DOI:
10.1039/C5SC04049C
(Edge Article)
Chem. Sci., 2016,
7, 2864-2869
Glutathione boosting the cytotoxicity of a magnetic platinum(IV) nano-prodrug in tumor cells†
Received
27th October 2015
, Accepted 20th January 2016
First published on 20th January 2016
Abstract
Superparamagnetic iron oxide nanoparticles (SPIONs) are potential vehicles for targeted drug delivery and viable contrast agents for magnetic resonance imaging (MRI). A PtIV prodrug (HSPt) derived from functionalization of cisplatin with hydroxyl and succinate is conjugated with a poly(ethylene glycol) (PEG)-modified SPION for cancer therapy and monitoring of therapeutic responses. The relaxivity of HSPt–PEG-SPIONs is larger than that of commercial contrast agent Feridex, and a tumor-selective negative contrast is observed in MRI in a magnetic field. HSPt–PEG-SPIONs can be dissociated and reduced into PtII species by glutathione (GSH). Instead of forming DNA–Pt crosslinks, the reduced product induces direct DNA single- or double-strand breaks, which is uncommon for Pt drugs. The cytotoxicity of HSPt–PEG-SPIONs is positively correlated with the GSH level of tumor cells, which is opposite to the scenario of current Pt drugs. HSPt–PEG-SPIONs are as cytotoxic as cisplatin against cancer cells but are almost nontoxic towards normal cells. Since the mechanism of action of the nanocomposite is different from the established paradigm for Pt drugs, it may become a special theranostic agent for cancer treatment.
Introduction
Cisplatin is a first-line chemotherapeutic drug against a variety of cancers.1 However, its application has been heavily conditioned by severe systemic toxicities like nephrotoxicity and neurotoxicity.2 In addition, the efficacy of cisplatin is limited because of inherent or acquired drug resistance.3 These defects mainly result from its indiscriminate body distribution and insufficient tumor accumulation, and also from its detoxification by sulfur-containing biomolecules.4 Targeted prodrug systems have been proven effective in minimizing the systemic toxicity and in maximizing the tumor accumulation of Pt drugs.5 SPIONs could guide drugs preferentially to the biological target through an external magnet and provide a strong negative contrast effect in T2-weighted MRI;6 and polymer-modified SPIONs possess some excellent properties, such as hydrophilicity, nontoxicity, and nonimmunogenicity, for drug delivery.7 Therefore, in the past few years we endeavoured to load PtII moieties onto SPIONs as theranostic agents for simultaneous therapy and diagnosis.8 However, PtII moieties are capable of reacting with bionucleophiles during the delivery, and hence could be toxic to normal tissues.
Octahedrally coordinated PtIV complexes are substantially more inert than PtII complexes and thus can avoid the undesirable side reactions in the blood plasma.9 Likewise, they are inactive towards nuclear DNA, so the reduction of PtIV complexes to kinetically labile PtII species is necessary to exert their cytotoxic effects. GSH is one of the intracellular reductants that activate PtIV complexes.10 Many studies have shown that cisplatin-based PtIV complexes can be reduced to reactive PtII species and bind DNA to form 1,2-d(GpG) DNA–Pt crosslinks akin to those formed by cisplatin.11 Meanwhile, the level of cellular GSH may decrease during the reduction, which is beneficial for attenuating its detoxifying effect on PtII species. For these potential merits, we decide to change the previous PtII pharmacophore to a PtIV complex.
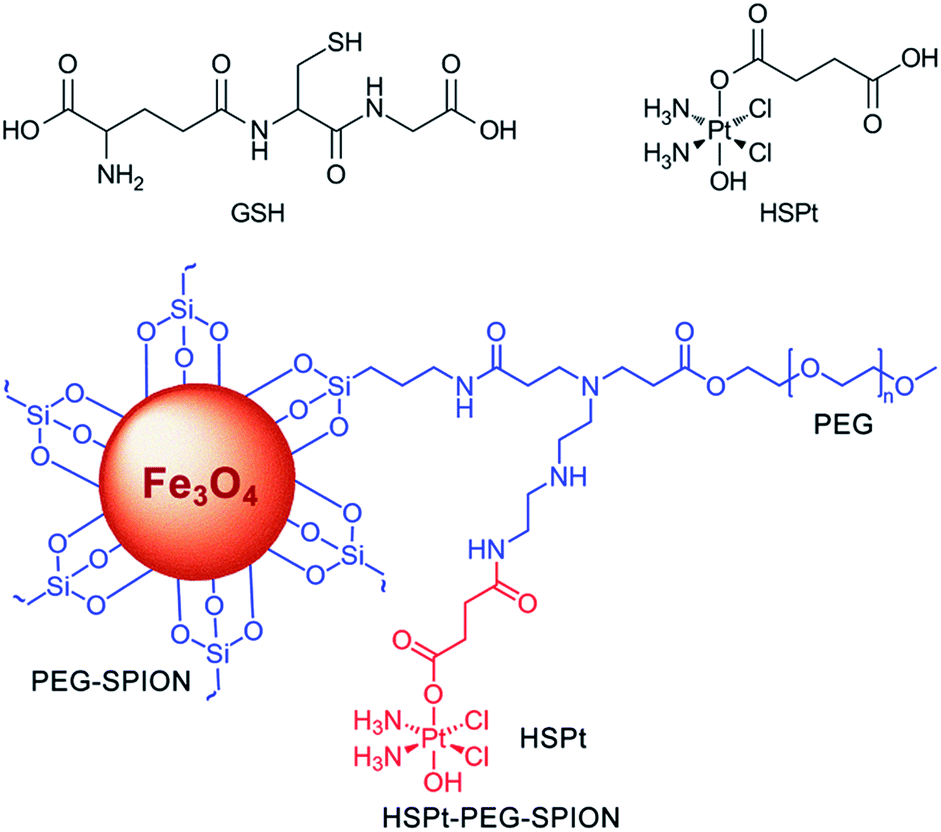
In this study, we continue to use SPIONs (Fe3O4) to design the targeted Pt prodrug system. In order to increase the stability and stealthiness in vivo, SPIONs are coated with PEG, which has been proven to improve the biocompatibility, blood circulation time, and immunotherapeutic efficacy of nanoparticles.12 A PtIV complex, c,t,c-[PtCl2(OH)(O2CCH2CH2CO2H)(NH3)2] (HSPt), is synthesized as the pharmacophore, which is an asymmetrically functionalized prodrug of cisplatin. HSPt is loaded onto the surface of the PEGylated SPIONs, forming HSPt–PEG-SPIONs (Fig. 1). This nanocomposite exhibits some unique properties in vitro, such as GSH-promoted cytotoxicity against tumor cells and nontoxicity towards normal cells. Its mechanism of action also differs from that of cisplatin. Moreover, it produces a significant negative contrast in MRI, and thus could be a potential theranostic agent for chemotherapy.
