
Charge–orbital synergistic engineering of TM@Ti3C2O1−xBx for highly selective CO2 electrochemical reduction†
Jiahe
Peng
ab,
Zuhao
Shi
 ab,
Jizhou
Jiang
c,
Peng
Zhang
d,
Jyh-Ping
Hsu
e and
Neng
Li
*abc
ab,
Jizhou
Jiang
c,
Peng
Zhang
d,
Jyh-Ping
Hsu
e and
Neng
Li
*abc
aState Key Laboratory of Silicate Materials for Architectures, Wuhan University of Technology, Wuhan 430070, China. E-mail: lineng@whut.edu.cn
bShenzhen Research Institute of Wuhan University of Technology, Shenzhen 518000, Guangdong, China
cSchool of Chemistry and Environmental Engineering, School of Environmental Ecology and Biological Engineering, Novel Catalytic Materials of Hubei Engineering Research Center, Wuhan Institute of Technology, Wuhan, 430205, China
dState Center for International Cooperation on Designer Low-Carbon & Environmental Materials (CDLCEM), School of Materials Science and Engineering, Zhengzhou University, Zhengzhou 450001, Henan, China
eDepartment of Chemical Engineering, National Taiwan University, Taipei, 10617, China
First published on 6th June 2023
Abstract
Inspired by MXene nanosheets and their regulation of surface functional groups, a series of Ti3C2-MXene-based single TM atom electrocatalysts with a doped boron (B) atom (TM@Ti3C2O2−xBx, TM is V, Cr, Mn, Fe, Co or Ni, x = 0.11) are proposed for achieving a high performance catalytic CO2 reduction reaction (CO2RR). The results reveal that the doped B atom involves in the adsorption reaction of CO2 molecules and CO intermediates in the CO2RR. The TM-to-C and B-to-C π-back bonding contribute to the activation of the CO2 molecules and CO intermediates in the CO2RR. Enough electrons from the single TM atom and B atom occupied orbitals can be injected into the CO2 molecules and *CO intermediates through direct bonding interactions, which effectively alleviates the difficulty of the first hydrogenation reaction step and further helps CO reduction towards CH4. The calculated values of ΔG for the first hydrogenation reaction and the formation of *CHO on Ti3C2O2−xBx are significantly smaller than those of other single-atom catalysts (SACs). Fe@Ti3C2O2−xBx is found to have the highest electrocatalytic activity with a limiting potential of ∼0.40 V and exhibits a high selectivity for obtaining CH4 through the CO2RR compared with the hydrogen evolution reaction. This work is expected to open a research path for engineering the charge–orbital state of the innate atoms of a substrate based on mechanistic insights, which guides the rational design of highly selective MXene-based CO2RR electrocatalysts.
 Neng Li | Congratulations on the 10th anniversary and best wishes to Materials Horizons. Our first paper was published in Materials Horizons, and then chosen as the outstanding paper in the in 2018, and selected as “highlighting the members of the Materials Horizons Advisory Board” in 2021. The best way to express our gratitude is to continue to interact closely with the community, and report our significant research advances, such as novel materials, and new reaction mechanisms. We are proud to have solid and excellent cooperation with such an esteemed journal. Here's to another decade, we would like to further contribute this esteemed journal. |
New conceptsAlthough the development of efficient atomic electrocatalysts to resolve activity and selectivity issues of the CO2 reduction reaction (CO2RR) continues to receive increasing attention, it is still challenging. Most of the previous works in this field have primarily focused on TM atoms and introducing a nonmetal atom to modulate the interplay between the TM active centers and multiple intermediates. Herein, we propose a novel mechanism of introducing a boron element, which takes the advantage of charge–orbital synergism to activate the TM atoms of dual atomic electrocatalysts for the CO2RR. The charge and orbital order controlling resulting from the combined effect of the single TM atoms and B atom can effectively activate CO2 molecules and CO intermediates in the CO2RR process. The TM-to-C, and B-to-C π-back bonding contribute to the activation of CO2 molecules and CO intermediates in the CO2RR. Based on this mechanism, we further design a series of TM atoms supported by a monolayer B-doped Ti3C2O2 (Ti3C2O2−xBx) for the CO2RR. Following a comprehensive screening strategy, Fe@Ti3C2O2−xBx are identified as promising catalysts with high activity and selectivity. Moreover, we identify the valence states of TM as a valid descriptor to evaluate the CO2RR performance on such catalysts, realizing a fast pre-screening of other potential dopants on the MXene-based CO2RR SACs. Our work thus provides a new direction for the rational design of MXene-based dual atomic electrocatalysts. |
1. Introduction
The rapid economic development and urbanization have already caused a sharp increase in the global concentration of carbon dioxide (CO2).1,2 The global warming resulting from the excessive consumption of fossil fuels not only poses a serious threat to global economic security but also affects resources, energy, ecology, and food security, among others. It also poses a severe challenge to human survival.3,4 In an attempt to alleviate the increase in the average global temperature, Paris Climate Change Conference signed the Paris Agreement.5–7 The electrochemical reduction of CO2 to produce carbon-based fuels and chemicals has great potential in solving current environmental problems and benefiting the economy of our society.8,9 However, the high chemical stability of CO2 making it hard to be activated at high conversion rates and selectivity is a crucial issue.10,11 In addition, the CO2RR pathway includes a chemical dehydration step leading to adsorption of CO,12–14 which may act as a poison blocking the surface and impeding further protonation steps because of the high desorption energy and a further reduction barrier.15–17In recent years, intense research has been carried out to discover efficient, selective and stable cathode catalysts. Copper-based materials have been widely regarded as efficient cathode materials for the CO2RR, producing diverse products, such as carbon monoxide (CO), formic acid (HCOOH), methanol (CH3OH), methane (CH4), ethanol (C2H5OH), and ethylene (C2H4).18–20 Intense optimization of the catalyst materials and reaction conditions has resulted in significant progress over the past few years.21,22 Irabien et al. found that the CuII atoms in the HKUST-1 metal–organic framework ([Cu3(μ6-C9H3O6)2]n) can promote the electrocatalytic conversion of CO2 to alcohols.23 The subsequent research studies have reported Cu-based bimetallic electrocatalysts through doping other transition metal atoms to further improve the CO2 conversion performance.24,25 Therefore, downsizing the metal nanostructures to singly dispersed metal atoms is highly desirable for maximizing the efficiency of catalytically active metal sites. Recently, single-atom catalysts (SACs) containing isolated metal atoms dispersed on solid substrates have attracted significant attention in the field of catalysis.
Single transition metal (TM) atoms anchored on a substrate are widely used as the active sites in the CO2RR.26,27 The excellent performance of transition metal-based catalysts can be ascribed to their occupied d orbitals, which can donate electrons to the antibonding orbitals of gas molecules.28–30 Being a nonmetal element, the boron (B) atom in a molecular catalyst with sp2 or sp3 hybridization has exhibited great potential for N2 fixation.31–33 The B atom also shows the ability to activate CO2 molecules in the CO2RR.34,35 Tang et al. found that the bare edge B on armchair boron nitride nanoribbons is a promising candidate for the catalysis of the CO2RR.36 In light of the above discussion, the active center of catalysts could be either a single TM atom coordinated by non-metal atoms or a B center. It can be inferred that if B serves as the coordination atom of TM, the active sites with the presence of both B and TM may provide stronger activation capacity.
The results of our previous study indicated that the combined effect of TM and B can effectively and selectively ameliorate the performance of electrochemical N2 reduction.37 Being an isoelectronic body of N2, CO is likely to be activated owing to the combined effect of TM and B atoms. Liu et al. successfully fabricated single atomic Fe sites anchored on B and N co-doped carbon (Fe-SA/BNC), which showed excellent CO2RR activity, achieving the highest faradaic efficiency of ∼94% at −0.7 V compared with RHE.38 However, there is a lack of an in-depth discussion on the mechanisms of CO2 activation. A detailed understanding of these mechanisms at the atomic scale is of great importance for clarifying the synergistic effect between TM and B atoms that contributes to the CO2RR. This is beneficial to developing a suitable catalyst with a high catalytic selectivity for the CO2RR.
In addition to the active center, the choice of substrate is another significant factor that can influence SACs’ performance.39,40 MXenes (e.g., Ti3C2Tx, Ti2CTx and Mo2CTx), having a general formula of Mn+1XnTx (n = 1 to 4) with M, X, and Tx being the early transition metals, C or N, and the surface functional groups (usually as –O, –OH or –F), respectively,41,42 have been considered as potential substrates for various applications,43,44 especially in the field of SACs.45–47 Compared with other supports (e.g., doped graphene, MoS2, and g-C3N4), MXenes have not only the highest metallic conductivity but also abundant surface functional groups for anchoring firmly single atoms.48 Recently, MXenes have been widely used as the substrates of SACs for the CO2RR.49 Zhao et al. have successfully demonstrated that a Pt@Ti3C2Tx-based SAC could readily capture CO2 or aniline, yielding value-added amides with a high conversion and selectivity.50 The subsequent study of Yang et al. has produced single atom Cu-immobilized Ti3C2Tx for an electrocatalytic CO2RR to produce methanol with a high faradaic efficiency of 59.1%.51 However, relevant theoretical analyses of the role of B in the MXene-based SACs’ CO2RR are very limited. The SACs of single atoms fixed on the Ti3C2Tx mixed functional group surface have been successfully synthesized experimentally.52,53
In particular, the B atom has been successfully introduced into Ti3C2Tx to replace the O functional group on its surface54 and the single Ru atom anchored on the B-doped Ti3C2Tx exhibited outstanding performance for the hydrogen evolution reaction (HER).55 Relevant reported results provide valuable information for studying the synergistic effect of TM and B atoms on the CO2RR performance of Ti3C2Tx-based SACs. Our previous theoretical analysis also showed that the CO2RR performance of MXene-based SACs can be efficiently improved through controlling of functional groups.56 It suggests that an in-depth study on the performance of the CO2RR through controlling of MXene-based SACs with a mixed functional group surface is necessary. Therefore, we discuss in this study the feasibility of anchoring low cost 3d single TM (TM is V, Cr, Mn, Fe, Co or Ni) atoms on the Ti3C2Tx's surface groups as a mixture of B and O (Ti3C2O2−xBx) to explore the synergistic effect of TM and B atoms on Ti3C2Tx-based SACs for the CO2RR. The analysis is based on DFT calculations.
As expected, the results of our computation show that the combined effect of single TM atoms and B atom markedly activated CO2 molecules and CO intermediates after adsorption. This can be attributed to the injection of adequate charge from the occupied p and d orbitals of B and TM, respectively. In addition, the introduction of the B atom into surface groups can significantly enhance the single atom anchoring capacity. The adsorbed CO2 can be further reduced to CH4 on the Fe@Ti3C2O2−xBx SAC with a rather low overpotential of 0.40 eV. The stability of TM@Ti3C2O2−xBx is systematically evaluated, and the results obtained reveal that the designed catalyst can be synthesized promisingly.
2. Computational details
All calculations were performed on the Vienna ab initio simulation package (VASP).57 The Perdew–Burke–Ernzerhof (PBE) functional in the generalized gradient approximation (GGA) was used to express electron exchange correlation58 and a projector augmented wave (PAW) method was applied to describe the pseudo-potentials.59 Dispersion-corrected DFT-D3 schemes were employed to describe possible van der Waals (VDW) interactions.60 The energy cut-off for the plane-wave basis was set at 450 eV. All configurations were based on a 3 × 3 × 1 supercell of Ti3C2Tx with a vacuum layer larger than 20 Å in the z direction to prevent the interaction between periodic images. The Brillouin zone in reciprocal space was sampled using the Monkhorst–Pack scheme with 5 × 5 × 1 k-point grids for geometry optimization and electronic structure calculations.All structures were fully relaxed until the forces became smaller than 0.01 eV Å−1; the convergence threshold in electronic relaxation was set at 10−5 eV using the conjugate gradient algorithm. The atomic charge was calculated through Bader's charge population analysis.61 The atomic structures were analyzed using the VESTA code.62 The effect of water was considered using the VASPsol code.63–65 The stability of catalysts was evaluated using ab initio molecular dynamics (AIMD) simulations carried out by placing MXene in a vacuum or liquid water and lasted for 10.0 ps with a time step of 1 fs in the NVT ensemble.
The binding energy (Eb) and cohesive energy (Ec) of the transition metals on Ti3C2Tx can be evaluated using
| Eb = ETM–MX − EMX − ETM | (1) |
| Ec = E(TMbulk)/N − ETM, | (2) |
| ΔEad = EX* − E* − EX, | (3) |
A computational hydrogen electrode (CHE) model proposed by Nørskov et al.68 was used to calculate the Gibbs free energy (G) of the reaction for the CO2RR elementary steps involving (H+ + e−) pair transfer. In the CHE model, H+ + e− ⇌ 1/2H2 (g) is in equilibrium with gaseous H2 at 0 V vs. the reversible hydrogen electrode (RHE) for all pH values at 1 bar of H2 pressure. The Gibbs free energy of each elementary step was calculated at 298.15 K using the following eqn (4):
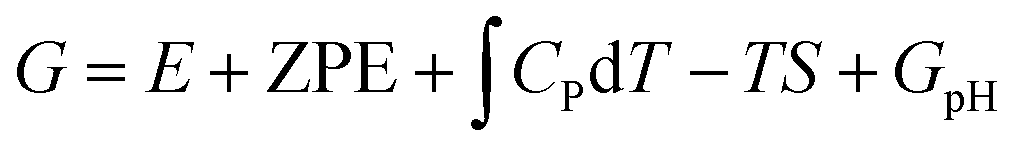 | (4) |
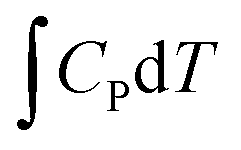 , and TS are the zero-point energy, the enthalpy changes from 0 to T K contributed by molecular vibrations, and the entropy correction based on the calculated vibrational frequencies, respectively. GpH represents the free energy correction for pH, defined as GpH = −kBT
, and TS are the zero-point energy, the enthalpy changes from 0 to T K contributed by molecular vibrations, and the entropy correction based on the calculated vibrational frequencies, respectively. GpH represents the free energy correction for pH, defined as GpH = −kBT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln[H+] = kBTln10 × pH, where kB is the Boltzmann constant.
ln[H+] = kBTln10 × pH, where kB is the Boltzmann constant.
In this work, the pH value was set at 0 for strongly acidic conditions.68 The dependence of the potential (U) on the RHE required for each protonation step is expressed as U = −ΔG/e, where ΔG is the free energy change along each CO2RR pathway. The limiting potential (UL) was obtained from the maximum ΔG(ΔGmax) of each elementary step. Since the Gibbs free energy of the O2, CO, and CO2 molecules in a gas-phase species cannot be calculated precisely using conventional PBE exchange–correlation function,14,69 the free energy of the experimental thermodynamic data was used as a ref. 70 and 71. The free energy of liquid-phase molecules (H2O and CH3OH) was calculated by considering them as an ideal gas and using their corresponding vapor fugacity (f) at μ(liquid) = μ(vapor).14
3. Results and discussion
3.1. Stability of single atoms embedded on Ti3C2O2−xBx
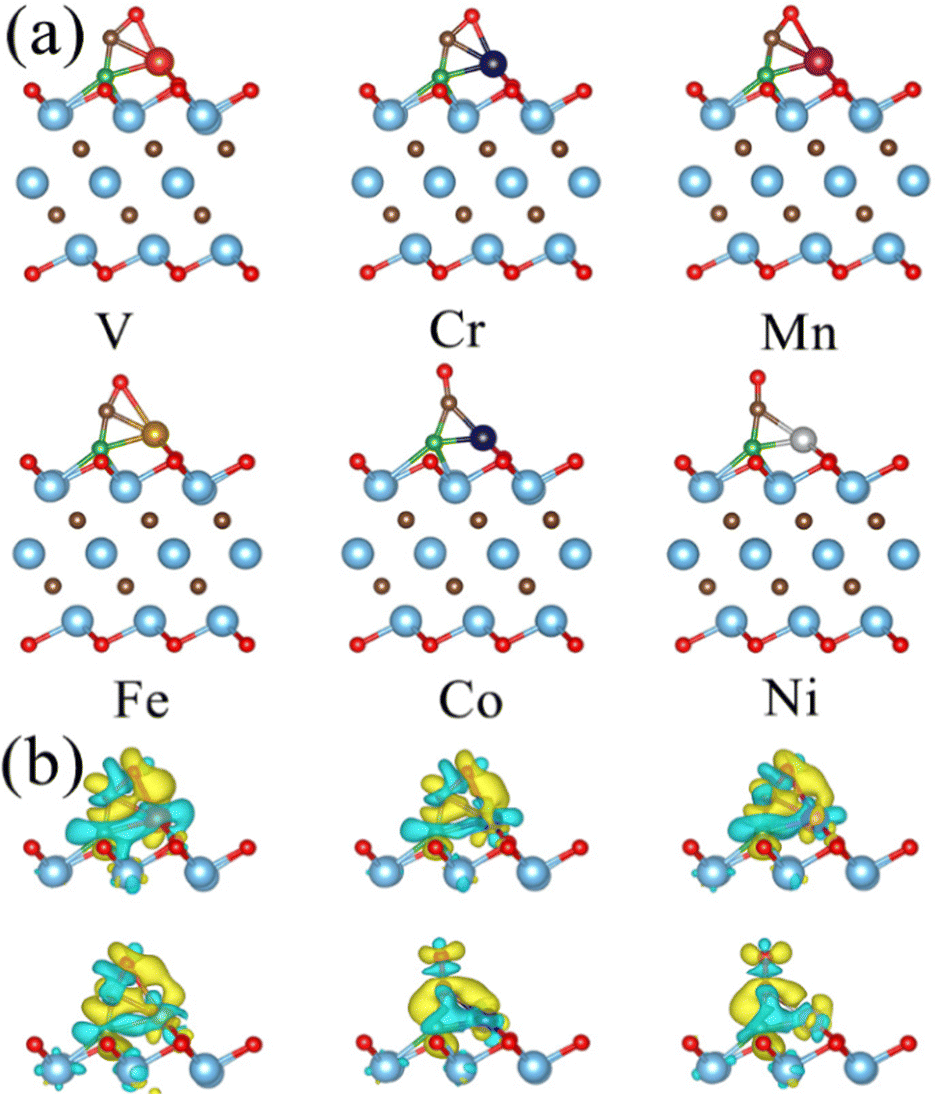
The diffusion and aggregation of the dispersed single atoms on a support can lower the activity and durability of SACs, whereas Ti3C2Tx can capture and stabilize the heteroatom effectively through creating tight bonds between the TM atoms and surface groups. In general, the hollow position consisting of three oxygen groups is the main site on pure Ti3C2Tx to fix single TM atoms.72,73 Therefore, we established a pure Ti3C2Tx with oxygen groups covering its surface (Ti3C2O2). As introduced previously, the B atom can replace the position of the surface oxygen functional group and be used as the coordination atom to fix the TM atom when doped in Ti3C2O2 (Ti3C2O2−xBx). As shown in Fig. 1a and b, two possible hollow sites (H1 and H2) might present to locate the TM on the surface of Ti3C2O2 and Ti3C2O2−xBx. Previous studies, both theoretical and experimental, have identified that the H1 site is a stable adsorption site for metal atoms (Fig. 1c and d).51,55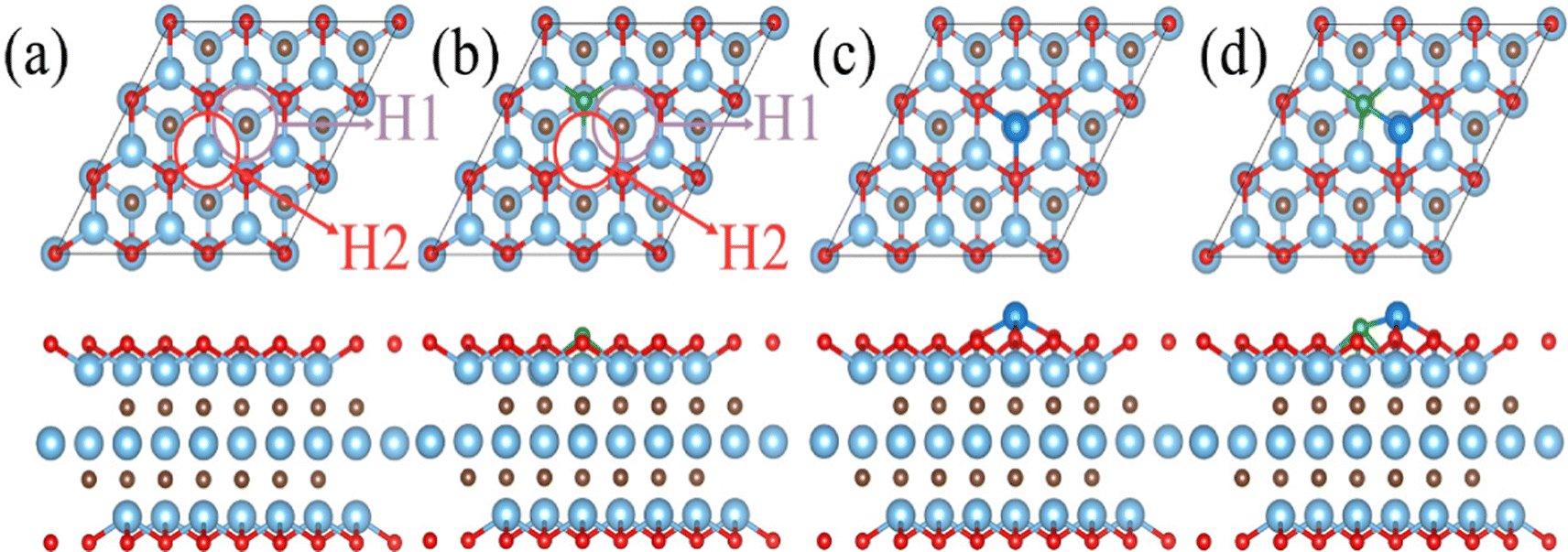 | ||
| Fig. 1 Optimized models of (a) pure Ti3C2O2 and (b) Ti3C2O2−xBx; color scheme: Ti, cyan; C, brown; O, red; B, green; and TM, blue. Circles represent the possible sites of TM on Ti3C2O2. Optimized structures of (c) pure Ti3C2O2 and (d) Ti3C2O2−xBx monolayer confining TM single atoms at the H1 site. | ||
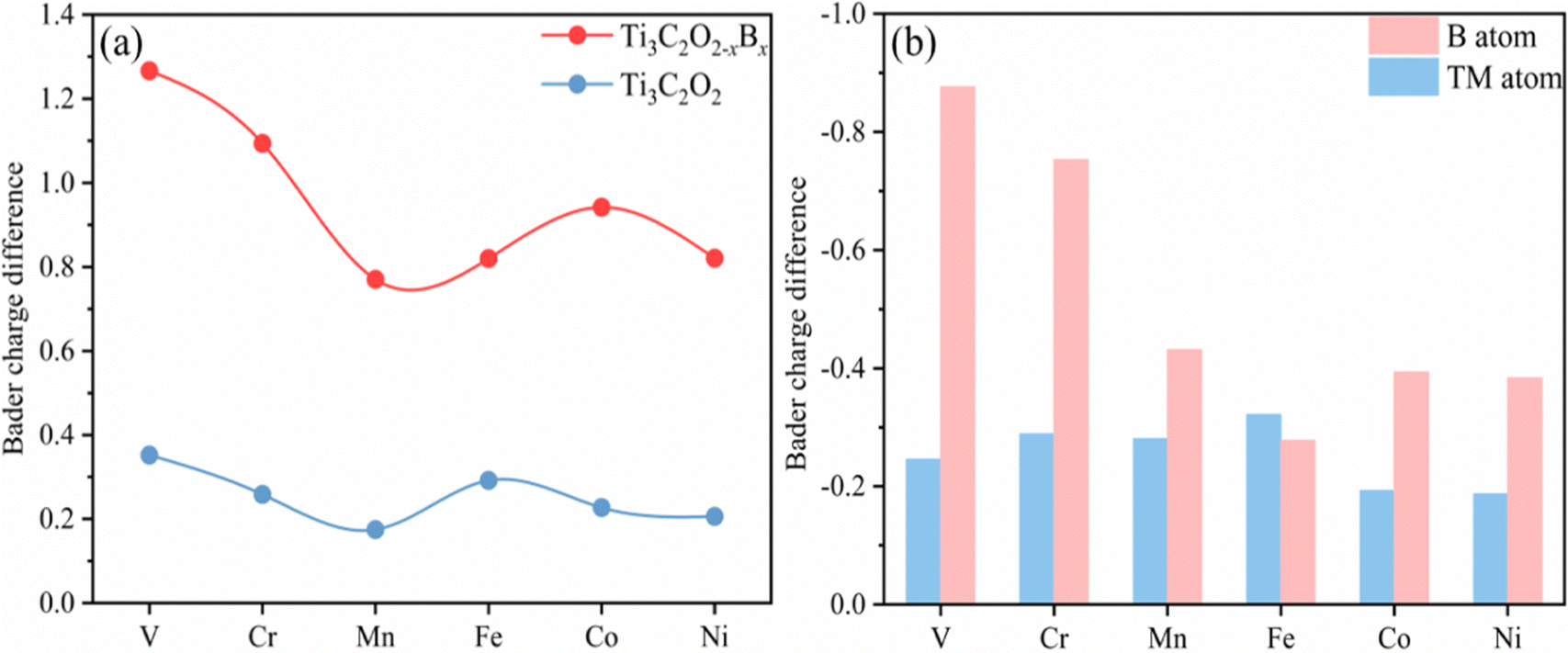
Thermodynamically, the aggregation of the dispersed TM atoms depends on the bond strength between the TM and the substrate as well as the recombination ability of metal atoms. Therefore, the stability of SACs can be measured using the values of Eb–Ecoh of the TM and substrate (eqn (1) and (2)): the smaller this value the more thermodynamically stable the structure of SACs (Table S1, ESI†). For all of the TM atoms, the value of Eb–Ecoh for TM@Ti3C2O2−xBx is lower than that for TM@Ti3C2O2, suggesting that the presence of B is advantageous for stabilizing TM atoms (Fig. 2a). This can be explained by the fact that B atoms bind TM atoms through substantial partial charge transferred from the latter to the former. When the B atom is doped into the Ti3C2O2 substrate, two electrons are transferred from the latter to the former, leaving the partial 2p orbital with lack of electrons. The empty 2p orbital helps the B atom gaining more electrons from the TM single atom than from O atom (Fig. 2a), leading to a thermodynamically more stable structure.
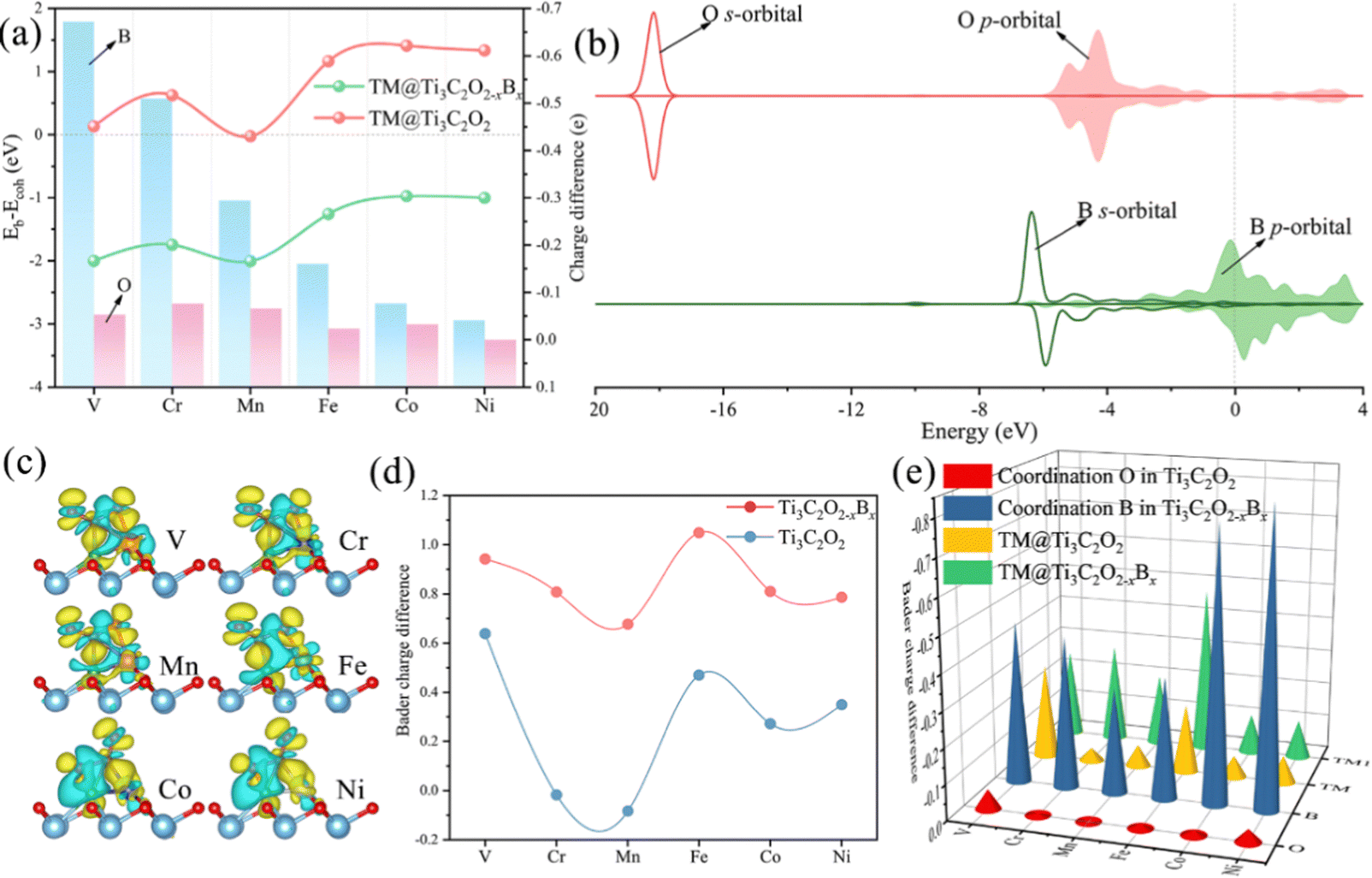 | ||
| Fig. 2 (a) The Eb–Ecoh values of TM@Ti3C2O2−xBx and TM@Ti3C2Tx (line chart), charge differences of B and O atoms before and after anchoring a single TM atom (histogram). (b) PDOSs (projected density of states) of the O or B surface atoms before fixing the TM single atoms. (c) Variation of charge density differences of TM@Ti3C2O2−xBx with a CO2 adsorption configuration. The yellow region denotes charge accumulation and the blue region indicates charge depletion, where the isosurface value is set to be 0.03 e Å−3. (d) Bader charge differences of the C atom in CO2 before and after adsorption on TM@Ti3C2O2−xBx and TM@Ti3C2Tx. (e) Bader charge differences of TM atoms, the coordination O atom in TM@Ti3C2O2 and the coordination B atom in TM@Ti3C2O2−xBx before and after CO2 adsorption. | ||
As shown in Fig. 2b, since the localized 2s orbital of the O atom is in the deep energy level, it is difficult for this orbital to participate in bonding. The 2p orbital of the O atom is almost below the Fermi level, implying that it is fully occupied. The O atom with a saturated electronic structure is not conducive for receiving electrons from TM atoms. Compared with the O atom, the 2s orbital of the B atom is localized in a shallower energy level that is adjacent to its 2p orbital. The 2s orbital of the B atom and the d orbitals of the TM atom have obvious overlaps in the energy level ranging from −6 to 0 eV, indicating intense interactions between the TM and B atoms (Fig. S1, ESI†).
A part of the 2s orbital of the B atom is at the delocalization state and slightly contributes to the electronic state at the Fermi level, demonstrating the hybridization of B s–p orbitals. The partial 2p orbital of the B atom is above the Fermi level and the unoccupied 2p orbital can accept electrons from TM atoms to form strong-polarized covalent TM–B bonds. In addition, we have calculated the area of the H1 site, which is defined as the triangular area enclosed by three oxygen atoms in Ti3C2O2 or two oxygen atoms and one boron atom in Ti3C2O2−xBx (Fig. S2, ESI†). The calculated area of the H1 site of Ti3C2O2−xBx (4.21 Å2) is larger than that of Ti3C2O2 (4.11 Å2). The larger area in the former provides more spacious H1 sites so that TM atoms can be effectively embedded into the flat substrate of Ti3C2O2−xBx. Since our results suggest that Ti3C2O2−xBx has an outstanding ability to fix TM single atoms, all the six kinds of metal are considered in the following CO2RR investigation.
3.2. CO2 electroreduction reaction on TM@Ti3C2O2−xBx SACs
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO) to a V-shaped (∠OCO < 180°) structure, where partially negatively charged CO2 is generated due to the injection of electrons into its antibonding
CO) to a V-shaped (∠OCO < 180°) structure, where partially negatively charged CO2 is generated due to the injection of electrons into its antibonding 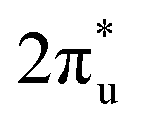 orbital. As shown in Fig. S3 (ESI†), CO2 binds to TM@Ti3C2O2−xBx (TM is V, Cr, Mn or Fe) through a B–C–TM–O configuration, but to TM@Ti3C2O2−xBx (TM is Co or Ni) through an O–B–C–TM configuration. The linear OCO is bent substantially and its C atom binds to the TM atom and the nearby B atom in both configurations. These bonding configurations indicate that the B atom can participate in the CO2 adsorption. In the case of TM@Ti3C2O2 (Fig. S3, ESI†), CO2 binds to TM (TM is V, Fe, Co or Ni) through a bidentate C–TM–O configuration with a bent structure, but binds to TM (TM is Cr or Mn) through a monodentate O–M configuration with a linear structure. This reveals that TM@Ti3C2O2 (TM is V, Fe, Co or Ni) also has the ability to activate CO2 molecules. However, for all the TM@Ti3C2O2 considered, CO2 molecules only bind to the TM atom, implying that the O atom of the functional groups does not participate in the CO2 activation directly. As can be seen in Table 1, the bending angle of the CO2 adsorbed on TM@Ti3C2O2−xBx is smaller than that on TM@Ti3C2O2, suggesting a more significant CO2 activation in the former.
orbital. As shown in Fig. S3 (ESI†), CO2 binds to TM@Ti3C2O2−xBx (TM is V, Cr, Mn or Fe) through a B–C–TM–O configuration, but to TM@Ti3C2O2−xBx (TM is Co or Ni) through an O–B–C–TM configuration. The linear OCO is bent substantially and its C atom binds to the TM atom and the nearby B atom in both configurations. These bonding configurations indicate that the B atom can participate in the CO2 adsorption. In the case of TM@Ti3C2O2 (Fig. S3, ESI†), CO2 binds to TM (TM is V, Fe, Co or Ni) through a bidentate C–TM–O configuration with a bent structure, but binds to TM (TM is Cr or Mn) through a monodentate O–M configuration with a linear structure. This reveals that TM@Ti3C2O2 (TM is V, Fe, Co or Ni) also has the ability to activate CO2 molecules. However, for all the TM@Ti3C2O2 considered, CO2 molecules only bind to the TM atom, implying that the O atom of the functional groups does not participate in the CO2 activation directly. As can be seen in Table 1, the bending angle of the CO2 adsorbed on TM@Ti3C2O2−xBx is smaller than that on TM@Ti3C2O2, suggesting a more significant CO2 activation in the former.
| Metal | Ti3C2O2−xBx | Ti3C2O2 | |||||
|---|---|---|---|---|---|---|---|
| BE (eV) | BD (Å) | BA (∠OCO, °) | BE (eV) | BD (Å) | BA (∠OCO, °) | ||
| TM–C | B–C | TM–C | |||||
| V | −1.49 | 2.12 | 1.72 | 128.7 | −0.62 | 2.01 | 140.7 |
| Cr | −1.03 | 2.10 | 1.74 | 130.5 | −0.65 | — | 179.7 |
| Mn | −0.52 | 2.15 | 1.81 | 136.5 | −0.48 | — | 179.4 |
| Fe | −0.73 | 2.48 | 1.62 | 126.5 | −0.27 | 2.06 | 151.9 |
| Co | −1.34 | 1.86 | 1.82 | 125.3 | −0.09 | 2.09 | 158.9 |
| Ni | −1.62 | 1.89 | 1.82 | 126.8 | −0.25 | 1.98 | 153.5 |
These phenomena we discussed above indicate that the doping of B facilitates the activation of CO2 through participating in its adsorption process. To understand the emerging role of the B atom in CO2 activation, we examine the electronic structures of TM@Ti3C2O2−xBx and TM@Ti3C2O2 through a Bader charge analysis. The images of the charge density difference of the CO2 adsorbed TM@Ti3C2O2−xBx are shown in Fig. 2c. As seen, charge accumulated in the CO2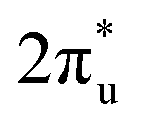 antibonding orbital around the C atom, leading to the activation of CO2 molecules. In contrast, charge depleted around B and TM atoms, confirming that the electrons involved in the activation of CO2 come from these atoms. To give more details of charge transfer density, Bader effective charge is investigated. When adsorbed on the surface, the C atom of CO2 in the case of TM@Ti3C2O2−xBx can gain more electrons than that in the case of TM@Ti3C2O2 (Fig. 2d).
antibonding orbital around the C atom, leading to the activation of CO2 molecules. In contrast, charge depleted around B and TM atoms, confirming that the electrons involved in the activation of CO2 come from these atoms. To give more details of charge transfer density, Bader effective charge is investigated. When adsorbed on the surface, the C atom of CO2 in the case of TM@Ti3C2O2−xBx can gain more electrons than that in the case of TM@Ti3C2O2 (Fig. 2d).
Generally, the strong electronegativity of oxygen shifts the focus of electronic clouds on both ends of CO2 molecules, yielding an electron-deficient carbon center. An injection of electron into the C atom is an effective way to activate CO2 molecules. The observation that TM@Ti3C2O2−xBx has a more activated CO2 is consistent with the results of calculations. Bader effective charge analysis (Fig. 2e) also shows that B and TM atoms can provide enough electrons for injection into CO2 molecules. In particular, in the cases of Co@Ti3C2O2−xBx and Ni@Ti3C2O2−xBx, most of the injected electrons come from the B atom. When CO2 molecules are activated on TM@Ti3C2O2, the Ti3C2O2 substrate can provide extra electrons to activate CO2, which is consistent with our previous result.56 But an indirect participation mode provides less electron than a direct participation.
The reaction activity of the doped B atom can be ascribed to its occupied 2p orbitals with s–p hybridization. Because the electronegativity of B is weaker than that of C, the electrons in the occupied orbitals of the B atom can transfer easily to the C atom of the CO2 molecule. In contrast, the full 2p-orbital occupation electronic structure of the O atom leads to a strong chemical stability. The stronger electronegativity of the O atom also makes it difficult for the C atom to grab its electrons. To gain insights into the chemisorption of CO2 on the active site of TM@Ti3C2O2−xBx, the bonding states of CO2 on TM@Ti3C2O2−xBx were investigated. According to the projected density of states (PDOS) for an isolated CO2 molecule (Fig. 3a–d and Fig. S4, ESI†), the empty orbital above the Fermi level is the antibonding 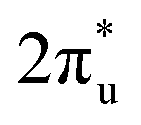 orbital.
orbital.
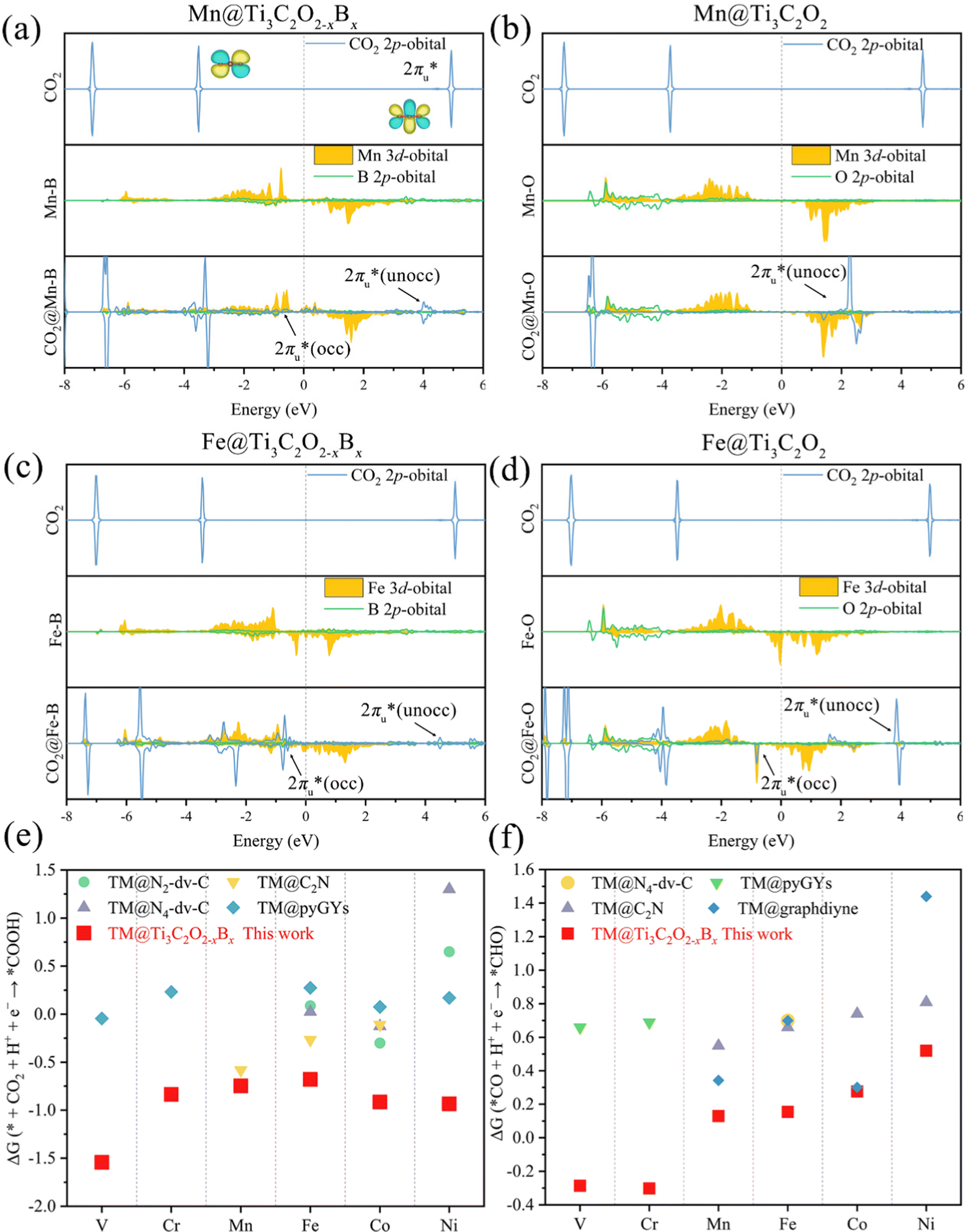 | ||
| Fig. 3 The PDOS of CO2 2p, TM 3d, and coordination atoms (the O atom in TM@Ti3C2O2 and the B atom in TM@Ti3C2O2−xBx) of the 2p orbital before and after CO2 adsorption on (a) Mn@Ti3C2O2−xBx, (b) Mn@Ti3C2O2, (c) Fe@Ti3C2O2−xBx and (d) Fe@Ti3C2O2. A comparison of the Gibbs free energy changes of (e) the 1st (CO2 + H+ + e− → *COOH) and (f) the 3rd (CO + H+ + e− → *CHO) hydrogenation reactions for TM@Ti3C2O2−xBx and other SACs (TM@N2-dv-C,74 TM@N4-dv-C,74 TM@C2N,75 TM@pyGYs,76 and TM@graphdiyne77) for the CO2 reduction. | ||
According to the PDOS of bare TM@Ti3C2O2−xBx, TM 3d overlaps with B 2p, indicating the bonding of TM and B atoms. After its adsorption on TM@Ti3C2O2−xBx, the antibonding 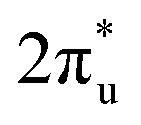 of CO2 splits into two parts: one above and one below the Fermi level. The states above the Fermi level are contributed by the unoccupied TM 3d-CO2
of CO2 splits into two parts: one above and one below the Fermi level. The states above the Fermi level are contributed by the unoccupied TM 3d-CO2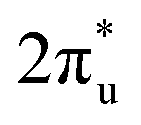 and B 2p-CO2
and B 2p-CO2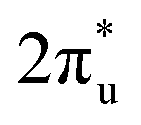 hybridization orbitals, while the overlapping of occupied TM 3d-CO2
hybridization orbitals, while the overlapping of occupied TM 3d-CO2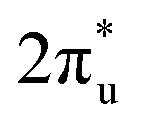 and B 2p-CO2
and B 2p-CO2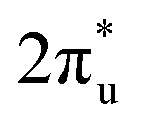 hybridization orbitals can be found at −2 to 0 eV, just below the Fermi level. After adsorbing on TM@Ti3C2O2, the antibonding
hybridization orbitals can be found at −2 to 0 eV, just below the Fermi level. After adsorbing on TM@Ti3C2O2, the antibonding 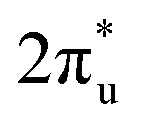 orbital of CO2 also splits into one part above the Fermi level and one part below it. Notably, the peak intensity of the unoccupied CO2
orbital of CO2 also splits into one part above the Fermi level and one part below it. Notably, the peak intensity of the unoccupied CO2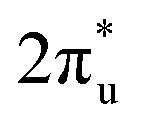 orbital when CO2 adsorbed on TM@Ti3C2O2−xBx decreases more sharply than that when CO2 adsorbed on TM@Ti3C2O2. This implies that TM@Ti3C2O2−xBx provides more electrons that can be injected into the
orbital when CO2 adsorbed on TM@Ti3C2O2−xBx decreases more sharply than that when CO2 adsorbed on TM@Ti3C2O2. This implies that TM@Ti3C2O2−xBx provides more electrons that can be injected into the 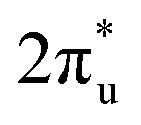 orbital, which is consistent with the results of Bader charge analysis.
orbital, which is consistent with the results of Bader charge analysis.
Besides CO2 activation ability, another key issue in CO2RR catalyst design is to obtain an energy-rich product (CH4), which is closely related to the adsorption Gibbs free energy of radical reaction intermediates and that of the elementary CO2 reduction reaction. CO2RR can be differentiated by the types of intermediates formed in the initial protonation steps of CO2: formate (*OCHO) and carboxyl (*COOH). The calculated Gibbs free energy changes (ΔG) for the formation of *COOH and *OCHO (*CO2 + H+ + e− → *COOH or *OCHO) on TM@Ti3C2O2−xBx are summarized in Fig. S5 (ESI†). This figure shows that the 1st hydrogenation (H+/e−) reactions are energetically prone to occur on the O atom rather than on the C atom, suggesting that the formation of *COOH is more energetically preferred than that of *OCHO. The calculated ΔG shown in Fig. 3e reveals that the ΔG of the first hydrogenation reaction to form *COOH on TM@Ti3C2O2−xBx is smaller than that in previous studies, illustrating the superior CO2 activation ability of TM@Ti3C2O2−xBx.
The relatively higher CO adsorption strength on TM@Ti3C2O2−xBx (Table S2, ESI†) results in poor activity for CO direct desorption under low electrode potentials. Due to the outstanding stability of CO, the hydrogenation reaction needs too much energy to reduce *CO. If the ΔG of the reaction *CO + H+ + e− → *CHO is larger than that of the reaction *CO → CO + *, CO is inclined to desorb rather than further hydrogenation. However, a high voltage is still needed to remove *CO from the surface if the adsorption of CO on the catalyst is strong. In general, the lower hydrogenation ΔG of the reaction *CO + H+ + e− → *CHO is necessary for the further CO reduction process. Therefore, the activation of the *CO intermediate is a key criterion to evaluate the performance of the CO2RR on SACs.
To verify the performance of TM@Ti3C2O2−xBx in *CO reduction, we calculated the ΔG of the reaction *CO + H+ + e− → *CHO, along with the ΔG of other SACs. Having examined the available results for the CO2RR of SACs, we can find that *CO + H+ + e− → *CHO is the potential-determining step in most of these studies. As shown in Fig. 3f, the calculated ΔG for the reaction *CO + H+ + e− → *CHO on TM@Ti3C2O2−xBx is smaller than the corresponding values of ΔG reported in previous studies, which verifies the superior CO activation ability of TM@Ti3C2O2−xBx. According to the adsorption configuration of *CO on TM@Ti3C2O2−xBx, the B atom also participates in the adsorption process (Fig. 4a), demonstrating its important role in CO activation. To gain insights into the combined effect of multi-active site TM and B atoms in ameliorating the CO reduction, the orbital and charge transfer of TM and B atoms during the adsorption of *CO is also investigated. The configuration of the *CO adsorbed on TM@Ti3C2O2 is used for comparison.
 | ||
| Fig. 4 (a) Optimized geometric structures of CO adsorbed on TM@Ti3C2O2−xBx. (b) Variation in the difference of charge density of TM@Ti3C2O2−xBx with a CO adsorption configuration. The yellow region denotes charge accumulation and the blue region indicates charge depletion, where the isosurface value is set to be 0.03 e Å−3. | ||
Accepting the lone-pair electrons from the σg orbital of CO and donating electrons to the antibonding 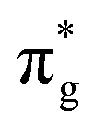 orbitals of CO are the main ways to weaken the C
orbitals of CO are the main ways to weaken the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O triple bond, known as the “acceptance–donation” process.78,79Fig. 4b and Fig. S6 (ESI†) illustrate the calculated charge density differences for the CO adsorption configuration on TM@Ti3C2O2−xBx and TM@Ti3C2O2. Charge accumulation and depletion can be observed for both CO molecules and B–TM atoms on TM@Ti3C2O2−xBx. This phenomenon is actually in perfect accordance with the “acceptance–donation” process mentioned above. A comparison between the area of the isosurface of CO adsorbed on TM@Ti3C2O2−xBx and that on TM@Ti3C2O2 reveals that the charge transfer in the former is more obvious than that in the latter.
O triple bond, known as the “acceptance–donation” process.78,79Fig. 4b and Fig. S6 (ESI†) illustrate the calculated charge density differences for the CO adsorption configuration on TM@Ti3C2O2−xBx and TM@Ti3C2O2. Charge accumulation and depletion can be observed for both CO molecules and B–TM atoms on TM@Ti3C2O2−xBx. This phenomenon is actually in perfect accordance with the “acceptance–donation” process mentioned above. A comparison between the area of the isosurface of CO adsorbed on TM@Ti3C2O2−xBx and that on TM@Ti3C2O2 reveals that the charge transfer in the former is more obvious than that in the latter.
To further elucidate the bonding nature, the PDOS of the adsorption of CO on TM@Ti3C2O2−xBx and that on TM@Ti3C2O2 are investigated (Fig. 5 and Fig. S7, ESI†). After adsorbing on TM@Ti3C2O2−xBx and TM@Ti3C2O2, the σg orbital of CO in a deep level obviously splits and moves to deeper energy states, indicating the interaction between the CO σg orbital and SACs. It can be seen that a part of the 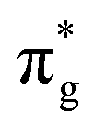 orbitals of CO unoccupied (unocc) shifts upward to the Fermi level and the other part moves downward to the Fermi level near −2 eV to form a
orbitals of CO unoccupied (unocc) shifts upward to the Fermi level and the other part moves downward to the Fermi level near −2 eV to form a 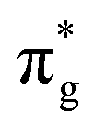 occupied (occ) orbital. Compared with that of TM@Ti3C2O2, the peak intensity of the unoccupied CO
occupied (occ) orbital. Compared with that of TM@Ti3C2O2, the peak intensity of the unoccupied CO 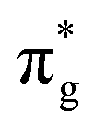 antibonding orbital decreases more sharply when CO is adsorbed on TM@Ti3C2O2−xBx, indicating the better CO activation performance of TM@Ti3C2O2−xBx. The Bader charge analysis shows that the C atom from CO can gain more electrons from TM@Ti3C2O2−xBx (Fig. 6a and b), which is consistent with the above PDOS analysis.
antibonding orbital decreases more sharply when CO is adsorbed on TM@Ti3C2O2−xBx, indicating the better CO activation performance of TM@Ti3C2O2−xBx. The Bader charge analysis shows that the C atom from CO can gain more electrons from TM@Ti3C2O2−xBx (Fig. 6a and b), which is consistent with the above PDOS analysis.
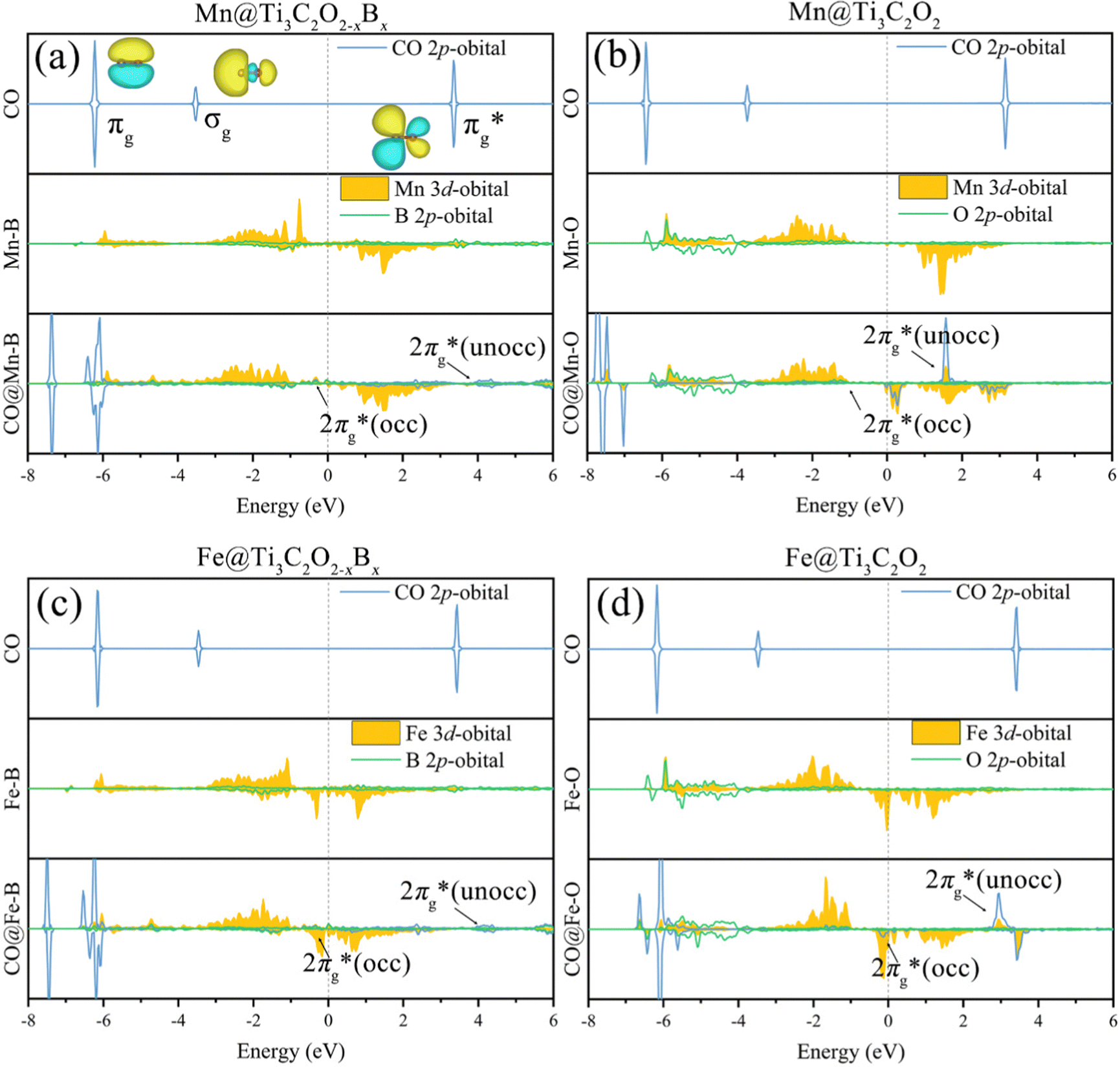 | ||
| Fig. 5 The PDOS of CO 2p, TM 3d, and coordination atoms (the O atom in TM@Ti3C2O2 and the B atom in TM@Ti3C2O2−xBx) of the 2p orbital before and after CO2 adsorption on (a) Mn@Ti3C2O2−xBx, (b) Mn@Ti3C2O2, (c) Fe@Ti3C2O2−xBx and (d) Fe@Ti3C2O2. | ||
 | ||
| Fig. 6 (a) Bader charge differences of the C atom in CO before and after adsorption on TM@Ti3C2O2−xBx and TM@Ti3C2Tx. (b) Bader charge differences of TM atoms and the B atom on TM@Ti3C2O2−xBx before and after CO adsorption. | ||
Owing to its weak electronegativity and not fully occupied orbital structure, the electrons in the occupied orbitals of the B atom can easily transfer to the empty orbitals of the CO2 molecule and CO intermediate. Based on the results gathered, it can be reasonably inferred that a combined effect between the single TM atom and B atom is present on the CO2RR (Fig. 7). The TM-to-C and B-to-C π-back bonding contribute to the activation of the CO2 molecules and CO intermediate. Enough electrons from the single TM atom and B atom can be injected into CO2 and CO molecules through a direct bonding interaction. Therefore, the B-doping strategy efficiently controls the charge–orbital state of the coordination atom on the surface of Ti3C2Tx-MXene, alleviating remarkably the difficulty of activating CO2 and CO molecules.
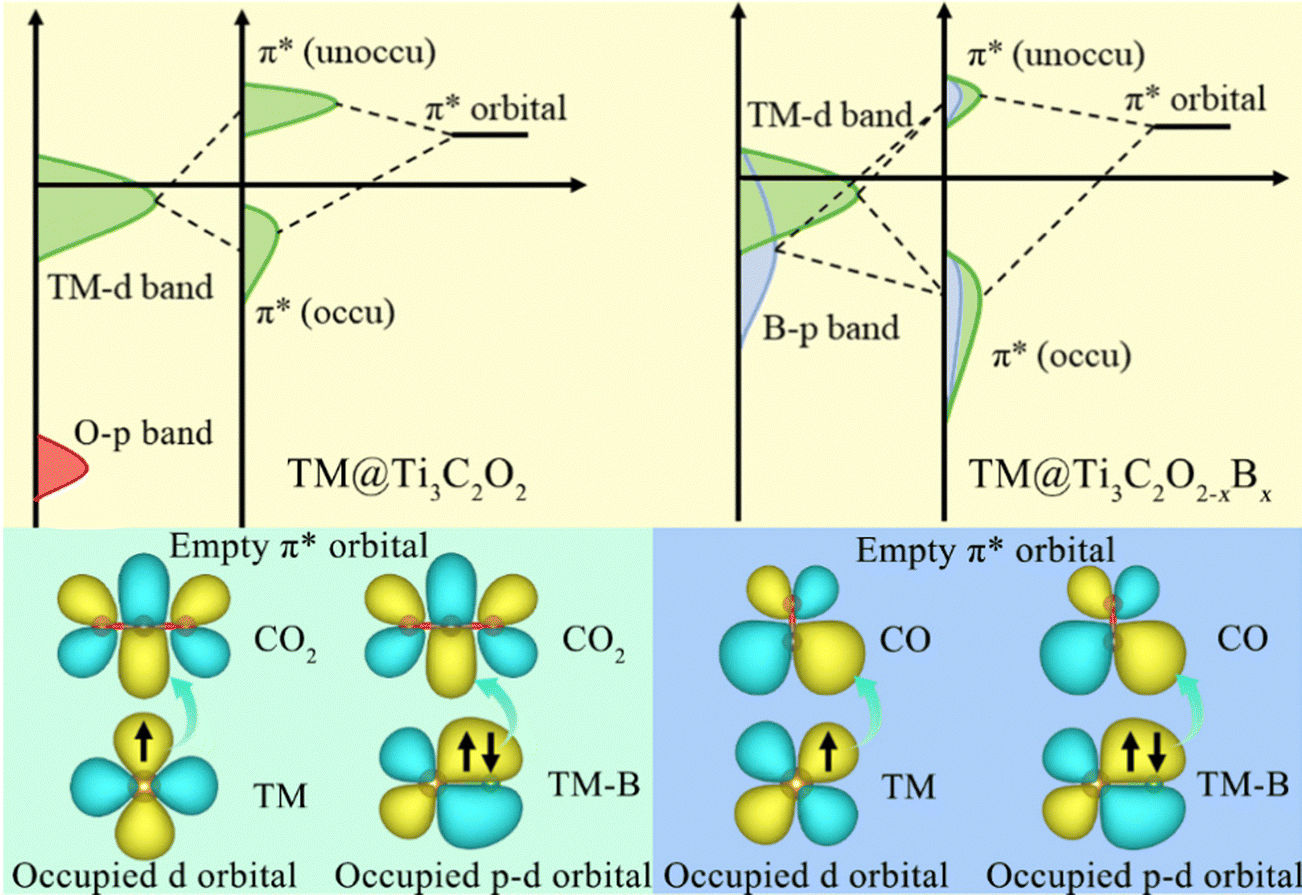 | ||
| Fig. 7 Schematic diagrams of CO2 molecule and CO intermediate activation mechanisms on TM@Ti3C2O2 and TM@Ti3C2O2−xBx. | ||
Fig. 8 shows the variation of the calculated free energy with the most favorable pathway for the reduction of CO2 on TM@Ti3C2O2−xBx SACs and the corresponding structures of the reaction intermediates are presented in Fig. S8 (ESI†). Owing to the excellent CO2 activation performance of TM@Ti3C2O2−xBx, the hydrogenation of the adsorbed CO2 to form *COOH is exothermic with a negative ΔG. The 3rd hydrogenation (H+/e−) reaction of *CO occurs, generating *CHO. *CO is more likely to undergo further reduction on all the TM@Ti3C2O2−xBx because the ΔG of the protonation step (*CO + H+ + e− → *CHO) is more negative than that of CO desorption. Therefore, we concluded that the production of CO can be effectively prohibited in the CO2 reduction. The small ΔG of the 3rd hydrogenation also implies that the adsorbed *CO, which is generally regarded as the poison on metallic electrodes, is less likely to poison the active sites in our case.
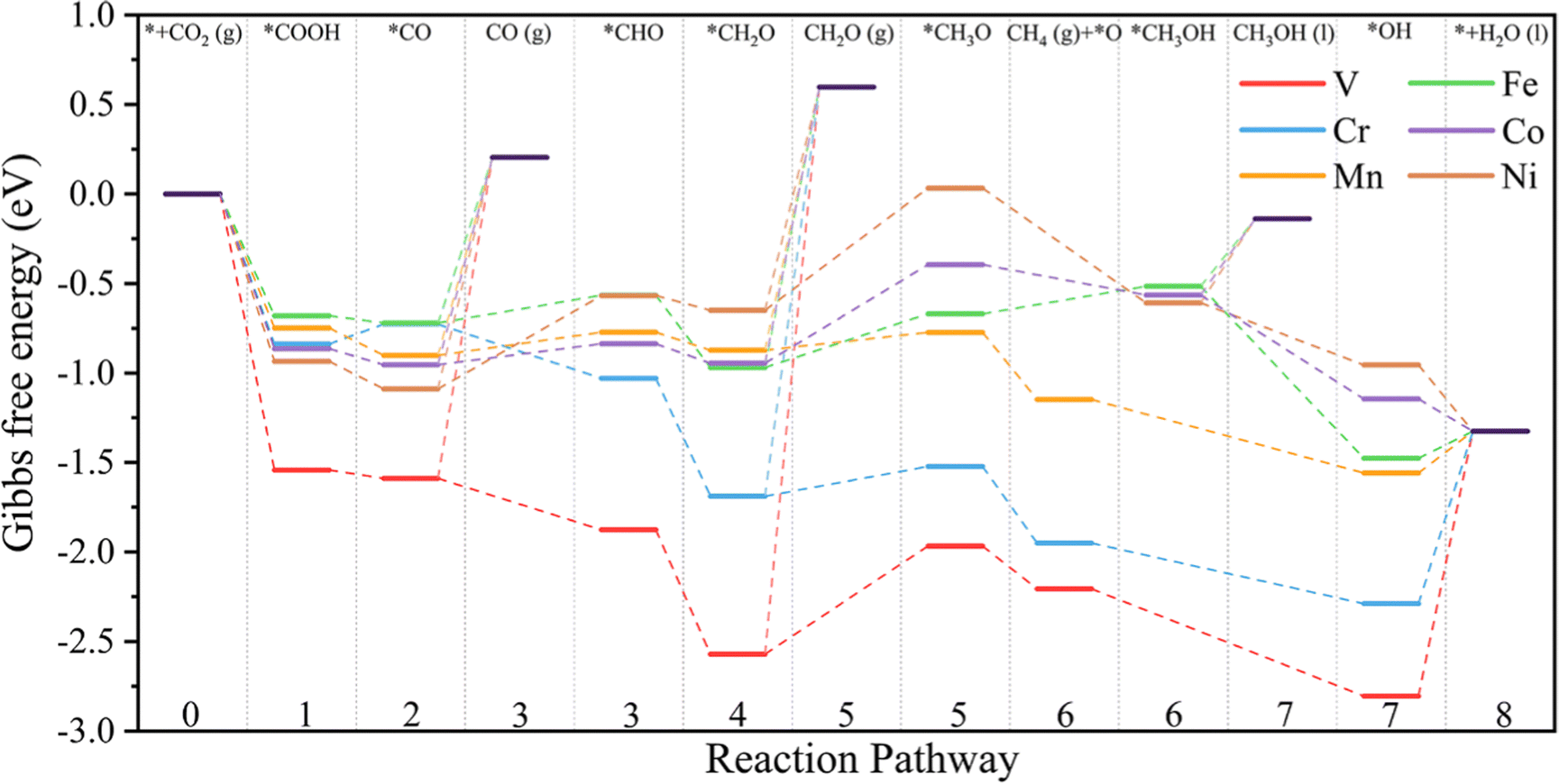 | ||
| Fig. 8 Variation of the Gibbs free energy for the reduction reaction of CO2 on TM@Ti3C2O2−xBx (TM is V, Cr, Mn, Fe, Co or Ni,) at 0 V with RHE. | ||
For the subsequent elementary reaction steps passing through the CHO* intermediate to generate CH4, it is worth mentioning that the desorption of CH2O from TM@Ti3C2O2−xBx is significantly endothermic due to their large negative adsorption energies (Table S2, ESI†), indicating that the production of methanol (CH2O) can be effectively prevented during the reduction of CO2 catalyzed by TM@Ti3C2O2−xBx. In addition, *CH3O is more likely to be further reduced to *O, releasing one CH4 molecule on TM@Ti3C2O2−xBx (TM is V, Cr or Mn). Therefore, the production of CH3OH can be avoided effectively. For the case of TM@Ti3C2O2−xBx (TM is Fe, Co or Ni), the desorption ΔG of CH3OH is appreciably large so that the desorption of CH3OH is difficult. After *CH3OH is formed on TM@Ti3C2O2−xBx, its C atom will be attacked by a proton to form CH4(g) and *OH. This suggests that CH4 is the primary reduction product on all the TM@Ti3C2O2−xBx examined, indicating its great selectivity for catalyzing the CO2 reduction to form CH4 with excellent abilities to limit the production of CO(g), CH2O(g), and CH3OH(l).
To assess the durability of TM@Ti3C2O2−xBx SACs in practical application, we have further investigated the protonation of remaining *OH as well as the release of the adsorbed H2O molecule (8th hydrogenation reaction). The results obtained show that the *OH on TM@Ti3C2O2−xBx (TM is V or Cr) is harder to be removed; the uphill values of ΔG are 1.47 and 0.96 eV, respectively. The positive ΔG indicates that *OH is more likely to occupy active sites by forming TM–OH, which hinders the next CO2 reduction process. For other TM@Ti3C2O2−xBx (TM is Mn, Fe, Co or Ni), the formation of *CHO (*CO + H+ + e− = *CHO) and *CH3O (*CH2O + H+ + e− = *CH3O) is both endothermic and the rest of the elementary steps are exothermic.
Except Mn@Ti3C2O2−xBx, the uphill value of ΔG for the formation of *CH3O on TM@Ti3C2O2−xBx (TM is Fe, Co or Ni) is larger than that for the formation of *CHO. Therefore, the formation of *CHO and *CH3O is the potential-determining step (PDS) for Mn@Ti3C2O2−xBx and TM@Ti3C2O2−xBx (TM is Fe, Co or Ni), respectively. The PDS and the values of UL predicted by DFT are summarized in Table 2. Based on the UL calculated from the primary PDS, it can be inferred that Mn@Ti3C2O2−xB and Fe@Ti3C2O2−xB have a higher activity for CH4 production, where their limiting potentials are 0.13 and 0.3 V, respectively, lower than those of other TM@Ti3C2O2−xBx.
| Catalyst | Potential determining step | U L (V) | Production |
|---|---|---|---|
| V@Ti3C2O2−xBx | *OH + H+ + e− → H2O (l) | 1.47 | CH4 |
| Cr@Ti3C2O2−xBx | *OH + H+ + e− → H2O (l) | 0.96 | CH4 |
| Mn@Ti3C2O2−xBx | *CO + H+ + e− → *CHO | 0.13 | CH4 |
| Fe@Ti3C2O2−xBx | *CH2O + H+ + e− → *CH3O | 0.30 | CH4 |
| Co@Ti3C2O2−xBx | *CH2O + H+ + e− → *CH3O | 0.55 | CH4 |
| Ni@Ti3C2O2−xBx | *CH2O + H+ + e− → *CH3O | 0.68 | CH4 |
We conclude that Mn@Ti3C2O2−xBx and Fe@Ti3C2O2−xBx have a remarkable selectivity and activity for the production of CH4(g). Considering the effect of the presence of water under real reaction conditions, we calculated the free energy diagram of the initial screening for Mn@Ti3C2O2−xB and Fe@Ti3C2O2−xB in the implicit solvent model using VASPsol (Fig. 9a and b). Notably, the levels of UL for Mn@Ti3C2O2−xB and Fe@Ti3C2O2−xB increased to 0.51 and 0.40 V, respectively. The overpotential of Fe@Ti3C2O2−xB (0.40 V) suggests that it can be a promising CO2RR catalyst for producing CH4. This level of limiting potentials is comparable or lower than those of the widely employed transition metal-based CO2RR electrocatalysts8,14,80 and other bimetallic electrocatalysts work on CO2 conversion to CH4 (Table S3, ESI†).
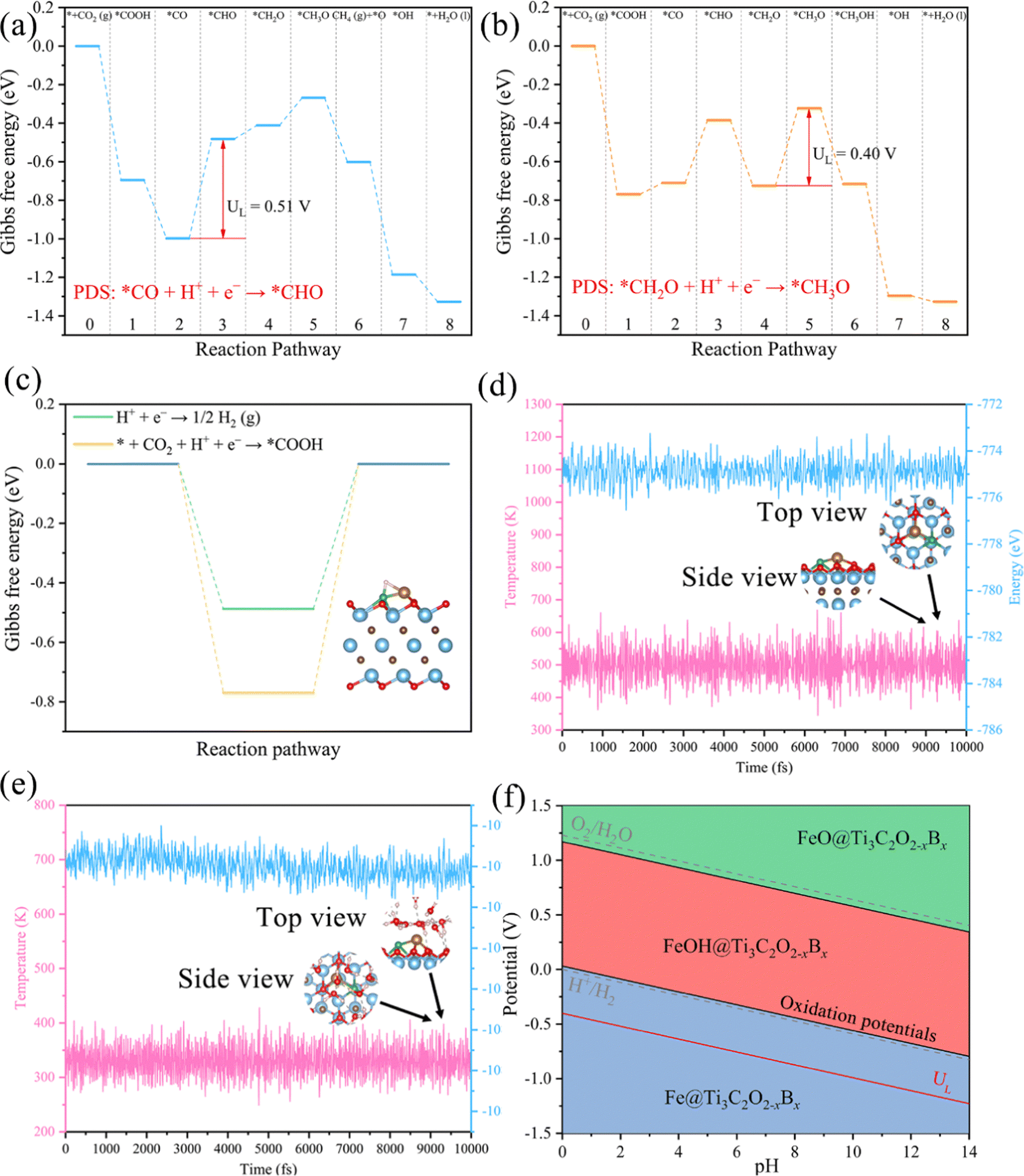 | ||
| Fig. 9 Free energy diagrams of the CO2RR on (a) Mn@Ti3C2O2−xB and (b) Fe@Ti3C2O2−xB after taking into account of solvation effects. (c) Variation of the Gibbs free energy changes (ΔG) for the first protonation step of the CO2RR with HER. Inset: the optimized geometric structures of H adsorbed on Fe@Ti3C2O2−xBx. (d) Variations of energy and temperature with the AIMD simulation time for Fe@Ti3C2O2−xB, where the AIMD simulation lasts for 10 ps at 500 K. (e) Variations of energy and temperature with the AIMD simulation time for Fe@Ti3C2O2−xB, an explicit solvent model is used to simulate the acidic aqueous solution. The AIMD simulation lasts for 10 ps at 330 K. (f) Calculated Pourbaix diagram of Fe@Ti3C2O2−xB. Gray dashed lines denote the water redox potentials. | ||
3.3. Stability of Fe@Ti3C2O2−xBx in solution and competition between the CO2RR and the HER
Apart from CO2 adsorption, the competitive adsorption of CO2RR and H on each active center is also a significant factor affecting CO2RR performance. The free energy of hydrogen adsorption (ΔGH) provides a measure for evaluating HER activity. A material is an optimal HER electrocatalyst if its ΔGH value is close to zero. However, a highly negative ΔGH indicates the excessively stable adsorption of H on the active sites, thereby preventing the CO2RR. Therefore, active sites with a large ΔGH are desirable for CO2RR electrocatalysts. Assuming a Volmer and Tafel mechanism, we calculated the ΔG for the formation of *COOH (* + CO2 + H+ + e− → *COOH) and that for the formation of *H (H+ + e− → *H) in the HER. According to Brønsted–Evans–Polanyi (BEP) relations, a reaction with a more negative ΔG is expected to encounter a smaller reaction barrier and, therefore, is more favored to occur.The adsorption sites for the H atom mainly locate on the bridge site between the TM and B atoms on Fe@Ti3C2O2−xBx (Fig. 9c). The calculated ΔGH of Fe@Ti3C2O2−xBx is −0.48 eV, which is more positive than the ΔG*COOH of the CO2RR (Fig. 9c). This suggests that Fe@Ti3C2O2−xBx contains inactive sites for the HER with a |ΔGH| of around 0.5 eV and exhibits a higher selectivity toward the CO2RR than the HER.
To assess the durability of MXene catalysts in practical applications, we investigated the stability of Fe@Ti3C2O2−xBxvia AIMD simulations. As seen in Fig. 9d, both the total energy and the temperature oscillate near the initial conditions and the geometric structures of Fe@Ti3C2O2−xBx are preserved well within 10 ps. An explicit solvent model is also applied to simulate the solution environment, and AIMD simulations are performed to study the thermal stability of Fe@Ti3C2O2−xBx in a solution at 330 K for 10 ns.
The results obtained reveal that the structure of Fe@Ti3C2O2−xBx can remain stable in an aqueous solution (Fig. 9e). Therefore, it can be inferred that Fe@Ti3C2O2−xBx can serve as an efficient CO2RR electrocatalyst under ambient conditions with a high stability. Further analysis of the Pourbaix diagram of Fe@Ti3C2O2−xBx (Fig. 9f) has also been carried out. It is a useful tool to identify the stable state of catalysts in water as a function of pH and applied potential. The oxidation potential of Fe@Ti3C2O2−xBx at pH 0 (black line) is less negative than the UL value of Fe@Ti3C2O2−xBx (red line), indicating that Fe@Ti3C2O2−xBx could possess superior electrochemical stability against surface oxidation under working conditions.
3.4. An activity descriptor for CO2 reduction on Ti3C2Tx-based SACs
It is beneficial to search an activity descriptor to establish a ‘‘volcano” relationship on catalytic behaviors so that we can further roughly estimate the effect of other potential dopants on the CO2RR performance. It is generally agreed that the electronic structures of catalytic centers would greatly influence the electron transfer and reaction energy in catalytic processes. Pan and coworkers have found that the valence state of TMs could be used as an activity descriptor, wherefore the limiting potential towards different products as a function of the valence state of TMs81 because the TM atoms can provide both empty d orbitals to accept electrons to form a coordination bond and lone electrons to form a covalent bond.82 Hence, the valence state of TMs would directly influence the bonding strength with the intermediates.The valence state of different TM atoms is defined as the number of electrons transferred from the TM atom to the substrate. Using the valence state of TMs as the descriptor, we find that a volcano trend is observed for TM@Ti3C2O2−xBx (Fig. 10a), meaning this descriptor is possible to estimate the catalytic performance of the TM@Ti3C2O2 system. Therefore, a brief calculation of N-doped Ti3C2O2-based SACs (TM@Ti3C2O2−xNx) is carried out to verify the accuracy of this descriptor. The adsorption configuration of CO2 and CO on TM@Ti3C2O2−xNx (Fig. S9, ESI†) indicates that the nitrogen atom is only used as the coordination atom to control the electronic structure of the TM. Generally, the potential-limiting step of CO2 reduction is the hydrogenation of *COOH to *CO or the hydrogenation of *CO to *CHO.83,84 As for the TM@Ti3C2O2 system studied in our work, *OH + H+ + e− → H2O(l) is the potential-limiting step on V@Ti3C2O2 and Cr@Ti3C2O2. Hence, we have calculated these key reaction steps on TM@Ti3C2O2−xNx and chosen the step with the largest reaction energy barrier as the potential-limiting step.
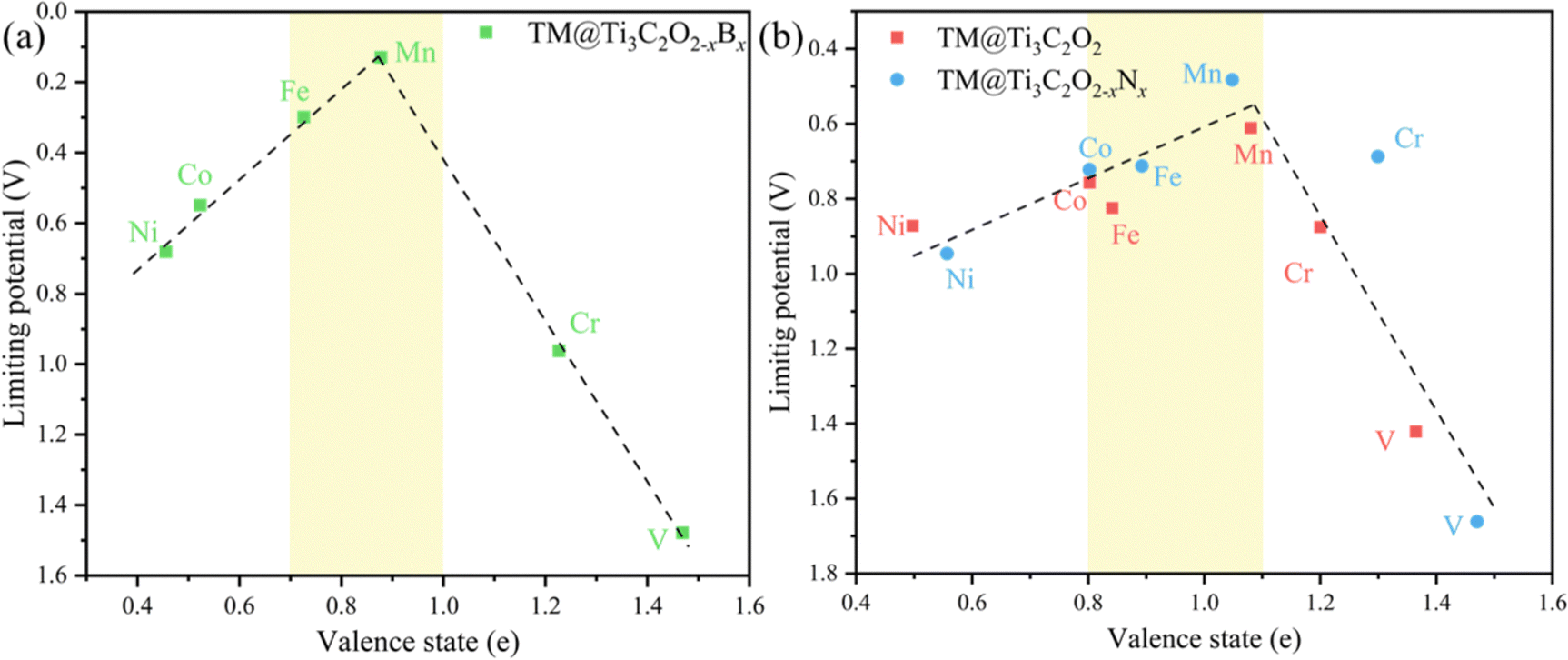 | ||
| Fig. 10 The limiting potential as a function of the valence state of TM atoms fixed on (a) Ti3C2O2−xBx and (b) Ti3C2O2, Ti3C2O2−xNx. | ||
The result shows that a volcano trend is observed with the optimal scope of the valence state ranging from ∼0.8 e to ∼1.1 e (Fig. 10b). We can see that the optimal scope of the valence state is different between TM@Ti3C2O2−xBx and TM@Ti3C2O2 (TM@Ti3C2O2−xBx). Because the boron atom can participate in CO2 reduction, the scaling relations between the adsorption strength of reaction intermediates have been broken. This simple descriptor well explains the catalytic performance of the TM@Ti3C2O2 and doped TM@Ti3C2O2 SACs and suggests that a certain moderate degree of electron transfer from TM atoms to the substrate will be expected to promote the overall catalytic activity. Before using this descriptor, we need to determine whether the coordination atoms participate in the reaction to ensure that the same linear relationship exists. Moreover, this descriptor can only roughly estimate the CO2RR performance of catalysts. A series of subsequent tests, such as the competition test, stability test and solvation effect should be considered to further screening out the most suitable catalysts.
4. Conclusions
In summary, by applying well-defined first-principles calculations, we have investigated the single-atom catalysts of Ti3C2O2−xBx monolayers for CO2 electrochemical reduction reactions. The results obtained suggest that the B atom not only serves as the coordination atom to anchor single TM atoms but also plays an important role in the CO2 electrochemical reduction process. The strategy of doping B regulates the charge–orbit distribution of the surface functional groups of Ti3C2O2. By analyzing charge transfer and DOS calculation results, the combined effect of the single TM atom and the adjacent B atom in Ti3C2O2−xBx on the CO2RR has been demystified. The empty orbitals in B atom hybridization help it in gaining more electrons from single TM atoms, forming strong-polarized covalent TM–B bonds, which is beneficial for anchoring single TM atoms on the Ti3C2O2 surface. The electrons provided by the occupied orbitals in B atom hybridization are effectively transferred to the π-antibonding orbitals of CO2 and CO, thereby helping single TM atoms to activate the CO2 molecules and *CO intermediate. Especially, the activation of the *CO intermediate induced by the combined effect effectively lowers the reaction energy barrier of the protonation step of *CO, facilitating further CO reduction towards CH4.Our results also demonstrated that the activated CO2 can be further reduced to CH4 by TM@Ti3C2O2−xBx, in which Fe@Ti3C2O2−xBx exhibits a better catalytic efficiency with a low limiting potential of ∼0.40 V. In addition, AIMD calculations reveal that Fe@Ti3C2O2−xBx has a high stability. Inspired by the successful preparation of Ru@Ti3C2O2−xBx SACs,55 the practical application of Fe@Ti3C2O2−xBx in CO2 capture and reduction can be reasonably anticipated in the near future. We conclude that our work provides atomic-level insights into the combined effect between the embedded single metal atom and the innate atom in a substrate and provides an important strategy for the design of MXenes as novel CO2RR catalysts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Guangdong Basic and Applied Basic Research Foundation (No. 2022A1515011303), the Central Government Guides Local Science and Technology Development Funds to Freely Explore Basic Research Projects (2021Szvup106), the Natural Science Fund for Distinguished Young Scholars of Hubei Province (No. 2020CFA087), the Basic Research Program of Shenzhen (No. JCYJ20190809120015163); and the Fundamental Research Funds for the Central Universities.References
- Ş. Kılkış, G. Krajačić, N. Duić, M. A. Rosen and M. D. A. Al-Nimr, Energy Convers. Manage., 2020, 225, 113410 CrossRef.
- D. Guan, J. Meng, D. M. Reiner, N. Zhang, Y. Shan, Z. Mi, S. Shao, Z. Liu, Q. Zhang and S. J. Davis, Nat. Geosci., 2018, 11, 551–555 CrossRef CAS.
- Z. Chu, M. Cheng and N. N. Yu, Technol. Forecase. Soc., 2021, 172, 121037 CrossRef.
- X. Zhao, Y. Shang and M. Song, Socio-Econ. Plan. Sci., 2020, 72, 100757 CrossRef.
- X. Zhao, X. Ma, B. Chen, Y. Shang and M. Song, Resour., Conserv. Recycl., 2022, 176, 105959 CrossRef CAS.
- M. Salvia, D. Reckien, F. Pietrapertosa, P. Eckersley, N.-A. Spyridaki, A. Krook-Riekkola, M. Olazabal, S. De Gregorio Hurtado, S. G. Simoes, D. Geneletti, V. Viguié, P. A. Fokaides, B. I. Ioannou, A. Flamos, M. S. Csete, A. Buzasi, H. Orru, C. de Boer, A. Foley, K. Rižnar, M. Matosović, M. V. Balzan, M. Smigaj, V. Baštáková, E. Streberova, N. B. Šel, L. Coste, L. Tardieu, C. Altenburg, E. K. Lorencová, K. Orru, A. Wejs, E. Feliu, J. M. Church, S. Grafakos, S. Vasilie, I. Paspaldzhiev and O. Heidrich, Renewable Sustainable Energy Rev., 2021, 135, 110253 CrossRef.
- Z. Liu, Z. Deng, G. He, H. Wang, X. Zhang, J. Lin, Y. Qi and X. Liang, Nat. Rev. Earth Environ., 2022, 3, 141–155 CrossRef.
- M. M. Ayyub and C. N. R. Rao, Mater. Horiz., 2021, 8, 2420–2443 RSC.
- Z. Sun, T. Ma, H. Tao, Q. Fan and B. Han, Chem, 2017, 3, 560–587 CAS.
- L. Dietz, S. Piccinin and M. Maestri, J. Phys. Chem. C, 2015, 119, 4959–4966 CrossRef CAS.
- X. Li, S. Wang, L. Li, X. Zu, Y. Sun and Y. Xie, Acc. Chem. Res., 2020, 53, 2964–2974 CrossRef CAS PubMed.
- D. W. DeWulf, T. Jin and A. J. Bard, J. Electrochem. Soc., 1989, 136, 1686 CrossRef CAS.
- Y. Hori, in Modern Aspects of Electrochemistry, ed. C. G. Vayenas, R. E. White and M. E. Gamboa-Aldeco, Springer New York, New York, NY, 2008, pp. 89–189 DOI:10.1007/978-0-387-49489-0_3.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
- W. Chen, J. Cao, W. Fu, J. Zhang, G. Qian, J. Yang, D. Chen, X. Zhou, W. Yuan and X. Duan, Angew. Chem., Int. Ed., 2022, 61, e202200190 CAS.
- Y.-J. Zhang, V. Sethuraman, R. Michalsky and A. A. Peterson, ACS Catal., 2014, 4, 3742–3748 CrossRef CAS.
- X. Chen, L. P. Granda-Marulanda, I. T. McCrum and M. T. M. Koper, Nat. Commun., 2022, 13, 38 CrossRef CAS PubMed.
- J. Albo and A. Irabien, J. Catal., 2016, 343, 232–239 CrossRef CAS.
- I. Merino-Garcia, J. Albo, J. Solla-Gullón, V. Montiel and A. Irabien, J. CO2 Util., 2019, 31, 135–142 CrossRef CAS.
- Y. Hori, A. Murata and R. Takahashi, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 2309–2326 RSC.
- J. Albo, G. Beobide, P. Castaño and A. Irabien, J. CO2 Util., 2017, 18, 164–172 CrossRef CAS.
- J. Albo, A. Sáez, J. Solla-Gullón, V. Montiel and A. Irabien, Appl. Catal., B, 2015, 176–177, 709–717 CrossRef CAS.
- J. Albo, D. Vallejo, G. Beobide, O. Castillo, P. Castaño and A. Irabien, ChemSusChem, 2017, 10, 1100–1109 CrossRef CAS PubMed.
- M. Perfecto-Irigaray, J. Albo, G. Beobide, O. Castillo, A. Irabien and S. Pérez-Yáñez, RSC Adv., 2018, 8, 21092–21099 RSC.
- I. Merino-Garcia, J. Albo, P. Krzywda, G. Mul and A. Irabien, Catal. Today, 2020, 346, 34–39 CrossRef CAS.
- Q. Zhang and J. Guan, Adv. Funct. Mater., 2020, 30, 2000768 CrossRef CAS.
- M. Li, H. Wang, W. Luo, P. C. Sherrell, J. Chen and J. Yang, Adv. Mater., 2020, 32, 2001848 CrossRef CAS PubMed.
- R. Wang, G. Liu, S. K. Kim, K. H. Bowen and X. Zhang, J. Energy Chem., 2021, 63, 130–137 CrossRef CAS.
- N. Li, X. Wang, X. Lu, P. Zhang and W.-J. Ong, Chem. – Eur. J., 2021, 27, 17900–17909 CrossRef CAS PubMed.
- J. Fu, L. Zhu, K. Jiang, K. Liu, Z. Wang, X. Qiu, H. Li, J. Hu, H. Pan, Y.-R. Lu, T.-S. Chan and M. Liu, Chem. Eng. J., 2021, 415, 128982 CrossRef CAS.
- C. Ling, X. Niu, Q. Li, A. Du and J. Wang, J. Am. Chem. Soc., 2018, 140, 14161–14168 CrossRef CAS PubMed.
- H. Yin, L.-Y. Gan and P. Wang, J. Mater. Chem. A, 2020, 8, 3910–3917 RSC.
- M.-A. Légaré, G. Bélanger-Chabot, R. D. Dewhurst, E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Science, 2018, 359, 896–900 CrossRef PubMed.
- M. Qu, G. Qin, J. Fan, A. Du and Q. Sun, Appl. Surf. Sci., 2021, 555, 149652 CrossRef CAS.
- Y. Jiao, A. Du, Z. Zhu, V. Rudolph, G. Q. Lu and S. C. Smith, Catal. Today, 2011, 175, 271–275 CrossRef CAS.
- S. Tang, X. Zhou, S. Zhang, X. Li, T. Yang, W. Hu, J. Jiang and Y. Luo, ACS Appl. Mater. Interfaces, 2019, 11, 906–915 CrossRef CAS PubMed.
- X. Chen, W.-J. Ong, X. Zhao, P. Zhang and N. Li, J. Energy Chem., 2021, 58, 577–585 CrossRef CAS.
- S. Liu, M. Jin, J. Sun, Y. Qin, S. Gao, Y. Chen, S. Zhang, J. Luo and X. Liu, Chem. Eng. J., 2022, 437, 135294 CrossRef CAS.
- H. Xu, Y. Zhao, Q. Wang, G. He and H. Chen, Coord. Chem. Rev., 2022, 451, 214261 CrossRef CAS.
- J. Li, Q. Guan, H. Wu, W. Liu, Y. Lin, Z. Sun, X. Ye, X. Zheng, H. Pan, J. Zhu, S. Chen, W. Zhang, S. Wei and J. Lu, J. Am. Chem. Soc., 2019, 141, 14515–14519 CrossRef CAS PubMed.
- N. Li, X. Chen, W.-J. Ong, D. R. MacFarlane, X. Zhao, A. K. Cheetham and C. Sun, ACS Nano, 2017, 11, 10825–10833 CrossRef CAS PubMed.
- J. Peng, X. Chen, W.-J. Ong, X. Zhao and N. Li, Chem, 2019, 5, 18–50 CAS.
- R. A. Soomro, S. Jawaid, Q. Zhu, Z. Abbas and B. Xu, Chin. Chem. Lett., 2020, 31, 922–930 CrossRef CAS.
- Y. Dong, H. Shi and Z.-S. Wu, Adv. Funct. Mater., 2020, 30, 2000706 CrossRef CAS.
- B. Huang, N. Li, W.-J. Ong and N. Zhou, J. Mater. Chem. A, 2019, 7, 27620–27631 RSC.
- Y. Meng, J.-X. Liang, C. Zhu, C.-Q. Xu and J. Li, Sci. China Mater., 2022, 65, 1303–1312 CrossRef CAS.
- G. Chen, M. Ding, K. Zhang, Z. Shen, Y. Wang, J. Ma, A. Wang, Y. Li and H. Xu, ChemSusChem, 2022, 15, e202102352 CAS.
- M. Zhang, C. Lai, B. Li, S. Liu, D. Huang, F. Xu, X. Liu, L. Qin, Y. Fu, L. Li, H. Yi and L. Chen, Small, 2021, 17, 2007113 CrossRef CAS PubMed.
- Y. Cui, Z. Cao, Y. Zhang, H. Chen, J. Gu, Z. Du, Y. Shi, B. Li and S. Yang, Small Sci., 2021, 1, 2100017 CrossRef CAS.
- D. Zhao, Z. Chen, W. Yang, S. Liu, X. Zhang, Y. Yu, W.-C. Cheong, L. Zheng, F. Ren, G. Ying, X. Cao, D. Wang, Q. Peng, G. Wang and C. Chen, J. Am. Chem. Soc., 2019, 141, 4086–4093 CrossRef CAS PubMed.
- Q. Zhao, C. Zhang, R. Hu, Z. Du, J. Gu, Y. Cui, X. Chen, W. Xu, Z. Cheng, S. Li, B. Li, Y. Liu, W. Chen, C. Liu, J. Shang, L. Song and S. Yang, ACS Nano, 2021, 15, 4927–4936 CrossRef CAS PubMed.
- W. Lin, Y.-R. Lu, W. Peng, M. Luo, T.-S. Chan and Y. Tan, J. Mater. Chem. A, 2022, 10, 9878–9885 RSC.
- H. Liu, Z. Hu, Q. Liu, P. Sun, Y. Wang, S. Chou, Z. Hu and Z. Zhang, J. Mater. Chem. A, 2020, 8, 24710–24717 RSC.
- Z. Chen, J. Cao, X. Wu, D. Cai, M. Luo, S. Xing, X. Wen, Y. Chen, Y. Jin, D. Chen, Y. Cao, L. Wang, X. Xiong and B. Yu, ACS Appl. Mater. Interfaces, 2022, 14, 12223–12233 CrossRef CAS PubMed.
- M. Bat-Erdene, M. Batmunkh, B. Sainbileg, M. Hayashi, A. S. R. Bati, J. Qin, H. Zhao, Y. L. Zhong and J. G. Shapter, Small, 2021, 17, 2102218 CrossRef CAS PubMed.
- N. Li, J. Peng, Z. Shi, P. Zhang and X. Li, Chin. J. Catal., 2022, 43, 1906–1917 CrossRef CAS.
- J. Hafner, J. Comput. Chem., 2008, 29, 2044–2078 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- E. Sanville, S. D. Kenny, R. Smith and G. Henkelman, J. Comput. Chem., 2007, 28, 899–908 CrossRef CAS PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- C. M. Gray, K. Saravanan, G. Wang and J. A. Keith, Mol. Simul., 2017, 43, 420–427 CrossRef CAS.
- F. Calle-Vallejo, R. F. de Morais, F. Illas, D. Loffreda and P. Sautet, J. Phys. Chem. C, 2019, 123, 5578–5582 CrossRef CAS.
- Q. Zhang and A. Asthagiri, Catal. Today, 2019, 323, 35–43 CrossRef CAS.
- L. Li, B. Li, Q. Guo and B. Li, J. Phys. Chem. C, 2019, 123, 14501–14507 CrossRef CAS.
- H. Dong, C. Liu, Y. Li and D.-E. Jiang, Nanoscale, 2019, 11, 11351–11359 RSC.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- F. Calle-Vallejo, J. I. Martínez, J. M. García-Lastra, M. Mogensen and J. Rossmeisl, Angew. Chem., Int. Ed., 2010, 49, 7699–7701 CrossRef CAS PubMed.
- Y. Li, H. Su, S. H. Chan and Q. Sun, ACS Catal., 2015, 5, 6658–6664 CrossRef CAS.
- D. R. Lide, CRC handbook of chemistry and physics, CRC Press, 2004 Search PubMed.
- Y. Gao, Y. Cao, Y. Gu, H. Zhuo, G. Zhuang, S. Deng, X. Zhong, Z. Wei, J. Chen, X. Pan and J.-G. Wang, Appl. Surf. Sci., 2019, 465, 911–918 CrossRef CAS.
- P. Li, J. Zhu, A. D. Handoko, R. Zhang, H. Wang, D. Legut, X. Wen, Z. Fu, Z. W. Seh and Q. Zhang, J. Mater. Chem. A, 2018, 6, 4271–4278 RSC.
- C. Guo, T. Zhang, X. Liang, X. Deng, W. Guo, Z. Wang, X. Lu and C.-M. L. Wu, Appl. Surf. Sci., 2020, 533, 147466 CrossRef CAS.
- X. Cui, W. An, X. Liu, H. Wang, Y. Men and J. Wang, Nanoscale, 2018, 10, 15262–15272 RSC.
- M. Wang, L. Kong, X. Lu and C.-M. Lawrence Wu, J. Mater. Chem. A, 2022, 10, 9048–9058 RSC.
- T. Liu, G. Wang and X. Bao, J. Phys. Chem. C, 2021, 125, 26013–26020 CrossRef CAS.
- A. Föhlisch, M. Nyberg, P. Bennich, L. Triguero, J. Hasselström, O. Karis, L. G. M. Pettersson and A. Nilsson, J. Chem. Phys., 2000, 112, 1946–1958 CrossRef.
- K. M. Gameel, I. M. Sharafeldin, A. U. Abourayya, A. H. Biby and N. K. Allam, Phys. Chem. Chem. Phys., 2018, 20, 25892–25900 RSC.
- X. Hong, K. Chan, C. Tsai and J. K. Nørskov, ACS Catal., 2016, 6, 4428–4437 CrossRef CAS.
- S. Zheng, C. Zuo, X. Liang, S. Li and F. Pan, J. Energy Chem., 2021, 56, 444–448 CrossRef CAS.
- L. Gong, D. Zhang, C.-Y. Lin, Y. Zhu, Y. Shen, J. Zhang, X. Han, L. Zhang and Z. Xia, Adv. Energy Mater., 2019, 9, 1902625 CrossRef CAS.
- Y. Ouyang, L. Shi, X. Bai, Q. Li and J. Wang, Chem. Sci., 2020, 11, 1807–1813 RSC.
- C. Li, X. Liu, F. Xu, D. Wu, H. Xu and G. Fan, Electrochim. Acta, 2022, 426, 140764 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3mh00503h |
| This journal is © The Royal Society of Chemistry 2023 |