
Rigid thermosetting epoxy/multi-walled carbon nanotube foams with enhanced conductivity originated from a flow-induced concentration effect†
Yu Xu,
Ying Li,
Jianjun Bao*,
Tao Zhou and
Aiming Zhang*
State Key Laboratory of Polymer Materials Engineering of China, Polymer Research Institute of Sichuan University, Chengdu, Sichuan 610065, China. E-mail: jjbao2000@sina.com; 1364680891@qq.com; Fax: +86-28-85402465; Tel: +86-28-85459500 Tel: +86-28-85402601
First published on 7th April 2016
Abstract
Herein, multi-walled carbon nanotube (MWCNT) reinforced rigid epoxy foams were innovatively prepared using expandable microspheres. A two-step technique, including prepolymerization and consecutive foaming, was employed to avoid the thermal degradation of epoxy due to the uncontrolled violent curing reaction. The effects of foaming on the MWCNT inter-connectivity, electrical percolation threshold, and through-plane electrical conductivity were investigated. Moderately foamed epoxy/MWCNT composites were confirmed to have better conductivity than their solid counterparts at the same MWCNT content. An effect we called flow-induced concentration increases the inter-connectivity by separating MWCNTs from a flowing matrix caused by thermally triggered expansion of microspheres, and changes the MWCNT distribution. For the foamed composites with 2 wt% microspheres, the electrical percolation threshold is approximately 0.3 wt% and the conductivity is obviously higher than that of the solid counterpart. Relationships between microstructures and conductivities were also discussed. The mechanical property and cellular structures of conductive foams obtained under different foaming temperatures, prepolymerization time, microsphere and MWCNT contents were respectively evaluated. All the results reveal that lightweight conductive epoxy foams with lower filler content and enhanced conductivity can be fabricated for applications in electronics, automobiles, and aerospace.
1. Introduction
Recent years have witnessed extensive applications of electrical conductive polymer composites (CPCs) in the electronics industries, such as capacitors,1 sensing devices,2 photoconductors3 and electromagnetic interference (EMI) shielding4 owing to their excellent erosion resistance, bargain price, tunable conductivity. Furthermore, light-weight and high-performance foamed CPCs are of great interest due to their low consumption of energy and raw material which are particularly desirable in missiles, automobiles, aviation and aerospace fields.5The carbonaceous fillers with great aspect ratio, carbon nanotubes (CNTs) and carbon nanofiber (CNF) for instance, are most often used to prepare CPC-based foams due to their low density, excellent mechanical performance, and outstanding electrical and thermal conductivities. Yang et al. ever reported CNF reinforced polystyrene foams which had almost similar percolation threshold to solid composites.6 A low density polyethylene foam with 5 wt% MWCNTs displayed a conductivity of 6.1 × 10−6 S cm−1.7 A kind of CNF/polypropylene (PP) foam exhibited higher conductivity than the solid composites at CNF concentration below 20 wt%.8 A 0.5 wt% graphene reinforced polyvinylidene fluoride foam showed a conductivity of 10−4 S m−1.9 It is said that 1.8 vol% graphene reinforced poly(methyl methacrylate) foam owned much higher conductivity of 3.11 S m−1.10 Besides, other conductive foams differing in their conductivities, such as CNTs/polylactide foams,11 MWCNTs/polycaprolactone foams,12 CNTs/polycarbonate,13 and PMI foam,14 were also studied before. Thermosetting polymers like polyurethane (PU) can also be foamed by the similar methods.15–17 A kind of conductive MWCNTs/PU foam with a density below 0.05 g cm−3 which exhibited a conductivity of around 4.3 × 10−5 S m−1 for a concentration of 2 wt% CNTs was prepared by Xu et al. via using water which could react with isocyanate to produce carbon dioxide.18 These works above seem imply that nearly all thermoplastic and thermosetting polymers can be used to fabricate conductive foams. However, PU is the only one which is extensively applied to fabricating conductive thermosetting foams.19 Despite their wide varieties and desirable attributes, all these foams above have common weaknesses of low strength or low heat resistance which limit their applications.
By contrast, expanded thermoset epoxy resin exhibits high stiffness, good mechanical property and resistance to high humidity, corrosion and heat. Foams obtained by reaction of polyepoxide, trialkylboroxine and an amine have outstanding thermal stability and are suitable to use at elevated temperature in the range of 204–316 °C.20 Syntactic epoxy foam consisting of rigid hollow particles and a polymer matrix can withstand the enormous hydrostatic pressure.21 The syntactic epoxy foam, with enough reinforcement of other fillers, such as hollow carbon microsphere, fiberglass mesh and short glass fiber, is verified to be of great use.22–24 Despite the importance of intrinsic attributes, conductive epoxy foams even including one containing conductive fillers have received surprisingly little attention and been virtually ignored. For my perspective, their main advantages are their abilities to perform in multi-functional applications combining the structural function under regular service conditions with other functions like EMI shielding.
One possible reason is that loading sufficient conductive fillers, MWCNTs for instance, will damage the foamability of epoxy blends.25 For traditional epoxy foams which use low boiling point liquids or chemical material as blowing agent, the key to successfully achieve epoxy foams with a nice and regular structure is to coordinate the curing exotherms, the viscosity changes and the boiling point or decomposition temperatures of blowing agent. Fast polymerization reactions undoubtedly produce large exotherms within a short time, resulting in a sudden viscosity rise and finally underdeveloped foams. Slow reactions give rise to small exotherms, long curing time, low foaming viscosity and consequent collapse of cellular structures. Adequate conductive fillers like MWCNTs always cause unfavorable viscosity increment during the foaming reaction, which forces us to design more difficult formulas and master very delicate operation skills.
In present work, a novel physical blowing agent Expancel is utilized to decouple the curing reaction and expansion mechanisms. This's an expandable polymer microsphere which has a liquid volatile hydrocarbon core encapsulated in a thermoplastic shell.26 The shell, upon heating, will soften and meanwhile the liquid core begins to vaporize and expands the thermoplastic shell (Fig. 1). It witnesses wide applications in shoe soles,27 wallpaper,28 leather,29 etc., due to its unique features including an attractive surface finish, no post-expansion or sink-marks, low density, wide applicable temperatures, good solvent resistance, and low cost.30 A polyvinyl chloride plastisol containing this kind of microsphere is confirmed to be suitable for use as underbody compound for vehicles.31 It is said that this microsphere can enable a uniform microcell of a silicone rubber composite.32 Here, since the cell expansion in foaming process is not such integrated with the curing reaction, using Expancel as a blowing agent gives great flexibility of formulation and wide latitude in material choices. Moreover, the preparation of conductive epoxy foam based on expandable microspheres has not been reported yet. Here we provide a facile approach to fabricate lightweight conductive MWCNTs/epoxy foams with well-controlled cellular structure.