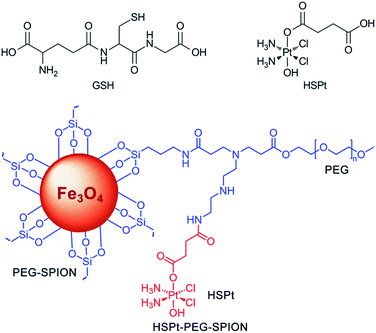 |
| | Fig. 1 Structures of GSH, HSPt and HSPt–PEG-SPIONs. | |
Results and discussion
Preparation and characterization
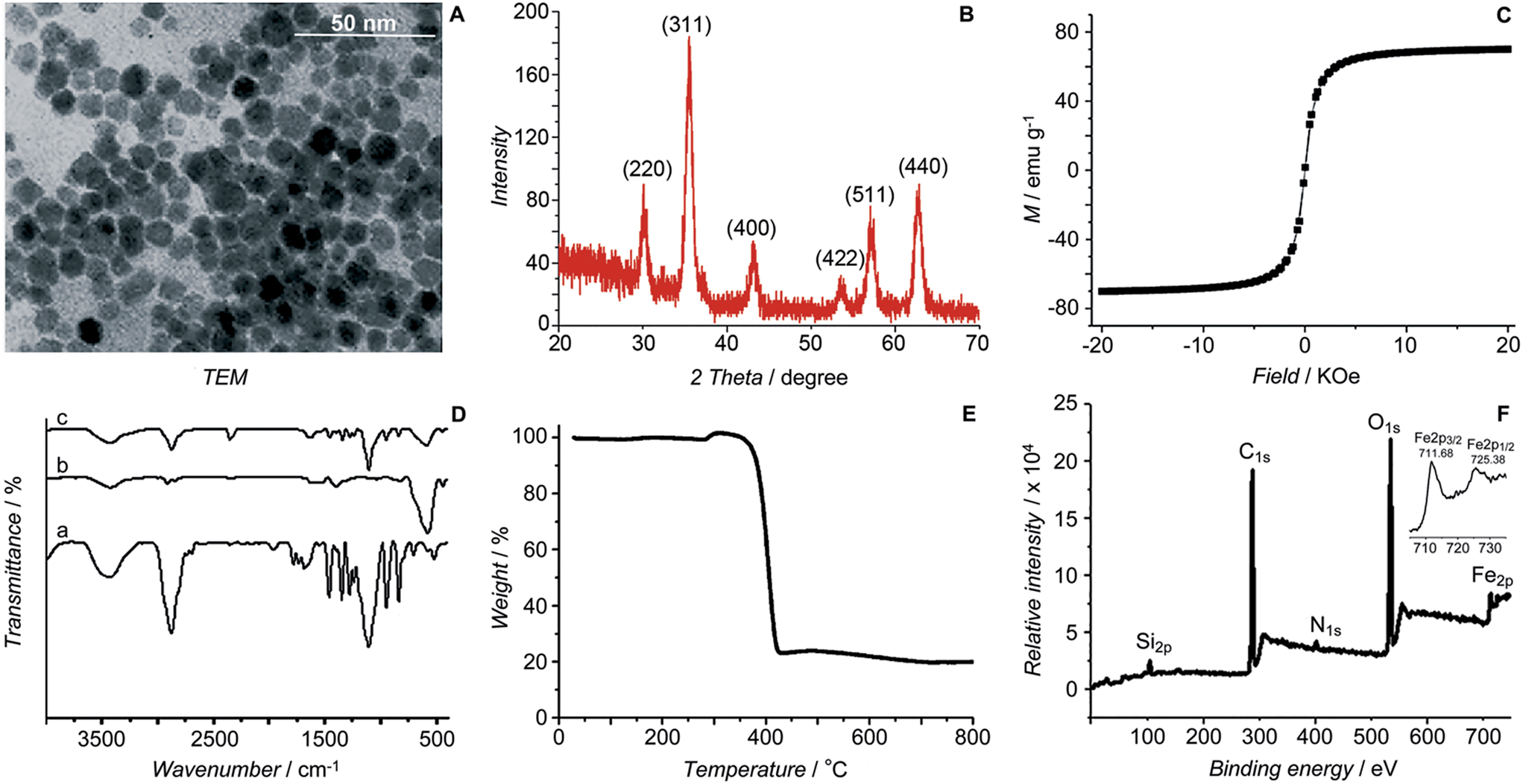
The initial SPIONs were synthesized referring to a reported method.13 PEG-SPIONs were obtained by modifying the SPIONs with silane-diethyltriamine-methoxy PEG, which contains an amine group in the chain. The TEM image shows that the size of the SPIONs is around 11 nm (Fig. 2A). The XRD spectrum of the SPIONs matches well with that from the JCPDS card (no. 01-1111) for the cubic-phase magnetite (Fig. 2B). The saturation magnetization (Ms) of the SPIONs is 70.05 emu g−1 at room temperature (Fig. 2C), which is lower than that of the bulk Fe3O4 (89 emu g−1) but is higher than that of the modified SPIONs.14 The absence of coercivity and remanence indicates that the SPIONs exhibit a superparamagnetic property at room temperature. The existence of the polymer on the surface of the SPIONs is confirmed by IR spectroscopy (Fig. 2D). Thermal gravitational analysis shows that the weight of the PEG-SPIONs decreases ca. 80% at 300–400 °C due to the evaporation of the surface polymer (Fig. 2E). In the XPS spectrum, the PEG-SPIONs exhibit Fe2p3/2 and Fe2p1/2 photoelectron peaks at 711.68 and 725.38 eV, but no obvious charge transfer satellite is observed near the Fe2p3/2 peak (Fig. 2F), suggesting that Fe is basically at a mixed oxidation state of +II and +III.15 The ICP-MS and ninhydrin assay show that the molar ratio of free amino groups to Fe in the PEG-SPIONs is 0.146.
