DOI:
10.1039/C5RA03979G
(Paper)
RSC Adv., 2015,
5, 42889-42902
Thermodynamic investigation of the interaction between the [(η6-p-cymene)Ru(benzaldehyde-N4-phenylthiosemicarbazone)Cl]Cl anticancer drug and ctDNA: multispectroscopic and electrochemical studies†
Received
6th March 2015
, Accepted 7th May 2015
First published on 8th May 2015
Abstract
In this contribution, the interaction between the [(η6-p-cymene)Ru(benzaldehyde-N4-phenylthiosemicarbazone)Cl]Cl anticancer drug and calf thymus DNA (ctDNA) was investigated systematically by using multispectroscopic and electrochemical methods along with viscosity measurements and a DNA melting technique. The absorption spectra of the drug with ctDNA showed a slight blue shift and weak hypochromic effect. The relative viscosity and the melting temperature of ctDNA all increased after the addition of the drug. Using berberine (BH) as a fluorescence probe, this drug statically quenched the fluorescence of the ctDNA–BH complex, and hydrogen bonding and van der Waals interactions played important roles in such an interaction. All the competitive experimental evidence indicated that the binding mode of the drug with ctDNA was intercalative binding. The influences of ionic strength, chemical denaturants, hyperthermia and pH on the binding of the drug with ctDNA were also investigated. The results of FT-IR and CD spectra indicated that the B-form conformation of ctDNA was constant even after the binding interaction of the drug with G–C base pairs of ctDNA. The results of electrochemical experiments further verified the intercalative interaction between ctDNA and the drug. All these results indicated that the biological activity of ctDNA was affected by the [(η6-p-cymene)Ru(benzaldehyde-N4-phenylthiosemicarbazone)Cl]Cl anticancer drug dramatically.
1. Introduction
It is well-known that deoxyribonucleic acid (DNA) contains genetic instructions for the development and function of living organisms. DNA is quite often the main target molecule that exists in many carcinogens, steroids and several classes of drugs. Consequently, the recognition and characterization of the interaction between small molecules and DNA have become much more important since such research can provide valuable information for the development of effective therapeutic agents in controlling DNA replication and gene expression.1–3 Therefore, the investigation of the interaction between DNA and small molecules is of biological importance, and especially drug molecules, have become an active and important subject in many research fields recently.4–6 It is widely reported that most small molecules can easily bind to DNA mainly through three different non-covalent modes: electrostatic attractions, groove-binding and intercalative binding.7 Electrostatic binding often take place on the surface of DNA, while groove-binding is related to the grooves in DNA double helix. In addition, intercalation involves the gap between the stacked base pairs being opened up and a planar aromatic molecule inserting into the bases above and below, which result in the lengthening and unwinding the helix of DNA.8–10 Although some active small molecules can interact with DNA directly, the detailed influencing factors among these interactions are quite complex. Thus, the investigation of the interaction modes and the kinetic mechanism between different kinds of small molecules and DNA are very significant and meaningful to the research of DNA structure after drug binding, also to the rational design and exploitation of novel efficient drugs targeted to DNA, and so forth.11,12
Due to their specific anticancer, antitumor, antiviral and antibacterial activities, Ru(II) arene complexes of thiosemicarbazone (TSC) with the half-sandwich-type structure have attracted great attention by biochemists and have been widely researched by chemists and medical scientists.13–16 Ru(II) arene complexes of TSC anticancer drugs exhibit higher antitumor activity and lower toxicity than platinum drugs, which make them have progressed into potential anticancer drugs of the near future.17–19 The bioactivity effects of some Ru(II) arene complexes are exerted by the inhibition of the replication and amplification of DNA, which lead to the death of cancer cells.20,21 Recently, [(η6-p-cymene)Ru(benzaldehyde-N4-phenylthiosemicarbazone)Cl]Cl is regarded as a novel anticancer drug with lower IC50 values for human cancer cell lines, which heralds its further potential application in medicinal chemistry and clinical medicine.22,23 Because of the potential biomedical applications of Ru(II) arene complexes with TSC ligands, more and more researchers are interested in the investigation of their interaction with biomacromolecules. The investigation of the binding interaction between these bioactive molecules and their target DNA has been an active area of the research, which can help us to deeper understand the anticancer mechanism of these drugs, together with directional design and efficient synthesis of novel and effective drugs against several diseases. Hence, the detailed investigations of the interaction between Ru(II) arene complexes with TSC ligands and DNA are very urgent and quite significant.
Until now, many techniques are applied to the research of interaction between different bioactive molecules and DNA, including spectroscopy,24–26 electrochemical measurements,27,28 viscosity measurement,29,30 capillary electrophoresis,31 atomic force microscope,32 second harmonic generation method33 and molecular docking.34 Among these approaches, the spectroscopic and electrochemical methods are regarded as common and effective strategies due to their sensitivity, convenience and reproducibility. In recent years, the application of spectroscopic techniques in conjunction with viscosity measurement have aroused increasing research interest,29,30 since they can provide information well beyond that obtained by conventional methods.
Herein, we have investigated the interaction between [(η6-p-cymene)Ru(benzaldehyde-N4-phenylthiosemicarbazone)Cl]Cl anticancer drug and calf thymus DNA (ctDNA) by using multispectroscopic and electrochemical methods along with viscosity measurement and DNA melting techniques. This study will be helpful to further understand the mechanism of the interaction between ctDNA and ruthenium(II) anticancer drugs as well as further provide valuable information of biological action of such ruthenium(II) anticancer drugs in organism. The knowledge obtained from such research is useful for the development of potential probes for DNA structure and new ruthenium(II) arene complexes with TSC reagents for cancers and other diseases.
2. Experimental
2.1 Reagents
[(η6-p-Cymene)Ru(benzaldehyde-N4-phenylthiosemicarbazone)Cl]Cl anticancer drug was synthesized and characterized according to the literature reported before.22 The structure of this drug was shown in Fig. 1. The stock solution of 1.0 × 10−3 mol L−1 drug was prepared by dissolving its crystals in DMSO. ctDNA (Sigma, USA) was dissolved in pH 7.4 Tris–HCl buffer and stored at 4 °C. The purity of ctDNA was verified by monitoring the ratio of absorbance at 260 nm to that at 280 nm. The concentration of ctDNA was determined spectrophotometrically using an extinction coefficient ε260 nm = 6600 L mol−1 cm−1.3 Berberine (BH), acridine orange (AO), ethidium bromide (EB) and all other reagents were of analytical reagent grade and used as received without further purification. Ultrapure water with a resistivity of 18.2 MΩ cm was produced by passing through a RiOs 8 unit followed by a Millipore-Q Academic purification set (Millipore, Bedford, MA, USA) and used throughout the whole experiments.
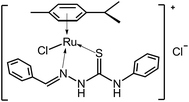 |
| | Fig. 1 The structure of [(η6-p-cymene)Ru(benzaldehyde-N4-phenylthiosemicarbazone)Cl]Cl anticancer drug. | |
2.2 Apparatus
UV-vis absorption spectra were measured on Cary 100 UV-vis spectrophotometer (Agilent, USA). Fluorescence spectra and intensities were performed on Perkin-Elmer LS-55 luminescence spectrometer (PerkinElmer, USA) equipped with thermostatic bath. FT-IR spectra were recorded on Nicolet iS10 spectrometer (Thermo, USA). CD spectra were recorded on Jasco J-810 automatic recording CD spectropolarimeter (Jasco, Japan). Viscosity measurements were carried out using viscosity meter (Yinhua Flowmeter Co. Ltd., China). Electrochemical experiments were performed on CHI-660E electrochemical workstation (Chenhua Instruments Inc., China). All pH measurements were made with a basic pH meter PB-10 (Sartorius Scientific Instruments Co., Ltd., China).
2.3 Procedures
2.3.1 UV-vis absorption measurements. UV-vis absorption spectra of ctDNA, drug as well as ctDNA–drug complex were measured from 200 nm to 500 nm in pH 7.4 Tris–HCl buffer at 25 °C. The solutions of the blank buffer and sample were placed in the reference and sample cuvettes, respectively. The concentration of ctDNA was 9.4 × 10−5 mol L−1, and the concentration of drug was increased from 0 to 2.4 × 10−5 mol L−1 at increments of 4.0 × 10−6 mol L−1, respectively. In order to reflect the interaction between drug and ctDNA, the absorbance values at 260 nm of ctDNA, drug as well as ctDNA–drug complex were recorded, and the absorbance value at 260 nm between ctDNA–drug complex was compared with the sum absorbance value of ctDNA and drug at the same wavelength.
2.3.2 Fluorescence spectra measurements. Fluorescence spectra of ctDNA–AO complex at the present of drug were recorded at 298 K with the excitation/emission slits of 1.5/1.5 nm. The excitation wavelength was set at 475 nm and the fluorescence intensity at 527 nm was recorded. The concentrations of ctDNA and AO were 4.7 × 10−5 mol L−1 and 7.65 × 10−6 mol L−1, respectively. The concentration of drug was increased from 0 to 8.0 × 10−5 mol L−1 at increments of 8.0 × 10−6 mol L−1.Fluorescence spectra of ctDNA–EB complex at the present of drug were recorded at 298 K with the excitation/emission slits of 3.0/3.0 nm. The excitation wavelength was set at 537 nm and the fluorescence intensity at 597 nm was recorded. The concentrations of ctDNA and EB were 4.7 × 10−5 mol L−1 and 2.7 × 10−5 mol L−1, respectively. The concentration of drug was increased from 0 to 8.0 × 10−5 mol L−1 at increments of 8.0 × 10−6 mol L−1.
Fluorescence spectra of ctDNA–BH complex at the present of drug were recorded at 298 K, 304 K and 310 K with the excitation/emission slits of 10.0/10.0 nm. The excitation wavelength was set at 350 nm and the fluorescence intensity at 530 nm was recorded. The concentrations of ctDNA and BH were 9.4 × 10−5 mol L−1 and 1.67 × 10−5 mol L−1, respectively. The concentration of drug was increased from 0 to 2.0 × 10−5 mol L−1 at increments of 2.0 × 10−6 mol L−1.
Fluorescence spectra of ctDNA–BH–drug system in the presence of NaCl, urea and GuHCl were recorded at 298 K with the excitation/emission slits of 10.0/10.0 nm. The excitation wavelength was set at 350 nm and the fluorescence intensity at 530 nm was recorded. The concentrations of ctDNA and BH were 9.4 × 10−5 mol L−1 and 1.67 × 10−5 mol L−1, respectively. The concentration of drug was increased from 0 to 2.0 × 10−5 mol L−1 at increments of 2.0 × 10−6 mol L−1. The concentrations of NaCl, urea and GuHCl were 0.15 mol L−1, 0.01 mol L−1 and 0.01 mol L−1, respectively.
Fluorescence spectra of ctDNA–BH–drug system at different denatured temperatures were recorded at 298 K with the excitation/emission slits at 10.0/10.0 nm. The excitation wavelength was at 350 nm and the fluorescence intensity at 530 nm was recorded. The native double-stranded ctDNA solution was heated in water bath for 30 min at 50 °C, 80 °C and 100 °C, and then the solution was allowed to cool to room temperature through two different cooling approaches: natural cooling and ice-water bath cooling. The concentrations of ctDNA and BH were 9.4 × 10−5 mol L−1 and 1.67 × 10−5 mol L−1, respectively. The concentration of drug was increased from 0 to 2.0 × 10−5 mol L−1 at increments of 2.0 × 10−6 mol L−1.
Fluorescence spectra of ctDNA–BH–drug system at different pH values were recorded at 298 K with the excitation/emission slits of 10.0/10.0 nm. The excitation wavelength was set at 350 nm and the fluorescence intensity at 530 nm was recorded. The concentrations of ctDNA and BH were 9.4 × 10−5 mol L−1 and 1.67 × 10−5 mol L−1, respectively. The concentration of drug was increased from 0 to 2.0 × 10−5 mol L−1 at increments of 2.0 × 10−6 mol L−1. The pH values of the solution changed from pH 2.4 to pH 10.4.
The appropriate blanks corresponding to buffer were subtracted to correct background of fluorescence. Titrations were performed manually by using trace syringes and each spectrum was the average of three scans.
2.3.3 FT-IR spectra measurements. FT-IR spectra of ctDNA and ctDNA–drug complex were recorded in the range of 800–1800 cm−1 with 128 interferograms to ensure good signal-to-noise ratio. The corresponding absorbance contributions of Tris–HCl buffer and drug were recorded and digitally subtracted with the same instrumental parameters. All spectra were taken via ATR method with a resolution of 4 cm−1. The subtraction of the reference spectrum from ctDNA spectrum was carried out in accord with the criteria that straight baseline was obtained between 2000 and 1750 cm−1.35 Blank windows, water and CO2 were subtracted from the spectra of ctDNA and ctDNA–drug complex. The difference spectra [(ctDNA–drug complex) – drug] were analyzed by OMNIC software, using sharp ctDNA band at 968 cm−1 as an internal reference.2,36 The relative intensity (Ir) of several peaks of ctDNA vibrations related to guanine, thymine, adenine and cytosine base pairs stretching vibration, and the phosphate groups stretching vibrations were measured. The plots of Ir versus drug/ctDNA molar ratios were obtained using Ir = Ii/I968, where Ii and I968 were the peak intensities at i cm−1 and 968 cm−1 for ctDNA alone and ctDNA–drug complex, respectively. The concentration of ctDNA was 3.0 × 10−2 mol L−1. The molar ratio of drug to ctDNA was changed from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 to 1:40.
:10 to 1:40.
2.3.4 CD spectra measurements. CD spectra were recorded from 220 nm to 320 nm in pH 7.4 Tris–HCl buffer at room temperature under constant nitrogen airflow. CD profiles were obtained employing scan speed of 500 nm min−1 and response time of 0.5 s. Each spectrum was the average of three successive scans and was corrected by buffer solution. Appropriate baseline corrections in CD spectra were made. The concentration of ctDNA was 4.5 × 10−5 mol L−1. The molar ratio of drug to ctDNA was increased from 0:1 to 10.7:1.
2.3.5 Viscosity measurements. Viscosity of ctDNA solution with different concentration of drug were measured in a viscometer that was kept at constant temperature of 25 ± 0.1 °C in a thermostatic water bath. Flow time measurements were performed by a digital stopwatch with a resolution of 0.01 s. At least five time records reproducible to 0.02 s were obtained, and the average value was used in the calculations. Data were presented as (η/η0)1/3 versus the molar ratio of [drug]/[ctDNA],37 where η and η0 were the viscosity of ctDNA in the presence and absence of drug, respectively. Viscosity values were calculated from the observed flow time of ctDNA containing solutions (t > 100 s) corrected for the flow time of buffer alone (t0), η = (t − t0)/t0. The concentration of ctDNA was 5.0 × 10−5 mol L−1, and the concentration of drug was increased from 0 to 6.65 × 10−5 mol L−1.
2.3.6 DNA melting studies. DNA melting experiments were carried out by monitoring the absorbance of ctDNA at 260 nm at different temperatures in the absence and presence of drug. The temperature was continuously monitored with a thermostatic bath. The absorbance was then plotted as a function of temperature ranging from 20 to 100 °C. The melting temperature of ctDNA was determined as the transition midpoint. The concentrations of ctDNA and drug were 1.0 × 10−4 mol L−1 and 6.0 × 10−5 mol L−1, respectively.
2.3.7 Electrochemical investigation. Cyclic voltammograms and electrochemical impedance spectra were performed on CHI-660E electrochemical workstation with conventional three-electrode electrochemical testing system. The working electrode was bare gold electrode (2 mm diameter), whereas Ag/AgCl electrode served as the reference electrode and platinum wire was used as the counter electrode. The electrolyte solution was 5.0 × 10−3 mol L−1 K3Fe(CN)6/K4Fe(CN)6 and 1.0 × 10−2 mol L−1 KCl. Bare gold electrode was successively polished by α-Al2O3 powder with the diameter of 1.0 μm, 0.3 μm and 0.05 μm, respectively. Subsequently, bare gold electrode was ultrasonically cleaned in acetone, 0.5 mol L−1 H2SO4 solution and ultrapure water, and then dried by nitrogen airflow. For ctDNA modified gold electrode preparation, 5 μL ctDNA solution (4.5 × 10−5 mol L−1) was dropped onto the surface of gold electrode. After 6 h incubation at 20 °C, the electrode was washed by ultrapure water, and then ctDNA modified gold electrode was obtained. For cyclic voltammograms and electrochemical impedance spectra measurements, different concentrations of drug were added into electrolyte solution and then the mixture was stirred for 5 min. The reaction system was at rest for 3 min before testing. Cyclic voltammograms were recorded at scan rate of 50 mV s−1 and electrochemical impedance spectra were measured within frequency range of 0.1–100 KHz. All measurements were repeated three times with different ctDNA modified gold electrode. The concentration of drug was increased from 0 to 9.6 × 10−6 mol L−1 for cyclic voltammograms measurements. The concentration of drug was increased from 0 to 4.32 × 10−5 mol L−1 for electrochemical impedance spectra measurements.