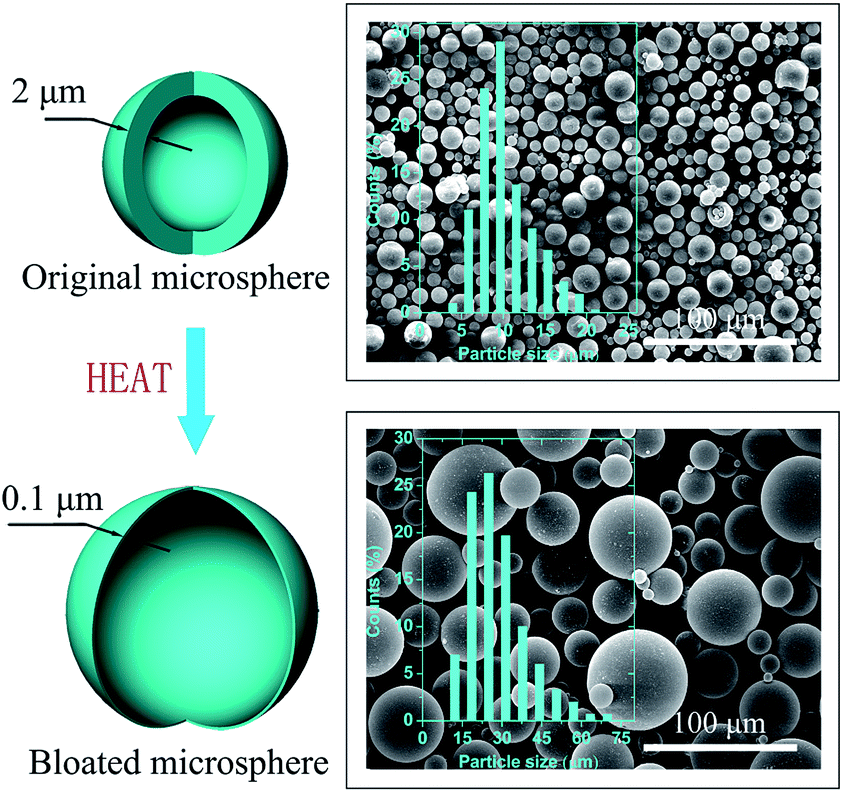 | ||
| Fig. 1 Schematic diagram for describing the expansion process of expandable microspheres, and SEMs of the original microspheres (upper right) and the bloated microspheres (bottom right). Inserts are the particle size distribution histograms. | ||
Herein, different contents of MWCNTs reinforced epoxy foams were prepared. The effect of foaming on the formation of electrically conductive networks was studied in detail. Interestingly, foamed composites reached a high conductivity value than the solids though the volume percentage of MWCNTs actually decreased after foaming. Limited works have been dedicated to this phenomenon. Xu18 has reported that the conductivity of a 2 wt% CNT/PU foam decreases gradually with the decrease of density. Further density decrease will led to a conductor–insulator transition which is called a weight-lightening limit for the foam composite with a given MWCNT loading. No enhancement in conductivity has been observed. It was found that foaming can improve the efficiency of electrical conduction of CNF reinforced PP foam prepared by a high pressure CO2 dissolution process.33 Foaming helps to break the aggregates and obtain a better CNF dispersion. The mechanism for the change in conductivity of foamed epoxy/MWCNT composite is examined and fully discussed. Based on our current acknowledge, foaming process here will lead to build more conductive networks not by obtaining a better dispersion but by inducing a better MWCNT concentration. This might be called “the flow-induced concentration effect”. The benefits of such an evaluation can lead to wider applications using light-weight conductive structural epoxy composites.
2. Experimental
2.1 Materials
Bisphenol-A type epoxy resin NPEL-128 was supplied by Nan Ya Plastics Co. (Taiwan). The hardener used, a kind of polyether amine (PEA) named Baxxodur™ EC 301, was bought from BASF SE (Germany). The chemical reaction of epoxy resin with PEA can be described by the equation in Fig. S1.† Based on the epoxy epoxide equivalent of NPEL-128 and the amine hydrogen equivalent weight of EC301, the optimum curing ratio of the resin to the hardener is 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :32 by weight. Industrial-grade MWCNTs, manufactured via chemical vapor deposition techniques through cracking the natural gas in the presence of Ni/γ-Al2O3 catalyst, were purchased from Chengdu Organic Chemicals Co. Ltd. (China). Expandable microspheres, Expancel™ 031DU40 (AkzoNobel, Sweden), were used as foaming agent. Detailed properties of MWCNTs, epoxy resin, PEA, and microspheres are available in ESI.†
:32 by weight. Industrial-grade MWCNTs, manufactured via chemical vapor deposition techniques through cracking the natural gas in the presence of Ni/γ-Al2O3 catalyst, were purchased from Chengdu Organic Chemicals Co. Ltd. (China). Expandable microspheres, Expancel™ 031DU40 (AkzoNobel, Sweden), were used as foaming agent. Detailed properties of MWCNTs, epoxy resin, PEA, and microspheres are available in ESI.†
2.2 Preparation of conductive MWCNTs/epoxy foams
The obstacle to maximize the conductivity of composite is mainly the easy agglomeration of MWCNTs due to the intermolecular van der Waals attraction. As shown in Fig. 2, the preparation process can be described as follows. | ||
| Fig. 2 Flow diagram for preparing conductive MWCNTs/epoxy foams (blue arrows) and their solid composites (red arrows). | ||
The MWCNTs and epoxy resin are firstly premixed for 5 min by a handheld dual-axis mixer at a speed of 200 rpm. Then, the MWCNTs/epoxy blends are transferred to a three-roll calendaring machine (S65, RUITONG Machinery Co. Ltd.) for further mixing at high shear rates to achieve better dispersion. The blends are passed through the tiny gap between two parallel rolls rotating at 500 rpm. This process will be repeated at least five times for each batch, which is accepted to be one of the simple and effective solutions by other researches to achieve a relatively better dispersion.34
Afterward, a certain amount of expandable microspheres and curing agent are respectively incorporated into the blends. The mixing is carried out again using the handheld dual-axis mixer rather than the three-roll calendaring machine for 10 min to avoid the pulverization of microsphere. Thus, the expandable precursors with well-dispersed MWCNTs are obtained.
