4t was determined by single crystal X-ray diffraction of a derivative. Of note, when aromatic benzaldehyde was allowed for the condensation with tryptamine derivatives 2a, the reaction did not lead to the formation of the related 7-membered ring, instead the generation of alkene 5 was observed under slightly varied conditions.
 | ||
| Scheme 1 Synthesis of the 7-membered azepino[4,5-b]indoles. | ||
Total synthesis of ngouniensines
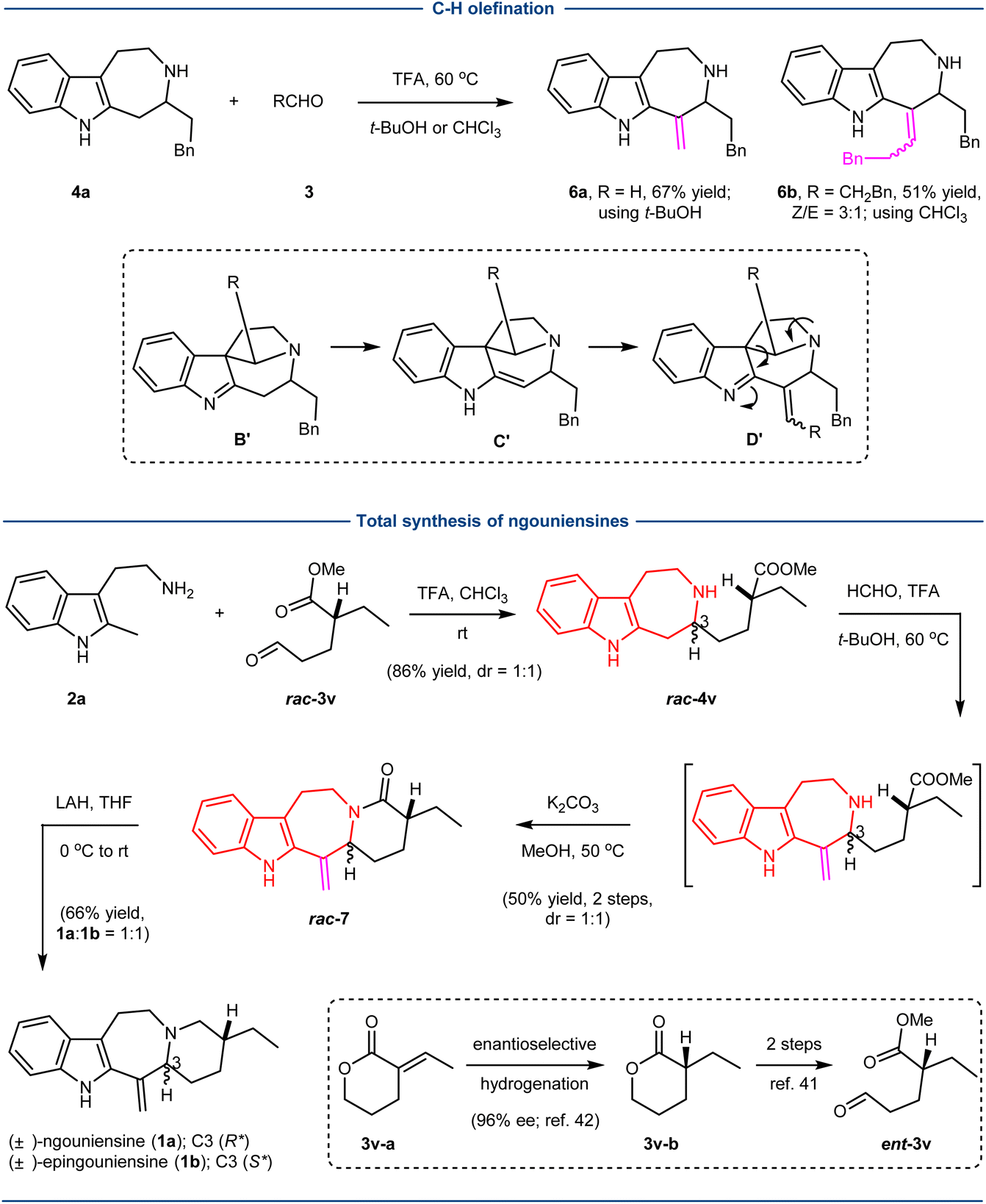
Next, we examined the possibility of a second C–H functionalization to introduce the exocyclic alkene as shown in the natural products ngouniensines using 4a as a model substrate (Scheme 2). We surmised that the further Pictet–Spengler cyclization of 4a with aldehydes would furnish the spiroindolenine B′, which could undergo imine/enamine tautomerization to generate C′. The subsequent aldol condensation would afford alkene D′, which finally could lead to the product 6 with an exocyclic alkene through the retro-Mannich reaction and hydrolysis of the resulting imine. After extensive optimization of reaction conditions, we could realize the desired transformation using trifluoroacetic acid (TFA) as the acid in either t-BuOH (6a; 67% yield) or CHCl3 (6b; 51% yield, Z/E = 3:1) at 60 °C.
 | ||
| Scheme 2 Total synthesis of ngouniensines. | ||
The total synthesis of ngouniensines was then realized based on the developed sequential C–H functionalization (Scheme 2). The first C–H functionalization involving the coupling of 2a and rac-3v41 successfully built up the azepino[4,5-b]indole core, affording rac-4v in 86% yield with 1:1 dr. The second C–H functionalization of rac-4v with formaldehyde rapidly installed the exocyclic conjugated alkene, delivering rac-7 upon subsequent amide formation with K2CO3. Finally, reduction of the amide group produced the natural products (±)-ngouniensines (1a:1b = 1:1). While the synthesis was carried out using rac-3v, the absolute chirality could be easily introduced by the known enantioselective hydrogenation (3v–a to 3v–b).42
Reaction mechanism
A plausible mechanism was proposed for the coupling of 2-alkyl tryptamines 2 and aliphatic aldehydes 3 (Scheme 3). The imine (A) formation between 2 and aldehyde 3 could trigger the Pictet–Spengler type cyclization (C3) to afford the spiroindolenine B, which underwent imine/enamine tautomerization to furnish C. Iminium D could be then generated from C and an excess of 3, and underwent either the concerted [3,3]-sigmatropic rearrangement or stepwise Mannich/retro Mannich pathway to furnish E. Finally, iminium hydrolysis would afford the 7-membered azepino[4,5-b]indoles 4. The formation of alkene 5 could be explained by the elimination reaction from 4 (or E), presumably due to the high stability of the conjugated system when R2 was an aromatic group.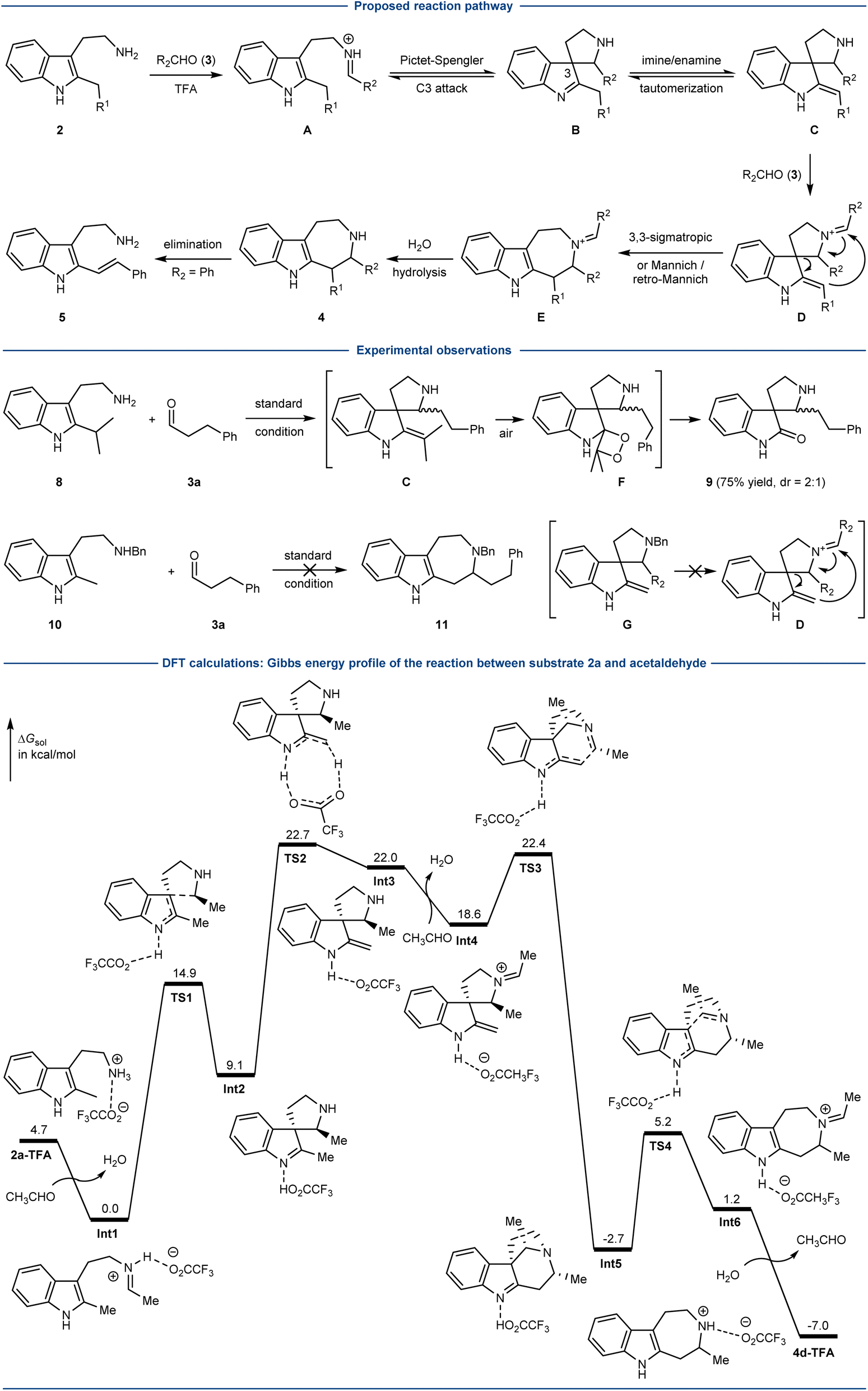 | ||
| Scheme 3 Reaction mechanism. | ||
While it was difficult to validate the proposed mechanism by the synthesis of a substrate resembling B or C due to its reversible transformation back to A under the acidic reaction conditions, some experimental observations supported the presence of intermediate C and the essential role of D. The reaction of bulky 2-iPr tryptamine 8 with aldehyde 3a afforded the oxindole 9 as the major product, presumably involving the oxidation of enamine C by oxygen (intermediate F).43,44 In another control study, the Bn-protected 2-Me-tryptamine 10 failed to give the corresponding product 11. According to the proposed reaction mechanism, this could be explained by the fact that the resulting G could not lead to the formation of intermediate D due to the presence of the Bn group.
To gain more insight, DFT calculations were carried out (Scheme 3). The reaction of substrate 2a and acetaldehyde was chosen as the model reaction, and calculated at the SMD(CHCl3)/PBE0-D3BJ/6-311+G(d,p)//PBE0-D3BJ/6-31+G(d,p) level. One of the key steps, the tautomerization of the spiroindolenine (Int2) to enamine (Int3) has an activation free energy of 13.6 kcal mol−1 (via TS2). The key ring forming event probably undergoes a Mannich/retro-Mannich pathway with the activation free energy of 3.8 kcal mol−1 (TS3) and 7.9 kcal mol−1 (TS4), respectively. The Gibbs free energy profile involving the concerted [3,3]-sigmatropic rearrangement pathway could not be located. Of note, a similar retro-Mannich reaction was previously reported by the group of You, involving an intermediate resembling Int5.25
Conclusions
In summary, we have developed an unprecedented approach for the synthesis of the azepino[4,5-b]indole skeleton by the direct C–H functionalization of 2-alkyl tryptamines. The synthetic utility has been demonstrated by the concise total synthesis of natural products ngouniensines, in which 2-Me-tryptamine undergoes sequential C–H functionalization with different aldehydes to afford the azepino[4,5-b]indole core and an exocyclic conjugated alkene, respectively. The unprecedented reaction mode, the direct and convergent manner, the operational simplicity and broad substrate scope of the approach should find further applications in the synthesis of azepino[4,5-b]indole containing molecules.Data availability
ESI† is available and includes the detailed synthetic procedure and characterization data of intermediates and final products. The X-ray crystallographic coordinates for structures reported in this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition numbers 2284681 (4a), 2284682 (4l) and 2284683 (4t).Author contributions
K. X., Z. S., P. C., and H. D. performed experiments and analyzed experimental data. Z. Y. and L.Z. directed the investigations and prepared the manuscript with contributions from all authors; all authors contributed to discussions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was made possible as a result of generous grants from the National Natural Science Foundation of China (22171161) and Key project at central government level: The ability establishment of sustainable use for valuable Chinese medicine resources (2060302).Notes and references
- E. D. Cox and J. M. Cook, Chem. Rev., 1995, 95, 1797–1842 Search PubMed.
- J. Stöckigt, A. P. Antonchick, F. Wu and H. Waldmann, Angew. Chem., Int. Ed., 2011, 50, 8538–8564 Search PubMed.
- J. M. Saya, E. Ruijter and R. V. A. Orru, Chem.–Eur. J., 2019, 25, 8916–8935 Search PubMed.
- C. Zheng and S.-L. You, Acc. Chem. Res., 2020, 53, 974–987 Search PubMed.
- A. Nash, X. Qi, P. Maity, K. Owens and U. K. Tambar, Angew. Chem., Int. Ed., 2018, 57, 6888–6891 Search PubMed.
- J. Bosch, M. L. Bennasar, E. Zulaica, G. Massiot and B. Massoussa, Tetrahedron Lett., 1987, 28, 231–234 Search PubMed.
- D. Passarella, M. Martinelli, N. Llor, M. Amat and J. Bosch, Tetrahedron, 1999, 55, 14995–15000 Search PubMed.
- K.-H. Lim and T.-S. Kam, Org. Lett., 2006, 8, 1733–1735 Search PubMed.
- Y. Du, H.-Y. Huang, H. Liu, Y.-P. Ruan and P.-Q. Huang, Synlett, 2011, 2011, 565–568 Search PubMed.
- S. Han and M. Movassaghi, J. Am. Chem. Soc., 2011, 133, 10768–10771 Search PubMed.
- R. A. Leal, D. R. Beaudry, S. K. Alzghari and R. Sarpong, Org. Lett., 2012, 14, 5350–5353 Search PubMed.
- T. Buyck, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2013, 52, 12714–12718 Search PubMed.
- B. Zhao, X.-Y. Hao, J.-X. Zhang, S. Liu and X.-J. Hao, Org. Lett., 2013, 15, 528–530 Search PubMed.
- H. Mizoguchi, H. Oikawa and H. Oguri, Nat. Chem., 2014, 6, 57–64 Search PubMed.
- S. Han, K. C. Morrison, P. J. Hergenrother and M. Movassaghi, J. Org. Chem., 2014, 79, 473–486 Search PubMed.
- S. C. Farrow, M. O. Kamileen, L. Caputi, K. Bussey, J. E. A. Mundy, R. C. McAtee, C. R. J. Stephenson and S. E. O'Connor, J. Am. Chem. Soc., 2019, 141, 12979–12983 Search PubMed.
- L. P. Cameron, R. J. Tombari, J. Lu, A. J. Pell, Z. Q. Hurley, Y. Ehinger, M. V. Vargas, M. N. McCarroll, J. C. Taylor, D. Myers-Turnbull, T. Liu, B. Yaghoobi, L. J. Laskowski, E. I. Anderson, G. Zhang, J. Viswanathan, B. M. Brown, M. Tjia, L. E. Dunlap, Z. T. Rabow, O. Fiehn, H. Wulff, J. D. McCorvy, P. J. Lein, D. Kokel, D. Ron, J. Peters, Y. Zuo and D. E. Olson, Nature, 2021, 589, 474–479 Search PubMed.
- T. Kaoudi, B. Quiclet-Sire, S. Seguin and S. Z. Zard, Angew. Chem., Int. Ed., 2000, 39, 731–733 Search PubMed.
- D. Orain, R. Denay, G. Koch and R. Giger, Org. Lett., 2002, 4, 4709–4712 Search PubMed.
- C. Ferrer and A. M. Echavarren, Angew. Chem., Int. Ed., 2006, 45, 1105–1109 Search PubMed.
- C. Ferrer, C. H. M. Amijs and A. M. Echavarren, Chem.–Eur. J., 2007, 13, 1358–1373 Search PubMed.
- P. Chen, L. Cao and C. Li, J. Org. Chem., 2009, 74, 7533–7535 Search PubMed.
- S. G. Stewart, C. H. Heath and E. L. Ghisalberti, Eur. J. Org Chem., 2009, 2009, 1934–1943 Search PubMed.
- J. E. Rixson, T. Chaloner, C. H. Heath, L. F. Tietze and S. G. Stewart, Eur. J. Org Chem., 2012, 2012, 544–558 Search PubMed.
- L. Huang, L.-X. Dai and S.-L. You, J. Am. Chem. Soc., 2016, 138, 5793–5796 Search PubMed.
- Y. Wang, C. Zheng and S.-L. You, Angew. Chem., Int. Ed., 2017, 56, 15093–15097 Search PubMed.
- L. Li, X.-M. Chen, Z.-S. Wang, B. Zhou, X. Liu, X. Lu and L.-W. Ye, ACS Catal., 2017, 7, 4004–4010 Search PubMed.
- S. Yorimoto, A. Tsubouchi, H. Mizoguchi, H. Oikawa, Y. Tsunekawa, T. Ichino, S. Maeda and H. Oguri, Chem. Sci., 2019, 10, 5686–5698 Search PubMed.
- H.-H. Li, S.-H. Ye, Y.-B. Chen, W.-F. Luo, P.-C. Qian and L.-W. Ye, Chin. J. Chem., 2020, 38, 263–268 Search PubMed.
- J. Chauhan, M. K. Ravva, L. Gremaud and S. Sen, Org. Lett., 2020, 22, 4537–4541 Search PubMed.
- R. Gurram, J. B. Nanubolu and R. S. Menon, Chem. Commun., 2021, 57, 635–638 Search PubMed.
- W. R. Gutekunst and P. S. Baran, Chem. Soc. Rev., 2011, 40, 1976–1991 Search PubMed.
- G. Massiot, M. Zèches, P. Thépenier, M.-J. Jacquier, L. Le Men-Olivier and C. Delaude, J. Chem. Soc. Chem. Commun., 1982, 768–769 Search PubMed.
- G. Massiot, P. Thépenier, M.-J. Jacquier, J. Lounkokobi, C. Mirand, M. Zèches, L. Le Men-Olivier and C. Delaude, Tetrahedron, 1983, 39, 3645–3656 Search PubMed.
- X. Xie and L. Zu, Synlett, 2018, 29, 1008–1013 Search PubMed.
- Y. Yu, G. Li, L. Jiang and L. Zu, Angew. Chem., Int. Ed., 2015, 54, 12627–12631 Search PubMed.
- G. Li, X. Xie and L. Zu, Angew. Chem., Int. Ed., 2016, 55, 10483–10486 Search PubMed.
- Y. Yu, J. Li, L. Jiang, J.-R. Zhang and L. Zu, Angew. Chem., Int. Ed., 2017, 56, 9217–9221 Search PubMed.
- H. Li, P. Cheng, L. Jiang, J.-L. Yang and L. Zu, Angew. Chem., Int. Ed., 2017, 56, 2754–2757 Search PubMed.
- B. Wei, P. Cheng and L. Zu, Org. Lett., 2022, 24, 7320–7322 Search PubMed.
- M. E. Kuehne and W. G. Bornmann, J. Org. Chem., 1989, 54, 3407–3420 Search PubMed.
- J.-Q. Li, X. Quan and P. G. Andersson, Chem.–Eur. J., 2012, 18, 10609–10616 Search PubMed.
- S. Yang, P. Li, Z. Wang and L. Wang, Org. Lett., 2017, 19, 3386–3389 Search PubMed.
- Q. Wang, Y. Qu, Q. Xia, H. Song, H. Song, Y. Liu and Q. Wang, Chem.–Eur. J., 2018, 24, 11283–11287 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2284681, 2284682 and 2284683. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc02802c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |