
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA MOF-derived Co3O4/nitrogen-doped carbon composite for chlorine-assisted production of ethylene oxide†
Tianlei
Li
a,
Hengzhou
Liu
a,
Jiaqi
Yu
b,
Yifu
Chen
 a,
Wenyu
Huang
b and
Wenzhen
Li
*a
a,
Wenyu
Huang
b and
Wenzhen
Li
*a
aDepartment of Chemical and Biological Engineering, Iowa State University, 618 Bissell Road, Ames, IA 50011, USA
bChemistry Department, Iowa State University, 1605 Gilman Hall, Ames, IA 50011, USA. E-mail: wzli@iastate.edu
First published on 6th February 2023
Abstract
Ethylene oxide (EO) is one of the most crucial materials in plastic industries. The traditional catalytic process requires high temperature and pressure to produce EO. A chlorine-assisted system has been reported to produce EO, but it required noble metal catalysts, which significantly increased the cost. In this work, a MOF-derived Co3O4/nitrogen-doped carbon composite (Co3O4/NC) prepared through a two-step calcination method exhibited remarkable chlorine evolution reaction (ClER) activity as compared with a commercial RuO2 catalyst, which can be attributed to the higher specific surface area and lower resistance of its porous structure and nitrogen-doped carbon. Furthermore, the Co3O4/NC maintained a stable potential and a high faradaic efficiency throughout the 10-hour electrolysis test.
Introduction
Ethylene oxide (EO), one of the most crucial chemicals for manufacturing consumer products and intermediates, was mass-produced with an annual production of over 34.5 Mt (2021).1 Taking great advantage of its strained ring structure, EO can participate in various addition reactions to open the ring. One major use of EO is its conversion to ethylene glycol,2 which is one of the most important precursors in the plastic industry. In addition, EO can be used to produce ethanol amines3 and various polymers with novel architectures.4 Patented by Lefort in 19355 and numerous follow-up studies,6–9 ethylene can be directly oxidized to EO on silver-based catalysts through thermal catalytic processes. However, relatively high temperature (220–280 °C) and high pressure (1–3 MPa) are required for this chemical transformation.10Alternatively, EO can be produced by a chlorohydrin process (RXN 2, 3 and 5).2 Powered by renewable electricity generated from solar and wind sources, chlorine (Cl2) can be in situ generated through an electrochemical process under ambient conditions (RXN 1). Then, EO can be produced from a chlorine-assisted process in the formation of ethylene chlorohydrin (RXN 2 and 3), followed by its chemical reaction with OH−, which gets accumulated at the cathode (RXN 4) to produce ethylene oxide via a dehydrochlorination process (RXN 5) (Fig. 1a). In the meantime, hydrogen is generated on the cathode.11 Iridium oxide (IrO2)12 and barium oxide-loaded IrO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 13 were reported to achieve EO production with a high faradaic efficiency (FE) of >70%. Besides, a dimensionally stable anode (DSA), made of ruthenium oxide and titanium oxide, has been proven to be an active and durable catalyst for the Cl2 evolution reaction (ClER) and is expected to be active for the chlorine-assisted EO generation process.14 However, all these catalysts are based on noble metals, which significantly increases the overall process costs. Exploration of low-cost, robust, non-noble metal catalysts is highly desirable. Although several works showed that non-noble cobalt oxide (Co3O4) is an active and durable catalyst in the ClER process,15–17 its performance is generally inferior to those of noble metal-based catalysts.
13 were reported to achieve EO production with a high faradaic efficiency (FE) of >70%. Besides, a dimensionally stable anode (DSA), made of ruthenium oxide and titanium oxide, has been proven to be an active and durable catalyst for the Cl2 evolution reaction (ClER) and is expected to be active for the chlorine-assisted EO generation process.14 However, all these catalysts are based on noble metals, which significantly increases the overall process costs. Exploration of low-cost, robust, non-noble metal catalysts is highly desirable. Although several works showed that non-noble cobalt oxide (Co3O4) is an active and durable catalyst in the ClER process,15–17 its performance is generally inferior to those of noble metal-based catalysts.
| 2Cl− + 2e− → Cl2 | (1) |
| Cl2 + H2O → HCl + HClO | (2) |
| HClO + C2H4 → Cl–C2H4–OH | (3) |
| 2H2O–2e− → H2 + 2OH− | (4) |
| Cl–C2H4–OH + OH− → C2H4O + Cl− + H2O | (5) |
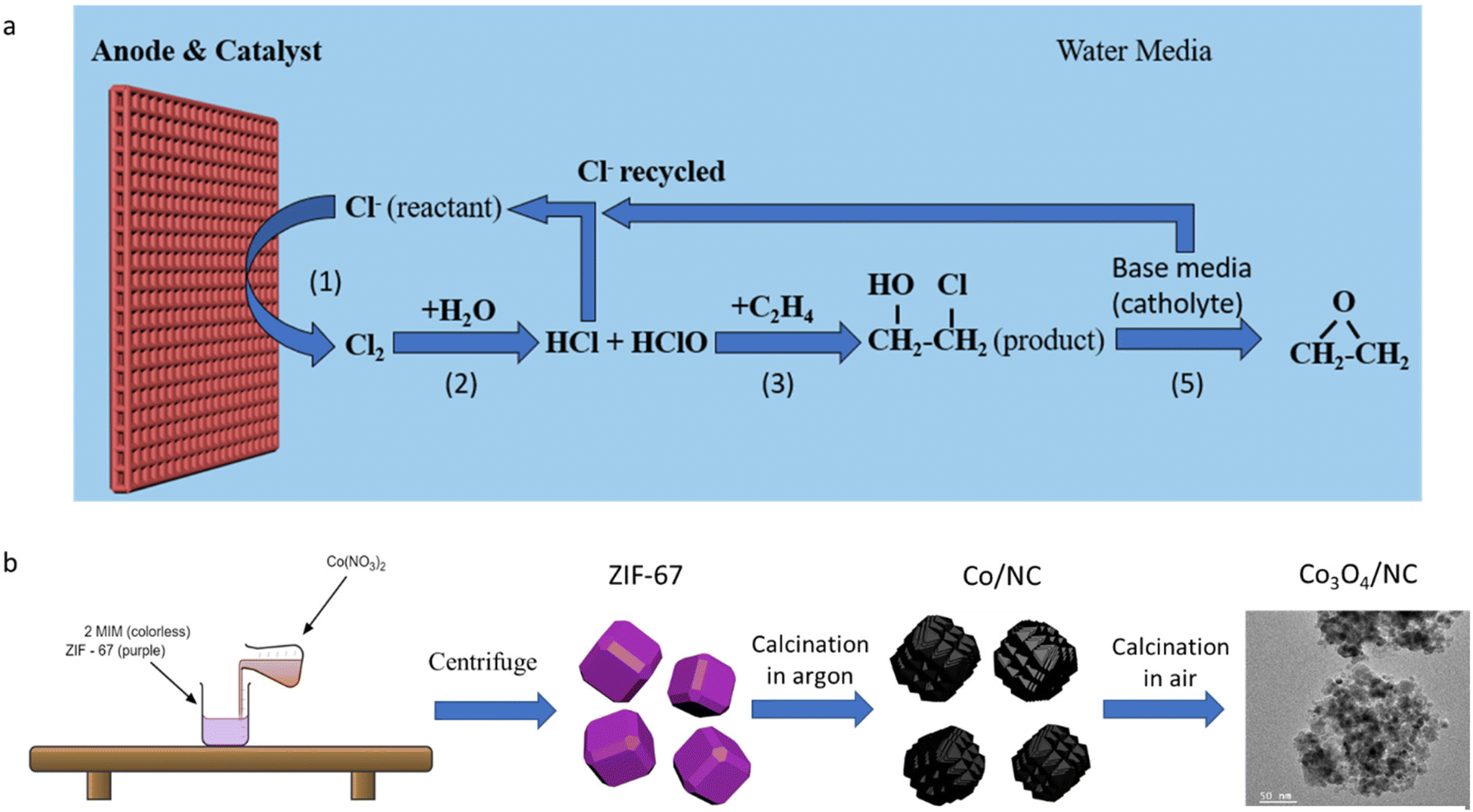 | ||
| Fig. 1 (a) Scheme of the chlorine-assisted EO generation process. (b) Synthesis process. | ||
In this work, a metal–organic framework (MOF)-derived porous cobalt oxide/nitrogen-doped carbon composite (Co3O4/NC) was studied for chlorine-assisted EO production. MOF-derived materials are well known for their unique porous structure that can offer a high specific area that is desirable for various applications, such as sensing, energy storage and catalysis.18–20 However, they have been rarely studied for the chlorine evolution reaction (ClER). Benefiting from the highly porous structure and carbonized organic linker of MOFs as a sacrificing template, Co3O4 catalysts with a high specific surface area and low charge transfer resistance were prepared. Compared to the generally used direct calcination in air to turn MOFs into metal oxides, we investigated a two-step calcination method and observed that the calcination method and temperature significantly influenced the structure and ClER activity. It was seen that the optimized catalyst exhibits comparable ClER catalytic activities to the commercial ruthenium oxide (RuO2) catalyst, while Co3O4 has a cost of a two orders of magnitude lower than RuO2. Finally, the Co3O4/NC exhibited long-term durability for chlorine-assisted EO production.
Results and discussion
Characterization of MOF-derived Co3O4
A cobalt containing zeolite imidazole framework (ZIF-67) was chosen as the sacrificing template due to its facile hydrothermal synthesis process and nitrogen-containing organic linker. MOF-derived Co3O4/NC samples were prepared from a two-step calcination treatment of ZIF-67 (Fig. 1b). The samples were named after the thermal treatment temperatures. For example, ZCO-700-300 (ZCO is short for ZIF-derived cobalt oxide) was obtained by calcinating ZIF-67 at 700 °C under argon for 1 hour, followed by calcination at 300 °C in air for 1 hour. The morphology and crystal structure of the pristine ZIF-67 after the hydrothermal synthesis were characterized by SEM (Fig. 2a) and XRD (Fig. 2d), showing a typical polyhedral morphology with a particle size of 150–200 nm.21 After the first calcination step at 700 °C under an argon atmosphere, SEM (Fig. 2b) showed a slight decrease in the particle size (∼100 nm). XRD (Fig. 2e) indicated the transformation of ZIF-67 into metallic Co, and the organic linker was converted to nitrogen-doped carbon (Fig. S1a†). The pore size distribution was well maintained after the first step calcination at 700 °C (Fig. S2a†). Therefore, the first step temperature was not optimized further. The metallic Co/NC can be further converted into desirable Co3O4/NC through the second calcination step in air, which shows a similar size to the sample after the first calcination step, as evidenced by SEM (Fig. 2c). The successful conversion of metallic Co into Co3O4 was also supported by the XRD patterns (Fig. 2f), and the XPS fitting spectra further suggested the existence of C–N bonds (Fig. S3†).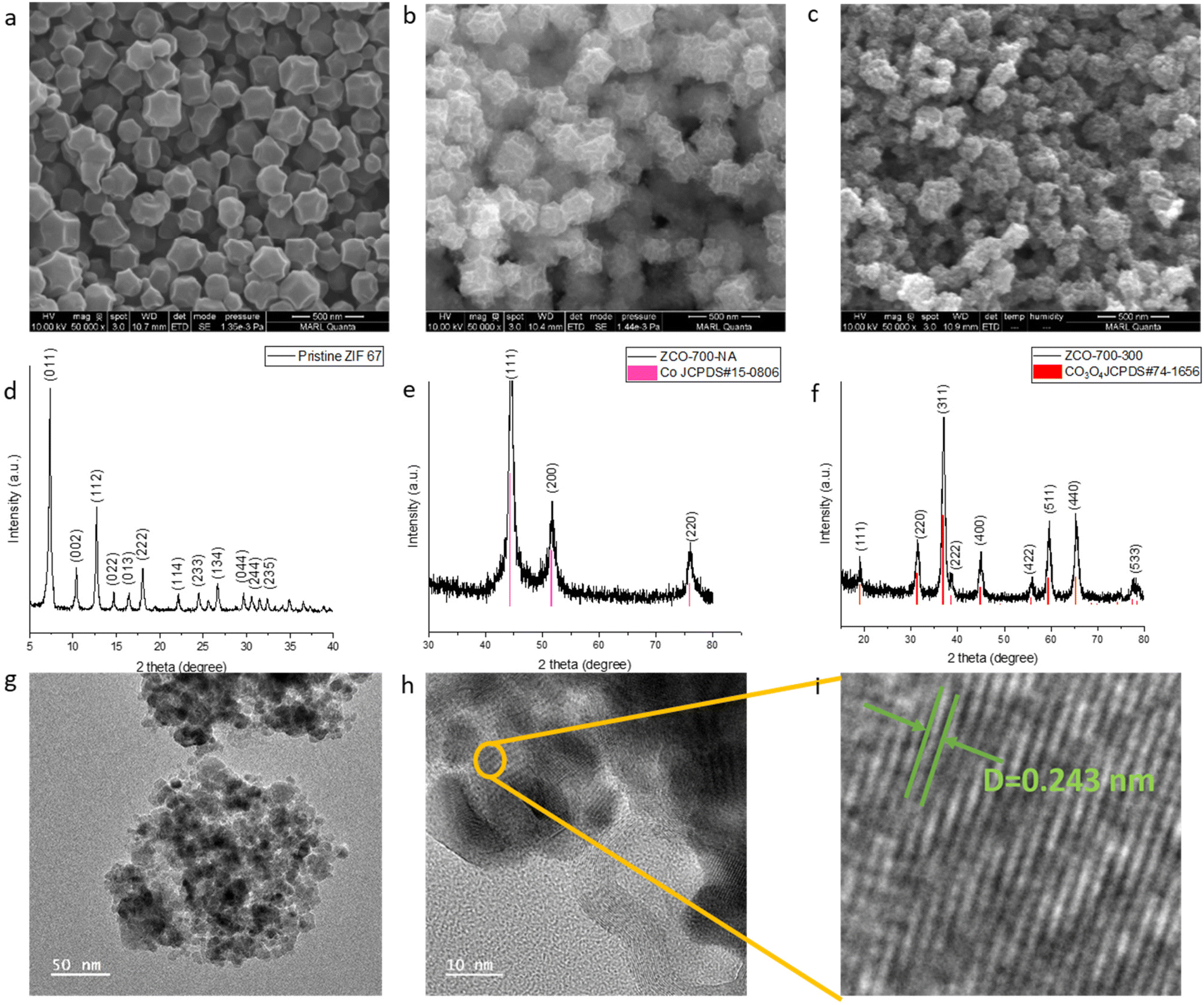 | ||
| Fig. 2 Characterization of the catalysts. SEM images and XRD patterns of the (a and d) as-synthesized ZIF-67, (b and e) ZCO-700-NA, and (c and f) ZCO-700-300. (g and h) TEM images of ZCO-700-300 at different magnifications. (i) A zoom-in image of h. | ||
As the temperature of the second thermal treatment largely affects the content species and porous structure (Fig. S1 and S2†), it is critical to optimize its operating temperature. An extremely high temperature could collapse the small pores of the MOF and remove nitrogen-doped carbon, but a relatively low temperature would not fully oxidize Co to Co3O4. We kept the first step calcination temperature at 700 °C under argon and optimized the second step calcination temperature in air. As suggested by the TGA and TPO curves (Fig. S4†), ZCO-700-NA (NA indicates that the second step has not been completed yet) starts to decompose at 250 °C, while ZIF-67 starts to decompose at 350 °C. As such, the second step of calcination was designed to be performed at 250, 300 and 350 °C. To compare the two-step method developed by us with the generally used one-step method, samples were prepared via one-step calcination at 350 and 400 °C in air. If the second step temperature is lower than 300 °C, Co can only be partially oxidized into Co3O4. For instance, as shown in the XRD patterns (Fig. S5†), ZCO-700-250 possesses both Co and Co3O4 phases, as compared to ZCO-700-300 with a solely Co3O4 phase (Fig. 2f). Further increasing the second step temperature to 350 °C would decompose all of the nitrogen-doped carbon, as shown in the TGA and TPO curves (Fig. S4†), which could undesirably increase the internal resistance of metal oxide catalysts. Energy dispersive spectroscopy (EDS) (Fig. S1†) and TGA (Table S1†) were performed to further confirm the species composition after calcination at different temperatures. EDS and TGA showed a similar trend: with an increase in the second step calcination temperature, a reduction in the amount of carbon and nitrogen was observed, along with an increase in the active content of Co3O4. With the second step calcination temperature over 300 °C (i.e., ZCO-700-300 and ZCO-700-350), the samples showed a similar oxygen to Co ratio, indicating that Co was fully oxidized to Co3O4, and 300 °C was the ideal temperature for the second calcination step.
To further understand how temperature affects the pore size distribution and surface area of ZCO samples, nitrogen physisorption was performed. The BET curves and pore size distributions are shown in Fig. S2.† All samples showed type IV hysteresis loops (Fig. S2a†), which indicates their mesoporous structures. As the second step calcination temperature increased, large-size pores appeared with an increase in their portion (Fig. S2b†). The BET surface area measured by nitrogen physisorption showed that the original pore structure in the ZIF-67 precursor collapsed during the calcination treatment, leading to a decrease in surface area (Table S1†). Therefore, the optimized second step calcination temperature was 300 °C, consistent with the EDS and TGA suggestions. The TEM image of ZCO-700-300 showed a d-spacing of 0.243 nm (Fig. 2i), which can be assigned to the Co3O4 (311) plane, in line with the XRD results. The above synthesis and characterization results showed the successful preparation of MOF-derived Co3O4.
Electrochemical measurements on MOF-derived Co3O4
Performance comparisons were carried out by linear scan voltammetry (LSV), as shown in Fig. 3a and S6.† ZCO-700-300 exhibited comparable performance to RuO2, and superior to commercial Co3O4 (C-Co3O4). At 100 mA cm−2, only a 0.04 V difference in the ClER was observed between RuO2 and ZCO-700-300. Similar kinetics between ZCO-700-300 and RuO2 was also supported by examining their Tafel slopes (Fig. 3b). The potential dependence of the ClER on ZCO-700-300 was also investigated. The FE of EO (FEEO) and current density increased by positively shifting the anodic potential, and the FEEO was maintained >70% at a potential of >1.22 V vs. Ag/AgCl (Fig. S7†). No active chlorine was detected in the absorbing vessel, and the ethylene chlorohydrin amount in the absorbing vessel was <1% in the anolyte. We reasoned that this was due to the facile reaction between ethylene and hypochlorous acid (HClO). Since HClO is consumed, the reversible reaction between chlorine gas and water will always favour the production of HCl and HClO, with negligible chlorine escaping from the anolyte. Furthermore, by comparing the samples prepared at different calcination temperatures, the sample with the second step calcination temperature of 300 °C showed a superior performance to other samples (Fig. 3c), consistent with our characterization studies which showed that Co was not only fully oxidized to Co3O4, but also maintained a better porous structure with an optimal nitrogen doped carbon content. EIS tests were further carried out on these samples to compare the charge transfer resistance (Fig. 3d). ZCO-700-300 showed the lowest charge transfer resistance, contributing to its good ClER activity. Although ZCO-700-250 possessed both high surface area and low charge transfer resistance, Co was not fully oxidized to Co3O4 and the remaining Co could transform to Co(OH)2 in the aqueous electrolyte under an anodic potential. XPS confirmed the presence of Co(OH)2 species in the ZCO-700-250 sample (Fig. S8†). It has been known that Co(OH)2 is an active oxygen evolution reaction (OER) catalyst,22 which is the main side reaction competing for the ClER. This can be the reason for the decreased activity and selectivity of the ClER on ZCO-700-250. At the second step calcination temperature of >300 °C, a lower ClER performance could be due to the decrease in surface area (Table S1†) and increase in internal resistance. Taken together, the optimized MOF-derived Co3O4/NC exhibited an outstanding performance for the ClER, and careful control over the second step calcination temperature at 300 °C is critical for optimizing the ClER activity and selectivity. The performance for the ClER reached the lowest potential at 100 mA cm−2 compared to other cobalt oxide catalysts and was even comparable to those of some noble metal catalysts, as shown in Table S2.†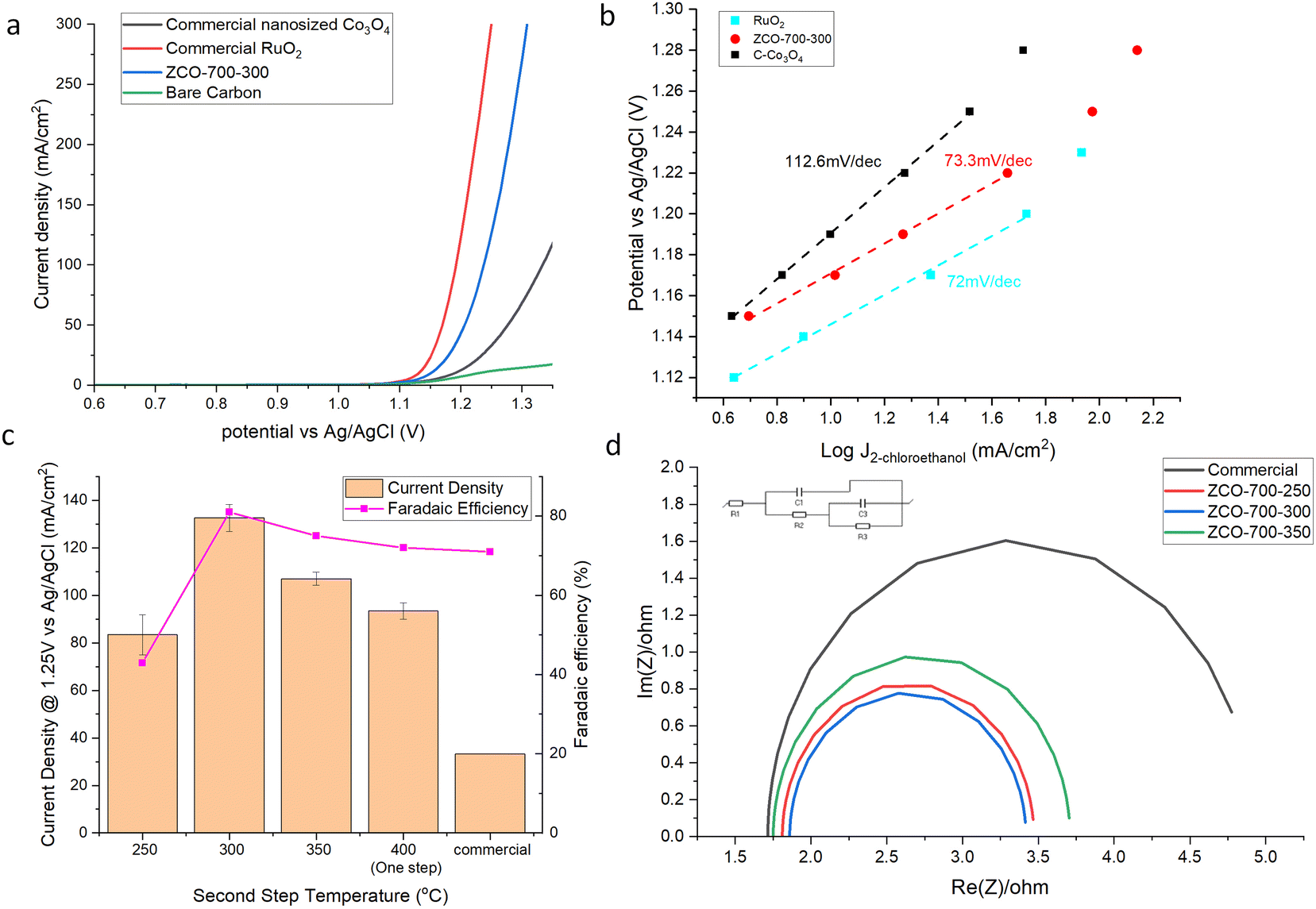 | ||
| Fig. 3 ClER performance of the (a) LSV test, (b) Tafel slope, (c) current density and FE of the samples prepared at different temperatures. (d) EIS test (equivalent circuit inserted). | ||
The intrinsic activity was also examined by comparing their area-specific activity. It should be noted that it remains challenging to quantify the Co3O4 surface area in ZCO-700-300, because either the nitrogen physisorption (i.e., BET) method or the double-layer capacitance method gives the total areas of both carbon and Co3O4. Instead, we used the chemical surface area (CSA) to calculate the area-specific activity of the ClER of our reported catalysts, and the intrinsic activities of ZCO-700-300, C–Co3O4, and C–Co3O4 physically mixed with the same nitrogen doped carbon in ZCO-700-300 (C–Co3O4–NC) were examined. As shown in Fig. S9,† C–Co3O4–NC showed a similar performance compared to C–Co3O4. The specific activity of ZCO-700-300 was about 50% better than those of C–Co3O4 and C–Co3O4–NC. The possible reason could be (1) the heterojunction formed between carbon and Co3O4 during MOF calcination, which could have facilitated the electron transfer and (2) Co3O4 particles in ZCO-700-300 were separated by carbon, which could have increased the utilization of the particle surface area. Decoupling of these contributing factors is under way in our lab.
Furthermore, the durability of Co3O4/NC was evaluated at a constant current density of 100 mA cm−2 for 10-hour electrolysis. The electrolyte was replaced by fresh electrolytes at each 2-hour interval to compensate for the consumption of the reactant Cl−. Electrolysis was carried out in both 0.2 M phosphate-buffered electrolyte at a pH of 6 and unbuffered 2 M KCl electrolyte. Buffered solution pH was set to 6 because it is the highest pH in which HClO is the dominant species.23 When the electrolyte pH is >6, the ClO− ion would be the dominant species, and it could not oxidize ethylene to ethylene chlorohydrin. Whether in the buffered or unbuffered system, ZCO-700-300 maintained a remarkably stable performance in terms of both low cell potential and high FE to EO (Fig. 4a). ICP-MS confirmed a trace number of cobalt ions in the buffered anolyte (equal to <0.002% of Co3O4 leaching). In the unbuffered system, although the anolyte pH continuously decreased and eventually formed a strongly acidic environment, we did not observe a significant dissolution of Co3O4 after 10-hour electrolysis (3.6% Co3O4 leaching obtained from ICP-OES results). The slow cobalt dissolution rate may benefit from effective protection of Co3O4 by the carbon residue.24,25 The slightly lower FEEO in the buffered system may be attributed to the use of the phosphate-buffered electrolyte, which can enhance the OER activity as phosphate has been reported to be able to facilitate the proton-coupled electron transfer process.26,27 In addition, in terms of thermodynamic potential, the acidic environment in the unbuffered system increased the OER equilibrium potential (e.g., 0.876 V at pH 6 and 1.171 V at pH 1), while the ClER potential remained unchanged (no proton participated, 1.36 V at all pH levels). Therefore, the standard reduction potential difference was reduced from 0.484 V to 0.136 V when the pH was changed from 6 to 1; this can also explain the slightly higher FEEO in the unbuffered electrolyte. To confirm the OER is the dominating side reaction, online gas chromatography (GC) was utilized to identify the gas product and NMR was employed to quantify soluble by-products. No signal of other C2H4 partial products was observed, indicating that O2 was the major side product (Fig. S10 and S11†). XPS was conducted to study the durability of ZCO-700-300 after 10-hour electrolysis. The peaks at 779.5 and 781.0 eV in the XPS fitting curve can be assigned to Co3+ and Co2+ in Co3O4, respectively. The overlapped XPS spectrum between the fresh catalyst (before the electrolysis) and spent catalyst (after 10-hour electrolysis) showed an unchanged valence state of Co (Fig. 4b and c), which indicated the robust durability of the Co3O4/NC catalyst.
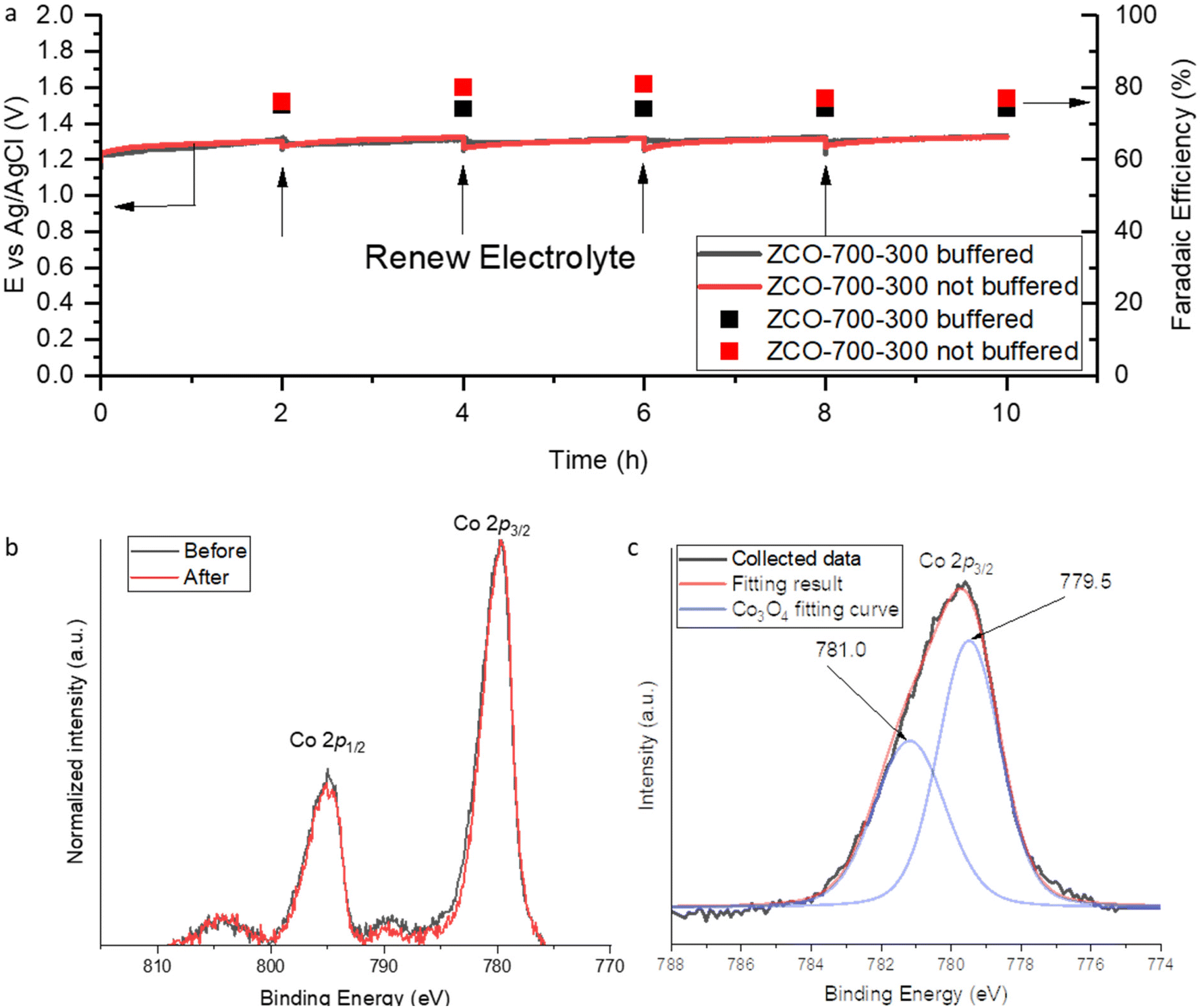 | ||
| Fig. 4 (a) CP test results (90% IR compensated). (b) XPS results before and after the durability test. (c) Co3O4 2p3/2 peak fitting curve. | ||
Conclusions
In summary, we have demonstrated that a MOF-derived Co3O4/NC through our self-developed two-step calcination method can serve as a highly active catalyst for the chlorine-assisted production of ethylene oxide. The effects of the calcination method and temperature on the ClER activity and FEEO on the ZCO samples were investigated and optimized. Benefitting from the porous structure with more exposed active sites and improved charge transfer, high ClER activity was achieved on the Co3O4/NC catalyst, which is comparable to a commercial RuO2 catalyst. In the 10-hour chronoamperometry test, the optimized Co3O4/NC catalyst exhibited remarkable stability in the buffered electrolyte, in terms of ClER activity, FEEO, and catalyst phase stability. The strategy for synthesizing MOF-derived Co3O4/NC materials could be used for the synthesis of efficient catalysts for other electrochemical reactions.Experimental methods
Materials and chemicals
All materials and chemicals were used as received without further purification. Platinum foil (25 × 25 × 0.025 mm, 99.99%) was purchased from Strem Chemicals Inc. Cobalt(II) nitrate hexahydrate (Co(NO3)2·6H2O, 98%), 2-methyl imidazole (2-MIM, 99%), ethylene oxide (EO, 50 mg mL−1 in methanol) and dimethylmalonic acid (DMMA, 99%) were purchased from Sigma-Aldrich. Potassium chloride (KCl, 99%) and ethylene glycol (EG) were purchased from Fisher Chemicals. An anion exchange membrane (AEM, Fumasep FAA-3–50) was purchased from Fuel Cell Store. Commercial Co3O4 was purchased from US Nano Research, Inc. Commercial RuO2 was purchased from Premetek Co.Preparation of the precursor ZIF-67
The precursor ZIF-67 was synthesized by a hydrothermal method.21 Typically, 0.45 g Co(NO3)2·6H2O was dissolved in 3 mL of deionized (DI) water, and 5.5 g of 2-methylimidazole (2-MIM) was dissolved in 20 mL of DI water separately. Then, cobalt nitrate solution was added into 2-MIM solution slowly under stirring. The solution was stirred for 6 hours at room temperature. The purple precipitate was collected by centrifuging at 8000 rpm for 15 min and washed with water and methanol, respectively. The obtained precipitate was dried in a vacuum oven at 80 °C for 24 hours.Preparation of ZCO samples
ZIF-derived cobalt oxide (ZCO) samples were prepared through a two-step calcination method (Fig. 1b). The first calcination step was conducted at 700 °C in a tube furnace under an argon atmosphere for 1 hour.28 The second step was performed in a muffle furnace under an air atmosphere for 1 hour at different temperatures of 250, 300, and 350 °C to study the temperature effect on the activity of the samples. To compare the effect of the calcination method, the samples made by the one-step calcination method were also prepared at 350 and 400 °C under an air atmosphere.Preparation of C–Co3O4–NC samples
ZIF-8 is a MOF with a similar structure to ZIF-67, which replaces Co with Zn as the metal source. ZIF-8 was chosen instead of ZIF-67, because it is easier to remove Zn, so the nitrogen-doped carbon (without metal) can be obtained. ZIF-8 was then synthesized with the same molar ratio of metal ions to the organic linker as ZIF-67 (reported in this work). The product was calcined in Ar at 700 °C for 1 hour with a temperature ramp of 5 °C min−1. The product after calcination is denoted as a Zn/NC composite, which should have a similar structure to ZCO-700-NA. Zn/NC was then soaked in 0.5 M H2SO4 overnight to remove Zn.29 The remaining Zn was <1 wt%, as confirmed by EDS. The ZIF-8 derived carbon was then mixed with commercial Co3O4 (C–Co3O4) with a 14:86 weight ratio, same as that for ZCO-700-300 according to the TGA results shown in Table S1.† The mixture was ground and dispersed in isopropanol alcohol and sprayed on 1 × 1 cm2 carbon paper to reach a Co3O4 loading of 1 mg cm−2. This final sample was named C–Co3O4–NC.
Preparation of electrodes
Commercial nanosized cobalt oxide (C–Co3O4), commercial ruthenium oxide (RuO2), and various self-prepared samples were loaded on hydrophilic carbon paper by spray coating. The spray ink was made of the active content and isopropanol (IPA) at a 5 mg mL−1 concentration. The amounts of catalyst were normalized to 1 mg cm−2 of Co3O4 or RuO2 according to the TGA results. Commercial samples were directly sprayed on carbon paper. Self-prepared samples were loaded by spray coating their precursor (ZIF-67 for the one-step method and ZCO-700-NA for the two-step method) on carbon paper, followed by calcination at the designated temperatures. This loading method would enhance the contact with carbon paper and prevent physical peel-off.Characterization of the samples
Scanning electron microscopy (SEM) images and energy dispersive spectroscopy (EDS) spectra were obtained using a FEI Quanta 250 FE-SEM. X-ray diffraction (XRD) data were collected from a Rigaku SmartLab diffractometer, with Cu Kα radiation (λ = 0.15418 nm) working at 40 kV and 44 mA. Transmission electron microscopy (TEM) images were captured using a JEOL 200 kV JSM-2100 scanning transmission electron microscope with a Gatan OneView 4K camera. X-ray photoelectron spectroscopy (XPS) was performed using a Kratos Amicus/ESCA 3400 instrument. Nitrogen physisorption was carried out using a Micromeritics ASAP 2020 surface area and porosity analyzer. It needs to be noted that BET measured both carbon and Co3O4 surface areas while carbon was inert in the ClER. Temperature-programmed oxidation was completed on the Micromeritics AutoChem II chemisorption analyzer. Leached cobalt during the durability test was determined using an inductively coupled plasma-optical emission spectrometer (ICP-OES) on a PerkinElmer Optima 8000 ICP-OES instrument and an Agilent Technologies 7700 inductively coupled plasma mass spectrometer (ICP-MS).Electrochemical tests
Electrochemical tests were conducted on a BioLogic VSP-300 electrochemical workstation. Measurements were carried out in a typical H-type cell with an additional absorbing vessel (Fig. S12†). An anode was prepared as described in Experimental section. The cathode was platinum foil. A saturated Ag/AgCl electrode was utilized as a reference electrode. The electrolyte was 2 M KCl solution at both the anode and cathode. An anion exchange membrane was used to separate the two compartments of this cell. Ethylene gas was purged into the anolyte through a gas diffuser. An additional compartment was added to capture the chlorine gas generated on the anode.Linear sweep voltammetry (LSV) was conducted to evaluate the activity performance of the ClER. The scanning rate of LSV was 5 mV s−1. A 90% IR compensation was used to eliminate the effect of solution resistance. Electrochemical impedance spectroscopy (EIS) was performed at 1.15 V vs. Ag/AgCl electrode to measure the charge transfer resistance. Chronopotentiometry (CP) mode was utilized to test the durability of the catalyst at 100 mA cm−2. The electrolyte was replaced with fresh electrolyte solution every 2 hours during the durability test to compensate for the consumption of chloride ions.
Product quantification
The amount of product was measured by nuclear magnetic resonance (NMR) using a Bruker NEO 400 instrument. An internal standard method was employed to quantify the product.17 Typically, the solution was made of 200 μL of D2O, 100 μL of 15 mM DMMA solution, and 400 μL of the anolyte. The quantity was obtained by calculating the peak area ratio of the product to the internal standard and comparing it to the calibration curve. The peak area was obtained using MestReNova software with the auto phase and baseline correction. A calibration curve generated from the known product concentration in 2 M KCl solution was used to quantify the product. An example of the NMR results and calibration curves for EG and EO are shown in the ESI (Fig. S11†). Faradaic efficiency was calculated as follows:where F is the Faraday constant, 96
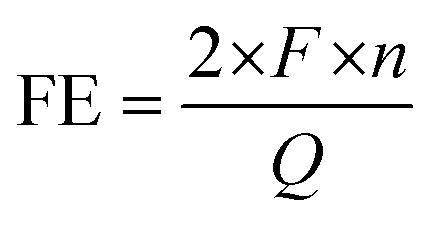 485.33 C mol−1; 2 is the number of electrons transferred when one ethylene molecule is oxidized to ethylene chlorohydrin; n is the total amount of product measured by NMR; and Q is the total amount of electrons transferred in the circuit during the whole reaction.
485.33 C mol−1; 2 is the number of electrons transferred when one ethylene molecule is oxidized to ethylene chlorohydrin; n is the total amount of product measured by NMR; and Q is the total amount of electrons transferred in the circuit during the whole reaction.
Active chlorine was tested using a WaterWorks free chlorine test strip. The lower detection limit was 25 ppm, corresponding to a 2 C charge. Typically, the experiment was carried out at 100 mA for 2 h, which was 720 C total charge. If the color change of the strip was not observed, we assumed that all active chlorine was consumed through reacting with ethylene.
Calculation of specific activity
Chemical surface area (CSA) is a good alternative to calculate the area-specific activity of the ClER of the reported catalysts to compare their intrinsic activity. By using the Debye–Scherrer equation, the average particle size of the metal nanoparticles could be estimated from the diffraction peak of an isolated crystal plane in XRD patterns. The average size of the Co3O4 nanoparticles obtained was 8 nm. The TEM image also shows that the Co3O4 particle size ranges from 5 to 13 nm. The specific surface area of Co3O4 thus can be calculated using the following equation.where S is the surface area of a spherical Co particle; m is the mass; ρ is the density of cobalt oxide (6.11 g cm−3); V is the volume of the particle, and r is the radius of the particle. The total specific surface area of Co3O4 in ZCO-700-300 would be 122.75 m2 g−1. Then the current density based on the geometric electrode surface area could be converted to the current density based on the specific surface area. The specific activity of ZCO-700-300 was 0.108 mA cm−2, which is 60% greater than the specific activity of commercial Co3O4 (0.067 mA cm−2). Although the surface area of the Co3O4 particles was not available for the ClER, the intrinsic activity of the MOF-derived Co3O4 with the commercial Co3O4 sample was calculated, based on the assumption that they exhibit same levels of catalyst utilization (= electrochemical active surface area/specific surface area).
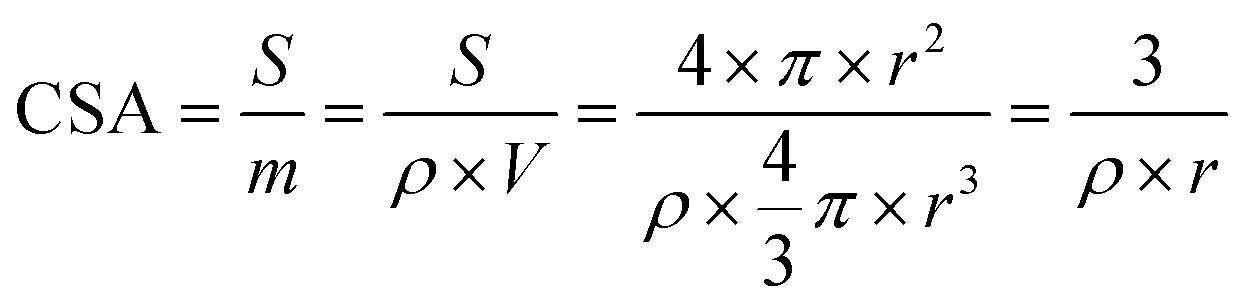
Author contributions
T. Li synthesized and characterized MOF-derived Co3O4 materials, and conducted most of the electrochemical tests. H. Liu helped with setting up electrochemical cells for the initial electrochemical tests and data analysis. J. Yu and W. Huang performed the ICP-MS tests and data collection and analysis. Y. Chen helped with the GC tests and some experimental design. W. Li proposed and supervised the research. T. Li, H. Liu and W. Li wrote the manuscript, and all authors discussed the results and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NSF grant (EFMA 2132200). W. Li is grateful to his Herbert L Stiles Faculty Fellowship, and ISU-PIRI grant. We acknowledge the fruitful discussions with Prof. Jean-Philippe Tessonnier, Dr Jungkuk Lee, Basil Rawah, Xiaopeng Liu, and Mohammad Albloushi. T. Li appreciates Dr Boote Brett, Dr Cady Sarah, Dr Warren Straszheim and Dr Arkady Ellern for their valuable training on TGA, NMR, SEM and EDS, and XRD, respectively. We thank Dr Patrick Johnston, Dr Tracy Stewart and Dr Dapeng Jing for their assistance with ICP, TEM, and XPS characterization studies, respectively.References
- S. Smith, Global and China Ethylene Oxide (EO) Industry Report, 2017–2021. Global Reports, 2017 Search PubMed.
- S. Rebsdat and D. Mayer, Ethylene Glycol, in Ullmann's Encyclopedia of Industrial Chemistry, 2000 Search PubMed.
- D. G. Weaver and J. Smart, Ethylene Oxide Derivatives. Glycols and Ethanolamines, Ind. Eng. Chem., 1959, 51(8), 894–900 CrossRef CAS.
- J. Herzberger, K. Niederer, H. Pohlit, J. Seiwert, M. Worm, F. R. Wurm and H. Frey, Polymerization of Ethylene Oxide, Propylene Oxide, and Other Alkylene Oxides: Synthesis, Novel Polymer Architectures, and Bioconjugation, Chem. Rev., 2016, 116(4), 2170–2243 CrossRef CAS PubMed.
- T. E. Lefort, Process for the production of ethylene oxide, US Pat., US1998878, 1935 Search PubMed.
- V. I. Bukhtiyarov, A. I. Nizovskii, H. Bluhm, M. Hävecker, E. Kleimenov, A. Knop-Gericke and R. Schlögl, Combined in situ XPS and PTRMS study of ethylene epoxidation over silver, J. Catal., 2006, 238(2), 260–269 CrossRef CAS.
- A. J. F. van Hoof, E. A. R. Hermans, A. P. van Bavel, H. Friedrich and E. J. M. Hensen, Structure Sensitivity of Silver-Catalyzed Ethylene Epoxidation, ACS Catal., 2019, 9(11), 9829–9839 CrossRef CAS.
- A. J. F. van Hoof, I. A. W. Filot, H. Friedrich and E. J. M. Hensen, Reversible Restructuring of Silver Particles during Ethylene Epoxidation, ACS Catal., 2018, 8(12), 11794–11800 CrossRef CAS PubMed.
- T. E. Jones, R. Wyrwich, S. Böcklein, E. A. Carbonio, M. T. Greiner, A. Y. Klyushin, W. Moritz, A. Locatelli, T. O. Menteş, M. A. Niño, A. Knop-Gericke, R. Schlögl, S. Günther, J. Wintterlin and S. Piccinin, The Selective Species in Ethylene Epoxidation on Silver, ACS Catal., 2018, 8(5), 3844–3852 CrossRef CAS.
- N. Platzer, Chemistry and Technology of Basic Organic and Petrochemical Synthesis. Volume 1 and Volume 2. N. N. Lebedev, MIR Publishers, Moscow, 1984,333 and 305 pp, J. Polym. Sci., Part C: Polym. Lett., 1986, 24(6), 293–293 CrossRef.
- G. Chen, X. Li and X. Feng, Upgrading Organic Compounds through the Coupling of Electrooxidation with Hydrogen Evolution, Angew. Chem., Int. Ed., 2022, 61(42), e202209014 CrossRef CAS PubMed.
- R. Leow Wan, Y. Lum, A. Ozden, Y. Wang, D.-H. Nam, B. Chen, J. Wicks, T.-T. Zhuang, F. Li, D. Sinton and H. Sargent Edward, Chloride-mediated selective electrosynthesis of ethylene and propylene oxides at high current density, Science, 2020, 368(6496), 1228–1233 CrossRef PubMed.
- Y. Li, A. Ozden, W. R. Leow, P. Ou, J. E. Huang, Y. Wang, K. Bertens, Y. Xu, Y. Liu, C. Roy, H. Jiang, D. Sinton, C. Li and E. H. Sargent, Redox-mediated electrosynthesis of ethylene oxide from CO2 and water, Nat. Catal., 2022, 5(3), 185–192 CrossRef CAS.
- N. Menzel, E. Ortel, K. Mette, R. Kraehnert and P. Strasser, Dimensionally Stable Ru/Ir/TiO2-Anodes with Tailored Mesoporosity for Efficient Electrochemical Chlorine Evolution, ACS Catal., 2013, 3(6), 1324–1333 CrossRef CAS.
- H. Ha, K. Jin, S. Park, K.-G. Lee, K. H. Cho, H. Seo, H.-Y. Ahn, Y. H. Lee and K. T. Nam, Highly Selective Active Chlorine Generation Electrocatalyzed by Co3O4 Nanoparticles: Mechanistic Investigation through in Situ Electrokinetic and Spectroscopic Analyses, J. Phys. Chem. Lett., 2019, 10(6), 1226–1233 CrossRef CAS PubMed.
- Q. Xiong, X. Zhang, Q. Cheng, G. Liu, G. Xu, J. Li, X. Ye and H. Gao, Highly selective electrocatalytic Cl− oxidation reaction by oxygen-modified cobalt nanoparticles immobilized carbon nanofibers for coupling with brine water remediation and H2 production, Nano Res., 2020, 14(5), 1443–1449 CrossRef.
- M. Chung, K. Jin, J. S. Zeng and K. Manthiram, Mechanism of Chlorine-Mediated Electrochemical Ethylene Oxidation in Saline Water, ACS Catal., 2020, 10(23), 14015–14023 CrossRef CAS.
- J. Li, M. Chen, D. A. Cullen, S. Hwang, M. Wang, B. Li, K. Liu, S. Karakalos, M. Lucero, H. Zhang, C. Lei, H. Xu, G. E. Sterbinsky, Z. Feng, D. Su, K. L. More, G. Wang, Z. Wang and G. Wu, Atomically dispersed manganese catalysts for oxygen reduction in proton-exchange membrane fuel cells, Nat. Catal., 2018, 1(12), 935–945 CrossRef CAS.
- L. Chen, J.-W. Ye, H.-P. Wang, M. Pan, S.-Y. Yin, Z.-W. Wei, L.-Y. Zhang, K. Wu, Y.-N. Fan and C.-Y. Su, Ultrafast water sensing and thermal imaging by a metal-organic framework with switchable luminescence, Nat. Commun., 2017, 8(1), 15985 CrossRef CAS PubMed.
- L. Chong, J. Wen, J. Kubal, F. G. Sen, J. Zou, J. Greeley, M. Chan, H. Barkholtz, W. Ding and D.-J. Liu, Ultralow-loading platinum-cobalt fuel cell catalysts derived from imidazolate frameworks, Science, 2018, 362(6420), 1276–1281 CrossRef CAS PubMed.
- J. Qian, F. Sun and L. Qin, Hydrothermal synthesis of zeolitic imidazolate framework-67 (ZIF-67) nanocrystals, Mater. Lett., 2012, 82, 220–223 CrossRef CAS.
- P. T. Babar, A. C. Lokhande, B. S. Pawar, M. G. Gang, E. Jo, C. Go, M. P. Suryawanshi, S. M. Pawar and J. H. Kim, Electrocatalytic performance evaluation of cobalt hydroxide and cobalt oxide thin films for oxygen evolution reaction, Appl. Surf. Sci., 2018, 427, 253–259 CrossRef CAS.
- D. Ghernaout, Disinfection and DBPs Removal in Drinking Water Treatment: A Perspective for a Green Technology, Int. J. Adv. Appl. Sci., 2018, 5, 108–117 CrossRef.
- D. Senthil Raja, P.-Y. Cheng, C.-C. Cheng, S.-Q. Chang, C.-L. Huang and S.-Y. Lu, In situ grown metal-organic framework-derived carbon-coated Fe-doped cobalt oxide nanocomposite on fluorine-doped tin oxide glass for acidic oxygen evolution reaction, Appl. Catal., B, 2022, 303, 120899 CrossRef CAS.
- V. Singh and T. C. Nagaiah, In situ incorporation of cobalt nanoclusters and nitrogen into the carbon matrix: a bifunctional catalyst for the oxygen depolarized cathode and chlorine evolution in HCl electrolysis, J. Mater. Chem. A, 2019, 7(16), 10019–10029 RSC.
- K. Jin, H. Seo, T. Hayashi, M. Balamurugan, D. Jeong, Y. K. Go, J. S. Hong, K. H. Cho, H. Kakizaki, N. Bonnet-Mercier, M. G. Kim, S. H. Kim, R. Nakamura and K. T. Nam, Mechanistic Investigation of Water Oxidation Catalyzed by Uniform, Assembled MnO Nanoparticles, J. Am. Chem. Soc., 2017, 139(6), 2277–2285 CrossRef CAS PubMed.
- Y. Surendranath, M. W. Kanan and D. G. Nocera, Mechanistic Studies of the Oxygen Evolution Reaction by a Cobalt-Phosphate Catalyst at Neutral pH, J. Am. Chem. Soc., 2010, 132(46), 16501–16509 CrossRef CAS PubMed.
- M. Liu, M. Zhang, P. Zhang, Z. Xing, B. Jiang, Y. Yu, Z. Cai, J. Li and J. Zou, ZIF-67-Derived Dodecahedral Co@N-Doped Graphitized Carbon Protected by a Porous FeS2 Thin-Layer as an Efficient Catalyst to Promote the Oxygen Reduction Reaction, ACS Sustainable Chem. Eng., 2020, 8(10), 4194–4206 CrossRef CAS.
- P. Liang, Q. Wang, J. Kang, W. Tian, H. Sun and S. Wang, Dual-metal zeolitic imidazolate frameworks and their derived nanoporous carbons for multiple environmental and electrochemical applications, Chem. Eng. J., 2018, 351, 641–649 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc04508g |
| This journal is © The Royal Society of Chemistry 2023 |