Once the mixing is finished, the precursors are quickly put into an air-circulating oven for prepolymerization at 45 °C and stirred at regular intervals. After a certain period of time, the precursors are soon poured into a pre-heated stainless steel mold with its internal surface sprayed a layer of release agent (DUDAO Chemical, China). Subsequently, the mold is moved to another air-circulating oven for foaming at a certain temperature and post-curing, and finally took out about 3 hours later. Thus, the conductive epoxy foam was obtained.
Formulations for preparing conductive epoxy/MWCNTs foams in our experiments were given in Table 1. As a comparison, the solid counterparts were also prepared in a similar way without adding microspheres (Group D in Table 1).
:32 and 45 °Ca
| Group | Parameters | ||||
|---|---|---|---|---|---|
| Number | tpre (min) | Tf (°C) | Expancel (wt%) | MWCNTs (wt%) | |
| a tpre, time of prepolymerization; Tf, foaming temperatures. | |||||
| A | A1-(1–5) | 90 | 80 | 0.5 | 0.5, 1, 2, 3, 4 |
| A2-(1–5) | 90 | 80 | 1 | 0.5, 1, 2, 3, 4 | |
| A3-(1–8) | 90 | 80 | 2 | 0.5, 1, 2, 3, 4, 5, 7, 0 | |
| A4-(1–5) | 90 | 80 | 3 | 0.5, 1, 2, 3, 4 | |
| A5-(1–5) | 90 | 80 | 4 | 0.5, 1, 2, 3, 4 | |
| A6-(1–5) | 90 | 80 | 5 | 0.5, 1, 2, 3, 4 | |
| B | B1-(1–2) | 80 | 80 | 2 | 1, 2 |
| B2-(1–2) | 90 | 80 | 2 | 1, 2 | |
| B3-(1–2) | 100 | 80 | 2 | 1, 2 | |
| B4-(1–2) | 105 | 80 | 2 | 1, 2 | |
| B5-(1–2) | 115 | 80 | 2 | 1, 2 | |
| C | C1-(1–2) | 90 | 60 | 2 | 1, 2 |
| C2-(1–2) | 90 | 70 | 2 | 1, 2 | |
| C3-(1–2) | 90 | 80 | 2 | 1, 2 | |
| C4-(1–2) | 90 | 90 | 2 | 1, 2 | |
| C5-(1–2) | 90 | 100 | 2 | 1, 2 | |
| D | D-(1–8) | 90 | — | — | 0.5, 1, 2, 3, 4, 5, 7, 0 |
2.3 Measurements and characterizations
The rheological tests of MWCNTs/epoxy blends were carried out on a HAAKE RARS III rheometer with 30 mm parallel plates. The gap size is maintained at 1 mm (volume of 7.065 cm3) per sample. Dynamic temperature tests (oscillatory temperature ramp) are performed from 30 to 80 °C at a ramp rate of 2 °C min−1 to observe the variations in viscosity with temperatures. The control variable of 12% strain is used with the sampling rate set at 1 point every 10 seconds at the frequency of 1 Hz. The post experiment step sets the temperature back to 30 °C.Samples consisting of 5–9 mg blends were placed in aluminum crucible and run on a NETZSCH DSC 204 series under nitrogen flow rate of 50 mL min−1. The sample was held at 30 °C for 3 min, then heated to 230 °C at a certain heating rate, and finally cooled down to 30 °C at 40 °C min−1. The areas under the exotherms were recorded to determine the reaction heat.
The foam structures were examined by a scanning electron microscopy (JEOL-7500F SEM, Japan) at an accelerating voltage of 5 kV. All samples were freeze-fractured after being submerged in liquid nitrogen for 10 min and the fractured surfaces were sputtered with a thin gold layer before observation.
The volume resistivity of the moderately conductive foams below 108 Ω cm was measured on a two-probe electrometer (UNI-T Technology, China), while those above 108 Ω cm were collected on a ZC90F ultrahigh resistance meter with a three-terminal fixture (TAIOU Electronics Co. Ltd., Shanghai). Circular copper sheet with a thickness of 2 mm and a diameter of 10 mm were fabricated using a mini lathe for resistivity measurements. The upper and bottom surfaces of the foam were respectively coated with copper paste to reduce contact resistance between the sample and the electrode. Each test was repeated at least 5 times, and the resistivity was calculated by the following eqn (1):
| ρ = (RS)/L | (1) |
Foam samples with a dimension of 30 × 30 × 15 mm3 were used for compression test which was carried out on a universal testing machine (4302 INSTRON, American) with a sensor of 30 kN at the test rate of 2 mm min−1. The compression strength and modulus at the compression strain of 10% were recorded to evaluate the mechanical property. The termination point of test was set at 15%. Each data shown in this paper was the average value after each test was repeated at least five times.
3. Results and discussion
3.1 The effect on the preparation of epoxy foam
 | ||
| Fig. 3 Optical images of foamed samples: (a) pure epoxy foam obtained by one-step method; (b) pure epoxy foam and (c) conductive epoxy/MWCNTs foam (sample A3-3) obtained by two-step method. The weight ratio of epoxy resin to PEA and the microsphere contents in these three samples are fixed at 100:32 and 2 wt%, respectively. In the two-step method, the prepolymerization temperature is 45 °C and the prepolymerization time is 90 min. | ||
In foaming process, heat of both internal curing reaction and external environment will promote the expansion of microspheres and increase the viscosity of system. The most idealized procedure is the increasing viscosity prevents microspheres from over-expansion and stabilizes the shape of bubbles. If system viscosity during foaming increases slowly, the expanding microspheres will continue to grow without restraint leading to rupturing and merging. The gas escapes from the rupturing microspheres, aggregates together and forms larger cavities. But as we mentioned above, a rapid curing will cause inner burning inside the foam.
