
Recent development of unimolecular micelles as functional materials and applications
Xiaoshan
Fan
a,
Zibiao
Li
*b and
Xian Jun
Loh
*bcd
aSchool of Chemistry and Chemical Engineering, Henan Normal University, China
bInstitute of Materials Research and Engineering (IMRE), A*STAR, 2 Fusionopolis Way, Innovis, #08-03, 138634, Singapore. E-mail: lizb@imre.a-star.edu.sg
cDepartment of Materials Science and Engineering, National University of Singapore, 9 Engineering Drive 1, Singapore 117576, Singapore
dSingapore Eye Research Institute, 11 Third Hospital Avenue, Singapore 168751, Singapore. E-mail: lohxj@imre.a-star.edu.sg; Tel: +65-65131612
First published on 5th September 2016
Abstract
Unimolecular micelles have attracted increasing attention due to their high functionality, encapsulation and site specific confinement capabilities in various applications. Compared to conventional supramolecular micelles, unimolecular micelles possess unique single molecular architectures which can maintain excellent stability when they are subjected to extreme surrounding environment changes such as high dilution and alterations in temperature, pH, ionic strength etc. In this review, the most recent advances in the design strategies of unimolecular micelles are presented with respect to different types of architectural polymers, including dendrimers, and hyperbranched, dendritic, star, brush-like and amphiphilic cyclic polymers. The diverse functions of these sophisticated materials endow a biosignificance in therapeutic agent delivery. And the use of unimolecular micelles as templates for inorganic nanoparticle preparation, catalysis, and energy harvesting are also summarized in this review. Finally, the challenges for the facile fabrication of unimolecular micelles and future perspectives are also discussed.
 Xiaoshan Fan | Xiaoshan Fan obtained his Ph.D. degree in the Department of Macromolecular Science at Fudan University in China in 2012 on the subject of the design and synthesis of polymers with complex architectures. In September of the same year, he went to Singapore and worked as a post-doctoral researcher in the National University of Singapore. He came back to China in 2015 and joined the School of Chemistry and Chemical Engineering of Henan Normal University. His current research interests focus on the synthesis of polymers and their applications in different areas. |
 Zibiao Li | Dr Zibiao Li obtained his Ph.D. in 2014 from the National University of Singapore. Currently, he works as a research scientist at the Institute of Materials Research and Engineering, A*STAR, Singapore. His research interests are focused on biodegradable and functional polymeric materials design, structural properties investigations and their hybrid fabrications for biomedical and consumer care applications. |
 Xian Jun Loh | Dr Xian Jun Loh is a polymer chemist working in the inter-disciplinary field of biomaterials. He is currently the Programme Manager of the A*STAR Personal Care Programme. He is concurrently the Programme Manager of the Consumer Care Programme at the Institute of Materials Research and Engineering (IMRE) and an Assistant Professor at the National University of Singapore (NUS). He is also an adjunct scientist at the Singapore Eye Research Institute. His main research interests are in the design of supramolecular and stimuli-responsive polymers and hydrogels for biomedical and personal care applications. Currently, he is the author and co-author of 121 journal papers, 14 patents, 20 book chapters and 4 books, publishing mainly in the area of biomaterials. |
1. Introduction
Amphiphiles can self-assemble into nano-sized micelles, also known as multimolecular micelles, of various morphologies in aqueous solution.1–16 During the past few decades, micelles have attracted tremendous attention owing to their applications in drug delivery, tissue engineering, diagnostic imaging, catalysis etc.17–25 However, conventional polymeric micelles represent thermodynamic aggregations of multiamphiphilic macromolecules above their critical micelle concentration (CMC). When these polymeric micelles are subjected to high dilution and alterations in other factors such as temperature, pH, and ionic strength, they disassemble into free polymeric chains.26,27 This inevitable drawback has heavily hindered their applications in different fields.1,28–32To overcome the thermodynamic instability issue of polymeric micelles, core and/or shell cross-linking approaches have been proposed. The crosslinked core–shell particles are often obtained by a two-step emulsion polymerization to result in the chemically stabilized micelles.33–37 To date, a library of core and/or shell cross-linked micelles with different compositions and architectures have been reported in the literature. For example, Leroux and co-workers recently reported core cross-linked micelles from eight-arm star-shaped polymers using a cellobiose-derived initiator.38 In this study, star polymers poly(glycidyl methacrylate) (PGMA) were firstly synthesized by ATRP, followed by hydrolysis to yield hydroxyl-rich PGOHMA. The partial cross-linking of these –OH groups allowed for the preparation of the stabilized micelles. In another study, Gong and his co-workers fabricated cross-linked nanovesicles from amphiphilic ABA triblock copolymers and embedded drugs into the hydrophobic bilayers for targeted therapeutic delivery.39,40 Shell and/or core cross-linking endows polymeric micelles with excellent structural stability. However, their biodegradability or drug release profiles are compromised after crosslinking.37,41,42 From certain respects, the crosslinked core–shell particles can be also considered as single macromolecules. However, this is beyond the scope of the current review since the stabilized particle structures are based on the aggregation of multiple polymer chains before cross-linking.
In addition to the cross-linking approach, design of unimolecular micelles provides an alternative strategy and opportunity to prepare stable polymeric micelles.43,44 Unimolecular micelles are defined as a class of single-molecule micelles with a distinct core and shell that are covalently bound together.45 Due to their unique architecture, unimolecular micelles show excellent stability regardless of the high dilution condition and other microenvironment changes, making them particularly attractive for the design of stable micelles for specific applications (Fig. 1).46 This review presents the most recent progress in the development of unimolecular micelles from various types of architectural materials. To highlight the structural uniqueness of these sophisticated materials, the diverse applications are summarized in three active domains of therapeutic agent delivery and catalysis and the use of unimolecular micelles as templates for the preparation of inorganic nanoparticles.
 | ||
| Fig. 1 Different behaviors of unimolecular and multimolecular micelles under dilution. Unimolecular micelles are stable and multimolecular micelles can fall apart. Reproduced with permission from American Chemical Society.46 | ||
2. Design strategies of unimolecular micelles
Unimolecular micelles are covalently bound molecular architectures that can be made from a variety of amphiphilic polymers. In this section, various types of architectural materials that have been applied in the fabrication of unimolecular micelles will be reviewed and the structural uniqueness mediated specific properties and functionalities will also be highlighted.2.1 Unimolecular micelles from amphiphilic dendrimers
Dendrimers are highly branched macromolecules that exhibit regular branching and structural symmetry. The presence of multiple terminal groups on the surface makes this type of monodisperse and globular polymers attractive in the design of unimolecular micelles to achieve high functionality and reactivity. In addition, the nanometer dimensions, discrete size and tailored structure of dendrimers have led to a wide range of diversified applications in scaffolds for drug delivery,47–55 carriers for gene transfection,56–60 biosensors61,62 and catalytic “nanoreactors”.23,63–67 Amphiphilic dendrimers, in addition to their characteristic dendritic properties, also possess different regions of contrasting polarity that can be easily used for unimolecular micelle fabrication. Unimolecular micelles made from amphiphilic dendrimers can maintain excellent stability under extremely high dilution. This unique property has made amphiphilic dendrimers appealing for many drug delivery applications because the hydrodynamic volumes are typically small enough to prevent accumulation in the spleen, liver and elsewhere yet large enough to slow renal filtration.68 Dendrimers can be synthesized through divergent or convergent approaches. In 1993, Fréchet and co-workers reported the first synthesis of amphiphilic dendrimers using a convergent approach.69 In this study, dendritic polyether macromolecules based on 3,5-dihydroxybenzyl alcohol building blocks were prepared and the presence of carboxyl end groups endowed good water-solubility to the as-formed unimolecular micelles (Fig. 2). When the resultant dendrimers were mixed with hydrophobic drugs, the drug concentration exhibited a linear relationship with the concentration of dendrimers even at very low concentrations of dendrimers, indicating the strong drug encapsulation capability of amphiphilic dendrimers. Since then, a large variety of amphiphilic dendrimers have been synthesized and used as hosts for encapsulation and delivery of different small drug molecules.70–72 As a typical example highlighted here, Meijer and his co-workers demonstrated a unique method for the preparation of drug-loading amphiphilic dendrimers consisting of an inner fifth generation poly(propylene imine).70,72 In this regard, the hydrophilic dye Rose Bengal was used as a guest molecule and sequestered into the internal cavities of the dendrimer. The drugs were then sealed by a rigid and bulky shell formed by the attachment of t-butoxycarbonyl (t-BOC)-protected phenylalanine groups onto each of the 64 chain ends. The following acid hydrolysis of the amino acid end groups could regenerate the “open shell” to programme the drug release in a controlled manner (Fig. 3).70,71 | ||
| Fig. 2 Structure of water-soluble unimolecular micelles made from dendritic polyether. Reproduced with permission from Royal Society of Chemistry.69 | ||
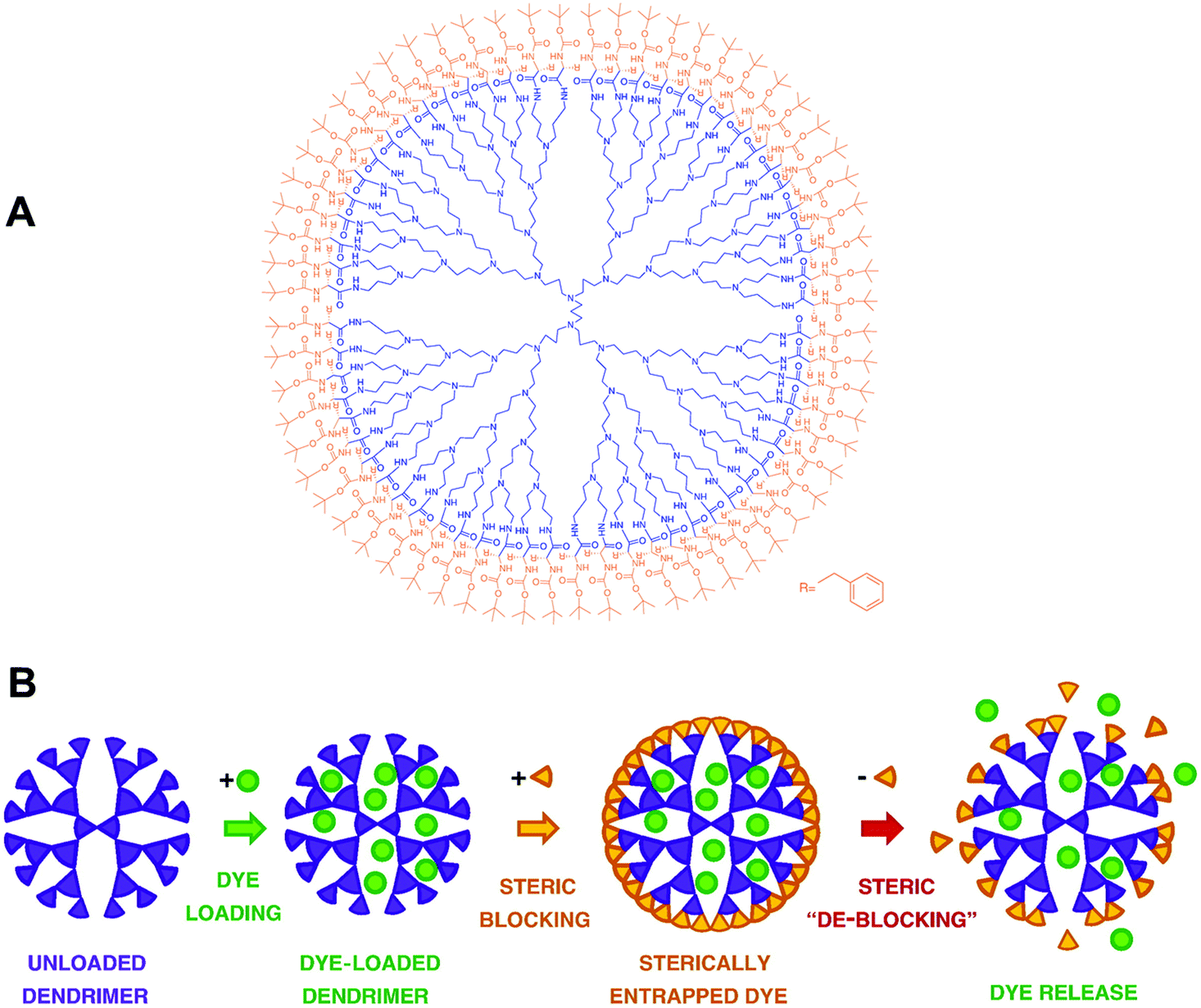 | ||
| Fig. 3 (A) Structure of the dendritic box with an inner fifth generation poly(propylene imine) dendrimer functionalized with t-BOC protected phenylalanine. (B) Schematic illustration showing the payload entrapment and release by modulation of the surface blocking groups in dendritic box. Reproduced with permission from American Association for the Advancement of Science and Elsevier.70,71 | ||
These studies represent the early concept of unimolecular micelle fabrication from dendrimers, in which the outer shell of the dendrimers consists of a single type of functional component or steric blocking group. Recently, dendrimers containing multiple distinct polymer chains have attracted tremendous interest in the fabrication of unimolecular micelles due to their versatile structural complexity and superior properties. Among these, poly(ethylene glycol) (PEG) was widely employed as a hydrophilic component in the outer shell (so-called PEGylation) of dendrimers to increase the water solubility and biocompatibility of the formed unimolecular micelles.73,74 In addition, biodegradable polyesters are extensively used as the dendrimer core, which can be further used as a reservoir to encapsulate hydrophobic drug molecules. More importantly, polyesters can be degraded into small molecules through hydrolysis and the degradation products can be easily excluded from the body at the end of its functional life.2,24,25,75–80 In one example, Shen et al. recently reported an efficient synthesis approach of aliphatic polyester derived dendrimers via the combination of a thiol/acrylate type click reaction and esterification. With the further endowed PEGylation effect, the obtained water-soluble and biocompatible unimolecular micelles exhibited a strong capability in encapsulation and controlled release of a hydrophobic anticancer drug (Fig. 4).81
 | ||
| Fig. 4 PEGylation of a fifth generation polyester dendrimer and the unimolecular micelles fabricated as drug carriers. Reproduced with permission from American Chemical Society.81 | ||
2.2 Unimolecular micelles from amphiphilic hyperbranched polymers