3. Results and discussion
3.1 Absorption spectra
It is well-known that hypochromism and hyperchromism are regarded as spectral characteristics for DNA double-helix structural variation when DNA reacts with other molecules. Usually, hypochromism originates from the DNA duplex stabilization by intercalative binding mode or electrostatic effect of small molecules, while hyperchromism originates from the DNA duplex breakage. So, the UV-vis absorption spectra of ctDNA–drug complex with various concentrations of [(η6-p-cymene)Ru(benzaldehyde-N4-phenylthiosemicarbazone)Cl]Cl anticancer drug in pH 7.4 Tris–HCl buffer at 25 °C were shown in Fig. 2. It was found that the maximum absorption peak of ctDNA was at around 260 nm, which came from the strong absorption of purine and pyrimidine bases in ctDNA.30 With the gradual addition of drug, the absorbance of ctDNA–drug complex increased dramatically, and the peak position of ctDNA–drug complex blue shifted slightly, suggesting that drug might interact with DNA and subsequently affect both the absorbance and peak position of DNA. Meanwhile, the sum absorbance value at 260 nm of ctDNA and drug was a little greater than the absorbance value at 260 nm of ctDNA–drug complex (Insert in Fig. 2), which indicated that a weak hypochromic effect existed after the interaction between ctDNA and drug. This phenomenon suggested that the intercalative binding might be the interaction mode during the interaction of ctDNA with drug.7,8 This speculation would be further supported from the other experiments described later on, which gave convincing evidence of the possible intercalative binding between ctDNA and drug.
 |
| | Fig. 2 UV-vis absorption spectra of ctDNA–drug complex with various concentrations of drug in pH 7.4 Tris–HCl buffer at 25 °C. Insert: Comparison of the absorbance value at 260 nm between ctDNA–drug complex (Square dots) and the sum absorbance value of ctDNA and drug at the same wavelength (Pentagram dots). c(ctDNA) = 9.4 × 10−5 mol L−1; c(drug)/(10−6 mol L−1), 1–7: from 0 to 24.0 at increments of 4.0. | |
3.2 Viscosity investigation
Viscosity experiment is an effective approach to decide the binding mode of other molecules with DNA. The interaction between other molecules and DNA has an effect on the hydrodynamic properties of DNA solution, and only the classical intercalation of other molecules with DNA base pairs can result in the elongation of DNA and increase the viscosity of DNA solution significantly.38,39 The influence of increasing concentration of drug on the relative viscosity of ctDNA at 25 °C was shown in Fig. 3A. The results showed that the relative viscosity of ctDNA increased significantly upon the continuous addition of drug, which indicated that the interaction mode between ctDNA and drug should be the intercalative binding.38,39 Since the viscosity cannot be measured accurately for the higher viscous mixture, the mixture can strongly adsorb on the glass inside the viscometer.40 In order to further confirm the binding mode, the slope value of the linear regression equation between the (η/η0)1/3 value and the lower molar ratio of [drug]–[ctDNA] (from 0 to 0.12) was calculated. As shown in Fig. 3A, the slope value was 0.99 that was near the slope of 1.0 displayed by an ideal intercalation, indicating that this drug was indeed intercalated into the double-helix of ctDNA and such classic intercalative binding led to the lengthening and unwinding of the ctDNA helix.38
 |
| | Fig. 3 (A) Effect of increasing concentration of drug on the relative viscosity of ctDNA at 25 °C. c(ctDNA) = 5.0 × 10−5 mol L−1; c(drug)/(10−6 mol L−1): 0, 1.5, 3.0, 4.5, 6.0, 13.5, 20.0, 26.5, 33.5, 40.0, 46.5, 53.0, 60.0 and 66.5. (B) Melting curves of ctDNA in the absence and presence of drug. c(ctDNA) = 1.0 × 10−4 mol L−1; c(drug) = 6.0 × 10−5 mol L−1. | |
3.3 DNA melting analysis
DNA melting temperature (Tm), which is the temperature at which half of double-stranded DNA is dissociated into single strands, is further evidence for the intercalation of other molecules into the double-helix of DNA. It is well-documented that the intercalation of other molecules with DNA can stabilize the natural structure of DNA and thus increase Tm value of about 5–8 °C.41 However, electrostatic interaction and groove-binding cause no obvious variation on Tm value of DNA. The melting curves of ctDNA in the absence and presence of drug were represented in Fig. 3B. The Tm values of ctDNA and ctDNA–drug complex were about 74.3 °C and 79.2 °C, respectively. The binding of drug with ctDNA resulted in the increase of Tm value by 4.9 °C, suggesting that the stabilization of ctDNA double-helix structure was increased after the intercalative interaction of drug with ctDNA.
3.4 Fluorescence spectroscopic investigation
3.4.1 Binding mode between drug and ctDNA. To obtain an insight into the binding mode of drug with ctDNA, the competitive binding between fluorescent probes and drug for ctDNA was investigated by using AO and EB as fluorescent probes. AO and EB, which are classic intercalating dyes for DNA, are employed as spectral probes to investigate the binding mode of drug with DNA in recent years.2,6,30 If drug intercalates into the double-helix of DNA, drug can compete with AO and EB for the intercalation sites of DNA, and then the fluorescence intensities of ctDNA–AO complex and ctDNA–EB complex can be significantly reduced. As indicated in Fig. 4, the fluorescence intensities of AO and EB all increased after the intercalative binding with DNA. However, after the addition of drug, the fluorescence intensities of both ctDNA–AO complex and ctDNA–EB complex decreased gradually with the increasing concentration of drug. Furthermore, 8.0 × 10−5 mol L−1 of drug presented a displacement effect of 21.0% for EB and 35.6% for AO, suggesting that drug substituted for some AO or EB molecules and dissociated AO or EB molecules from the ctDNA–AO or ctDNA–EB system into the solution. These results reconfirmed that the binding mode between drug and ctDNA was the classic intercalative binding.
 |
| | Fig. 4 (A) Fluorescence spectra of ctDNA–AO complex with various concentrations of drug. The insert corresponds to the Stern–Volmer plot. (B) Fluorescence spectra of ctDNA–EB complex with various concentrations of drug. The insert corresponds to the Stern–Volmer plot. c(ctDNA) = 4.7 × 10−5 mol L−1; c(AO) = 7.65 × 10−6 mol L−1; c(EB) = 2.7 × 10−5 mol L−1; c(drug)/(10−6 mol L−1), 1−11: from 0 to 80.0 at increments of 8.0. | |
3.4.2 Competitive interaction of drug and BH with ctDNA. Since the endogenous fluorescence of ctDNA is very weak, a reliable method of investigating the binding interaction of small molecules to ctDNA is the fluorescence quenching method by using fluorescent probe. In order to understand the interaction pattern of drug with ctDNA more clearly, the competitive binding experiments were carried out using BH as fluorescent probe. BH is one of the most typical intercalators and emits intense fluorescence after the addition of ctDNA. It is reported that the enhanced fluorescence of ctDNA–BH complex can be efficiently quenched after the addition of other molecules.6 The extent of the fluorescence quenching of ctDNA–BH complex is used to determine the extent of the binding between other molecules and ctDNA.The fluorescence spectra of ctDNA–BH complex with various concentrations of drug were shown in Fig. 5. It was obviously that BH exhibited original weak fluorescence around 530 nm in the absence of ctDNA. However, the fluorescence intensity of BH around 530 nm increased gradually after the addition of ctDNA, which indicated that the fluorescence of the system increased after the intercalative binding between ctDNA and BH. Furthermore, the fluorescence intensity of ctDNA–BH complex at 530 nm was quenched regularly with the increasing concentration of drug, and the fluorescence intensity of ctDNA–BH complex decreased dramatically when higher concentration of drug was present. Since 2.0 × 10−5 mol L−1 of drug presented a displacement effect of 38.7% for BH, suggesting that some BH molecules were replaced by drug and released into aqueous medium. These results confirmed the competitive binding interaction between drug and BH with ctDNA, and also suggested that drug probably bound to ctDNA by intercalative mode.
 |
| | Fig. 5 Fluorescence spectra of ctDNA–BH complex with various concentrations of drug. The insert corresponds to the Stern–Volmer plot. c(ctDNA) = 9.4 × 10−5 mol L−1; c(BH) = 1.67 × 10−5 mol L−1; c(drug)/(10−6 mol L−1), 1–11: from 0 to 20.0 at increments of 2.0. | |
It is widely reported that the Ru(II) arene complexes can intercalate between DNA base-pairs mainly through the planarity of the arene group,42,43 so the arene group or this drug may play important role in the intercalative binding between drug and ctDNA. Furthermore, the TSC ligand exhibited almost no influence on the fluorescence intensity of ctDNA–BH complex (Fig. S1†), but the Ru(II) arene complex without TSC ligand showed much stronger quenching effect on the fluorescence of ctDNA–BH complex (Fig. S2 and S3†), indicating that the Ru(II) arene complex without TSC ligand played vital role in such intercalative binding. Therefore, it can be speculated that the combination of TSC ligand and the Ru(II) arene complex dramatically enhance the intercalative binding ability of this drug with ctDNA, which makes this drug easily displace some classical ctDNA intercalators, such as AO, EB and BH.
3.4.3 Quenching mechanism. The fluorescence quenching plot of ctDNA–BH complex by drug (insert in Fig. 5) illustrated that the quenching of ctDNA–BH complex by drug was in good agreement with the classical Stern–Volmer equation:44| |
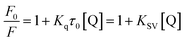 | (1) |
in the above equation, F0 and F are the fluorescence intensities of ctDNA–BH complex in the absence and presence of drug, respectively. Kq is the bimolecular quenching constant and the maximum value of Kq is 2.0 × 1010 L mol−1 s−1 for dynamic quenching. τ0 is the average fluorescent lifetime of biomolecule and the value of τ0 is about 1.0 × 10−8 s. KSV is the Stern–Volmer quenching constant and [Q] is the concentration of drug, respectively. Therefore, the KSV value can be determined by linear regression plot of F0/F versus [Q].The fluorescence quenching mechanism is usually classified as static quenching and/or dynamic quenching, which can be distinguished easily by the different on temperature dependence. Quenching constants decrease with the increment of temperature for static quenching while the reverse is true for dynamic quenching. Static quenching depends upon complex formation and higher temperatures can decrease the stability of complex, therefore the quenching constants decrease with increasing temperature. On the other hand, the increase in temperature will result in larger diffusion coefficients, and thus increase the values of dynamic quenching constant. So, the fluorescence intensities of ctDNA–BH complex at 530 nm with different concentration of drug were measured at 298 K, 304 K, and 310 K, respectively. The fluorescence quenching data were plotted according to Stern–Volmer equation. As shown in Fig. 6A, the results agreed well with the Stern–Volmer equation at lower concentrations. The perfect linear relationship indicated that only one type of quenching process was occurred, either static or dynamic quenching.45 The Stern–Volmer quenching constants KSV at three different temperatures were listed in Table 1. The calculated KSV values showed that increasing temperature resulted in a decrease of KSV values, which was attributed to the static quenching mechanism. Moreover, the calculated Kq values were obtained to be 3.08 × 1012 L mol−1 s−1, 2.75 × 1012 L mol−1 s−1, and 2.39 × 1012 L mol−1 s−1 at 298 K, 304 K, and 310 K, respectively, which were much greater than the maximum value of bimolecular dynamic quenching constant (2.0 × 1010 L mol−1 s−1). The higher Kq values noticed in the present investigation revealed that the quenching mechanism was static quenching.44
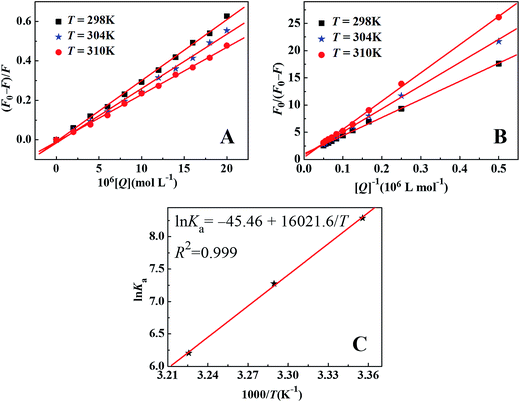 |
| | Fig. 6 Stern–Volmer plots of ctDNA–BH–drug system (A), modified Stern–Volmer plots of ctDNA–BH–drug system (B), and van't Hoff plot of ctDNA–BH–drug system (C). | |
Table 1 Stern–Volmer quenching constants KSV, bimolecular quenching constants Kq, modified Stern–Volmer quenching constants Ka and relative thermodynamic parameters for the interaction between drug and ctDNA–BH complex at three different temperatures
| T (K) |
KSV (104 L mol−1) |
Kq (1012 L mol−1 s−1) |
R2a |
S.D.b |
Ka (104 L mol−1) |
R2a |
ΔH (kJ mol−1) |
ΔG (kJ mol−1) |
ΔS (J mol−1 K−1) |
R2a |
S.D.b |
| R2 is the correlation coefficient. S.D. is standard deviation. |
| 298 |
3.08 |
3.08 |
0.999 |
0.009 |
3.20 |
0.999 |
−133.2 |
−20.56 |
−377.9 |
0.999 |
0.015 |
| 304 |
2.75 |
2.75 |
0.998 |
0.012 |
1.92 |
0.998 |
−18.30 |
0.998 |
0.035 |
| 310 |
2.39 |
2.39 |
0.999 |
0.007 |
0.73 |
0.999 |
−16.03 |
0.999 |
0.028 |
For static quenching process, fluorescence quenching data can be used to calculate the association constant through the modified Stern–Volmer equation:44
| |
 | (2) |
In the present case, fa is the mole fraction of solvent accessible fluorophore, Ka is the modified Stern–Volmer quenching constant for the accessible fluorophores and [Q] is the concentration of drug, respectively. The dependence of F0/ΔF on the reciprocal value of quencher concentration [Q]−1 is linear with the slope equaling the value of (fa Ka)−1, and the value of fa−1 is fixed on the ordinate. The modified Stern–Volmer quenching constant Ka can be calculated from the quotient of the ordinate fa−1 and the slope (fa Ka)−1. The plots of the modified Stern–Volmer equation were shown in Fig. 6B, and the corresponding modified Stern–Volmer quenching constants Ka were also listed in Table 1. The decreasing trend of Ka with increasing temperature was in accord with KSV's dependence on temperature as discussed above, which further confirmed that the fluorescence quenching of ctDNA–BH complex by drug was mainly arisen from static quenching by complex formation.
3.4.4 Binding forces investigation. Usually, the molecular forces contributing to biomolecules interactions with small molecules, which may contain hydrophobic force, van der Waals interactions, multiple hydrogen bonds, electrostatic interactions and so on, can be elucidated through the variation of the thermodynamic parameters calculated from the van't Hoff plots.46 If there is no significant variation in temperature, the enthalpy change (ΔH) does not change obviously in the temperature range, both the enthalpy change (ΔH) and the entropy change (ΔS) can be estimated from the van't Hoff equation:| |
 | (3) |
where Ka is the modified Stern–Volmer quenching constant and R is the gas constant, respectively. As shown in Fig. 6C, a good linear relationship between ln Ka and 1/T existed. The values of ΔH and ΔS were calculated according to the slopes and the intercepts as shown in Fig. 6C. Furthermore, the value of free energy change (ΔG) can be obtained from the following equation:The values of ΔH, ΔS and ΔG were all incorporated in Table 1. The negative values of ΔG indicated that the binding process of drug to ctDNA−BH was spontaneous. The negative ΔH and the negative ΔS values of the interactions of drug and ctDNA–BH suggested that the hydrogen bonding and van der Waals interactions played major roles in such binding reaction.47 Thus, both hydrogen bonding and van der Waals interactions played predominant roles in the binding of drug to ctDNA and contributed to the stability of ctDNA–drug complex.
3.4.5 Effect of ionic strength. Since this anticancer drug carries positive charge and ctDNA has negative polyphosphate skeleton, the influence of ionic strength on ctDNA–drug binding interaction has been studied. It is reported that high concentration of strong electrolyte NaCl may screen the electrostatic repulsion between consecutive phosphate groups, prompt the helix to shrink and thereby hinder the electrostatic interaction between other molecules and DNA.48 Herein, to facilitate the comparison of the influence of NaCl on the binding interaction between drug and ctDNA, the quenching constants of ctDNA–BH–drug system in the absence and presence of NaCl were analyzed using the Stern–Volmer method and the modified Stern–Volmer method (Fig. 7), respectively. The corresponding results were shown in Table 2. These results indicated that both the Stern–Volmer quenching constants KSV and the modified Stern–Volmer quenching constant Ka decreased slightly in the presence of NaCl, which eliminated the strong electrostatic interaction between drug and ctDNA and further confirmed the intercalative binding of drug with ctDNA.
 |
| | Fig. 7 Stern–Volmer plots (A) and modified Stern–Volmer plots (B) of ctDNA–BH–drug system in the absence and presence of NaCl. c(ctDNA) = 9.4 × 10−5 mol L−1; c(BH) = 1.67 × 10−5 mol L−1; c(drug)/(10−6 mol L−1): from 0 to 20.0 at increments of 2.0. c(NaCl) = 0.15 mol L−1. | |
Table 2 Stern–Volmer quenching constants KSV and modified Stern–Volmer quenching constants Ka of ctDNA−BH−drug system in the absence and presence of NaCl, urea or GuHCl
| System |
KSV (104 L mol−1) |
R2a |
S.D.b |
Ka (104 L mol−1) |
R2a |
S.D.b |
| R2 is the correlation coefficient. S.D. is standard deviation. |
| Blank |
3.08 |
0.999 |
0.009 |
3.20 |
0.999 |
0.015 |
| With NaCl |
2.91 |
0.998 |
0.012 |
3.15 |
0.999 |
0.016 |
| With urea |
2.94 |
0.997 |
0.022 |
2.35 |
0.999 |
0.173 |
| With GuHCl |
2.71 |
0.997 |
0.018 |
2.77 |
0.998 |
0.241 |
3.4.6 Effect of chemical denaturation. In comparison with ionic strength, chemical denaturants are more effective in disturbing the structure of DNA and the noncovalent interactions between other molecules and DNA.3 To investigate the effects of chemical denaturants on the binding interaction between drug and ctDNA, urea and GuHCl were introduced into ctDNA–BH–drug system. The quenching constants of ctDNA–BH–drug system in the absence and presence of chemical denaturants were analyzed using the Stern–Volmer method and the modified Stern–Volmer method (Fig. 8), respectively. The corresponding results were also displayed in Table 2. Both the Stern–Volmer quenching constants KSV and the modified Stern–Volmer quenching constant Ka decreased partially in the presence of urea and GuHCl, indicating the weakened intercalating ability of drug with ctDNA. These results suggested that both urea and GuHCl were able to make double-stranded ctDNA separate into single-stranded DNA and then liberate drug from the double-helix of ctDNA.
 |
| | Fig. 8 Stern–Volmer plots (A) and modified Stern–Volmer plots (B) of ctDNA–BH–drug system in the absence and presence of urea or GuHCl. c(ctDNA) = 9.4 × 10−5 mol L−1; c(BH) = 1.67 × 10−5 mol L−1; c(drug)/(10−6 mol L−1): from 0 to 20.0 at increments of 2.0. c(urea) = c(GuHCl) = 0.01 mol L−1. | |
3.4.7 Effect of thermal denaturation. Further support for the intercalative binding of drug with ctDNA is obtained through thermal denaturation experiment. To investigate the effects of hyperthermia on the binding interaction between drug and ctDNA, the native double-stranded ctDNA solution was heated in water bath for 30 min at 50 °C, 80 °C and 100 °C, and then the solution was allowed to cool to room temperature through two different cooling approaches: natural cooling and ice-water bath cooling. Fig. 9 showed the Stern–Volmer plots and the modified Stern–Volmer plots of ctDNA–BH–drug system at different denatured temperatures, respectively. The corresponding results exhibited in Table 3 showed that both the Stern–Volmer quenching constants KSV and the modified Stern–Volmer quenching constant Ka decreased with the increasing temperature, since higher temperature turned more double-stranded DNA to single-stranded DNA. These single-stranded DNA inhibited the intercalating interaction between drug and adjacent base pairs of double-stranded DNA. The modified Stern–Volmer quenching constants obtained in natural cooling DNA system were slight higher than the relevant one in ice-water bath cooling DNA system. Because of the different extent of DNA renaturation under different cooling approaches, the degree of DNA renaturation under natural cooling approach was higher than the degree of DNA renaturation under ice-water bath cooling approach.
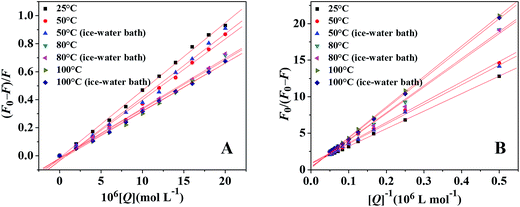 |
| | Fig. 9 Stern–Volmer plots (A) and modified Stern–Volmer plots (B) of ctDNA–BH–drug system at different denatured temperatures. c(ctDNA) = 9.4 × 10−5 mol L−1; c(BH) = 1.67 × 10−5 mol L−1; c(drug)/(10−6 mol L−1): from 0 to 20.0 at increments of 2.0. | |
Table 3 Stern–Volmer quenching constants KSV and modified Stern–Volmer quenching constants Ka of ctDNA–BH–drug system at different denatured temperatures
| T |
KSV (104 L mol−1) |
R2a |
S.D.b |
Ka (104 L mol−1) |
R2a |
S.D.b |
| R2 is the correlation coefficient. S.D. is standard deviation. |
| 25 °C |
3.08 |
0.999 |
0.015 |
3.20 |
0.999 |
0.066 |
| 50 °C |
2.90 |
0.996 |
0.028 |
3.06 |
0.998 |
0.025 |
| 50 °C (ice-water bath) |
2.78 |
0.995 |
0.030 |
2.98 |
0.999 |
0.015 |
| 80 °C |
2.28 |
0.998 |
0.015 |
1.06 |
0.999 |
0.023 |
| 80 °C (ice-water bath) |
2.25 |
0.998 |
0.016 |
0.96 |
0.997 |
0.040 |
| 100 °C |
2.20 |
0.998 |
0.016 |
0.06 |
0.999 |
0.099 |
| 100 °C (ice-water bath) |
2.19 |
0.999 |
0.011 |
0.05 |
0.999 |
0.015 |
3.4.8 Effect of pH. Solution pH plays important role in the stability of double-stranded structures of DNA, so the effects of pH values on the binding interaction between drug and ctDNA are investigated. Fig. 10 presented the Stern–Volmer plots and the modified Stern–Volmer plots of ctDNA–BH–drug system at different pH values, respectively. The corresponding results listed in Table 4 indicated that the Stern–Volmer quenching constant KSV was the lowest and the modified Stern–Volmer quenching constant Ka was the highest at pH 7.4. In addition, the Stern–Volmer quenching constant KSV and the modified Stern–Volmer quenching constant Ka in strong acid solution changed much bigger than the others, because the strong acid disturbed the stability of double-stranded structures of ctDNA and weakened the ability of drug intercalating to ctDNA.
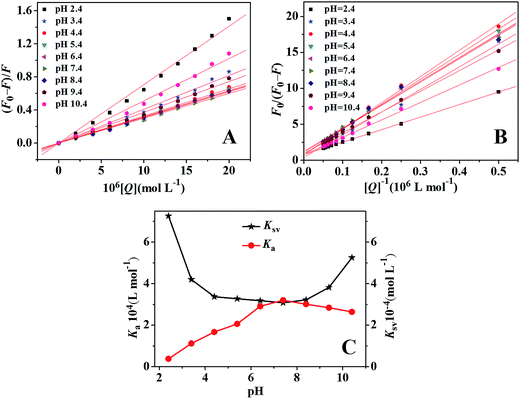 |
| | Fig. 10 Stern–Volmer plots (A) and modified Stern–Volmer plots (B) of ctDNA–BH–drug system at different pH values. (C) Plot of quenching constants versus pH value. c(ctDNA) = 9.4 × 10−5 mol L−1; c(BH) = 1.67 × 10−5 mol L−1; c(drug)/(10−6 mol L−1): from 0 to 20.0 at increments of 2.0. | |
Table 4 Stern–Volmer quenching constants KSV and modified Stern–Volmer quenching constants Ka of ctDNA–BH–drug system at different pH values
| pH |
KSV (104 L mol−1) |
R2a |
S.D.b |
Ka (104 L mol−1) |
R2a |
S.D.b |
| R2 is the correlation coefficient. S.D. is standard deviation. |
| 2.4 |
7.26 |
0.997 |
0.047 |
0.38 |
0.999 |
0.030 |
| 3.4 |
4.20 |
0.996 |
0.026 |
1.12 |
0.997 |
0.034 |
| 4.4 |
3.37 |
0.997 |
0.021 |
1.67 |
0.998 |
0.032 |
| 5.4 |
3.27 |
0.999 |
0.011 |
2.06 |
0.998 |
0.023 |
| 6.4 |
3.18 |
0.998 |
0.015 |
2.91 |
0.997 |
0.035 |
| 7.4 |
3.08 |
0.998 |
0.013 |
3.20 |
0.997 |
0.036 |
| 8.4 |
3.22 |
0.998 |
0.012 |
3.01 |
0.995 |
0.048 |
| 9.4 |
3.82 |
0.997 |
0.020 |
2.84 |
0.999 |
0.014 |
| 10.4 |
5.25 |
0.996 |
0.039 |
2.64 |
0.998 |
0.017 |
3.5 Conformation change investigation
3.5.1 CD spectroscopy. CD spectroscopy is an effective technique to investigate the chiral conformation of DNA under the influence of other molecules. It has been reported that the variation in CD signals of DNA may be assigned to the corresponding changes in DNA structure after the interaction with other molecules.3,49 In order to reveal the influence of drug on the chiral conformation of ctDNA, the CD spectra of ctDNA and ctDNA–drug complexes with different molar ratios of drug to ctDNA were illustrated in Fig. 11A. The ctDNA exhibited a CD spectrum characteristic of the right-handed B-form helicity with a negative band of 246 nm and the base-stacking with a positive band of 277 nm in UV region (Fig. 11A, curve a).3 However, this drug had almost no CD signals in the same UV region. With increasing concentration of drug, the intensity of negative band increased significantly while the intensity of positive band decreased slightly without any remarkable shift in the band position. Therefore, it was deduced that more changes existed in the right-handed B-form helicity than in the base stacking due to the intercalative interaction of drug with ctDNA. However, the right-handed B-form helicity of ctDNA was still constant.
 |
| | Fig. 11 (A) CD spectra of ctDNA, drug and ctDNA–drug complex with various concentrations of drug. c(ctDNA) = 4.5 × 10−5 mol L−1. The molar ratios of drug to ctDNA were 0:1 (a), 1.3:1 (b), 5.3:1 (c) and 10.7:1 (d), respectively. (B) FT-IR spectrum of ctDNA, and the difference FT-IR spectra between ctDNA–drug complex and drug with different molar ratios of drug to ctDNA. c(ctDNA) = 3.0 × 10−2 mol L−1. The molar ratios of drug to ctDNA were 1:10 (a), 1:20 (b) and 1:40 (c), respectively. (C) Intensity ratio variations for several ctDNA vibrations as a function of drug concentration. | |
3.5.2 FT-IR spectroscopy. FT-IR spectroscopy is another efficient method to directly analyze the variation of the chiral conformation of DNA under the influence of other molecules. The variations in FT-IR spectra of DNA can be used to study the specific binding site of other molecules with DNA. So, the FT-IR spectrum of ctDNA, and the difference FT-IR spectra between ctDNA–drug complex and drug with different molar ratios of drug to ctDNA were shown in Fig. 11B. The changes of vibration intensity in spectra were shown in Fig. 11C. It is reported that ctDNA exhibits phosphate stretching vibrations, deoxyribose stretching vibrations and nitrogenous bases stretching vibrations.30 As shown in Fig. 11B, the bands at 1226 cm−1 and 1087 cm−1 of ctDNA were ascribed to the phosphate asymmetric and symmetric vibrations, respectively. Furthermore, the frequency position at 968 cm−1 was corresponded to the deoxyribose C–C and C–O stretching vibrations, and the frequency position at 835 cm−1 was corresponded to the phosphodiester mode that was regarded as a marker band for the B-form of ctDNA.50 With the increasing concentration of drug, the bands of both phosphate stretching vibrations and deoxyribose stretching vibrations had almost no shift (Fig. 11B), and the intensities of these bands varied slightly (Fig. 11C), suggesting that the chiral conformation of ctDNA kept constant in the range of drug concentration. Meanwhile, the band of deoxyribose C–C and C–O stretching vibrations also kept constant upon ctDNA–drug interaction, so this band was used as an internal reference for data analysis.In addition, the bands at 1716 cm−1, 1650 cm−1, 1608 cm−1 and 1496 cm−1 were attributed to guanine, thymine, adenine and cytosine stretching vibration of ctDNA, respectively.50 The changes in the position and intensity of these bands can be used to analyze the binding of drug with four nitrogenous bases. As shown in Fig. 11C, with increasing concentration of drug, the thymine, adenine and cytosine stretching vibration bands shifted from 1650 cm−1 to 1647 cm−1, from 1608 cm−1 to 1606 cm−1, and from 1496 cm−1 to 1506 cm−1, respectively, which were ascribed to the interactions of drug with thymine, adenine and cytosine of ctDNA. Cytosine stretching vibration band had a bigger red-shift, suggesting the stronger interaction between drug and cytosine of ctDNA. Furthermore, the different level increases in the intensities of four bases were observed at higher concentration of drug. Considering changes in both position and intensity of different vibration bands, drug was more prone to interact with G–C base pairs of ctDNA as reported previously.20
3.6 Electrochemical investigation
Cyclic voltammetry can be used to directly analyze the interaction between drug and ctDNA. As indicated in Fig. 12A, a good electrochemical response of K3Fe(CN)6/K4Fe(CN)6 existed on ctDNA modified gold electrode. With the increasing concentration of drug, the redox peak currents decreased significantly and the redox peak potentials shifted slightly, due to the reduction of diffusion coefficient of electrolyte K3Fe(CN)6/K4Fe(CN)6 after the interactive binding of drug with ctDNA. According to values of redox peak currents with different concentrations of drug, the relationship between the reciprocal of the current drop and the reciprocal of drug concentration can be described with the Langmuir equation:51| |
 | (5) |
herein, ΔIp is the current drop, ΔIpmax is the maximum of the current drop, Ka is the binding equilibrium constant, and c is the concentration of drug, respectively. According to the equation and the slope of curve inserted in Fig. 12A, the binding equilibrium constant Ka was calculated to be 6.4 × 104 L mol−1, which was a little higher than the value obtained by spectroscopic methods.
 |
| | Fig. 12 (A) Cyclic voltammograms of bare gold electrode and ctDNA modified gold electrode with various concentrations of drug. c(drug)/(10−6 mol L−1), 1−9: from 0 to 9.6 at increments of 1.2. Insert was the linear relationship between the reciprocal of the current drop and the reciprocal of drug concentration. (B) Electrochemical impedance spectra of ctDNA modified gold electrode with various concentrations of drug. c(drug)/(10−6 mol L−1), 1–10: from 0 to 43.2 at increments of 4.8. Insert was the linear relationship between c/Rct and c. | |
Electrochemical impedance spectroscopy is also used to investigate the interface properties of surface-modified electrodes. The Rct is associated with the semicircle diameter at higher frequency in the Nyquist plot of impedance spectroscopy, and its value increases with the increase of the surface binding on electrode. As illustrated in Fig. 12B, with the continuous addition of drug, the Rct values increased dramatically due to the intercalative interaction between drug and ctDNA. Furthermore, the affinity constant from the data of electrochemical impedance spectroscopy can be calculated according to the Langmuir isotherm model:52
| |
 | (6) |
According to the above equation, the affinity constant Ka can be calculated through the linear plot of c/Rct as a function of c. As shown in the insert of Fig. 12B, the affinity constant Ka, which was calculated from the slope divided by the intercept, was about 3.9 × 104 L mol−1. This value was almost the same with the value measured from the fluorescence titration. All these electrochemical results further validated the intercalative interaction between drug and ctDNA.
4. Conclusions
The interaction of [(η6-p-cymene)Ru(benzaldehyde-N4-phenylthiosemicarbazone)Cl]Cl anticancer drug with ctDNA was investigated by fluorescence spectroscopy, UV-vis absorption, FT-IR, CD spectroscopy and electrochemical methods, coupled with viscosity measurement and DNA melting technique. The results of UV-vis absorption spectra, viscosity measurement, DNA melting and fluorescence spectra indicated that this drug intercalated into the double-helix structure of ctDNA. Furthermore, the results obtained from the analysis of fluorescence spectra exhibited the competitive interaction between drug and AO, EB and BH with ctDNA, which further confirmed the intercalative binding mode between drug and ctDNA. In addition, the fluorescence quenching mechanism of ctDNA–BH complex by this drug was static quenching. Both negative ΔH and negative ΔS values suggested that the interaction was driven mainly by hydrogen bonding and van der Waals interactions. The results of FT-IR spectra and CD spectra revealed that drug bound to G–C base pairs of ctDNA, and such binding interaction did not alter the B-form conformation of ctDNA. The electrochemical experimental results further confirmed the intercalative interaction between this anticancer drug and ctDNA. These results made a better understanding on the interaction between ruthenium(II) arene anticancer drug and ctDNA at the molecular level, which was expected to provide useful information for further understanding the pharmacological mechanism of such ruthenium(II) arene anticancer drugs.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21203035, 21261005, 21403039), the Guangxi Natural Science Foundation (2013GXNSFCA019005), the Scientific Research Foundation of Guangxi Provincial Education Department (ZD2014081), the Innovation Project of Guangxi Graduate Education (YCSZ2014186) and Guangxi Colleges and Universities Key Laboratory of Synthetic and Natural Functional Molecular Chemistry, Guangxi Teachers Education University.
References
- S. Kashanian, Z. Shariati, H. Roshanfekr and S. Ghobadi, DNA binding studies of 3,5,6-trichloro-2-pyridinol pesticide metabolite, DNA Cell Biol., 2012, 31, 1341–1348 CrossRef CAS PubMed.
- Y. Li, G. W. Zhang, J. H. Pan and Y. Zhang, Determination of metolcarb binding to DNA by spectroscopic and chemometrics methods with the use of acridine orange as a probe, Sens. Actuators, B, 2014, 191, 464–472 CrossRef CAS PubMed.
- Nucleic Acids in Chemistry and Biology, ed. G. M. Blackburn and M. J. Gait, Oxford University Press, New York, 2nd edn, 1996 Search PubMed.
- N. H. Campbell, D. L. Smith, A. P. Reszka, S. Neidle and D. O'Hagan, Fluorine in medicinal chemistry: β-fluorination of peripheral pyrrolidines attached to acridine ligands affects their interactions with G-quadruplex DNA, Org. Biomol. Chem., 2011, 9, 1328–1331 CAS.
- Y. Akiyama, Q. Ma, E. Edgar, A. Laikhter and S. M. Hecht, Identification of strong DNA binding motifs for bleomycin, J. Am. Chem. Soc., 2008, 130, 9650–9651 CrossRef CAS PubMed.
- X. L. Li, Y. J. Hu, H. Wang, B. Q. Yu and H. L. Yue, Molecular spectroscopy evidence of berberine binding to DNA: Comparative binding and thermodynamic profile of intercalation, Biomacromolecules, 2012, 13, 873–880 CrossRef CAS PubMed.
- J. E. Coury, L. McFail-Isom, L. D. Williams and L. A. Bottomley, A novel assay for drug-DNA binding mode, affinity, and exclusion number: Scanning force microscopy, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 12283–12286 CrossRef CAS.
- J. B. Chaires, A thermodynamic signature for drug–DNA binding mode, Arch. Biochem. Biophys., 2006, 453, 26–31 CrossRef CAS PubMed.
- M. J. Hannon, Supramolecular DNA recognition, Chem. Soc. Rev., 2007, 36, 280–295 RSC.
- R. Martinez and L. Chacon-Garcia, The search of DNA-intercalators as antitumoral drugs: what it worked and what did not work, Curr. Med. Chem., 2005, 12, 127–151 CrossRef CAS.
- E. Froehlich, J. S. Mandeville, C. M. Weinert, L. Kreplak and H. A. Tajmir-Riahi, Bundling and aggregation of DNA by cationic dendrimers, Biomacromolecules, 2011, 12, 511–517 CrossRef CAS PubMed.
- C. L. Tong, G. H. Xiang and Y. Bai, Interaction of paraquat with calf thymus DNA: A terbium(III) luminescent probe and multispectral study, J. Agric. Food Chem., 2010, 58, 5257–5262 CrossRef CAS PubMed.
- M. Adams, Y. Q. Li, H. Khot, C. D. Kock, P. J. Smith, K. Land, K. Chibale and G. S. Smith, The synthesis and antiparasitic activity of aryl- and ferrocenyl-derived thiosemicarbazone ruthenium(II)-arene complexes, Dalton Trans., 2013, 42, 4677–4685 RSC.
- A. K. Singh, D. S. Pandey, Q. Xu and P. Braunstein, Recent advances in supramolecular and biological aspects of arene ruthenium(II) complexes, Coord. Chem. Rev., 2014, 270–271, 31–56 CrossRef CAS PubMed.
- Q. Zhou, P. Y. Li, R. M. Lu, Q. Q. Qian, X. L. Lei, Q. Xiao, S. Huang, L. F. Liu, C. S. Huang and W. Su, Synthesis, X-ray diffraction study and cytotoxicity of a cationic p-cymene ruthenium chloro complex containing a chelating semicarbazone ligand, Z. Anorg. Allg. Chem., 2013, 639, 943–946 CrossRef CAS PubMed.
- L. F. Liu, P. Y. Li, Q. Q. Qian, X. L. Lei, Y. X. Huang, Q. Zhou, S. Huang, Q. Xiao and W. Su, Synthesis, structure, anticancer activity of 2-formylthiophene thiosemicarbazones and their organometallic ruthenium complexes, Chin. J. Org. Chem., 2013, 33, 854–859 CrossRef CAS.
- F. A. Beckford, D. Dourth, M. Shaloski Jr, J. Didion, J. Thessing, J. Woods, V. Crowell, N. Gerasimchuk, A. Gonzalez-Sarrías and N. P. Seeram, Half-sandwich ruthenium-arene complexes with thiosemicarbazones: synthesis and biological evaluation of [(η6-p-cymene)Ru(piperonal thiosemicarbazones)Cl]Cl complexes, J. Inorg. Biochem., 2011, 105, 1019–1029 CrossRef CAS PubMed.
- T. Stringer, B. Therrien, D. T. Hendricks, H. Guzgay and G. S. Smith, Mono- and dinuclear (η6-arene) ruthenium(II) benzaldehyde thiosemicarbazone complexes: synthesis, characterization and cytotoxicity, Inorg. Chem. Commun., 2011, 14, 956–960 CrossRef CAS PubMed.
- B. Demoro, R. F. M. de Almeida, F. Marques, C. P. Matos, L. Otero, J. C. Pessoa, I. Santos, A. Rodríguez, V. Moreno, J. Lorenzo, D. Gambino and A. I. Tomaz, Screening organometallic binuclear thiosemicarbazone ruthenium complexes as potential anti-tumour agents: cytotoxic activity and human serum albumin binding mechanism, Dalton Trans., 2013, 42, 7131–7146 RSC.
- P. Hanczyc, P. Lincoln and B. Norden, Interactions of binuclear ruthenium(II) complexes with oligonucleotides in hydrogel matrix: Enantioselective threading intercalation into GC context, J. Phys. Chem. B, 2013, 117, 2947–2954 CrossRef CAS PubMed.
- A. Kurzwernhart, W. Kandioller, E. A. Enyedy, M. Novak, M. A. Jakupec, B. K. Keppler and C. G. Hartinger, 3-Hydroxyflavones vs. 3-hydroxyquinolinones: Structure–activity relationships and stability studies on RuII (arene) anticancer complexes with biologically active ligands, Dalton Trans., 2013, 42, 6193–6202 RSC.
- W. Su, Q. Zhou, Y. M. Huang, Q. Y. Huang, L. N. Luo, Q. Xiao, S. Huang, C. S. Huang, R. Chen, Q. Q. Qian, L. F. Liu and P. Y. Li, Synthesis, crystal and electronic structure, anticancer activity of ruthenium(II) arene complexes with thiosemicarbazones, Appl. Organomet. Chem., 2013, 27, 307–312 CrossRef CAS PubMed.
- W. Su, Q. Q. Qian, P. Y. Li, X. L. Lei, Q. Xiao, S. Huang, C. S. Huang and J. G. Cui, Synthesis, characterization, and anticancer activity of a series of ketone-N4-substituted thiosemicarbazones and their ruthenium(II) arene complexes, Inorg. Chem., 2013, 52, 12440–12449 CrossRef CAS PubMed.
- Z. G. Chen, Z. Wang, J. H. Chen, X. Chen, J. H. Wu, Y. Y. Wu and J. Y. Liang, Resonance light scattering technique as a new tool to determine the binding mode of anticancer drug oridonin to DNA, Eur. J. Med. Chem., 2013, 66, 380–387 CrossRef CAS PubMed.
- K. Wu, W. B. Hu, Q. Luo, X. C. Li, S. X. Xiong, P. J. Sadler and F. Y. Wang, Competitive binding sites of a ruthenium arene anticancer complex on oligonucleotides studied by mass spectrometry: ladder-sequencing versus Top-Down, J. Am. Soc. Mass Spectrom., 2013, 24, 410–420 CrossRef CAS PubMed.
- M. Uemura, Y. Yoshikawa, M. Chikuma and S. Komeda, A circular dichroism study uncovers a two-step interaction of antitumor azolato-bridged dinuclear platinum(II) complexes with calf thymus DNA, Metallomics, 2012, 4, 641–644 RSC.
- A. Erdem and G. Congur, Impedimetric detection of in situ interaction between anti-cancer drug bleomycin and DNA, Int. J. Biol. Macromol., 2013, 61, 295–301 CrossRef CAS PubMed.
- C. L. Bian, Q. X. Zeng, L. J. Yang, H. Y. Xiong, X. H. Zhang and S. F. Wang, Voltammetric studies of the interaction of rutin with DNA and its analytical applications on the MWNTs–COOH/Fe3O4 modified electrode, Sens. Actuators, B, 2011, 156, 615–620 CrossRef CAS PubMed.
- G. W. Zhang and Y. D. Ma, Spectroscopic studies on the interaction of sodium benzoate, a food preservative, with calf thymus DNA, Food Chem., 2013, 141, 41–47 CrossRef CAS PubMed.
- X. Y. Zhou, G. W. Zhang and L. H. Wang, Binding of 8-methoxypsoralen to DNA in vitro: Monitoring by spectroscopic and chemometrics approaches, J. Lumin., 2014, 154, 116–123 CrossRef CAS PubMed.
- A. A. Ouameur and H. A. Tajmir-Riahi, Structural analysis of DNA interactions with biogenic polyamines and cobalt(III) hexamine studied by Fourier transform infrared and capillary electrophoresis, J. Biol. Chem., 2004, 79, 42041–42054 CrossRef PubMed.
- X. M. Hou, X. H. Zhang, K. J. Wei, C. Ji, S. X. Dou, W. C. Wang, M. Li and P. Y. Wang, Cisplatin induces loop structures and condensation of single DNA molecules, Nucleic Acids Res., 2009, 37, 1400–1410 CrossRef CAS PubMed.
- B. Doughty, Y. Rao, S. W. Kazer, S. J. J. Kwok, N. J. Turro and K. B. Eisenthal, Binding of the anti-cancer drug daunomycin to DNA probed by second harmonic generation, J. Phys. Chem. B, 2013, 117, 15285–15289 CrossRef CAS PubMed.
- N. Shahabadi, S. M. Fili and F. Kheirdoosh, Study on the interaction of the drug mesalamine with calf thymus DNA using molecular docking and spectroscopic techniques, J. Photochem. Photobiol., B, 2013, 128, 20–26 CrossRef CAS PubMed.
- A. C. Dong, P. Huang and W. S. Caughey, Protein secondary structures in water from second-derivative amide I infrared spectra, Biochemistry, 1990, 29, 3303–3308 CrossRef CAS.
- C. D. Kanakis, P. A. Tarantilis, M. G. Polissiou, S. Diamatoglou and H. A. Tajmir-Riahi, An overview of DNA and RNA bindings to antioxidant flavonoids, Cell Biochem. Biophys., 2007, 49, 29–36 CrossRef CAS PubMed.
- S. Kashanian and J. E. N. Dolatabadi, In vitro study of calf thymus DNA interaction with butylated hydroxyanisole, DNA Cell Biol., 2009, 28, 535–540 CrossRef CAS PubMed.
- G. Cohen and H. Eisenberg, Viscosity and sedimentation study of sonicated DNA–proflavine complexes, Biopolymers, 1969, 8, 45–55 CrossRef CAS PubMed.
- S. Satyanarayana, J. C. Dabrowiak and J. B. Chaires, Neither Δ- nor ˄-tris(phenanthroline)ruthenium(II) binds to DNA by classical intercalation, Biochemistry, 1992, 31, 9319–9324 CrossRef CAS.
- H. C. M. Yau, H. L. Chan and M. S. Yang, Determination of mode of interactions between novel drugs and calf thymus DNA by using quartz crystal resonator, Sens. Actuators, B, 2002, 81, 283–288 CrossRef CAS.
- C. V. Kumar, R. S. Turner and E. H. Asuncion, Groove binding of a styrylcyanine dye to the DNA double helix: the salt effect, J. Photochem. Photobiol., A, 1993, 74, 231–238 CrossRef CAS.
- H. K. Liu, J. A. Parkinson, J. Bella, F. Y. Wang and P. J. Sadler, Penetrative DNA intercalation and G-base selectivity of an organometallic tetrahydroanthracene RuII anticancer complex, Chem. Sci., 2010, 1, 258–270 RSC.
- M. S. Deshpande, A. A. Kumbhar, A. S. Kumbhar, M. Kumbhakar, H. Pal, U. B. Sonawane and R. R. Joshi, Ruthenium(II) complexes of bipyridine-glycoluril and their interactions with DNA, Bioconjugate Chem., 2009, 20, 447–459 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer, New York, 3rd edn, 2006 Search PubMed.
- N. Shahabadi, M. Maghsudi, Z. Kiani and M. Pourfoulad, Multispectroscopic studies on the interaction of 2-tert-butylhydroquinone (TBHQ), a food additive, with bovine serum albumin, Food Chem., 2011, 124, 1063–1068 CrossRef CAS PubMed.
- D. Leckband, Measuring the forces that control protein interactions, Annu. Rev. Biophys. Biomol. Struct., 2000, 29, 1–26 CrossRef CAS PubMed.
- D. P. Ross and S. Subramanian, Thermodynamics of protein association reactions: forces contributing to stability, Biochemistry, 1981, 20, 3096–3102 CrossRef.
- D. Sarkar, P. Das, S. Basak and N. Chattopadhyay, Binding interaction of cationic phenazinium dyes with calf thymus DNA: A comparatives study, J. Phys. Chem. B, 2008, 112, 9243–9249 CrossRef CAS PubMed.
- P. Lincoin, E. Tuite and B. Norden, Short-circuiting the molecular wire: Cooperative binding of Δ-[Ru(phen)(2)dppz](2+) and Δ-[Rh(phi)(2)bipy](3+) to DNA, J. Am. Chem. Soc., 1997, 119, 1454–1455 CrossRef.
- F. Arjmand, S. Parveen, M. Afzal and M. Shahid, Synthesis, characterization, biological studies (DNA binding, cleavage, antibacterial and topoisomerase I) and molecular docking of copper(II) benzimidazole complexes, J. Photochem. Photobiol., B, 2012, 114, 15–26 CrossRef CAS PubMed.
- S. Huang, F. W. Zhu, Q. Xiao, Q. Zhou, W. Su, H. N. Qiu, B. Q. Hu, J. R. Sheng and C. S. Huang, Combined spectroscopy and cyclic voltammetry investigates the interaction between [(η6-p-cymene)Ru(benzaldehyde-N(4)-phenylthiosemicarbazone)Cl]Cl anticancer drug and human serum albumin, RSC Adv., 2014, 4, 36286–36300 RSC.
- D. L. Li, X. Q. Zou, Q. Shen and S. J. Dong, Kinetic study of DNA/DNA hybridization with electrochemical impedance spectroscopy, Electrochem. Commun., 2007, 9, 191–196 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra03979g |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
