To balance the heat release and the adequate increasing of system viscosity, we established a “two-step method” which introduces a prepolymerization procedure, that is the expandable MWCNTs/epoxy blends are firstly pre-polymerized at low temperatures (45 °C for instance) for a given period of time and then foamed at given temperature (90 °C). Fig. 3b and c respectively exhibit the optical photographs of pristine epoxy foam and conductive epoxy foam obtained via two-step method. It's pretty exciting to see that the foam in image (b) has a snowy white section without any motley, and no cavities can be observed with the unaided eye. The foam in image (c) exhibits a perfect appearance, too. These two foams process a perfect and desirable cellular structure without any defects. The prepolymerization technique makes it easier to provide optimal force to restrict the growth of expandable microspheres and control the expanding epoxy. It has a great positive effect on epoxy foam preparation.
 | ||
| Fig. 4 DSC analysis: (a) non-isothermal curing thermograms for the epoxy/PEA system at the heating rates of 5, 10, 15 and 20 °C min−1; (b) fitting curves corresponding to the characteristic temperatures obtained from DSC curves in (a). The weight proportion of epoxy to PEA is 100:32. | ||
To determine the optimal curing temperature, the To, Tp and Te are respectively plotted against the heating rates in Fig. 4b. Linear relationships between these characteristic temperatures and heating rates can be clearly observed. The curing characteristic temperature To, Tp and Te at zero heating rate are 70.7 °C, 106.9 °C and 157.75 °C, which respectively correspond to the gel temperature, curing temperature and post curing temperature.
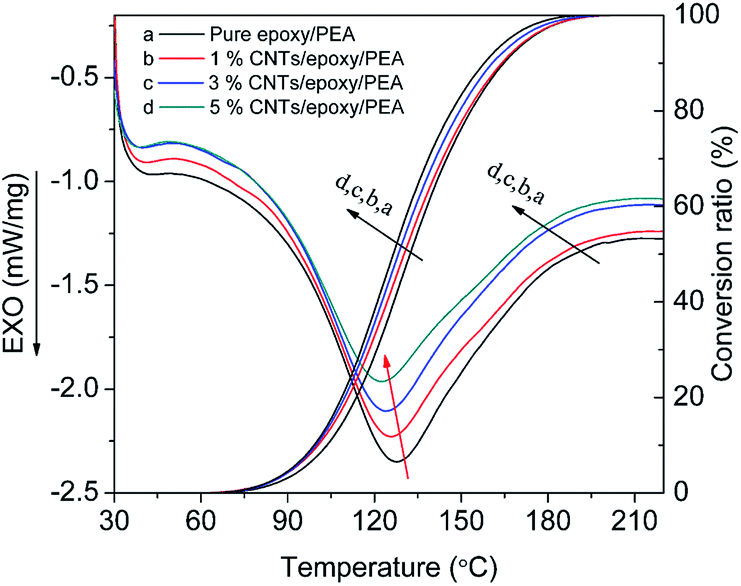 | ||
| Fig. 5 Non-isothermal DSC thermograms and conversion ratio curves at the heating rate of 10 °C min−1 for the neat epoxy/PEA system and epoxy/PEA systems containing 1 wt%, 3 wt%, and 5 wt% MWCNTs. | ||
The characteristic temperatures and total curing enthalpies corresponding to the non-isothermal DSC thermograms in Fig. 5 are summarized in Table 2. Both the To and Tp decrease with the increase of MWCNT loading, indicating that the catalytic effect is intensified. The total curing enthalpies (ΔH) of the MWCNTs/epoxy/PEA systems calculated by the total area under the exothermic peak are all smaller than that of the neat epoxy/PEA system. For example, the ΔH of the 3 wt% MWCNTs/epoxy/PEA is 429.6 J g−1, which is obviously less than that (436.4 J g−1) of the neat epoxy/PEA. This could be because of the reduction of epoxy concentration in the composite system and the decreased overall curing degree.35
| Sample | To (°C) | Tp (°C) | ΔH (J g−1) |
|---|---|---|---|
| Neat epoxy/PEA | 85.3 | 127.4 | 436.4 |
| 1 wt% MWCNTs | 82.0 | 125.1 | 432.3 |
| 3 wt% MWCNTs | 80.7 | 122.8 | 429.6 |
| 5 wt% MWCNTs | 79.9 | 121.3 | 403.9 |
The curing extent can be described by conversion ratio which is calculated by the eqn (2) below:
| Ψ = ΔHt/ΔH | (2) |
3.2 The conductivity of MWCNTs/epoxy foams
A series of MWCNTs/epoxy foams with different MWCNT contents are prepared and their electrical conductivities are compared with those of the solid parts. The preparation procedures of the foams studied here consist of the prepolymerization at 45 °C for 90 min and the following foaming process at 80 °C. Fig. 6a shows the relationship between the electrical resistivity of two sets of samples (Groups A3 and D in Table 1) and the MWCNT contents at room temperature. The phenomenon that the resistivity declines with the increasing MWCNT content is clearly observed both in the foams and their solid counterparts. For the solids, a decrease in resistivity from approximately 4.91 × 1014 Ω cm to approximately 1.50 × 107 Ω cm is clearly observed in the MWCNT loading range of 0.1–1.5 wt%, which can be considered as the percolation zone. Thus, the percolation threshold is estimated as 0.8 wt%. The resistivity decreases slowly as the MWCNT loading exceeds 1.5 wt%. According to the percolation threshold theory, electrical conductivity of a composite follows a power law relationship below:| σ = ρ−1 ∝ (m − mc,σ)βc,σ | (3) |
 | ||
| Fig. 6 Comparative analyses of conductive foams and their solid counterparts: (a) electrical resistivity of the prepared MWCNTs/epoxy foams (Group A3-(1–8) in Table 1) and their solid counterparts (Group D in Table 1) at the MWCNT loading range of 0–7 wt%; (b) a power law dependence of electrical conductivity of solid composites on the reduced MWCNT loading; (c) SEM image of solid sample D3; (c) SEM image of sample A3-1. | ||
As shown in Fig. 6c, the well dispersed MWCNTs are embedded throughout the resin matrix which confirms the effectiveness of our mixing method. In image (d), the obtained conductive foam exhibits spherical closed-cell structures which have the cell sizes ranging from 10 to 200 μm. The foamed conductive epoxy composite is composed of four phases, namely the solid polymer matrix, the MWCNTs, the microsphere shells, and the gaseous phase. The length of MWCNTs here is about 10–30 μm.
