
Asymmetric side-chain engineering in semiconducting polymers: a platform for greener processing and post-functionalization of organic electronics†
Madison
Mooney‡
a,
Audithya
Nyayachavadi‡
a,
Angela
Awada
a,
Ekaterini
Iakovidis
a,
Yunfei
Wang
 b,
Mei-Nung
Chen
c,
Yuzi
Liu
d,
Jie
Xu
d,
Yu-Cheng
Chiu
c,
Xiaodan
Gu
b and
Simon
Rondeau-Gagné
*a
b,
Mei-Nung
Chen
c,
Yuzi
Liu
d,
Jie
Xu
d,
Yu-Cheng
Chiu
c,
Xiaodan
Gu
b and
Simon
Rondeau-Gagné
*a
aDepartment of Chemistry and Biochemistry, University of Windsor, Ontario, Canada N9B 3P4. E-mail: srondeau@uwindsor.ca
bSchool of Polymer Science and Engineering, The University of Southern Mississippi, Hattiesburg, MS 39406, USA
cDepartment of Chemical Engineering, National Taiwan University of Science and Technology, Taipei 106, Taiwan
dNanoscience and Technology Division, Argonne National Laboratory, Lemont, IL, USA
First published on 23rd December 2022
Abstract
Organic semiconducting polymers are a powerful platform for the design of next-generation technologies due to their excellent optoelectronic properties and solution processability, allowing access to low-cost and scalable manufacturing techniques such as spin-coating, slot-die coating and roll-to-roll printing. However, their extended π-conjugation results in low solubility, requiring the use of toxic halogenated solvents to generate thin films and devices. Furthermore, accessible post-functionalization of semiconductors toward the development of multifunctional devices and sensors remains a challenge due to limited solid-state chemistry for alkyl side chains. In this work, an asymmetric side-chain engineering approach was used to introduce terminal hydroxyl moieties alongside traditional solubilizing branched alkyl chains into an isoindigo-based polymer. The hydroxyl moieties led to significantly improved processability in alcohol-based solvents without sacrificing electronic performance in thin film organic field-effect transistors. Solid state morphologies of the thin films processed from both alcohol-based and traditional halogenated solvents were further characterized using atomic force microscopy and grazing incidence wide angle X-ray scattering. Additionally, Cryo-EM was utilized in order to characterize the role of asymmetric side-chain functionality in solution state aggregation. The versatility of this design was further probed using fluorescein isothiocyanate to directly functionalize the asymmetric polymer in thin film. This facile solid-state post-functionalization further demonstrates asymmetric side-chain engineering to be a viable approach toward the development of sustainably manufactured multifunctional electronics.
Introduction
Organic semiconducting polymers (SPs) are increasingly important materials for the development of new technologies due to their excellent electronic properties, which have recently demonstrated top performance comparable to amorphous silicon.1,2 Their mechanical properties, including conformability and low Young's moduli compared to other electroactive materials, are also particularly favorable for the development of conformable, wearable and stretchable electronics.3,4 Moreover, SPs are solution-processable, allowing for the use of low-cost production and manufacturing techniques such as printing that remain inaccessible to silicon-based devices.5 Another important feature of SPs is the potential for post-functionalization through careful selection and incorporation of chemical motifs, allowing for the development of multifunctional organic electronics.6,7 It is therefore unsurprising that these materials have been explored for applications in many emerging technologies, including skin-like electronics, large area flexible and plastic photovoltaics, conformable solid-state batteries, and high-precision biological and chemical sensors.8–13While these attributes are highly sought after, the mass production and commercialization of organic electronics is currently restrained by two major factors. First, due to their extended π-conjugation, current high-performance semicrystalline rigid donor–acceptor polymers are largely processed in toxic halogenated solvents that severely impede the sustainability of future technologies.14 These include high-boiling point aromatic (chlorobenzene, 1,2,4-trichlorobenzene, toluene), halogenated (chloroform, dichloromethane), and other harmful or toxic solvents. Beyond environmental concerns, the limited solubility of these materials also limits their use in manufacturing methods such as printing that require fine-tuning of solvents and additive optimization to formulate usable inks.15 Second, it is still challenging to include functional groups to SPs, which are designed often only with large aliphatic side chains for solubility. The incorporation of desirable functional groups (azides, alcohols, amines, etc.) that can be used for chemical functionalization tends to render materials insoluble at high concentrations while also sacrificing regioregularity, making batch-to-batch variation a significant obstacle for electronic performance.16
Among the various strategies for functional group incorporation to control different properties of SPs, asymmetric side-chain engineering is an increasingly relevant approach which has already shown promising results for the fine-tuning of solid-state morphology, mechanical robustness, processability, and electronic performance of semiconducting materials.17–20 It is notable that, compared to other molecular engineering strategies, asymmetric side-chain engineering maintains high conjugation lengths in SPs while offering greater synthetic versatility as the mismatch in design allows for functional group incorporation at larger concentrations that would likely render symmetrical designs insoluble.19,21 Asymmetric side-chain design is also advantageous in terms of regularity and decreased batch-to-batch variation in comparison to recently reported statistical copolymerization designs.22 Liu et al. reported one of the first asymmetrically alkylated semiconductors for applications in organic electronic (organic field-effect transistors, OFETs) devices through a combination of linear and branched alkyl chains for naphthalene diimide (NDI) based SPs.23 The asymmetric materials demonstrated electron mobilities over a magnitude greater than its fully symmetric branched-chain counterpart, which was directly attributed to closer packing of the molecular backbones afforded by the asymmetric linear alkyl chains. More recently, Gumyusenge et al. reported the synthesis of an asymmetric diketopyrrolopyrrole (DPP) based polymer containing a siloxane-terminated alkyl chain and a branched alkyl chain. The material was melt processed at 115 °C in a blend with 5% mass of an amorphous nonconjugated polymer to produce OFET devices with hole mobilities as high as 1.0 cm2 V−1 s−1.24 Whiles these designs showed great promises toward the preparation of new advanced semiconducting polymers, the asymmetric design strategy often does not include new functional groups, nor significantly change the polarity of the side chains. Therefore, fine-tuning of the design of asymmetric SPs can positively impact many more properties than what have been previously reported, such as greener processability, mechanical robustness, and post-functionality of the materials.
Herein, we report the synthesis and characterization of an asymmetric isoindigo-based SP containing branched alkyl and linear hydroxyl side chains. This new design strategy resulted in a polymer that is processable in green alcohol-based solvents due to the regioregular hydroxyl side chain.25 After complete solid-state characterization by atomic force microscopy (AFM), grazing incidence wide angle X-ray scattering (GIWAXS) and optical spectroscopies, the material was directly used in the fabrication of OFETs with both conventional halogenated and eco-friendly solvents, providing hole mobility values typical of established side-chain engineered isoindigo-based SPs.26,27 Importantly, performance was not impacted by the exposure of the hydroxyl side chains or use of greener solvents for processing. In fact, a slight increase in performance was observed in the greener processing conditions. Compared to previously reported asymmetric side chain designs, the use of terminal alcohol side chains in combination with traditionally solubilizing branched alkyl chains also confers unique opportunities for organic electronic design. Unlike polyethylene glycol or siloxane asymmetric sidechains, terminal alcohol chains are commercially available and bio-sourced, resulting in greener and sustainable sourcing of materials as well as fewer synthetic steps to achieve target materials of interest. Additionally, a myriad of readily cleavable and solubilizing protecting groups exist for terminal hydroxyl motifs, allowing for good solubility of monomers in organic solvents and the ability to polymerize into high molecular weight polymers, sometimes a problem seen with the use of polar side chains.12,28,29 Importantly, the presence of terminal alcohols offers an accessible route for post-functionalization in thin films, providing a versatile platform for the development of multifunctional OFETs in a greener manner that are tailored to specific applications through rational selection of motifs.
To demonstrate the potential of this system as a platform for multifunctional organic electronics, the asymmetric SP (in thin film) was functionalized with fluorescein isothiocyanate (FITC), a contemporary fluorescent probe that is used for detection of various analytes including phosphates, antibodies, and apoptotic cells.30–32 Polymer thin films were exposed to a solution of FITC probe to form covalent thiocarbamate linkages upon solid-state reaction with terminal alcohols, confirmed through excitation–emission spectroscopy. Thus, the utilization of terminal alcohol side chains in combination with solubilizing branched chains provides a novel and facile route toward greener processed multifunctional organic electronics. This approach also opens new avenues for the design and synthesis of advanced conjugated polymers with various functional groups, particularly sought after to access novel soft electronics and electrochemical sensors.
Results and discussion
Synthesis and solid-state properties
The synthetic pathway to the poly(isoindigo)-based random copolymers a-P(iIT)-TBS and a-P(iIT)-OH is shown in Scheme 1. Compound 1 was synthesized via a one-pot alkylation with both the tert-butyldimethylsilyl (TBS) protected polar side chain and branched alkyl side chain added simultaneously to produce the symmetrically alkylated derivatives of both side chains as well as the target asymmetric product. The resulting mixture of products was separated using column chromatography to afford compound 1 in moderate yields (30%). The asymmetric monomer was then polymerized with commercially available 2,5-bis(trimethylstannyl)thiophene through Stille polycondensation to produce a-P(iIT)-TBS. The polymer was purified through successive Soxhlet extractions. Upon purification of a-P(iIT)-TBS, bearing a tert-butyldimethylsilyl-protected hydroxyl side chain, the polymer was deprotected under mild acidic conditions to access a-P(iIT)-OH with asymmetric polar and nonpolar side chains. A detailed experimental procedure for the synthesis of a-P(iIT)-TBS and a-P(iIT)-OH can be found in the ESI.† | ||
| Scheme 1 Synthetic pathway to asymmetric polymer a-P(iIT)-OH. | ||
To confirm the successful deprotection of the hydroxyl moiety to afford a-P(iIT)-OH, Fourier-transform infrared (FTIR) spectroscopy was utilized in the solid state to characterize the polymer before and after acid treatment with the results depicted in Fig. 1 and S1.† As shown in Fig. 1a, the appearance of a clear peak, indicated by an arrow at 3400 cm−1, associated with intermolecular hydrogen bonding of the alcohol moieties confirms the removal of TBS. To further confirm the structure of the polymer, 1H NMR spectroscopy at 100 °C in deuterated 1,1,2,2-tetrachloroethane-d2 (TCE) was performed. As shown in Fig. S2,† variable-temperature NMR spectroscopy confirmed that the acid-catalyzed removal of TBS was successful. Upon confirmation of their chemical structures, the synthesized polymers were characterized through various methods, with the results summarized in Table 1. a-P(iIT)-TBS was found to have a molecular weight of 36.9 kDa as measured by high-temperature gel-permeation chromatography (GPC). This observation of good molecular weights for a linear side chain design indicates that this strategy can lead to high molecular weight species without sacrificing solubility during the polymerization process directly afforded by side-chain asymmetry.33 As shown in Fig. S3 and S4,† the HOMO/LUMO energy levels and bandgap were evaluated by UV-vis spectroscopy and cyclic voltammetry. Additionally, thermogravimetric analysis (TGA) (Fig. S5†) was utilized to determine the thermal decomposition temperatures (measured at 5% weight loss). Notably, upon deprotection of the alcohol moieties, the degradation temperature increased from 283 to 358 °C. This difference in decomposition temperature can be directly attributed to the decomposition of the TBS side chains. As previously reported, the network of hydrogen bonds that is afforded by the terminal hydroxyl moieties can also play a role in the enhanced thermal stability.34,35
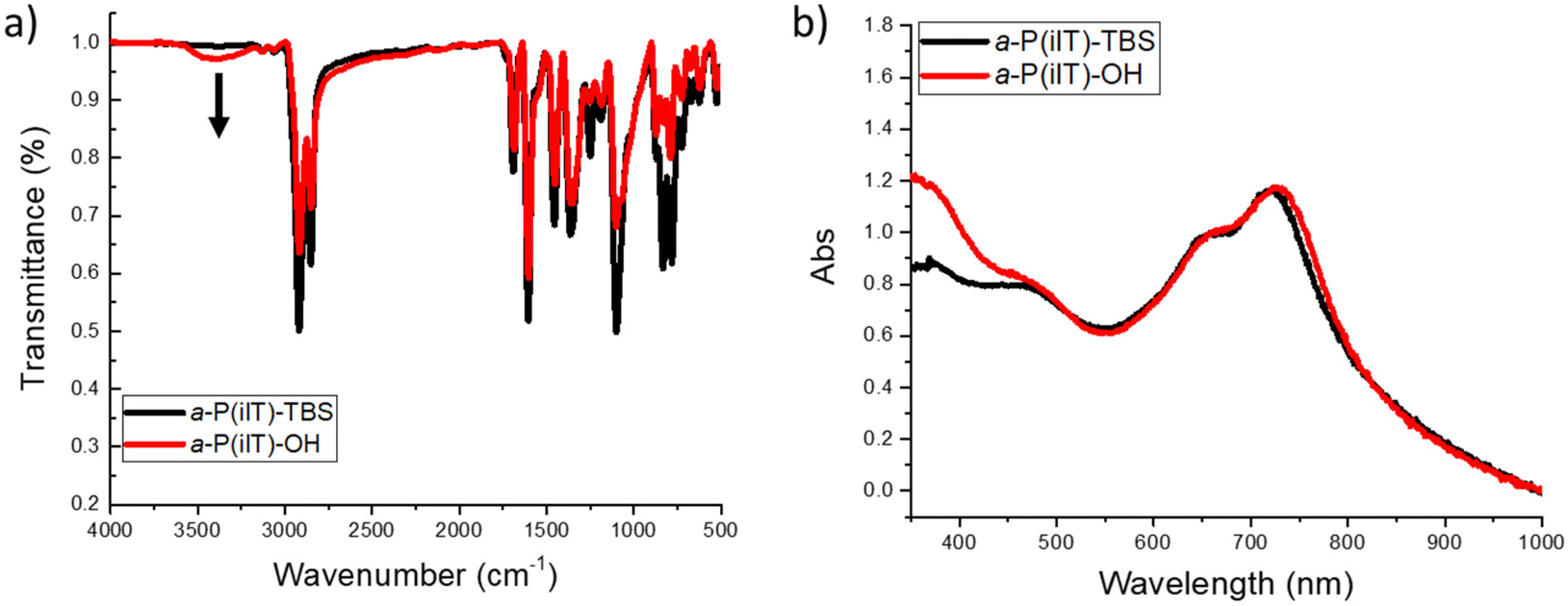 | ||
| Fig. 1 Fourier-Transform infrared (FTIR) spectra of (a) a-P(iIT)-TBS and a-P(iIT)-OH in the solid-state, and (b) UV-vis spectra of a-P(iIT)-TBS and a-P(iIT)-OH thin films casted on SiO2. | ||
M
n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a,b (kDa) a,b (kDa) |
M
wa,b (kDa) |
Đ |
λ
max (film)d (nm) |
E
optge (eV) |
HOMOf (eV) | LUMOg (eV) |
T
dh (°C) |
|
|---|---|---|---|---|---|---|---|---|
| a Number-average molecular weight and weight-average molecular weight estimated by high-temperature gel permeation chromatography in 1,2,4-trichlorobenzene at 180 °C using polystyrene as standard. b Molecular weight data was collected for the TBS-protected polymer only since the deprotection constitutes a side-chain post-modification. c Dispersity defined as Mw/Mn. d Absorption maxima in thin film. e Calculated by the following equation: gap = 1240/λonset of polymer film. f Calculated from cyclic voltammetry (potentials vs. Ag/AgCl) using 0.1 M TBAPF6 in CH3CN as electrolyte where EHOMO = −4.38 eV − (OxOnset). g Estimated from calculated Eg and HOMO. h Estimated from thermogravimetry analysis (TGA) at 5% mass loss. | ||||||||
| a-P(iIT)-TBS | 36.9 | 107.2 | 2.8 | 719 | 1.45 | −5.42 | −3.97 | 283 |
| a-P(iIT)-OH | 724 | 1.44 | 358 | |||||
To further probe for the influence of the hydroxyl moieties on the optoelectronic properties of the conjugated polymer, UV-vis spectroscopy was performed on thin films (Fig. 1b) before and after material deprotection. A broad absorption band centered at λ = 680 nm was observed for both a-P(iIT)-TBS and a-P(iIT)-OH, which can be attributed to the donor–acceptor charge transfer in the π-conjugated backbone. This confirms that the deprotection of the hydroxyl moieties does not negatively affect the π-conjugation of the polymers. Notably, a slight bathochromic shift is observed upon deprotection, which can be directly attributed to the presence of hydrogen bonding promoted aggregation in a-P(iIT)-OH.
Solid-state characterization of the two polymers were further investigated using grazing-incidence wide-angle X-ray scattering (GIWAXS) and atomic force microscopy (AFM) to observe the molecular packing, crystallite characteristics and thin film morphologies of a-P(iIT)-TBS and a-P(iIT)-OH in both toxic halogenated and greener solvents. Since the hydroxyl groups exposed in a-P(iIT)-OH significantly impact the solubility of the polymer, thin films of both polymers processed in several different solvents were characterized. A typical, high boiling point chlorinated solvent (chlorobenzene), and two greener solvent systems (o-anisole and a 20% v/v blend of o-anisole in n-butanol) were used to generate thin films.36 These solvents were selected for several reasons. Chlorobenzene was chosen as a standard halogenated solvent for comparison to the greener processing conditions because it is commonly used in OFET fabrication (which these films would later be used for). o-Anisole was chosen because, like chlorobenzene, it is a high-boiling aromatic solvent but it is significantly less toxic and environmentally harmful, making it a greener alternative with similar solubilizing properties. Finally, the 20% v/v o-anisole/n-butanol blend was chosen due to previous reports of this combination for use in the fabrication of OFETs with polar hydroxyl-containing carbohydrate side chains.37 While the hydroxyl side chains of a-P(iIT)-OH was observed to be fairly soluble in n-butanol alone, the resulting film quality was suboptimal due to the presence of large aggregates. With the addition of 20% o-anisole by volume, the resulting thin films were more uniform, owing to o-anisole that helped to break up the π–π interactions in solution while n-butanol alone only interacted with the hydroxyl side chains.
1D sector-averaged profiles (both out-of-plane and in-plane directions) obtained from GIWAXS are shown in Fig. 2. Both polymers show relatively low crystallinity in the solid-state as demonstrated by the lack of higher order lamellar spacing reflections in the qz direction. These observations are consistent with other SP designs that possess asymmetric side-chain engineering motifs that disrupt solid-state packing due to their lack of symmetry parallel to the polymer backbone direction.38 The deprotected a-P(iIT)-OH showed no higher order reflections peak in all 3 solvent systems tested. This indicates that despite the lack of the bulky TBS protecting group combined to the presence of hydrogen bonding that can increase thin film crystallinity, the isotropic nature of hydroxyl-based hydrogen bonding in combination with the asymmetric side chain chemical structure promotes an amorphous morphology independent of solvent processing conditions.
 | ||
| Fig. 2 Grazing-incidence wide-angle X-ray scattering (GIWAXS) scattering profile slices of a-P(iIT)-TBS (a) in-plane and (b) out-of-plane, and a-P(iIT)-OH (c) in-plane and (d) out-of-plane. | ||
Atomic force microscopy (AFM) was used to investigate the surface microstructures of a-P(iIT)-TBS and a-P(iIT)-OH and probe for the influence of the asymmetric hydroxyl motif on the solid-state morphology (Fig. 3 and S6†). Thin films of the polymers were cast in chlorobenzene, anisole and anisole/n-BuOH solvent systems. Both the protected and deprotected polymers showed relatively smooth surfaces, with a-P(iIT)-OH showing increased surface roughness in comparison to the a-P(iIT)-TBS, which can be attributed to the presence of hydrogen bonding driven aggregation domains.12 Notably, the addition of o-anisole as an additive in n-BuOH during spin-coating for a-P(iIT)-TBS and a-P(iIT)-OH resulted in opposing influences on thin film roughness (Fig. 3c and f), with a-P(iIT)-TBS showing an increase in surface roughness and a-P(iIT)-OH showing reduced surface roughness when processed from this solvent mixture. This trend is opposite to the results obtained when both polymers were in o-anisole, which suggests that the addition of n-BuOH assists in disrupting the solution-state aggregation and improves the effective solubility of the hydrogen bond containing polymer.
 | ||
| Fig. 3 Atomic force microscopy (AFM) height images with root-mean-square (RMS) surface roughness for a-P(iIT)-OH thin films casted from (a) chlorobenzene, (b) o-anisole and (c) 20% v/v o-anisole/n-BuOH; a-P(iIT)-TBS thin films casted from (d) chlorobenzene, (e) o-anisole and (f) 20% v/v o-anisole/n-BuOH. Scale bar is 200 nm. | ||
Greener fabrication of OFETs
After assessing the physical and solid-state properties of the new asymmetric isoindigo-based polymers, thin films of a-P(iIT)-TBS and a-P(iIT)-OH were used as active layers in OFETs to evaluate their electronic properties. Bottom-gate top-contact devices were fabricated using chlorobenzene, o-anisole and 20% v/v o-anisole/n-butanol. The experimental procedure for the fabrication of these devices can be found in the ESI.† Briefly, a-P(iIT)-TBS and a-P(iIT)-OH were each dissolved in the desired solvents (4 mg mL−1) before being spin-coated at 1000 rpm onto an octyltrimethoxysilane (OTS) functionalized Si/SiO2 wafer.39 Physical vapor deposition was then used to deposit gold electrodes onto the substrate. Fig. S7 and S8† show the measured transfer and output curves of the two polymers processed in various solvents with the results summarized in Table 2. While some output curves exhibit non-ideal saturation behaviour at lower VDS, potentially attributed to charge injection resulting in minor contact resistance, extracted mobilities are reliable since the transfer curve measurements were made using a −60VDS bias, within the saturation region of the output curves (Fig. S7†).40 The charge carrier mobility was extracted from the transfer curves using linear fitting of the IDS1/2vs. VGS curves in the saturation regime using the following equation: IDS(sat) = (WC/2L) μsat (VG − Vth).2 It is important to note that, while average mobilities may be lower than some recently reported isoindigo-based polymers, the field-effect mobilities found are comparable to previously reported values for similar materials designed with various functional side chains.37,41,42| Polymer | Solvent | Thicknessa (nm) | μ aveh/μmaxh [10−4 cm2 V−1 s−1] | I ON/IaveOFF | V aveth [V] |
|---|---|---|---|---|---|
| a Thickness confirmed by AFM. | |||||
| a-P(iIT)-TBS | Chlorobenzene | 40–50 | 1.88 ± 0.15/2.04 | 103 | −31 |
| o-Anisole | 40–50 | 1.07 ± 0.13/1.22 | 103 | −25 | |
| 20% v/v o-anisole/n-BuOH | 40–50 | — | — | — | |
| a-P(iIT)-OH | Chlorobenzene | 40–50 | 1.75 ± 0.21/2.06 | 103 | −32 |
| o-Anisole | 40–50 | 2.01 ± 0.08/2.12 | 103 | −27 | |
| 20% v/v o-anisole/n-BuOH | 40–50 | 2.49 ± 0.43/2.90 | 103 | −34 | |
Devices prepared with a-P(iIT)-TBS achieved an average mobility of 1.88 × 10−4 cm2 V−1 s−1 when processed in chlorobenzene. In agreement with the results obtained from GIWAXS and AFM analysis, the performance of a-P(iIT)-TBS decreased when processed in greener solvents, with an average mobility of 1.07 × 10−4 cm2 V−1 s−1 when processed in o-anisole and a thin film of too poor quality to measure any electronic performance when processed in a 20% v/v o-anisole/n-BuOH mixture. Importantly, device performance did not decrease upon removal of the protecting TBS groups to access the polar alcohol side chains. a-P(iIT)-OH demonstrated the highest average mobility of all devices tested (2.49 × 10−4 cm2 V−1 s−1) when processed in 20% v/v o-anisole/n-BuOH. When processed in o-anisole, the average mobility decreased slightly to 2.01 × 10−4 cm2 V−1 s−1, though it remained greater than the highest average mobility of a-P(iIT)-TBS. The average performance of a-P(iIT)-OH only dropped slightly below that of a-P(iIT)-TBS when processed in chlorobenzene (1.75 × 10−4 cm2 V−1 s−1). Notably, the transfer curves of these devices (Fig. S8†) appear to show some ambipolarity, indicating potential electron transport, which has been previously demonstrated in other isoindigo-based polymers.42,43 Unfortunately, no measurable charge mobility was observed. Charge mobilities for all devices remained in the range of 10−4 cm2 V−1 s−1, independently of the solvent used, which confirm that the asymmetric design of π-conjugated semiconducting polymers can be a tool to achieve enhanced solubility in greener solvent without sacrificing device performance.
Investigation of solution-state aggregation
Asymmetric side-chain engineering of SPs allows for the regioregular incorporation of new functional groups into conjugated materials. This can have profound effects on aggregation and bulk physical properties.44 To further probe for the influence of the hydroxyl moieties on polymer aggregation in solution, cryo-electron microscopy (cryo-EM) was utilized, and the results are depicted in Fig. 4 and S9.† Samples were prepared with 4 mg mL−1 solutions of the polymers in 20% v/v o-anisole/n-BuOH that were flash frozen at −170 °C, upon which images were taken of the polymer solution aggregates. Interestingly, as shown in Fig. 4b,a-P(iIT)-OH demonstrated long-range ordered aggregation. In comparison, a-P(iIT)-TBS was observed to form short-cluster aggregations in 20% v/v o-anisole/n-BuOH. These observations indicate that minor changes in chemical structure such as side chain polarity and the introduction of hydrogen bonding can drastically alter the aggregation properties of asymmetric SPs, thus potentially influencing solid state material properties. Aggregation was also probed by UV-Vis spectroscopy in solution (Fig. S10†). In contrast to what was observed by cryo-EM, the UV-vis spectrum for a-P(iIT)-TBS was found to have a more intense 0–0 vibration peak, centered at λ = 700 nm, which can be correlated to increased aggregation in solution resulting from poor solubility of a-P(iIT)-TBS in the solvent blend, thus resulting in the formation of short cluster aggregates in comparison to the elongated longer range aggregate morphology a-P(iIT)-OH. These results indicate that a careful design of SP through side chain engineering (polarity, functional groups, structures, etc.) combined with a meticulous selection of the solvent systems used in processing have important impacts on aggregation in solution. The asymmetric design introduced in this work is, therefore, a potential platform for controlling solution state aggregation, and ultimately, performance in solution-processed organic electronics.45 | ||
| Fig. 4 Cryo-electron microscopy images of (a) a-P(iIT)-TBS and (b) a-P(iIT)-OH in 20% v/v o-anisole/n-BuOH. Scale bar is 200 nm. | ||
Post-functionalization of asymmetric SPs
Functionalization of conjugated polymers to incorporate new functional groups or recognition moieties is a particularly important process for the development of emerging electronics, including biosensors and organic electrochemical transistors (OECTs).46,47 Ideally, a post-functionalization reaction would be highly efficient (high yield/high degree of functionalization), would not require the addition of catalyst or additives, and could be performed directly on thin films (solid-state) to avoid detrimental effects on solid-state morphology and device architecture. In recent years, various important post-functionalization strategies for high-performance semiconducting polymers have been developed.48 Particularly, post-functionalization through Huigsen 1,3-dipolar cycloaddition (click chemistry) has been shown to be notably efficient and versatile.49,50 The presence of a regioregular free hydroxyl moiety on a-P(iIT)-OH opens new avenues for post-functionalization not previously explored. Among others, the addition of cyanate, isocyanate, thiocyanate or thioisocyanate to an alcohol is particularly intriguing, as this reaction is highly efficient and spontaneous. Moreover, a-P(iIT)-OH, due to its asymmetric design, can maintain a good solubility in common organic solvent without sacrificing regioregularity, which is not always possible in more traditional symmetric designs. This novel post-functionalization technique has a lot of potential for designing sensing materials and devices specific analytes and/or receptor–ligand interactions, particularly for biosensing applications,51–54 including but not limited to the detection and determination of DNA concentrations, and printable immunosensors for point-of-care biosensor applications.55,56To evaluate the new asymmetric design for facile solid-state post-functionalization, a-P(iIT)-OH was reacted with fluorescein isocyanate (FITC) to afford the functionalized a-P(iIT)-O-C(S)-NH-Fluo (Fig. 5). FITC is a widely used fluorescent probe for a variety applications, particularly for the labelling and detection of biological molecules.57 This water-soluble probe has an absorption of 495 nm and upon excitation, emits a yellow green color at an emission maximum of 525 nm, clearly distinct from the non-functionalized semiconducting polymer, thus facilitating its detection through fluorescence emission spectroscopy.58 A detailed procedure for the functionalization can be found in the ESI† and is depicted in Fig. 5. Briefly, P(iIT)-OH was deposited on a silicon wafer through spin-coating and submerged in a solution of fluorescein isocyanate (FITC) and dibutyltin dilaurate in DMSO for 24 hours. The thin films were then rinsed extensively with DMSO to remove any non-covalently bonded FITC before characterization by fluorescence emission spectroscopy. It is important to note that, while the formation of thiocarbamates is spontaneous, dibutyltin dilaurate was used to improve the reaction kinetics to minimize reaction time. Results of the FITC-functionalization are depicted in Fig. 6. As expected, a-P(iIT)-O-C(S)-NH-Fluo showed an increase intensity in fluorescence emission at λem = 525 nm, which can be directly attributed to the FITC attached to the conjugated polymer. A 500–700 nm emission window was selected to prevent overlap of the fluorescence emission of Isoindigo based polymers, which have been reported to have emission at approximately 500 nm.26 In contrast, control spectra recorded of a-P(iIT)-OH and a-P(iIT)-OH+FITC (without the use of dibutyltin dilaurate as catalyst) did not show any significant fluorescence in this region, thus confirming the successful post-functionalization of the asymmetric conjugated polymer in thin film.47 While not being quantitative, this result demonstrates that the terminal alcohol in a-P(iIT)-OH can serve as an effective anchoring group for post-functionalization with various optically active materials for potential applications in sensing and bio-imaging.
 | ||
| Fig. 5 Schematic of the functionalization of a-P(iIT)-OH (thin film) with fluorescein isothiocyanate (FITC) to afford a-P(iIT)-O-C(S)-NH-Fluo. | ||
 | ||
| Fig. 6 Fluorescence emission studies for a-P(iIT)-OH (control, black line), a-P(iIT)-OH dipped in fluorescein solution (control, red line) and fluorescein-functionalized a-P(iIT)-O-C(S)-NH-Fluo (green line) thin films on SiO2 in the solid state. All samples were excited at 495 nm and monitored for fluorescence emission between 500–700 nm. | ||
To further investigate the efficiency of the FITC functionalization, X-ray photoelectron spectroscopy (XPS) was used to characterize the thin films of both a-P(iIT)-OH and a-P(iIT)-O-C(S)-NH-Fluo. The general surveys and high-resolution carbon spectra are depicted in Fig. S11.† In the deconvoluted high-resolution spectrum of a-P(iIT)-O-C(S)-NH-Fluo, the appearance of a binding energy corresponding to O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at Ebind = 289.1 eV was observed, attributed to FITC (Fig. S11b†). This peak, attributed to the carboxylic acid functional group on FITC does not appear in the a-P(iIT)-OH spectrum (Fig. S11a†). Quantification of the degree of functionalization was conducted by comparing the nitrogen atom percentages in the general surveys of the pristine and functionalized films (Fig. S11c and d†). Since these measurements were done on thin films deposited onto OTS-modified Si/SiO2 substrates, nitrogen was chosen because this atom is only present in the polymer and FITC. With the addition of FITC, the number of nitrogen atoms per repeat unit increases from two to three, indicating that a complete functionalization should result in a theoretical ratio of 1.5. Upon evaluation of the films by XPS, an experimental ratio of nitrogen composition of 1.53 was measured. It is important to mention that this method of quantification does not account for non-covalently bonded FITC that may remain on the surface of the functionalized thin film, accounting for the slight discrepancy in the theoretical versus calculated nitrogen percentages. Nonetheless, these results indicate a good degree of functionalization and further confirm that this novel design approach is promising for post-functionalization of semiconducting polymers in thin films.
O at Ebind = 289.1 eV was observed, attributed to FITC (Fig. S11b†). This peak, attributed to the carboxylic acid functional group on FITC does not appear in the a-P(iIT)-OH spectrum (Fig. S11a†). Quantification of the degree of functionalization was conducted by comparing the nitrogen atom percentages in the general surveys of the pristine and functionalized films (Fig. S11c and d†). Since these measurements were done on thin films deposited onto OTS-modified Si/SiO2 substrates, nitrogen was chosen because this atom is only present in the polymer and FITC. With the addition of FITC, the number of nitrogen atoms per repeat unit increases from two to three, indicating that a complete functionalization should result in a theoretical ratio of 1.5. Upon evaluation of the films by XPS, an experimental ratio of nitrogen composition of 1.53 was measured. It is important to mention that this method of quantification does not account for non-covalently bonded FITC that may remain on the surface of the functionalized thin film, accounting for the slight discrepancy in the theoretical versus calculated nitrogen percentages. Nonetheless, these results indicate a good degree of functionalization and further confirm that this novel design approach is promising for post-functionalization of semiconducting polymers in thin films.
Conclusions
In summary, an isoindigo-based -conjugated polymer was synthesized using asymmetric side chain engineering to introduce terminal hydroxyl moieties. This approach was used as a platform to demonstrate several key advantages of the asymmetric design, including greener processing and facile post-functionalization. Prepared through one-pot asymmetric alkylation and Stille polycondensation, a-P(iIT)-TBS and a-P(iIT)-OH were evaluated for their solid-state morphology and optoelectronic properties using various characterizations such as UV–Vis spectroscopy, AFM, and GIWAXS. The presence of the terminal hydroxyl group was shown to have a profound effect on the solubility of a-P(iIT)-OH in the non-halogenated 20% v/v o-anisole/n-butanol solvent blend while maintaining good electronic performance, which was not possible for a-P(iIT)-TBS. These observations were further investigated using Cryo-EM, which showed that minor changes in the side chain polarity of the asymmetric design are capable of showing significantly different aggregation morphologies when viewed in solution. Furthermore, thin film coupling with an FITC probe using dibutyltin dilaurate demonstrated that the terminal hydroxyl groups are capable of solid-state post-functionalization towards the development of multifunctional organic electronics. Overall, these findings show that asymmetric side-chain engineering is a facile approach toward sustainable organic electronics.Author contributions
S. R.-G., M. M. and A. N. conceived and directed the project. M. M., A. N., A. A. and E. I. performed the synthesis and basic characterizations of the materials through GPC and optical spectroscopy. M.-N. C.; Y.-C. C., Y. W. and X. G. performed the X-ray scattering measurements and AFM imaging. A. N., Y. L. and J. X. performed the cryo-EM experiments. M. M. and E. I. fabricated OFET devices. A. N., M. M. and A. A. performed the post-functionalization of the materials and their characterization. X. G., J. X., and Y.-C. C. helped with the discussion. M. M., A. N. and S.-R. G. wrote the manuscript, and all the coauthors commented on the manuscript.All authors contributed to the manuscript. All authors have given approval to the final version of the manuscript.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by NSERC through a Discovery Grants (RGPIN-2022-04428) and the NSERC Green Electronics Network (GreEN) (NETGP 508526-17). S. R.-G. also acknowledge the Canada Foundation for Innovation (CFI), the Ontario Research Fund, and the University of Windsor for financial support. M. M. and A. N. thank NSERC for financial support through a Canada Graduate Postgraduate Scholarship – Doctoral. Y. W., X. G. thank the financial support from the U.S. Department of Energy, Office of Science, Office of Basic Energy Science under award number of SC0019361 for supporting the scattering portion of this work. Y.-C. C. acknowledges the financial support from the Ministry of Science and Technology in Taiwan (MOST 111-2628-E-011-008-MY3). Work performed at the Center for Nanoscale Materials, a U.S. Department of Energy, Office of Science User Facility, was supported by the U.S. DOE, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. The authors thank Jean-François Morin and his group (U. Laval) for MS measurements.References
- L. Luo, W. Huang, C. Yang, J. Zhang and Q. Zhang, Recent advances on π-conjugated polymers as active elements in high performance organic field-effect transistors, Front. Phys., 2021, 3, 1–32 Search PubMed.
- S. Holliday, J. E. Donaghey and I. McCulloch, Advances in charge carrier mobilities of semiconducting polymers used in organic transistors, Chem. Mater., 2014, 26, 647–663 CrossRef CAS.
- M. U. Ocheje, B. P. Charron, A. Nyayachavadi and S. Rondeau-Gagné, Flexible Printed Electron., 2017, 4, 043002 CrossRef.
- D. J. Lipomi, H. Chong, M. Vosgueritchian, J. Mei and Z. Bao, Toward mechanically robust and intrinsically stretchable organic solar cells: Evolution of photovoltaic properties with tensile strain, Sol. Energy Mater. Sol. Cells, 2012, 107, 355–365 CrossRef CAS.
- B. Schmatz, Z. Yuan, A. W. Lang, J. L. Hernandez, E. Reichmanis and J. R. Reynolds, Aqueous Processing for Printed Organic Electronics: Conjugated Polymers with Multistage Cleavable Side Chains, ACS Cent. Sci., 2017, 3, 961–967 CrossRef CAS PubMed.
- J. Rivnay, R. M. Owens and G. G. Malliaras, The rise of organic bioelectronics, Chem. Mater., 2014, 26, 679–685 CrossRef CAS.
- D. Wang, Q. Guo, H. Gao, Z. Yang, H. Cao, W. He and H. Wang, Facile synthesis of functional poly(vinylene sulfide)s containing donor-acceptor chromophores by a double click reaction, RSC Adv., 2016, 6, 59327–59332 RSC.
- J. Kim, J. H. Kim and K. Ariga, Redox-Active Polymers for Energy Storage Nanoarchitectonics, Joule, 2017, 1, 739–768 CrossRef CAS.
- W. W. Bao, R. Li, Z. C. Dai, J. Tang, X. Shi, J. T. Geng, Z. F. Deng and J. Hua, Diketopyrrolopyrrole (DPP)-Based Materials and Its Applications: A Review, Front. Chem., 2020, 8, 1–6 CrossRef PubMed.
- Q. Liu, S. E. Bottle and P. Sonar, Developments of Diketopyrrolopyrrole-Dye-Based Organic Semiconductors for a Wide Range of Applications in Electronics, Adv. Mater., 2020, 32, 1–46 Search PubMed.
- J. Lauko, P. H. J. Kouwer, P. Kasak and A. E. Rowan, Tunable properties based on regioselectivity of 1,2,3-triazole units in axially chiral 2,2′-linked 1,1′-binaphthyl-based copolymers for ions and acid responsiveness, Eur. Polym. J., 2018, 108, 191–198 CrossRef CAS.
- J. Yao, C. Yu, Z. Liu, H. Luo, Y. Yang, G. Zhang and D. Zhang, Significant Improvement of Semiconducting Performance of the Diketopyrrolopyrrole-Quaterthiophene Conjugated Polymer through Side-Chain Engineering via Hydrogen-Bonding, J. Am. Chem. Soc., 2016, 138, 173–185 CrossRef CAS PubMed.
- J. W. Rumer and I. McCulloch, Organic photovoltaics: Crosslinking for optimal morphology and stability, Mater. Today, 2015, 18, 425–435 CrossRef CAS.
- M. Mooney, A. Nyayachavadi and S. Rondeau-Gagné, J. Mater. Chem. C, 2020, 8, 14645–14664 RSC.
- M. M. Ling and Z. Bao, Thin film deposition, patterning, and printing in organic thin film transistors, Chem. Mater., 2004, 16, 4824–4840 CrossRef CAS.
- M. Arslan and M. A. Tasdelen, Polymer nanocomposites via click chemistry reactions, Polymers, 2017, 10, 499 CrossRef PubMed.
- Y. Ji, C. Xiao, Q. Wang, J. Zhang, C. Li, Y. Wu, Z. Wei, X. Zhan, W. Hu, Z. Wang, R. A. J. Janssen and W. Li, Asymmetric Diketopyrrolopyrrole Conjugated Polymers for Field-Effect Transistors and Polymer Solar Cells Processed from a Nonchlorinated Solvent, Adv. Mater., 2016, 28, 943–950 CrossRef CAS PubMed.
- U. K. Aryal, J. Lee, K. Kranthiraja, S. S. Reddy, V. G. Sree, T. Park, M. Song and S. H. Jin, The effect of irregularity from asymmetric random π-conjugated polymers on the photovoltaic performance of fullerene-free organic solar cells, Polym. Chem., 2019, 10, 4407–4412 RSC.
- Y. C. Lin, F. H. Chen, Y. C. Chiang, C. C. Chueh and W. C. Chen, Asymmetric Side-Chain Engineering of Isoindigo-Based Polymers for Improved Stretchability and Applications in Field-Effect Transistors, ACS Appl. Mater. Interfaces, 2019, 11, 34158–34170 CrossRef CAS PubMed.
- Z. Tang, X. Xu, R. Li, L. Yu, L. Meng, Y. Wang, Y. Li and Q. Peng, Asymmetric Siloxane Functional Side Chains Enable High-Performance Donor Copolymers for Photovoltaic Applications, ACS Appl. Mater. Interfaces, 2020, 12, 17760–17768 CrossRef CAS PubMed.
- J. Lee, S. A. Park, S. U. Ryu, D. Chung, T. Park and S. Y. Son, Green-solvent-processable organic semiconductors and future directions for advanced organic electronics, J. Mater. Chem. A, 2020, 8, 21455–21473 RSC.
- S. Lv, L. Li, Y. Mu and X. Wan, Side-chain engineering as a powerful tool to tune the properties of polymeric field-effect transistors, Polym. Rev., 2021, 61, 520–552 CrossRef CAS.
- J. Ma, Z. Zhao, Y. Guo, H. Geng, Y. Sun, J. Tian, Q. He, Z. Cai, X. Zhang, G. Zhang, Z. Liu, D. Zhang and Y. Liu, Improving the Electronic Transporting Property for Flexible Field-Effect Transistors with Naphthalene Diimide-Based Conjugated Polymer through Branching/Linear Side-Chain Engineering Strategy, ACS Appl. Mater. Interfaces, 2019, 11, 15837–15844 CrossRef CAS PubMed.
- A. Gumyusenge, X. Zhao, Y. Zhao and J. Mei, Attaining Melt Processing of Complementary Semiconducting Polymer Blends at 130 °c via Side-Chain Engineering, ACS Appl. Mater. Interfaces, 2018, 10, 4904–4909 CrossRef CAS PubMed.
- C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster and H. F. Sneddon, Updating and further expanding GSK's solvent sustainability guide, Green Chem., 2016, 13, 3879–3890 RSC.
- M. Gsänger, D. Bialas, L. Huang, M. Stolte and F. Würthner, Organic Semiconductors based on Dyes and Color Pigments, Adv. Mater., 2016, 28, 3615–3645 CrossRef PubMed.
- H. Gu, S. Ming, K. Lin, S. Chen, X. Liu, B. Lu and J. Xu, Isoindigo as an electron−deficient unit for high−performance polymeric electrochromics, Electrochim. Acta, 2018, 260, 772–782 CrossRef CAS.
- D. Orain, J. Ellard and M. Bradley, Protecting groups in solid-phase organic synthesis., J. Comb. Chem., 2002, 4, 1–16 CrossRef CAS PubMed.
- M. U. Ocheje, B. P. Charron, Y. H. Cheng, C. H. Chuang, A. Soldera, Y. C. Chiu and S. Rondeau-Gagné, Amide-Containing Alkyl Chains in Conjugated Polymers: Effect on Self-Assembly and Electronic Properties, Macromolecules, 2018, 51, 1336–1344 CrossRef CAS.
- A. K. Patri, I. J. Majoros and J. R. Baker, Dendritic polymer macromolecular carriers for drug delivery, Curr. Opin. Chem. Biol., 2002, 6, 466–471 CrossRef CAS PubMed.
- F. Le Guern, V. Mussard, A. Gaucher, M. Rottman and D. Prim, Fluorescein derivatives as fluorescent probes for ph monitoring along recent biological applications, Int. J. Mol. Sci., 2020, 21, 1–23 Search PubMed.
- A. E. Caprifico, E. Polycarpou, P. J. S. Foot and G. Calabrese, Biomedical and Pharmacological Uses of Fluorescein Isothiocyanate Chitosan-Based Nanocarriers, Macromol. Biosci., 2021, 21, 1–27 CrossRef PubMed.
- I. Song, H. W. Kim, J. Ahn, M. Han, S. K. Kwon, Y. H. Kim and J. H. Oh, Non-halogenated solution-processed ambipolar plastic transistors based on conjugated polymers prepared by asymmetric donor engineering, J. Mater. Chem. C, 2019, 7, 14977–14985 RSC.
- S. Matsumura, A. R. Hlil, C. Lepiller, J. Gaudet, D. Guay, Z. Shi, S. Holdcroft and A. S. Hay, New Carbazole-Based Conjugated Polymers Containing Pyridylvinyl Thiophene Units for Polymer Solar Cell Applications: Morphological Stabilization Through Hydrogen Bonding, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7207–7224 CrossRef.
- M. Hayashi and F. Tournilhac, Thermal stability enhancement of hydrogen bonded semicrystalline thermoplastics achieved by combination of aramide chemistry and supramolecular chemistry, Polym. Chem., 2017, 8, 461–471 RSC.
- R. K. Henderson, C. Jiménez-González, D. J. C. Constable, S. R. Alston, G. G. A. Inglis, G. Fisher, J. Sherwood, S. P. Binks and A. D. Curzons, Expanding GSK's solvent selection guide - Embedding sustainability into solvent selection starting at medicinal chemistry, Green Chem., 2011, 13, 854–862 RSC.
- M. Mooney, Y. Wang, A. Nyayachavadi, S. Zhang, X. Gu and S. Rondeau-Gagné, Enhancing the Solubility of Semiconducting Polymers in Eco-Friendly Solvents with Carbohydrate-Containing Side Chains, ACS Appl. Mater. Interfaces, 2021, 13, 25175–25185 CrossRef CAS PubMed.
- H. C. Yen, Y. C. Lin and W. C. Chen, Modulation of the Hydrophilicity on Asymmetric Side Chains of Isoindigo-Based Polymers for Improving Carrier Mobility-Stretchability Properties, Macromolecules, 2021, 54, 1665–1676 CrossRef CAS.
- G. C. Yuan, Z. Xu, C. Gong, Q. J. Cai, Z. S. Lu, J. S. Shi, F. J. Zhang, S. L. Zhao, N. Xu and C. M. Li, High performance organic thin film transistor with phenyltrimethoxysilane- modified dielectrics, Appl. Phys. Lett., 2009, 94, 111 Search PubMed.
- M. Waldrip, O. D. Jurchescu, D. J. Gundlach and E. G. Bittle, Contact Resistance in Organic Field-Effect Transistors: Conquering the Barrier, Adv. Funct. Mater., 2020, 30, 1904576 CrossRef CAS.
- N. M. Randell, C. L. Radford, J. Yang, J. Quinn, D. Hou, Y. Li and T. L. Kelly, Effect of Acceptor Unit Length and Planarity on the Optoelectronic Properties of Isoindigo-Thiophene Donor-Acceptor Polymers, Chem. Mater., 2018, 30, 4864–4873 CrossRef CAS.
- R. Stalder, S. R. Puniredd, M. R. Hansen, U. Koldemir, C. Grand, W. Zajaczkowski, K. Müllen, W. Pisula and J. R. Reynolds, Ambipolar Charge Transport in Isoindigo-Based Donor-Acceptor Polymers, Chem. Mater., 2016, 28, 1286–1297 CrossRef CAS.
- T. Lei, J.-H. Dou, Z.-J. Ma, C.-H. Yao, C.-J. Liu, J.-Y. Wang and J. Pei, Ambipolar Polymer Field-Effect Transistors Based on Fluorinated Isoindigo: High Performance and Improved Ambient Stability, J. Am. Chem. Soc., 2012, 134, 20025–20028 CrossRef CAS PubMed.
- M. Chang, G. T. Lim, B. Park and E. Reichmanis, Control of molecular ordering, alignment, and charge transport in solution-processed conjugated polymer thin films, Polymers, 2017, 9, 23–31 CrossRef.
- S. R. Forrest, The path to ubiquitous and low-cost organic electronic appliances on plastic, Nature, 2004, 428, 911–918 CrossRef CAS PubMed.
- S. Inagi and T. Fuchigami, Electrochemical post-functionalization of conducting polymers, Macromol. Rapid Commun., 2014, 35, 854–867 CrossRef CAS PubMed.
- A. Salinas-Castillo, M. Camprubí-Robles and R. Mallavia, Synthesis of a new fluorescent conjugated polymer microsphere for chemical sensing in aqueous media, Chem. Commun., 2010, 46, 1263–1265 RSC.
- H. J. Kim, M. Skinner, H. Yu, J. H. Oh, A. L. Briseno, T. Emrick, B. J. Kim and R. C. Hayward, Water Processable Polythiophene Nanowires by Photo-Cross-Linking and Click-Functionalization, Nano Lett., 2015, 15, 5689–5695 CrossRef CAS PubMed.
- S. Tane and T. Michinobu, Cu(I)-catalyzed azide–alkyne cycloaddition synthesis and fluorescent ion sensor behavior of carbazole-triazole-fluorene conjugated polymers, Polym. Int., 2020, 2–12 Search PubMed.
- A. Marrocchi, A. Facchetti, D. Lanari, S. Santoro and L. Vaccaro, Click-chemistry approaches to π-conjugated polymers for organic electronics applications, Chem. Sci., 2016, 7, 6298–6308 RSC.
- L. Bai, C. G. Elósegui, W. Li, P. Yu, J. Fei and L. Mao, Biological Applications of Organic Electrochemical Transistors: Electrochemical Biosensors and Electrophysiology Recording, Front. Chem., 2019, 7, 313 CrossRef CAS PubMed.
- N. Yousefi, C. Caba, A. Hu, M. Mooney, S. Zhang, A. D. Agostinis, M. Mirhassani, M. J. Ahamed, Y. Tong and S. Rondeau-Gagné, Building a Versatile Platform for the Detection of Protein–Protein Interactions Based on Organic Field-Effect Transistors, ACS Appl. Electron. Mater., 2022, 4, 4972–4981 CrossRef CAS.
- E. Macchia, K. Manoli, B. Holzer, C. Di Franco, M. Ghittorelli, F. Torricelli, D. Alberga, G. F. Mangiatordi, G. Palazzo, G. Scamarcio and L. Torsi, Single-molecule detection with a millimetre-sized transistor, Nat. Commun., 2018, 9, 3223 CrossRef PubMed.
- M. Magliulo, D. Altamura, C. Di Franco, M. V. Santacroce, K. Manoli, A. Mallardi, G. Palazzo, G. Scamarcio, C. Giannini and L. Torsi, Structural and Morphological Study of a Poly(3-hexylthiophene)/Streptavidin Multilayer Structure Serving as Active Layer in Ultra-Sensitive OFET Biosensors, J. Phys. Chem. C, 2014, 118, 15853–15862 CrossRef CAS.
- K. Ditte, T. A. Nguyen Le, O. Ditzer, D. I. Sandoval Bojorquez, S. Chae, M. Bachmann, L. Baraban and F. Lissel, Rapid Detection of SARS-CoV-2 Antigens and Antibodies Using OFET Biosensors Based on a Soft and Stretchable Semiconducting Polymer, ACS Biomater. Sci. Eng. DOI:10.1021/acsbiomaterials.1c00727.
- M. L. Hammock, O. Knopfmacher, B. D. Naab, J. B.-H. Tok and Z. Bao, Investigation of Protein Detection Parameters Using Nanofunctionalized Organic Field-Effect Transistors, ACS Nano, 2013, 7, 3970–3980 CrossRef CAS PubMed.
- H. H. Cho, J. H. Choi, S. Y. Been, N. Kim, J. M. Choi, W. Kim, D. Kim, J. J. Jung, J. E. Song and G. Khang, Development of fluorescein isothiocyanate conjugated gellan gum for application of bioimaging for biomedical application, Int. J. Biol. Macromol., 2020, 164, 2804–2812 CrossRef CAS PubMed.
- Y. Zhang, S. Li, H. Zhang and H. Xu, Design and Application of Receptor-Targeted Fluorescent Probes Based on Small Molecular Fluorescent Dyes, Bioconjugate Chem., 2021, 32, 4–24 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2py01244h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |