
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanoresponsive diselenide-crosslinked microgels with programmed ultrasound-triggered degradation and radical scavenging ability for protein protection†
Tetiana
Kharandiuk
 *ab,
Kok Hui
Tan
ab,
Wenjing
Xu
ab,
Fabian
Weitenhagen
ab,
Susanne
Braun
ab,
Robert
Göstl
a and
Andrij
Pich
*abc
*ab,
Kok Hui
Tan
ab,
Wenjing
Xu
ab,
Fabian
Weitenhagen
ab,
Susanne
Braun
ab,
Robert
Göstl
a and
Andrij
Pich
*abc
aDWI – Leibniz Institute for Interactive Materials, Forckenbeckstr. 50, 52056 Aachen, Germany. E-mail: pich@dwi.rwth-aachen.de; kharandiuk@dwi.rwth-aachen.de
bInstitute of Technical and Macromolecular Chemistry, RWTH Aachen University, Worringerweg 1, 52074 Aachen, Germany
cAachen Maastricht Institute for Biobased Materials (AMIBM), Maastricht University, Brightlands Chemelot Campus, Urmonderbaan 22, 6167 RD Geleen, The Netherlands
First published on 22nd August 2022
Abstract
In the context of controlled delivery and release, proteins constitute a delicate class of cargo requiring advanced delivery platforms and protection. We here show that mechanoresponsive diselenide-crosslinked microgels undergo controlled ultrasound-triggered degradation in aqueous solution for the release of proteins. Simultaneously, the proteins are protected from chemical and conformational damage by the microgels, which disintegrate to water-soluble polymer chains upon sonication. The degradation process is controlled by the amount of diselenide crosslinks, the temperature, and the sonication amplitude. We demonstrate that the ultrasound-mediated cleavage of diselenide bonds in these microgels facilitates the release and activates latent functionality preventing the oxidation and denaturation of the encapsulated proteins (cytochrome C and myoglobin) opening new application possibilities in the targeted delivery of biomacromolecules.
1 Introduction
Ultrasound-based technologies are clinically established for their use in diagnostic and therapeutic medical applications1 and can be applied locally with a sufficient penetration depth in biological tissues. The FDA approves modes for ultrasound application in medical therapies with frequencies between 20 kHz and 4.6 MHz.2 Its clinical abundance and instrumental simplicity have rendered ultrasound an attractive stimulus for the development of drug delivery applications, such as rooted in polymer mechanochemistry (sonopharmacology)3 or the remote-controlled degradation of drug carriers.4 Recently, also photoacoustic effects induced by pulsed lasers have been used for drug delivery and therapeutic purposes.5,6 In this context, protein therapeutics constitute a particularly delicate cargo.7 Ultrasound may affect the chemical structure and architecture of proteins due to the formation of reactive oxygen species (ROS) and local “hot spots” that lead to denaturing.8 In addition, ultrasound-induced shear forces rapidly obliterate the secondary structure by force-induced unfolding if the protein is not specifically engineered to withstand this.9 This may have serious implications not only for the delivery of functional protein payloads, but also for important physiological processes and the local biological environment. Therefore, the establishment of new ultrasound-triggered drug delivery concepts requires the optimization and adaption of the drug carriers to ensure programmed release of the payload combined with the reduced risk of damage for both biomacromolecules and cells.Responsive microgels are soft, porous network structures made of crosslinked polymers, which form stable colloidal dispersions that are stable in aqueous solutions.10–13 They can be customized by modifying the network structure or functional groups to respond to external stimuli, such as pH,14 temperature,15 light,16 redox agents,17 or ionic strength.18 These features render them suitable materials for many applications in catalysis,19 filtration and separation,20 sensors,21 antibacterial coatings,22 and drug delivery.23 Microgels based on poly(N-vinylcaprolactam) (PVCL) are popular due to their chemical stability, biocompatibility, and temperature-responsive properties with a Volume Phase Transition Temperature (VPTT) close to physiological 32 °C rendering them an ideal material for biomedical applications.24–28 Importantly for drug delivery applications, they can be tailored to exhibit stimuli-triggered degradation.29,30 The most straightforward strategy to achieve this is the incorporation of degradable crosslinkers that yield water-soluble polymer chains of small hydrodynamic size after degradation, which are safely removed by renal clearance.30,31 Therefore, crosslinkers containing cleavable disulfides,32 esters,33 hydrazines,34 or host–guest complexes35 were successfully incorporated into microgels inducing degradation by pH, enzymes, or redox triggers, such as H2O2 or glutathione (GSH). Among these stimuli,32,33,36–39 the ultrasound-induced degradation of microgels is not well explored. Up to today, only few examples have been reported that uses shear force in solution to cleave force-responsive molecular motifs (mechanophores) with PVCL-based microgels crosslinked by fluorogenic Diels–Alder adducts.40–42 These works revealed the surprisingly facile mechanical degradability of microgels through force-induced covalent bond scission.43–46 Yet, microgels have not been used to activate other latent mechanochemical functionalities by force.
Selenium (Se) is gaining popularity in research due to its unique redox properties and formation of dynamic chemical bonds.47,48 Se has been utilized in its molecular form49 or incorporated into polymer structures50 to explore, e.g., catalysis,51 drug delivery,52 antioxidants,53 and anticancer properties.54 Notably, the thermal bond dissociation energy of the Se–Se bond (172 kJ mol−1) is considerably lower than that of the C–C bond (346 kJ mol−1), yet high enough to be stable under ambient conditions.55 The degradation of diselenide bonds is commonly triggered by redox processes52 and light.56 However, the mechanically induced cleavage of the diselenide bond has received little attention up to now,57,58 though it can be regarded as homologous to the established disulfide mechanophore.3,59–63 Degradable crosslinkers containing non-covalent bonds, such as based on H-bonding, π–π stacking, or van der Waals interactions, could be interesting alternatives for covalent crosslinkers, since they exhibit even lower dissociation energies (100, 50, and 5 kJ mol−1 respectively)64 and can provide higher sensitivity to sonication treatment, thus, they can be used at milder conditions. However, non-covalent bonds have lower stability at ambient conditions, which limits the applicability of such microgels. Therefore, SeSe covalent crosslinkers provide higher stability at ambient conditions while having a lower bond dissociation energy (172 kJ mol−1)55 compared to conventional SS crosslinkers (240 kJ mol−1).65
Here we demonstrate for the first time that the integration of mechanoresponsive diselenide crosslinks into microgels allows synthesizing colloidal carriers with dual function: ultrasound-triggered release and protection of proteins. The biomedical advantage of diselenide groups is their ability to undergo transformation to seleninic acid groups upon homolytic cleavage and oxidation in aqueous solutions. These chemical groups are capable of scavenging free radicals and thus reduce the risk of oxidation and tissue damage.
2 Results and discussion
A series of PVCL-based microgels with integrated SeSe crosslinks was synthesized using a diselenide crosslinker (see ESI† for synthesis and characterization details) through precipitation polymerization in a H2O/DMSO mixture using 2,2′-azobis(2-methylpropionamidine) dihydrochloride (AAPH) as initiator and cetyltrimethylammonium bromide (CTAB) as surfactant (Fig. 1). DMSO was used to enhance the solubility of the hydrophobic crosslinker. To investigate the influence of diselenide content on the degradation process under ultrasonication, four microgel batches, varied in diselenide content (0.5, 1.5, 3.0, and 5.0 mol%) were synthesized. In addition, three microgel batches with fixed diselenide content (1.5 mol%) were synthesized with varied CTAB contents (0.1, 0.5, and 1.0 wt%) to study the effect of microgel size on the mechanochemical degradation. As a control, PVCL microgels with only N,N′-methylenebis(acrylamide) (BIS) crosslinker (1.5 mol%) were also synthesized.The synthesized SeSe-crosslinked PVCL microgels (SeμG) were characterized by Raman spectroscopy and dynamic light scattering (DLS) (Fig. 1). After the incorporation of the SeSe crosslinker into the PVCL microgels, an increase in the intensity of the diselenide Raman band at 290 cm−1 was observed (Fig. 1b and c). DLS measurements indicated that RH increased with a higher content of diselenide crosslinking. Increasing the crosslinker concentration in the reaction mixture increased the incorporation efficacy of the precipitated polymer chains into growing microgels and finally caused the larger sizes. Expectedly, RH reduced with a higher amount of CTAB due to more efficient stabilization and increased number of microgel nuclei resulting in smaller sizes of the final microgel particles. Electrophoretic mobility (EM) measurements were conducted to examine the surface charge of the SeμG (Fig. S1†). All microgels showed positive EM values due to the positively-charged AAPH used in the synthesis of the microgels. Moreover, all Se1.5 microgels synthesized using CTAB showed EM values that are close to the EM value of Se1.5 without CTAB indicating that the free surfactant was successfully removed after purification by dialysis against water.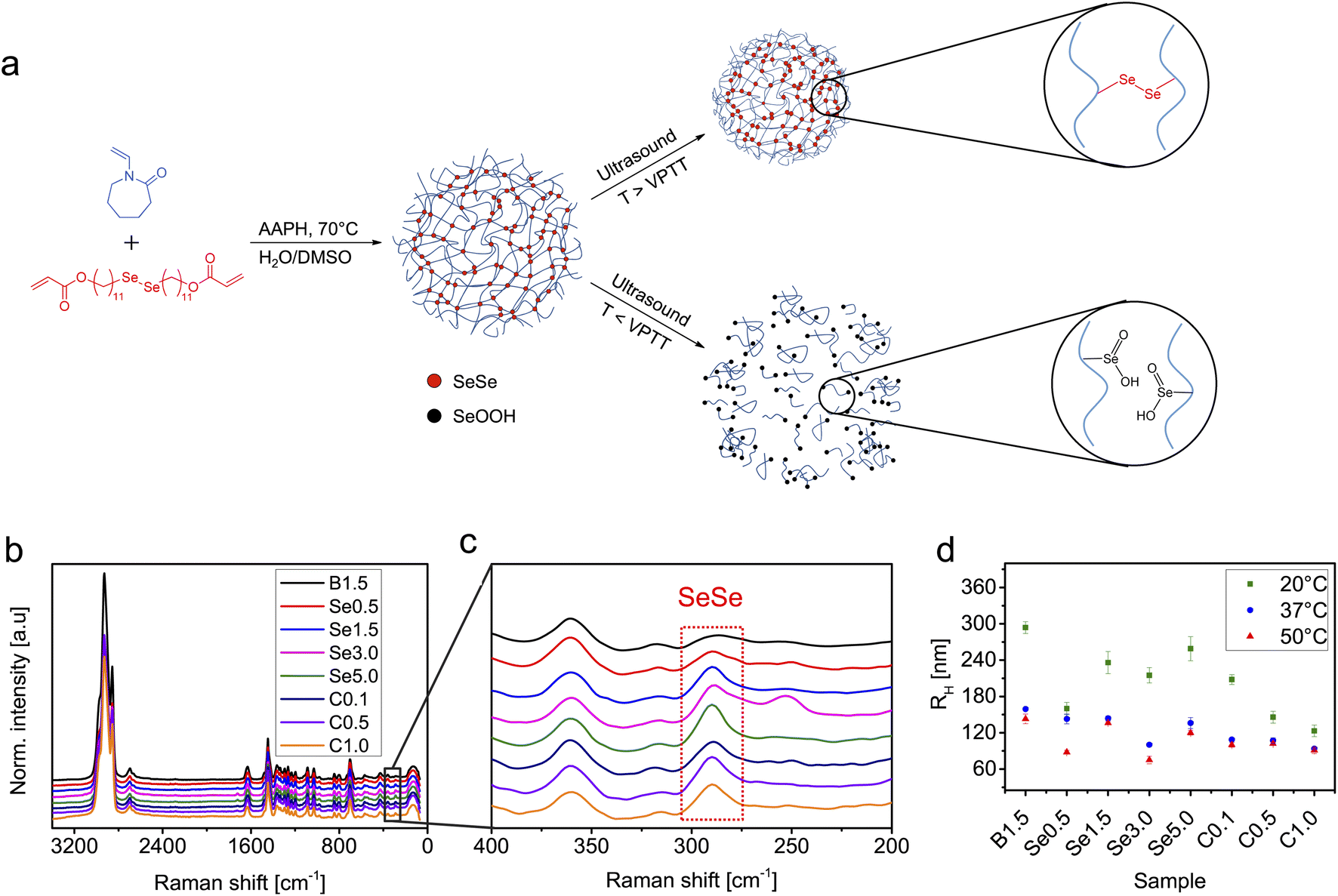 | ||
Fig. 1 (a) Synthesis of diselenide-crosslinked PVCL microgels (SeμG) by precipitation polymerization. Upon ultrasonication at a temperature above the VPTT, SeμG structure remains intact whilst at a temperature below the VPTT the microgel network is destroyed and diselenide crosslinks are transformed into seleninic acid side groups included into water-soluble PVCL chains. (b) Raman spectra, (c) the inset between 400 and 200 cm−1 of (b), and (d) hydrodynamic radii (RH) of SeSe-crosslinked PVCL microgels. B: BIS content in mol%; Se: SeSe content in mol%; C: CTAB content in wt%. Raman spectra were normalized to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O signal of PVCL at 1630 cm−1. O signal of PVCL at 1630 cm−1. | ||
In a proof of concept degradation experiment, PVCL microgels with either BIS crosslinker (1.5 mol%) or SeSe crosslinker (1.5 mol%) were treated with ultrasound using an immersion probe sonicator at 20 kHz in an ice bath at 30% amplitude for 10 min. In the case of B1.5, the microgel solution remains turbid but a clear solution was obtained for Se1.5 (Fig. 2). This indicated that the microgel structure of B1.5 remained almost intact but in the case of Se1.5, the microgel had completely degraded to a water-soluble polymer.
 | ||
| Fig. 2 Photographs before and after 10 min ultrasonication at 30% amplitude and 20 kHz in an ice bath of (a) B1.5 and (b) Se1.5 microgels. STEM images of (c) B1.5 and (d) Se1.5 microgels after ultrasonication. | ||
Further investigations on the ultrasound-triggered degradation of the SeμG were conducted with samples exhibiting variable SeSe contents, sizes, as well as sonication parameters. Fig. 3 and S2† depict the DLS results for the microgels with different SeSe contents and sizes treated with ultrasound for 10 min. Microgel samples were analyzed directly after sonication (Fig. S2†) and after dialysis (Fig. 3). Dialysis allowed to remove polymer chains and their aggregates, formed as a result of microgel degradation, and to determine the size of intact microgels, that remained after sonication without depletion interactions. The RH of microgels with different diselenide contents gradually decreased along with the sonication time with no clear trend observed (Fig. S2a†). After dialysis of the sonicated solutions, the resulting data were more easily interpreted. The RH values decreased significantly after ultrasonication for 1 min (Fig. 3a). This was caused by the absence of depletion interactions of smaller microgels and agglomeration provoked by the increasing amount of short polymer chains produced.40 The ultrasound power intensity IP was controlled by adjusting the amplitude. In Fig. S2b and 3b† Se1.5 microgels were sonicated in an ice bath at 20, 30, and 40% amplitude. The degradation increased with a higher amplitude as indicated by the reduction of RH. After dialysis, RH decreased significantly after 1 min sonication (Fig. 3b).
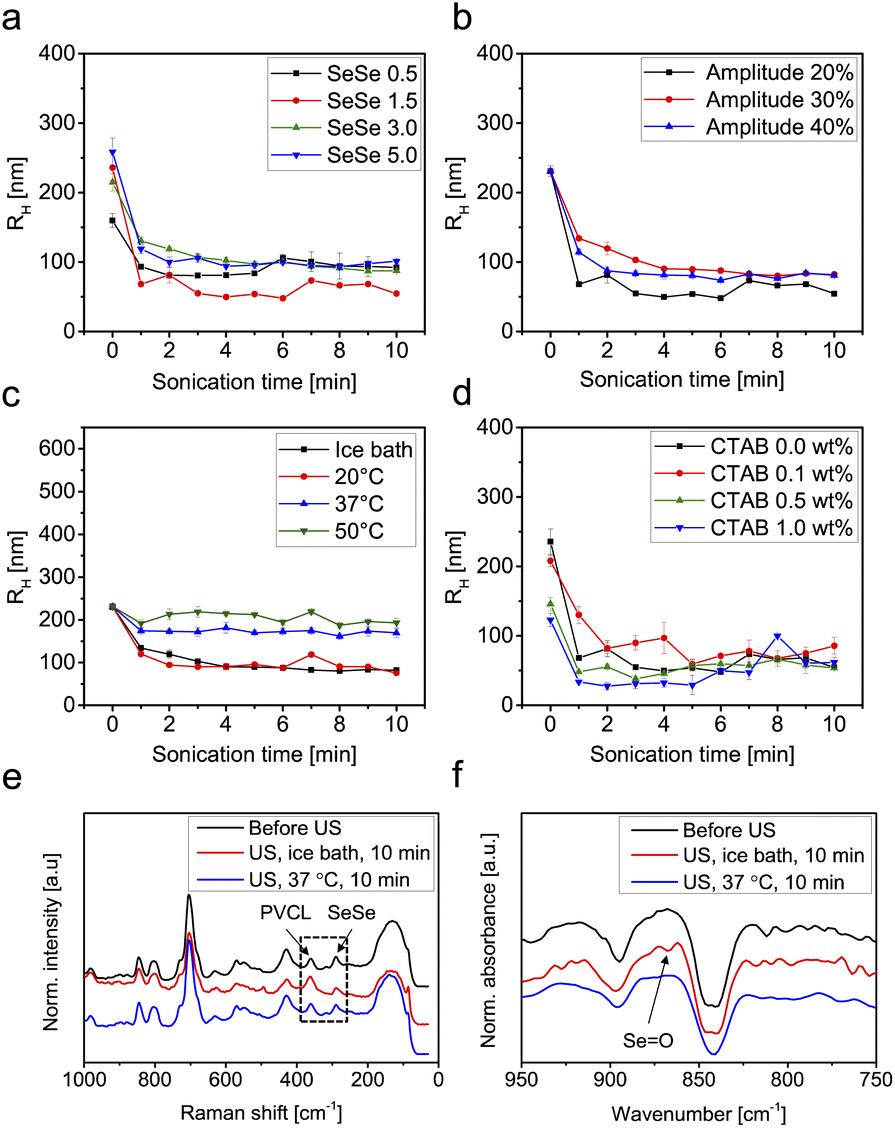 | ||
| Fig. 3 R H over the course of sonication and after dialysis of (a) SeμG with 0.5, 1.5, 3.0, and 5.0 mol% diselenide contents, (b) Se 1.5 mol% with 20, 30, and 40% amplitude, (c) Se 1.5 mol% at different temperatures, and (d) Se 1.5 mol% synthesized with 0, 0.1, 0.5, and 1.0 wt% of CTAB. Sonication experiments of (a), (b), and (d) were performed in an ice bath, and (a), (c), and (d) were performed at 30% amplitude. Power intensity for 30% amplitude was IP = 1.82 W cm−2. (e) Raman and (f) IR spectra of Se1.5 before and after 10 min ultrasonication in an ice bath at 37 °C. | ||
Since PVCL microgels possess thermo-responsive behaviour that induces swelling–deswelling transitions at temperatures below and above the VPTT (32 °C),66 Se1.5 microgels were sonicated for 10 min with 30% amplitude in an ice bath, at 20, 37, and 50 °C. Below the VPTT, RH decreased with increasing sonication time indicating degradation of microgels. However, above the VPTT, an increase of RH was observed indicating agglomeration of the microgels (Fig. S2c†). The samples were purified by dialysis and the RH was re-measured to eliminate the depletion interaction effect. DLS results show that at a temperature below the VPTT, RH of Se1.5 microgels decreased from 231 nm (before sonication) to 103 nm (after sonication) in an ice bath and 90 nm at 20 °C whilst at a temperature above the VPTT, the radii decreased to 171 nm at 37 °C and 218 nm at 50 °C. These results indicated that the deswelling of the microgels served as a mechanical protection mechanism against shear force inhibiting the scission of diselenide crosslinks. This suggestion can be supported by the findings of Lendlein and coworkers,67 who showed that microgels are mechanically fragmented through ultrasonication only in the case of maximum observed swelling, where the polymer chains are expanded. Above the VPTT the polymer chains collapsed and the microgels are no longer responsive to ultrasound (microgel fragmentation was not observed). In addition, the effect of microgel radii on the degradation was investigated. Se1.5 microgels synthesized with 0.1, 0.5, and 1.0 wt% CTAB were irradiated with ultrasound for 10 min in an ice bath at 30% amplitude (Fig. S2d and 3d†). The results show that regardless of the microgels' initial radii, the degradation behaviour does not show significant differences. All microgels degraded to approximately the same radii after 10 min sonication.
STEM images were captured to visualize the morphology changes of the microgels before and after sonication. Fig. 4 depicts the STEM images of Se0.5, 1.5, 3.0, and 5.0 before and after ultrasonication. The microgels were subsequently degraded to polymer clusters after 10 min of sonication. In the case of Se0.5, some microgel structures remained (Fig. 4a′). For higher diselenide contents, the microgels degraded completely to polymer fragments after ultrasonication (Fig. 4b′–d′). Next, STEM images of Se1.5 that were subjected to ultrasonication at different amplitudes were captured (Fig. S3†). Upon sonication with 20% amplitude, the microgel size decreased significantly but the structure remained intact whilst from 30% onwards the microgels degraded completely as indicated by the formation of polymer clusters due to the drying effect on the TEM grid (Fig. S3†). In the case of ultrasonication at different temperatures (Fig. S4†) two individual trends were observed. Below the VPTT, the microgels were fully degraded and formed polymer clusters while at a temperature above the VPTT, the microgel structure remained intact (Fig. S4†).
 | ||
| Fig. 4 STEM images of SeμG before sonication of (a) 0.5, (b) 1.5, (c) 3.0, and (d) 5.0 mol% diselenide content. The images after 10 min sonication (a′), (b′), (c′), and (d′) correspond to (a), (b), (c), and (d), respectively. Sonication was carried out in an ice bath at 30% amplitude for 10 min. | ||
After the sonication experiments, the obtained degradation products (PVCL chains) were analysed by FTIR and Raman spectroscopy to confirm the cleavage of SeSe bonds. As demonstrated in Fig. 3e and f, SeSe bonds were transformed to seleninic acid groups. A similar effect was detected in our previous work after treatment of SeSe-containing microgels with H2O268 and in publication of other research groups.69 This observation can be rationalized by the oxidative species formed by sonication in water.70
To explore the possibility of using SeμG as carriers for protein delivery, we loaded a microgel sample with 1.5 mol% SeSe crosslinks with the model proteins cytochrome C (cytC, loading content 0.24 mg mg−1 microgel) and myoglobin (Mb, loading content 0.33 mg mg−1 microgel). Both proteins exhibit comparable molar masses (12 and 17 kDa, respectively) rendering them small enough to penetrate into the microgel network.71 Moreover, both contain an iron moiety in their structure.72,73 Free proteins and microgels loaded with proteins were subjected to sonication at 0 °C and 37 °C (cytC) as well as 50 °C (Mb) followed by the evaluation of the protein structure using UV-vis and CD spectroscopy. There are several ways how ultrasound can negatively affect the integrity of proteins: disruption of H-bonds or covalent bonds in protein by mechanical force and formation of reactive oxygen species (ROS), which can cause oxidation of proteins. Proteins can undergo free radical oxidation through the following mechanisms:74 oxidation of the active metal site (e.g., Fe2+ to Fe3+); oxidation of the peptide chain;75,76 oxidation of side chains (glutamyl, aspartyl, proline);77 oxidation of amino acids.76,78 Iron ions can play an important role in the free radical oxidation of proteins, since it can be involved in the initiation of the process by donating electrons. However, this is not the case when iron content is low (e.g., when iron is present only as active site of the protein). Fe2+ also participates in the mechanism of peptide chain cleavage, caused by the attack of hydroxyl radicals on the α-carbon atom of any amino-acid residue. This attack leads to the formation of alkyl radicals and subsequently alkyl peroxy radicals, which is followed by Fe2+ oxidation to Fe3+ and cleavage of the peptide bond. Change of the iron oxidation state can be a good indication of ongoing oxidation processes of proteins. As shown in the CD spectra, pure cytC did not undergo denaturation under the applied ultrasonication conditions as indicated by the retained α-helix structure (Fig. S5 and Table S1†). However, the UV-vis spectra presented in Fig. 5a indicated that during the sonication Fe(III) of native cytC was reduced to Fe(II) as reflected in the increase of the peak intensity at 551 nm.72 This process was accelerated at elevated temperatures. In the case of native Mb, the iron moiety was Fe(II) and thus could only undergo oxidation under the chosen conditions (Fig. 5e). As a result of the sonication treatment of the myoglobin, the oxidation state of Fe did not change – no formation of the peak typical for Fe(III) in the area of 530 nm79 was observed (Fig. 5c). However, ultrasound caused structural changes to Mb: the intensity of the Soret band at 408 nm significantly decreased indicating alterations in the coordination sphere of the iron.80 Structural changes of the Mb were particularly well inferred from the CD spectra (Fig. S6 and Table S2†). Mb completely lost its α-helix structure (from 79.3% in native Mb to 0% in ultrasonicated pure protein) converting to β-sheet and random coil (56.9% and 43.2% respectively). Increasing the sonication temperature to 50 °C caused further transformations of the β-sheet conformation to β-turn and random coil (Table S2†). These results were in a good agreement with the literature80 where the authors concluded that Mb was more sensitive to mechanical force due to its more rigid and “fragile” tertiary structure, caused by higher content of the α-helix conformation in its structure compared to cytC.
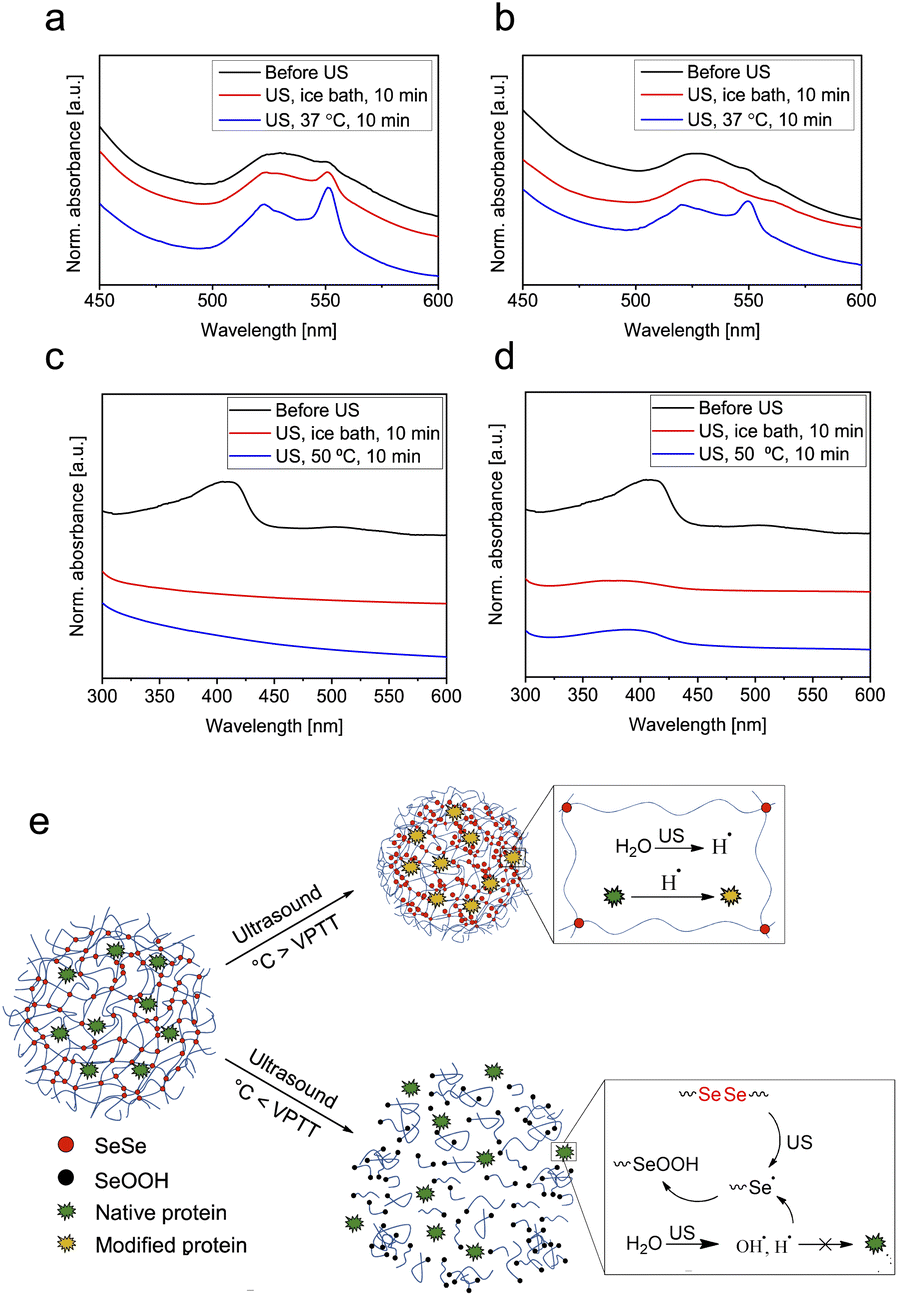 | ||
| Fig. 5 UV-vis spectra of (a) pure cytC in water after 10 min ultrasonication in an ice bath at 37 °C and (b) cytC-loaded Se1.5 before and after 10 min ultrasonication at the same conditions. (c) Pure Mb in pH 4 buffer solution after 10 min ultrasonication in an ice bath at 50 °C and (d) Mb-loaded Se1.5 before and after 10 min ultrasonication under the same conditions. (e) Scheme of cytC-loaded diselenide-crosslinked microgels sonicated below and above the VPTT demonstrating inhibition of the reduction of cytC by the radical species produced from the ultrasonication of water. | ||
Contrarily, the sonication of cytC and Mb encapsulated within microgels under similar conditions (i.e., above VPTT of the microgels) led to reduced protein degradation. The UV-vis spectra depicted in Fig. 5b indicated that after sonication at 37 °C the reduction of the Fe(III) in the cytC structure was significantly suppressed compared to the free protein (Fig. 5a). Similar phenomena were observed for Mb where alterations in iron coordination were reduced (Fig. 5d), especially at 50 °C. Degradation of the α-helix structure to β-sheet, β-turn, and random coil conformation was also reduced: after sonication at 0 °C, the α-helix content in Mb remained at 54.6% and after sonication at 50 °C at 24.1% (Table S2†). Similar to above, the microgels did not decompose significantly upon sonication above VPTT and thus, we reason, provided protection to the protein by hydrodynamic shielding.
Protein degradation was even further suppressed when the protein-loaded microgels were sonicated below the VPTT of the microgels. For cytC, reduction of Fe(III) to Fe(II) was completely inhibited accompanied by the release of the protein into aqueous solution. Alongside, the tertiary structure of Mb was only partially deteriorated from 79.3% to 54.6% α-helix and 27.5% β-turn conformation (Table S2†). We hypothesized that under these conditions the SeSe bonds underwent mechanochemical bond scission and the resulting Se radicals efficiently scavenged free radicals in the vicinity of the protein (Fig. 5e). However, due to the low concentration of Se-moieties in the microgels and the short lifetime and low stability of alkyl selenol radicals it was not possible to prove the formation of Se radicals by instrumental analysis, such as electron paramagnetic resonance (EPR) spectroscopy (Fig. S12†). We suggest, that Se radicals were formed as a result of sonication and immediately transformed into SeOOH moieties. The formation of SeOOH could also not be observed by 77Se-NMR due to the low Se-content of the microgels and the low sensitivity of 77Se-NMR measurements (Fig. S13†). However, the SeOOH-group was detected by XPS spectroscopy (Fig. S10†). To further prove this hypothesis, we performed control experiment with PVCL microgels crosslinked with BIS (B1.5%). From the UV-vis spectra of pure cytC protein and cytC loaded in B1.5% we observed identical reduction behavior, while for cytC in Se1.5 microgels inhibition of Fe(III) reduction can be clearly seen (Fig. S11†). Thereby, protective properties for the iron moieties of cytC and Mb were provided similar to the action of selenocysteine in the glutathione peroxidase which protects cells from oxidative stress.81 Contrarily, when protein-loaded microgels were sonicated above the VPTT of the microgels, the SeSe bonds were only partially cleaved thus resulting in diminished reduction suppression of the Fe(III) moiety of cytC. Yet, microgels in the deswollen state acted as conformational stabilizers. Comparing the conformational structure of myoglobin after sonication at 50 °C (Table S2†) with and without microgel, we observed that when loaded within microgels the protein partially preserved its α-helix structure (24.1% compared to 0% without microgel).
To investigate the conformation stabilizing properties of the microgels, proteins α-chymotrypsin (α-Chy, loading content 0.32 mg mg−1 microgel) and albumin (Alb, loading content 0.32 mg mg−1 microgel) (molar mass 25 and 66 kDa)82,83 without iron moieties were subjected to sonication at 0 °C and 50 °C (Fig. S6 and S7, Table S3 and S4†). These proteins differed by their structure: Alb contained predominantly α-helix, while α-Chy exhibited mostly β-sheets. Sonication at 0 °C did not cause significant structural changes of α-Chy (Fig. S6 and Table S3†) contrary to Alb, whose α-helix content reduced from 64.8% to 41.9% (Fig. S8 and Table S4†). Increasing the temperature to 50 °C led to complete destruction of the Alb α-helix structure (Table S4†) and partial degradation of α-Chy (Table S3†). These results additionally corroborate the higher sensitivity of α-helix proteins to mechanical force.
Eventually, protein-loaded microgels were sonicated at the same temperatures below VPTT of the microgels as pure Alb and α-Chy. From the results (Tables S3 and S4) we can infer no benefit of the radical scavenging properties for the preservation of Alb and α-Chy secondary structures. However, the sonication at 50 °C showed that microgels stabilized the conformational structure of both proteins to a certain degree, where the effect on α-Chy was larger than that of Alb. However, the destructive effects of high temperatures in combination with sonication was not fully compensated by Se-microgel protection. We hypothesized that due to the large size both α-Chy and Alb could not penetrate deeply into the microgel interior and were localized on the microgel surface, which might substantially contribute to the poor protecting effect. Further studies of the microgel structure (monomer nature, crosslinking nature, and density) on the conformational stabilization of different types of proteins need to be performed to better understand these effects.
3 Conclusions
In this work, series of PVCL microgels with varied amounts of SeSe crosslinks and sizes were synthesized through precipitation polymerization. The incorporation of diselenide to the PVCL microgels was validated by Raman spectroscopy. After the incorporation of diselenide crosslinkers, the microgels retained their temperature-responsive behavior as indicated by DLS. We successfully showed that due to the low bond dissociation energy, diselenide-crosslinked microgels were efficiently and more quickly degraded by ultrasound compared to BIS-crosslinked control microgels. Our experimental data indicated that the degradation of the diselenide-crosslinked microgels depended on multiple parameters. Firstly, 30% amplitude (IP = 1.82 W cm−2) was required to induce a complete degradation over 10 min sonication. At temperatures below the VPTT (swollen state), the microgels degraded under action of ultrasound, but at temperatures above the VPTT (deswollen state) the degradation by ultrasound was inhibited. For different diselenide contents and microgel sizes no significant effect was discerned regarding their degradation behavior. We demonstrated that diselenide-crosslinked microgels protected proteins form oxidative damage and mechanical force during the sonication process. Due to these dual protection properties, the microgels acted as radical scavenger and thereby protected proteins (in particular cytC) form oxidation of the Fe-moiety. Moreover, the diselenide-crosslinked microgels acted as conformational stabilizers of the proteins (in particular Mb) upon sonication above the VPTT (32 °C).Our experimental results highlight the potential of SeSe-crosslinked microgels as efficient carriers for the delivery and controlled release of peptides and proteins in vivo triggered by ultrasound. In addition to the fast and efficient ultrasound-triggered degradation mechanism, Se-containing functional groups present in the degradation products could reduce the local concentration of reactive oxygen species thus protecting proteins from chemical damage and denaturation minimizing the risk of cell apoptosis and tissue damage.
Data availability
Experimental data is stored in a data base of DWI – Leibniz Institute for Interactive Materials and can be provided upon request.Author contributions
K. H. Tan performed the experiments, designed the synthesis procedure of diselenide crosslinker, performed 1H NMR, electrophoretic mobility of microgels, UV, STEM measurements and wrote the manuscript, W. Xu performed the experiments, F. Weitenhagen performed synthesis of microgels, sonication experiments of microgels, DLS-measurements. S. Braun performed Raman and FTIR measurements, T. Kharandiuk performed sonication experiments with protein-loaded microgels, UV, CD measurements, and wrote the manuscript. R. Göstl and A. Pich conceived the project, discussed the results, and wrote the manuscript. All authors provided input and feedback for manuscript preparation. All authors approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Deutsche Forschungsgemeinschaft for financial support within the Collaborative Research Center SFB 985 “Functional Microgels and Microgel Systems” (No. 191948804). R. G. is grateful for support by a Freigeist-Fellowship of the Volkswagen Foundation (No. 92888). Parts of the analytical investigations were performed at the Center for Chemical Polymer Technology CPT, which was supported by the European Commission and the federal state of North Rhine-Westphalia (No. 300088302). We thank Bea Becker for the FTIR and Raman spectroscopy measurements, Tobias Seitz and Prof. Dr Sonja Herres-Pawlis for EPR measurements, Dr Meike Emondts for performing 77Se NMR measurements and Joachim Roes for XPS measurements.References
- V. Paefgen, D. Doleschel and F. Kiessling, Front. Pharmacol., 2015, 6, 1–197 CrossRef PubMed.
- D. L. Miller, N. B. Smith, M. R. Bailey, G. J. Czarnota, K. Hynynen and I. R. S. Makin, J. Med. Ultrasound, 2012, 31, 623–634 CrossRef PubMed.
- S. Huo, P. Zhao, Z. Shi, M. Zou, X. Yang, E. Warszawik, M. Loznik, R. Göstl and A. Herrmann, Nat. Chem., 2021, 13, 131–139 CrossRef CAS.
- J. L. Paris, M. V. Cabañas, M. Manzano and M. Vallet-Regí, ACS Nano, 2015, 9, 11023–11033 CrossRef CAS PubMed.
- Z. Wang, M. Zhan and X. Hu, Chem.–Eur. J., 2022, 28, e202200042 CrossRef CAS PubMed.
- Z. Wang, M. Zhan, W. Li, C. Chu, D. Xing, S. Lu and X. Hu, Angew. Chem., Int. Ed., 2021, 60, 4720–4731 CrossRef CAS PubMed.
- B. Leader, Q. J. Baca and D. E. Golan, Nat. Rev. Drug Discovery, 2008, 7, 21–39 CrossRef CAS PubMed.
- P. Tharkar, R. Varanasi, W. S. F. Wong, C. T. Jin and W. Chrzanowski, Front. Bioeng. Biotechnol., 2019, 7, 324 CrossRef PubMed.
- Y. Zhou, S. Huo, M. Loznik, R. Göstl, A. J. Boersma and A. Herrmann, Angew. Chem., Int. Ed., 2021, 60, 1493–1497 CrossRef CAS PubMed.
- F. A. Plamper and W. Richtering, Acc. Chem. Res., 2017, 50, 131–140 CrossRef CAS.
- G. Agrawal and R. Agrawal, Polymers, 2018, 10, 418 CrossRef PubMed.
- A. Pich and W. Richtering in Chemical Design of Responsive Microgels. Adv. Polym. Sci., Springer, Berlin, Heidelberg, Germany, 2010, vol. 234, pp. 1–37 Search PubMed.
- T. Shu, Q. Shen, X. Zhang and M. J. Serpe, Analyst, 2020, 145, 5713–5724 RSC.
- A. Ahiabu and M. J. Serpe, ACS Omega, 2017, 2, 1769–1777 CrossRef CAS PubMed.
- L. A. Lyon, Z. Meng, N. Singh, C. D. Sorrell and A. st. John, Chem. Soc. Rev., 2009, 38, 865 RSC.
- L. E. Theune, J. Buchmann, S. Wedepohl, M. Molina, J. Laufer and M. Calderón, J. Controlled Release, 2019, 311–312, 147–161 CrossRef CAS PubMed.
- Y. Gao and C.-M. Dong, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 1067–1077 CrossRef CAS.
- H. Kobayashi and R. G. Winkler, Sci. Rep., 2016, 6, 19836 CrossRef CAS PubMed.
- D. Kleinschmidt, M. S. Fernandes, M. Mork, A. A. Meyer, J. Krischel, M. V. Anakhov, R. A. Gumerov, I. I. Potemkin, M. Rueping and A. Pich, J. Colloid Interface Sci., 2020, 559, 76–87 CrossRef CAS PubMed.
- M. Wiese, T. Lohaus, J. Haussmann and M. Wessling, J. Membr. Sci., 2019, 588, 117190 CrossRef CAS.
- M. A. Lifson, J. A. Carter and B. L. Miller, Anal. Chem., 2015, 87, 7887–7893 CrossRef CAS PubMed.
- M. Kather, M. Skischus, P. Kandt, A. Pich, G. Conrads and S. Neuss, Angew. Chem., Int. Ed., 2017, 56, 2497–2502 CrossRef CAS PubMed.
- Y. Ji, L. Winter, L. Navarro, M.-C. Ku, J. S. Periquito, M. Pham, W. Hoffmann, L. E. Theune, M. Calderón and T. Niendorf, Cancers, 2020, 12, 1380 CrossRef CAS PubMed.
- G. Agrawal and R. Agrawal, Small, 2018, 14, 1801724 CrossRef.
- N. A. Cortez-Lemus and A. Licea-Claverie, Prog. Polym. Sci., 2016, 53, 1–51 CrossRef CAS.
- J. Liu, A. Debuigne, C. Detrembleur and C. Jérôme, Adv. Healthc. Mater., 2014, 3, 1941–1968 CrossRef CAS PubMed.
- J. Ramos, A. Imaz and J. Forcada, Polym. Chem., 2012, 3, 852–856 RSC.
- P. Kodlekere and A. Pich, ChemNanoMat, 2018, 4, 889–896 CrossRef CAS.
- H.-Q. Wu and C.-C. Wang, Langmuir, 2016, 32, 6211–6225 CrossRef CAS PubMed.
- X. Zhang, S. Malhotra, M. Molina and R. Haag, Chem. Soc. Rev., 2015, 44, 1948–1973 RSC.
- K. M. Morrissey, S. L. Stocker, M. B. Wittwer, L. Xu and K. M. Giacomini, Annu. Rev. Pharmacol. Toxicol., 2013, 53, 503–529 CrossRef CAS.
- Y. Wang, J. Nie, B. Chang, Y. Sun and W. Yang, Biomacromolecules, 2013, 14, 3034–3046 CrossRef CAS PubMed.
- D. Kehren, C. M. Lopez, S. Theiler, H. Keul, M. Möller and A. Pich, Polymer, 2019, 172, 283–293 CrossRef CAS.
- F. P. Garcia, M. Rippe, M. V. P. Companhoni, T. F. Stefanello, B. Louage, S. van Herck, L. Sancey, J.-L. Coll, B. G. de Geest, C. Vataru Nakamura and R. Auzély-Velty, Biomater. Sci., 2018, 6, 1754–1763 RSC.
- S.-H. Jung, S. Schneider, F. Plamper and A. Pich, Macromolecules, 2020, 53, 1043–1053 CrossRef CAS.
- G. Agrawal, R. Agrawal and A. Pich, Part. Part. Syst. Charact., 2017, 34, 1700132 CrossRef.
- D. Klinger and K. Landfester, Polymer, 2012, 53, 5209–5231 CrossRef CAS.
- S. Bian, J. Zheng and W. Yang, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1676–1685 CrossRef CAS.
- Z. Deng and S. Liu, J. Controlled Release, 2020, 326, 276–296 CrossRef CAS PubMed.
- E. Izak-Nau, D. E. Demco, S. Braun, C. Baumann, A. Pich and R. Göstl, ACS Appl. Polym. Mater., 2020, 2, 1682–1691 CrossRef CAS.
- E. Izak-Nau, S. Braun, A. Pich and R. Göstl, Adv. Sci., 2022, 9, 2104004 CrossRef CAS PubMed.
- M. F. Schulte, E. Izak-Nau, S. Braun, A. Pich, W. Richtering and R. Göstl, Chem. Soc. Rev., 2022, 51, 2939–2956 RSC.
- R. T. O'Neill and R. Boulatov, Nat. Rev. Chem., 2021, 5, 148–167 CrossRef.
- Y. Chen, G. Mellot, D. van Luijk, C. Creton and R. P. Sijbesma, Chem. Soc. Rev., 2021, 50, 6659–6660 RSC.
- M. Stratigaki and R. Göstl, ChemPlusChem, 2020, 85, 1095–1103 CrossRef CAS PubMed.
- E. Izak-Nau, D. Campagna, C. Baumann and R. Göstl, Polym. Chem., 2020, 11, 2274–2299 RSC.
- H. J. Reich and R. J. Hondal, ACS Chem. Biol., 2016, 11, 821–841 CrossRef CAS PubMed.
- S. Ji, W. Cao, Y. Yu and H. Xu, Angew. Chem., Int. Ed., 2014, 53, 6781–6785 CrossRef CAS PubMed.
- G. Hua and J. D. Woollins, in Selenium and Tellurium Reagents, De Gruyter, 2019, pp. 151–206 Search PubMed.
- J. Xia, T. Li, C. Lu and H. Xu, Macromolecules, 2018, 51, 7435–7455 CrossRef CAS.
- F. V. Singh and T. Wirth, Catal.: Sci. Technol., 2019, 9, 1073–1091 RSC.
- H. Ye, Y. Zhou, X. Liu, Y. Chen, S. Duan, R. Zhu, Y. Liu and L. Yin, Biomacromolecules, 2019, 20, 2441–2463 CrossRef CAS PubMed.
- D. Bhowmick and G. Mugesh, Org. Biomol. Chem., 2015, 13, 10262–10272 RSC.
- S. Pan, J. Yang, S. Ji, T. Li, S. Gao, C. Sun and H. Xu, CCS Chem., 2020, 2, 225–235 CrossRef CAS.
- N. K. Kildahl, J. Chem. Educ., 1995, 72, 423 CrossRef CAS.
- C. Sun, L. Wang, B. Xianyu, T. Li, S. Gao and H. Xu, Biomaterials, 2019, 225, 119514 CrossRef CAS PubMed.
- Q. Wu, Y. Yuan, F. Chen, C. Sun, H. Xu and Y. Chen, ACS Macro Lett., 2020, 9, 1547–1551 CrossRef CAS PubMed.
- J. Xia, P. Zhao, S. Pan and H. Xu, ACS Macro Lett., 2019, 8, 629–633 CrossRef PubMed.
- Z. Shi, Q. Song, R. Göstl and A. Herrmann, Chem. Sci., 2021, 12, 4184 RSC.
- F. Wang and C. E. Diesendruck, Chem. Commun., 2020, 56, 2143–2146 RSC.
- Z. Shi, J. Wu, Q. Song, R. Göstl and A. Herrmann, J. Am. Chem. Soc., 2020, 142, 14725–14732 CrossRef CAS PubMed.
- U. F. Fritze, S. L. Craig and M. von Delius, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 1404–1411 CrossRef CAS.
- T. J. Kucharski, Z. Huang, Q.-Z. Yang, Y. Tian, N. C. Rubin, C. D. Concepcion and R. Boulatov, Angew. Chem., Int. Ed., 2009, 48, 7040–7043 CrossRef CAS PubMed.
- B. B. Gerbelli, S. V. Vassiliades, J. E. U. Rojas, J. N. B. D. Pelin, R. S. N. Mancini, W. S. G. Pereira, A. M. Aguilar, M. Venanzi, F. Cavalieri, F. Giuntini and W. A. Alves, Macromol. Chem. Phys., 2019, 220, 1970027 CrossRef.
- A. Fava, A. Iliceto and E. Camera, J. Am. Chem. Soc., 1957, 79, 833–838 CrossRef CAS.
- A. Imaz and J. Forcada, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2510–2524 CrossRef CAS.
- R. Vukićević, A. T. Neffe, K. Luetzow, B. F. Pierce and A. Lendlein, Macromol. Rapid Commun., 2015, 36, 1891–1896 CrossRef PubMed.
- K. H. Tan, W. Xu, S. Stefka, D. E. Demco, T. Kharandiuk, V. Ivasiv, R. Nebesnyi, V. S. Petrovskii, I. I. Potemkin and A. Pich, Angew. Chem., Int. Ed., 2019, 58, 9791–9796 CrossRef CAS PubMed.
- N. Ma, Y. Li, H. Xu, Z. Wang and X. Zhang, J. Am. Chem. Soc., 2010, 132, 442–443 CrossRef CAS PubMed.
- K. Makino, M. M. Mossoba and P. Riesz, J. Phys. Chem., 1983, 87, 1369–1377 CrossRef CAS.
- W. Xu, A. A. Rudov, R. Schroeder, I. v. Portnov, W. Richtering, I. I. Potemkin and A. Pich, Biomacromolecules, 2019, 20, 1578–1591 CrossRef CAS PubMed.
- T. Kondo, V. Mišík and P. Riesz, Ultrason. Sonochem., 1996, 3, S193–S199 CrossRef CAS.
- S. Takamiya, Y. Yu, M. E. Cavaleante, K. Murayama, H. Taka, S. Tateno, T. Takeuchi and T. Aoki, Mol. Biochem. Parasitol., 1996, 79, 61–70 CrossRef CAS PubMed.
- V. I. Lushchak, Biochem, 2007, 72, 809–827 CAS.
- E. R. Stadtman, Annu. Rev. Biochem., 1993, 62, 797–821 CrossRef CAS PubMed.
- E. R. Stadtman and R. L. Levine, Amino Acids, 2003, 25, 207–218 CrossRef CAS PubMed.
- O. I. Aruoma, J. Am. Oil Chem. Soc., 1998, 75, 199–212 CrossRef CAS PubMed.
- R. T. Dean, S. Fu, R. Stocker and M. J. Davies, Biochem. J., 1997, 324, 1–18 CrossRef CAS PubMed.
- W. D. Butt and D. Keilin, Proc. R. Soc. London, Ser. B, 1962, 156, 429–458 CAS.
- M. Barteri, M. Fioroni and M. C. Gaudiano, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1996, 1296, 35–40 CrossRef.
- D. Bhowmick, S. Srivastava, P. D'Silva and G. Mugesh, Angew. Chem., Int. Ed., 2015, 54, 8449–8453 CrossRef CAS PubMed.
- A. Hoffmann, J. Bredno, M. Wendland, N. Derugin, P. Ohara and M. Wintermark, Transl. Stroke Res., 2011, 2, 106–111 CrossRef CAS PubMed.
- S. K. Lam and T. B. Ng, Phytomedicine, 2010, 17, 621–625 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc03153a |
| This journal is © The Royal Society of Chemistry 2022 |