The similar improvements in conductivity are found in the conductive foams with different microsphere contents. The relationship between the conductivity and the microsphere contents is discussed in detail. As shown in Fig. 7, there are apparent declines in the resistivity of the foams when the microsphere contents increase from 0.5 to 2 wt% at every test MWCNT loading. For example the resistivity value of 0.5 wt% MWCNTs/epoxy foam drops from 5.14 × 106 Ω cm to 5.57 × 105 Ω cm, namely about 1 orders of magnitude. But when the microsphere contents are further increased from 3% to 5 wt%, the conductivities of foams deteriorate. Thus, the foams with 2 wt% of microspheres exhibit the best conductivity.
 | ||
| Fig. 7 Electrical resistivity of the conductive epoxy foams with different MWCNT loading as a function of microsphere contents. The samples studied here correspond to Group A1-(1–5), A2-(1–5), A3-(1–5), A4-(1–5) and A5-(1–5), respectively. | ||
Morphology study can be used to reveal what happens in the foaming process. The microstructures of samples with 1–4 wt% microspheres (A2-3, A3-3, A4-3, and A5-3) are respectively examined by SEM. The results in Fig. 8 show that the global cellular structures of the conductive foams deteriorate unacceptably and the dispersion of the MWCNTs in the cell walls becomes inhomogeneous as the dosage of the microsphere continuingly increases. In the foam composites with 1 wt% (a and e) microspheres, the MWCNTs exhibit definite non-uniform distribution. In the foam composites with 2 wt% (b and f) and 3 wt% (c and g) microspheres, it can be clearly observed that the MWCNTs are enriched in cell wall between two spheres. As we know, the foam with 2 wt% microspheres happens to show the best conductivity, these two phenomena must be interrelated. This forced enrichment of MWCNTs has led to build more conductive tunnels in the foams. In the foam composites with 4 wt% microspheres (d and h), huge porous structures and breakup cell walls are formed due to the over-foaming process. The disruption of cell structure in foams will damage the continuity of conductive pathway and cause conductivity deterioration.
 | ||
| Fig. 8 High magnification SEM images of the microstructure of MWCNTs/epoxy foams with different microsphere contents: (a and e) 1 wt%; (b and f) 2 wt%; (c and g) 3 wt%, and (d and h) 4 wt%. Images (a–d) are the global structures, and those in (c and d) are the local structures. The samples in image (a and e), (b and f), (c and g) and (d and h) correspond to Group A2-3, A3-3, A4-3, and A5-3, respectively. | ||
Previous works have proved that the dispersion in MWCNTs/polymer composites includes two aspects: (1) disentanglement of CNT bundles or agglomerates, which is so-called nanoscopic dispersion, and (2) uniform distribution of individual CNTs or their agglomerates throughout the composites, which is more of a micro and macroscopic dispersion.40 Based on the SEM graphs, it can be concluded that, during the gradual curing reaction of epoxy blends, the foaming process has changed the uniform distribution of individual CNTs or their agglomerates. We think this redistribution is induced by the resin flow due to the thermally triggered microsphere expansion. The reason could be the notable relative motion between the MWCNTs and the resin.
Fig. 9 shows the dependence of the steady shear viscosity with temperatures for the neat epoxy and the epoxy blends containing 1 wt%, 2 wt%, 3 wt% and 4 wt% MWCNTs. It can be observed that the viscosity of the epoxy blends at various temperatures all increases significantly due to the introduction of MWCNTs. For example, the viscosity of neat epoxy is 4.67 Pa s at 30 °C, while that of the 1 wt% MWCNTs/epoxy blend suddenly jumps to 71.70 Pa s, which is elevated 15 times. When the dosage of MWCNTs further increases to 4 wt%, the viscosity of the blend finally reaches 1178.87 Pa s, which is nearly 252 times higher than that of the neat epoxy. In the flow field, the entangled MWCNTs rub against the epoxy resin in rapid motion, which significantly deteriorates the fluidity of the whole blends. As a result, a progressive viscosity enhancement is observed. These data imply the strong resistance originated from the MWCNTs to the flowing resin. The aggregates of MWCNTs interact with each other to form interconnected networks and offer further resistance to the deformation and flow of blends.41,42 Additionally at 80 °C, the viscosity of neat epoxy, 1 wt% MWCNTs/epoxy blend and 4 wt% MWCNTs/epoxy blend are 0.113 Pa s, 7.15 Pa s and 75.16 Pa s, respectively. That is to say it has nearly a 70-fold increase in viscosity for the blend with 1 wt% MWCNTs and 750-fold increase for the blend with 4 wt% MWCNTs, illustrating that the contribution of MWCNTs to viscosity increase with the elevated temperature. In other words, the relative motion between the MWCNTs and the resin becomes more notable at high temperature than that at low temperature.
 | ||
| Fig. 9 The variation of steady shear viscosity with temperature for the neat epoxy resin and epoxy suspensions containing 1 wt%, 2 wt%, 3 wt% and 4 wt% of MWCNTs. | ||
The distribution of MWCNTs in the foams is described by a schematic diagram in Fig. 10. After heated, the microspheres begin to expand and meanwhile produce expansion forces near the external surface to extrude the surrounding MWCNTs/epoxy blend. At low dosage (a), the microspheres cannot enable sufficient flowing movement of the blends and will not do much for the distribution of MWCNTs. When the dosage of microspheres increases, the swelling process is too strong to be neglected. The blends surrounding the microsphere are forced to quickly move toward the other microsphere nearby. Early in solidification, the high-fluidness resin moves so faster that the entangled MWCNTs can't catch up. Consequently, the MWCNTs are left behind and “concentrated”. This phenomenon is vividly called a “flow-induced concentration effect” by us. Obviously, this concentration effect will be strong at the fast-flowing area in the blends. At the cell wall between any nearby expanding microspheres, the epoxy resin is pushed away quickly and few resins are left behind. So these concentration phenomena can be seen very clearly at the cell wall zone. At areas away from expanding microspheres, the flowing speed of the blend gradually declines and the MWCNTs maintain their location. This macroscopic redistribution of MWCNTs caused by the expanding microspheres throughout the nanocomposites is beneficial to increase the amount of effective conductive pathway. As the dosage of microspheres further increases (c), the matrix surrounding the microspheres is violently squeezed by the stronger swelling forces. The MWCNTs in per unit volume intensively decrease and the break-up of the cell-strut network has even occurred, which leads to the substantial declines in quantity of conductive pathway. It is not difficult to understand why the conductivity of the MWCNTs/epoxy foam has declined as the microsphere loading further increases.
 | ||
| Fig. 10 Schematic diagram for illustrating the “flow-induced concentration effect” that happens in the conductive MWCNTs/epoxy foams. | ||
Based on this explanation, it can be predicted that different prepolymerization extent and foaming temperatures can also change the conductivities of the foams as well. In Fig. 11a, the electrical resistivity of the foam is plotted against the prepolymerization time. The resistivity of the foam fluctuates around 105 Ω cm after pre-polymerizing for 80 min, and then has a small decrease within the next 10 min, illustrating that the conductivity of the foams is slightly improved. As the prepolymerization time further extends from 90 to 115 min, the resistivity increases sharply and jumps to about 1010 Ω cm, which is nearly 5 orders of magnitude higher than that of the foam with a 80 min of prepolymerization. The precursors with a long prepolymerization time have high viscosity. The high viscosity can prevent the expansion of microspheres and the flow of epoxy resin, which leads to nearly no concentration effect. The expansion of foam simply decreases the volume percentage of MWCNTs and causes the conductor–insulator transition. The resulted foam composite has a smaller conductivity even than the solid counterparts.
 | ||
| Fig. 11 Electrical resistivity of MWCNTs/epoxy foams as a function of: (a) prepolymerization time and (b) foaming temperatures. The samples studied in (a) and (b) correspond to Groups B(1–5)-1 and Groups C(1–5)-1, respectively. | ||
Variation in foaming temperatures can also create diverse cellular structures and consequently result in performance differences. In Fig. 11b, the resistivity of the foam is plotted against the foaming temperature. The samples studied in Fig. 11b correspond to Groups C(1–5)-1, respectively. Noteworthy, as the foaming temperature increases from 60 to 90 °C, the resistivity sharply jumps from about 7.38 × 107 to about 9.01 × 104 Ω cm. High foaming temperature will produce more expansion flow and cause stronger concentration effect. The further increase of foaming temperature fails to sustain such a desirable trend. A over-foaming leads to a minor increase in resistivity instead. Therefore, the optimum foaming temperature for the foam with a better conductivity is unquestionably located in 80–90 °C.
3.3 Mechanical properties
Compression strength is commonly used to evaluate the mechanical properties of MWCNTs/epoxy foams. The influence of prepolymerization time on the compression property is firstly discussed and shown in Fig. 12a. At early stage of prepolymerization, the compression strength fluctuates in the range of 2.49–4.59 MPa, and the compression modulus fluctuates in the range of 71.01–72.96 MPa. As the value of time is longer than 100 min, the compression properties increase rapidly along the prepolymerization time. Long prepolymerization time will significantly increase the epoxy resin viscosity and result in the formation of small bubbles and a higher density (Fig. S3, S6b, and Table S5†). In Fig. 12b, the compression properties of the foams gradually decline with the increasing foaming temperature in the temperature range below 100 °C which was caused by the gradual increase in cell size and decrease of foam density (Fig. S4, S6c, and Table S5†).26,43 The microsphere content shows a similar effect on the compression property. As shown in Fig. 12c, the MWCNTs/epoxy foam with 1 wt% microspheres exhibits the best compression properties including compression strength of 10.21 MPa and modulus of 184.0 MPa. It's easy to understand that high microsphere contents certainly form more voids and thinner cell walls in the matrix which consequently lower the densities (Fig. S5, S6d, and Table S5†).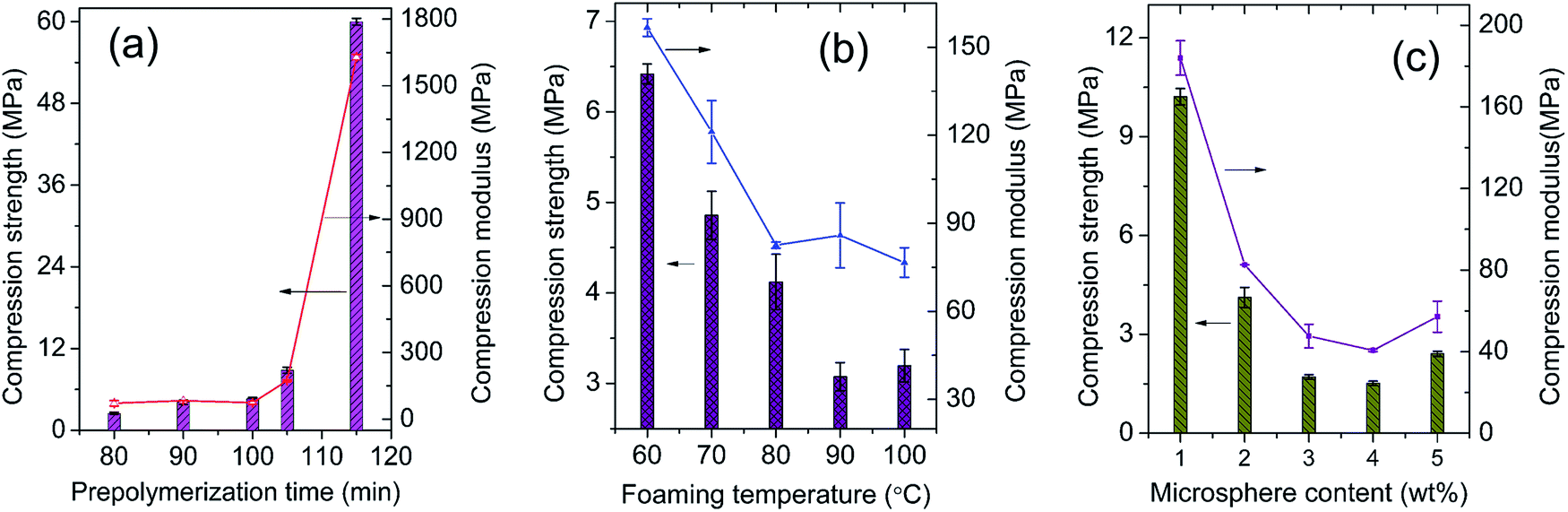 | ||
| Fig. 12 Effects on the compression properties of conductive MWCNTs/epoxy foams: (a) prepolymerization time, (b) microsphere contents, and (c) foaming temperature. The preparation formula of the foams is listed in Table 1. The samples in images (a and c) correspond to the Group A at 2 wt% MWCNTs, Group B(1–5)-2, and Group C(1–5)-2, respectively. | ||
Previous works have ever confirmed that there is a power-law relationship between the compression property and the foam density, which can be expressed as follows:
| υ = mρα | (4) |
| ξ = nρβ | (5) |
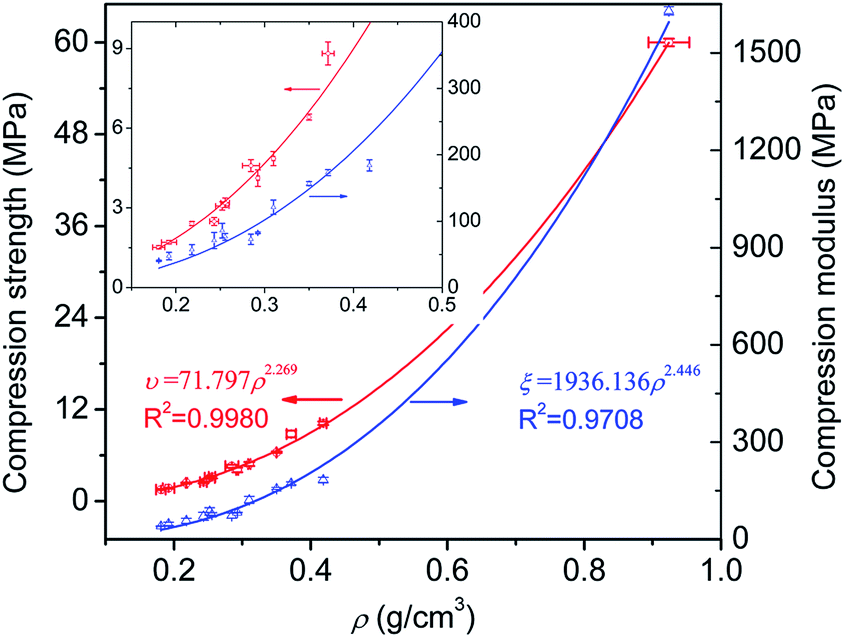 | ||
| Fig. 13 Power-law dependence of the compressive strength and modulus on foam densities. | ||
The MWCNT content can also affect the compression property (Fig. S7a†). The compression strength has a little decrease at MWCNT loading below 1 wt% and then raises quickly with further increase in MWCNT content. The rising viscosity caused by enhanced MWCNT loading results in small cell-structure, high foam density and increase of mechanical property. Different to those foams with same MWCNT content, the foams discussed here show no power-law dependence between compression properties and foam densities (Fig. S7b†). There are two reasons that can explain the changes in mechanical properties.45,46 One is that the agglomerates originated in the flow-induced high MWCNT concentration can cause cracks to initiate and propagate easily. The other is that the weak bonding between MWCNTs and the surrounding matrix causes the MWCNTs to be easily pulled out. In this work, the foaming process forces the MWCNTs to flow and agglomerate in the cell walls, which cause the decrease in compression properties of the foams with low MWCNT content (−1 wt%). But the enhanced viscosity due to the high MWCNT concentration results in more small cells and high foam densities (Fig. S2, S6a and Table S5†). The enhancement in compression properties caused by the growing foam density is larger than the decline caused by the agglomerates and weak bonding between the MWCNTs and the matrix.
Overall, the conductive epoxy foam with compression strength above 7 MPa can be easily obtained by controlling the prepolymerization time, microsphere contents, MWCNT dosages, and other preparation conditions. This light-weight and conductive foam can be used as an example to develop more foamed epoxy composites with high performance which will exhibit superiority in electronics, weaponry, aerospace, etc.
4. Conclusions
Conductive MWCNTs/epoxy foams using the expandable microsphere were prepared via a two-step foaming technique. The conductivity and mechanical property of the foams obtained by controlling different preparation parameters were investigated in detail. Based on the comprehensive analysis, the following conclusions are obtained:(1) Foamed epoxy/MWCNT composites can be perfectly prepared by a two-step technique using expandable microspheres. For the 1 wt% MWCNTs/epoxy foam with 2 wt% microspheres, the conductivity is about 6 orders of magnitude larger than that of solid counterparts. The foaming changes the percolation region of solid composite from 0.1–1.5 wt% to 0.1–0.5 wt% and finally improves the electrical conductivity.
(2) This enhancement in conductivity is ascribed to the flow-induced concentration effect. There is notable relative motion between MWCNTs and epoxy resin. During the resin flow caused by microsphere expansion, the MWCNTs are concentrated, especially in cell wall structure. Foaming changes the uniform redistribution of MWCNTs but improves the MWCNT inter-connectivity in the matrix.
(3) The optimum conditions to prepare conductive epoxy foams includes 90 min of prepolymerization, 1–4 wt% MWCNTs, 1–3 wt% microspheres and foaming at 80 °C. Insufficient prepolymerization, high dosages of microspheres and foaming temperatures all bring more break-ups and ruptured bubbles in the foams, which sacrifice the mechanical property and conductivity. Excessive prepolymerization, low dosages of microspheres or foaming temperatures cannot produce enough expansion forces to redistribute the MWCNTs, which cannot effectively promote the conductivity.
Acknowledgements
The authors highly appreciate the financial support provided by the National Natural Science Foundation of China (Grant No. 51573118).Notes and references
- H. Gao and K. Lian, RSC Adv., 2014, 4, 33091–33113 RSC.
- Y. Yang, X. Yang, W. Yang, S. Li, J. Xu and Y. Jiang, RSC Adv., 2014, 4, 42546–42553 RSC.
- L. Cao, H. Chen, M. Wang, J. Sun, X. Zhang and F. Kong, J. Phys. Chem. B, 2002, 106, 8971–8975 CrossRef CAS.
- Y. Xu, W. Xu and J. Bao, J. Polym. Res., 2014, 21, 1–8 CAS.
- Y. Xu, Y. Li, W. Xu and J. Bao, J. Mater. Sci.: Mater. Electron., 2015, 26, 1159–1171 CrossRef CAS.
- Y. Yang, M. C. Gupta, K. L. Dudley and R. W. Lawrence, Adv. Mater., 2005, 17, 1999–2003 CrossRef CAS.
- R. Rizvi, J.-K. Kim and H. Naguib, Smart Mater. Struct., 2009, 18, 104002 CrossRef.
- M. Antunes, M. Mudarra and J. I. Velasco, Carbon, 2011, 49, 708–717 CrossRef CAS.
- V. Eswaraiah, V. Sankaranarayanan and S. Ramaprabhu, Macromol. Mater. Eng., 2011, 296, 894–898 CrossRef CAS.
- H.-B. Zhang, Q. Yan, W.-G. Zheng, Z. He and Z.-Z. Yu, ACS Appl. Mater. Interfaces, 2011, 3, 918–924 CAS.
- D. Wu, Q. Lv, S. Feng, J. Chen, Y. Chen, Y. Qiu and X. Yao, Carbon, 2015, 95, 380–387 CrossRef CAS.
- J.-M. Thomassin, C. Pagnoulle, L. Bednarz, I. Huynen, R. Jerome and C. Detrembleur, J. Mater. Chem., 2008, 18, 792–796 RSC.
- X. Zhi, H.-B. Zhang, Y.-F. Liao, Q.-H. Hu, C.-X. Gui and Z.-Z. Yu, Carbon, 2015, 82, 195–204 CrossRef CAS.
- J. Ling, W. Zhai, W. Feng, B. Shen, J. Zhang and W. G. Zheng, ACS Appl. Mater. Interfaces, 2013, 5, 2677–2684 CAS.
- M. M. Bernal, I. Molenberg, S. Estravis, M. A. Rodriguez-Perez, I. Huynen, M. A. Lopez-Manchado and R. Verdejo, J. Mater. Sci., 2012, 47, 5673–5679 CrossRef CAS.
- D. X. Yan, K. Dai, Z. D. Xiang, Z. M. Li, X. Ji and W. Q. Zhang, J. Appl. Polym. Sci., 2011, 120, 3014–3019 CrossRef CAS.
- D. Chen, J. Yang and G. Chen, Composites, Part A, 2010, 41, 1636–1638 CrossRef.
- X. B. Xu, Z. M. Li, L. Shi, X. C. Bian and Z. D. Xiang, Small, 2007, 3, 408–411 CrossRef CAS PubMed.
- M. Antunes and J. I. Velasco, Prog. Polym. Sci., 2014, 39, 486–509 CrossRef CAS.
- H. H. Chen, US Pat., 3025249, 1962.
- X. Wu, Y. Wang, X. Yang, J. Yu, L. Wang, S. Hou and P. Jiang, RSC Adv., 2015, 5, 61204–61217 RSC.
- G. Sui, X. Li, M. Zhu, X. Tang, Q. Zhang and X. Yang, RSC Adv., 2015, 63, 50919–50928 Search PubMed.
- L. Wang, J. Zhang, X. Yang, C. Zhang, W. Gong and J. Yu, Mater. Des., 2014, 55, 929–936 CrossRef CAS.
- R. Huang and P. Li, Composites, Part B, 2015, 78, 401–408 CrossRef CAS.
- D.-X. Yan, P.-G. Ren, H. Pang, Q. Fu, M.-B. Yang and Z.-M. Li, J. Mater. Chem., 2012, 22, 18772–18774 RSC.
- L. Wang, X. Yang, J. Zhang, C. Zhang and L. He, Composites, Part B, 2014, 56, 724–732 CrossRef CAS.
- R. Erb, H. J. Kim and M. Grott, US Pat., 7073277, 2006.
- J. N. Biancella, J. M. Biancella and K. M. Szilagyi, US Pat., 8367176, 2013.
- D. K. Robinson, J. J. Erickson and M. Redwood, US Pat., 6179879, 2001.
- W. E. Cohrs and R. E. Gunderman, US Pat., 4108806, 1978.
- L. Malmbom and L. Lysell, US Pat., 5629364, 1997.
- A. Nakamura and Y. C. Tsuji, US Pat., 5246973, 1993.
- A. Ameli, P. Jung and C. Park, Carbon, 2013, 60, 379–391 CrossRef CAS.
- I. D. Rosca and S. V. Hoa, Carbon, 2009, 47, 1958–1968 CrossRef CAS.
- K. Zhao, J. Wang, X. Song, C. Liang and S. Xu, Thermochim. Acta, 2015, 605, 8–15 CrossRef CAS.
- M. Yin, J. Koutsky, T. Barr, N. Rodriguez, R. Baker and L. Klebanov, Chem. Mater., 1993, 5, 1024–1031 CrossRef CAS.
- D. Puglia, L. Valentini, I. Armentano and J. Kenny, Diamond Relat. Mater., 2003, 12, 827–832 CrossRef CAS.
- L. Guadagno, L. Vertuccio, A. Sorrentino, M. Raimondo, C. Naddeo, V. Vittoria, G. Iannuzzo, E. Calvi and S. Russo, Carbon, 2009, 47, 2419–2430 CrossRef CAS.
- G. Hu, C. Zhao, S. Zhang, M. Yang and Z. Wang, Polymer, 2006, 47, 480–488 CrossRef CAS.
- J. Li, P. C. Ma, W. S. Chow, C. K. To, B. Z. Tang and J. K. Kim, Adv. Funct. Mater., 2007, 17, 3207–3215 CrossRef CAS.
- S. Rahatekar, K. Koziol, S. Butler, J. Elliott, M. Shaffer, M. Mackley and A. Windle, J. Rheol., 2006, 50, 599–610 CrossRef CAS.
- Z. Fan and S. G. Advani, J. Rheol., 2007, 51, 585–604 CrossRef CAS.
- N. Gupta, R. Ye and M. Porfiri, Composites, Part B, 2010, 41, 236–245 CrossRef.
- Q. Li, L. Chen, X. Li, J. Zhang, X. Zhang, K. Zheng, F. Fang, H. Zhou and X. Tian, Composites, Part A, 2016, 82, 214–225 CrossRef CAS.
- Y. S. Song and J. R. Youn, Carbon, 2005, 43, 1378–1385 CrossRef CAS.
- X. Chen, L. Zhang, M. Zheng, C. Park, X. Wang and C. Ke, Carbon, 2015, 82, 214–228 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra02310j |
| This journal is © The Royal Society of Chemistry 2016 |