
Bimetallic core–shell nanocrystals: opportunities and challenges
Chenxiao
Wang†
 a,
Yifeng
Shi†
b,
Dong
Qin
c and
Younan
Xia
*abd
a,
Yifeng
Shi†
b,
Dong
Qin
c and
Younan
Xia
*abd
aSchool of Chemistry and Biochemistry, Georgia Institute of Technology, Atlanta, Georgia 30332, USA. E-mail: younan.xia@bme.gatech.edu
bSchool of Chemical and Biomolecular Engineering, Georgia Institute of Technology, Atlanta, Georgia 30332, USA
cSchool of Materials Science and Engineering, Georgia Institute of Technology, Atlanta, Georgia 30332, USA
dThe Wallace H. Coulter Department of Biomedical Engineering, Georgia Institute of Technology and Emory University, Atlanta, Georgia 30332, USA
First published on 19th June 2023
Abstract
With mastery over the colloidal synthesis of monometallic nanocrystals, a combination of two distinct metals with intricate architectures has emerged as a new direction of innovation. Among the diverse architectures, the one with a core–shell structure has attracted the most scientific endeavors owing to its merits of high controllability and variability. Along with the new hopes arising from the addition of a shell composed of a different metal, there comes unexpected complications for the surface composition, hindering both structural understanding and application performance. In this Focus article, we present a brief overview of the opportunities provided by the bimetallic core–shell nanocrystals, followed by a discussion of the technical challenge to elucidate the true composition of the outermost surface. Some of the promising solutions are then highlighted as well, aiming to inspire future efforts toward this frontier of research.
 Chenxiao Wang | Chenxiao Wang received his BS in chemistry in 2018 from Wuhan University. He is pursuing his PhD degree under the tutelage of Xia in the School of Chemistry and Biochemistry at Georgia Tech. His research interests include the synthesis of bi- and multi-metallic nanocrystals and study of their thermal and catalytic properties. |
 Yifeng Shi | Yifeng Shi is a postdoctoral fellow in the Department of Electrical Engineering and Computer Sciences at University of California, Berkeley. She received her BE degree from Sichuan University in 2017 and her PhD degree from Georgia Tech in 2022 under the supervision of Xia. Her graduate work focused on the synthesis and structure modifications of noble-metal nanocrystals for (electro)catalytic applications. Her current research interests include the self-assembly of nanocrystals using DNA nanotechnology for the next-generation optical and electronic devices. |
 Dong Qin | Dong Qin holds a PhD degree in physical chemistry from University of Pennsylvania (1996) and an MBA degree from the University of Washington (2003). After a postdoctoral stint with George M. Whitesides at Harvard University (1996–1997), she held administrative positions as an Associate Director of Center for Nanotechnology at the University of Washington (2002–2007) and an Associate Dean for Research in the School of Engineering and Applied Science at Washington University in St. Louis (2007–2011). Since 2012, she has held a faculty position in the School of Materials Science and Engineering at Georgia Tech. She served as an Associate Editor of Nanoscale from 2021–2023. |
 Younan Xia | Younan Xia received his PhD in physical chemistry from Harvard University in 1996 (with George M. Whitesides). He started as an Assistant Professor of Chemistry at the University of Washington (Seattle) in 1997 and then joined the Department of Biomedical Engineering at Washington University in St. Louis in 2007 as the James M. McKelvey Professor. Since 2012, he has held the position of Brock Family Chair and Georgia Research Alliance Eminent Scholar at Georgia Tech. He served as an Associate Editor of Nano Letters from 2002–2019. |
1. Introduction
The design and synthesis of bimetallic nanocrystals have received considerable interest owing to their enriched and/or enhanced properties relative to their monometallic counterparts. By simply bringing together two distinct metals, the properties of the resultant nanocrystals can be vastly expanded as a result of the possible variations and synergistic effects.1,2 It is well-documented that the performance of bimetallic nanocrystals in a wide variety of applications can be augmented by manipulating the elemental composition, in addition to the spatial distributions of the two elements in the crystal lattice.3,4Among various types of bimetallic nanocrystals, including those featuring a core–shell, core–satellite, Janus, intermetallic, and alloy structure, core–shell nanocrystals have been most extensively explored owing to the well-developed protocols for their syntheses, as well as their tunable properties arising from the adjustment of the shell thickness.5,6 As suggested by its name, a core–shell nanocrystal is supposed to consist of a core comprising one metal and a complete shell made of another metal (Fig. 1). The immediate merits and advantages offered by the core–shell structure include: (i) an avenue to produce cost-effective catalysts by replacing the bulk of a catalytic particle with an inexpensive and abundant metal;7,8 (ii) the ability to process noble metals with high cohesive energies into nanocrystals with well-defined surface structures by replicating the atomic structures of the core;9,10 (iii) the capability to access new crystal phases for the metal in the shell by templating against the atomic packing of the metal in the core;11–13 and (iv) an opportunity to enhance the properties of the shell metal by leveraging its electronic and/or geometric interactions with the core, and such enhancement will be particularly strong if the shell thickness is controlled below six atomic layers.14
 | ||
| Fig. 1 Atomic model of a bimetallic nanocube featuring an ideal core–shell structure. | ||
Although core–shell nanocrystals have been extensively explored in the literature, most of the reports failed to answer the following two questions: (i) what is the elemental composition of the outermost layer and (ii) how will the surface composition change during the storage or utilization of such bimetallic nanocrystals? Herein, we start with a brief overview of the opportunities offered by bimetallic core–shell nanocrystals, followed by a discussion about the issues faced by this class of nanomaterials and perspective on the potential solutions.
2. Opportunities: two is better than one
With two distinct metals configured in a well-defined core–shell structure, a number of unique ways are provided to expand the structures, properties, and applications of the constituent metals. For example, by simply switching the order of metals used for the core and the shell, one can obtain surfaces with completely different compositions and catalytic properties.15–17 By coating the surface of a nanocrystal made of a plasmonic metal (e.g., Ag, Au, and Cu) with a catalytic metal (e.g., Pd, Pt, Rh, Ru, and Ir), one can access a bifunctional nanomaterial with both plasmonic and catalytic properties.18 By coating the surface of a nanocrystal made of a reactive metal with a less reactive metal, one can greatly enhance the chemical stability of the original nanocrystal.17,19 Moreover, by controlling the electronic coupling and/or lattice mismatching between the core and shell metals, one can augment the catalytic activity and/or selectivity of the shell metal.20 Here we highlight some of the recent progress in the development of bimetallic core–shell nanocrystals with novel structures, properties, and applications.Prior to any specific discussion of representative case studies, it is worth pointing out that, despite the major research efforts on the surface-dependent properties of bimetallic core–shell nanocrystals, it is the interplay between the core and the shell that plays the most important role in shaping these surface properties. In stark contrast to the misconception that such interplay is merely geometrical, the influence of the core metal on the outermost surface is far more significant and sophisticated, as notably embodied in recent reports about synergistic catalysis. It has been established that, through both geometric21 and ligand22 effects arising from core–shell interaction, the spatial and electronic distributions of the atoms in the outermost layer are critical to enhancing the catalytic performance toward various reactions. For instance, M@Pt core–shell nanocrystals have achieved great success in boosting the activity of Pt toward the oxygen reduction reaction (ORR) on the basis of per Pt atom. It is traditionally believed that the compressive strain in M@Pt core@shell catalysts can weaken the Pt–O binding on the Pt(111) surface and thereby increase the ORR activity.23–25 Taking the Pd@Pt core–shell icosahedra as an example,26 under the lateral confinement imposed by both tensile strain and twin boundaries on the Pd icosahedral core, the Pt shell would evolve into a compressed structure. Such induced strain, combined with the ligand effect, would lead to d-band center shift for the outermost layer and thus destabilization of the adsorbed OH group, enhancing the activity toward oxygen reduction. As shown in Fig. 2(A), when the surface has a net compressive strain of −6.1%, the calculated specific activities of Pd@Pt(111) icosahedra with different Pt shell thicknesses are all much higher than that of the pure Pt(111) surface. The trend of these calculation results agrees with the experimental data, demonstrating the potential of core–shell nanocrystals in studying structure–property relationships and developing high-performance catalysts. However, the influence of strain on the ORR catalytic performance of Pt has been proven to be much more complicated and case-specific with no generic rule of thumb. For example, a study on biaxially strained PtPb@Pt hexagonal nanoplates demonstrated that the large tensile strain of 7.5% on Pt(110) planes of the nanoplates can also increase the ORR activity and activate the low-coordinated surface Pt atoms.27 The bidirectional control of strain was further explored in a recent study through the expansion and shrinkage of Pd@Pt nanocubes as a result of phosphorization and dephosphorization. It was reported that the strain–activity correlation for the methanol oxidation reaction and hydrogen evolution reaction followed an M-shaped curve and a volcano-shaped curve, respectively.28 The different dependences of these two electrocatalytic reactions on the surface strain highlight the importance to screen and identify the optimal lattice strain for a specific combination of electrocatalyst and reaction. To sum up, strain engineering through the formation of core–shell nanocrystals provides a powerful means to tune the adsorption energy of intermediate species on the catalyst surface and thus augment the catalytic performance.