Hyperbranched polymers are another type of materials that have been widely used for unimolecular micelle preparation. In contrast to the multi-step synthesis of precisely defined dendrimers, hyperbranched polymers can be synthesized in an easier approach, such as a convenient one-step synthesis on a large scale and proper yields with high purities. Feasible techniques including polycondensation of ABx monomers, self-condensation vinyl polymerization, ring-opening polymerization of latent ABx type cyclic monomers and copolymerization of two complementary monomers have been reported for the successful synthesis of different hyperbranched polymers.82–87 Similar to dendrimers, strategies developed for preparing amphiphilic hyperbranched polymers include the incorporation of hydrophobic components into a hydrophilic scaffold and modifying the scaffold by attaching different functional polymer arms onto the surface.88,89 For example, Emrick et al. reported the synthesis of amphiphilic hyperbranched polymers consisting of a branched polyphenylene backbone and carboxylic acid end groups. The obtained polymers could form unimolecular micelles in basic aqueous medium due to the presence of multiple carboxylates on the periphery.90On the other hand, attaching polymer arms onto a hyperbranched core is another efficient approach to yield amphiphilic hyperbranched polymers. Because of the easy control of polymer size and functionality, hyperbranched polymers produced by this method have recently received significant attention in the design of functional unimolecular micelles.91–93 For example, Song and co-workers recently reported an amphiphilic hyperbranched polymer, H40-PLLA-block-mPEG, consisting of a Boltorn H40 (H40) core, an inner poly(L-lactide) shell and an outer PEG shell. Aliphatic polyester H40 was selected as the core of a hyperbranched polymer due to its good biodegradability and biocompatibility. A DLS study showed that the obtained amphiphilic H40-PLLA-block-mPEG formed unimolecular micelles with hydrophobic H40-PLA as the micelle core and hydrophilic MPEG as the shell. The diameter of the unimolecular micelles was in the range of 11–17 nm.91 The same research group later went on to develop a Folate-conjugated amphiphilic hyperbranched block copolymer (H40-PLA-b-MPEG/PEG-FA) and used it as a carrier for tumor-targeted drug delivery.92 Cellular uptake and cytotoxicity studies showed that DOX-loaded H40-PLA-b-MPEG/PEG-FA micelles had a greater cellular uptake when compared to DOX-loaded H40-PLAb-MPEG micelles, indicating enhanced cytotoxicity against 4T1 tumor cells. Recently, Malmström et al. reported the fabrication of an intelligent unimolecular micelle system from amphiphilic hyperbranched dendritic-linear polymers (HBDLPs) (Fig. 5).94 In this regard, the high molecular weight and core–shell structured HBDLPs were synthesized through a combination of self-condensing vinyl copolymerization (SCVCP) and atom transfer radical polymerization (ATRP). Cleavable disulfide bonds were also introduced either in the backbone or in pendant groups of the hyperbranched core of the HBDLPs to make the materials sensitive to Redox. By triggered reductive degradation, the HBDLPs showed up to 7-fold decrease in molecular weight and this could further result in an instant release of the conjugated drugs.
 | ||
| Fig. 5 Schematic illustration of the synthetic route to the unimolecular micelles with (a) backbone-cleavable disulfide bonds and (b) azide-functional cleavable pendant disulfide bonds. Reproduced with permission from American Chemical Society.94 | ||
2.3 Unimolecular micelles from amphiphilic dendrimer-like polymers
Dendrimer-like are a new class of macromolecules built from narrowly polydipersed polymer chains, which is different from the construction of dendrimers using different types of monomers.95 In addition, the dendrimer-like are superior to dendrimers in terms of synthesis feasibility and structural accuracy control in functional materials.96 For example, a few generations of dendrimer-like can offer a 3D highly branched globular structure with comparable properties to the dendrimer analogy. Also, the particle size of dendritic polymer mediated self-assembly can be easily controlled by tuning the length of polymeric building blocks. Moreover, the flexible interior cavities in a dendrimer-like core can significantly improve the drug-loading capacity compared with rigid cores in dendrimers. In recent years, although amphiphilic dendrimer-like have attracted increasing attention, studies on using this type of materials as unimolecular micelles and applications have not been extensively reported. For instance, Hirao and He's groups have recently done some excellent studies on the design consideration and synthesis of amphiphilic dendrimer-like polymers, yet understanding the self-assembling properties in aqueous solution was not explored.97–100 Recently, our group reported the synthesis of a well-defined amphiphilic dendritic copolymer, POSS-(G3-PLLA-b-PEO-COOH)8, with a hydrophobic third generation dendritic PLLA core and a hydrophilic PEO shell functionalized with carboxylic groups (Fig. 6).101 The DLS and TEM results revealed that POSS-(G3-PLLA-b-PEO-COOH)8 existed as stable core–shell unimolecular micelles in aqueous solution with a uniform size distribution. The mean size was in the range of 99.9–102.5 nm. The in vitro release profile indicates that DOX encapsulated within the unimolecular micelles was released over a sustained time period in a slow and steady release manner. The POSS-(G3-PLLA-b-PEO-COOH)8 unimolecular micelles can be considered as promising candidates for controlled hydrophobic cancer drug delivery due to their biocompatibility and biodegradability, excellent stability and controlled release ability.101 | ||
| Fig. 6 Schematic illustration of the synthetic route to the core–shell structural amphiphilic copolymer POSS-(G3-PLLA-b-PEO-COOH)8 and the unimolecular micelle formation formed from the copolymer. Reproduced with permission from Royal Society of Chemistry.101 | ||
In a similar approach, we recently demonstrated the fabrication of unimolecular micelles that possess a pH-induced “breathing” feature (Fig. 7).102 In this study, a hybrid copolymer, POSS-(PAA-(PLLA-PEG)4)8, in which eight linear-dendritic like arms poly(acrylic acid)-(poly(L-lactide)-poly(ethylene glycol))4 (PAA-(PLLA-PEG)4) are grafted onto an oligomericsilsequioxane core, was synthesized. The unimolecualr micelles were made from POSS-(PAA-(PLLA-PEG)4)8 copolymers in aqueous solution, which was composed of a biocompatible PEG outer corona, a biodegradable hydrophobic PLA layer in the middle and inner hydrophilic PAA cavities. Interestingly, the formed micelles can “breathe”, namely, the size of the micelles changes as pH values vary. The unique architecture and features make this novel amphiphilic dendritic polymer a promising material in controlled drug delivery due to its ability for encapsulation of both hydrophobic and hydrophilic drugs.102
 | ||
| Fig. 7 Unimolecular micelle formed from the hybrid POSS-(PAA-(PLLA-OH)4)8 copolymer and its pH-dependent size change behavior. Reproduced with permission from Royal Society of Chemistry.102 | ||
2.4 Unimolecular micelles from amphiphilic star polymers
Star polymers are a kind of macromolecules containing a central core from which multiple chains emanate.103–105 Due to the facile synthesis, tunable feature and accessible core cavity, well-defined amphiphilic star polymers have also been used as alternative materials for unimolecular micelle fabrication and their applications in a range of fields have been explored. Typically, star polymers can be synthesized via three main strategies: (1) core-first,106 (2) coupling onto,107 and (3) arm-first.108 Specifically, amphiphilic star polymers are further classified into two categories by considering the different compositions of the arms, i.e., homoarm amphiphilic star polymers and miktoarm amphiphilic star polymers.109 Homoarm amphiphilic star polymers consist of arms with similar molecular weights and identical chemical composition, in which the structures of the arm could be linear, rod-coil or linear-dendritic like (Fig. 8). In contrast, miktoarm amphiphilic star polymers have two or more arm species with different chemical compositions and/or molecular weights. | ||
| Fig. 8 Categories of amphiphilic star polymers: (A) homoarm star polymers with block copolymer arms; (B) homoarm star polymers with rod-coil like arms; (C) homoarm star polymers with linear-dendritic like arms; (D) miktoarm star polymers with two types of homopolymer arms. | ||
Amphiphilic star block copolymers of vinyl ethers were first reported by Kanaoka via living cationic polymerization, where the arm chain consists of hydrophilic polyalcohol and hydrophobic poly(t-butyl vinyl ether) segments.110 The solubility characteristics of these polymers are primarily dependent on the properties of the outer segments and showed a clear difference from those of the corresponding linear block copolymers. During the past few decades, the rapid development of living polymerizations combined with other synthetic techniques has diversified a library of amphiphilic star block copolymers with different compositions and complex structures.111–113 For example, Pang et al. recently reported the synthesis of an amphiphilic 21-arm star block copolymer, poly(acrylicacid)-b-polystyrene (PAA-b-PS), by a sequential ATRP of t-butyl acrylate and styrene using 21Br-β-CD as a macroinitiator (Fig. 9). The 21 substitutable hydroxyl groups on the outer surface of β-CD could provide the capability of making a core with 21 initiation sites to form 21-arm, star-like block copolymers. The well-defined star-like PAA-b-PS diblock copolymers were composed of hydrophilic PAA blocks as the core and hydrophobic PS blocks as the shell with a narrow molecular weight distribution and good control of the molecular weight of each block.114 The unimolecular micelles formed from amphiphilic star-like PAA-b-PS had an average diameter of 19 nm and possessed monodisperse size distribution. With the increase of the molecular weight of PAA-b-PS, the average diameter of unimolecular micelles increased to ∼65 nm, indicating a tunable micelle size through the control of the compositions of amphiphilic star block copolymers.114
 | ||
| Fig. 9 Schematic representation of the synthetic route to a novel amphiphilic 21-arm, star-like diblock copolymer PAA-b-PS by sequential ATRP and its unimolecular micelle structure in aqueous solution. Reproduced with permission from American Chemical Society.114 | ||
In another aspect, amphiphilic star polymers with miktoarm can be prepared by attaching different types of polymer arms with desired functionalities to the same core. For example, Tsitsilianis et al. reported the synthesis of a series of amphiphilic miktoarm star polymers consisting of differing numbers of poly(ethylene oxide) (PEO) and polystyrene (PS) arms by sequential anionic living copolymerization.115 The obtained results showed that the functionality of the star copolymers was influenced mainly by the molar ratio of divinyl benzene per living end and the molecular weight of the linear PS precursor. The amphiphilic behavior of the star-shaped copolymers also exhibited strong association phenomena in both water and THF solution. Recently, Wang and co-workers synthesized a series of amphiphilic A4B4miktoarmstar polymers by mechanisms transformation combining with a thiol–ene reaction.90,116 In this design, to avoid the introduction of two types of initiating sites simultaneously, the authors introduced the first class of active hydroxyl groups by designing an amikto-initiator containing the same number of active hydroxyl groups and allyl groups, and then the second class of active hydroxyl groups at core position was introduced by transformation of relative inert allyl groups through thiol–ene reaction. Hydrophilic arms poly(ethylene oxide) and hydrophobic arms poly(ε-caprolactone) (PCL), polystyrene (PS) or poly(tert-butyl acrylate) were selected to prepare amphiphilic A4B4 star-shaped copolymers (PEO)4(PCL)4, (PEO)4(PS)4, and (PEO)4(PtBA)4, respectively. In addition, amphiphilic dendrimer-like star polymers are another special kind of star polymers which consist of a star-shaped core and a dendron shell. The development of dendrimer-like star polymers has shown superior properties in the combination of rapid synthesis of star polymers with large size and multiple peripheral functional groups of dendrimers. As a typical example highlighted here, Zhu et al. recently reported the synthesis of functionalized amphiphilic dendrimer-like star polymers with a hydrophobic star-shaped poly(L-lactide) (PLLA) core and a hydrophilic poly(amidoamine) (PAMAM) dendron shell (Fig. 10).117 During the synthesis, a carboxylic acid-functionalized PLLA star polymer was first obtained by the ROP of L-lactide followed by functionalization with succinic anhydride. 1-, 2-, and 3-generation PAMAM dendrons with a primary amine at the dendron root and benzyl ester protections at the periphery were then prepared via a divergent method. Through amide coupling between the carboxylic acid-terminated PLLA star polymer and six PAMAM dendrons, amphiphilic DLSPs were successfully synthesized. To further enhance the bioactivity and bioconjugation capability, different functional groups including carboxylic acid, primary amine, and triethylene glycol functional groups were introduced onto the surface of the PAMAM shell, respectively. The nature of different surface groups could result in large differences in thermal behaviors. The functionalized amphiphilic dendrimer-like star polymers exhibited a unique unimolecular micelle (14–28 nm) behavior in aqueous solution and could greatly enhance the water solubility of a hydrophobic DOX from 0.0625 up to 0.272 mg mL−1, indicating their potential use in controlled hydrophobic drug delivery.117
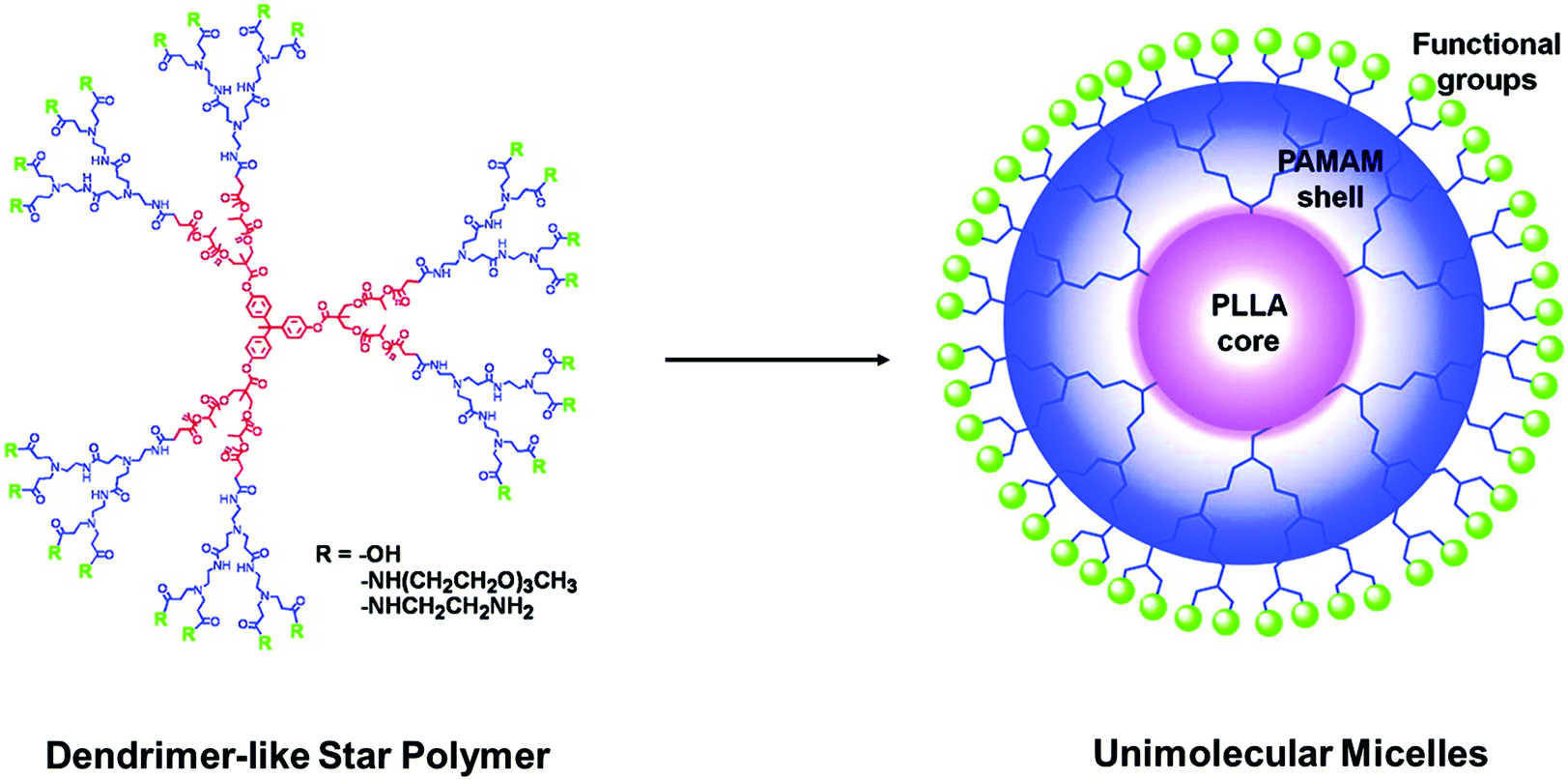 | ||
| Fig. 10 Structures of the amphiphilic dendrimer-like star polymer with a hydrophobic star-shaped poly(L-lactide) (PLLA) core and a hydrophilic poly(amidoamine) (PAMAM) dendron shell and the formed unimolecular micelles. Reproduced with permission from American Chemical Society.117 | ||
2.5 Unimolecular micelles from other types of amphiphilic polymers
Amphiphilic brush-shaped and cyclic polymers have also been used in the fabrication of stable unimolecular micelles. Brush-shaped polymers are a special category of synthetic macromolecules that contain multiple side chains grafting from a backbone, including star-graft,118,119 block-graft,120–125 V-shaped graft,126,127 heterograft and heterograft block structure.128–130 The three main strategies utilized to synthesize amphiphilic graft copolymers are “grafting through”, “grafting onto”, and “grafting from”.24,25,131 Despite the wide use of core–shell unimolecular micelles in many applications, unimolecular micelle formation from amphiphilic brush-shaped polymers has not been extensively explored. Recently, the investigation of brush-shaped unimolecular micelles has begun to attract increasing interest from researchers. In one example, Gong and co-workers reported multifunctional unimolecular micelles based on a brush-shaped amphiphilic block copolymer for both tumor-targeted drug delivery and noninvasive PET imaging (Fig. 11).132 The backbone of the brush-shaped amphiphilic block copolymer was poly(2-hydroxyethyl methacrylate) (PHEMA) and the side chains were poly(L-lactide)-poly(ethylene glycol) (PLLA-PEG). The results demonstrated that DOX-loaded micelles exhibited a uniform size distribution and the release of DOX from the unimolecular micelles was over a sustained time period in a pH-dependent manner. When CD105-targeting antibodies (TRC105) were conjugated onto unimolecular micelles, the cellular uptake of the unimolecular micelles was much higher in CD105-positive cells than that in non-targeted micelles. A much higher level of tumor accumulation was also demonstrated in 4T1 murine breast tumor-bearing mice treated with 64Cu-labeled targeted micelles when compared with those treated with non-targeted ones based on the PET imaging and bio-distribution studies. These multifunctional tumor-targeting unimolecular micelles with pH-controllable drug release profiles and PET imaging capability are promising drug/agent nanocarriers for targeted cancer theranostics.132 | ||
| Fig. 11 Schematic illustration of the multifunctional unimolecular micelles fabricated from brush-shaped amphiphilic block copolymers for cancer therapy. Reproduced with permission from American Chemical Society.132 | ||
In addition to amphiphilic brush-shaped polymers, unimolecular micelles formed from cyclic polymers are also drawing increasing attention. The “endless” topology and confined conformational freedom afford cyclic polymers a number of different properties in both solution and bulk when compared with their linear counterparts, including reduced viscosity and glass transition temperatures, lower hydrodynamic volume and higher refractive index.133–135 In addition, the cyclic conformation also demonstrated increased circulation lifetimes for soluble drug carriers in the bloodstream and improved targeting to tumor tissue.136 During the few past decades, the fast development of living polymerization techniques and emergence of highly efficient coupling reactions have provided more approaches to synthesize cyclic polymers and lined up with a platform to investigate their different applications. For example, some single macromolecular amphiphilic cyclic polymers such as “sun-shaped” and dendronized cyclic polymers have been previously reported (Fig. 12). In one particular example, Huang and his co-workers reported a “sun-shaped” amphiphilic polymer consisting of a hydrophilic PEO ring and PS lateral chains (c-PEO-g-PS).137 Subsequent studies demonstrated that micelles of the amphiphilic cyclic polymer c-PEO-g-PS can be used as a carrier to efficiently transfer dye molecules from the water to organic phase.
 | ||
| Fig. 12 Illustration of different single amphiphilic cyclic polymers (A) grafted with homoarms, (B) grafted with block arms and (C) grafted with dendritic arms. | ||
The intriguing architecture also brought some novel characteristics to amphiphilic cyclic polymers. Recently, Williams et al. reported the synthesis of cyclic amphiphilic graft copolymers with a hydrophobic polycarbonate backbone and hydrophilic poly(N-acryloylmorpholine) (PNAM) side arms via a combination of ROP, cyclization via copper-catalyzed azide–alkyne cycloaddition (CuAAC), and reversible addition–fragmentation chain transfer (RAFT) polymerization (Fig. 13).138 The obtained cyclic graft copolymers were able to form unimolecular micelles when dispersed in water and the as-formed particles size could be finely tuned by the variation of PNAM arm length. However, significant differences in solution conformations, loading capabilities and morphologies of cyclic graft copolymers were observed in comparison with linear graft copolymer analogues. For example, at short PNAM arm lengths, cyclic graft copolymers exhibited larger particle dimensions and greater loading capacities than the equivalent linear graft copolymers. When the PNAM arm length was increased, the assemblies of cyclic and linear graft copolymer particles were observed in different morphologies, i.e., the cyclic graft copolymer particles switched from a spherical to a cylindrical conformation as the PNAM arm length increased whereas the linear graft copolymer particles remained spherical. Moreover, investigation of the thermo-responsive properties of the graft copolymers showed that the linear graft copolymer exhibited a cloud point temperature of 47 °C, whereas the cloud point temperature for the equivalent cyclic graft copolymer was 67 °C, indicating significant difference in thermo-responsiveness of the polymers. The subtle changes in polymer architecture dramatically influenced the materials’ nanostructure and properties, which is a critical consideration for the future development of these materials as drug carriers.138
 | ||
| Fig. 13 Illustration of cyclic graft copolymer unimolecular micelles and their different thermo-responsive properties with linear polymer analogues. Reproduced with permission from American Chemical Society.138 | ||
3. Applications of unimolecular micelles
Unimolecular micelles have certain structural advantages and unique properties that make them attractive candidates for many applications. In this section, the most investigated biomedical applications in using unimolecular micelles as drug carriers for controlled release or targeted delivery will be summarized. The utilization of unimolecular micelles in catalysis and as a template for the preparation of inorganic particles will also be discussed.3.1 Unimolecular micelles for drug delivery
Since Duncan and Kopecek introduced the concept of using functional polymers for drug delivery in the 1980s, micelles have been extensively studied for controlled release and targeted delivery of different therapeutic and diagnostic drug molecules.76,139–141 The inner hydrophobic core of the micelles can be used to solubilize hydrophobic drugs through hydrophobic interactions, electrostatic interactions and hydrogen bonding, while the outer hydrophilic corona could endow the micelles with excellent biocompatibility and physiological stability.142–145 However, conventional polymeric micelles represent thermodynamic aggregations of multiamphiphilic macromolecules above their CMC. When they are introduced into the bloodstream, polymeric micelles might disassemble into free polymeric chains when they are subjected to high dilution and alterations in other factors such as temperature, pH and ionic strength.26,27 The disruption of micelle structures could lead to the burst release of entrapped drugs, which may cause serious toxicity issues due to the potentially large fluctuations in drug concentrations.94,146 This in vivo thermodynamic instability issue of polymeric micelles brings increasing interest to apply unimolecular micelles as carriers for drug delivery.147–149 The use of unimolecular micelle for loading drugs can be basically accomplished in two different ways: physically encapsulating the drug within the scaffold of the unimolecular micelles or covalently conjugating the drug onto the scaffold. By comparison, the former has been widely used due to its relatively easier performance. However, the latter can efficiently avoid the drugs “escaping” from unimolecular micelles before they reach the target therapeutic sites. In one example, Yao et al. developed a series of generation-3.0 PAMAM-g-poly[3-dimethyl(methacryloyloxyethyl) ammonium propanesulfonate] (PAMAM3.0-g-PDMAPS) based unimolecular micelles with PAMAM3.0 as a hydrophobic core and zwitterionic PDMAPS segments as a hydrophilic shell to stabilize the unimolecular micelles.150 As shown in DLS results, in PAMAM3.0-g-PDMAPS unimolecular micelle the sizes were in the range of 6.5 to 8.5 nm and they exhibited excellent stability upon dilution in the complex biological microenvironment. In addition, the presence of zwitterionic PDMAPS in the shell layer suppressed the non-specific protein adsorption, and micelle aggregation was thus prevented. There are good characteristics of micelles of prolonged circulation time and improved accumulation at tumor sites (Fig. 14). Moreover, the anticancer drug DOX was successfully encapsulated in both the PAMAM3.0 core via hydrophobic interactions and the PDMAPS shell layer via hydrogen bonds. It showed that drug leakage from PAMAM3.0-g-PDMAPS unimolecular micelles was prevented at pH 7.4, while a rapid release rate of drug was achieved in the acidic tumour microenvironment. The proliferation of cancer cells was significantly inhibited when the DOX loaded PAMAM3.0-g-PDMAPS unimolecular micelles were incubated with cells, showing their great potential application as a nanocarrier to deliver anticancer drugs.150 | ||
| Fig. 14 Schematic illustration of anti-biofouling of PAMAM3.0-g-PDMAPS unimolecular micelles and their intracellular release of DOX triggered by the acidic microenvironment in cancer cells. Reproduced with permission from Royal Society of Chemistry.150 | ||
In the design of unimolecular micelles as drug carriers, materials with good biocompatibility and biodegradability are highly desirable. For example, PEG and its derivatives have been widely used to build the outer shell of the unimolecular micelles because PEG is one of the most promising synthetic polymers approved by the U.S. Food and Drug Administration (FDA) for direct use in the biomedical field.151–155 Degradability is another critical requirement to be considered for the development of a drug delivery carrier. This is because the molecular weight of the unimolecular micelles is generally very high, which is beneficial for a long circulation time in bloodstream and passive targeting on cancer tissues.149,156 However, unimolecular micelles are facing great challenges in their exclusion from the body after use owing to their larger volume. Therefore, biodegradable polymers are suitable candidates for the construction of the inner core of unimolecular micelles, in which the materials can be easily degraded in vivo into small molecules and removed from the body by renal filtration.24,75,76 Pan et al. recently introduced novel unimolecular micelles formed from hyperbranched star copolymers consisting of disulfide as reduction-responsive linkage and PEG in the shell for the stabilization of the micelles in drug delivery.157 It showed that the diameters of camptothecin (CPT)-loaded unimolecular micelles were 3.56–6.08 nm and were easily triggered by mild acidic pH at 6.0 and 5.0, respectively. These unimolecular micelles were successfully internalized by the tumor cells and released CPT within the cells. When the hyperbranched star copolymer was treated with DTT, the molecular weight of the products was significantly decreased from 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 to 2100 g mol−1 due to cleavage of the disulfide linkage in the branching units. Therefore, the unimolecular micelles containing the anticancer drug CPT are redox-sensitive, and the degraded products may be easily cleaned after drug delivery. More importantly, through the design of highly reactive bromine groups on the surface of hyperbranched polymers, a variety of targeting groups or bioactive compounds were linked to their surface, forming various targeting drug delivery systems and improved blood compatibility.157 In a similar fashion, biodegradable unimolecular micelles for glutathione-mediated intracellular drug delivery were developed based on an amphiphilic hyperbranched multiarm copolymer (H40-star-PLA-SS-PEP) with disulfide linkages between the hydrophobic polyester core and hydrophilic arms.158 Cell culture results demonstrated that H40-star-PLA-SS-PEP micelles exhibited a faster drug release in glutathione monoester (GSH-OEt) pretreated Hela cells than that in the non-pretreated cells, showing a potential to improve the antitumor efficacy of hydrophobic chemotherapeutic drugs.
300 to 2100 g mol−1 due to cleavage of the disulfide linkage in the branching units. Therefore, the unimolecular micelles containing the anticancer drug CPT are redox-sensitive, and the degraded products may be easily cleaned after drug delivery. More importantly, through the design of highly reactive bromine groups on the surface of hyperbranched polymers, a variety of targeting groups or bioactive compounds were linked to their surface, forming various targeting drug delivery systems and improved blood compatibility.157 In a similar fashion, biodegradable unimolecular micelles for glutathione-mediated intracellular drug delivery were developed based on an amphiphilic hyperbranched multiarm copolymer (H40-star-PLA-SS-PEP) with disulfide linkages between the hydrophobic polyester core and hydrophilic arms.158 Cell culture results demonstrated that H40-star-PLA-SS-PEP micelles exhibited a faster drug release in glutathione monoester (GSH-OEt) pretreated Hela cells than that in the non-pretreated cells, showing a potential to improve the antitumor efficacy of hydrophobic chemotherapeutic drugs.
Cell uptake via the enhanced permeation and retention (EPR) effect is the main way by which unimolecular micelles deliver the encapsulated drugs to the therapeutic sites. Due to the clearance by the reticuloendothelial system (RES) and the lack of tumor targeting ability, the accumulation of unimolecular micelles in tumor tissues only by RES is poor, inevitably leading to the decreased therapeutic effect and undesirable side effects. Accordingly, in order to achieve better therapeutic efficacy, unimolecular micelles conjugated with target moieties have been developed in recent years. The target moieties on the surface can help unimolecular micelles to reach the tumor sites effectively and thereby increase the EPR effect. Neuroendocrine (NE) cancers have been a serious issue to cause significant patient morbidity. There are no curative treatments for NE cancers and their metastases except surgery, emphasizing the urgent need to develop alternative therapies. Recently, Gong and his co-workers developed multifunctional unimolecular micelles for targeted NE cancer therapy (Fig. 15).159 In this study, the unimolecular micelles were built from a multi-arm star amphiphilic block copolymer, poly(amidoamine)-poly(valerolactone)-poly(ethylene glycol), conjugated with KE108 peptide and Cy5 dye (PAMAM-PVL-PEG-KE108/Cy5). The unimolecular micelles formed a spherical core–shell structure in aqueous solution and exhibited a uniform size distribution and excellent stability. With the presence of KE108 peptide as the targeting moiety to NE tumor, the cellular uptake of the unimolecular micelles was dramatically increased in NE cancer cells overexpressing SSTRs compared to that of non-targeted micelles. This is because of the strong binding affinity of KE108 to all five subtypes of somatostatin receptors (SSTR 1–5). Moreover, the KE108-conjugated unimolecular micelles exhibited the greatest tumor accumulation due to their passive targeting and active targeting capabilities. The efficient anticancer efficacy without detectable systemic toxicity could offer a promising approach for targeted NE cancer therapy.159 By using a cRGD peptide conjugated star amphiphilic block copolymer, Boltron® H40 (H40, a 4th generation hyperbranched polymer)-biodegradable photo-luminescent polymer (BPLP)-poly(ethylene glycol) (H40-BPLP-PEG-cRGD), the same research group also demonstrated self-fluorescent unimolecular micelles of excellent stability in aqueous solutions, high drug loading level, pH-controlled drug release, and passive and active tumor-targeting abilities, thereby making them a promising candidate for many potential biomedical applications including tumor-targeted drug delivery and bioimaging.160 In a similar strategy, unimolecular micelles formed from 20-arm hyperbranched amphiphilic H40-(PMA-Hyd-DOX-co-PCL)-MPEG/PEG-FA copolymers were also reported for targeted delivery of anticancer drugs to tumor cells.161
 | ||
| Fig. 15 (A) Schematic illustration of the multifunctional unimolecular micelles formed by the multi-arm star amphiphilic block copolymer PAMAM-PVL-PEG-OCH3/Cy5/KE108 for targeted NE cancer therapy. (B) Schematic illustration of the passive and active tumor targeting capabilities exhibited by the multifunctional unimolecular micelles after intravenous injection. Reproduced with permission from Elsevier.159 | ||
In an another report, Liu and co-workers fabricated self-reporting theranostic drug nanocarriers based on a novel unimolecular micelle system consisting of hyperbranched cores conjugated with reduction-activated camptothecin prodrugs and a magnetic resonance (MR) imaging contrast agent (Gd complex), and hydrophilic coronas functionalized with guanidine residues.9 Upon cellular internalization, the reductive milieu-actuated release of anticancer drugs in the active form, the activation of therapeutic efficacy (>70-fold enhancement in cytotoxicity), and the turn-on of MR imaging (∼9.6-fold increase in T1 relaxivity) were simultaneously achieved in the simulated cytosol milieu. In addition, guanidine decorated unimolecular micelles exhibited extended blood circulation with a half-life of up to ∼9.8 h and excellent tumor cell penetration potency. The hyperbranched chain topology thus provides a novel theranostic polyprodrug platform for synergistic imaging/chemotherapy and enhanced tumor uptake (Fig. 16).9 Recently, Zhu and his-coworkers synthesized a series of well-defined core–shell unimolecular micelles with a conjugated polymeric core and flexible hydrophilic arms.162–166 These unimolecular micelles exhibited excellent fluorescence performance and good biocompatibility in aqueous solution, providing a new platform for effective diagnosis and biological applications including tumor imaging, real-time drug release monitoring, protein staining, gene transfection, and bacterial detection using fluorescent probes.167–170
 | ||
| Fig. 16 Schematic illustration of polyprodrug unimolecular micelles with hyperbranched cores conjugated with DOTA (Gd) and reductive milieu-cleavable camptothecin prodrugs and hydrophilic coronas functionalized with guanidine residues. Reproduced with permission from American Chemical Society.9 | ||
3.2 Unimolecular micelles as a nanocontainer for catalysis
Catalysts such as metal-based nanoparticles and other types of small molecules are becoming more and more important in organic chemistry. However, due to some intractable issues such as the challenge in separation of small molecular catalysts from targeted products and the heterogeneous problem of the traditional catalyst system, the wide use of these catalysts in industrialized organic reactions is significantly limited.171 Organic polymers with well-defined nanostructure have provided a promising approach to overcome these intrinsic limitations. Compared with micrometer-scaled polymer beads, the larger surface area of nanometer-sized polymers could facilitate small molecules to rapidly diffuse in and out of the polymeric supports.172 Linear polymers stand out for their easy synthesis and they have been employed as soluble macromolecular supports for catalysts.173 However, the loading capacities are relatively low due to the lack of persistent shape and the limited number of functional groups in linear polymers.174 Recently, unimolecular micelles with core–shell structure were explored for catalysis applications due to their many advantages, including a narrow nanoparticle size distribution and a high metal loading capacity.172,175 The cavities within the inner core could provide vast space to accommodate the catalyst molecules and increase the loading capacity, whereas the hydrophilic outer shell could be used to enhance the solubility of catalyst-loaded particles in reaction media and further prevented the particles from aggregation.176–178 In addition, unimolecular micelles maintain the structural integrity under high dilution, which is beneficial for the prevention of the undesired catalyst escape from the core to the solution.176 Moreover, the highly branched structure in unimolecular micelles can provide many end groups for further functionalization.179Recently, Nabid and his co-workers reported the preparation of a H40-PCL-PEG unimolecular micelle system and employed it as a micellar catalyst for Heck reaction in water.180 In this study, H40 functionalized with PCL as a hydrophobic core provided anchoring sites for palladium nanoparticles, and the presence of hydrophilic PEG chains in the outer shell made the unimolecular micelles uniformly dispersed in water. The activity and efficiency of the catalyst were evaluated in a Heck reaction. It was found that the catalyst and substrates were concentrated in nanosize sites and they were efficient catalysts in cross-coupling reactions of aryl iodides, bromides and also chlorides with olefinic compounds in short reaction time and high yields. In addition, the catalysts were reusable without the loss of activity after an easy recycling process such as extraction, dialysis or ultra-centrifugation.180 In another report, Fréchet et al. reported a one-pot scalable method for quick access to a wide range of hydrophobic or water-incompatible catalysts by using the modular star polymer (PS(N3))-PEG.181 The amphiphilic star polymer consisted of a hydrophobic functional PS core with azide groups and outer water-soluble PEG arms (Fig. 17). The azide groups in the cores could allow for the “clickable” facile attachment of various alkyne-containing “payloads”, including organo catalysts, metal complexes, and dyes for quick access to a variety of polymer catalysts. A model Knoevenagel condensation between benzaldehyde and ethyl cyanoacetate was used to probe the viability of the material as a catalyst support in water. It showed that the rate of the uncatalyzed reaction was negligible at room temperature. However, (PS(9))-PEG exhibited higher activity than L-proline and the functional PEG. In addition, (PS(9))-PEG could be used multiple times and there was no significant loss of activity after recycling.181 The same research group also reported the fabrication of a unimolecular catalytic system from dendrimers consisting of tetradecyl-substituted benzyl bromide as the terminal unit and methyl ester functionalized diphenol as the repeat unit.182 In this design, the high polarity of the inner core was combined with the low polarity of the outer corona to form unimolecular dendritic reverse micelles which can provide a suitable nano-environment for catalytic reactions involved a polar transition state. The catalytic capability of the unimolecular micelles was investigated by the SN2 alkylation of pyridine with CH3I. It was found that the 4th generation dendrimer possessed the highest activity. However, incomplete conversions were also observed when the reactions were performed at low dendrimer concentrations. This is possibly due to the product inhibition effect since the polar alkylated pyridinium salts have a high affinity for the dendritic core.182
 | ||
| Fig. 17 Schematic illustration showing the “clickable” attachment of polymer catalysts to the unimolecular core of the modular star polymer (PS(N3))-PEG. Reproduced with permission from American Chemical Society.181 | ||
Recently, unimolecular micelles formed from amphiphilic dendrimers containing 27 triethylene glycol termini and 9 intradendritic triazole rings were also explored as a catalytic nanoreactor (Fig. 18).183 The designed system can accelerate the CuI-catalyzed alkyne–azidecycloaddition (CuAAC) “click” reactions of various substrates in water. For example, it considerably facilitated the catalysis by [Cu(hexabenzyltren)]Br (0.1% vs. substrate) of the CuAAC reactions in water and the catalytic efficiency was nearly quantitative in the presence of this micelle nanoreactor. Moreover, the unimolecular micelle nanoreactor was recycled and demonstrated for repeated use with no loss or decomposition.183 In synergy with this effect, the presence of intradendritic triazole ligands in the nanoreactor also activated the catalyzed CuAAC reactions with down to 4 ppm of commercial CuSO4·5H2O and sodium ascorbate, leading to exceptional TONs up to 510 000. In addition, the reaction with hydrophobic biomolecules was also successfully performed in water at 30 °C and quantitative yields were obtained. Based on these promising results, the authors envisioned that this fully recyclable catalytic nanoreactor could allow a considerable decrease of the copper catalyst amount to industrially tolerable residues, and open the route to future biomedical and cosmetic applications.183–185
 | ||
| Fig. 18 Dendritic unimolecular micelles as a nanoreactor and a ligand for CuAAC catalysis. Reproduced with permission from American Chemical Society.183 | ||
3.3 Unimolecular micelles as a template for the preparation of inorganic nanoparticles
Due to their intriguing optical, electronic and catalytic properties, inorganic metal and semiconductor nanoparticles hold great promise in many applications ranging from optoelectronics and sensors to catalysis and medicine.186–192 Since these unique properties are closely related to the dimension of inorganic nanoparticles, it is of critical importance to precisely control the size, shape and polydispersity of these nanostructure materials.193 To date, many synthetic approaches have been developed for the preparation of various types of metal and semiconductor nanoparticles with desired shape, compositions and morphologies.194–196 Among these methods, unimolecular micelles directed template-synthesis of inorganic nanoparticles is a relatively new approach but has become an active area of research due to the easy control in the size and composition of the materials.197 This section will summarize the recent progress in the synthesis of inorganic nanoparticles by using unimolecular micelles as templates.Recently, Minko and co-workers reported a unimolecular micelle system constituted by the star-shaped PS7-P2VP7 polysterene/poly(2-vinylpyridine) block copolymer and applied it for the deposition of nanosized palladium clusters.198 Unimolecular micelles with core–shell structures were obtained with noticeable segregation into the collapsed core and the extended shell formed by stretched polymer arms of P2VP and PS in water and toluene, respectively. Metallization of P2VP arms of the copolymer could result in the localization of 1–3 nm palladium clusters in the outer shell of unimolecular micelles, forming star-like structures with metallized arms. With the presence of palladium clusters, the obtained organic–inorganic nanocomposite exhibited significantly improved contrast for AFM imaging.198 In another report, Schubert et al. demonstrated the preparation of gold nanoparticles templated into the PEO core of unimolecular micelles formed from PEO-b-PCL star-block copolymers.196 In this study, a low molecular weight five-arm PEO core was selected in the construction of the unimolecular template to control the production of gold nanoparticles of small size. During the synthesis, the PEO core was soaked with KAuCl4 in DMF and gold nanoparticles were subsequently obtained by reduction with NaBH4. It showed that monodisperse spherical gold nanoparticles with an average size of approximately 3 nm could be formed in the PEG core. In contrast, the presence of coronal PCL with different chain lengths was beneficial to the long-term stability against aggregation of the gold nanoparticles, indicating the role of PCL blocks as a stabilizing component for the nanoparticles.196 In a similar strategy, the corePEGDMA-(P2VP-b-PS)n amphiphilic star copolymer was also applied as a unimolecular template for the synthesis of Au nanoparticles in toluene.199 In this regard, the AuCl4− anion precursors would selectively complex with the inner blocks of P2VP in the star copolymers to confine the formation of Au nanoparticles within the polymer interior. When the reducing agent N2H4 was added into the solution, Au nanoparticles were thus deposited within the micelle core. The outer PS shell provided protection of the synthesized Au nanoparticle and enabled the easy dispersion of the particles in toluene.196 When corePDVB-(PAA-b-PMMA)n (DVB = divinylbenzene) polymers were used as unimolecular templates, the selective binding between carboxylic acid groups in the inner PAA blocks and Ag cations was also successfully demonstrated for the synthesis of Ag nanoparticles.200
Zhang et al. recently reported multifunctional polymer unimolecular micelles, which are used as templates to fabricate stable gold nanoparticles in a one-step reaction without the addition of external reductants (Fig. 19).201 In this study, the unimolecular micellar nanoreactors were made from the 21-arm star-like block copolymer β-cyclodextrin-(poly(lactide))-poly(2-(dimethylamino)ethylmethacrylate)-poly[oligo(2-ethyl-2-oxazoline)methacrylate]21 (β-CD-(PLA-PDMAEMA-PEtOxMA)21), in which both β-CD and PLA formed the hydrophobic core of the unimolecular micelles and EtOxMA with short side chains formed the shell. The tertiaryamine groups of the PDMAEMA block could act as a reducing agent to reduce the AuCl4− precursor to zero-valent gold in aqueous solution, and these gold atoms combined mutually to form the final Au nanoparticles. It showed that the sizes and morphologies of the gold nanoparticles were well controlled by adjusting the PDMAEMA length and the concentrations of the star-like polymer and HAuCl4. Significantly different from pervious methods, the in situ generation of Au nanoparticles in this study could avoid the use of organic solvents and other reducing reagents. Together with the stabilization behaviors and low cytotoxicity, the gold nanoparticles developed in this study could adapt to further biological applications.201
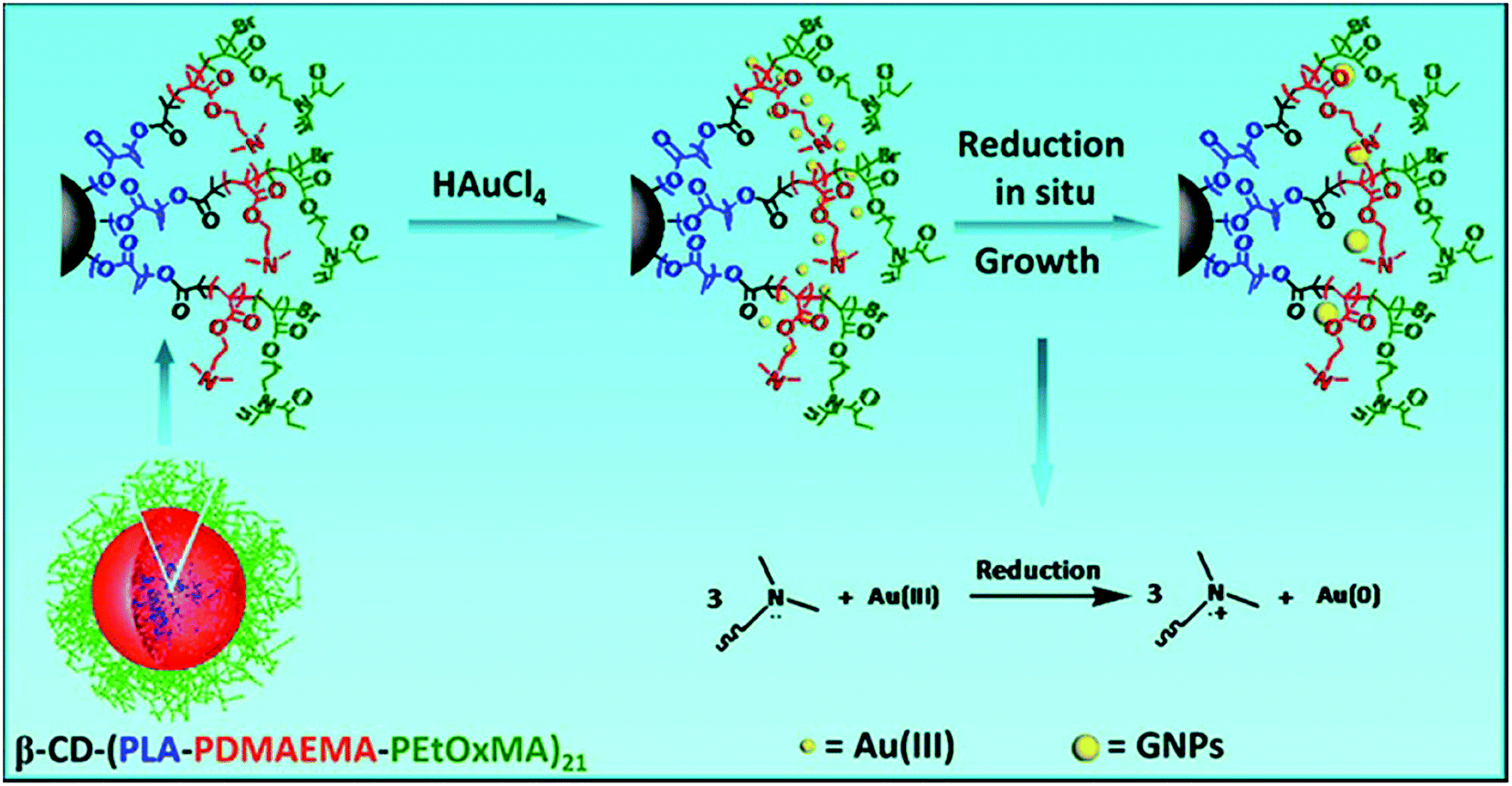 | ||
| Fig. 19 Schematic representation of using the star-like block copolymer β-CD-(PLA-PDMAEMA-PEtOxMA)21 as a unimolecular micelle template for the synthesis and stabilization of gold nanoparticles. Reproduced with permission from Wiley.201 | ||
Pang et al. reported a versatile unimolecular template mediated synthesis of a large variety of functional nanocrystals with precisely controlled dimensions, compositions and architectures including core–shell and hollow nanostructures.202 The unimolecular micelles formed from a new class of copolymers such as a series of polymers such as 21-arm β-CD-P4VP-PAA-b-PS, β-CD-PAA-b-PS, and β-CD-PAA-b-PEO were structurally stable and can overcome the intrinsic instability of linear block copolymer micelles. The permanent connection between the nanoparticles and the respective hydrophobic or hydrophilic polymer chains rendered these polymers soluble in either organic or aqueous environments, respectively. This could further facilitate the easy synthesis of various sizes and architectures of metallic, ferroelectric, magnetic, semiconductor and luminescent colloidal nanoparticles.202
In addition to controlling the size and morphology of the inorganic nanoparticles, utilization of a unimolecular template to tune the spatial distribution of inorganic nanoparticles has also been explored. Liu and co-workers recently reported the conjugation of Au nanoparticles onto the surface of thiol-functionalized thermo-sensitive H40-PNIPAM unimolecular micelles for the fabrication of satellite-like nanostructures.203 Due to the covalent linkage between surface thiol groups and gold nanoparticles, the obtained hybrid nanostructure is highly stable. In addition, the unimolecular micelle templates were thermo-sensitive H40-PNIPAM polymers and exhibited reversible swelling and shrinkage in response to external temperatures. For example, the average size of the unimolecular micelles of H40-PNIPAM220 decreased from 140 to ∼100 nm when the solution temperature was elevated from 25 to 40 °C. The shrinkage of PNIPAM corona would concomitantly decrease the average distance between gold nanoparticles on the micelle surfaces (Fig. 20). This would further induce a dramatic red-shift of the maximum of the surface plasmon band from 524 to 535 nm by the enhanced inter-particle coupling effect.203
 | ||
| Fig. 20 (A) Schematic illustration of the two-step preparation of the hybrid unimolecular micelle surface decorated with Au nanoparticles and (B) the thermo-tunable spatial distance between Au nanoparticles attached at the unimolecular micelle surface. Reproduced with permission from American Chemical Society.203 | ||
3.4. Unimolecular micelles for energy harvesting and storage
In addition to the applications mentioned above, some types of unimolecular micelles have also shown potential in energy-harvesting applications.204–207 For example, the unique characteristics of highly compacted dendritic unimolecular micelles such as globular shape, uniform size and controlled arrangements of different functional groups have made them promising in energy harvesting and storage applications.208 When an energy-transfer or similar electronic link is introduced between the periphery and the unimolecular micelle core, a light-harvesting antenna having controlled intramolecular energy transfer by geometry and chromophore selection can be prepared. Through this design, a useful form of light amplification can be achieved and a large array of terminal donor chromophores could collect photons and transfer the energy through space to an acceptor unit located in the core.206,207 In such applications, the size of the materials is of great importance to determine the distances over energy transfer. With rational design, it is expected to achieve highly efficient transfer of energy and avoid the close neighbor interactions which might further lead to unwanted interchromophoric phenomena.209 Recently, Aida and coworkers demonstrated that multiple peripheral chromophores could funnel their energy to a central chromophore in a dendritic structure. In this study, a very large array of 28 zinc porphyrin donor chromophores radially emanating from a central free-based porphyrin acceptor was designed.210 The combination of unimolecular micelles with moieties capable of charge separation and electron injection might lessen the current high dependence on energy from fossil fuels in the future.4. Conclusion and future perspectives
Compared to the multimolecular micelles in dynamic equilibrium in solution, unimolecular micelles are covalently reinforced core–shell nanostructured materials which show excellent stability to environmental changes such as pH, temperature, ionic strength, dilution etc. The unimolecular micelle formation does not rely on self-assembly. Thus, developing these sophisticated materials could overcome many challenges of using micelles as delivery carriers in biomedical applications. For example, the premature disassociation of the self-assembled multimolecular micelles during circulation in the bloodstream can cause a burst release of highly toxic drugs and further cause potential systemic toxicity and insufficient tumor-targeting ability. Another advantage is the highly branched structure in unimolecular micelles, which can provide many possibilities for further functionalization to adapt to specific applications. This review summarizes the latest developments of unimolecular micelles from different types of single macromolecular amphiphilic polymers, including dendrimers and hyperbranched, dendritic, star, brush-like and cyclic polymers. The current progress in using these sophisticated materials for different applications is also covered, mainly in the area of therapeutic drug delivery, catalysis, preparation of inorganic nanoparticles and energy harvesting.The development of different synthetic methodologies has manifested a rapid progress in the field of unimolecular research. For example, controlled living radical polymerization not only provides access to a library of unimolecular amphiphilic polymers with well-controlled structures, tunable sizes and variable compositions, but also enables efficient functionalization of various polymer end groups. However, there still are some challenges for clinical applications of using unimolecular micelles as drug delivery carriers: (1) the amphiphilic polymers used for the preparation of unimolecular micelles are commonly in complex architectures, in which multiple step synthesis and purification processes are involved. This has greatly limited their large scale applications. (2) In drug delivery, there is another issue that physically loaded drug molecules might diffuse from the micelles before they reach the tumor site, leading to a decreased drug efficacy. Although conjugating the drugs to the unimolecular micelles through cleavable linkers has been proposed, adding new functionality inevitably elevates the complexity and the rate of the drug release from the conjugated micelles is difficult to control.
