
Synthesis of 4-aminobenzoic acid esters of polyethylene glycol and their use for pegylation of therapeutic proteins†
Abstract
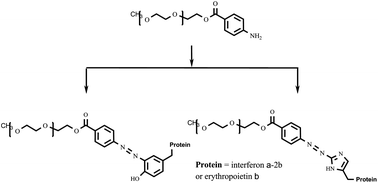
As the majority of reagents commonly used for protein pegylation have certain disadvantages concerning their reactivity, stability, convenience and selectivity of pegylation reaction, etc., the search for new pegylation reagents is of current interest. In the present paper, 4-aminobenzoic acid esters of polyethylene glycol are considered as promising pegylation reagents for chemical modification of molecules of biologically active proteins to prepare their conjugates characterized by improved therapeutic properties. These reagents are highly reactive and stable and make it possible to perform histidine- and tyrosine-targeted pegylation of protein chains. The convenient technique for the synthesis of 4-aminobenzoic acid esters of polyethylene glycol was optimized to prepare these pegylation reagents with actual quantitative loading of the polymer with the functional group (the residue of 4-aminobenzoic acid). The efficiency of the synthesized compounds was shown in the pegylation of such proteins as interferon α-2b and erythropoietin β and it was found that the activity of the prepared interferon α-2b conjugate is comparable with those of its commercial analogs such as Pegintron® and Pegasys®.
 Please wait while we load your content...
Please wait while we load your content...