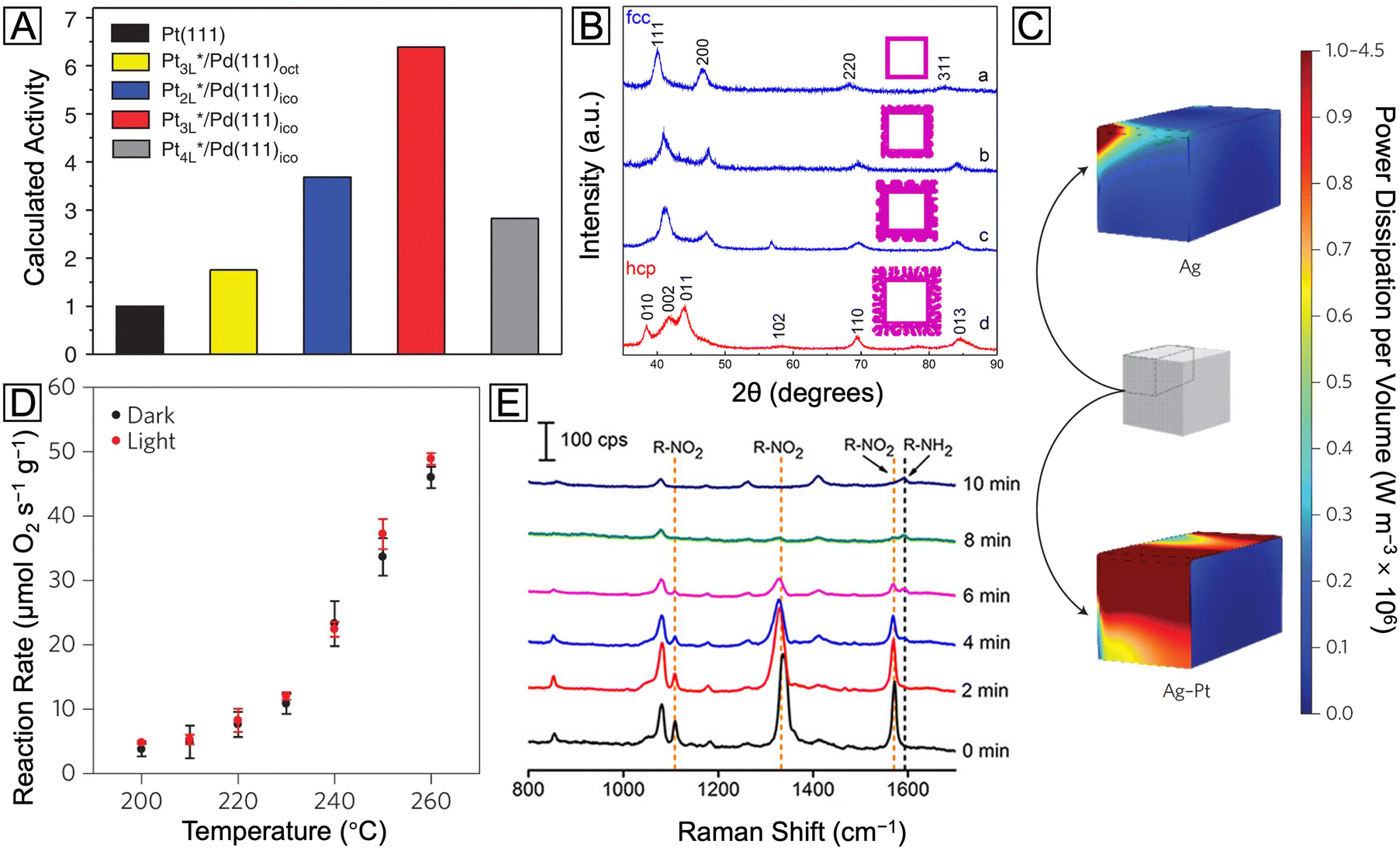 | ||
| Fig. 2 (A) Calculated relative specific activity of Pd@PtnL nanocrystals at 0.9 V (vs. reversible hydrogen electrode) toward oxygen reduction. Reproduced from ref. 26 with permission. Copyright 2015 Springer Nature. (B) XRD patterns of the Ru nanocages after the Pd cores had been etched away from Pd@Ru nanocrystals. Reproduced from ref. 13 with permission. Copyright 2016 American Chemical Society. (C) Heat maps of the power dissipation per volume at the LSPR peaks for the Ag nanocube (455 nm) and Ag@Pt nanocube (460 nm), respectively. (D) Reaction rate vs. temperature for preferential CO oxidation in excess H2 on the Ag@Pt core–shell nanocubes under light off and on conditions, respectively. Reproduced from ref. 18 with permission. Copyright 2017 Springer Nature. (E) SERS spectra recorded during the reduction of 4-NTP to 4-ATP as catalyzed by the Ag@Pd–Ag nanocubes. Reproduced from ref. 42 with permission. Copyright 2015 American Chemical Society. | ||
Besides the enhancement of existing properties of the shell metal, the interaction between the core and the shell offers an effective way to address synthetic challenges, as well as opportunities to develop nanocrystals with novel shapes and crystal structures. A notable example can be found in the development of Pd@Ru nanocrystals.13,29–31 As a noble metal high in cohesive energy, Ru is known to form very small (typically, <4 nm) particles during a solution-phase synthesis, making it a challenge to prepare nanocrystals with well-defined and controllable facets.32 By introducing Pd nanocrystals with well-defined shapes as seeds, the deposition of Ru can be switched from an island to a layer-by-layer growth mode. Through careful optimization of the growth conditions, ultrathin Ru shells with a smooth surface have been deposited on Pd cubic, octahedral, and icosahedral seeds in a layer-by-layer fashion.13,29,30 A similar strategy has also been successfully applied to the preparation of Pd@Pt,33 Pd@Rh34 and Pd@Ir35 nanocrystals. It is worth noting that, owing to the relatively small lattice mismatch between Pd and Ru, the well-defined Pd seeds could serve as a template to control not only the geometrical shape but also the crystal structure of the Ru shell. In the as-obtained Pd@Ru nanocubes, Ru exhibited the same face-centered cubic (fcc) phase as the Pd core, distinct from the intrinsic hexagonal close-packed (hcp) structure of bulk Ru. Significantly, the new phase could be well retained even when the Pd core was etched away, generating Ru nanocages with a wall thickness of up to six atom layers and fcc phase, as confirmed by the characteristic X-ray diffraction (XRD) pattern in Fig. 2(B).13 A recent study also showed that when the fcc metal in the core takes an anisotropic shape (e.g., Pd nanoplate), the deposition mode and final crystal structure of the Ru shell is also dependent on the lateral size of the nanoplate due to the interplay between the surface and bulk energies of the shell and the core.36
Indeed, the aforementioned templating effect from the core metal, by virtue of its subtlety and controllability, offers a great opportunity to modify the shell metal. Nevertheless, one should never overlook the power arising from a simple combination of two metals with distinct properties. For instance, covering plasmonic Ag nanocubes with a thin shell made of catalytic Pt creates a novel plasmonic catalyst.18 As an immediate benefit from the Pt shell, a much greater fraction of energy dissipation would occur through absorption on the surface when the core–shell nanocubes were illuminated with visible light, as compared with the counterpart made of pure Ag. This optical surface enhancement via non-plasmonic Pt covering is demonstrated by the sharp contrast in intensity for the simulated heat map in Fig. 2(C). The mechanism can be attributed to the fact that the Pt shell provides a fast and critical plasmon decay channel via the formation of energetic electron–hole pairs. These energetic charge carriers could be further used to enhance the selective oxidation of CO by O2 in excess H2, as demonstrated by the significant increase in the amount of reacted O2 under visible light (Fig. 2(D)). Taken together, controlling the energy flow through the decay of the localized surface plasmon resonance (LSPR) can become an efficient way to extract charge carriers for multiple applications, and several related studies have contributed to the understanding of this topic through both experimental and theoretical studies.37–39