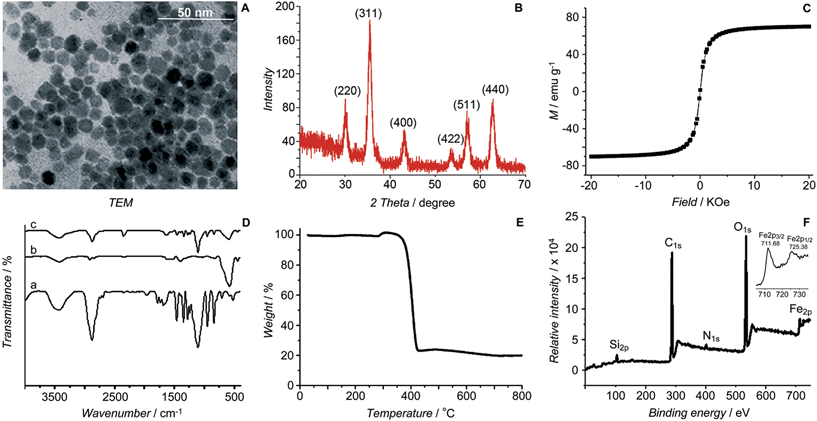 |
| | Fig. 2 Characterization of SPIONs and PEG-SPIONs. (A) TEM image of SPIONs; (B) X-ray diffraction patterns of SPIONs; (C) field-dependent magnetization curve of SPIONs at 300 K; (D) IR spectra of (a) silane-diethyltriamine-methoxy PEG, (b) SPION/oleic acid/oleylamine, and (c) PEG-SPIONs: 3200–3700 cm−1 (νOH, νNH2), 2885 cm−1 (νC–H), 1688 cm−1 (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1539 and 1410 cm−1 (νCOO), 1284 cm−1 (τCH2, νSi–C), 1201 cm−1 (ρOCH3), 1113 cm−1 (νSi–O–R), 839 cm−1 (ωNH2), 588 cm−1 (νFe–O); (E) TGA spectrum of PEG-SPIONs; (F) XPS spectra of PEG-SPIONs and Fe2p. O), 1539 and 1410 cm−1 (νCOO), 1284 cm−1 (τCH2, νSi–C), 1201 cm−1 (ρOCH3), 1113 cm−1 (νSi–O–R), 839 cm−1 (ωNH2), 588 cm−1 (νFe–O); (E) TGA spectrum of PEG-SPIONs; (F) XPS spectra of PEG-SPIONs and Fe2p. | |
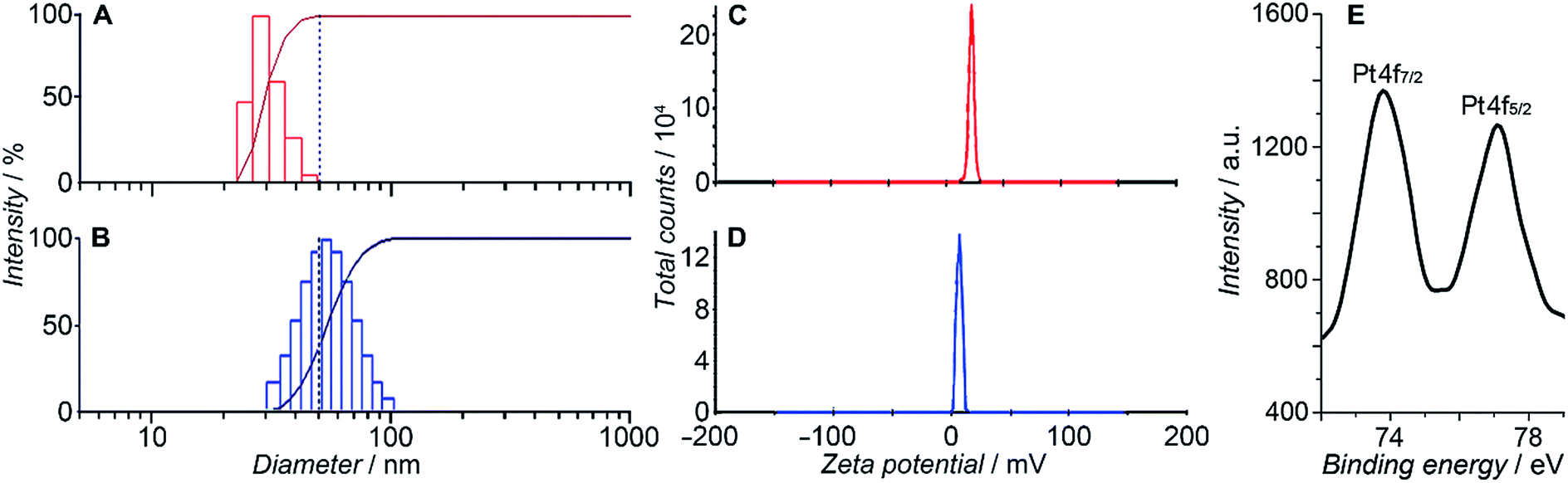
HSPt was derived from cisplatin by oxidation with hydrogen peroxide and substitution with succinate, successively. Owing to the existence of an amino group in the polymer and a carboxyl group in HSPt, HSPt–PEG-SPIONs were readily formed via EDC/NHS coupling chemistry. After conjugation, the average hydrodynamic diameter of the PEG-SPIONs increased from 31.60 ± 3.50 to 47.80 ± 5.20 nm (Fig. 3A and B), whereas the zeta potential decreased from +18.10 ± 1.30 to +1.77 ± 0.50 mV (Fig. 3C and D) due to the shielding of amine groups. These changes suggest that HSPt has bonded with the PEG-SPIONs. The weak positive potential may help the HSPt–PEG-SPIONs pass through the cell membrane (negative inside). Actually, the cellular uptake of Fe and Pt is indeed enhanced after the conjugation (see Table S1†). In the XPS spectrum of the HSPt–PEG-SPIONs (Fig. 3E), two new peaks are observed at 73.68 and 77.08 eV as compared with that of the PEG-SPIONs (Fig. 2F), which are assignable to the photoelectron peaks of Pt4f7/2 and Pt4f5/2, respectively, and are consistent with the reported binding energies for PtIV species.16 The maximum loading ratio of Pt to Fe (w/w) for the HSPt–PEG-SPIONs is 0.20 (see Table S2†).
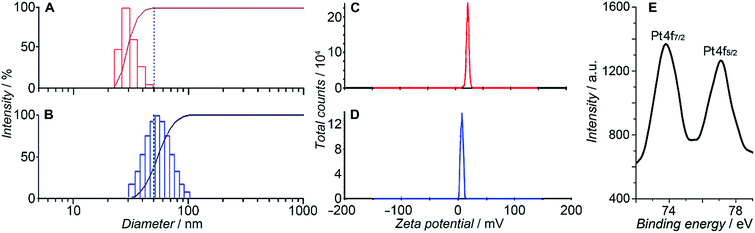 |
| | Fig. 3 Size distribution of (A) PEG-SPIONs and (B) HSPt–PEG-SPIONs determined by dynamic light scattering; zeta potential of (C) PEG-SPIONs and (D) HSPt–PEG-SPIONs; and (E) Pt4f XPS spectrum of HSPt–PEG-SPIONs. | |
Transverse relaxivity (r2) and in vitro MRI
Transverse relaxivity (r2) represents the efficiency of a contrast agent in shortening the proton relaxation time.17 As Fig. 4A shows, the T2 relaxation rate (1/T2) of the HSPt–PEG-SPIONs increases linearly with the Fe concentration and the r2 value is calculated to be 228.96 mM−1 s−1, which is significantly higher than that of the T2-weighted MRI contrast agent Feridex (SPION, r2 ≈ 100 mM−1 s−1) used in clinics,18 and is also higher than that of similar nanocomposites.19Fig. 4B reveals an explicit Fe concentration-dependent darkening effect of HSPt–PEG-SPION aqueous suspensions. The in vitro imaging potential was examined by testing the contrast effect in HeLa cells. As Fig. 4C shows, the MR images of the cells after incubation with HSPt–PEG-SPIONs present an enhanced contrast in comparison with that of the control. The signal intensity is negatively correlated with the concentration of Fe. These results indicate that HSPt–PEG-SPIONs can effectively shorten the T2 relaxation time, enter into tumor cells and produce negative contrast in MRI.
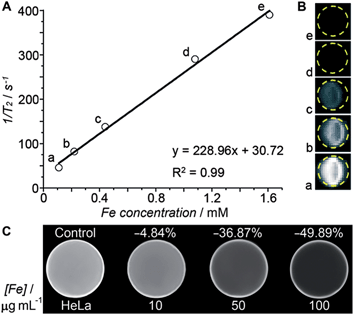 |
| | Fig. 4 (A) The relaxation rate (1/T2) versus the concentration of HSPt–PEG-SPIONs (in terms of Fe), (B) T2-weighted MR images of aqueous HSPt–PEG-SPION suspensions at different Fe concentrations, and (C) T2-weighted MR images of HeLa cells after incubation with different concentrations of HSPt–PEG-SPIONs (in terms of Fe) at 37 °C for 18 h. | |
In vivo MRI
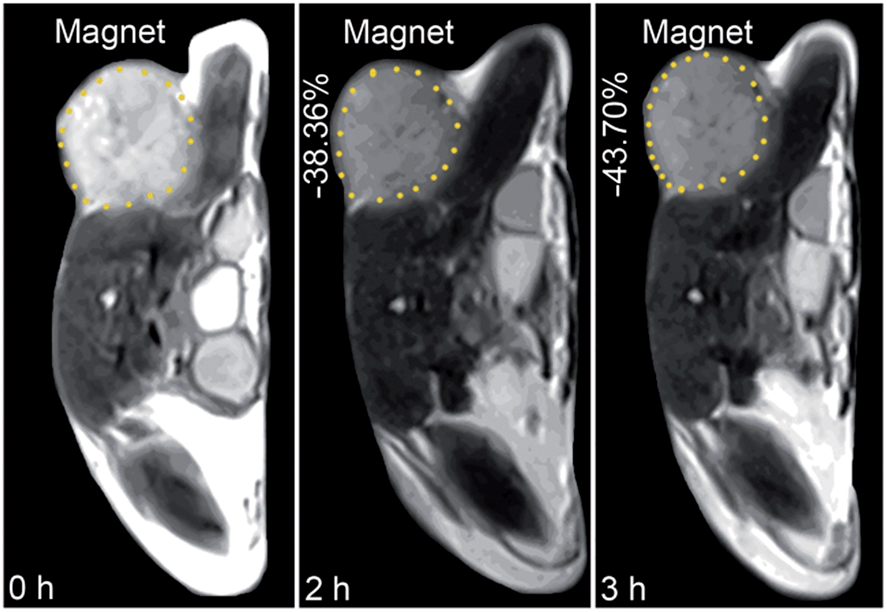
The in vivo tumor-specific accumulation and imaging potential of the HSPt–PEG-SPIONs were evaluated under an external magnetic field in B6 mice bearing implanted RM1 murine prostate cancer. Fig. 5 shows the T2-weighted MR images acquired before and after the intravenous injection of the HSPt–PEG-SPIONs. A noticeable darkening effect is observed in the circled tumor area after the injection, indicating that a substantial amount of HSPt–PEG-SPIONs has accumulated within the cancer tissue. Evidently, the negative contrast enhancement is attributed to the high relaxivity (r2) of the HSPt–PEG-SPIONs. The results show that HSPt–PEG-SPIONs can target tumor tissues with the help of external magnet and achieve real-time imaging of tumor tissues during the therapy.
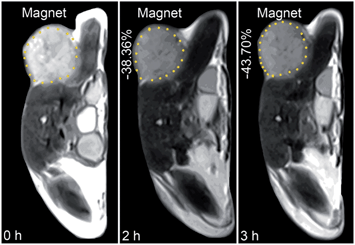 |
| | Fig. 5 MR images of B6 mice bearing implanted RM1 murine prostate cancer before and after the injection of HSPt–PEG-SPIONs (3 mg Fe per kg) in a magnetic field. | |
Drug release
The release of PtII species from HSPt–PEG-SPIONs in the presence of GSH was monitored by ICP-MS (see Fig. S1†), HPLC and identified by ESI-MS. Peaks assignable to [Pt(NH3)2Cl(OH) + Na]+ and [Pt(NH3)2Cl2(OH)(O2CCH2CH2CO2)]− are observed in the HPLC at 6 h after the reaction (see Fig. S2 and Table S3†), indicating that GSH can facilitate the dissociation of HSPt–PEG-SPIONs and reduce the PtIV prodrug into PtII species. As compared with the spectrum recorded at the start point, the intensity of HSPt and polymer peaks increased, while that of the GSH peak decreased markedly. It is known that intracellular GSH can react with cisplatin to form a Pt–GS or Pt–GS–Pt adduct, which can diminish the amount of reactive PtII species available for DNA and hence undermine the cytotoxicity of cisplatin.20 In this case, we only detected the [Pt(NH3)2(GS)H2O]+ adduct, which still maintains some reactivity towards DNA. The results imply that the depletion of GSH was mainly caused by the dissociation and reduction of HSPt–PEG-SPIONs, and the detoxification of PtII species by GSH is limited in this system.
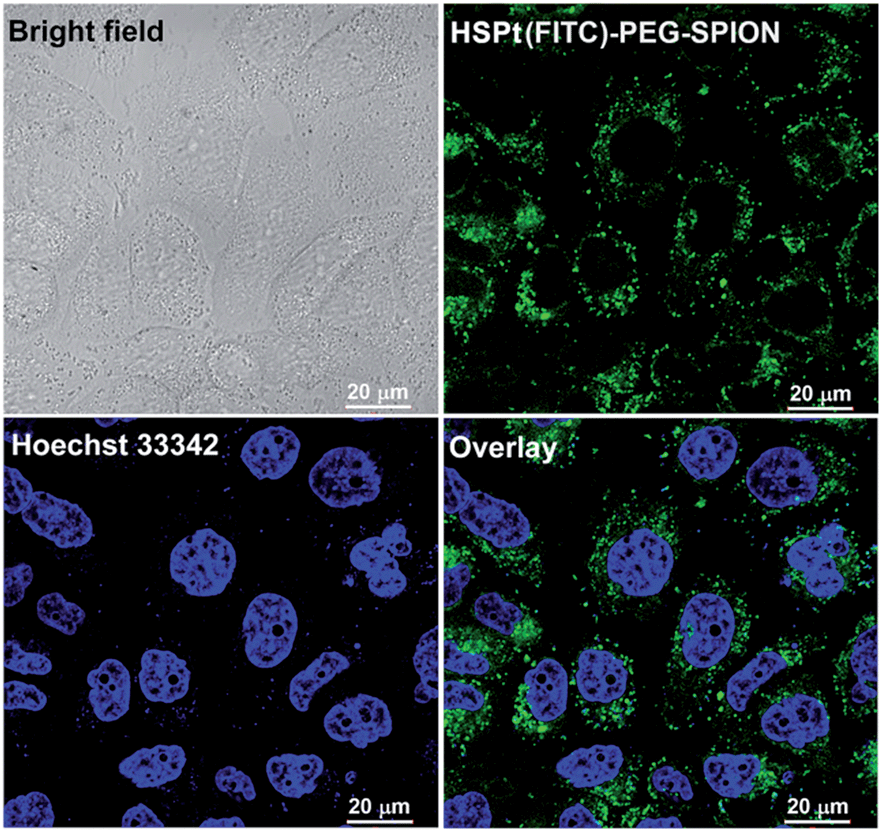
The dissociation of the HSPt–PEG-SPIONs in HeLa cells was monitored by confocal fluorescence microscopy. Due to the existence of surface amine groups, fluorescein isothiocyanate (FITC) could readily be conjugated onto PEG-SPIONs along with HSPt, forming the hybrid composite HSPt(FITC)–PEG-SPION; meanwhile, the fluorescence of FITC was quenched due to the Förster resonance energy transfer mechanism (see Fig. S3†).19 However, the fluorescence recovered as the composite was internalized by the cells (Fig. 6versus S4†), showing that FITC- or HSPt-polymer moieties can become detached from the SPIONs in the presence of cellular GSH. The co-localization image after nucleolar staining with Hoechst 33342 revealed that the fluorescent FITC-polymer species or HSPt-polymer species appear only outside the nuclei, suggesting that further dissociation is required if the Pt unit is aimed at nuclear DNA.
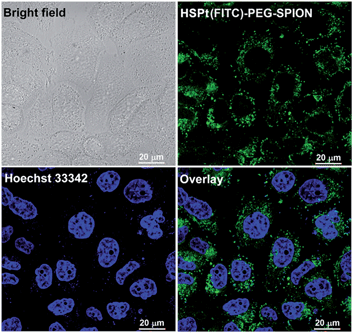 |
| | Fig. 6 Photomicrographs of HSPt(FITC)–PEG-SPIONs (20 μM Pt) in HeLa cells observed under a confocal microscope after incubation at 37 °C for 12 h. | |
Interaction with DNA
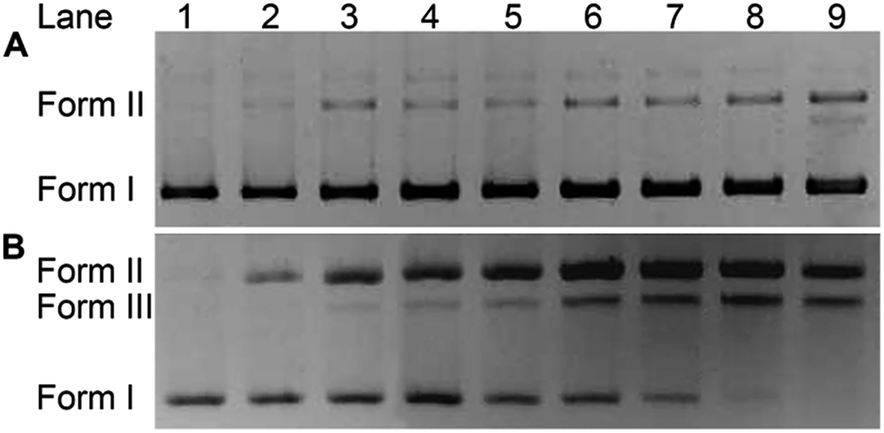
The impact of HSPt–PEG-SPIONs on DNA was studied by agarose gel electrophoresis. As Fig. 7 shows, without GSH, HSPt–PEG-SPIONs only slightly damage the supercoiled DNA (form I), producing a finite amount of nicked DNA (form II); while cisplatin retards the migration rate of form I due to the unwinding of the DNA duplex induced by Pt binding.21 With GSH, HSPt–PEG-SPIONs effectively cleave form I into form II and further into linear DNA (form III); and the cleavage efficiency is positively related to the concentration of GSH (see Fig. S5†). The appearance of linear DNA demonstrates that some double-strand breaks (DSBs) have occurred. By contrast, cisplatin and HSPt only cause mild damage to DNA in the presence of GSH (see Fig. S6†). The results again suggest that the reduction product is not cisplatin, which is in accordance with the above HPLC results. It is believed that the cytotoxicity of cisplatin primarily originates from its covalent binding to DNA and subsequent inhibiting of DNA transcription.9 Since the reduced PtII species here do not induce DNA inter- and intra-strand crosslinks as cisplatin does, we suppose the DNA strand breaks, especially the DSBs, play a key role in the bioactivity of HSPt–PEG-SPIONs. In fact, the formation of DSBs represents the most lethal form of DNA damage, because unrepaired or misrepaired DSBs can lead to cell death and genomic instability.22 In current chemotherapeutic agents, only bleomycin induces direct DSBs, whereas other agents such as cisplatin induce DSBs by indirect routes.23 Direct DNA DSBs induced by Pt anticancer drugs were never reported before. We presumed that some reactive oxygen species (ROS) might be responsible for the cleavage and hence conducted a verification using common radical scavengers such as DMSO for ˙OH, KI for H2O2, and NaN3 for 1O2 (see Fig. S7†). Contrary to our assumption, the cleavage activity of the HSPt–PEG-SPIONs did not reduce markedly, suggesting that ROS may not be a determinant in the process. Thus the cleavage mechanism is not yet clear.
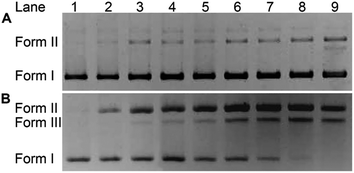 |
| | Fig. 7 Agarose gel electrophoretic patterns of supercoiled pUC19 plasmid DNA (20 ng μL−1) in the presence of HSPt–PEG-SPIONs (A) without or (B) with GSH (2 mM) after incubation in buffer (50 mM Tris–HCl, 50 mM NaCl, pH 7.4) at 37 °C for 16 h. Lane 1, DNA control; lanes 2–9, DNA + HSPt–PEG-SPIONs (6, 12, 24, 30, 36, 42, 48, 54 μM, respectively). | |
Cytotoxicity
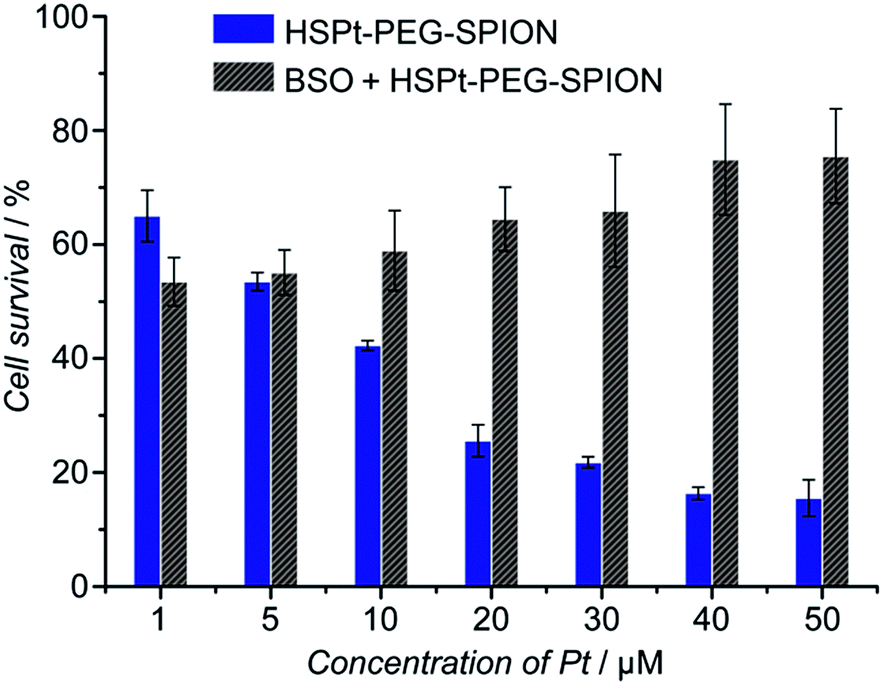
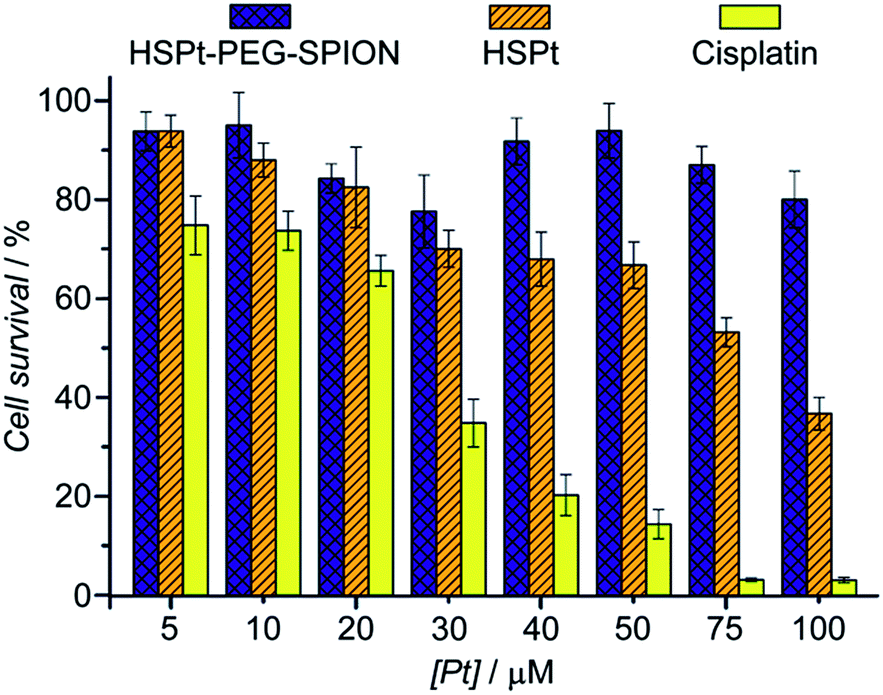
The cytotoxicity of the HSPt–PEG-SPIONs against human non-small lung cancer A549, human cervical cancer HeLa, and human gastric cancer SGC-7901 cell lines was tested by MTT assays. Meanwhile, the concentration of GSH in these cell lines before and after the drug treatment was measured by a GSH assay kit. The inhibition efficiency of the HSPt–PEG-SPIONs is comparable to that of cisplatin towards A549 and HeLa cell lines, but is inferior to that of cisplatin towards the SGC-7901 cell line (see Fig. S8 and Table S4†). The half-maximal inhibitory concentrations (IC50) of the HSPt–PEG-SPIONs and the cellular GSH concentrations are listed in Table 1. The results indicate that the IC50 values decrease with the concentration of GSH, that is, intracellular GSH can boost the cytotoxicity. In order to further confirm the promotive effect of GSH, the A549 cell line was pre-treated with buthionine sulfoximine (BSO), which is an inhibitor of intracellular GSH synthesis.24 After the treatment, the cytotoxicity of the HSPt–PEG-SPIONs was retested against this cell line. As Fig. 8 shows, the cytotoxicity was dramatically reduced because of the decrease in GSH, thus verifying the promotive effect of GSH on the cytotoxicity of the HSPt–PEG-SPIONs.
Table 1 IC50 values (μM) of HSPt–PEG-SPIONs at 48 h and intracellular concentrations of GSH (nmol per mg protein) before and after the treatment with HSPt–PEG-SPIONs
| Cells |
IC50 |
GSH before treatment |
GSH after treatment |
GSH depletion |
| A549 |
6.55 ± 3.84 |
48.85 ± 4.29 |
25.06 ± 1.44 |
48.7% |
| HeLa |
10.34 ± 4.56 |
40.73 ± 2.56 |
25.47 ± 2.67 |
37.5% |
| SGC |
37.52 ± 2.43 |
22.08 ± 2.79 |
16.35 ± 2.37 |
26.0% |
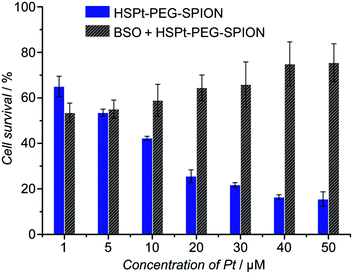 |
| | Fig. 8 The concentration-dependent cytotoxicity of HSPt–PEG-SPIONs against the A549 cell line at 48 h before and after the pre-treatment with BSO (200 μM) for 24 h. | |
It is known that GSH negatively influences the therapeutic efficacy of PtII drugs through detoxification due to its chelation with PtII centers.20 For PtIV prodrugs, GSH usually shows a biphasic action involving reduction of PtIV and sequent chelation with PtII. However, as we demonstrated above, chelation of PtII was not observed except a limited coordination with the reduced PtII species. Therefore, the cytotoxicity of the HSPt–PEG-SPIONs is positively related to the concentration of intracellular GSH or to the depletion of GSH, as high levels of GSH are beneficial to the reduction of HSPt and the cleavage of DNA. These results imply that HSPt–PEG-SPIONs may have an advantage for overcoming the tumor resistance to cisplatin induced by GSH. It should be clarified that the depletion of cellular GSH not only resulted from the reduction of the PtIV prodrug, but also resulted from the consumption for scavenging the ROS generated through DNA damage25 and for forming the Pt–GS complex with PtII species. Therefore, the percentage of GSH depletion in Table 1 appears to be quite significant.
Finally, the biocompatibility of the HSPt–PEG-SPIONs was examined on HL-7702 normal liver cells by an MTT assay. More than 80% of the cells are alive even when the concentration reaches 100 μM, while only fractional cells are alive at this concentration for cisplatin (Fig. 9). Therefore, HSPt–PEG-SPIONs are almost nontoxic to normal liver cells.
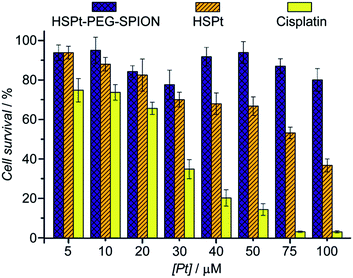 |
| | Fig. 9 Viability of HL-7702 normal liver cells after incubation with HSPt–PEG-SPIONs, HSPt and cisplatin, respectively, at 37 °C for 48 h. Data are expressed as mean (%) ± standard deviation (S.D.) of at least three independent assays. | |
Conclusions
We have prepared a SPION-based nanocomposite loaded with a PtIV prodrug as a potential targeted theranostic agent for cancer treatment. In addition to magnetic-targeting and antiproliferative properties, the composite also showed low toxicity to normal cells. The foremost findings are the significant MR contrast enhancement, unique DNA damaging mode and increased cytotoxicity in the presence of GSH. The former guarantees the applicability of the nanocomposite as an MR contrast agent for real-time imaging, and the latter two reveal a novel mechanism of action for PtIV prodrugs loaded on nanoparticles. The role of GSH in cisplatin resistance has been studied extensively in both cell lines and cancer tissues, and an increase in the GSH level has been correlated with cisplatin resistance in ovarian, cervical, lung, embryonal and bladder cancer cell lines due to increased inactivation of the drug.3 The results of this study indicate that the basic function of intracellular GSH for nano-based PtIV prodrugs is different from that for PtII drugs, which has never been perceived so far. The exceptional cleavage mode of DNA and distinct action of GSH may suggest that the cisplatin resistance related to DNA repair and GSH detoxification could be conquered by functionalized PtIV prodrugs.
Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (Grants 21271101, 31570809, 21131003, 91213305, 91413116, 21361140352, 81172009) and the National Basic Research Program of China (Grant 2015CB856300).
Notes and references
-
X. Y. Wang and Z. J. Guo, New Trends and Future Developments of Platinum-Based Antitumor Drugs, in Bioinorganic Medicinal Chemistry, ed. E. Alessio, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim, 2011, pp. 97–149 Search PubMed.
- N. A. G. dos Santos, M. A. C. Rodrigues, N. M. Martins and A. C. dos Santos, Arch. Toxicol., 2012, 86, 1233–1250 CrossRef PubMed.
- B. Köberle, M. T. Tomicic, S. Usanova and B. Kaina, Biochim. Biophys. Acta, Rev. Cancer, 2010, 1806, 172–182 CrossRef PubMed.
- H. Burger, W. J. Loos, K. Eechoute, J. Verweij, R. H. J. Mathijssen and E. A. C. Wiemer, Drug Resist. Updates, 2011, 14, 22–34 CrossRef CAS PubMed.
- X. Y. Wang and Z. J. Guo, Chem. Soc. Rev., 2013, 42, 202–224 RSC.
- R. Hao, R. J. Xing, Z. C. Xu, Y. L. Hou, S. Gao and S. H. Sun, Adv. Mater., 2010, 22, 2729–2742 CrossRef CAS PubMed.
- L. Douziech-Eyrolles, H. Marchais, K. Hervé, E. Munnier, M. Soucé, C. Linassier, P. Dubois and I. Chourpa, Int. J. Nanomed., 2007, 2, 541–550 Search PubMed.
-
(a) J. Z. Wang, X. Y. Wang, Y. J. Song, J. Wang, C. L. Zhang, C. J. Chang, J. Yan, L. Qiu, M. M. Wu and Z. J. Guo, Chem. Sci., 2013, 4, 2605–2612 RSC;
(b) J. Z. Wang, X. Y. Wang, Y. J. Song, C. C. Zhu, J. Wang, K. Wang and Z. J. Guo, Chem. Commun., 2013, 49, 2786–2788 RSC;
(c) R. M. Xing, X. Y. Wang, C. L. Zhang, J. Z. Wang, Y. M. Zhang, Y. Song and Z. J. Guo, J. Mater. Chem., 2011, 21, 11142–11149 RSC.
- J. J. Wilson and S. J. Lippard, Chem. Rev., 2014, 114, 4470–4495 CrossRef CAS PubMed.
- E. Wexselblatt and D. Gibson, J. Inorg. Biochem., 2012, 117, 220–229 CrossRef CAS PubMed.
- C. F. Chin, D. Y. Q. Wong, R. Jothibasu and W. H. Ang, Curr. Top. Med. Chem., 2011, 11, 2602–2612 CrossRef CAS PubMed.
-
(a) K. Hervé, L. Douziech-Eyrolles, E. Munnier, S. Cohen-Jonathan, M. Soucé, H. Marchais, P. Limelette, F. Warmont, M. L. Saboungi, P. Dubois and I. Chourpa, Nanotechnology, 2008, 19, 465608 CrossRef PubMed;
(b) J. Hernández-Gil, M. Cobaleda-Siles, A. Zabaleta, L. Salassa, J. Calvo and J. C. Mareque-Rivas, Adv. Healthcare Mater., 2015, 4, 1034–1042 CrossRef PubMed.
- S. H. Sun and H. Zeng, J. Am. Chem. Soc., 2002, 124, 8204–8205 CrossRef CAS PubMed.
- Y. Ding, Y. Hu, L. Y. Zhang, Y. Chen and X. Q. Jiang, Biomacromolecules, 2006, 7, 1766–1772 CrossRef CAS PubMed.
- M. Descostes, F. Mercier, N. Thromat, C. Beaucaire and M. Gautier-Soyer, Appl. Surf. Sci., 2000, 165, 288–302 CrossRef CAS.
- H. H. Xiao, H. Q. Song, Y. Zhang, R. G. Qi, R. Wang, Z. G. Xie, Y. B. Huang, Y. X. Li, Y. Wu and X. B. Jing, Biomaterials, 2012, 33, 8657–8669 CrossRef CAS PubMed.
- H. Yang, C. X. Zhang, X. Y. Shi, H. Hu, X. X. Du, Y. Fang, Y. B. Ma, H. X. Wu and S. P. Yang, Biomaterials, 2010, 31, 3667–3673 CrossRef CAS PubMed.
- Y.-X. J. Wang, S. M. Hussain and G. P. Krestin, Eur. J. Radiol., 2001, 11, 2319–2331 CrossRef CAS PubMed.
- Z. Y. Cheng, Y. L. Dai, X. J. Kang, C. X. Li, S. S. Huang, H. Z. Lian, Z. Y. Hou, P. G. Ma and J. Lin, Biomaterials, 2014, 35, 6359–6368 CrossRef CAS PubMed.
- X. Y. Wang and Z. J. Guo, Anti-Cancer Agents Med. Chem., 2007, 7, 19–34 CrossRef.
- M. V. Keck and S. J. Lippard, J. Am. Chem. Soc., 1992, 114, 3386–3390 CrossRef CAS.
- S. Grudzenski, A. Raths, S. Conrad, C. E. Rübe and M. Löbrich, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14205–14210 CrossRef CAS PubMed.
- W. M. Bonner, C. E. Redon, J. S. Dickey, A. J. Nakamura, O. A. Sedelnikova, S. Solier and Y. Pommier, Nat. Rev. Cancer, 2008, 8, 957–967 CrossRef CAS PubMed.
- E. Desideri, G. Filomeni and M. R. Ciriolo, Autophagy, 2012, 8, 1769–1781 CrossRef CAS PubMed.
- M. A. Kang, E.-Y. So, A. L. Simons, D. R. Spitz and T. Ouchi, Cell Death Dis., 2012, 3, e249 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary Fig. S1–S8, Tables S1–S4 and experimental procedures. See DOI: 10.1039/c5sc04049c |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a