In future, more simple yet effective synthetic methodologies are urgently required for the facile fabrication of well-defined single macromolecular amphiphilic polymers. In addition, multifunctional unimolecular micelles with the combination of controlled release and specific targeting ability should have better therapeutic efficacy in cancer therapy. The drug loaded micelles reach the specific tumor site via targeting affinity followed by a fast release of the payload triggered by intracellular stimulation. However, integrating multiple functions into one domain is still challenging because of increasing synthetic difficulties and lower controllability.
References
- Z. Li, P. L. Chee, C. Owh, R. Lakshminarayanan and X. J. Loh, RSC Adv., 2016, 6, 28947–28955 RSC.
- A. A. Karim, Q. Dou, Z. Li and X. J. Loh, Chem. – Asian J., 2016, 11, 1300–1321 CrossRef PubMed.
- T. A. Tockary, K. Osada, Y. Motoda, S. Hiki, Q. X. Chen, K. M. Takeda, A. Dirisala, S. Osawa and K. Kataoka, Small, 2016, 12, 1193–1200 CrossRef CAS PubMed.
- T. B. Ren, Q. M. Liu, H. Lu, H. M. Liu, X. Zhang and J. Z. Du, J. Mater. Chem., 2012, 22, 12329–12338 RSC.
- J. Z. Du, L. Fan and Q. M. Liu, Macromolecules, 2012, 45, 8275–8283 CrossRef CAS.
- C. C. Zhou, M. Z. Wang, K. D. Zou, J. Chen, Y. Q. Zhu and J. Z. Du, ACS Macro Lett., 2013, 2, 1021–1025 CrossRef CAS.
- R. Salva, J. F. Le Meins, O. Sandre, A. Brulet, M. Schmutz, P. Guenoun and S. Lecommandoux, ACS Nano, 2013, 7, 9298–9311 CrossRef CAS PubMed.
- M. Massignani, C. LoPresti, A. Blanazs, J. Madsen, S. P. Armes, A. L. Lewis and G. Battaglia, Small, 2009, 5, 2424–2432 CrossRef CAS PubMed.
- X. Hu, G. Liu, Y. Li, X. Wang and S. Liu, J. Am. Chem. Soc., 2015, 137, 362–368 CrossRef CAS PubMed.
- Z. Li, B. H. Tan, T. Lin and C. He, Prog. Polym. Sci., 2016 DOI:10.1016/j.progpolymsci.2016.1005.1003.
- A. A. Karim and X. J. Loh, Soft Matter, 2015, 11, 5425–5434 RSC.
- D. Kai, Z. W. Low, S. S. Liow, A. A. Karim, H. Y. Ye, G. R. Jin, K. Li and X. J. Loh, ACS Sustainable Chem. Eng., 2015, 3, 2160–2169 CrossRef CAS.
- X. J. Loh, J. del Barrio, T. C. Lee and O. A. Scherman, Chem. Commun., 2014, 50, 3033–3035 RSC.
- X. J. Loh, S. J. Ong, Y. T. Tung and H. T. Choo, Polym. Chem., 2013, 4, 2564–2574 RSC.
- X. J. Loh, S. J. Ong, Y. T. Tung and H. T. Choo, Mater. Sci. Eng., C, 2013, 33, 4545–4550 CrossRef CAS PubMed.
- X. J. Loh, J. Appl. Polym. Sci., 2013, 127, 992–1000 CrossRef CAS.
- J. P. Lin, J. Q. Zhu, T. Chen, S. L. Lin, C. H. Cai, L. S. Zhang, Y. Zhuang and X. S. Wang, Biomaterials, 2009, 30, 108–117 CrossRef CAS PubMed.
- K. S. Soppimath, L. H. Liu, W. Y. Seow, S. Q. Liu, R. Powell, P. Chan and Y. Y. Yang, Adv. Funct. Mater., 2007, 17, 355–362 CrossRef CAS.
- N. Rapoport, Prog. Polym. Sci., 2007, 32, 962–990 CrossRef CAS.
- Y. C. Tan, K. Hettiarachchi, M. Siu and Y. P. Pan, J. Am. Chem. Soc., 2006, 128, 5656–5658 CrossRef CAS PubMed.
- H. G. Cui, Z. Y. Chen, S. Zhong, K. L. Wooley and D. J. Pochan, Science, 2007, 317, 647–650 CrossRef CAS PubMed.
- A. Harada and K. Kataoka, Prog. Polym. Sci., 2006, 31, 949–982 CrossRef CAS.
- E. N. Savariar, S. V. Aathimanikandan and S. Thayumanavan, J. Am. Chem. Soc., 2006, 128, 16224–16230 CrossRef CAS PubMed.
- Z. Li and X. J. Loh, Chem. Soc. Rev., 2015, 44, 2865–2879 RSC.
- Z. Li, J. Yang and X. J. Loh, NPG Asia Mater., 2016, 8, e265 CrossRef CAS.
- R. Savic, T. Azzam, A. Eisenberg and D. Maysinger, Langmuir, 2006, 22, 3570–3578 CrossRef CAS PubMed.
- J. Gong, M. Chen, Y. Zheng, S. Wang and Y. Wang, J. Controlled Release, 2012, 159, 312–323 CrossRef CAS PubMed.
- H. Ye, C. Owh, S. Jiang, C. Ng, D. Wirawan and X. Loh, Polymer, 2016, 8, 130 Search PubMed.
- S. S. Liow, A. A. Karim and X. J. Loh, MRS Bull., 2016, 41, 557–566 CrossRef CAS.
- S. S. Liow, Q. Dou, D. Kai, A. A. Karim, K. Zhang, F. Xu and X. J. Loh, ACS Biomater. Sci. Eng., 2016, 2, 295–316 CrossRef CAS.
- L. Jiang, S. S. Liow and X. J. Loh, Polym. Chem., 2016, 7, 1693–1700 RSC.
- J. Y. Zheng, M. J. Tan, P. Thoniyot and X. J. Loh, RSC Adv., 2015, 5, 62314–62318 RSC.
- H. Kawaguchi, Prog. Polym. Sci., 2000, 25, 1171–1210 CrossRef CAS.
- K. Landfester, Angew. Chem., Int. Ed., 2009, 48, 4488–4507 CrossRef CAS PubMed.
- C. J. Hawker and K. L. Wooley, Science, 2005, 309, 1200–1205 CrossRef CAS PubMed.
- A. M. Nystrom and K. L. Wooley, Acc. Chem. Res., 2011, 44, 969–978 CrossRef CAS PubMed.
- C. F. van Nostrum, Soft Matter, 2011, 7, 3246–3259 RSC.
- H. Gao, M. C. Jones, J. Chen, R. E. Prud'homme and J. C. Leroux, Chem. Mater., 2008, 20, 3063–3067 CrossRef CAS.
- X. Q. Yang, J. J. Grailer, I. J. Rowland, A. Javadi, S. A. Hurley, V. Z. Matson, D. A. Steeber and S. Q. Gong, ACS Nano, 2010, 4, 6805–6817 CrossRef CAS PubMed.
- X. Yang, J. J. Grailer, I. J. Rowland, A. Javadi, S. A. Hurley, D. A. Steeber and S. Gong, Biomaterials, 2010, 31, 9065–9073 CrossRef CAS PubMed.
- U. Kedar, P. Phutane, S. Shidhaye and V. Kadam, Nanomed. Nanotechnol. Biol. Med., 2010, 6, 714–729 CrossRef CAS PubMed.
- M. Talelli, C. J. F. Rijcken, W. E. Hennink and T. Lammers, Curr. Opin. Solid State Mater. Sci., 2012, 16, 302–309 CrossRef CAS.
- Y. Zhou, K. Jiang, Q. Song and S. Liu, Langmuir, 2007, 23, 13076–13084 CrossRef CAS PubMed.
- S. Luo, J. Xu, Z. Zhu, C. Wu and S. Liu, J. Phys. Chem. B, 2006, 110, 9132–9139 CrossRef CAS PubMed.
- J. Xu, S. Luo, W. Shi and S. Liu, Langmuir, 2006, 22, 989–997 CrossRef CAS PubMed.
- M. C. Lukowiak, B. N. S. Thota and R. Haag, Biotechnol. Adv., 2015, 33, 1327–1341 CrossRef CAS PubMed.
- R. Esfand and D. A. Tomalia, Drug Discovery Today, 2001, 6, 427–436 CrossRef CAS PubMed.
- H. T. Chen, M. F. Neerman, A. R. Parrish and E. E. Simanek, J. Am. Chem. Soc., 2004, 126, 10044–10048 CrossRef CAS PubMed.
- U. Boas and P. M. H. Heegaard, Chem. Soc. Rev., 2004, 33, 43–63 RSC.
- M. J. Liu, K. Kono and J. M. J. Frechet, J. Polym. Sci., Polym. Chem., 1999, 37, 3492–3503 CrossRef CAS.
- E. R. Gillies and J. M. J. Frechet, Drug Discovery Today, 2005, 10, 35–43 CrossRef CAS PubMed.
- S. Sevenson and D. A. Tomalia, Adv. Drug Delivery Rev., 2012, 64, 102–115 CrossRef.
- L. J. Twyman, A. E. Beezer, R. Esfand, M. J. Hardy and J. C. Mitchell, Tetrahedron Lett., 1999, 40, 1743–1746 CrossRef CAS.
- M. J. Liu, K. Kono and J. M. J. Frechet, J. Controlled Release, 2000, 65, 121–131 CrossRef CAS PubMed.
- I. J. Majoros, A. Myc, T. Thomas, C. B. Mehta and J. R. Baker, Biomacromolecules, 2006, 7, 572–579 CrossRef CAS PubMed.
- S. E. Stiriba, H. Frey and R. Haag, Angew. Chem., Int. Ed., 2002, 41, 1329–1334 CrossRef CAS.
- C. Dufes, I. F. Uchegbu and A. G. Schatzlein, Adv. Drug Delivery Rev., 2005, 57, 2177–2202 CrossRef CAS PubMed.
- L. Qin, D. R. Pahud, Y. Ding, A. U. Bielinska, J. F. Kukowska-Latallo, J. R. Baker Jr. and J. S. Bromberg, Hum. Gene Ther., 1998, 9, 553–560 CrossRef CAS PubMed.
- A. U. Bielinska, A. Yen, H. L. Wu, K. M. Zahos, R. Sun, N. D. Weiner, J. R. Baker and B. J. Roessler, Biomaterials, 2000, 21, 877–887 CrossRef CAS PubMed.
- D. Joester, M. Losson, R. Pugin, H. Heinzelmann, E. Walter, H. P. Merkle and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1486–1490 CrossRef CAS PubMed.
- D. W. Chang, G.-J. Sohn, L. Dai and J.-B. Baek, Soft Matter, 2011, 7, 8352–8357 RSC.
- N. Wimmer, R. J. Marano, P. S. Kearns, E. P. Rakoczy and I. Toth, Bioorg. Med. Chem. Lett., 2002, 12, 2635–2637 CrossRef CAS PubMed.
- E. Murugan, R. L. Sherman, H. O. Spivey and W. T. Ford, Langmuir, 2004, 20, 8307–8312 CrossRef CAS PubMed.
- J.-J. Lee, W. T. Ford, J. Moore and Y. Li, Macromolecules, 1994, 27, 4632–4634 CrossRef CAS.
- T. Habicher, F. Diederich and V. Gramlich, Helv. Chim. Acta, 1999, 82, 1066–1095 CrossRef CAS.
- M. Enomoto and T. Aida, J. Am. Chem. Soc., 1999, 121, 874–875 CrossRef CAS.
- M. A. Azagarsamy, P. Sokkalingam and S. Thayumanavan, J. Am. Chem. Soc., 2009, 131, 14184–14185 CrossRef CAS PubMed.
- M. Longmire, P. L. Choyke and H. Kobayashi, Nanomedicine, 2008, 5, 703–717 CrossRef PubMed.
- C. J. Hawker, K. L. Wooley and J. M. J. Fréchet, J. Chem. Soc., Perkin Trans. 1, 1993, 1287–1297, 10.1039/P19930001287.
- J. Jansen, E. M. M. Debrabandervandenberg and E. W. Meijer, Science, 1994, 266, 1226–1229 CAS.
- Y. Wang and S. M. Grayson, Adv. Drug Delivery Rev., 2012, 64, 852–865 CrossRef CAS PubMed.
- J. Jansen, E. W. Meijer and E. M. M. Debrabandervandenberg, J. Am. Chem. Soc., 1995, 117, 4417–4418 CrossRef CAS.
- F. M. Veronese and G. Pasut, Drug Discovery Today, 2005, 10, 1451–1458 CrossRef CAS PubMed.
- A. Kolate, D. Baradia, S. Patil, I. Vhora, G. Kore and A. Misra, J. Controlled Release, 2014, 192, 67–81 CrossRef CAS PubMed.
- Z. Li, D. Yuan, G. Jin, B. H. Tan and C. He, ACS Appl. Mater. Interfaces, 2016, 8, 1842–1853 CAS.
- Z. Li and B. H. Tan, Mater. Sci. Eng., C, 2014, 45, 620–634 CrossRef CAS PubMed.
- X. Fan, Z. Wang, D. Yuan, Y. Sun, Z. Li and C. He, Polym. Chem., 2014, 5, 4069–4075 RSC.
- Z. Li and J. Li, J. Phys. Chem. B, 2013, 117, 14763–14774 CrossRef CAS PubMed.
- Z. Li, Z. Zhang, K. L. Liu, X. Ni and J. Li, Biomacromolecules, 2012, 13, 3977–3989 CrossRef CAS PubMed.
- Z. Li, H. Yin, Z. Zhang, K. L. Liu and J. Li, Biomacromolecules, 2012, 13, 3162–3172 CrossRef CAS PubMed.
- X. P. Ma, Z. X. Zhou, E. L. Jin, Q. H. Sun, B. Zhang, J. B. Tang and Y. Q. Shen, Macromolecules, 2013, 46, 37–42 CrossRef CAS.
- Y. H. Kim, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 1685–1698 CrossRef CAS.
- C. J. Hawker, J. M. Frechet, R. B. Grubbs and J. Dao, J. Am. Chem. Soc., 1995, 117, 10763–10764 CrossRef CAS.
- M. Suzuki, A. Ii and T. Saegusa, Macromolecules, 1992, 25, 7071–7072 CrossRef CAS.
- M. Suzuki, S. Yoshida, K. Shiraga and T. Saegusa, Macromolecules, 1998, 31, 1716–1719 CrossRef CAS.
- M. Jikei, S. H. Chon, M. Kakimoto, S. Kawauchi, T. Imase and J. Watanebe, Macromolecules, 1999, 32, 2061–2064 CrossRef CAS.
- T. Emrick, H.-T. Chang and J. M. Frechet, Macromolecules, 1999, 32, 6380–6382 CrossRef CAS.
- X. Wang, L. Li, W. He and C. Wu, Macromolecules, 2015, 48, 7327–7334 CrossRef CAS.
- J. Liu, W. Huang, Y. Pang and D. Yan, Chem. Soc. Rev., 2015, 44, 3942–3953 RSC.
- Y. H. Kim and O. W. Webster, J. Am. Chem. Soc., 1990, 112, 4592–4593 CrossRef CAS.
- M. Prabaharan, J. J. Grailer, S. Pilla, D. A. Steeber and S. Gong, Macromol. Biosci., 2009, 9, 515–524 CrossRef CAS PubMed.
- M. Prabaharan, J. J. Grailer, S. Pilla, D. A. Steeber and S. Gong, Biomaterials, 2009, 30, 3009–3019 CrossRef CAS PubMed.
- X. Li, Y. Qian, T. Liu, X. Hu, G. Zhang, Y. You and S. Liu, Biomaterials, 2011, 32, 6595–6605 CrossRef CAS PubMed.
- C. Porsch, Y. Zhang, M. I. Montañez, J.-M. Malho, M. A. Kostiainen, A. M. Nyström and E. Malmström, Biomacromolecules, 2015, 16, 2872–2883 CrossRef CAS PubMed.
- M. Z. Yin, K. Ding, R. A. Gropeanu, J. Shen, R. Berger, T. Weil and K. Mullen, Biomacromolecules, 2008, 9, 3231–3238 CrossRef CAS PubMed.
- Y. Wang, G. Qi and J. He, ACS Macro Lett., 2016, 5, 547–551 CrossRef CAS.
- J. L. Six and Y. Gnanou, Macromol. Symp., 1995, 95, 137–150 CrossRef CAS.
- A. Hirao, T. Watanabe, K. Ishizu, M. Ree, S. Jin, K. S. Jin, A. Deffieux, M. Schappacher and S. Carlotti, Macromolecules, 2009, 42, 682–693 CrossRef CAS.
- H.-S. Yoo, T. Watanabe, Y. Matsunaga and A. Hirao, Macromolecules, 2011, 45, 100–112 CrossRef.
- H. F. Zhang, J. P. He, C. Zhang, Z. H. Ju, J. Li and Y. L. Yang, Macromolecules, 2012, 45, 828–841 CrossRef CAS.
- X. S. Fan, Z. G. Hu and G. W. Wang, RSC Adv., 2015, 5, 100816–100823 RSC.
- X. Fan, Z. Wang and C. He, J. Mater. Chem. B, 2015, 3, 4715–4722 RSC.
- M. Kaupp, K. Hiltebrandt, V. Trouillet, P. Mueller, A. S. Quick, M. Wegener and C. Barner-Kowollik, Chem. Commun., 2016, 52, 1975–1978 RSC.
- M. Hu, Y. Shen, L. Zhang and L. Qiu, Biomacromolecules, 2016, 17, 1026–1039 CrossRef CAS PubMed.
- P. Chmielarz, S. Park, A. Sobkowiak and K. Matyjaszewski, Polymer, 2016, 88, 36–42 CrossRef CAS.
- S. Angot, K. S. Murthy, D. Taton and Y. Gnanou, Macromolecules, 1998, 31, 7218–7225 CrossRef CAS.
- H. Gao and K. Matyjaszewski, Macromolecules, 2006, 39, 4960–4965 CrossRef CAS.
- J. Xia, X. Zhang and K. Matyjaszewski, Macromolecules, 1999, 32, 4482–4484 CrossRef CAS.
- H. Gao and K. Matyjaszewski, Prog. Polym. Sci., 2009, 34, 317–350 CrossRef CAS.
- S. Kanaoka, M. Sawamoto and T. Higashimura, Macromolecules, 1991, 24, 5741–5745 CrossRef CAS.
- K. Mortensen and M. Annaka, ACS Macro Lett., 2016, 5, 224–228 CrossRef CAS.
- C. Geyik, M. Ciftci, B. Demir, B. Guler, A. B. Ozkaya, Z. P. Gumus, F. B. Barlas, D. O. Demirkol, H. Coskunol and S. Timur, Polym. Chem., 2015, 6, 5470–5477 RSC.
- C. Yang, S. Q. Liu, S. Venkataraman, S. J. Gao, X. Ke, X. T. Chia, J. L. Hedrick and Y. Y. Yang, J. Controlled Release, 2015, 208, 93–105 CrossRef CAS PubMed.
- X. Pang, L. Zhao, M. Akinc, J. K. Kim and Z. Lin, Macromolecules, 2011, 44, 3746–3752 CrossRef CAS.
- C. Tsitsilianis, D. Papanagopoulos and P. Lutz, Polymer, 1995, 36, 3745–3752 CrossRef CAS.
- Q. Q. Guo, C. Y. Liu, T. T. Tang, J. Huang, X. G. Zhang and G. W. Wang, J. Polym. Sci., Polym. Chem., 2013, 51, 4572–4583 CrossRef CAS.
- W. Cao and L. Zhu, Macromolecules, 2011, 44, 1500–1512 CrossRef CAS.
- X. L. Luo, G. W. Wang, X. C. Pang and J. L. Huang, Macromolecules, 2008, 41, 2315–2317 CrossRef CAS.
- G. Koutalas, H. Iatrou, D. J. Lohse and N. Hadjichristidis, Macromolecules, 2005, 38, 4996–5001 CrossRef CAS.
- J.-Z. Du, L.-Y. Tang, W.-J. Song, Y. Shi and J. Wang, Biomacromolecules, 2009, 10, 2169–2174 CrossRef CAS PubMed.
- W. Yuan, J. Yuan, F. Zhang, X. Xie and C. Pan, Macromolecules, 2007, 40, 9094–9102 CrossRef CAS.
- G. Cheng, A. Böker, M. Zhang, G. Krausch and A. H. E. Müller, Macromolecules, 2001, 34, 6883–6888 CrossRef CAS.
- J.-Z. Du, L.-Y. Tang, W.-J. Song, Y. Shi and J. Wang, Biomacromolecules, 2009, 10, 2169–2174 CrossRef CAS PubMed.
- W. Z. Yuan, J. Y. Yuan, F. B. Zhang, X. M. Xie and C. Y. Pan, Macromolecules, 2007, 40, 9094–9102 CrossRef CAS.
- G. L. Cheng, A. Boker, M. F. Zhang, G. Krausch and A. H. E. Muller, Macromolecules, 2001, 34, 6883–6888 CrossRef CAS.
- G. W. Wang, X. S. Fan and J. L. Huang, J. Polym. Sci., Polym. Chem., 2010, 48, 3797–3806 CrossRef CAS.
- M. R. Xie, J. Y. Dang, H. J. Han, W. Z. Wang, J. W. Liu, X. H. He and Y. Q. Zhang, Macromolecules, 2008, 41, 9004–9010 CrossRef CAS.
- D. Neugebauer, Y. Zhang, T. Pakula and K. Matyjaszewski, Polymer, 2003, 44, 6863–6871 CrossRef CAS.
- M. Hans, H. Keul, A. Heise and M. Moeller, Macromolecules, 2007, 40, 8872–8880 CrossRef CAS.
- Y. Xia, B. D. Olsen, J. A. Kornfield and R. H. Grubbs, J. Am. Chem. Soc., 2009, 131, 18525–18532 CrossRef CAS PubMed.
- Z. Li and L. Wu, in Polyhydroxyalkanoates (PHAs): Biosynthesis, Industrial Production and Applications in Medicine, ed. W. Linping, Nova Science Publishers, USA, 2014, ch. 17, pp. 181–217 Search PubMed.
- J. T. Guo, H. Hong, G. J. Chen, S. X. Shi, T. R. Nayak, C. P. Theuer, T. E. Barnhart, W. B. Cai and S. Q. Gong, ACS Appl. Mater. Interfaces, 2014, 6, 21769–21779 CAS.
- H. R. Kricheldorf, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 251–284 CrossRef CAS.
- E. Minatti, R. Borsali, M. Schappacher, A. Deffieux, V. Soldi, T. Narayanan and J. L. Putaux, Macromol. Rapid Commun., 2002, 23, 978–982 CrossRef CAS.
- M. Schappacher and A. Deffieux, J. Am. Chem. Soc., 2008, 130, 14684–14689 CrossRef CAS PubMed.
- N. Nasongkla, B. Chen, N. Macaraeg, M. E. Fox, J. M. Fréchet and F. C. Szoka, J. Am. Chem. Soc., 2009, 131, 3842–3843 CrossRef CAS PubMed.
- Z. Jia, Q. Fu and J. Huang, Macromolecules, 2006, 39, 5190–5193 CrossRef CAS.
- R. J. Williams, A. Pitto-Barry, N. Kirby, A. P. Dove and R. K. O'Reilly, Macromolecules, 2016, 49, 2802–2813 CrossRef CAS PubMed.
- R. Duncan and J. Kopecek, Adv. Polym. Sci., 1984, 57, 51–101 CrossRef CAS.
- Z. Li, B. H. Tan, G. Jin, K. Li and C. He, Polym. Chem., 2014, 5, 6740–6753 RSC.
- Z. Li, D. Yuan, X. Fan, B. H. Tan and C. He, Langmuir, 2015, 31, 2321–2333 CrossRef CAS PubMed.
- M. Ferrari, Nat. Rev. Cancer, 2005, 5, 161–171 CrossRef CAS PubMed.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS PubMed.
- C. Oerlemans, W. Bult, M. Bos, G. Storm, J. F. W. Nijsen and W. E. Hennink, Pharm. Res., 2010, 27, 2569–2589 CrossRef CAS PubMed.
- C. Kiparissides and O. Kammona, Can. J. Chem. Eng., 2013, 91, 638–651 CrossRef CAS.
- I. N. Kurniasih, H. Liang, J. P. Rabe and R. Haag, Macromol. Rapid Commun., 2010, 31, 1516–1520 CrossRef CAS PubMed.
- Z. Xu, S. Liu, H. Liu, C. Yang, Y. Kang and M. Wang, Chem. Commun., 2015, 51, 15768–15771 RSC.
- S. J. Tabatabaei Rezaei, M. R. Nabid, H. Niknejad and A. A. Entezami, Polymer, 2012, 53, 3485–3497 CrossRef CAS.
- R. Jaskula-Sztul, W. Xu, G. Chen, A. Harrison, A. Dammalapati, R. Nair, Y. Cheng, S. Gong and H. Chen, Biomaterials, 2016, 91, 1–10 CrossRef CAS PubMed.
- Y. Wang, L. Li, J. Li, B. Yang, C. Wang, W. Fang, F. Ji, Y. Wen and F. Yao, RSC Adv., 2016, 6, 17728–17739 RSC.
- I. Rashkov, N. Manolova, S. Li, J. Espartero and M. Vert, Macromolecules, 1996, 29, 50–56 CrossRef CAS.
- S. Tanodekaew, R. Pannu, F. Heatley, D. Attwood and C. Booth, Macromol. Chem. Phys., 1997, 198, 927–944 CrossRef CAS.
- T. Fujiwara, M. Miyamoto, Y. Kimura and S. Sakurai, Polymer, 2001, 42, 1515–1523 CrossRef CAS.
- C. Heald, S. Stolnik, K. Kujawinski, C. De Matteis, M. Garnett, L. Illum, S. Davis, S. Purkiss, R. Barlow and P. Gellert, Langmuir, 2002, 18, 3669–3675 CrossRef CAS.
- S. Ben-Shabat, N. Kumar and A. J. Domb, Macromol. Biosci., 2006, 6, 1019–1025 CrossRef CAS PubMed.
- S. Movassaghian, O. M. Merkel and V. P. Torchilin, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2015, 7, 691–707 CrossRef CAS PubMed.
- L. Qiu, Q. Liu, C.-Y. Hong and C.-Y. Pan, J. Mater. Chem. B, 2016, 4, 141–151 RSC.
- J. Liu, Y. Pang, W. Huang, X. Huang, L. Meng, X. Zhu, Y. Zhou and D. Yan, Biomacromolecules, 2011, 12, 1567–1577 CrossRef CAS PubMed.
- G. Chen, R. Jaskula–Sztul, A. Harrison, A. Dammalapati, W. Xu, Y. Cheng, H. Chen and S. Gong, Biomaterials, 2016, 97, 22–33 CrossRef CAS PubMed.
- G. Chen, L. Wang, T. Cordie, C. Vokoun, K. W. Eliceiri and S. Gong, Biomaterials, 2015, 47, 41–50 CrossRef CAS PubMed.
- X. Yang, J. J. Grailer, S. Pilla, D. A. Steeber and S. Gong, Bioconjugate Chem., 2010, 21, 496–504 CrossRef CAS PubMed.
- Y. Zhou and D. Yan, Chem. Commun., 2009, 1172–1188 RSC.
- Y. Zhou, W. Huang, J. Liu, X. Zhu and D. Yan, Adv. Mater., 2010, 22, 4567–4590 CrossRef CAS PubMed.
- F. Qiu, D. Wang, Q. Zhu, L. Zhu, G. Tong, Y. Lu, D. Yan and X. Zhu, Biomacromolecules, 2014, 15, 1355–1364 CrossRef CAS PubMed.
- F. Qiu, C. Tu, Y. Chen, Y. Shi, L. Song, R. Wang, X. Zhu, B. Zhu, D. Yan and T. Han, Chem. – Eur. J., 2010, 16, 12710–12717 CrossRef CAS PubMed.
- F. Qiu, C. Tu, R. Wang, L. Zhu, Y. Chen, G. Tong, B. Zhu, L. He, D. Yan and X. Zhu, Chem. Commun., 2011, 47, 9678–9680 RSC.
- F. Qiu, Y. Huang and X. Zhu, Macromol. Chem. Phys., 2016, 217, 266–283 CrossRef CAS.
- F. Qiu, D. Wang, R. Wang, X. Huan, G. Tong, Q. Zhu, D. Yan and X. Zhu, Biomacromolecules, 2013, 14, 1678–1686 CrossRef CAS PubMed.
- Y. Zhuang, Q. Zhu, C. Tu, D. Wang, J. Wu, Y. Xia, G. Tong, L. He, B. Zhu and D. Yan, J. Mater. Chem., 2012, 22, 23852–23860 RSC.
- Y. Zhuang, Y. Su, Y. Peng, D. Wang, H. Deng, X. Xi, X. Zhu and Y. Lu, Biomacromolecules, 2014, 15, 1408–1418 CrossRef CAS PubMed.
- B. Helms and J. M. Frechet, Adv. Synth. Catal., 2006, 348, 1125–1148 CrossRef CAS.
- B. Helms and J. M. J. Fréchet, Adv. Synth. Catal., 2006, 348, 1125–1148 CrossRef CAS.
- K. E. Price, B. P. Mason, A. R. Bogdan, S. J. Broadwater, J. L. Steinbacher and D. T. McQuade, J. Am. Chem. Soc., 2006, 128, 10376–10377 CrossRef CAS PubMed.
- T. J. Dickerson, N. N. Reed and K. D. Janda, Chem. Rev., 2002, 102, 3325–3344 CrossRef CAS PubMed.
- H. Gao, Macromol. Rapid Commun., 2012, 33, 722–734 CrossRef CAS PubMed.
- J. Zou, Y. Zhao and W. Shi, J. Phys. Chem. B, 2006, 110, 2638–2642 CrossRef CAS PubMed.
- R. M. Crooks and M. Zhao, Adv. Mater., 1999, 11, 217–220 CrossRef.
- V. Chechik and R. M. Crooks, J. Am. Chem. Soc., 2000, 122, 1243–1244 CrossRef CAS.
- S. Aryal, M. Prabaharan, S. Pilla and S. Gong, Int. J. Biol. Macromol., 2009, 44, 346–352 CrossRef CAS PubMed.
- M. R. Nabid and Y. Bide, Appl. Catal., A, 2014, 469, 183–190 CrossRef CAS.
- V. Rodionov, H. Gao, S. Scroggins, D. A. Unruh, A.-J. Avestro and J. M. J. Fréchet, J. Am. Chem. Soc., 2010, 132, 2570–2572 CrossRef CAS PubMed.
- M. E. Piotti, F. Rivera, R. Bond, C. J. Hawker and J. M. J. Fréchet, J. Am. Chem. Soc., 1999, 121, 9471–9472 CrossRef CAS.
- C. Deraedt, N. Pinaud and D. Astruc, J. Am. Chem. Soc., 2014, 136, 12092–12098 CrossRef CAS PubMed.
- C. Deraedt and D. Astruc, Acc. Chem. Res., 2014, 47, 494–503 CrossRef CAS PubMed.
- C. Wang, R. Ciganda, L. Salmon, D. Gregurec, J. Irigoyen, S. Moya, J. Ruiz and D. Astruc, Angew. Chem., Int. Ed., 2016, 55, 3091–3095 CrossRef CAS PubMed.
- S. A. Maier, M. L. Brongersma, P. G. Kik, S. Meltzer, A. A. Requicha and H. A. Atwater, Adv. Mater., 2001, 13, 1501–1505 CrossRef CAS.
- S. Chen and Y. Yang, J. Am. Chem. Soc., 2002, 124, 5280–5281 CrossRef CAS PubMed.
- T. A. Taton, C. A. Mirkin and R. L. Letsinger, Science, 2000, 289, 1757–1760 CrossRef CAS PubMed.
- A. Y. Nazzal, L. H. Qu, X. G. Peng and M. Xiao, Nano Lett., 2003, 3, 819–822 CrossRef CAS.
- C. T. Campbell, S. C. Parker and D. E. Starr, Science, 2002, 298, 811–814 CrossRef CAS PubMed.
- M. Zhao and R. M. Crooks, Angew. Chem., Int. Ed., 1999, 38, 364–365 CrossRef CAS.
- M.-C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS PubMed.
- H. Gao, Macromol. Rapid Commun., 2012, 33, 722–734 CrossRef CAS PubMed.
- L. M. Bronstein and Z. B. Shifrina, Chem. Rev., 2011, 111, 5301–5344 CrossRef CAS PubMed.
- B. A. Rozenberg and R. Tenne, Prog. Polym. Sci., 2008, 33, 40–112 CrossRef CAS.
- M. Filali, M. A. R. Meier, U. S. Schubert and J.-F. Gohy, Langmuir, 2005, 21, 7995–8000 CrossRef CAS PubMed.
- H. Xu, J. Xu, Z. Zhu, H. Liu and S. Liu, Macromolecules, 2006, 39, 8451–8455 CrossRef CAS.
- G. Gorodyska, A. Kiriy, S. Minko, C. Tsitsilianis and M. Stamm, Nano Lett., 2003, 3, 365–368 CrossRef CAS.
- J. H. Youk, M.-K. Park, J. Locklin, R. Advincula, J. Yang and J. Mays, Langmuir, 2002, 18, 2455–2458 CrossRef CAS.
- K. Ishizu, T. Furukawa and H. Yamada, Eur. Polym. J., 2005, 41, 2853–2860 CrossRef CAS.
- N. Yao, W. Lin, X. Zhang, H. Gu and L. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 186–196 CrossRef CAS.
- X. Pang, L. Zhao, W. Han, X. Xin and Z. Lin, Nat. Nanotechnol., 2013, 8, 426–431 CrossRef CAS PubMed.
- H. Xu, J. Xu, X. Jiang, Z. Zhu, J. Rao, J. Yin, T. Wu, H. Liu and S. Liu, Chem. Mater., 2007, 19, 2489–2494 CrossRef CAS.
- J. M. Serin, D. W. Brousmiche and J. M. Fréchet, J. Am. Chem. Soc., 2002, 124, 11848–11849 CrossRef CAS PubMed.
- G. M. Stewart and M. A. Fox, J. Am. Chem. Soc., 1996, 118, 4354–4360 CrossRef CAS.
- J. M. Fréchet, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3713–3725 CrossRef.
- W. Wu, W. Wang and J. Li, Prog. Polym. Sci., 2015, 46, 55–85 CrossRef CAS.
- J. M. Frechet, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4782–4787 CrossRef CAS PubMed.
- A. Adronov and J. M. Fréchet, Chem. Commun., 2000, 1701–1710 RSC.
- M. S. Choi, T. Aida, T. Yamazaki and I. Yamazaki, Angew. Chem., Int. Ed., 2001, 113, 3294–3298 CrossRef.
| This journal is © The Royal Society of Chemistry 2016 |