Another fascinating optical property of plasmonic metal nanocrystals is their shape- and size-dependent LSPR, which contributed to the signal enhancement of surface enhanced Raman spectroscopy (SERS).40,41 When the surface of the plasmonic nanocrystal is covered by a shell made of catalytic metal, SERS offers a way to achieve in situ monitoring of chemical reactions. Fig. 2(E) shows a series of SERS spectra recorded during the reduction of 4-nitrothiophenol (4-NTP) catalyzed by Ag@Pd–Ag nanocubes.42 Here only the Pd atoms on the surface are able to catalyze the reaction, while the Ag atoms in the core and on the surface serve to report the evolution of 4-NTP into 4-aminothiophenol (4-ATP) in situ. The progressive disappearance of three characteristic vibrational bands of 4-NTP in these spectra, as well as the concomitant emergence of the phenol-ring vibrational band of 4-ATP, clearly demonstrates the excellent catalytic performance of these dual-functional core–shell nanocubes, as well as the remarkable SERS detection capability.
3. Challenges: elucidating the surface composition and its dynamics
It is clear from the previous discussion that the exact surface composition and structure are both vital to the catalytic performance of core–shell nanocrystals. In order to maximize the synergistic effect and dual-functionality on the surface, the shell has to be thin enough so that the influences (e.g., strain and electronic coupling) from the core metal will not dissipate too much before reaching the surface. However, accompanied by the infiltration of such influence is often the interpenetration of matter, that is, the shell is seldom composed of the pure shell metal, but rather a mix of both core and shell metals due to interdiffusion and/or co-reaction. The intermixing may be further intensified during a non-ideal synthetic process. As schematically illustrated in Fig. 3, the natural occurrence of atomic intermixing is inevitable in all three chemical approaches to core–shell nanocrystals. Among them, seed-mediated growth is the most versatile and best controlled as it separates the deposition of the shell from the synthesis of the core (nanocrystal seed). However, the possible galvanic replacement reaction between the seed and a precursor to the second metal can give rise to the formation of bimetallic alloy shells and complicate the surface composition.43 Fortunately, this detrimental side reaction can be compressed by adding a strong reducing agent to ensure that the reduction rate far outpaces the rate of galvanic replacement.44 Nevertheless, the interdiffusion between the atoms in the core and the shell cannot be avoided or neglected, especially when the synthesis is conducted at elevated temperatures. As evidenced by a variety of studies, most of the reported “core–shell” nanocrystals could be transformed into nanocages through chemical etching, suggesting the presence of atom-wide channels of the core metal through the shell.45 The mixing between the core and the shell will become more significant when the approach is switched to one-pot synthesis. In this case, it is critical to program the reduction kinetics of the two metal precursors in the same reaction mixture to ensure their sequential reduction.46 The coexistence of both precursors often results in a greater extent of surface alloying during the synthesis. For example, in the case of Pd@Pt octahedral nanocrystals prepared through one-pot synthesis, the mixing between Pd and Pt atoms in the shell can be directly visualized through the scanning transmission electron microscopy (STEM) image and energy-dispersive X-ray spectroscopy (EDX) line scanning.47 As for the third approach built on dealloying, it typically involves treatment (e.g., chemical etching,48,49 electrochemical dissolution,25,50,51 and thermal annealing48,52) of alloy nanocrystals to selectively leach out one of the metals. The complexity of the alloy makes it hard to completely etch away all the undesired atoms on the surface so the surface of the alloy nanocrystals should still stay in the form of an alloy, only enriched with the less reactive metal. Depending on the final applications, the formation of an alloy can be either a desired53 or undesired54 outcome. Nevertheless, surface alloying increases the complexity in analyzing the structure–property relationship of the bimetallic nanostructures and is unavoidable in the fabrication of core–shell nanocrystals.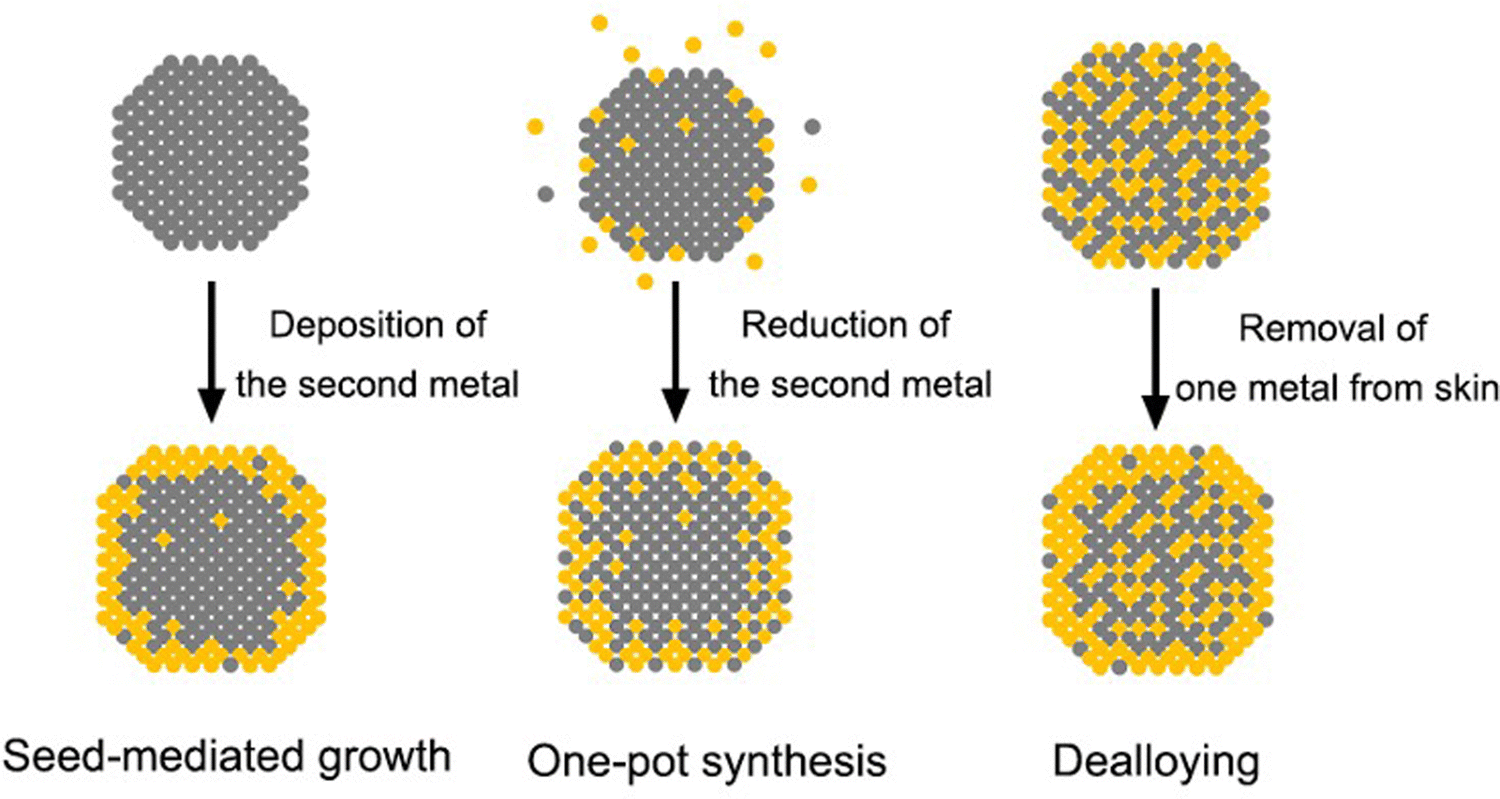 | ||
| Fig. 3 Schematics of the three main approaches to the synthesis of core–shell nanocrystals. | ||
Besides the complexity brought by the synthetic approach, the dynamic changes induced by the reaction environment pose even more uncertainties to the surface composition.17 In the context of catalytic applications, most nanocrystals work at elevated temperature and/or under specific gases, both of which can cause dramatic changes to the surface composition. Fig. 4(A)–(F) illustrates the interdiffusion between the core and the shell when a Pd@Pt nanocube was heated to different temperatures.19 At room temperature, the Pd core and the Pt shell could be clearly distinguished by their different contrasts (Fig. 4(A) and (D)). After heating at 500 °C for 60 min, the cubic shape became rounded, along with a less distinguishable border between the core and the shell, indicating the occurrence of interdiffusion at elevated temperatures (Fig. 4(B) and (E)). When the temperature was further increased to 800 °C, the nanocube completely lost its core–shell structure and evolved into a round particle (Fig. 4(C) and (F)). In this study, the nanocrystal was heated under vacuum. If a certain gas environment is involved in the heating process, changes to the surface composition will become even more significant. Fig. 4(G) and (H) shows a visual presentation of the immigration of atoms in a PtPb@Pt nanoplate when it was exposed to CO at 300 °C.55 The CO stripped out the Pb atoms from the core and induced the aggregation of Pt at the corner, transforming the original core–shell nanoplate into a Janus particle.
 | ||
| Fig. 4 (A)–(C) Changes to the surface composition of a single Pd@Pt core–shell nanocube upon heating to different temperatures: (A) room temperature, (B) 500 °C for 60 min, and (C) right after the temperature was increased to 800 °C, with corresponding atomic models shown in (D)–(F). Reproduced from ref. 19 with permission. Copyright 2017 American Chemical Society. (G), (H) STEM images of the PtPb@Pt nanoplate (G) before CO treatment and (H) after CO treatment at 300 °C, respectively. Reproduced from ref. 55 with permission. Copyright 2019 Wiley-VCH. | ||
The atomic diffusion, interfacial mixing, and phase segregation of core–shell nanocrystals motivate people to develop highly sensitive and surface-specific tools to understand the surface in terms of composition, structure, and dynamics under different conditions. To this end, X-ray photoelectron spectroscopy (XPS) is the most utilized technique to obtain the information about surface composition. Since it can also provide the information on the chemical state of a metal, XPS has become especially suitable for analyzing core–shell structures whose shell metal could potentially be oxidized.56,57 However, the detection depth of XPS inevitably brings up information from the subsurface regions, making it very challenging to analyze the elemental distribution at the outermost surface of a nanocrystal.58 On the other hand, high-resolution transmission electron microscopy (HRTEM), a non-surface-specific means of characterization as well, likewise suffers from the difficulty and confusion in data interpretation caused by undesired signals from the interior of a nanocrystal. Even with a direct elemental analysis tool such as 2D EDX mapping, the projection of the particles could still cause confusion to the 3D spatial distribution of each element.59 Taken together, there is an urgent need to develop new techniques for true surface elemental analysis.
A number of techniques have been developed in recent years to help elucidate the surface composition of metal nanostructures. Here we only introduce several representative ones that seem to hold the solution to this critical yet thorny issue, hoping to inspire more future endeavors aiming at the core–shell structure. To begin with, 3D electron tomography (ET) offers a viable platform for the visualization of nanoparticle morphology/composition in three dimensions.60 It is especially suitable for imaging anisotropic core–shell nanocrystals and the dynamic evolution of their surface. A recent study combining in situ heating and fast electron tomography on Au@Ag nanocrystals with different shapes, sizes, and surface defects has shed light on how these features affect the thermal stability of such core–shell nanocrystals.61 It was demonstrated that the shape of the core and the interfacial crystal plane between the core and the shell did not play a significant role in the intermixing of the core and shell upon heating. On the other hand, the presence of twin boundaries would greatly increase the atomic diffusion and intermixing between the two metals owing to the formation of distortion and vacant sites (Fig. 5(A)). It is worth mentioning that 3D electron tomography can be used to reveal the difference in the degree of alloying between 92% and 96%, which would be challenging for the typical 2D EDX mapping. The quantitative compositional analysis using 3D ET was achieved by assigning the two voxel intensities at the lower and higher maxima to Ag and Au, respectively (see the two peaks at 0 s in Fig. 5(B), left panel).62 The relative atomic content of Au in each voxel was then calculated using a simple equation involving the intensity of the voxel and the two intensities corresponding to pure Au and Ag. Finally, the degree of alloying was obtained by comparing the standard deviation value of voxel intensities at a certain time step and the standard deviation values of voxel intensities in the initial core–shell and in the perfectly alloyed particle. In this way, the alloying process can be tracked and quantified from the 3D reconstruction data with high precision (Fig. 5(B), right panel). However, 3D ET also requires the sample to be exposed to the electron beam for a longer time than regular electron microscopy imaging, which might cause shape deformation in the sample. This is especially an issue for in situ studies as the damage from the beam can hardly be separated from the change induced by the in situ heating/reaction environment.
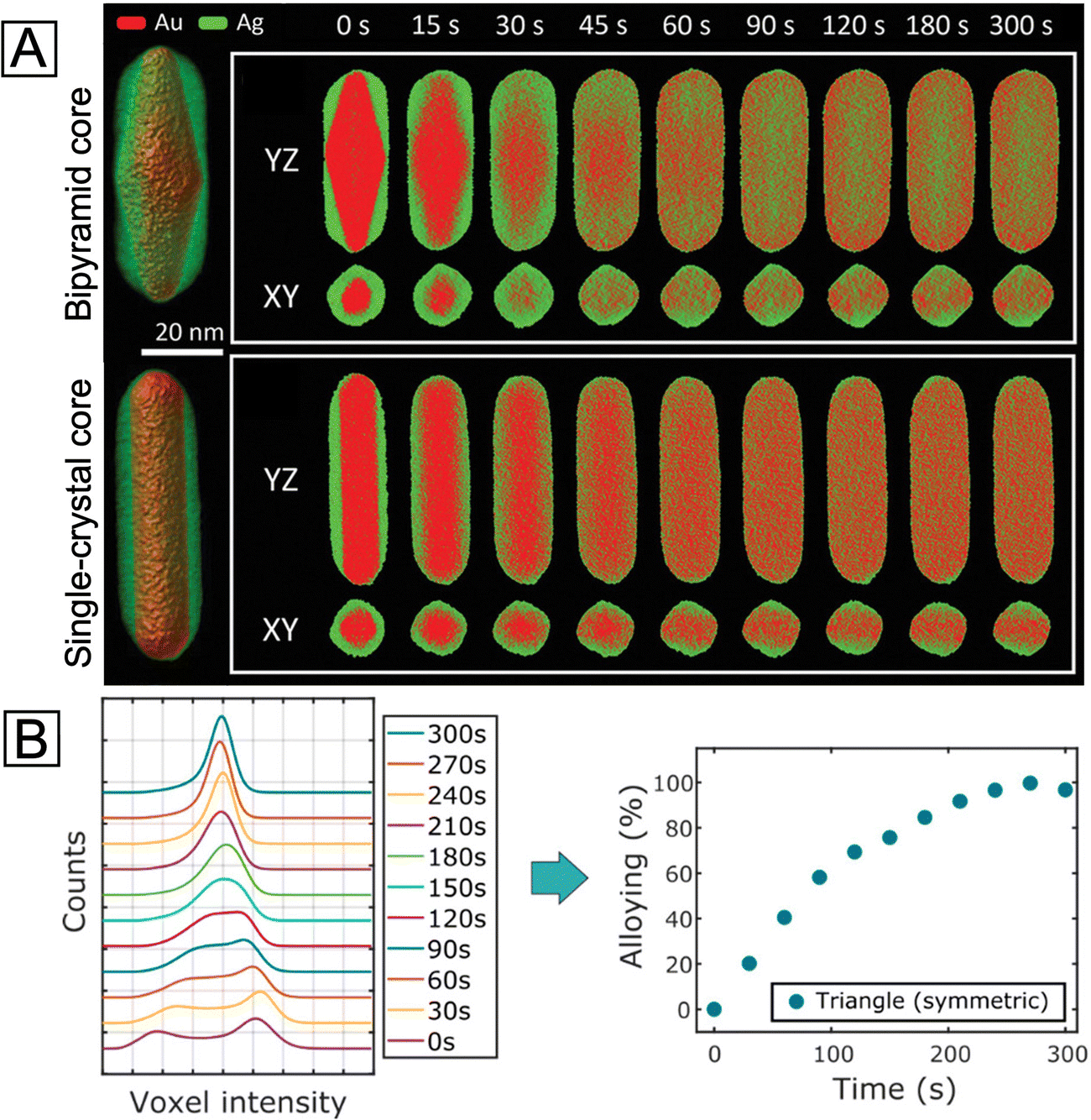 | ||
| Fig. 5 (A) Left: Visualization of 3D reconstructions for the two Au@Ag nanorods with bipyramid and single-crystal Au cores, respectively. Right: The corresponding YZ- and XY-slices of elemental distributions at different stages of alloying at 450 °C. Reproduced from ref. 61 with permission. Copyright 2021 Wiley-VCH. (B) Left: Evolution of the histogram of voxel intensities in the reconstruction of a symmetric Ag@Au nanotriangle during alloying. Right: Progress of alloying for the symmetric nanotriangle during heating at 450 °C, estimated from the spread of histograms of voxel intensities in the reconstruction. Reproduced from ref. 62 with permission. Copyright 2019 American Chemical Society. | ||
In terms of direct probing of the surface feature, scanning tunneling microscopy (STM), one of the most powerful surface imaging techniques, has been extensively used to investigate the atomic-scale surface morphology of bimetallic systems.63 In particular, these reports about the incorporation of hetero-metal atoms into single-crystal substrates have unmasked some essential information about the geometry and local electronic structure of near-surface alloys, providing valuable references for comprehending the subtlest surface structures of core–shell nanocrystals. For instance, a fine analysis of the STM images revealed the temperature dependence of Pd's incorporation into the Au{111} surface.64 As shown in Fig. 6(A), at a deposition temperature of 460 K, Pd adatoms were fully coordinated below the Au surface (imaged as three-lobed depressions marked as red triangles in Fig. 6(A), right panel), yielding stable alloys. At a temperature of 380 K, however, a portion of Pd adatoms appeared on the surface (imaged as protrusions in Fig. 6(B), right panel). At an even lower temperature of 290 K, most of the Pd atoms grew into islands on top of the Au surface (shown in Fig. 6(C)), with the substitutional Pd atoms on the Au surface serving as nucleation sites. Such direct microscopy knowledge is of great importance to the rational synthesis of core–shell nanocrystals with desired surface compositions. To further probe the interaction of bimetallic catalysts with gas-phase adsorbates,65 the co-adsorption of H2 and CO molecules on the Pt single atom/Cu(111) alloy substrate was interrogated using STM, as shown in Fig. 6(D)–(F). With the removal of mobile H adatoms (Fig. 6(D)) by increasing the bias, some dark immobile depressions remained on the surface (Fig. 6(E)), indicating strong binding with the underlying metal atoms. As some individual bias pulses were locally delivered to these spots, the dark depressions disappeared and turned into stationary protrusions at the same locations (Fig. 6(F)). Hence these strong-binding adsorbates were identified as CO molecules selectively adsorbed on the Pt sites, unmasking the preferential Pt poisoning by CO during H2 dissociation. Altogether, these two case studies demonstrated the power of this in situ manipulation and imaging techniques, especially in providing atom-scale structural basis for both comprehension and optimization of a bimetallic surface.
 | ||
| Fig. 6 (A)–(C) Side-view schematics and corresponding STM images showing the temperature dependence of Pd's incorporation (A), (B) in the subsurface and surface layers and (C) on the surface as adatom islands. Reproduced from ref. 64 with permission. Copyright 2010 American Chemical Society. (D)–(F) STM images showing the co-adsorption of H and CO on a Pt–Cu(111) “single-atom alloy” surface and STM tip-induced adsorbate removal to reveal the binding sites beneath. Reproduced from ref. 65 with permission. Copyright 2016 American Chemical Society. | ||
It is worth emphasizing that the success of probe-based techniques is not limited to microscopy, and it has been extended to the spectroscopic domain. As an alternative to microscopy, probe-based spectroscopy has recently shown distinctive power in the statistical surface analysis of metal nanocrystals. Their capability in terms of in situ and real-time detection at large-scale, together with atomic-level sensitivity, makes it possible to elucidate the surface composition, as well as the growth kinetics, of core–shell nanocrystals through spectroscopic analysis. Among various types of spectroscopic techniques, SERS is an especially remarkable representative. With tactfully selected molecular probes, both the species and configurations of metal adatoms on a heterometal surface can be unraveled by following the chemisorption-induced Raman shifts. For example, according to recent reports, the stretching frequency of the N–C bond (νNC) in isocyanide molecules is sensitive to not only the metal atoms to which they are bound, but more subtly the configurations of the binding.66 When the –NC group of the 2,6-dimethylphenyl isocyanide (2,6-DMPI) molecule binds to one, two, and three adjacent Pd atoms in the atop, bridge, and hollow configurations (Fig. 7(A)), respectively, the π-back-donation of Pd will weaken the N–C bond and result in a series of redshifted νNC bands (designated νNC(Pd)-atop, νNC(Pd)-bridge, and νNC(Pd)-hollow, respectively). On the contrary, binding to Ag atoms will lead to a blue shift. Since bridge and hollow configurations are only possible when Pd atoms exist as clusters, the nucleation of Pd on the Ag surface can be tracked by analyzing the corresponding SERS peaks. Fig. 7(B) shows a preliminary demonstration of 2,6-DMPI as a characteristic reporter for such a growth process. When a relatively small amount of Pd precursor was added into the suspension of Ag nanocubes, only the peak of νNC(Pd)-atop emerged in the spectrum, implying that Pd was deposited as individual atoms. As the Pd supply was increased, the peaks corresponding to νNC(Pd)-bridge and νNC(Pd)-hollow started to appear, indicating the formation of Pd clusters. With further in situ dynamic tracking, the roles played by reaction temperature and precursor species in the heterogeneous nucleation of Pd could also be investigated. It is worth pointing out that, on {111} facets (i.e., corners) of Ag nanocubes, the hollow configuration is characteristic while the bridge configuration is prohibited, as illustrated in Fig. 7(A). While the diffusion pathway of Pd adatoms was not clearly resolved by the facet-dependent SERS peaks in this case study, such facet-configuration relationship offers a good opportunity to study the growth mechanism. Optimistically speaking, further development of the detection strategy and resolving power may enable the distinction of both facets and configurations at the same time. By then, one would be able to observe the dynamic evolution of core–shell nanocrystals through in situ SERS analysis.
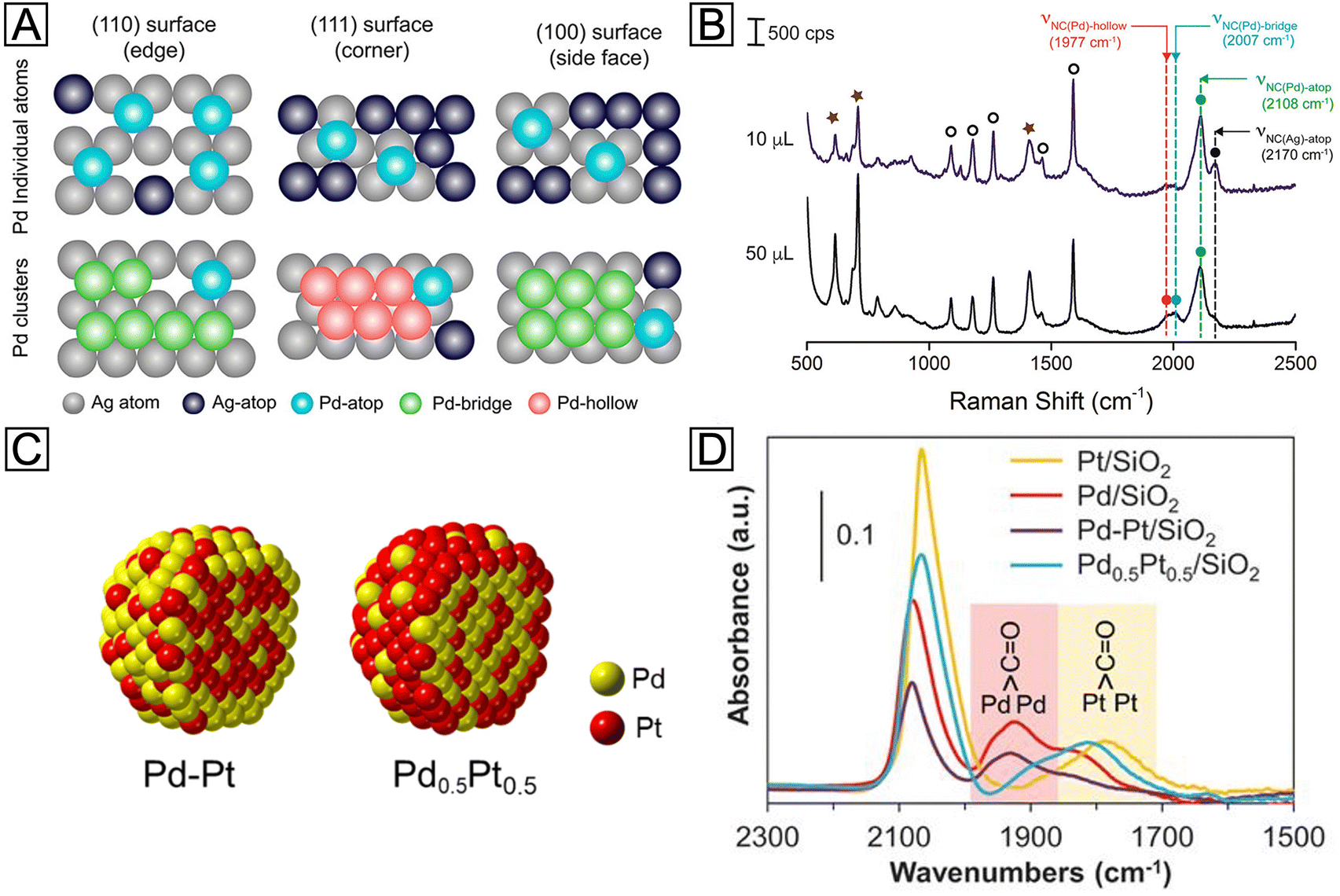 | ||
| Fig. 7 (A) Schematic illustrations showing the different binding sites and configurations for Pd adatoms on the surface of an Ag nanocube. (B) Ex situ SERS spectra recorded from 2,6-DMPI-functionalized samples prepared from Ag nanocubes, ascorbic acid, polyvinylpyrrolidone, and different volumes of Na2PdCl4. Reproduced from ref. 66 with permission. Copyright 2018 American Chemical Society. (C) Atomic models showing the distributions of Pt and Pd atoms in Pd–Pt and Pd0.5Pt0.5 bimetallic nanoparticles. (D) IR spectra of CO molecules adsorbed on Pd/SiO2, Pt/SiO2, Pd–Pt/SiO2, and Pd0.5Pt0.5/SiO2 catalysts, respectively. Reproduced from ref. 70 with permission. Copyright 2018 American Association for the Advancement of Science. | ||
In addition to isocyanide molecules, CO is another promising candidate for probing the surface of metal-based materials. Being isoelectronic to the –NC group, CO binds to a transition metal (TM) in a similar way. As established by the extensive literature pertaining to its chemisorption, the adsorption strength of CO varies across the TM series, leading to vibrational shifts in varying degrees.67 By taking advantage of such differences, CO can be a simple and yet powerful probe for probing the surface of noble-metal nanocrystals. In an early study on single-crystal substrates, the thickness-dependent surface strain of Pt layers on Ru(0001) was successfully detected by infrared spectroscopy (IR), with CO as a probe.68 When the thickness of Pt was increased from 1.3 monolayers to 2.5 monolayers, the IR frequency showed a quite different trend of shifting during the thermal desorption of CO. A more recent study has also successfully detected the Pt atom distribution on the Cu(111) surface as a function of Pt coverage and deposition temperature.69 These examples suggest that the geometric effect caused by the shell thickness can be readily interpreted using CO as a probe molecule, making it feasible to optimize the catalytic activity of core–shell nanocrystals. Moreover, such characterization is not limited to single-crystal surfaces. In a report on Pd–Pt bimetallic nanoparticles, for example, CO was employed as an IR probe to detect the relative content of these two metals on the surface.70 Two types of bimetallic particles were synthesized through sequential adsorption and co-adsorption, designated as Pd–Pt and Pd0.5Pt0.5, respectively (Fig. 7(C)). By comparing the intensities of νCO(Pd)-bridge and νCO(Pt)-bridge peaks, as shown in Fig. 7(D), the Pd–Pt and Pd0.5Pt0.5 exhibited Pd-like and Pt-like spectral features, respectively, suggesting their respective Pd- and Pt-rich surfaces. Besides adsorption energies, νTM-CO is also found to be strongly affected by lattice strain. Thus, the application of IR strain detection in the nanoscale may provide a long-sought guidance for the rational design and precise synthesis of core–shell nanocrystals.
4. Concluding remarks
Thanks to the efforts over the past two decades, great progress has been made with regard to the employment of core–shell structures to enhance the performance of metallic materials, as well as to tackle synthetic challenges. The successful preparation and application of a vast set of bimetallic core–shell nanocrystals exemplify the great opportunities they are providing while suggesting more achievements to be accomplished in this field. Nevertheless, the mystery of their surface composition is like a fickle dark cloud hanging over researchers, obstructing new insights into their structure–property relationships. To attain rational synthesis of bimetallic core–shell nanocrystals, it is safe to say that elucidation of their surface composition and dynamic changes is a rite of passage. Due to the excessive penetration depth of electrons, some electron-based microscopic and spectroscopic techniques do not work well on this matter. On the contrary, many probe-based techniques seem to offer a powerful means to resolve relevant issues. We believe that the techniques highlighted in this Focus article, namely ET, STM, SERS, and IR, hold great promise in revealing the characteristics of the greatest concern regarding the surface of bimetallic core–shell nanocrystals. We should also admit, however, that all four tools (and other relevant ones, too) are deficient to a certain extent. For ET, despite its capability to understand the 3D configuration of a core–shell structure, the considerably increased beam exposure might alter the morphology and composition of the nanocrystal. For scanning probe microscopy techniques like STM, the stringent conditions for testing make in situ and real-time observation out of the question, while for molecular probe-based spectroscopic methods including SERS and IR, only statistical information can be obtained and the variations among individual particles is diminished. Therefore, future improvements and innovations are eagerly anticipated to relieve such poignant blindness on the surface.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported in part by a number of research grants from NSF, including CHE-2002653 and CHE-2105602. It has also been sponsored by the start-up funds from Georgia Tech. We are grateful to our co-workers and collaborators for their invaluable contributions to this project.References
- K. D. Gilroy, A. Ruditskiy, H. C. Peng, D. Qin and Y. Xia, Chem. Rev., 2016, 116, 10414–10472 CrossRef CAS PubMed.
- M. Zhou, C. Li and J. Fang, Chem. Rev., 2021, 121, 736–795 CrossRef CAS PubMed.
- D. Wang and Y. Li, Adv. Mater., 2011, 23, 1044–1060 CrossRef CAS PubMed.
- F. Calvo, Phys. Chem. Chem. Phys., 2015, 17, 27922–27939 RSC.
- R. Ghosh Chaudhuri and S. Paria, Chem. Rev., 2012, 112, 2373–2433 CrossRef CAS PubMed.
- H. Zou, Z. Luo, X. Yang, Q. Xie and Y. Zhou, J. Mater. Sci., 2022, 57, 10912–10942 CrossRef CAS.
- W. Wang, Z. Chen, Y. Shi, Z. Lyu, Z. Cao, H. Cheng, M. Chi, K. Xiao and Y. Xia, ChemCatChem, 2020, 12, 5156–5163 CrossRef CAS.
- X. Yang, Z. Liang, S. Chen, M. Ma, Q. Wang, X. Tong, Q. Zhang, J. Ye, L. Gu and N. Yang, Small, 2020, 16, 2004727 CrossRef CAS PubMed.
- N. Eom, M. E. Messing, J. Johansson and K. Deppert, ACS Nano, 2021, 15, 8883–8895 CrossRef CAS PubMed.
- S. E. Habas, H. Lee, V. Radmilovic, G. A. Somorjai and P. Yang, Nat. Mater., 2007, 6, 692–697 CrossRef CAS PubMed.
- Z. Fan and H. Zhang, Acc. Chem. Res., 2016, 49, 2841–2850 CrossRef CAS PubMed.
- Y. C. Yao, D. S. He, Y. Lin, X. Q. Feng, X. Wang, P. Q. Yin, X. Hong, G. Zhou, Y. Wu and Y. D. Li, Angew. Chem., Int. Ed., 2016, 55, 5501–5505 CrossRef CAS PubMed.
- M. Zhao, L. Figueroa-Cosme, A. O. Elnabawy, M. Vara, X. Yang, L. T. Roling, M. Chi, M. Mavrikakis and Y. Xia, Nano Lett., 2016, 16, 5310–5317 CrossRef CAS PubMed.
- X. Wang, L. Figueroa-Cosme, X. Yang, M. Luo, J. Liu, Z. Xie and Y. Xia, Nano Lett., 2016, 16, 1467–1471 CrossRef CAS PubMed.
- N. V. Long, M. Ohtaki, T. D. Hien, J. Randy and M. Nogami, Electrochim. Acta, 2011, 56, 9133–9143 CrossRef.
- Y. Yan, H. Shan, G. Li, F. Xiao, Y. Jiang, Y. Yan, C. Jin, H. Zhang, J. Wu and D. Yang, Nano Lett., 2016, 16, 7999–8004 CrossRef CAS PubMed.
- P. Zhai, Y. Shi, Q. Wang, Y. Xia and K. Ding, Nanoscale, 2021, 13, 18498–18506 RSC.
- U. Aslam, S. Chavez and S. Linic, Nat. Nanotechnol., 2017, 12, 1000–1005 CrossRef CAS PubMed.
- M. Vara, L. T. Roling, X. Wang, A. O. Elnabawy, Z. D. Hood, M. Chi, M. Mavrikakis and Y. Xia, ACS Nano, 2017, 11, 4571–4581 CrossRef CAS PubMed.
- S. Xue, G. Chen, F. Li, Y. Zhao, Q. Zeng, J. Peng, F. Shi, W. Zhang, Y. Wang, J. Wu and R. Che, Small, 2021, 17, e2100559 CrossRef PubMed.
- P. Moseley and W. A. Curtin, Nano Lett., 2015, 15, 4089–4095 CrossRef CAS PubMed.
- T. Bligaard and J. K. Nørskov, Electrochim. Acta, 2007, 52, 5512–5516 CrossRef CAS.
- X. Zhang and G. Lu, J. Phys. Chem. Lett., 2014, 5, 292–297 CrossRef CAS PubMed.
- M. Mavrikakis, B. Hammer and J. K. Nørskov, Phys. Rev. Lett., 1998, 81, 2819–2822 CrossRef.
- P. Strasser, S. Koh, T. Anniyev, J. Greeley, K. More, C. Yu, Z. Liu, S. Kaya, D. Nordlund, H. Ogasawara, M. F. Toney and A. Nilsson, Nat. Chem., 2010, 2, 454–460 CrossRef CAS PubMed.
- X. Wang, S.-I. Choi, L. T. Roling, M. Luo, C. Ma, L. Zhang, M. Chi, J. Liu, Z. Xie, J. A. Herron, M. Mavrikakis and Y. Xia, Nat. Commun., 2015, 6, 7594 CrossRef PubMed.
- L. Bu, N. Zhang, S. Guo, X. Zhang, J. Li, J. Yao, T. Wu, G. Lu, J.-Y. Ma, D. Su and X. Huang, Science, 2016, 354, 1410–1414 CrossRef CAS PubMed.
- T. He, W. Wang, F. Shi, X. Yang, X. Li, J. Wu, Y. Yin and M. Jin, Nature, 2021, 598, 76–81 CrossRef CAS PubMed.
- M. Zhao, A. O. Elnabawy, M. Vara, L. Xu, Z. D. Hood, X. Yang, K. D. Gilroy, L. Figueroa-Cosme, M. Chi, M. Mavrikakis and Y. Xia, Chem. Mater., 2017, 29, 9227–9237 CrossRef CAS.
- M. Zhao, L. Xu, M. Vara, A. O. Elnabawy, K. D. Gilroy, Z. D. Hood, S. Zhou, L. Figueroa-Cosme, M. Chi, M. Mavrikakis and Y. Xia, ACS Catal., 2018, 8, 6948–6960 CrossRef CAS.
- A. Janssen, Q. N. Nguyen, Z. Lyu, V. Pawlik, C. Wang and Y. Xia, J. Phys. Chem. C, 2023, 127, 1280–1291 CrossRef CAS.
- S. H. Joo, J. Y. Park, J. R. Renzas, D. R. Butcher, W. Huang and G. A. Somorjai, Nano Lett., 2010, 10, 2709–2713 CrossRef CAS PubMed.
- S. Xie, S.-I. Choi, N. Lu, L. T. Roling, J. A. Herron, L. Zhang, J. Park, J. Wang, M. J. Kim, Z. Xie, M. Mavrikakis and Y. Xia, Nano Lett., 2014, 14, 3570–3576 CrossRef CAS PubMed.
- S.-I. Choi, A. Young, S. R. Lee, C. Ma, M. Luo, M. Chi, C.-K. Tsung and Y. Xia, Nanoscale Horiz., 2019, 4, 1232–1238 RSC.
- X. Xia, J. Zhang, N. Lu, M. J. Kim, K. Ghale, Y. Xu, E. McKenzie, J. Liu and H. Ye, ACS Nano, 2015, 9, 9994–10004 CrossRef CAS PubMed.
- A. Janssen, Z. Lyu, M. Figueras-Valls, H.-Y. Chao, Y. Shi, V. Pawlik, M. Chi, M. Mavrikakis and Y. Xia, Nano Lett., 2022, 22, 3591–3597 CrossRef CAS PubMed.
- S. Chavez, U. Aslam and S. Linic, ACS Energy Lett., 2018, 3, 1590–1596 CrossRef CAS.
- W. Hong, J. Lan, H. Li, Z. Yan, Y. Li, H. Jiang and M. Chen, Opt. Laser Technol., 2022, 146, 107565 CrossRef CAS.
- M. S. Sajna, G. Vimal and K. K. Sadasivuni, Top. Catal., 2022 DOI:10.1007/s11244-021-01552-8.
- W. Jiang, B. Q. L. Low, R. Long, J. Low, H. Loh, K. Y. Tang, C. H. T. Chai, H. Zhu, H. Zhu, Z. Li, X. J. Loh, Y. Xiong and E. Ye, ACS Nano, 2023, 17, 4193–4229 CrossRef CAS PubMed.
- Q. Li, H. Huo, Y. Wu, L. Chen, L. Su, X. Zhang, J. Song and H. Yang, Adv. Sci., 2023, 10, 2202051 CrossRef CAS PubMed.
- J. Li, J. Liu, Y. Yang and D. Qin, J. Am. Chem. Soc., 2015, 137, 7039–7042 CrossRef CAS.
- J. Chen, J. M. McLellan, A. Siekkinen, Y. Xiong, Z.-Y. Li and Y. Xia, J. Am. Chem. Soc., 2006, 128, 14776–14777 CrossRef CAS PubMed.
- Y. Yang, J. Liu, Z.-W. Fu and D. Qin, J. Am. Chem. Soc., 2014, 136, 8153–8156 CrossRef CAS PubMed.
- M. Zhao, X. Wang, X. Yang, K. D. Gilroy, D. Qin and Y. Xia, Adv. Mater., 2018, 30, 1801956 CrossRef PubMed.
- P. Hou, H. Liu, J. Li and J. Yang, CrystEngComm, 2015, 17, 1826–1832 RSC.
- M. Zhou, H. Wang, A. O. Elnabawy, Z. D. Hood, M. Chi, P. Xiao, Y. Zhang, M. Mavrikakis and Y. Xia, Chem. Mater., 2019, 31, 1370–1380 CrossRef CAS.
- J. Li, Z. Xi, Y.-T. Pan, J. S. Spendelow, P. N. Duchesne, D. Su, Q. Li, C. Yu, Z. Yin, B. Shen, Y. S. Kim, P. Zhang and S. Sun, J. Am. Chem. Soc., 2018, 140, 2926–2932 CrossRef CAS PubMed.
- X. Lyu, Y. Jia, X. Mao, D. Li, G. Li, L. Zhuang, X. Wang, D. Yang, Q. Wang, A. Du and X. Yao, Adv. Mater., 2020, 32, 2003493 CrossRef CAS PubMed.
- S. Koh and P. Strasser, J. Am. Chem. Soc., 2007, 129, 12624–12625 CrossRef CAS PubMed.
- X. Lu, M. Ahmadi, F. J. DiSalvo and H. D. Abruña, ACS Catal., 2020, 10, 5891–5898 CrossRef CAS.
- K. J. J. Mayrhofer, V. Juhart, K. Hartl, M. Hanzlik and M. Arenz, Angew. Chem., Int. Ed., 2009, 48, 3529–3531 CrossRef CAS PubMed.
- G. González-Rubio, P. Díaz-Núñez, W. Albrecht, V. Manzaneda-González, L. Bañares, A. Rivera, L. M. Liz-Marzán, O. Peña-Rodríguez, S. Bals and A. Guerrero-Martínez, Adv. Opt. Mater., 2021, 9, 2002134 CrossRef.
- J. E. S. van der Hoeven, J. Jelic, L. A. Olthof, G. Totarella, R. J. A. van Dijk-Moes, J.-M. Krafft, C. Louis, F. Studt, A. van Blaaderen and P. E. de Jongh, Nat. Mater., 2021, 20, 1216–1220 CrossRef CAS PubMed.
- Q. Wang, Z. L. Zhao and M. Gu, Small, 2019, 15, 1903122 CrossRef CAS PubMed.
- K. Li, X. Ma, S. He, L. Wang, X. Yang, G. Zhang, S. Guan, X. Qu, S. Zhou and B. Xu, ACS Biomater. Sci. Eng., 2022, 8, 540–550 CrossRef CAS PubMed.
- P. Weber, D. J. Weber, C. Dosche and M. Oezaslan, ACS Catal., 2022, 12, 6394–6408 CrossRef CAS.
- C. S. Bonifacio, S. Carenco, C. H. Wu, S. D. House, H. Bluhm and J. C. Yang, Chem. Mater., 2015, 27, 6960–6968 CrossRef CAS.
- B. Goris, L. Polavarapu, S. Bals, G. Van Tendeloo and L. M. Liz-Marzán, Nano Lett., 2014, 14, 3220–3226 CrossRef CAS PubMed.
- K. Jenkinson, L. M. Liz-Marzán and S. Bals, Adv. Mater., 2022, 34, 2110394 CrossRef CAS PubMed.
- M. Mychinko, A. Skorikov, W. Albrecht, A. Sanchez-Iglesias, X. Zhuo, V. Kumar, L. M. Liz-Marzan and S. Bals, Small, 2021, 17, e2102348 CrossRef PubMed.
- A. Skorikov, W. Albrecht, E. Bladt, X. Xie, J. E. S. van der Hoeven, A. van Blaaderen, S. Van Aert and S. Bals, ACS Nano, 2019, 13, 13421–13429 CrossRef CAS PubMed.
- M. Salmeron and B. Eren, Chem. Rev., 2021, 121, 962–1006 CrossRef CAS PubMed.
- A. E. Baber, H. L. Tierney and E. C. H. Sykes, ACS Nano, 2010, 4, 1637–1645 CrossRef CAS PubMed.
- J. Liu, F. R. Lucci, M. Yang, S. Lee, M. D. Marcinkowski, A. J. Therrien, C. T. Williams, E. C. H. Sykes and M. Flytzani-Stephanopoulos, J. Am. Chem. Soc., 2016, 138, 6396–6399 CrossRef CAS PubMed.
- Y. Wu and D. Qin, J. Am. Chem. Soc., 2018, 140, 8340–8349 CrossRef CAS PubMed.
- S. E. Mason, I. Grinberg and A. M. Rappe, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 161401 CrossRef.
- A. Schlapka, M. Lischka, A. Groß, U. Käsberger and P. Jakob, Phys. Rev. Lett., 2003, 91, 016101 CrossRef CAS.
- D. Moline, M. Inagaki, E. Kazuma, Y. Kim and M. Trenary, J. Phys. Chem. C, 2023, 127, 9796–9806 CrossRef.
- K. Ding, D. A. Cullen, L. Zhang, Z. Cao, A. D. Roy, I. N. Ivanov and D. Cao, Science, 2018, 362, 560–564 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |