
Clarification of Li deposition behavior on anodes with a porous interlayer in Li-free all-solid-state batteries†
Se Hwan
Park‡
 ,
Dayoung
Jun‡
,
Ji Eun
Jung
,
Seong Gyu
Lee
,
Gyu Hyun
Lee
and
Yun Jung
Lee
*
,
Dayoung
Jun‡
,
Ji Eun
Jung
,
Seong Gyu
Lee
,
Gyu Hyun
Lee
and
Yun Jung
Lee
*
Department of Energy Engineering, Hanyang University, Seoul 04763, Republic of Korea. E-mail: yjlee94@hanyang.ac.kr
First published on 3rd October 2022
Abstract
In Li-free all-solid-state batteries (ASSBs) with a sulfide solid electrolyte (SE), the reactivity between Li and sulfide renders the separation of these two components desirable. A porous interlayer coated on the anode may limit this contact of SE with Li. However, a lack of understanding regarding Li deposition makes the composite anode design difficult. Herein, we identify the factors determining the morphology and location of Li deposits in Li-free ASSBs with a porous interlayer based on the thermodynamics of Li electrodeposition and the kinetics of Li movement through the interlayer. We find the adhesion work as one determining factor for the location of the main Li deposits. We explore several kinetic factors including the pore size of the interlayer, temperature, scaffold materials, and surface modification with Ag nanoparticles. The influence of the pore size on Li deposition suggests that Li moves through the interlayer via Coble creep, with smaller pores, higher temperatures, and a lithiophilic surface facilitating this process. We propose a scenario for pore-size-dependent Li deposition based on diffusional creep, inhomogeneity, and resulting stress within the interlayer. This work increases the fundamental understanding of Li deposition behavior and opens new avenues for the development of highly reversible Li-free ASSBs.
1. Introduction
Li-free or anodeless all-solid-state batteries (ASSBs) are promising next-generation energy storage devices due to their high energy density and safety. The relatively high mechanical strength of solid electrolytes (SEs) can suppress dendritic Li growth and guarantee the safe operation of ASSBs while using Li metal as the anode active material.1,2 Because cathode active materials are solely used as the Li source in Li-free ASSBs and no Li metal is involved in the initial battery assembly process, the energy density of the entire cell can be significantly increased.3–5 Furthermore, the absence of Li metal during cell assembly can streamline the production process.6 For Li-free ASSBs to achieve high-capacity retention over many cycles, high reversibility is particularly required during repeated Li deposition and stripping processes within the anode because the Li supply is very limited without excess in Li-free ASSBs.7,8 However, inhomogeneous Li deposition, unstable interfacial contact, and side reactions between the Li metal and SE can lead to premature cell failure.9–11 Moreover, though modeling suggested that SEs (especially inorganic SEs) have sufficiently high mechanical strength for the physical suppression of dendritic Li growth,1 surface inhomogeneities in SEs and the resulting uneven local current density may lead to dendritic Li growth and internal short circuits.12–14As in cells employing a liquid electrolyte, composite anodes with a diverse range of architectures and materials can potentially suppress irregular Li deposition in Li-free ASSBs. Composite anodes usually contain porous architecture but, despite the intense research interest, the development of composite anodes for Li-free ASSBs has been challenged due to the following restrictions. First, unlike liquid electrolytes, the SE cannot infiltrate the pores to deliver Li ions and retract to accommodate the volume expansion of Li. Therefore, the scaffold of porous architecture itself should deliver Li ions to fill the pores with Li deposits.15–17 Second, because SEs (especially sulfide SEs) suffer from the side reactions with reductive Li metal, the Li electrodeposits and SE need to be physically separated.3,18
In a previous study, Li-free ASSBs employing a silver–carbon (Ag–C) composite anode exhibited a high energy density and excellent cyclability.3 A porous composite layer composed of carbon and Ag nanoparticles (NPs) was coated on a stainless steel (SUS) current collector (CC). When the full cell was charged, and Li was plated onto the anode, a dense Li metal layer was deposited between the Ag–C composite layer and the SUS CC. Despite the much faster electron conduction compared to that of Li ions through the Ag–C composite layer, the metallic Li was plated not on the surface of the SE but between the Ag–C layer and the SUS CC (and also in the Ag–C layer). As a result, the Ag–C interlayer significantly reduced the direct contact between the Li metal deposits and the SE, suppressing the side reactions. However, the principal mechanisms underlying this Li deposition behavior and the factors that determine the location of Li deposits have not yet been clarified.
In situ transmission electron microscopy (TEM) observations of electrochemical Li deposition and stripping in carbon tubules with an inner diameter of 100 nm have been used to elucidate the mechanisms underlying Li deposition into the porous composite anodes.19 The Li advanced and retracted in carbon tubules during Li deposition and stripping mainly via diffusional Coble creep. In these studies,19,20 it was expected that the pore size of the porous architecture and the Li-conducting properties of the scaffold material should meet certain criteria to enable Coble creep-based Li movement, but this was not proven through systematic experiments.
In the present study, we analyzed Li deposition behavior in Li-free ASSBs with a porous interlayer coated CC and identified the factors that determine the morphology and location of the Li deposits. Of the various possible locations for Li deposits within composite anodes, the porous interlayer–CC interface is preferred in order to suppress undesirable side reactions between the SE and deposited Li. We analyzed the location and morphology of the Li deposits based on both the thermodynamics of Li electrodeposition and the kinetics of Li movement in ASSBs through the porous interlayer. The work of adhesion or interfacial energy was identified as one determining factor for the location of the main Li deposits in the energetics of the Li deposition in ASSBs with porous interlayer. In this study, the main Li layer was deposited at the porous layer–CC interface when the SE–porous layer contact formed at a pressure of 380 MPa, suggesting that the work of adhesion between the SE and the porous interlayer is higher than that between the porous layer and the CC.
However, to be deposited at the porous layer–CC interface, Li needs to pass through the porous interlayer with sufficient mobility of Li (either Li ions or atoms). Many factors can affect the kinetics of Li movement, and we investigated several of these in the present study, including the pore size of the composite anode, temperature, scaffold materials, and surface modification with Ag NPs. The pore size dependency of Li movement through the interlayer indicated that the Li advanced/retracted through the porous interlayer via diffusional Coble creep. Several factors that facilitate the kinetics of diffusional Coble creep were explored; smaller pores, higher temperatures, and a lithiophilic surface. The composite interlayer fabricated with carbon spheres of an average diameter of 75 nm, which had the smallest average pore size, demonstrated the most significant improvement in cyclability during repeated Li deposition and stripping because the main Li deposit layer was at the SE–porous layer interface, while the interlayer with larger size exhibited partial or complete contact between the deposited Li and the SE layer. Surface modification of the carbon spheres with Ag NPs in composite anodes led to denser and more homogeneous Li deposits compared to those without Ag. This was ascribed to the highly lithiophilic nature of Ag and Li–Ag alloys, which provides thermodynamically stable Li deposition sites and facilitated Li diffusion. The results of the present study enhance the fundamental understanding of Li deposition behavior in Li-free ASSBs with a porous interlayer on the CC and thus motivates further development of highly reversible Li-free ASSBs.
2. Experimental procedure
2.1 Material synthesis
The carbon spheres were fabricated via the carbonization of polymer spheres that had been synthesized using sol–gel polymerization with resorcinol and formaldehyde as precursors. The particle size was controlled by varying the solvent composition and precursor concentration or by adding a stabilizer (only for CS3). A mixture of deionized (DI) water and ethanol at different ratios was prepared as a solvent (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 in 84 ml for CS1, 8:3 in 200 ml for CS2, and 5:2 in 336 ml for CS3). For CS3, 1200 mg of Pluronic® F127 (Sigma Aldrich) was added to further reduce the particles. Subsequently, ammonium hydroxide (Daejung Chemicals) and resorcinol (Sigma Aldrich) were added to the prepared solution, followed by 1 h of stirring (0.3 ml of ammonium hydroxide and 600 mg of resorcinol for CS1 and CS2, and 1.2 ml of ammonium hydroxide and 2400 mg of resorcinol for CS3). Finally, polymerization was initiated with the addition of formaldehyde solution (Sigma Aldrich, 0.9 ml for CS1 and CS2 and 3.6 ml for CS3), and the solutions were stirred overnight. All processes were carried out at room temperature. The resulting suspensions were washed with DI water three times and ethanol two times via centrifugation and dried overnight at 80 °C in a vacuum oven. To carbonize the polymer spheres, the obtained powders were annealed under an Ar atmosphere with the following temperature profile: heating to 350 °C at a ramping rate of 1 °C min−1, maintaining the temperature at 350 °C for 2 h, heating to 600 °C at a ramping rate of 1 °C min−1, and maintaining at 600 °C for 4 h.
:9 in 84 ml for CS1, 8:3 in 200 ml for CS2, and 5:2 in 336 ml for CS3). For CS3, 1200 mg of Pluronic® F127 (Sigma Aldrich) was added to further reduce the particles. Subsequently, ammonium hydroxide (Daejung Chemicals) and resorcinol (Sigma Aldrich) were added to the prepared solution, followed by 1 h of stirring (0.3 ml of ammonium hydroxide and 600 mg of resorcinol for CS1 and CS2, and 1.2 ml of ammonium hydroxide and 2400 mg of resorcinol for CS3). Finally, polymerization was initiated with the addition of formaldehyde solution (Sigma Aldrich, 0.9 ml for CS1 and CS2 and 3.6 ml for CS3), and the solutions were stirred overnight. All processes were carried out at room temperature. The resulting suspensions were washed with DI water three times and ethanol two times via centrifugation and dried overnight at 80 °C in a vacuum oven. To carbonize the polymer spheres, the obtained powders were annealed under an Ar atmosphere with the following temperature profile: heating to 350 °C at a ramping rate of 1 °C min−1, maintaining the temperature at 350 °C for 2 h, heating to 600 °C at a ramping rate of 1 °C min−1, and maintaining at 600 °C for 4 h.
For Ag NP decoration, 90 mg of polymer spheres were dispersed in 150 ml of DI water and ethanol solution (14:1 volume ratio) using tip sonication. Following this, 70.9 mg of silver nitrate was added to the polymer sphere dispersion. After stirring for 10 min, 50 ml of 1 M KOH (Sigma Aldrich) solution and 1 M ethylene glycol (Sigma Aldrich) solution were added dropwise. After stirring for 3 h at 80 °C, the dispersion was washed with DI water three times and ethanol two times using centrifugation and dried overnight at 80 °C. The final carbonization process was identical to that for the carbon spheres.
2.2 Cell fabrication
The composite anodes were fabricated by casting the slurry containing the carbon spheres or Ag NP-decorated carbon spheres and polyvinylidene fluoride (PVDF, Sigma Aldrich) binder in NMP (Daejung Chemicals) at a weight ratio of 9:1 on SUS 316 foil and drying in an oven at 120 °C. The composite anode was punched into a circular shape with a diameter of 10 mm. Lithium foil (Honjo, 400 μm) and Li3PS4 (LPS, NEI corporation) were used as the counter electrode and solid-state electrolyte, respectively. Initially, LPS powder was pelletized under a pressure of 270 MPa in a PEEK mold with an inner diameter of 10 mm. The prepared composite anode was placed on one side of the LPS pellet and pressed at 380 MPa. The Li foil was assembled on the other side of the pellet and pressed to an operating pressure of 5 MPa. All cells were assembled in an Ar-filled glove box (O2 and H2O levels < 0.1 ppm) and operated under constant pressure and temperature (a pressure of 5 MPa and a temperature of 30, 45, or 60 °C). Galvanostatic Li deposition and stripping were conducted using a WonaTech WBCS 3000 automatic battery cycler. Li metal was deposited on the composite anode to a specific capacity (0.5, 1, or 3 mA h cm−2) and stripped to 1 V (vs. Li/Li+) under a constant areal current density of 0.5 mA cm−2. Prior to the initial Li deposition, the anodes were pre-cycled from 0 to 1 V 5 times with a current density of 0.2 mA cm−2 to activate the anode.
2.3 Characterization
The morphology of the prepared materials and Li deposits was observed using SEM (Nova Nano SEM 450) with an EDS detector. The morphology and crystallinity were further characterized by high-resolution TEM (JEM 2100F, JEOL) with an EDS detector. The pore size distribution of the composite anodes was identified using mercury porosimetry (Autopore V 9600). The surface properties of the carbon spheres were analyzed using Raman spectroscopy (DXR-3xi, Thermo Fisher Scientific). Electrochemical impedance was measured using EIS (BioLogic, SP-200) in the frequency range from 7 MHz to 100 mHz.3. Results and discussion
3.1 Energetics of Li electrodeposition
In Li-free ASSBs with a composite anode consisting of a porous interlayer coated on the CC, metallic Li can be plated within the pores of the interlayer, at the SE–porous layer interface, or at the porous layer–CC interface. In some cases, Li deposition can take place at several places simultaneously. The location and morphology of Li deposits in ASSBs are affected by both the thermodynamics of Li electrodeposition and the kinetics of Li movement. In a recent study, a model was proposed to assess the change in Gibbs free energy with the deposition of Li onto a metal CC in Li-free ASSBs in consideration of the volume free energy of Li (i.e., the change in the Gibbs free energy for the reduction of Li ions) and the work associated with the creation of new interfaces and the removal of interface due to delamination following Li deposition.21 The work associated with removal of the interface is related to the adhesion energy of the interfaces involved. However, Li deposition in ASSBs induces stress, and ASSBs are generally under applied pressure, thus these mechanical factors should be included in thermodynamic calculations, as shown in eqn (1):| ΔG = ΔGr − eΦ + Wads,overall + (Pexternal − σLim)ΩLi | (1) |
Here, ΔGr is volume Gibbs free energy change of Li electrodeposition (Li+ + e− → Li), Φ is the overpotential, Wads,overall is the overall work associated with the adhesion energy related to Li deposition, Pexternal is the pressure applied to Li deposits, σLim is the stress of the deposited Li, and ΩLi is the molar volume of the deposited Li. Considering the energetics of Li deposition, Li is preferably deposited in regions where the compressive stress is minimized. Therefore, in the initial stages of deposition, Li is deposited within the free volume of the porous layer if the kinetics support the sufficient movement of Li for Li deposition within the pores. Once the free volume is filled with Li deposits, subsequent Li deposition should open the existing interfaces and form new ones. Because each interface has a different adhesion energy, the overall work associated with this delamination and formation of new interfaces varies depending on the Li deposition site. The overall work associated with adhesion energies for each deposition location can be estimated as follow:
For Li deposition at the interface of the SE and porous layer, delamination of the SE–porous layer interface and the formation of new Li–SE and Li–porous layer interfaces occur:
| Wads,overall = WSE–Li·porous layer = Wads,SE–porous layer − Wads,Li–SE − Wads,Li–porous layer |
For Li deposition at the interface of the porous layer and the CC, delamination of the porous layer–CC interface and the formation of new Li–porous layer and Li–CC interfaces occur:
| Wads,overall = WCC–Li–porous layer = Wads,CC–porous layer − Wads,Li–CC − Wads,Li–porous layer |
Here, Wads is the work of adhesion (i.e., the work required to open the interface) of the individual contacts. Because Li would be deposited where the change in the Gibbs free energy is minimized (and if the kinetics supports this), Li is deposited where Wads,overall is lowest (also if kinetics supports). When we focus on the interfaces being delaminated, the one with lower adhesion work has the lower overall adhesion work. In shorts, when Li deposition requires existing interfaces to be opened, that with the lower adhesion energy is preferred.
To verify that the location of Li deposition is strongly influenced by the energetics, we controlled Wads,SE–porous layer by changing the pressure for forming SE–porous layer interfacial contact in cell fabrication process and observed the change in location of the main Li deposits. A composite anode consisting of carbon spheres (diameter of ∼75 nm) on the CC (thickness of carbon interlayer ∼5 μm) was used, and the Li was plated at a curtailing capacity of 3 mA h cm−2 under uniaxial pressure of 5 MPa (details of the experimental conditions are provided in the following section). First, the interface with lower adhesion energy between SE–porous layer and porous layer–CC interface was identified by peel-off test using sticky tape. Peel-off tests were carried out with the anodes–SE pellet assembly after forming the SE–porous layer interfacial contact by applying a pressure of 5 or 380 MPa. As shown in Fig. S1a–c,† the anode was completely removed from the SE pellet when the SE–porous layer interface was formed at 5 MPa, indicating that SE–porous layer interface has lower adhesion energy than that of porous layer–CC interface (Wads,SE–porous layer < Wads,CC–porous layer). In contrast, when the pressure forming interface increased to 380 MPa, only CC was delaminated leaving the porous layer on the surface of SE pellet (Fig. S1d–f†). This confirmed that SE–porous layer interface has higher adhesion energy than that of porous layer–CC (Wads,SE–porous layer > Wads,CC–porous layer). As we conjectured, Li metal was mainly deposited at the interface with lower adhesion energy (Fig. S2†). Within the cell with high adhesion energy between the SE and the porous interlayer where the SE–porous layer interface was formed at a pressure of 380 MPa, Li metal was mainly deposited at the interface between the porous layer and the CC (Fig. S2a†). In contrast, within the cell with a low adhesion energy between the SE and the porous interlayer where interface was formed at a pressure of 5 MPa, Li metal was deposited at the interface between the SE and the porous layer (Fig. S2b†). This verified that Li metal is preferentially deposited within a composite anode at locations where the Gibbs free energy is minimized. Based on the energetics of Li deposition, we found that the main Li deposition layer occurs between the porous carbon interlayer and the CC if the interfacial contact between the carbon interlayer and the SE is sufficiently strong. In our experiment, when the SE–porous layer contact was formed at a pressure of 380 MPa, the work of adhesion between SE–porous layer is higher than that between the porous layer–CC. However, to pass through the porous carbon interlayer, kinetics should provide sufficient mobility for Li. There can be several factors affecting mobility of Li in the porous anode. In the following section, we clarify the fundamental mechanisms underlying Li movement in a porous anode and the factors that affect Li mobility and determine Li deposition behavior.
3.2 Kinetic factors: Li deposition behavior depending on the pore size of the carbon layer
To study Li deposition behavior into composite anodes depending on the pore size, we synthesized the carbon spheres with different diameters and fabricated a porous anode by layering these spheres. The carbon spheres were fabricated via the carbonization of polymer beads that were synthesized by a sol–gel polymerization method using resorcinol and formaldehyde as precursors. The diameters of the polymer beads were controlled by varying the concentrations of the precursors and the solvent ratio or by adding a triblock copolymer (details of the synthesis process are provided in the Methods section). As shown in Fig. 1a–c and S3a–c,† carbon spheres with diameters of 1 μm (CS1, Fig. 1a and S3a†), 350 nm (CS2, Fig. 1b and S3b†), and 75 nm (CS3, Fig. 1c and S3c†) were synthesized. High-resolution TEM images revealed that all of the carbon spheres were largely amorphous (Fig. S3d–f†). The Raman spectra of the carbon spheres (Fig. 1d) revealed that they had similar ID/IG ratios (0.83, 0.89, and 0.86 for CS1, CS2, and CS3, respectively), confirming that there was no noticeable difference in their defective nature of the carbon spheres. Because all of the carbon spheres were synthesized from the same precursors and had similar surface properties, composite anodes fabricated from the three types of carbon sphere were suitable for the analysis of Li deposition behavior exclusively depending on the pore size. The composite anodes were fabricated by casting the slurry containing the carbon spheres on an SUS CC, and the cast carbon interlayers have thickness of around 5 μm. The pore size distribution of the prepared electrodes was analyzed using mercury porosimetry. As shown in Fig. 1e, the pore diameters decreased as the size of the carbon spheres decreased. The average pore diameters were 433.3, 104.7, and 30.9 nm for CS1, CS2, and CS3, respectively. Furthermore, peel-off tests confirmed that all the porous layers have higher adhesion energy with SE than CC when a pressure of 380 MPa was applied to form SE–porous layer interfacial contact (Fig. S1†). This result means that Li deposition at the porous layer–CC interface is thermodynamically favorable for all the cells with CS anodes. In following experiments, we applied 380 MPa to form strong adhesion between SE and the porous interlayer.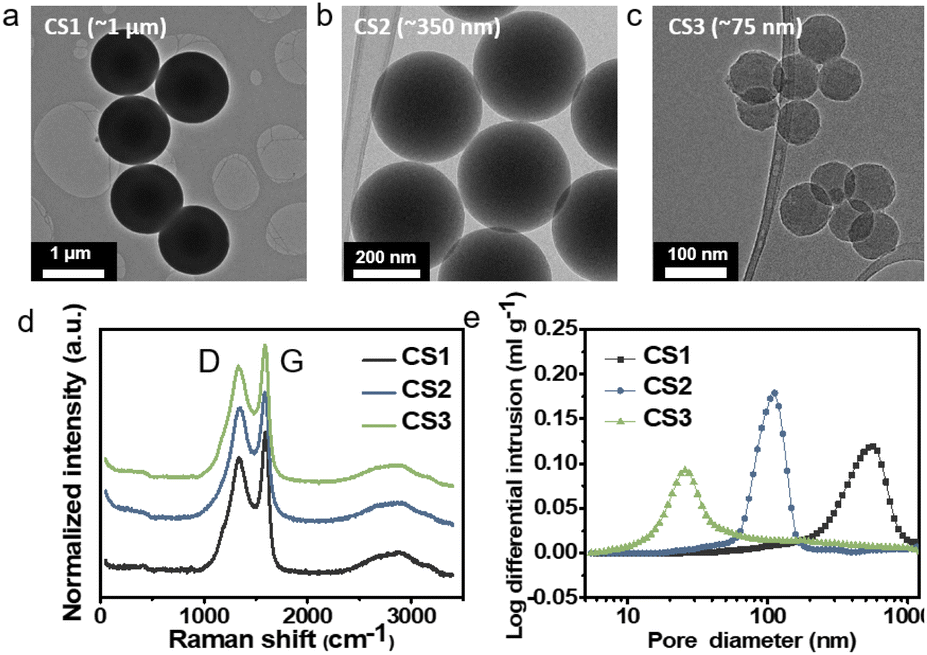 | ||
| Fig. 1 TEM images of (a) CS1, (b) CS2, and (c) CS3. (d) Raman spectra of CS1, CS2, and CS3. (e) Pore size distribution of CS1, CS2, and CS3 electrodes obtained from mercury porosimetry. | ||
To investigate the Li deposition behavior depending on the pore size of the interlayer, Li metal was electrochemically plated onto the composite anodes at a current density of 0.5 mA cm−2 and a curtailing capacity of 3 mA h cm−2 (Fig. S4a†). Lithium thiophosphate (Li3PS4, hereafter LPS) was employed as the SE, and Li metal foil was used as the counter electrode. The cell was operated under a uniaxial pressure of 5 MPa and a temperature of 60 °C. Fig. 2a–c and d–f present cross-sectional and top-view scanning electron microscopy (SEM) images of the cells after Li plating, respectively. The top-view SEM images were obtained after removing the CC as shown in Fig. S4b.† Additional SEM images with boundaries marked for clear distinction and different magnifications are presented in Fig. S5 and S6.†Fig. 2g–i schematically illustrate the morphology of the Li deposits in the CS composite anodes plated to a capacity of 3 mA h cm−2 based on the information obtained from the top-view and cross-sectional SEM images.
 | ||
| Fig. 2 (a–c) Cross-sectional and (d–f) top-view SEM images and (g–i) schematic illustrations of the Li in CS composite anodes deposited to a capacity of 3 mA h cm−2. (a, d, and g) for CS1, (b, e, and h) for CS2, and (c, f, and i) for CS3. Top-view SEM images of Li deposits on the CS3 composite anode at capacities of (j) 0.5 and (k) 1 mA h cm−2. | ||
The location and morphology of the Li deposits varied depending on the pore size of the CS layer. For the cell with the CS1 composite anode, which had the largest average pore size, the Li metal layer was plated between the CS1 layer and the SE (Fig. 2a, S5a and d†), and CS1 layer on the CC side maintained its pristine flat architecture without deformation (Fig. 2d). When voids of the CS1 layer on the CC side were observed in detail (Fig. S6a and b†), most of the voids remained empty, but only a small number of voids were filled with Li deposits (the yellow square marked in Fig. S5a†). For the CS2 composite anode (Fig. 2b), Li deposits grew from SE surface and crawled up the CS2 layer after splitting the CS2 layer (see also Fig. S5b and e†). Some regions of the CS2 layer were thus located below the Li deposits and others above. The top-view SEM image obtained using secondary electron (SE) (Fig. 2e) and backscattered electron (BSE) modes (Fig. S6c†) clearly show that the Li deposits grew toward the CC after splitting the CS2 layer. The carbon appeared brighter compared to the Li in the BSE-mode SEM images. For easier understanding, the Li is colored yellow and the carbon colored grey in the SEM image in Fig. S6d.† In the magnified image of the CS2 layer above the Li deposits on the CC side (Fig. S6e†), only a small proportion of the voids in the CS2 interlayer was filled with Li deposits (the blue square marked in Fig. S6f†) and most voids remained empty, as with CS1.
Unlike the Li deposits with CS1 and CS2, which were in direct contact with the SE, in the CS3 composite anode, the Li metal completely passed through the CS3 layer and was plated between this layer and the CC (Fig. 2c, S5c and f†). The CS3 layer thus blocked direct contact between the main Li layer and the SE. The voids in the CS3 layer on the CC side were completely filled with Li deposits (Fig. S6h†). Only Li deposits were exposed in the top-view SEM image (Fig. 2f). Interestingly, rather than forming a flat surface morphology, the Li deposits exhibited a rough surface with ridge lengths of a few tens of micrometers (the dashed yellow line in Fig. S6g†) and plateaus spreading out from the ridges (the red arrow in Fig. S6g†).
During Li deposition on a composite anode, the carbon spheres electrochemically react with Li ions and become lithiated carbon above 0 V prior to the reduction of the Li ions to metallic Li. The voltage profiles of initial activation cycle prior to the initial Li deposition (current density of 0.2 mA cm−2 from 0 to 1 V) confirmed the lithiation of carbon spheres above 0 V (Fig. S7†). The Li ion diffusivity of lithiated carbon is in the range of 10−11 to 10−6 cm2 s−1 depending on the Li ion diffusion pathway and morphology of carbon materials.22 Compared to the other allying materials such as Si,23,24 Ge25 and Sn26 (10−13 to 10−11, 10−12 to 10−10, and 10−16 to 10−13 cm2 s−1 for lithiated Si, Ge, and Sn, respectively), lithiated carbon shows much higher diffusivity. Thus, some Li ions can be transported through the carbon spheres. However, electron transport through the conductive carbon spheres is much faster than that of Li ions. Therefore, most of the Li ions must be reduced at the interface of SE–carbon layers, and only small proportion of the Li ions may be reduced on the surface of the carbon spheres away from the SE, presumably due to the overpotential. For the three types of carbon composite anode in the present study, we assume that all of the carbon spheres would have similar Li ion diffusivity because they all had the same chemical composition and surface properties. Regardless of the diameter of the spheres, the Li ions were mostly reduced at the interface of the SE–carbon layer. However, the morphology and location of the Li deposits differed depending on the diameter of carbon spheres. We believe that this difference originates from variation in the kinetic behavior and the mobility of Li in pores of different sizes. Because Li movement was enhanced in pores of a smaller size, the mechanism of Li movement in small pores is considered via diffusional creep.27,28
As briefly mentioned in the introduction, previous studies have reported that Li metal was deformed via diffusional Coble creep within carbon tubules with a width of 100 nm during Li deposition and stripping.19 On the other hand, in another study, Li metal was deformed via different mechanisms during uniaxial press tests when the diameter of the Li metal pillar was larger than 0.98 μm.29 These Li pillars were deformed by displacive plasticity, which occurs under higher stress than does Coble creep. However, systematic analysis of the change in deformation mechanisms based on the size of Li metal on a scale of a few hundred nanometers has not yet been reported, presumably due to the challenges associated with the sample preparation, transfer, and testing of Li metal. However, compressive tests have been applied to Sn pillars with diameters from 450 to 130 nm.28 The deformation mechanism for Sn pillars switched from displacive plasticity to diffusional Coble creep when the size of the Sn pillars decreased from 450 to 130 nm. Because Sn has a similar homologous temperature to that of Li (T/TM, 0.66 and 0.60 for Li and Sn, respectively), it is expected that both metals would show similar deformation behavior.20
In Coble creep, the deformation occurs via interfacial diffusion. The strain rate of Li due to a diffusional Coble creep is given by eqn (2),28,30
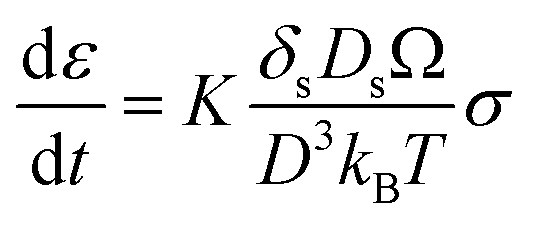 | (2) |
K is the dimensionless constant, δs is the nominal surface layer thickness, Ds is the surface diffusivity of Li, Ω is the atomic volume of Li, kB is the Boltzmann's constant, T is the absolute temperature, σ is the applied stress, and D is the average grain diameter. Considering the previous research that Li electrodeposits within the carbon tubules are single crystal,19 the average grain size of Li deposits in porous anode may be reasonably regarded as the pore size of the anode. Therefore, the strain rate of Li could be inversely proportional to the average pore diameter of the interlayer. Accordingly, plastic deformation via diffusional Coble creep is likely to occur more rapidly when the composite anode has smaller pores. In pores larger than the critical limit, deformation into the pores occurs only when the stress level reaches a level sufficient for displacive plasticity to occur. In the present study, it appeared that the Coble creep was hindered in the CS1 composite anode due to its large pore diameter (average 473.3 nm; see Fig. 1e). Therefore, metallic Li was deposited at the location where the Li ions were reduced. In contrast, in the CS3 composite anode, the Li deposits readily entered the pores due to its high strain rate originating from the small CS3 anode pore diameter. Therefore, Li readily moved from the SE surface to the CC side through the pores in the CS3 layer and was deposited at the interface between the CC and the CS3 layer.
Fig. 2j and k present SEM images of the Li deposits on the CS3 composite anode for two deposition capacities (0.5 mA h cm−2 and 1 mA h cm−2, respectively). With deposition at 0.5 mA h cm−2, the Li deposits started to protrude from the CS3 layer, exhibiting a ridge-like morphology with a width of less than 50 nm covering only a small area of the CS3 layer surface. Because the carbon interlayer was not completely homogeneous in terms of the pore size, Li diffusivity, and pressure, the movement of Li deposits would presumably be facilitated in certain regions of the CS3 layer and emerge from those points. When the deposition capacity was increased to 1 mA h cm−2 (Fig. 2k) and 3 mA h cm−2 (Fig. 2f), the ridge-like Li deposits grew further while maintaining their acute peaks, and Li spreads from the ridge of mountain range. This morphology evolution during Li deposition in the CS3 composite anode is analogous to the formation of a volcanic mountain range, indicating that Li behaves like a fluid within the CS3 composite layer and was deposited between CS3 and the SUS CC mainly via diffusional Coble creep.
For the CS2 composite anode, the inhomogeneity of the pore size, Li diffusivity, and pressure produced a complicated, unique deposition morphology. As described above, Li deposits split the CS2 layer; however, rather than positioning completely within the CS2 layer, some regions of the CS2 layer were located below the Li deposits, while others were above them. The proposed scenario for the formation of this morphology is schematically presented in Fig. S8.† During initial plating, Li may diffuse into some regions of the CS2 layer with higher Li mobility (for example, in areas with a smaller pore size), maintaining the intimate contact between the SE and CS2 layer. However, if the strain rate in the immediate vicinity of this region is too low to allow the Li to advance toward the CC (Fig. S8†) due to inhomogeneity in the CS2 layer (for example, lower Li mobility due to a larger pore size), Li metal will be plated directly on the SE surface and separate the SE–CS2 interface. The region of the CS2 layer where diffusional creep occurred will then be pressed toward the SE by the external pressure, and the region where Li is directly electrodeposited on the surface of SE will be pushed toward the CC by the deposited Li metal. The two stresses applied to the CS2 layer act in opposite directions (Fig. S8b†), acting as a tearing stress; if they are higher than the cohesion energy of the CS2 layer, this layer will be broken up and crevices can form (Fig. S8c†). Once the CS2 layer contains crevices, Li deposits will preferentially grow along the free surface of these crevices to the CC (Fig. S8d†).
To evaluate the relationship between Li deposition behavior and electrochemical performance, half-cells with carbon-based composite anodes were cycled. Fig. 3a presents the initial voltage profiles for Li deposition and stripping at a current density of 0.5 mA cm−2 and a capacity limit of 3 mA h cm−2. The coulombic efficiency (CE) decreased from CS3 to CS1, suggesting that the CE decreased as the contact area between the Li deposits and SE increased. The cell with the CS3 anode, where physical contact between the main Li deposits and the SE was blocked by the carbon layer, produced the highest CE (81.9%), while that with the CS2, where a portion of the Li deposits was in contact with the SE, had a CE of 72.1%. The cell with CS1, in which the entire Li layer was in contact with the SE had a CE of 60.8%. This is presumably because the plated Li metal was consumed by the side reactions that occurred at the interface of SE–Li deposits. In the inset of Fig. 3b, the CS2 cell produced the highest overpotential (112.5 mV) compared to the CS1 and CS3 cells (109.2 and 105.3 mV, respectively), presumably because splitting the CS2 layer requires additional energy to overcome the binding energy of the polymeric binder between the carbon spheres. The CS3 cell had a lower overpotential compared to that of CS1. We ascribed this to the stronger adhesion between the carbon layer and SE than that between the carbon layer and CC, as indicated by the energetics analysis above.
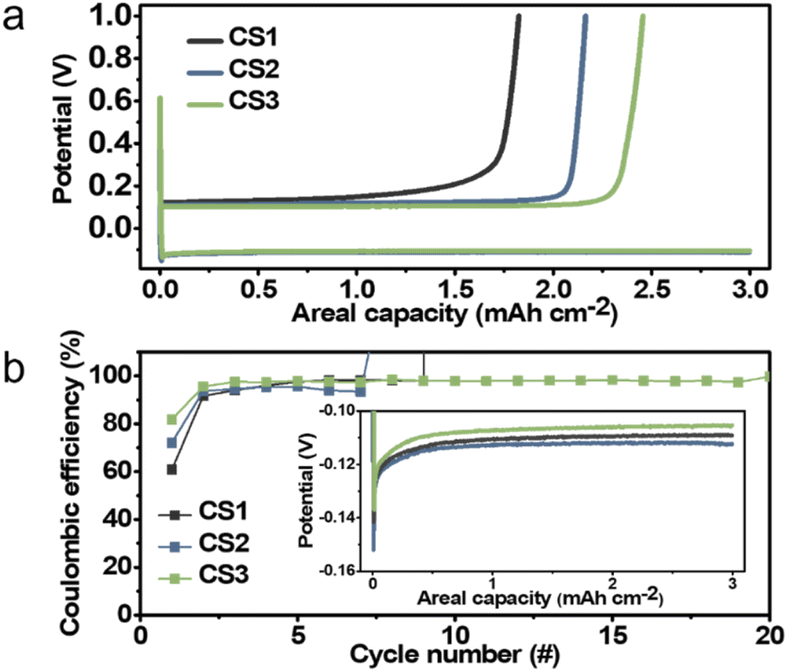 | ||
| Fig. 3 (a) Initial Li deposition and stripping voltage profiles for the CS1, CS2, and CS3 composite anodes. (b) Variation in the coulombic efficiency during repeated Li plating and stripping of the CS1, CS2, and CS3 composite anodes. (Inset) Enlarged voltage profiles of the initial Li deposition. | ||
In the repeated Li deposition/stripping tests, the CS3 cell ran for more than 20 cycles without short-circuiting, whereas the CS1 and CS2 cells failed significantly earlier in their 9th and 7th cycle, respectively. The voltage profiles of CS1 and CS2 cells when short-circuiting occurs are displayed in Fig. S9.† The overpotential suddenly dropped to below 5 mV when short-circuiting occurs. This early failure may be due to the unstable interface and side reactions between the Li and SE. In particular, repeated separation of the interface between the SE and carbon layer and the accumulation of by-products in the CS1 and CS2 cells may have increased the interfacial resistance and led to inhomogeneous Li deposition. This could lead to a higher local current density, eventually resulting in dendritic Li growth into the SE pellet and a short circuit, which is the main cause of early failure. In contrast, the interface between the CS3 interlayer and SE remained stable without separation, and side reactions were suppressed because the CS3 interlayer separates the main Li deposits from the SE. The results of electrochemical impedance spectroscopic (EIS) analysis summarized in Fig. S10† were in line with the cycling test results. All of the cells exhibited similar resistance before cycling (232, 258, and 261 Ω cm2 for CS1, CS2, and CS3, respectively); however, after 5 cycles, CS3 had the lowest resistance (866, 1246, and 666 Ω cm2 for CS1, CS2, and CS3, respectively). The resistance of CS2 cell significantly increased after cycling because the crevice formed within CS2 layer during Li deposition were maintained after Li stripping (Fig. S11b and c†) while CS1 (Fig. S11a†) and CS3 (Fig. S11d†) layer maintained their pristine architecture without cracks. The presence of crevice in the CS2 interlayer remarkably hindered transport pathway and increased resistance compared to that of other CS layers.
3.3 Kinetic factors: temperature-dependent Li deposition behavior in CS3 composite anodes
Metallic Li was plated onto the CS3 composite anode at 45 and 30 °C to observe the effect of temperature on the morphology evolution of the Li deposits. The surface diffusivity of Li is given by eqn (3),31| Ds = 0.014e−6.54Tm/T | (3) |
The morphology of Li plated at 45 °C was similar to that deposited at 60 °C. As shown in Fig. 4a and b (see also Fig. S12a and b†), the entire Li deposit was plated between the CS3 layer and CC with a ridge-like surface morphology on the CC side. This suggests that the diffusion and strain rate at 45 °C was sufficiently high for Li to advance through the pores in the CS3 layer. However, at 30 °C, as shown in Fig. 4c and d (see also Fig. S12c and d†), the morphology of the Li deposits was similar to that of CS2 at 60 °C (Fig. 2b and e). Due to the limited surface diffusion at a low temperature, Li mobility in the CS3 layer would be insufficient to completely penetrate the CS3 layer. Similar to the Li deposition within the CS2 composite anode at 60 °C, the inhomogeneity in the CS3 layer presumably produced local differences in Li mobility and deposition, leading to a morphology similar to that of the CS2 composite anode at 60 °C.
 | ||
| Fig. 4 (a and c) Cross-sectional and (b and d) top-view SEM images of the Li deposited CS3 composite anode plated to a capacity of 3 mA h cm−2 at (a and b) 45 °C and (c and d) 30 °C. | ||
3.4 Kinetic factors: Li deposition onto composite anode composed of other materials
Li mobility through the interlayer not only depends on the temperature but also on the interlayer materials due to the different surface diffusivity of Li on the interlayer materials. To assess the effect of different interlayer materials, Li metal was deposited on composite anodes composed of commercial carbon black SUPER C65 (SC65) with a diameter of around 40 nm (Fig. S13a†), lithium titanate (Li4Ti5O12, LTO) nanopowder with a diameter of around 200 nm (Fig. S14a†), Cu particles with a diameter of around 100 nm (Fig. S15a†), and Ni particles with a diameter of around 300 nm (Fig. S15d†).Because SC65 has a smaller diameter than CS3 (75 nm), even though SC65 carbon is more defective than CS3 with larger ID/IG ratio of 1.06 (Fig. S16†), we expected that diffusional Coble creep would occur in the SC65 composite anode. As shown in Fig. S13b–d,† Li deposits on SC65 exhibited a similar morphology to that of CS3. All of the Li deposits plated at the SC65–CC interface and exhibited a ridge-like morphology. As shown in Fig. S14g† When the Li metal was plated onto the LTO, the Li deposits also exhibited a similar morphology to that of CS3 (Fig. S14b and g†) despite the similar pore diameter of LTO with that of CS2 (Fig. S17,† 94.2 nm for LTO and 104.7 nm for CS2). The voids in the LTO composite anode were filled with Li deposits (Fig. S14c†) and a ridge-like morphology was observed during Li plating (Fig. S14e–g†). However, when Cu and Ni NPs were employed as composite anode components, the Li deposition behavior was quite different from those of CS anodes. Though the average pore diameters of metal interlayers are similar to that of CS3 (Fig. S17,† 42.4, 29.6, and 30.9 nm for Cu, Ni, and CS3, respectively), the horizontal Li layer was plated on the surface of the SE below the metal particle interlayer (Fig. S15b for Cu and Fig. S15e† for Ni). The location and morphology of main Li deposits layers were similar to that of CS1 which has much larger pore diameter than metal interlayer. However, the Li deposition behavior within interlayer was similar to that of CS3, filling the entire voids of metal interlayer (Fig. S15c and f†).
We ascribe this Li deposition behavior to the surface Li mobility on the NPs of the composite layer. Because the Li deposits were in contact with the particles, their diffusion and deformation were significantly influenced by the surface of the anode materials. Carbon and LTO, which are used as electrode active materials in Li-ion batteries, have meaningful Li diffusivity.19,32,33 In particular, the lithiophilic surface of LTO may facilitate surface diffusion of Li, enabling fast diffusional creep along the interface despite its pore size similar to that of CS2. On the other hand, Cu and Ni are relatively inert with respect to Li and have a different crystal structure to that of Li, leading to a large overpotential for Li nucleation on them.35 Therefore, relatively sluggish Li movement was likely on the surface of Cu and Ni, despite similar pore size with CS3. Thus, Li deposits can only advance to the interlayer and further penetration toward CC was hindered due to the suppressed creep.
3.5 Kinetic factors: Li deposition behavior onto a composite anode with Ag-decorated carbon spheres
To further expedite Li movement through the interlayer by facilitating its surface diffusion, we decorated the three types of carbon sphere with highly lithiophilic Ag NPs and denoted the results as Ag–CS1, Ag–CS2, and Ag–CS3. Ag forms an alloy with Li and is soluble in Li (∼9 at%@145.5 °C).34,35 The Ag-decorated carbon spheres were synthesized by the carbonization of Ag-decorated polymer spheres (for more detail, see the Methods section). TEM images (Fig. 5) and corresponding energy dispersive spectrometry (EDS) mapping images (Fig. S18†) showed that Ag NPs with diameters of around 20 nm were evenly embedded on the surface of the carbon spheres. | ||
| Fig. 5 TEM images of (a) Ag–CS1, (b) Ag–CS2, and (c) Ag–CS3. | ||
Li metal was plated onto the Ag–CS composite anodes under the same experimental conditions as used for the CS composite anodes (i.e., a current density of 0.5 mA cm−2 at a capacity of 3 mA h cm−2 with an operating temperature of 60 °C and a uniaxial pressure of 5 MPa). Fig. 6a–f (Fig. 6a–c for SE mode, and Fig. 6d–f for BSE mode) and Fig. 6g–i present cross-sectional and top-view SEM images, respectively, after Li deposition onto the Ag–CS composite anodes. SEM images with marked boundaries and EDS elemental mapping images are also displayed in Fig. S19.† The location of the Li deposits depending on the pore size of Ag–CS composite anodes exhibited a similar trend to that without Ag. Li was plated on the surface of the SE in the cell with the Ag–CS1 composite anode (Fig. 6a, d, g and S19a†). In the cell with Ag–CS2, the Li deposits split the Ag–CS2 layer, crawling from the surface of the SE to the CC side along the crevices in the Ag–CS2 (Fig. 6b, e, h, S19b and S20†). In the Ag–CS3 cell, Li deposits were formed between the Ag–CS3 layer and the CC maintaining the intimate contact between the Ag–CS3 layer and the SE (Fig. 6c, f, i, and S19c†).
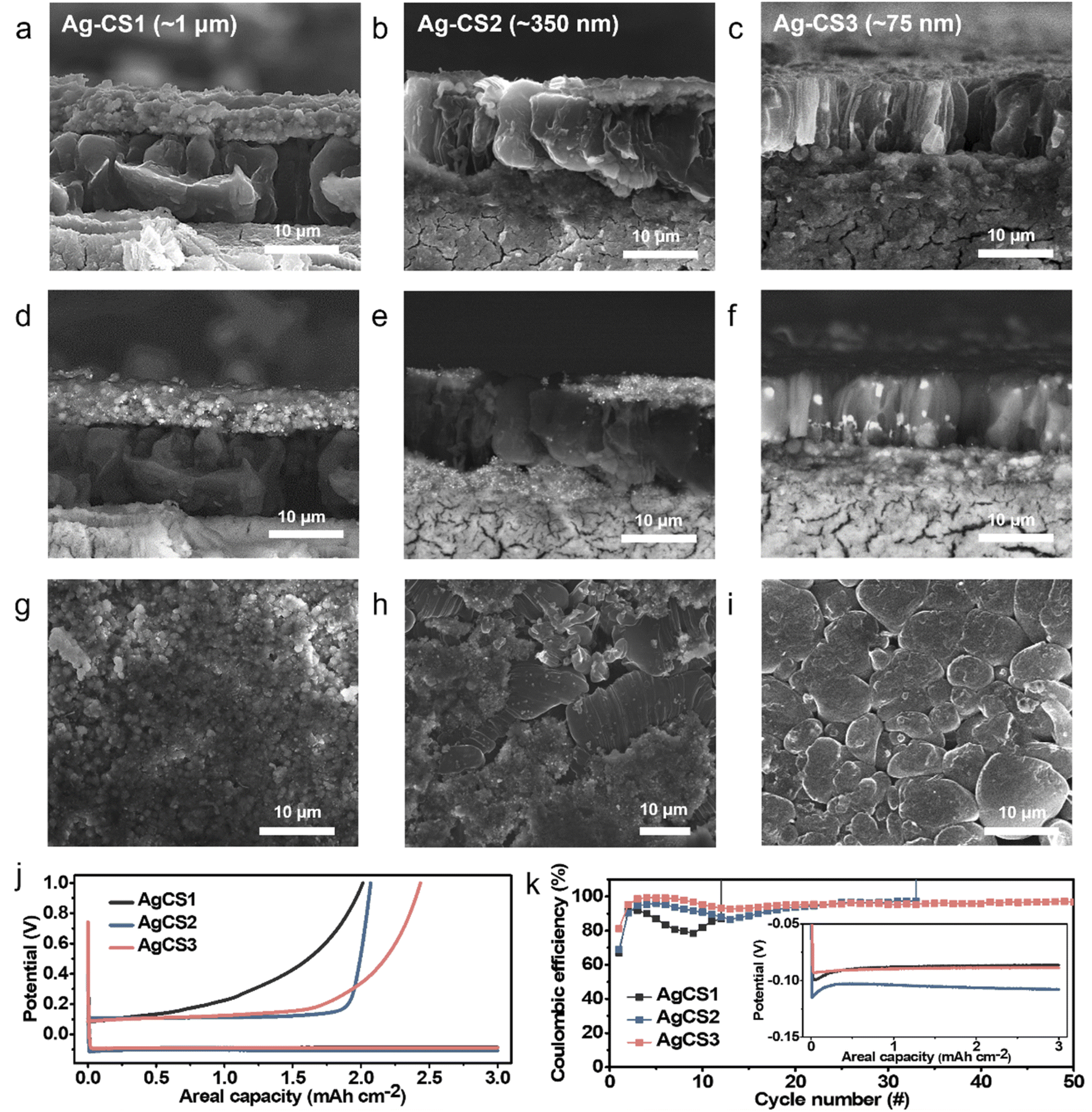 | ||
| Fig. 6 Cross-sectional SEM images obtained from (a–c) secondary electron mode and (d–f) backscattered electron mode, and (g–i) top-view SEM images of Li deposited Ag–CS composite anodes plated to a capacity of 3 mA h cm−2: (a, d and g) Ag–CS1, (b, e, and h) Ag–CS2, and (c, f, and i) CS3, respectively. (j) Initial Li deposition and stripping voltage profiles for the Ag–CS composite anodes. (k) Variation in the coulombic efficiency during the repeated Li plating and stripping of the Ag–CS composite anodes. (Inset) Enlarged voltage profiles for the initial Li deposition. | ||
However, the microstructure of the Li deposits in the Ag–CS composite anodes demonstrated clear differences with that of the CS composite anodes. In contrast to the CS1 and CS2 cells, in which most of the voids in the carbon layer remained empty after Li deposition, all of the voids in the Ag–CS1 and Ag–CS2 composite layers were filled with Li deposits (Fig. S21a and b†). Furthermore, for all Ag–CS cells, the Li deposits exhibited a vertically aligned rod-shaped morphology with a diameter of 5–10 μm, which is notably different from the single cluster Li deposits in the CS composite anodes. In particular, within the Ag–CS3 cell, the Li deposits were densely packed and had an even thickness, while the CS3 cell had a ridge-and-plateau morphology and an inhomogeneous plane distribution (Fig. 2c and i). The distribution of the Ag differed depending on the pore size. In the BSE-mode SEM images (Fig. 6d–f), the Ag NPs appeared to be much brighter compared to the Li and carbon. Within the Ag–CS1 and Ag–CS2 cells, Ag particles were only observed in the Ag–CS interlayer, not in the main Li deposits. However, according to the corresponding EDS mapping images in Fig. S19a and b,† Ag was detected in both the Ag–CS interlayer and Li deposits, but the intensity in the Li deposits was significantly lower than in the interlayer. It seems that only a limited amount of Ag had dissolved and diffused into the Li deposits from the interlayer in the Ag–CS1 and CS2 anodes. However, in the Ag–CS3 cell, Ag particles were observed in both the Ag–CS3 interlayer and Li deposits in BSE-mode SEM analysis (Fig. 6f). In addition, Ag was uniformly detected throughout the Li deposits and Ag–CS3 interlayer in the EDS mapping images (Fig. S19c†). The boundary between the Li deposits and Ag–CS3 was not clear in the EDS mapping images of Ag. Similar to the CS3 interlayer, it was assumed that Li moved through the Ag–CS3 interlayer via diffusional Coble creep.
The distinctive Li deposition behavior in the Ag–CS composite anodes compared to that in the CS anodes was attributed to the solubility of Ag within Li and the highly lithiophilic surface of Ag and Li–Ag alloys. It has been widely reported that these features strongly facilitate Li deposition kinetics by reducing the nucleation overpotential,35–37 boosting Li adatom diffusion,38 and reducing the exchange current density.39 Due to the synergistic effect of increased diffusivity and decreased exchange current density, Li can diffuse further before deposition and establish a more thermodynamically stable morphology. The improved Li diffusivity facilitates Li movement within the Ag–CS1 and Ag–CS2 composite layers, which was impossible within the CS1 and CS2 layers due to their large pores. As a result, Li can fill the empty voids within the Ag–CS1 and Ag–CS2 composite layers. We also concluded that the rod-shaped morphology of the Li deposits originated from preferential Li nucleation around the Ag particles. The reduced nucleation overpotential for Li on the Ag surface would allow preferential Li nucleation around the Ag particles, and higher Li adatom diffusion would generate a thermodynamically more stable uniform and rounded morphology. The Ag particle presumably worked as a seed for Li growth, and Li rods could vertically grow from it. The morphology of the Li deposits in the Ag–CS3 composite anode at the lower plating capacity of 1 mA h cm−2 supported the proposed Li growth mechanism. As shown in Fig. S22a–d,† the Li deposits had a spherical morphology after Li deposition of 1 mA h cm−2. The Ag particles appeared brighter compared to Li in the SEM images obtained in BSE mode. In the enlarged SEM images of the small Li particles with a diameter of hundreds of nanometers (Fig. S22h and i† corresponding to the blue square marked in Fig. S22e and f,† respectively), the Li deposits were wrapped around the Ag particles. This suggests that Li was initially grown on the Ag particles forming rounded, spherical particles and developed into vertically aligned rods as deposition continued. When SC65 carbon was decorated with Ag particles, it demonstrated similar results to those for Ag–CS3 (Fig. S23†).
The electrochemical performance of the Ag–CS3 composite anodes was assessed using repeated Li deposition and stripping tests. The Ag–CS composite anodes were tested under the same experimental process as that employed for the CS composite anode. After the first Li deposition–stripping cycle, the CE of Ag–CS1, Ag–CS2, and Ag–CS3 was 67.1, 69.0, and 81.2%, respectively (Fig. 6j). Similar to the CS composite anodes, CE increased as the contact area between the main Li deposits and SE decreased. Consistent with previous reports,35–37 Ag incorporation significantly reduced the nucleation overpotential. The Ag–CS1, Ag–CS2, and Ag–CS3 anodes had a nucleation overpotential of 12.9, 12.2, and 4.3 mV, respectively (inset of Fig. 6k), while CS1, CS2, and CS3 had 32.4, 39.5, 31.7 mV, respectively (inset of Fig. 4b). In particular, the cell with the Ag–CS3 composite anode had a notably lower nucleation overpotential because the effect of Ag on Li nucleation was maximized in Ag–CS3 due to Li movement through the interlayer containing Ag via Coble creep. In the cycling tests, all of the cells employing a Ag–CS composite anode operated for a higher number of cycles before short-circuiting compared to the cells without Ag (Fig. 6k). Of the Ag–CS composite anodes, Ag–CS3 produced the most stable performance, reaching more than 50 cycles with average CE of 96.0% without short-circuiting. EIS analysis confirmed that the incorporation of Ag NPs in the composite anode suppressed the increase in resistance during the cycles (Fig. S24†). The resistance increased by 10.0, 62.3, and 2.0% for Ag–CS1, Ag–CS2, and Ag–CS3, respectively, after 5 cycles compared to the pristine state. These increases were significantly lower than those for the composite anodes without Ag (371, 483, and 255% for CS1, CS2, and CS3, respectively). Furthermore, after 10 cycles of repeated Li deposition and stripping, Ag–CS3 had a lower resistance than Ag–CS1 and Ag–CS2 (323, 526, and 258 Ω cm2 for Ag–CS1, Ag–CS2, and Ag–CS3, respectively).
4. Conclusions
In this study, the factors determining the morphology and location of Li deposits in Li-free ASSBs with a porous interlayer on the CC were identified by analyzing the Li deposition behavior. To suppress undesirable side reactions between the SE and deposited Li, the main Li deposit layer at the porous interlayer–CC interface is preferable to that at the SE–porous interlayer interface. The location and morphology of the Li deposits in the ASSBs with a porous interlayer on the CC are affected by both the thermodynamics of Li electrodeposition and the kinetics of Li movement. Based on our analysis of the energetics of Li electrodeposition, we found that the interfacial energy is one factor determining the location of the main Li deposits in ASSBs with a porous interlayer on the CC. In our system, the work of adhesion between the SE and the porous interlayer at a pressure of 380 MPa was higher than that for the porous layer and CC; consequently, the main Li layer was deposited at the porous layer–CC interface. However, deposition at the porous layer–CC interface requires the sufficient mobility of Li (either as ions or atoms) from the SE through the interlayer.We thus explored several kinetic factors in this study, including the pore size of the composite anode, temperature, scaffold materials, and surface modification with Ag NPs. Based on the observed Li deposition behavior depending on the pore size of the interlayer, diffusional Coble creep was identified as the mechanism for Li movement through the porous layer. Therefore, Li movement was facilitated by the presence of smaller pores, higher temperatures, and lithiophilic scaffold materials. We proposed a scenario to interpret the Li deposition behavior depending on the pore size based on diffusive creep, inhomogeneity in the porous interlayer, and the resulting stress within the interlayer. The main Li deposit layer was observed at the SE–porous layer interface for the interlayer with the smallest average pore size (CS3), while the interlayers with larger pore sizes exhibited partial or complete contact between the deposited Li and the SE layer. As a result, the CS3 cell exhibited the most improved cyclability during repeated Li deposition and stripping. Ag NPs coated on the surface of the carbon spheres within the composite anode provided thermodynamically stable Li deposition sites and facilitated Li diffusion due to the highly lithiophilic nature of Ag and Li–Ag alloys. The Ag (and alloys) behaved as seeds for Li growth. In particular, within the Ag–CS3 cell, denser and more homogeneous Li deposits were produced compared to that without Ag. The synergistic effects of homogeneous Li deposition and stable anode/SE interfaces led to stable Li deposition and stripping.
This work extends our fundamental understanding of the Li deposition mechanisms in Li-free ASSBs, especially those with a porous interlayer, and proposes a thermodynamic and kinetic interpretation of the factors determining the location and morphology of Li deposits. These results provide useful guidelines for the development of highly reversible anodes for ASSBs.
Author contributions
S. H. P.: conceptualization, methodology, formal analysis, data curation, investigation writing – original draft, writing – review & editing. D. J.: conceptualization, methodology, formal analysis, data curation, investigation, writing – review & editing. J. E. J.: investigation, methodology, formal analysis. S. G. L.: investigation, methodology, formal analysis. G. H. L.: methodology, investigation. Y. J. L.: supervision, project administration, conceptualization, methodology, writing – review & editing, funding acquisition. S. H. P. and D. Y. J. equally contributed to this work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Basic Science Research Program of the National Research Foundation of Korea (NRF), Korean Ministry of Science & ICT (MSIT) (Grant No. NRF-2021R1A2C2006170), and the Engineering Research Center of Excellence (ERC) Program supported by the NRF, MSIT (Grant No. NRF-2017R1A5A1014708). SEM, TEM, and Raman spectroscopy analyses were conducted at the Hanyang LINC+ Analytical Equipment Center (Seoul).References
- C. Monroe and J. Newman, J. Electrochem. Soc., 2003, 510, A1377 CrossRef.
- G. Liu, W. Weng, Z. Zhang, L. Wu, J. Yang and X. Yao, Nano Lett., 2020, 20, 6660–6665 CrossRef CAS PubMed.
- Y.-G. Lee, S. Fujiki, C. Jung, N. Suzuki, N. Yashiro, R. Omoda, D.-S. Ko, T. Shiratsuchi, T. Sugimoto and S. Ryu, Nat. Energy, 2020, 5, 299–308 CrossRef CAS.
- N. Suzuki, N. Yashiro, S. Fujiki, R. Omoda, T. Shiratsuchi, T. Watanabe and Y. Aihara, Adv. Energy Sustainability Res., 2021, 2, 2100066 CrossRef.
- P. Oh, J. Yun, J. H. Choi, K. S. Saqib, T. J. Embleton, S. Park, C. Lee, J. Ali, K. Ko and J. Cho, Angew. Chem., Int. Ed., 2022, 61, e202201249 CAS.
- S. H. Park, D. Jun, G. H. Lee, S. G. Lee and Y. J. Lee, J. Mater. Chem. A, 2021, 9, 14656 RSC.
- H. Wang, Y. Liu, Y. Li and Y. Cui, Electrochem. Energy Rev., 2019, 2, 509–517 CrossRef CAS.
- Z. Xie, Z. Wu, X. An, X. Yue, J. Wang, A. Abudula and G. Guan, Energy Storage Mater., 2020, 32, 386–401 CrossRef.
- J. Wu, L. Shen, Z. Zhang, G. Liu, Z. Wang, D. Zhou, H. Wan, X. Xu and X. Yao, Electrochem. Energy Rev., 2021, 4, 101–135 CrossRef CAS.
- J. Wu, S. Liu, F. Han, X. Yao and C. Wang, Adv. Mater., 2020, 33, 2000751 CrossRef.
- Z. Liu, D. Guo, W. Fan, F. Xu and X. Yao, ACS Mater. Lett., 2022, 4, 1516–1522 CrossRef CAS.
- T. Krauskopf, R. Dippel, H. Hartmann, K. Peppler, B. Mogwitz, F. H. Richter, W. G. Zeier and J. Janek, Joule, 2019, 3, 2030–2049 CrossRef CAS.
- P. Barai, A. T. Ngo, B. Narayanan, K. Higa, L. A. Curtiss and V. Srinivasan, J. Electrochem. Soc., 2020, 167, 100537 CrossRef CAS.
- M. Nagao, A. Hayashi, M. Tatsumisago, T. Kanetsuku, T. Tsuda and S. Kuwabata, Phys. Chem. Chem. Phys., 2013, 15, 18600–18606 RSC.
- X. Xing, Y. Li, S. Wang, H. Liu, Z. Wu, S. Yu, J. Holoubek, H. Zhou and P. Liu, ACS Energy Lett., 2021, 6, 1831–1838 CrossRef CAS.
- S. Xu, D. W. McOwen, C. Wang, L. Zhang, W. Luo, C. Chen, Y. Li, Y. Gong, J. Dai, Y. Kuang, C. Yang, T. R. Hamann, E. D. Wachsman and L. Hu, Nano Lett., 2018, 18, 3926–3933 CrossRef CAS PubMed.
- C. Yang, L. Zhang, B. Liu, S. Xu, T. Hamann, D. McOwen, J. Dai, W. Luo, Y. Gong, E. D. Wachsman and L. Hu, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 3770–3775 CrossRef CAS PubMed.
- D. Xie, S. Chen, Z. Zhang, J. Ren, L. Yao, L. Wu, X. Yao and X. Xu, J. Power Sources, 2018, 389, 140–147 CrossRef CAS.
- Y. Chen, Z. Wang, X. Li, X. Yao, C. Wang, Y. Li, W. Xue, D. Yu, S. Y. Kim, F. Yang, A. Kushima, G. Zhang, H. Huang, N. Wu, Y.-W. Mai, J. B. Goodenough and J. Li, Nature, 2020, 578, 251–255 CrossRef CAS.
- Z. Wang, X. Li, Y. Chen, K. Pei, Y.-W. Mai, S. Zhang and J. Li, Chem, 2020, 6, 11 Search PubMed.
- M. J. Wang, E. Carmona, A. Gupta, P. Albertus and J. Sakamoto, Nat. Commun., 2020, 11, 1–9 CrossRef.
- K. Persson, V. A. Sethuraman, L. J. Hardwick, Y. Hinuma, Y. S. Meng, A. van der Ven, V. Srinivasan, R. Kostecki and G. Ceder, J. Phys. Chem. Lett., 2010, 1, 1176 CrossRef CAS.
- N. Ding, J. Xu, Y. X. Yao, G. Wegner, X. Fang, C. H. Chen and I. Lieberwirth, Solid State Ionics, 2009, 180, 222 CrossRef CAS.
- J. Xie, N. Imanishi, T. Zhang, A. Hirano, Y. Takeda and O. Yamamoto, Mater. Chem. Phys., 2010, 120, 421 CrossRef CAS.
- B. Laforge, L. Levan-Jodin, R. Salot and A. Billard, J. Electrochem. Soc., 2008, 155, A181 CrossRef CAS.
- J. Xie, N. Imanishi, A. Hirano, Y. Takeda, O. Yamamoto, X. B. Zhao and G. S. Cao, Solid State Ionics, 2010, 181, 1611 CrossRef CAS.
- S. H. Park, D. Jun, G. H. Lee, S. G. Lee, J. E. Jung, K. Y. Bae, S. Son and Y. J. Lee, Adv. Sci., 2020, 2203130 Search PubMed.
- L. Tian, J. Li, J. Sun, E. Ma and Z.-W. Shan, Sci. Rep., 2013, 3, 2113 CrossRef PubMed.
- C. Xu, Z. Ahmad, A. Aryanfar, V. Viswanathan and J. R. Greer, Proc. Natl. Acad. Sci., 2017, 114, 57–61 CrossRef CAS.
- R. L. Coble, J. Appl. Phys., 1963, 34, 1697 CrossRef.
- D.-G. Xie, Z.-J. Wang, J. Sun, J. Li, E. Ma and Z.-W. Shan, Nat. Commun., 2015, 14, 899–903 CAS.
- B. Yan, M. Li, X. Li, Z. Bai, J. Yang, D. Xiong and D. Li, J. Mater. Chem. A, 2015, 3, 11773–11781 RSC.
- M. Wilkening, R. Amade, W. Iwaniak and P. Heitjans, Phys. Chem. Chem. Phys., 2007, 9, 1239–1246 RSC.
- J. Gu, C. Shen, Z. Fang, J. Yu, Y. Zheng, Z. Tian, L. Shao, X. Li and K. Xie, Front. Chem., 2019, 7, 572 CrossRef CAS PubMed.
- K. Yan, Z. Lu, H.-W. Lee, F. Xiong, P.-C. Hsu, Y. Li, J. Zhao, S. Chu and Y. Cui, Nat. Energy, 2016, 1, 1–8 Search PubMed.
- S. Kim, C. Jung, H. Kim, K. E. Thomas-Alyea, G. Yoon, B. Kim, M. E. Badding, Z. Song, J. Chang, J. Kim, D. Im and K. Kang, Adv. Energy Mater., 2020, 10, 1903993 CrossRef CAS.
- S. Zhang, G. Yang, Z. Liu, S. Weng, X. Li, X. Wang, Y. Gao, Z. Wang and L. Chen, ACS Energy Lett., 2021, 6, 4118–4126 CrossRef CAS.
- I. T. Røe and S. K. Schnell, J. Mater. Chem. A, 2021, 9, 11042–11048 RSC.
- Y. Liu, X. Xu, M. Sadd, O. O. Kapitanova, V. A. Krivchenko, J. Ban, J. Wang, X. Jiao, Z. Song and J. Song, Adv. Sci., 2021, 8, 2003301 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ta06466a |
| ‡ S. H. P. and D. Y. J. equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2022 |