
A new cost-effective Ru-chloramphenicol base derivative catalyst for the asymmetric transfer hydrogenation/dynamic kinetic resolution of N-Boc α-amino-β-ketoesters and its application to the synthesis of the chiral core of vancomycin†
Abstract
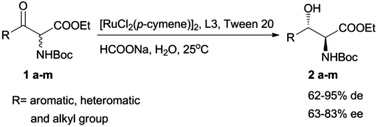
Herein we describe the application of a series of newly developed Ru-chloramphenicol base derivative complexes as catalysts for the highly diastereo- and enantioselective transfer hydrogenation of N-Boc α-amino-β-ketoesters for the asymmetric synthesis of anti-N-Boc-β-hydroxy-α-amino esters. This report highlights the utility of this catalytic methodology for the preparation of pharmaceutical compounds bearing a N-Boc α-amino-β-hydroxy substructure with two stereocenters.
 Please wait while we load your content...
Please wait while we load your content...

